
A high-throughput liquid biopsy for rapid rare cell separation from large-volume samples†
Yaoping
Liu
a,
Tingyu
Li
a,
Mingxin
Xu
b,
Wei
Zhang
c,
Yan
Xiong
d,
Ligong
Nie
c,
Qi
Wang
*b,
Haichao
Li
*c and
Wei
Wang
 *ae
*ae
aInstitute of Microelectronics, Peking University, 100871, Beijing, China. E-mail: w.wang@pku.edu.cn
bDepartment of Respiratory Medicine, The Second Affiliated Hospital of Dalian Medical University, 116027, Dalian, China. E-mail: wqdlmu@163.com
cDepartment of Respirology, No. 1 Hospital of Peking University, Beijing, 100034, China. E-mail: lhch91767@sina.com
dDepartment of Pathology, No. 1 Hospital of Peking University, Beijing, 100034, China
eNational Key Laboratory of Science and Technology on Micro/Nano Fabrication, 100871, Beijing, China
First published on 5th December 2018
Abstract
Liquid biopsy techniques for rare tumor cell separation from body fluids have shown enormous promise in cancer detection and prognosis monitoring. This work established a high-throughput liquid biopsy platform with a high recovery rate and a high cell viability based on a previously reported 2.5D micropore-arrayed filtration membrane. Thanks to its high porosity (>40.2%, edge-to-edge space between the adjacent micropores <4 μm), the achieved filtration throughputs can reach >110 mL min−1 for aqueous samples and >17 mL min−1 for undiluted whole blood, only driven by gravity with no need for any extra pressure loading. The recoveries of rare lung tumor cells (A549s) spiked in PBS (10 mL), unprocessed BALF (10 mL) and whole blood (5 mL) show high recovery rates (88.0 ± 3.7%, 86.0 ± 5.3% and 83.2 ± 6.2%, respectively, n = 5 for every trial) and prove the high performance of this platform. Successful detection of circulating tumor cells (CTCs) from whole blood samples (5 mL) of lung cancer patients (n = 5) was demonstrated. In addition, it was both numerically and experimentally proved that a small edge-to-edge space was significant to improve the viability of the recovered cells and the purity of the target cell recovery, which was reported for the first time to the best of the authors' knowledge. This high-throughput technique will expand the detecting targets of liquid biopsy from the presently focused CTCs in whole blood to the exfoliated tumor cells (ETCs) in other large-volume clinical samples, such as BALF, urine and pleural fluid. Meanwhile, the technique is easy to operate and ready for integration with other separation and analysis tools to fulfill a powerful system for practical clinical applications of liquid biopsy.
Introduction
Liquid biopsy has been one of the key tools in the precision medicine of cancer.1 Liquid biopsy techniques for the separation of rare tumor cells from body fluids have shown enormous promise in sensitive cancer detection and dynamic prognosis monitoring.2–6 Particularly, the involvement of microtechnology, utilizing micrometer-scale structures to realize precise manipulation at a single-cell level, has demonstrated a great advantage in rare cell separation in the last decade.7–19 The previously reported microtechniques for liquid biopsy include microfiltration (micropore,20–30 microweir,10,29,31 micropost7,29,32–34), cell-affinity micro-chromatography,7,13,35–38 microvortexing,39–41 hydrodynamic microfluidics,15,16,36,42 inertial microfluidics,43–48 physical property-based separation,32,49–52etc. These reported techniques have shown excellent cell recovery capabilities but suffer from a low throughput (the flow rate of the sample it can realize).2,4,5,12,14,18,53 The obtained throughput of the present microtechniques, 0.2–2 mL min−1, could be sufficient for the separation of circulating tumour cells (CTCs) from blood (∼7.5 mL), the present main detecting targets of liquid biopsy. Nevertheless, the detection of exfoliated tumor cells (ETCs) from other body fluids of a large volume from several milliliters to even hundreds of milliliters, such as bronchoalveolar lavage fluid (BALF, 40–80 mL),54 urine (50–100 mL),55 pleural fluid (500–1000 mL),56etc., is of higher value because of its advantage to obtain more information about tumor primary sites. The key to fulfilling the liquid biopsy of ETCs from large-volume samples is the breakthrough of the sample handling throughput.4,5,11,14,18,57 Among the developed techniques, the most promising one to achieve a high throughput in mL min−1 along with a high recovery rate is the use of micropore-arrayed filtration membrane based devices.2,4,5,12,14 The principle of micropore-arrayed filtration based cell separation is dependent on the size differences between the target cells and background cells and is schematically shown in Fig. 1. Various filtrations have been developed, however, the throughput of presently existing micropore-arrayed filtrations is still low (∼2 mL min−1) even under an extra pressure loading and in high demand for improvement to facilitate the detection of ETCs from large-volume body fluids. The requirement for extra pressure and yet a low throughput is due to low porosity, i.e. a large supporting structure (edge-to-edge space between the adjacent micropores), required to guarantee the mechanical stability of the filtration membrane. An extra pressure loading (i.e. a large hydrodynamic stress acting on cells during filtration) would cause damage to cells and thereby reduce the viability of recovered cells dramatically.12,20,24 The cell viability was improved via a 3D double-layer micropore array from Zheng et al., with the space between adjacent micropores on the bottom membrane as a support and thus lower the stress experienced by the trapped cells on the top membrane.21 However, its wide applicability is still questionable because of the complexity of the preparation process, together with the limitation of low throughput. In addition, the large edge-to-edge space caused a high non-specific adhesion of background competitor cells, which happens when the space is larger than half of the diameter of the cell, as previously reported.12,18 The poor viability of recovered cells and the high non-specific adhesion of background cells will degrade down-stream analysis, such as cell culturing, drug screening, gene sequencing, etc. A performance comparison of previously reported typical filtration methods is shown in Table 1.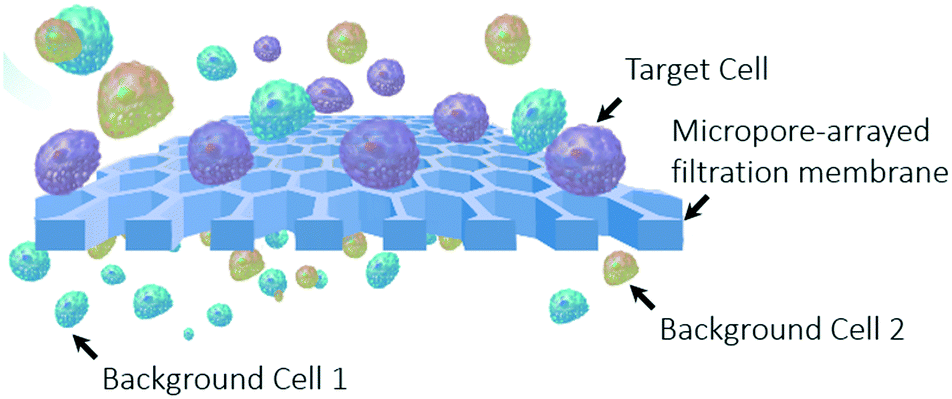 | ||
| Fig. 1 A schematic illustration of the principle of micropore-arrayed filtration based cell separation. | ||
| Filtration structure | Diameter (μm) | Space (μm) | Porosity | Recovery rate/detection sensitivity | Purity | Throughput (medium) | Drive mode | Group/company |
|---|---|---|---|---|---|---|---|---|
| 2D (monolayer) Parylene C micropore array20 | 10 | 10 | 5.6% | 87.3 ± 7.0–89.1 ± 7.0% | N/A | 1 mL/<10 min (diluted blood) | Pump (∼3.45 kPa) | Prof. S. Zheng |
| 3D (double-layer) Parylene C micropore array21 | 8 | 12 | 6.96% | 86.5 ± 5.3% | N/A | 1 mL/5 min (diluted blood) | Pump (∼3.45 kPa) | Prof. S. Zheng |
| Track etched pore22 | 7.5 ± 0.36 | Randomly distributed | N/A | 74–91.2% | N/A | ∼2 mL min−1 (diluted blood) | Vacuum | Screen cell |
| 6.5 ± 0.33 | ||||||||
| SU-8 membrane23 | 5–8 | N/A | 6.25–16% | >82.44% | 1.7 ± 1.4% | 5 mL min−1 (diluted blood) | Pump (0.1–14 Psi) | Creatv MicroTech |
| 3D palladium micropocket array24 | 8 | 26 | 5.02% | >85% | N/A | 2–2.5 mL min−1 (treated blood) | Aspiration (18 Pascal) | Prof. H. Nakanishi |
| PEGDA conical hole25 | 5.5–8 | 25 | 3.25–5.88% | 85–95% | N/A | 0.2 mL min−1 (diluted blood) | Pump | MesoBioSystem |
| PDMS membrane26 | 9.1 ± 0.2 | ∼9 | ∼25% | >90% | N/A | 10 mL h−1 (whole blood) | Pump | Prof. C. Jia |
| Silicon micropore array27 | 5 | 14 | 5.4% | >59% | N/A | ∼2.3 mL min−1 (whole blood) | Pump (aspiration) | VyCAP; Prof. L. Terstappen |
| PET microcavity28 | 8–11 | 60 | <1.38% | 76–94% | N/A | 0.2 mL min−1 (whole blood) | Pump (vacuum) | Prof. T. Matsunaga |
| Track etched cylindrical pores29 | 8 | Randomly distributed | N/A | >80% | N/A | N/A (diluted blood) | Pump (vacuum) | ISET, RareCell diagnostics |
| 2.5D micropore array | 8–12 | <4 | >40.2% | 80–96% | 2.8 ± 0.1–15.6 ± 6.3% | >17 mL min−1 (whole blood) | Gravity only | The present work |
| >110 mL min−1 (aqueous sample) |
Given the above, the key challenge for transferring liquid biopsy techniques to practical clinical applications is to realize a high recovery rate, a high viability of recovered cells and a low non-specific adhesion of background cells (non-target recovery) at a high throughput. And increasing the porosity (decreasing the edge-to-edge space) of micropore arrays is extremely important to solve the above challenge. The limitations of a relatively low porosity (large edge-to-edge space) for the previously reported micropore-arrayed filtration membrane are mainly ascribed to the fabrication difficulties and mechanical strength issue. Our previous works have reported a large area 2.5-dimensional (2.5D) honeycombed micropore array with high porosity and high mechanical strength, key parameters for a robust and reliable liquid biopsy, prepared via the Parylene C molding technique.58,59 A high-throughput filtration of phosphate buffered saline (PBS) was realized, and high size precision and separation efficiency were demonstrated by carefully-designed filtrations of microparticles in PBS solution and target cells spiked in urine.60 In this work, based on the previously developed 2.5D micropore arrays, we established a high-throughput liquid biopsy platform with a high recovery rate, a high cell viability and good purity, as listed in Table 1. To demonstrate the performance of this platform in liquid biopsy, recoveries of rare lung tumor cells (A549s) spiked in large-volume PBS, unprocessed BALF and whole blood were tested. Then the detection of CTCs from whole blood samples of lung cancer patients was also performed. In addition, a 3D numerical simulation and the corresponding experiments were carried out to investigate two features, cell viability and non-specific adhesion of background cells, which are critical for down-stream analysis.
