
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper-catalysed photoinduced decarboxylative alkynylation: a combined experimental and computational study†‡
Yu
Mao
,
Wenxuan
Zhao
,
Shuo
Lu
,
Lei
Yu
,
Yi
Wang
 *,
Yong
Liang
*,
Shengyang
Ni
* and
Yi
Pan
*,
Yong
Liang
*,
Shengyang
Ni
* and
Yi
Pan
State Key Laboratory of Coordination Chemistry, Jiangsu Key Laboratory of Advanced Organic Materials, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210023, China. E-mail: yiwang@nju.edu.cn; yongliang@nju.edu.cn
First published on 29th April 2020
Abstract
Redox-active esters (RAEs) as alkyl radical precursors have demonstrated great advantages for C–C bond formation. A decarboxylative cross-coupling method is described to afford substituted alkynes from various carboxylic acids using copper catalysts CuCl and Cu(acac)2. The photoexcitation of copper acetylides with electron-rich NEt3 as a ligand provides a general strategy to generate a range of alkyl radicals from RAEs of carboxylic acids, which can be readily coupled with a variety of aromatic alkynes. The scope of this cross-coupling reaction can be further expanded to aliphatic alkynes and alkynyl silanes using a catalytic amount of preformed copper-phenylacetylide. In addition, DFT calculations revealed the favorable reaction pathway and that the bidentate acetylacetonate ligand of the copper intermediate plays an important role in inhibiting the homo-coupling of the alkyne.
Introduction
Carbon–carbon triple bonds are versatile intermediates for diverted transformations.1 Serving as both nucleophiles and electrophiles, alkynes are broadly employed in organic synthesis, materials science,2 chemical biology,3 and drug discovery.4 Among many metal-catalysed cross-coupling strategies for the construction of the Csp–C bond, the Sonogashira reaction was the most renowned approach to access alkyl substituted alkynes (Scheme 1A).5 Under basic conditions, copper acetylides generated in situ from Cu(I) salts and terminal alkynes undergo metal-catalysed cross-coupling reactions.6 However, the Sonogashira reaction usually requires functionalised electrophiles with adequate leaving groups and Pd complexes as catalysts.7 The development of mild and general cross-coupling strategies for copper acetylides without precious catalysts is still a great challenge and in high demand.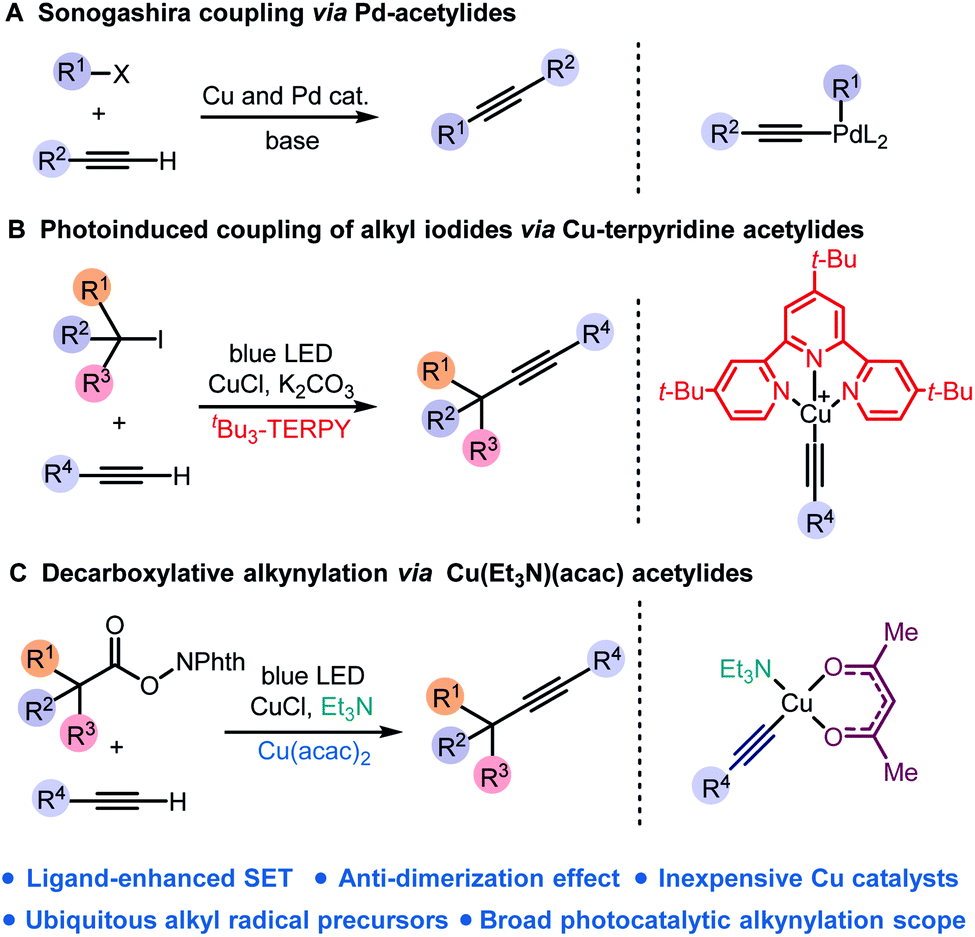 | ||
| Scheme 1 (A) Sonogashira coupling; (B) photoinduced coupling of alkyl iodides via Cu-terpyridine acetylides; (C) decarboxylative alkynylation via Cu(Et3N)(acac) acetylides. | ||
Copper has merged into the recent evolution of visible-light photoredox catalysis which led to a paradigm shift in organic synthesis.8 Recently, photoexcitation of copper complexes has been applied to the formation of carbon–carbon and carbon–heteroatom bonds.9 Fu and Peters discovered a photoinduced copper-mediated C–N coupling reaction.10 Reiser developed difunctionalisation of alkenes and alkynes using Cu(dap)Cl2 as a photocatalyst.11 Hwang discovered copper acetylide complexes as a photosensitiser for various coupling reactions.12 Recently, Lalic achieved the coupling products of alkyl iodides and terminal alkynes via photoexcited Cu(I)-acetylide combined with a substituted terpyridine ligand (Scheme 1B).13
In comparison with alkyl halides, inexpensive carboxylic acids are widely accessible and used for decarboxylative reactions.14 Notably, recent methods using redox-active esters for decarboxylative cross-coupling reactions have been developed;15 in particular, decarboxylative alkynylation of RAEs has been reported by Weix,15e Baran,15f and Chen15t using prefunctionalised alkynyl sources, including alkynyl bromides, alkynyl zinc reagents and alkynyl benziodoxolone. Fu developed a photocatalysed decarboxylative cross-coupling reaction using terminal alkynes and redox-active esters of amino-acids, but only a limited scope of substrates was demonstrated.15s
Drawing inspiration from photosensitive copper acetylide as an intermediate for sp3–sp bond formation, we envisioned RAEs of carboxylic acids to be widely accessible surrogates of alkyl iodides for cross-coupling of terminal alkynes. In the presence of a weak base, photoexcitation of copper acetylide would trigger the decarboxylative fragmentation of RAEs followed by cross-coupling to provide alkylated alkynes. Herein, we report a photoinduced decarboxylative alkynylation of redox-active esters using inexpensive copper catalysts (Scheme 1C).
Results and discussion
To determine the suitable conditions for copper acetylide-initiated radical alkylation of alkynes, we first tried the cross-coupling of a cyclohexanecarboxylic acid derived redox-active ester and 4-methylphenylacetylene under Lalic's conditions. However, the reaction resulted in no desired cross-coupling product and only a homo-coupling product was generated (Scheme 2A). We speculated that the excited copper acetylide with the tri-tBu-TERPY ligand could not efficiently transfer an electron to the redox-active ester, making alkyl radical generation unsuccessful. To test this hypothesis, we calculated the thermodynamics of the electron transfer processes from the excited copper species to isopropyl iodide and the isobutyric acid derived redox-active ester, respectively.16 As shown in Scheme 2B, electron transfer from the Cu–terpyridine complex to alkyl iodide is exergonic, with a reaction free energy of −4.4 kcal mol−1. In contrast, the process for the redox-active ester is endergonic (5.5 kcal mol−1), inhibiting the subsequent alkyl radical formation. Therefore, choosing competent ligands is essential to activate redox-active esters for the realisation of copper-catalysed decarboxylative alkynylation.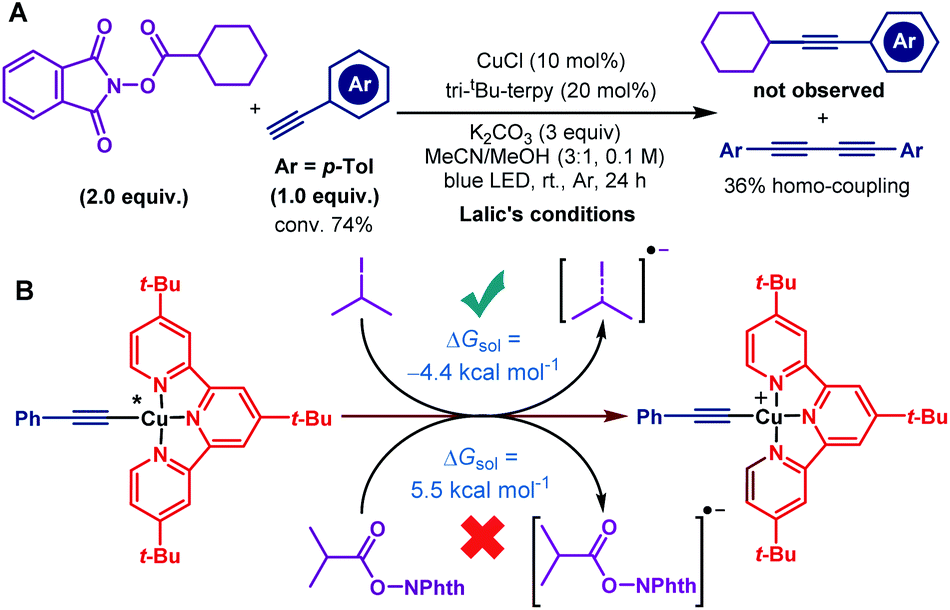 | ||