Materials and methods
Numerical simulation
A 3D numerical simulation was carried out to understand well the significance of a small edge-to-edge space in increasing the cell viability and decreasing the non-target cell contamination in micropore-arrayed filtration. In numerical simulation, the fluid flow during filtration is set as a laminar flow because of the small Reynolds number (∼1.2 × 10−2). Thus micropore-arrayed filtration could be described via the Navier–Stokes equation for incompressible fluid. A non-slip boundary condition (no normal velocity) is set on the surface of the filtration membrane. Then, a laminar flow model with a constant flow rate boundary condition was built and solved via COMSOL Multiphysics, as shown in Fig. 2a and b. In the numerical model, differently sized (8 μm and 15 μm) rigid microspheres were used to mimic the cells which went through and were trapped in the micropores, respectively. Meanwhile, it was assumed that the micropore-arrayed membrane was rigid, i.e. no deformation (negligible deformation has been verified experimentally in our previous work59). The diameters of the micropore and the edge-to-edge space were set as 12 μm as an example. Since the present simulation is focused on the influence of the edge-to-edge space size, different edge-to-edge sizes (2 μm, 4 μm, 8 μm and 16 μm) were compared in terms of cell viability and non-target cell contamination. The diameter is the length of the diagonal for a hexagon micropore unless specifically stated. The values of flow rates on the boundary condition for different edge-to-edge spaces were set according to the previously experimentally measured data for PBS solution.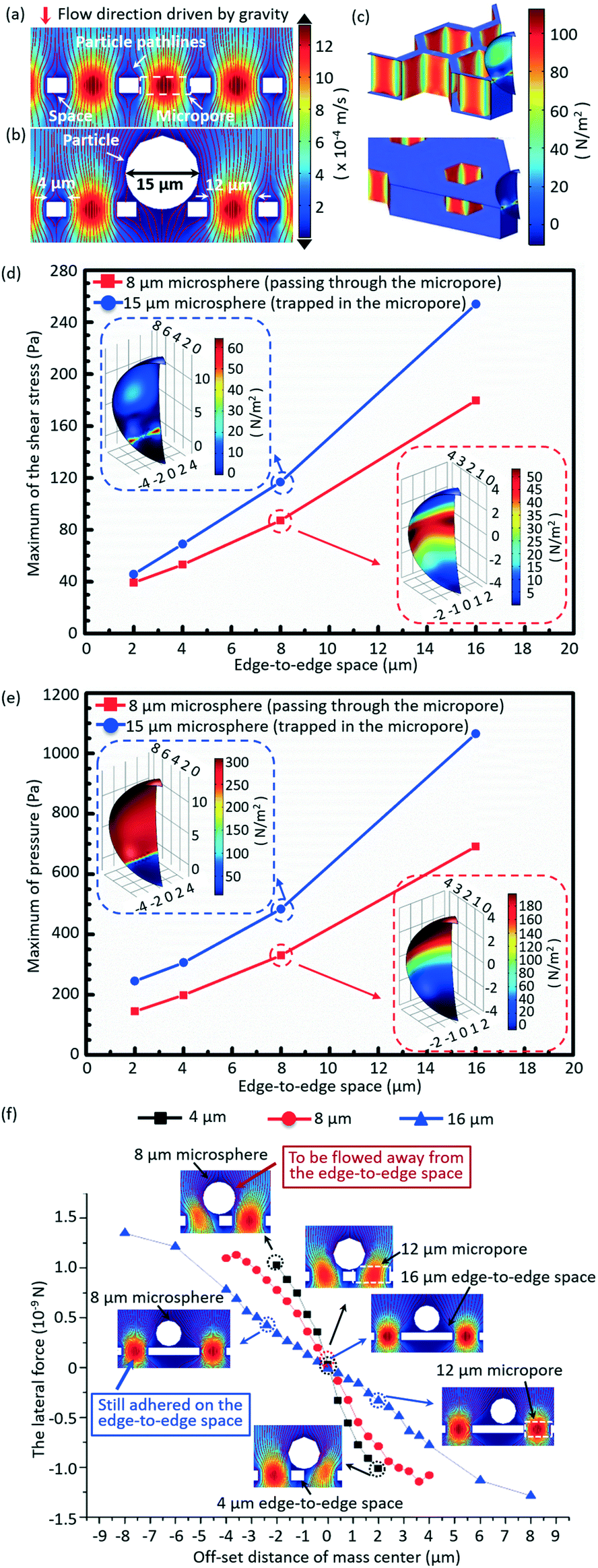 | ||
| Fig. 2 The simulated distributions of the fluid and velocity field under gravity-driven flow without (a) and with (b) the 15 μm microsphere trapped in the micropores, respectively. (c) The distributions of total stresses acting on the 15 μm microspheres trapped in the micropore arrays with edge-to-edge spaces of 2 μm (top) and 16 μm (bottom). The calculated shear stress (d) and pressure (e) acting on the microspheres (15 μm and 8 μm) during filtration through micropore arrays with differently sized edge-to-edge spaces. (f) The numerically simulated lateral hydrodynamic force variations with the offset distance of the mass center of an 8 μm microsphere on the top surface of membranes with different edge-to-edge spaces (4 μm, 8 μm and 16 μm). | ||
To investigate the influence of the edge-to-edge space size on the cell viability, the hydraulic pressures and shear stresses applied on the differently sized (8 μm and 15 μm) microspheres during the micropore-arrayed filtrations with differently sized spaces were numerically calculated. Additionally, the lateral forces applied on the 8 μm microspheres located on the differently sized edge-to-edge spaces during filtrations, critical to eliminate the non-specific adhesion (decrease non-target cell contamination), were numerically calculated.