| Scheme 2 (A) Initial experimental test and (B) computational interpretation. | ||
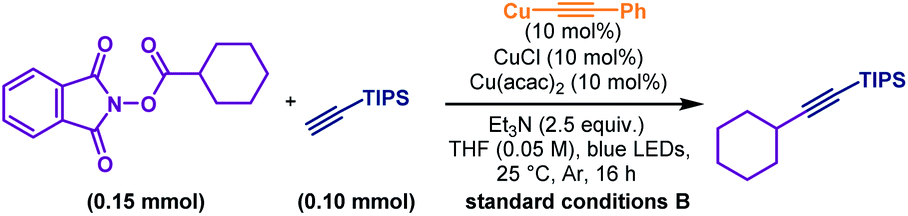
After a comprehensive survey of metal salts, ligands and bases (Table 1 and also see the ESI‡), the optimised reaction conditions have been selected as alkyne 1 (1.0 equiv.), RAE 2 (1.5 equiv.) with 10 mol% each of Cu(acac)2 and CuCl, and Et3N (2.5 equiv.) in THF under the irradiation of 90 W blue LEDs. With 95% conversion of 2, the desired decarboxylative alkynylation product 3 in 72% and 22% of the homo-coupling byproduct were afforded (Table 1, entry 1). In the catalytic system, CuCl could be replaced by copper powder to give similar results (entry 2). When Cu(acac)2 was replaced by other transition metal salts, Fe(acac)2 provided low yield of the cross-coupling product (entry 3). Co(acac)2 and Ni(acac)2 demonstrated similar results to Cu(acac)2 (entries 4 & 5). Using Zn(acac)2, the product 3 could only be obtained in moderate yield with a considerable amount of homo-coupling product (entry 6). The previously reported terpyridine ligand was found incompatible with this decarboxylative alkynylation protocol, and only 8% of the product was detected with 72% of the homo-coupling product (entry 7). In the absence of Cu(acac)2, low yield of 3 was obtained with a large amount of the homo-coupling product (entry 8). These results demonstrated that the presence of the acetylacetonate anion is crucial mainly for inhibiting the homo-coupling of the alkyne. Further control reactions illustrated that in the absence of the Cu(I) salt, this cross-coupling reaction could not proceed (entry 9). By removing Et3N or switching to K2CO3, the reaction failed to proceed (entries 10 & 11). Using a different light source, 20 W compact fluorescent light, afforded slightly lower yield (entry 12). The reaction was completely shut down in darkness (entry 13). In the attempts for non-aryl substituted alkynes, no reaction was detected under the standard conditions A, primarily for the unmatched energy transfer between visible light and unconjugated copper acetylides (Table 2, entry 2). Further study revealed that by simply adding a catalytic amount of phenyl copper acetylide as the photocatalyst, the alkylated ethynylsilane was generated in 95% yield (entry 1). Other components in the conditions are proven to be necessary. No reaction occurred or very low yield was obtained without CuCl, Cu(acac)2, Et3N or light (entries 3–6). Thus, the standard conditions A and B were selected to explore the scope of aryl and non-aryl alkynes, respectively.