System package for filtration and throughput measurement
The micropore-arrayed filtration membranes used in this work were prepared with the Parylene C molding technique, and the detailed information could be found in our previous work.58 Before the filtration operation, the prepared membranes were packaged with our newly (2nd generation) home-designed Teflon holders and commercially available ($1.5 for each piece) Nd–Fe–B strong ring magnets, shown in Fig. 3d and e. The grade of the magnet is N35 according to the international grades of neodymium, with the magnetic force distributed along the direction of thickness. The highest working temperature of the magnet is 80 °C. The sample loading was schematically illustrated with rhodamine B, as shown in Fig. 3d. Different pore diameters/spaces were investigated in the filtration throughput measurement, cases 1–4, as listed in Table 2. And >3 experiments for each case were performed.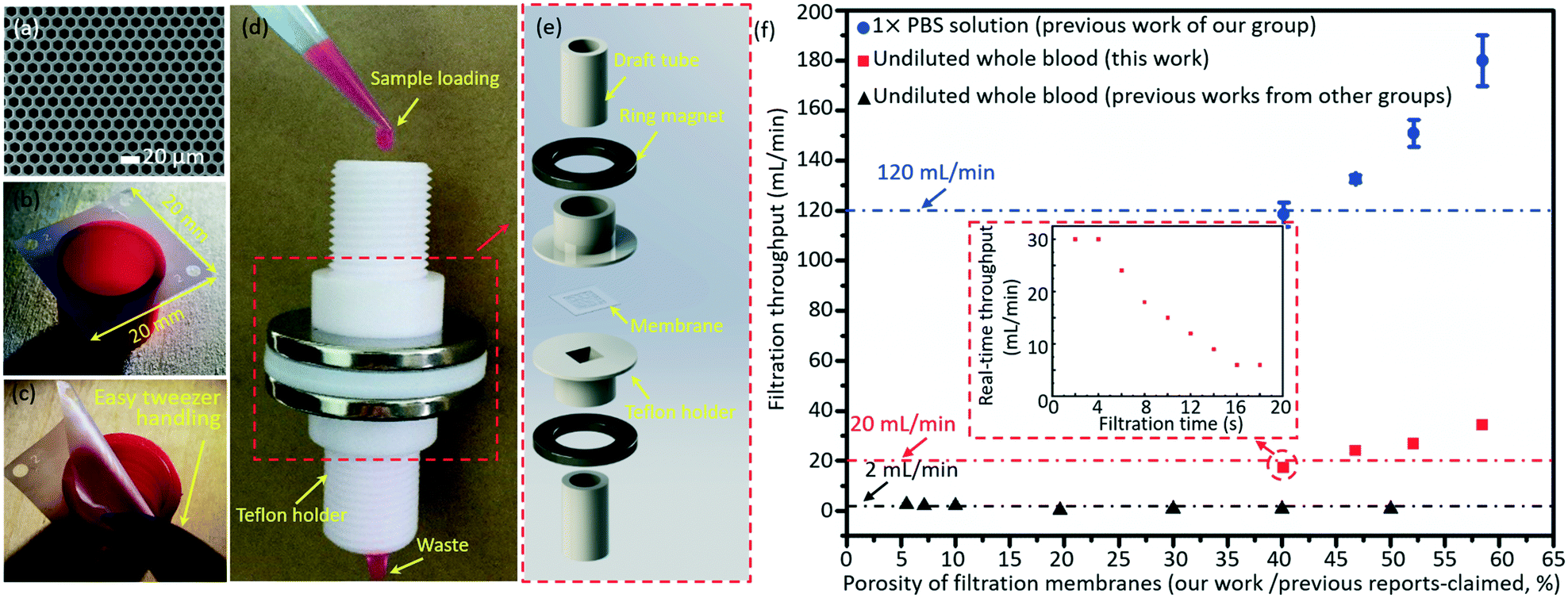 | ||
| Fig. 3 A typical SEM image (a) and optical photo (b) of the prepared micropore-arrayed membrane. (c) Easy manual handling with tweezers shows that the prepared 2.5D Parylene C filtration membrane has strong mechanical strength. (d) The package system with home-made Teflon holders and commercial ring magnets. (e) The expanded view of package details. (f) The filtration throughputs of PBS solution (our previous work60) and undiluted whole blood (this work and previous work from other groups20,21,23–28); the inset shows the real-time throughput vs. filtration time plot for one typical whole blood filtration through the micropore array of case 1 as listed in Table 2. | ||
| Case no. | Design value in mask (μm) | Measured value after preparation (μm) | Porosity (%) | ||||
|---|---|---|---|---|---|---|---|
| d | d′ | s | d | d′ | s | ||
| 1 | 8 | 6.93 | 4 | 7.51 ± 0.13 | 6.50 ± 0.11 | 4.46 ± 0.09 | 40.19 |
| 2 | 10 | 8.66 | 4 | 9.13 ± 0.09 | 7.90 ± 0.08 | 4.69 ± 0.24 | 46.79 |
| 3 | 12 | 10.40 | 4 | 11.21 ± 0.11 | 9.71 ± 0.10 | 4.77 ± 0.15 | 52.14 |
| 4 | 15 | 12.99 | 4 | 14.52 ± 0.20 | 12.57 ± 0.17 | 4.43 ± 0.06 | 58.46 |
| 5 | 12 | 10.40 | 2 | 11.42 ± 0.17 | 9.89 ± 0.15 | 2.46 ± 0.11 | 70.33 |
| 6 | 12 | 10.40 | 8 | 11.3 ± 0.15 | 9.79 ± 0.13 | 8.57 ± 0.21 | 31.93 |
| 7 | 12 | 10.40 | 16 | 11.39 ± 0.18 | 9.86 ± 0.16 | 16.46 ± 0.17 | 15.50 |
The average throughputs were calculated as the ratio of the operated volume to the filtration time length extracted from the recorded videos of the whole filtration process. The total volumes of samples for the throughput calculation were 10 mL for PBS solution and 5 mL for whole blood. A movie displaying the filtration of PBS and whole blood is shown in Video S1 of the ESI.†
Experimental verifications of cell viability and non-specific background adhesion
Besides the numerical simulation, the cell viability and non-specific adhesion of background cells were also verified via carefully designed experiments. 10 mL PBS (10010001, Gibco, Thermo Fisher) solutions containing around 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 A549s (cultured lung tumor cells, ordered from the National Infrastructure of Cell Line Resource, Beijing, China) spiked via serial dilution were filtrated through the micropore arrays with different edge-to-edge spaces (cases 3 and 5–7 as listed in Table 2) with every case repeated 3 times. After the filtrations, in situ staining of Calcein-AM (labelling for live cells, green fluorescence) and ethidium homodimer-1 (EthD-1, labelling for dead cells, red fluorescence) from a commercially available live/dead kit (L3224, Invitrogen™, Thermo Fisher) was performed, along with the disassembly and sealing of a membrane between a glass slide and a coverslip with ProLong™ Diamond Antifade Mountant (P36961, Invitrogen™, Thermo Fisher). Then the membrane with cells was observed under a fluorescence microscope (CKX53, Olympus) to count the live cells and dead cells separately. The short-term viability of recovered cells was further analysed based on the counting results. Besides, a control group was also set with the cells stored under the same condition for the same period as the filtration groups in the viability experiments. Moreover, to further investigate the long-term viability of cells after filtrations, in situ culture for proliferation directly after filtration through micropore arrays of case 3 listed in Table 2 was conducted. The cells on the filtration membrane after culture for 72 h were observed under a laser scanning confocal microscope (TCS SP5 X, Leica) to check the cell morphology and adhesion/spreading status. Experiments for each case were repeated more than 3 times.
000 A549s (cultured lung tumor cells, ordered from the National Infrastructure of Cell Line Resource, Beijing, China) spiked via serial dilution were filtrated through the micropore arrays with different edge-to-edge spaces (cases 3 and 5–7 as listed in Table 2) with every case repeated 3 times. After the filtrations, in situ staining of Calcein-AM (labelling for live cells, green fluorescence) and ethidium homodimer-1 (EthD-1, labelling for dead cells, red fluorescence) from a commercially available live/dead kit (L3224, Invitrogen™, Thermo Fisher) was performed, along with the disassembly and sealing of a membrane between a glass slide and a coverslip with ProLong™ Diamond Antifade Mountant (P36961, Invitrogen™, Thermo Fisher). Then the membrane with cells was observed under a fluorescence microscope (CKX53, Olympus) to count the live cells and dead cells separately. The short-term viability of recovered cells was further analysed based on the counting results. Besides, a control group was also set with the cells stored under the same condition for the same period as the filtration groups in the viability experiments. Moreover, to further investigate the long-term viability of cells after filtrations, in situ culture for proliferation directly after filtration through micropore arrays of case 3 listed in Table 2 was conducted. The cells on the filtration membrane after culture for 72 h were observed under a laser scanning confocal microscope (TCS SP5 X, Leica) to check the cell morphology and adhesion/spreading status. Experiments for each case were repeated more than 3 times.
Separation of rare cells spiked in large-volume samples
Separation of rare cells spiked in large-volume samples (PBS, BALF and whole blood) was conducted to demonstrate the performance of the invented liquid biopsy platform. First, the recoveries of different numbers (≤3, ∼10, 100, 1000, and 10000) of A549s spiked in 10 mL PBS (i.e. at concentrations of ≤3/10 mL, ∼1/mL, 10/mL, 100/mL, and 1000/mL) were tested with the proposed micropore-arrayed filtration based platform and the vertical centrifugation (i.e. the usually used approach for cell recovery in cellular experiments of biological labs or clinical labs), in parallel. The addition of A549s for numbers higher than 100 was realized via serial dilution, while the addition of ≤3 and ∼10 A549s was via operation with a micropipette and a precise manipulator (KITE-R, WPI, Inc.) under a stereomicroscope. Then, to test the sensitivity of our platform to rare cells in a complex background, we used healthy BALF (10 mL) and whole blood (5 mL) spiked with 10 A549s. In this section, the A549s were pre-labelled with live fluorescence for the convenience of observation and counting under a fluorescence microscope to obtain the recovery rate. For the prelabelling, the trypsinized A549s were incubated with Cell Tracker Red/Green (C34552/C2925, Invitrogen™, Thermo Fisher) of 5 μM and Hoechst 33342 (H1399, Invitrogen™, Thermo Fisher) of 1 μg mL−1, at room temperature for 30 min in PBS. After incubation, the cells were washed with PBS three times for 5 minutes each and re-suspended in PBS for cell addition (100, 1000, 10000) via serial dilutions. Meanwhile, for the addition of ≤3 and ∼10 cells, the labelled A549s were re-suspended in the F-12K medium (21127022, Gibco™, Thermo Fisher) containing 10% FBS (fetal bovine serum, 10099133, Gibco™, Thermo Fisher) to guarantee cell viability during the relatively slow micropipette precise operation.