|
|
||||
|---|---|---|---|---|
| Entry | Deviation from the reaction conditions | Convention of 2 | Homocoupling of 2a | Yield of 3a |
| a Crude yields determined by 1H NMR spectroscopy using dibromomethane as an internal standard. b 69% isolated yield. c Copper powder treated with hydrochloric acid. | ||||
| 1 | None | 95% | 22% | 72%b |
| 2c | CuCl to Cu powder | 92% | 16% | 67% |
| 3 | Cu(acac)2 to Fe(acac)2 | 83% | 36% | 12% |
| 4 | Cu(acac)2 to Co(acac)2 | 90% | 18% | 68% |
| 5 | Cu(acac)2 to Ni(acac)2 | 98% | 18% | 74% |
| 6 | Cu(acac)2 to Zn(acac)2 | 87% | 32% | 36% |
| 7 | Cu(acac)2 to tBu3-TERPY | 91% | 72% | 8% |
| 8 | Without Cu(acac)2 | 86% | 62% | 21% |
| 9 | Without CuCl | 0% | 0% | 0% |
| 10 | Without Et3N | 0% | 0% | 0% |
| 11 | Et3N to K2CO3 | 0% | 0% | 0% |
| 12 | Blue LED to 20 W CFL | 92% | 10% | 68% |
| 13 | In darkness | 0% | 0% | 0% |
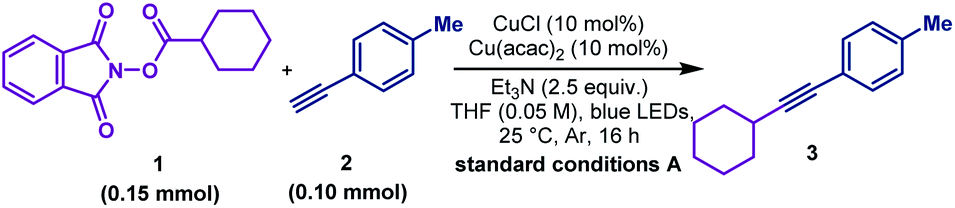
To explore the scope of this decarboxylative alkynylation, a range of primary, secondary and tertiary redox-active esters underwent radical cross-coupling sequence with 4-methylphenylacetylene under mild irradiation conditions (Scheme 3A). Carboxylic acids bearing cyclic hydrocarbons and heterocycles were tolerated in this transformation (3–9 & 11–17). Highly strained alkynyl cyclic products were obtained from the corresponding commercially available carboxylic acids (11–15), Notably, the α-oxygen and nitrogen substituted carboxylic acids (16–17) that were considered inert in radical reactions can also provide moderate yields. To demonstrate the utility of this cross-coupling protocol, we applied the standard conditions to a range of natural and pharmacal products (Scheme 3A). Searic acid (19), arachidonic acid (20), L-glutamic acid (21), pinonic acid (22), chloroambucil (23) and dehyrocholic acid (24) derived esters were compatible to afford alkynylated derivatives.
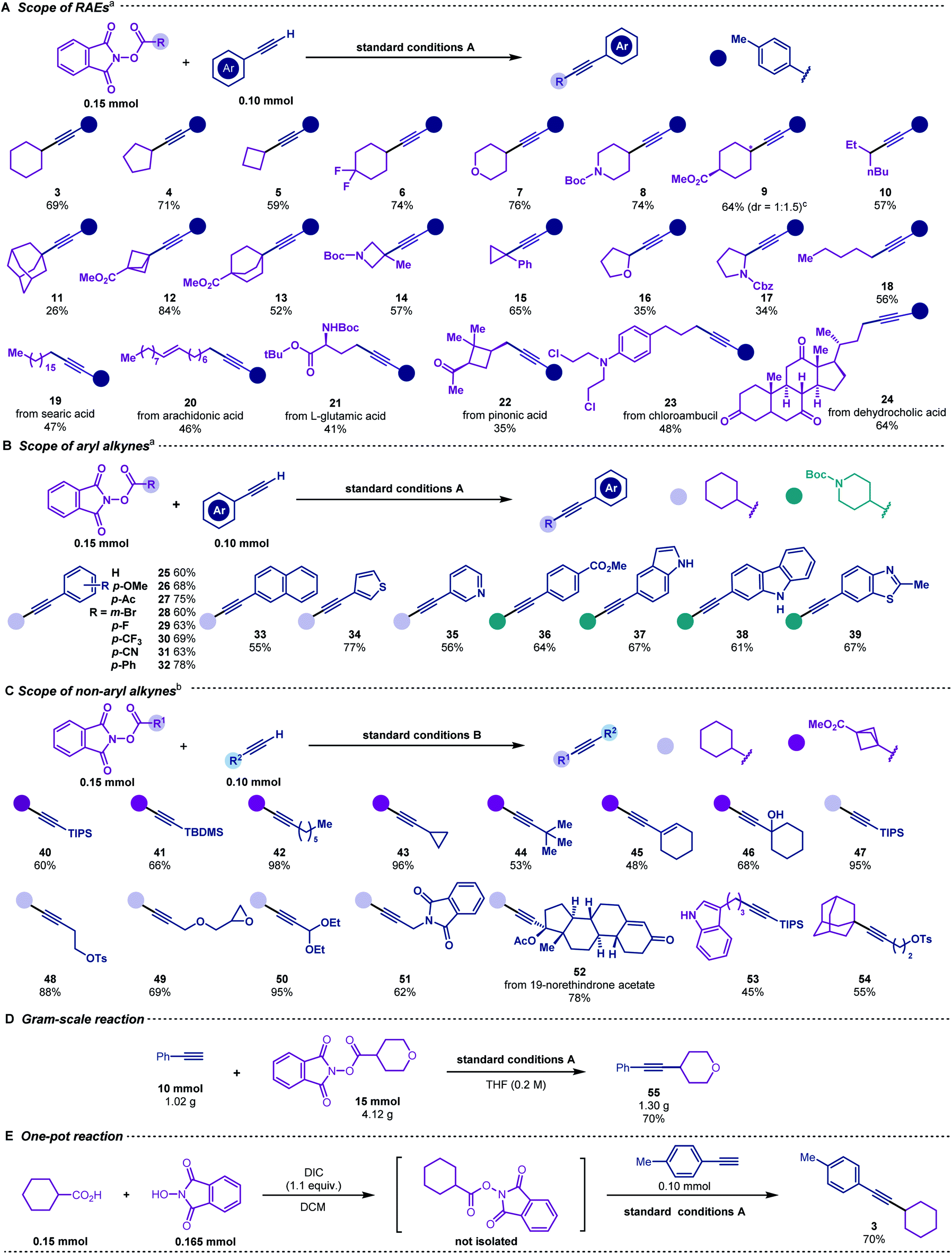 | ||
| Scheme 3 (A–C) Substrate scope of the decarboxylative alkynylation; (D) gram-scale reaction; (E) one-pot reaction. [a] The reaction was conducted under standard conditions A; [b] the reaction was conducted under standard conditions B; [c] the ratios of diastereomers were determined by 1H NMR. | ||
We next employed cyclohexyl and piperidinyl substituted esters to examine the scope of aryl alkynes (Scheme 3B). Both electron-donating and electron-withdrawing groups were tolerated under the same conditions (25–32 & 36). Naphthyl (33), thiophene (34), pyridyl (35), indoyl (37), carbazoyl (38) and benzothiozyl (39) groups were applicable for generating the desired products in good yields. Notably, no alkylation occurred for unprotected N–H (37 & 38).