Every cell separation experiment was repeated more than 3 times for ≥10 cells/10 mL fluids and no less than 6 times for ≤3 cells/10 mL. The micropore diameter and edge-to-edge space for all the used filtration membranes in this section are those of case 2, as listed in Table 2.
Detection of CTCs in clinical samples
To validate the use of our liquid biopsy platform in real clinical applications, we processed whole blood samples (n = 5) from patients with lung cancer. The cancer stage/type and other information about the patients are listed in Table 3. Blood samples from healthy donors (n = 3) were collected and operated as negative controls to verify the specificity of the present capturing and staining protocol. 5 mL blood samples were collected into vacutainer tubes (BD Vacutainer) containing the anticoagulant K2EDTA and were processed within 6 hours after blood drawing.| Patient no. | Sex | Age | TNM | Tumour position | Pathology type |
|---|---|---|---|---|---|
| 1 | Female | 30 | PT1M0N0 | Superior lobe of right lung | Microinvasive adenocarcinoma |
| 2 | Female | 53 | PTxM0N0 | Superior lobe of left lung | Moderately differentiated adenocarcinoma |
| PT1M0N0 | Superior lobe of right lung | Microinvasive adenocarcinoma | |||
| 3 | Female | 40 | PT1M0N0 | Middle lobe of right lung | Moderately differentiated adenocarcinoma |
| 4 | Female | 71 | PT1M0N0 | Inferior lobe of left lung | Moderately differentiated adenocarcinoma |
| 5 | Male | 82 | PTxM0N0 | Superior lobe of right lung | Adenocarcinoma |
The filtration of the whole blood was followed by loading of 5 mL PBS to wash the blood cells off from the membrane. After filtration, the cells on the membranes were fixed with 4% (w/v) paraformaldehyde (PFA, FB002, Invitrogen™, Thermo Fisher) for 15 minutes and subsequently, permeabilized with 0.1% (v/v) Triton X-100 (85111, Thermo Fisher) in PBS for 10 minutes at room temperature, with each step followed by PBS washing for 5 minutes × 3 times. Cells were then blocked with 1 μg mL−1 IgG (206195, Abcam) in PBS for 30 minutes at room temperature. After blocking of nonspecific binding sites, without a PBS washing step, cells were incubated with primary antibodies, anti-cytokeratin 7 (PA5-29033, Thermo Fisher) and anti-CD45 (MA5-12795, Thermo Fisher), at 4 °C overnight. The membranes were washed with PBS (5 minutes × 3 times) and then incubated with appropriately matched Alexa Fluor-conjugated secondary antibodies (A-11001 and A-21428, Invitrogen™, Thermo Fisher) to identify cells. After immunofluorescence staining, the membranes were washed with PBS three times for 5 minutes each and sealed with ProLong™ Diamond Antifade Mountant with DAPI (P36962, Invitrogen™, Thermo Fisher) between a glass slide and a coverslip. The mounted membranes were stored at 4 °C in the dark. The image acquisition and quantification of the stained cells were conducted under a fluorescence microscope.
The micropore diameter and edge-to-edge space for all the filtration membranes used in this section are those of case 2, as listed in Table 2.
All experiments involving whole blood samples were performed in compliance with the relevant laws and institutional guidelines (Good Clinical Practice (GCP) of China), approved by committees and signed by the chairman of the committee (Baowei Zhang) of the Peking University No. 1 Hospital. Informed consent was obtained for any experimentation with human subjects.
Results and discussion
Numerical simulation
The simulated velocity field near the micropores during filtration is displayed in Fig. 2a and b, without and with a microsphere trapped, respectively. Fig. 2c shows the typical distributions of the hydrodynamic stress acting on the microsphere trapped in the micropores with edge-to-edge spaces of 2 μm and 16 μm during filtrations. The extracted maximum values of shear stress and pressure acting on the 15 μm (trapped in the micropores) and 8 μm (going through the micropores) microspheres during filtration through the micropore arrays with different edge-to-edge spaces are shown in Fig. 2d and e, respectively. The inserted images are the distributions of shear stress and pressure acting on the 8 μm and 15 μm microspheres, with the center of the microsphere and the height of the top surface of the membrane set as the zero point of the x/y and z axes, respectively. Fig. 2d and e reveal that both the shear stress and pressure acting on the 8 μm and 15 μm microspheres significantly increase as the size of the edge-to-edge space enlarges, during the filtration through micropores of a certain size. The increment of shear stress and pressure would cause more damage to cells, and thereby a lower viability. Therefore, the size of the edge-to-edge space should be designed as small as possible to obtain a high cell viability, which is critical for culture and proliferation for down-stream analysis. In this work, design of 4 μm edge-to-edge space was chosen in overall consideration of the filtration throughput, fabrication efficiency and mechanical strength (ease of operation).Fig. 2f shows the variations of numerically simulated lateral forces with the offset distance of the microsphere (8 μm) mass center on the top surface of membranes with differently sized edge-to-edge spaces (4 μm, 8 μm and 16 μm). The middle point of the edge-to-edge space is set as the zero point. During the filtration, when the offset distance of the mass center is 2 μm, the lateral stress acting on the 8 μm microsphere is 1.12 × 10−9 N for the 4 μm edge-to-edge space, far more than that under the condition of the 16 μm edge-to-edge space (0.31 × 10−9 N), implying that the lateral force dramatically decreased with the increase of the edge-to-edge space. The greater lateral force the microsphere experienced, the higher the instability of the mass center, and the easier it is for it to be flowed away from the edge-to-edge space and filtrated through the sideward micropores, resulting in lower non-specific adhesion of background cells. The low non-specific adhesion of background cells is critical to improve the purity of the target cell recovery.
In short, the numerical results prove that a small edge-to-edge space is significant to attain high viability of recovered cells and low non-specific adhesion of background cells.
The filtration system set-up and achieved throughput
The Parylene C micropore arrayed membrane was prepared with a previously reported molding technique, and a typical scanning electronic microscope (SEM) image and optical photo of the filtration membrane (case 3 as listed in Table 2) are displayed in Fig. 3a and b, respectively. As Fig. 3c shows, the present large-area (20 mm × 20 mm) Parylene C membrane with high porosity could be operated manually with ease, revealing its strong mechanical strength and excellent reliability, critical for a robust and reliable liquid biopsy. The Parylene C filtration membrane was packaged with the home-made Teflon holders and commercially available ring magnets for filtration operation shown in Fig. 3d and e. The present package (the so-called 2nd generation system), based on ring magnet-assisted sealing, is very simple but of an adequate strength and is also more operation-friendly and elegant compared to the previously reported system based on PMMA gadgets and screw turning (the 1st generation one60). The sample loading and filtration totally driven by gravity is displayed in Fig. 3d, with Rhodamine B as an example, for convenience of observation. Fig. 3e shows the expanded view of the package system.After packaging, the filtration throughputs of undiluted whole blood were measured through micropore arrays of different porosities (cases 1–4 as listed in Table 2), as shown in Fig. 3f. The error bars stand for the standard deviations between different filtrations. The throughput displayed in Fig. 3f is the calculated average value, i.e. the ratio of the filtrated volume of PBS (10 mL) and whole blood (5 mL) to the total time length used during the whole filtration process. In the actual filtration of biological samples, the real-time throughput decreased with the filtration time due to the fouling of biological contents. The real-time throughput vs. filtration time plot for one typical whole blood filtration through the micropore array of case 1 as listed in Table 2 is shown in the inset of Fig. 3f. The real-time throughput decreased with time, which is around 30 mL min−1 and 6 mL min−1 in the beginning 4 s and last 4 s, respectively. Filtration of 5 mL whole blood could be finished within 18 s (i.e. the average filtration throughput is around 17 mL min−1). Throughputs as high as 34 mL min−1 (obtained at a porosity of 58.46%, case 4 as listed in Table 2) for the undiluted whole blood were achieved, only driven by gravity with no need for any extra pressure loading. The high throughput, benefiting from the high porosity (>40.2%, i.e. edge-to-edge space <4 μm) and a large filtering area, is the highest recorded value among the reported values, as far as the authors know. The high filtration throughput demonstrates that the present filtration based system possesses a strong capacity to fulfill liquid biopsy of rare tumor cells from large-volume samples in real clinical applications.
Experimental verifications of cell viability and non-specific background adhesion
The short-term viability tested via the live/dead assay is shown in Fig. 4a. The viability of cells trapped on the micropores after filtration decreases dramatically as the size of the edge-to-edge space increases. The viability of recovered cells on the filtration membrane of case 5 as listed in Table 2 (edge-to-edge space at 2.46 ± 0.11 μm) is as high as 92.5 ± 3.8%, comparable to that of the control group (96.8 ± 3.3%), while it is reduced to only 33.2 ± 4.7% for the filtration membrane of case 7 as listed in Table 2 (edge-to-edge space at 16.46 ± 0.17 μm, most cases of the presently existing filtration membranes). The experimental results correspond with the conclusions of numerical simulation. For case 3 as listed in Table 2 (edge-to-edge space at 4.77 ± 0.15 μm), the short-term viability is 84.3 ± 5.1%. Moreover, in situ culture of the recovered cells on the filtration membrane of case 3 was carried out, and the well-presented adhesion and spreading, as shown in the confocal images (Fig. 4b and c), further confirmed the long-term viability of recovered cells with the invented filtration system. Its compatibility for in situ culture and proliferation widens its applicability in down-stream analysis, like drug screening, genetic sequencing, etc. Meanwhile, culture of tumor cells from real clinical samples still needs more careful investigations due to their intrinsic complexity and heterogeneity.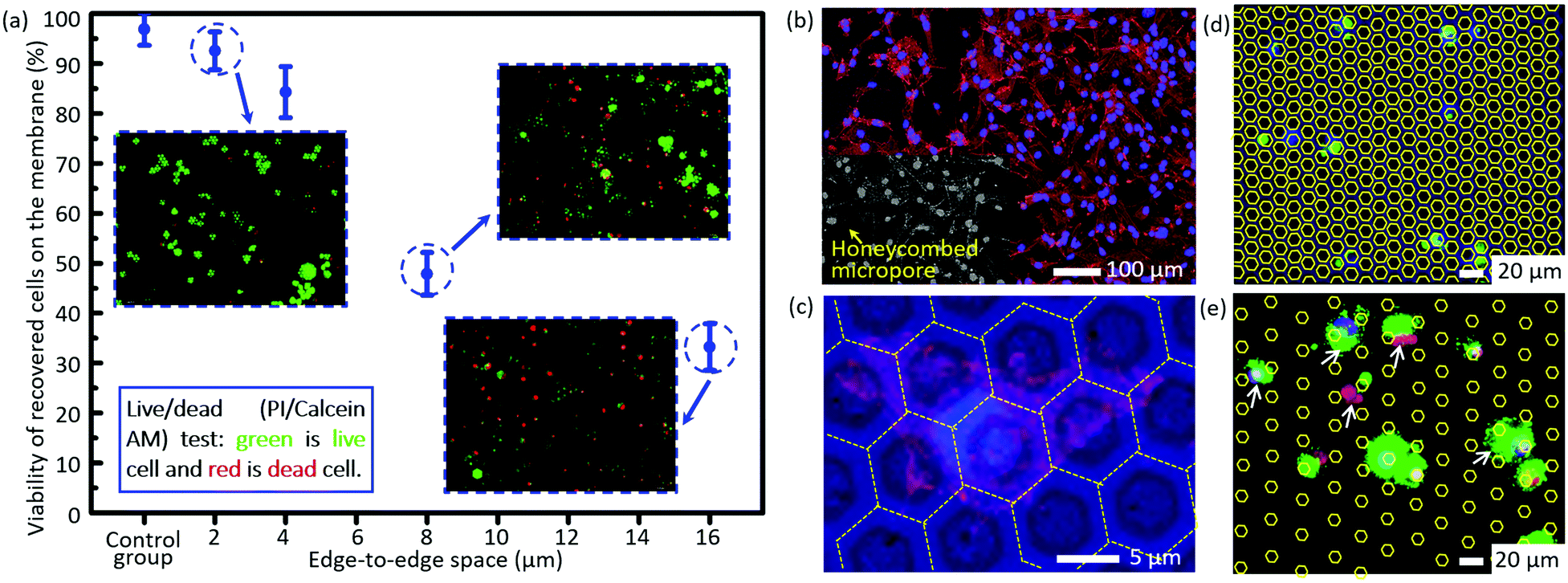 | ||
| Fig. 4 (a) The viability of the recovered cells on the membranes with differently sized edge-to-edge spaces after filtration, via staining with a live/dead kit (green for live cells and red for dead cells). (b) The confocal fluorescence images of the in situ cultured A549 cells for 72 h after filtration. (c) The magnification of (b) for a detailed view of cell adhesion/spreading on the membrane. The images of non-specifically adhered cells on the membranes with an edge-to-edge space of 4.77 ± 0.15 μm and 16.46 ± 0.17 μm (cases 3 and 7 as listed in Table 2) (d) and (e), respectively. The white arrows show the non-specifically adhered cells (non-target cell contamination) on the edge-to-edge space. The contours of the micropores in (d) and (e) are outlined with yellow lines for convenience of location searching. | ||
There is almost no non-specifically adhered background competitor cell on the membrane of case 3 as listed in Table 2 (edge-to-edge space at 4.77 ± 0.15 μm) after filtration, while the number of background cells on the membrane of case 7 as listed in Table 2 (edge-to-edge space at 16.46 ± 0.17 μm) reveals an obvious increase, as shown in Fig. 4d and e, which well verified the prediction from numerical simulation in the preceding part of this paper. The lower non-specific background adhesion of background cells should be attributed to the larger lateral stress acting on the cells adhered on the smaller edge-to-edge space and thus the higher probability to be flowed away/filtrated through the sideward micropores. Furthermore, the elimination of non-specifically adhered cells smaller than the diameter of micropores means a small dispersion in the size of trapped cells on the membrane, namely, a high size resolution in the filtration based separation, and this is coincident with our previously reported high size resolution in the microparticle separation.60
In a word, a small edge-to-edge space (high porosity) is significant to increase the viability of recovered cells and lower the non-specific adhesion of background cells besides improving the filtration throughput, which has been proved well by a 3D numerical simulation and corresponding experiments.