With a slight variation in the optimised conditions, we further expanded the scope of alkynes for this decarboxylative alkynylation reaction. Using a catalytic amount of copper-phenylacetylide as the photocatalyst, the alkylations of non-aryl alkynes could proceed smoothly using LED light (Scheme 3C). To examine the scope of aliphatic alkynes, bicyclic (40–46), cyclohexyl (47–52), linear (53) and adamantane (54) substituted carboxylic acids were attempted under conditions B. Both linear and cyclic alkyl tethered alkynes transformed into the alkylated products (42–44). Terminal alkynes bearing silyl (40, 41, 47 and 53), alkenyl (45), hydroxyl (46), epoxyl (49), acetal (50) and amide (51) groups were susceptible to the reaction conditions to achieve the corresponding products in good to excellent yields. Contraception drug 19-morethindrone acetate was also subjected to the standard conditions to furnish the alkylated derivative in 78% yield (52).
Scalability was also evaluated, and the reaction was found to proceed smoothly to deliver product 55 on a gram-scale in 70% yield (Scheme 3D). In addition, the redox-active ester could be prepared in situ and afford the cross-coupling product 3 in a one-pot fashion (Scheme 3E).
To gain insight into the mechanism and understand the ligand effects in this reaction, computational studies are conducted using DFT calculations.16 The reaction between an aromatic alkyne (Ar = Ph) and the redox-active ester of carboxylic acid (R = i-Pr) is chosen as the model system. Organometallic intermediates and transition states with various coordination numbers of NEt3 are optimised, with the lowest state shown in this study. The reaction pathways are summarised in Scheme 4. First, Cu(I) acetylide is generated with the assistance of NEt3. Excitation with blue light promotes Cu(I) acetylide to its triplet state. Subsequently, excited Cu(I) acetylide transfers an electron to activate N-acyloxyl phthalimide into its radical anion, accompanied by its oxidation to the Cu(II) acetylide cation (Scheme 4A).17 In contrast with tBu3-TERPY (endergonic by 5.5 kcal mol−1, Scheme 2B), the NEt3 ligand enables exergonic reduction of the redox-active ester (−12.3 kcal mol−1, Scheme 4A), which can be attributed to the electron-rich nature of NEt3 in stabilising cationic Cu(II) species. The reaction of propyne as a model of aliphatic alkyne is also investigated. Compared to phenyl acetylide, excitation of methyl substituted Cu(I) acetylide Int3-Me is more endergonic (75.1 kcal mol−1, Scheme 4A), which is outside the blue light energy range (λ = 400 nm, hν = 71.5 kcal mol−1). Therefore, a catalytic amount of Cu(I) phenylacetylide (Int3) is required to facilitate the photoredox process, in accordance with experimental findings (Scheme 3C). In the case of non-aryl alkynes as the substrate, after the photoexcitation of catalyst Int3 and subsequent electron transfer to the redox-active ester, an exchange reaction (Scheme 4A) between the cationic Cu(II) species Int5 and Cu(I) methylacetylide (Int3-Me) will occur to regenerate photocatalyst Int3 and to form Int5-Me as the precursor for the alkynylation of aliphatic alkynes.16
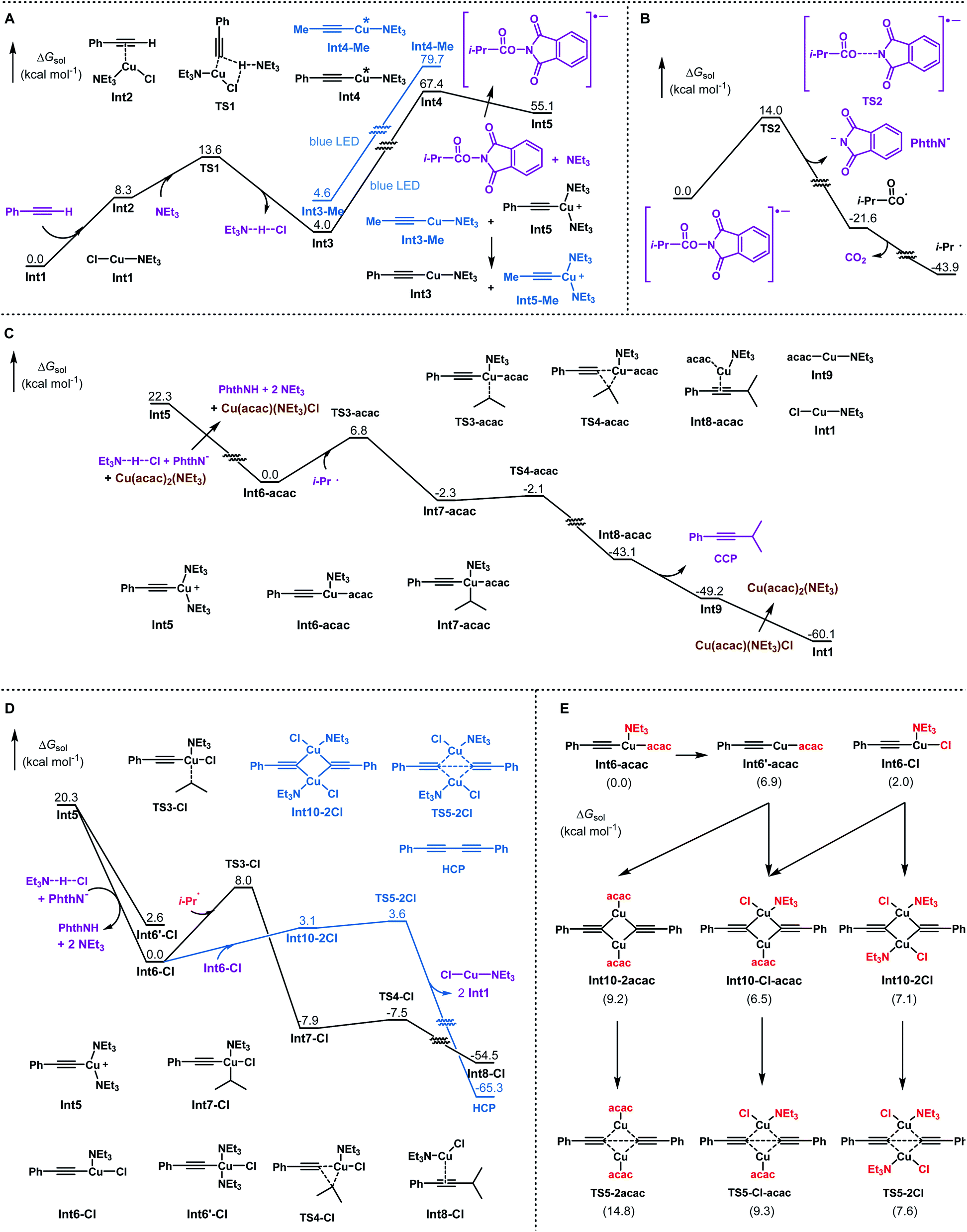 | ||
| Scheme 4 DFT calculations of the reaction pathways. Numbers associated with each molecule are relative Gibbs free energy in THF. Note that in each part the zero points of energy are different. (A) Formation, excitation, and electron transfer of Cu(I) acetylide. (B) Fragmentation of the N-acyloxyl phthalimide radical anion. (C) Ligand exchange, radical addition to Cu(II) acetylide, and reductive elimination to generate the cross-coupling product. (D) Competing pathways without the participation of Cu(acac)2. (E) Homo-coupling side reaction pathways start from Int6-acac. | ||
The subsequent fragmentation of the N-acyloxyl phthalimide radical anion leads to the iso-butyryloxy radical, which undergoes fast decarboxylation to afford the iso-propyl radical (Scheme 4B).18 Then the Cu(II) acetylide cation undergoes ligand exchange with NEt3·HCl and Cu(acac)2 to generate intermediate Int6-acac. The addition of the iso-propyl radical to Int6-acac forms the Cu(III) intermediate8,19 with an activation barrier of 6.8 kcal mol−1, followed by a facile reductive elimination to produce the cross-coupling product and the regeneration of the Cu catalyst (Scheme 4C).20 The overall rate-limiting step after photoexcitation is the fragmentation of the N-acyloxyl phthalimide radical anion (14.0 kcal mol−1, Scheme 4B), consistent with experimental results that reactions proceed smoothly at room temperature.