Separation of rare cells spiked in large-volume samples with high recovery rates
The results of separation of rare A549s spiked in large volume samples (PBS, BALF and undiluted whole blood) are shown in Fig. 5. The error bars stand for the standard deviations between different filtrations. Fig. 5a displays the recovery rates of rare spiked A549s from 10 mL PBS solution via filtrations and centrifugation. The present technology realized a much higher recovery rate (86.2 ± 4.8%) of rare cells spiked in 10 mL PBS solution at a low cell concentration (∼100 cells per mL), compared to the traditional centrifugation strategy (9.63 ± 1.45%). When the cell concentration was lower (∼10 cells per mL and 1 cell per mL), the centrifugation failed to recover any cells even at an ultimate condition in regular cell biological experiments (3500 rpm for 20 minutes), while the present filtration method still had a high recovery rate of 86.7 ± 6.8% and 88.0 ± 3.7%, respectively. For the case of ≤3 spiked A549 cells in 10 mL PBS, the developed micropore-arrayed filtration still demonstrated a high positive detection rate (83.3%), a key index in clinical analysis, as shown in Fig. 5b, along with a recovery rate of 55.6 ± 38.9%. To test the sensitivity of the invented platform for real clinical samples with a complex background, the separation of rare tumour cells spiked in unprocessed healthy BALF and whole blood was performed. As shown in Fig. 5c, the results show high recovery rates (86 ± 5.3% and 83.2 ± 6.2%) for rare cell separation from large-volume fluids (∼10 A549s in 10 mL BALF and 5 mL whole blood, respectively, n = 5 for every trial), although these clinical samples consisted of complex compositions. The fluorescence and SEM images of recovered A549s from PBS are displayed in Fig. 5d. The images of recovered A549s on the membranes from BALF and whole blood are shown in Fig. 5e and f, respectively. The results here revealed that the established filtration based platform demonstrates a powerful capability in rapid and sensitive rare cell separation from large-volume samples for practical applications of liquid biopsy. As shown in Table 1, the present 2.5D micropore arrayed filtration membrane has an overwhelming advantage in terms of the operation throughput, meanwhile the recovery rate is competitive compared to existing filtration approaches.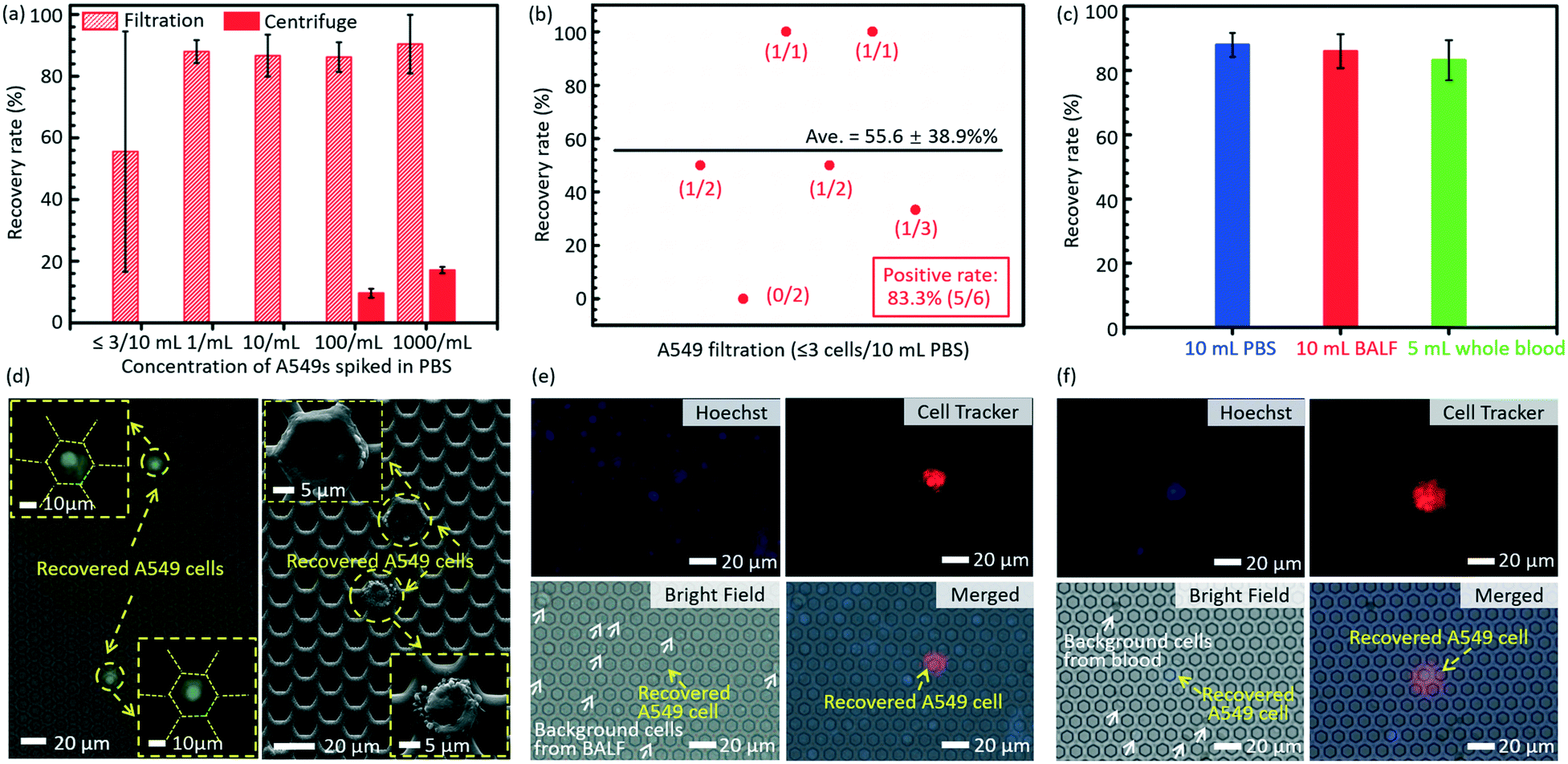 | ||
| Fig. 5 (a) The recovery rates of cell separation with the present filtration membrane and the traditional centrifuge, in parallel. (b) The positive detection rate of ≤3 spiked A549 cells in 10 mL PBS solution. (c) The recovery rates of 10 spiked A549 cells from 10 mL PBS (blue), 10 mL unprocessed BALF (red) and 5 mL undiluted whole blood (green). (d) Fluorescence and SEM images of recovered A549 cells from PBS (1000/10 mL) on the membrane after filtration. The images of recovered A549 cells on the membrane from BALF (e) and whole blood (f), respectively. | ||
Detection of CTCs in clinical samples
As a clinical validation of this technique, we tested the invented high-throughput liquid biopsy platform with blood samples collected from four patients with lung cancers. Immunostaining of CK and CD45 was used to confirm the presence of CTCs and white blood cells (WBCs), respectively. Cells with a staining pattern of CK+/CD45−/DAPI+ were identified as CTCs, while those with CK−/CD45+/DAPI+ were identified as WBCs. 3 negative controls with whole blood samples from healthy donors were performed and the results indicated that the present capturing and staining protocol has specificity to the CTCs in the lung cancer patients. Typical fluorescence images are shown in Fig. 6b based on this immunostaining detection criterion. The detection of CTCs from all of the four whole blood samples was all positive, although there was variance in the counts of verified CTCs for different lung cancer patients, as shown in Fig. 6a. Some CTC clusters were also observed on the membrane from the blood sample of one patient.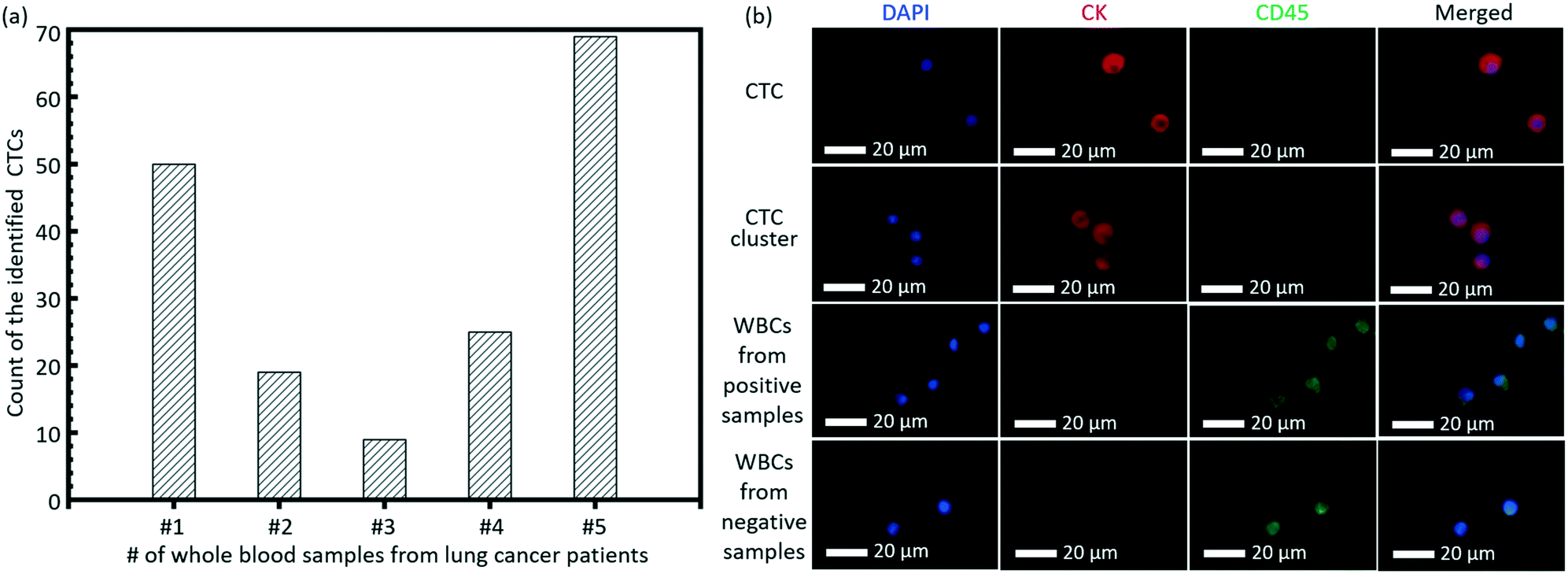 | ||
| Fig. 6 (a) The count of the identified CTCs from the whole blood samples of 5 lung patients. (b) Typical images of the CTC, CTC cluster and WBCs on the membrane. | ||
Besides the detection sensitivity, purity is always a challenging issue to physical property based separation approaches, such as the size based micropore-arrayed filtration in this work. The purity of CTCs on the filtration membrane of case 2 as listed in Table 2 is calculated based on the counts of recovered CTCs and WBCs, with a quantitative value of 13.3 ± 8.7%. The background cells, a cause of poor purity, on the filtration membrane are attributed to two sources. The first source is the WBCs' diameter being larger than that of the micropore. Since the size of CTCs has an overlap with that of WBCs, it is unavoidable to capture WBCs larger than the micropore to ensure as many CTCs as possible are captured, which causes poor purity of the present size based CTC separations. The other background cells are the WBCs smaller than the micropore to be captured (on the edge-to-edge spaces) via non-specific adhesion, even if they should have theoretically passed through the micropores because of having a smaller diameter. This non-specific contamination problem exists in almost all the previously reported micropore-arrayed filtrations listed in Table 1. Meanwhile, in this work, according to the numerical simulation and cultured cell experiments, benefiting from a small edge-to-edge space, our filtration membrane could strain away as many WBCs smaller than the micropore diameter as possible, i.e. have a relatively low number of non-target cells (smaller than the micropore diameter). For the whole blood filtration through the micropore array with a diameter of around 12 μm (cases 3 and 7 as listed in Table 2), when the edge-to-edge space is 4.77 ± 0.15 μm, the count of captured WBCs smaller than the pore diameter was around 21, while that for the case of the 16.46 ± 0.17 μm edge-to-edge space was 176, in the total filtration area (1.13 cm2). From the above, it is the small edge-to-edge space in the present 2.5D micropore-array that brings about better purity of target cell recovery, compared to the other previously reported filtration approaches listed in Table 1.