To elucidate the crucial role of Cu(acac)2 in the success of this reaction, we investigated several possible competing pathways that may exist in this system. Since the homo-coupling of alkyne is found to be the major side reaction, this competing pathway is also calculated for both with and without Cu(acac)2. As shown in Scheme 4D, radical addition barriers are only slightly higher without Cu(acac)2. The homo-coupling pathway via a bi-copper complex21Int10-2Cl has much lower activation free energy (transition state TS5-2Cl, 3.6 kcal mol−1) than that of the radical addition via the transition state TS3-Cl (8.0 kcal mol−1), which will lead to the cross-coupling product. This is in agreement with experimental findings that homo-coupling products will dominate with CuCl as the only catalyst. Scheme 4E shows homo-coupling pathways starting from Int6-acac. Three possible bi-copper complexes can be formed with different ligands. However, their reductive elimination barriers (8–15 kcal mol−1) are all higher than those for the iso-propyl radical addition to Int6-acac (6.8 kcal mol−1, Scheme 4C). To rationalise this result, we noticed that the bidentate nature22 of the acetylacetonate ligand causes Int6-acac to be four-coordinated. Unlike the three-coordinated Int6-Cl intermediate, it is required for Int6-acac to dissociate the NEt3 ligand prior to the bi-copper complex formation, which is endergonic by 6.9 kcal mol−1 (Scheme 4E). Meanwhile, the ligand exchange to generate Int6-Cl is also endergonic. As a result, three possible combinations of Int6′-acac and Int6-Cl are all energetically disfavored, making the relative dominance of the two pathways (homo-coupling versus cross-coupling) reverse. Therefore, the bidentate acetylacetonate ligand of the copper intermediate elevates the homo-coupling barriers and remarkably promotes the formation of cross-coupling products.
Based on the above experimental and computational evidence, a plausible mechanism is suggested (Scheme 5). First, in the presence of NEt3, Cu(I) complex A is formed which reacts with the alkyne to afford photosensitive copper(I) acetylide B. The photoexcitation of B furnishes copper acetylide C, which undergoes the SET process with a redox-active ester to afford a radical anion of the RAE and Cu(II)-acetylide D. Subsequent decarboxylative fragmentation provides an alkyl radical. Ligand exchange between Cu(II)-acetylide D and Cu(acac)2(NEt3) gives the key copper intermediate E. Finally, the addition of the alkyl radical to E gives Cu(III) intermediate F, followed by reductive elimination to furnish the cross-coupling product and G. Hence, several critical factors are pointed out for the realisation of the cross-coupling reaction. First, a conjugated aromatic alkyne is required for the photoexcitation of Cu(I) acetylides within the visible light energy range. Second, the electron-rich ligand NEt3 promotes the electron transfer from copper acetylides to redox-active esters. Finally, the bidentate acetylacetonate ligand effectively intervenes in the generation of bi-copper complexes, which prohibits the formation of undesired homo-coupling byproducts.
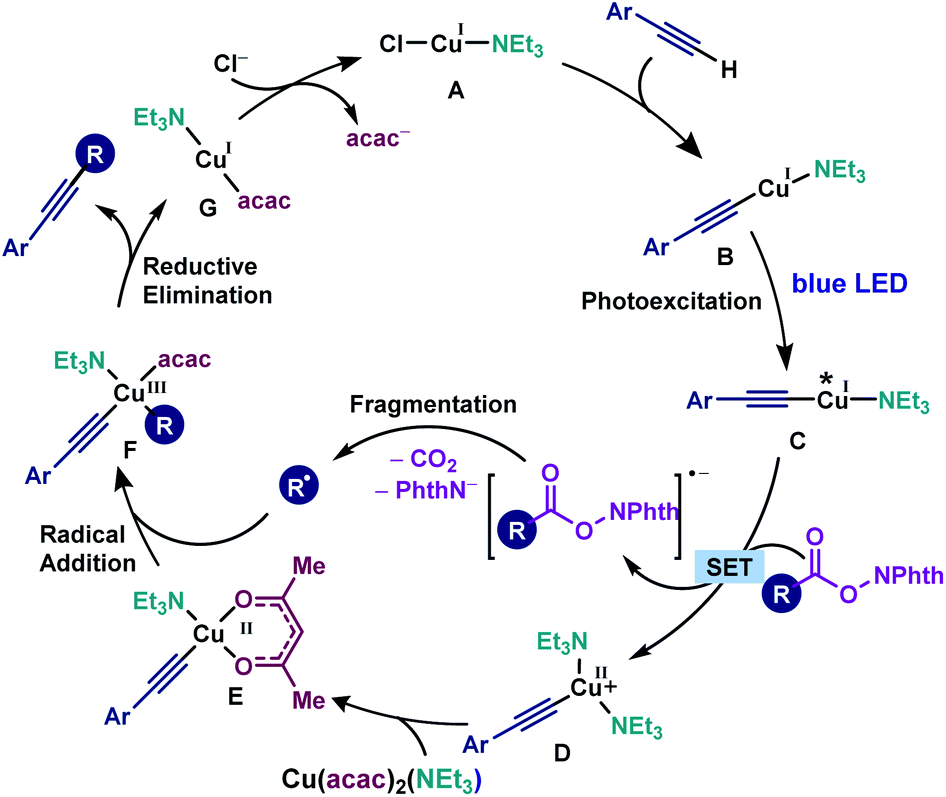 | ||
| Scheme 5 The catalytic cycle for the copper-catalysed photoinduced decarboxylative alkynylation and the important role of NEt3 and acetylacetonate ligands in the reaction mechanism. | ||
Experimental
Standard conditions for decarboxylative alkynylation with aryl alkynes (conditions A)
In a glovebox, redox active esters (0.15 mmol, 1.5 equiv.), cuprous chloride (10 mol%), cupric acetylacetonate (10 mol%), terminal alkyne (0.1 mmol, 1.0 equiv.), triethylamine (2.5 equiv.) and anhydrous THF (2 mL) were added. A tube was set between two lamps (10 cm away from the lamp, 90 W each) and the mixture was stirred at room temperature (with a fan to cool down the reaction) for 16 h. The solvent was removed under vacuum. The crude product was purified by column chromatography or preparative TLC to afford the corresponding alkyne (silica, 0–100% EtOAc/petroleum ether).Standard conditions for decarboxylative alkynylation with non-aryl alkynes (conditions B)*