The current liquid biopsy platform demonstrated the successful detection of CTCs at a clinically friendly throughput; it was validated with lung cancer patients, and further clinical trials are still on-going. It is in the early stage of development but is easy to operate and ready for integration with other separation and analysis tools to achieve further system optimization and performance improvement.
Conclusions
In this study, a high-throughput liquid biopsy platform is established for rapid and sensitive rare cell separation from large-volume samples based on a micropore-arrayed filtration membrane. Thanks to its high porosity (>40.2%, i.e. edge-to-edge space between the adjacent micropores <4 μm), the present micropore-arrayed membrane can achieve a high throughput, >110 mL min−1 for aqueous samples and >17 mL min−1 for undiluted whole blood, only driven by gravity with no need for any extra pressure loading, which are the highest recorded values among the reported values, as far as we know. Besides the high throughput, the present technique realized a much higher recovery rate (86.2 ± 4.8%) of low-concentration (∼100 cells per mL) tumor cells spiked in 10 mL PBS solution, compared to the traditional centrifugation strategy (9.63 ± 1.45%). When the cell concentration was lower (∼10 cells per mL and 1 cell per mL), centrifugation failed to recover any cells even at an ultimate condition in regular cell biological experiments (3500 rpm for 20 minutes), while the present method still had a high recovery rate of 86.7 ± 6.8% and 88.0 ± 3.7%, respectively. For the cases of ≤3 cells/10 mL cell concentration, the developed micropore-arrayed filtration still demonstrated a high positive rate (83.3%), along with a recovery rate of 55.6 ± 39.0%. In addition, the capabilities for the recovery of rare (∼10) tumor cells spiked in unprocessed 10 mL BALF (n = 5) and 5 mL whole blood (n = 5) of the present technique were validated, with high rates of 86 ± 5.29% and 83.2 ± 6.23%, respectively. Then the successful detection of CTCs in blood samples from lung cancer patients (n = 5) was demonstrated. In addition, a 3D numerical simulation and the corresponding experiments show that the developed platform features a high viability (>84.3 ± 5.1%) and compatibility of in situ culture of recovered cells, benefiting from a small edge-to-edge space, which are critical for down-stream analysis, such as drug screening, gene sequencing, etc. The advantage of high-throughput will break the bottleneck in real clinical applications of the presently existing liquid biopsy techniques and expand the detecting targets of liquid biopsy from the presently studied CTCs in whole blood (∼7.5 mL) to ETCs in other large-volume clinical samples, such as BALF, urine, pleural fluid, etc. With its ease of operation, this platform is ready to be integrated with other separation and analysis tools to fulfill a powerful system for liquid biopsy of various body fluids in clinical practice in the coming precision medicine era.Statement of contributions
Y. Liu conceived all aspects of this study, performed the device preparation, filtration system set-up, simulation and cell separation from PBS, participated in all other experiments and prepared the manuscript. T. Li helped perform experiments on BALF and whole blood with spiked cells, as well as prepare some of the figures. M. Xu helped perform experiments on whole blood samples from lung patients. W. Zhang helped in the collection of BALF samples and in the conceptualization and validation of clinically relevant experiments, together with Y. Xiong and L. Nie. Q. Wang, H. Li and W. Wang designed the experiments and revised the manuscript. All the authors reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant No. 81611540352, 81471750 and 81330060), the Beijing Natural Science Foundation (Grant No. 4172028), and a Seeding Grant for Medicine and Information Sciences (2016-MI-04) awarded by Peking University.References
- C. Alix-Panabieres and K. Pantel, Clin. Chem., 2013, 59, 110–118 CrossRef CAS PubMed.
- S. K. Arya, B. Lim and A. R. A. Rahman, Lab Chip, 2013, 13, 1995–2027 RSC.
- L. L. Cai, H. M. Ye, L. M. Zheng, R. S. Ruan and C. M. Tzeng, Curr. Drug Targets, 2014, 15, 965–972 CAS.
- R. Harouaka, Z. Kang, S. Y. Zheng and L. Cao, Pharmacol. Ther., 2014, 141, 209–221 CrossRef CAS PubMed.
- B. Hong and Y. Zu, Theranostics, 2013, 3, 377–394 CrossRef CAS PubMed.
- T. Schlange and K. Pantel, Pharmacogenomics, 2016, 17, 183–186 CrossRef CAS PubMed.
- S. Nagrath, L. V. Sequist, S. Maheswaran, D. W. Bell, D. Irimia, L. Ulkus, M. R. Smith, E. L. Kwak, S. Digumarthy, A. Muzikansky, P. Ryan, U. J. Balis, R. G. Tompkins, D. A. Haber and M. Toner, Nature, 2007, 450, 1235–1239 CrossRef CAS PubMed.
- K. Avid and C. H. L. Paul, Curr. Pharm. Biotechnol., 2016, 17, 810–821 Search PubMed.
- A. A. Bhagat, H. Bow, H. W. Hou, S. J. Tan, J. Han and C. T. Lim, Med. Biol. Eng. Comput ., 2010, 48, 999–1014 CrossRef PubMed.
- G. E. Hvichia, Z. Parveen, C. Wagner, M. Janning, J. Quidde, A. Stein, V. Muller, S. Loges, R. P. Neves, N. H. Stoecklein, H. Wikman, S. Riethdorf, K. Pantel and T. M. Gorges, Int. J. Cancer, 2016, 138, 2894–2904 CrossRef CAS PubMed.
- E. S. Park, S. P. Duffy and H. Ma, Methods Mol. Biol., 2017, 1634, 21–32 CrossRef CAS PubMed.
- L. Yu, S. R. Ng, Y. Xu, H. Dong, Y. J. Wang and C. M. Li, Lab Chip, 2013, 13, 3163–3182 RSC.
- T. F. Didar and M. Tabrizian, Lab Chip, 2010, 10, 3043–3053 RSC.
- M. E. Warkiani, L. Wu, A. K. P. Tay and J. Han, Annu. Rev. Biomed. Eng., 2015, 17, 1–34 CrossRef CAS PubMed.
- S. Yan, J. Zhang, D. Yuan and W. Li, Electrophoresis, 2017, 38, 238–249 CrossRef CAS PubMed.
- W. Zhigang and H. Klas, Micro Nanosyst., 2009, 1, 181–192 CrossRef.
- M. T. Gabriel, L. R. Calleja, A. Chalopin, B. Ory and D. Heymann, Clin. Chem., 2016, 62, 571–581 CrossRef CAS PubMed.
- D. R. Gossett, W. M. Weaver, A. J. Mach, S. C. Hur, H. T. Tse, W. Lee, H. Amini and D. Di Carlo, Anal. Bioanal. Chem., 2010, 397, 3249–3267 CrossRef CAS PubMed.
- M. X. Zhou, H. Zheng, Z. B. Wang, R. Li, X. R. Liu, W. K. Zhang, Z. H. Wang, H. P. Li, Z. W. Wei and Z. Y. Hu, Theranostics, 2017, 7, 4710–4721 CrossRef CAS PubMed.
- S. Zheng, H. Lin, J. Q. Liu, M. Balic, R. Datar, R. J. Cote and Y. C. Tai, J. Chromatogr. A, 2007, 1162, 154–161 CrossRef CAS PubMed.
- S. Zheng, H. K. Lin, B. Lu, A. Williams, R. Datar, R. J. Cote and Y. C. Tai, Biomed. Microdevices, 2011, 13, 203–213 CrossRef PubMed.
- I. Desitter, B. S. Guerrouahen, N. Benali-Furet, J. Wechsler, P. A. Janne, Y. Kuang, M. Yanagita, L. Wang, J. A. Berkowitz, R. J. Distel and Y. E. Cayre, Anticancer Res., 2011, 31, 427–441 Search PubMed.
- D. L. Adams, P. X. Zhu, O. V. Makarova, S. S. Martin, M. Charpentier, S. Chumsri, S. H. Li, P. Amstutz and C. M. Tang, RSC Adv., 2014, 4, 4334–4342 RSC.
- A. Yusa, M. Toneri, T. Masuda, S. Ito, S. Yamamoto, M. Okochi, N. Kondo, H. Iwata, Y. Yatabe, Y. Ichinosawa, S. Kinuta, E. Kondo, H. Honda, F. Arai and H. Nakanishi, PLoS One, 2014, 4(1–11), e88821 CrossRef PubMed.
- Y. D. Tang, J. Shi, S. S. Li, L. Wang, Y. E. Cayre and Y. Chen, Sci. Rep., 2014, 4(1–7), 6052 CAS.
- X. Y. Fan, C. P. Jia, J. Yang, G. Li, H. J. Mao, Q. H. Jin and J. L. Zhao, Biosens. Bioelectron., 2015, 71, 380–386 CrossRef CAS PubMed.
- S. de Wit, G. van Dalum, A. T. M. Lenferink, A. G. J. Tibbe, T. J. N. Hiltermann, H. J. M. Groen, C. J. M. van Rijn and L. W. M. M. Terstappen, Sci. Rep., 2015, 5(1–10), 12270 CrossRef CAS PubMed.
- M. Hosokawa, T. Hayata, Y. Fukuda, A. Arakaki, T. Yoshino, T. Tanaka and T. Matsunaga, Anal. Chem., 2010, 82, 6629–6635 CrossRef CAS PubMed.
- M. G. Krebs, J. M. Hou, R. Sloane, L. Lancashire, L. Priest, D. Nonake, T. H. Ward, A. Backen, G. Clack, A. Hughes, M. Ranson, F. H. Blackhall and C. Dive, J. Thorac. Oncol., 2012, 7, 306–315 CrossRef PubMed.
- J. Ko, N. Bhagwat, S. S. Yee, T. Black, C. Redlinger, J. Romeo, M. O'Hara, A. Raj, E. L. Carpenter, B. Z. Stanger and D. Issadore, Lab Chip, 2017, 17, 3086–3096 RSC.
- T. Gerhardt, S. Woo and H. S. Ma, Lab Chip, 2011, 11, 2731–2737 RSC.
- L. Pang, S. F. Shen, C. Ma, T. T. Ma, R. Zhang, C. Tian, L. Zhao, W. M. Liu and J. Y. Wang, Analyst, 2015, 140, 7335–7346 RSC.
- S. J. Tan, R. L. Lakshmi, P. F. Chen, W. T. Lim, L. Yobas and C. T. Lim, Biosens. Bioelectron., 2010, 26, 1701–1705 CrossRef CAS PubMed.
- A. F. Sarioglu, N. Aceto, N. Kojic, M. C. Donaldson, M. Zeinali, B. Hamza, A. Engstrom, H. Zhu, T. K. Sundaresan, D. T. Miyamoto, X. Luo, A. Bardia, B. S. Wittner, S. Ramaswamy, T. Shioda, D. T. Ting, S. L. Stott, R. Kapur, S. Maheswaran, D. A. Haber and M. Toner, Nat. Methods, 2015, 12, 685–691 CrossRef CAS PubMed.
- W. Zhao, R. Cheng, B. D. Jenkins, T. Zhu, N. E. Okonkwo, C. E. Jones, M. B. Davis, S. K. Kavuri, Z. Hao, C. Schroeder and L. Mao, Lab Chip, 2017, 17, 3097–3111 RSC.
- X. Jiang, K. H. K. Wong, A. H. Khankhel, M. Zeinali, E. Reategui, M. J. Phillips, X. Luo, N. Aceto, F. Fachin, A. N. Hoang, W. Kim, A. E. Jensen, L. V. Sequist, S. Maheswaran, D. A. Haber, S. L. Stott and M. Toner, Lab Chip, 2017, 17, 3498–3503 RSC.
- E. Reategui, N. Aceto, E. J. Lim, J. P. Sullivan, A. E. Jensen, M. Zeinali, J. M. Martel, A. J. Aranyosi, W. Li, S. Castleberry, A. Bardia, L. V. Sequist, D. A. Haber, S. Maheswaran, P. T. Hammond, M. Toner and S. L. Stott, Adv. Mater., 2015, 27, 1593–1599 CrossRef CAS PubMed.
- W. A. Zhao, C. H. Cui, S. Bose, D. G. Guo, C. Shen, W. P. Wong, K. Halvorsen, O. C. Farokhzad, G. S. L. Teo, J. A. Phillips, D. M. Dorfman, R. Karnik and J. M. Karp, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 19626–19631 CrossRef CAS PubMed.
- R. Khojah, R. Stoutamore and D. Di Carlo, Lab Chip, 2017, 17, 2542–2549 RSC.
- E. Sollier, D. E. Go, J. Che, D. R. Gossett, S. O'Byrne, W. M. Weaver, N. Kummer, M. Rettig, J. Goldman, N. Nickols, S. McCloskey, R. P. Kulkarni and D. Di Carlo, Lab Chip, 2014, 14, 63–77 RSC.
- S. C. Hur, A. J. Mach and D. Di Carlo, Biomicrofluidics, 2011, 5(1–10), 022206 CrossRef PubMed.
- L. R. Huang, E. C. Cox, R. H. Austin and J. C. Sturm, Science, 2004, 304, 987–990 CrossRef CAS PubMed.
- M. G. Lee, J. H. Shin, C. Y. Bae, S. Choi and J. K. Park, Anal. Chem., 2013, 85, 6213–6218 CrossRef CAS PubMed.
- J. M. Martel, K. C. Smith, M. Dlamini, K. Pletcher, J. Yang, M. Karabacak, D. A. Haber, R. Kapur and M. Toner, Sci. Rep., 2015, 5(1–12), 11300 CrossRef CAS PubMed.
- A. A. Bhagat, H. W. Hou, L. D. Li, C. T. Lim and J. Han, Lab Chip, 2011, 11, 1870–1878 RSC.
- S. Shen, C. Ma, L. Zhao, Y. Wang, J. C. Wang, J. Xu, T. Li, L. Pang and J. Wang, Lab Chip, 2014, 14, 2525–2538 RSC.
- M. E. Warkiani, G. Guan, K. B. Luan, W. C. Lee, A. A. Bhagat, P. K. Chaudhuri, D. S. Tan, W. T. Lim, S. C. Lee, P. C. Chen, C. T. Lim and J. Han, Lab Chip, 2014, 14, 128–137 RSC.
- M. E. Warkiani, B. L. Khoo, D. S. Tan, A. A. Bhagat, W. T. Lim, Y. S. Yap, S. C. Lee, R. A. Soo, J. Han and C. T. Lim, Analyst, 2014, 139, 3245–3255 RSC.
- D. R. Gossett, H. T. K. Tse, S. A. Lee, Y. Ying, A. G. Lindgren, O. O. Yang, J. Y. Rao, A. T. Clark and D. Di Carlo, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7630–7635 CrossRef CAS PubMed.
- H. Choi, K. B. Kim, C. S. Jeon, I. Hwang, S. Lee, H. K. Kim, H. C. Kim and T. D. Chung, Lab Chip, 2013, 13, 970–977 RSC.
- P. R. C. Gascoyne and S. Shim, Cancers, 2014, 6, 545–579 CrossRef PubMed.
- A. Venkatanarayanan, T. E. Keyes and R. J. Forster, Anal. Chem., 2013, 85, 2216–2222 CrossRef CAS PubMed.
- E. Dahl, A. Jung, J. Fassunke, M. Hummel, R. Penzel, W. Dietmaier and S. Lassmann, Pathologe, 2015, 36, 92–97 CrossRef CAS PubMed.
- S. A. Ahrendt, J. T. Chow, L. H. Xu, S. C. Yang, C. F. Eisenberger, M. Esteller, J. G. Herman, L. Wu, P. A. Decker, J. Jen and D. Sidransky, J. Natl. Cancer Inst., 1999, 91, 332–339 CrossRef CAS PubMed.
- J. H. Appel, H. Ren, M. L. Y. Sin, J. C. Liao and J. Chae, Analyst, 2016, 141, 652–660 RSC.
- H. T. K. Tse, D. R. Gossett, Y. S. Moon, M. Masaeli, M. Sohsman, Y. Ying, K. Mislick, R. P. Adams, J. Rao and D. Di Carlo, Sci. Transl. Med., 2013, 5, 163–212 Search PubMed.
- C. W. T. Shields, K. A. Ohiri, L. M. Szott and G. P. Lopez, Cytometry, Part B, 2017, 92, 115–125 CrossRef PubMed.
- Y. Liu, W. Wang, W. Wu, H. Li, F. Yang and W. Wang, 18th International Conference on Solid-State Sensors, Actuators and Microsystems (Transducers 2015), 2015, pp. 1767–1769 Search PubMed.
- W. Z. Dai, Y. P. Liu, Y. T. Fu, H. Xu, F. Su, J. Wu and W. Wang, 19th International Conference on Solid-State Sensors, Actuators and Microsystems (Transducers 2017), 2017, pp. 1215–1217 Search PubMed.
- H. X. Y. Liu, W. Dai, H. Li and W. Wang, Microsyst. Nanoeng., 2018, 4(13), 1–12 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8lc01048j |
| This journal is © The Royal Society of Chemistry 2019 |