In a glovebox, redox active esters (0.15 mmol, 1.5 equiv.), cuprous chloride (10 mol%), cupric acetylacetonate (10 mol%), and copper phenylacetylide (10 mol%) were mixed in a sealed culture tube. Terminal alkyne (0.1 mmol, 1.0 equiv.), triethylamine (2.5 equiv.) and anhydrous THF (2 mL) were added in sequence. A tube was set between two lamp (10 cm away from the lamp, 90 W each) and the mixture was stirred at room temperature (with a fan to cool down the reaction) for 16 h. The solvent was removed under vacuum. The crude product was purified by column chromatography or preparative TLC to afford the corresponding alkyne (silica, 0–100% EtOAc/petroleum ether). *Note: a small amount (5%) of the alkylation product of phenylacetylene was observed in the crude reaction mixture.General procedure for decarboxylative alkynylation with aryl alkynes (gram scale)
Redox active esters (15 mmol, 1.5 equiv.), cuprous chloride (10 mol%) and cupric acetylacetonate (10 mol%) were mixed in a sealed flask with an argon balloon. Then anhydrous THF (50 mL) was added. Terminal alkyne (10 mmol, 1.0 equiv.) and triethylamine (2.5 equiv.) were added in sequence. A tube was set between two lamps (10 cm away from the lamp, 90 W each) and the mixture was stirred at room temperature (with a fan to cool down the reaction) for 16 h. The solvent was removed under vacuum. The crude product was purified by column chromatography to afford the corresponding alkyne (silica, 0–100% EtOAc/petroleum ether).General procedure for in situ activation of carboxylic acids in decarboxylative alkynylation
A culture tube was charged with carboxylic acid (0.15 mmol, 1 equiv.) and N-hydroxyphthalimide (0.165 mmol, 1.1 equiv.). Dichloromethane was added (1.5 mL, 0.1 M), and the mixture was stirred vigorously. Then, N,N′-diisopropylcarbodiimide (DIC, 0.165 mmol, 1.1 equiv.) was added dropwise via a syringe, and the mixture was stirred until the acid was consumed (determined by TLC). After consumption of all starting materials, the solvent was removed on a rotary evaporator at 35 °C under reduced pressure and dried on a high-vacuum line for at least 5 minutes to remove residual CH2Cl2. Next, the tube was moved into a glovebox. Cuprous chloride (0.01 mmol, 10 mol%) and cupric acetylacetonate (0.01 mmol, 10 mol%) were mixed in a sealed tube. Terminal alkyne (0.1 mmol, 1.0 equiv.), triethylamine (0.25 mmol, 2.5 equiv.) and anhydrous THF (2 mL) were added in sequence. A tube was set between two lamps (10 cm away from the lamp, 90 W each) and the mixture was stirred at room temperature (with a fan to cool down the reaction) for 16 h. The solvent was removed under vacuum. The crude product was purified by column chromatography or PTLC to afford the corresponding alkyne (silica, 0–100% EtOAc/petroleum ether).Conclusions
In summary, we have developed a decarboxylative cross-coupling method to access alkyl substituted alkynes. The photoexcitation of copper acetylide complexes provides a feasible strategy to generate a range of alkyl radicals from readily available carboxylic acid derivatives without the use noble metal photocatalysts, which can be successfully applied in the copper-catalysed cross-coupling reaction with a broad range of alkynes. In addition, DFT calculations revealed that NEt3 and acetylacetonate ligands are crucial for the catalytic cycle. Further study of RAE-initiated cross-coupling is underway in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Nos. 21772085, 21803030, and 21971107), the Fundamental Research Funds for the Central Universities, the National Thousand Young Talents Program, the Jiangsu Innovation & Entrepreneurship Talents Plan, and the NSF of Jiangsu Province (BK20170631) in China. The numerical calculations in this paper have been done on the computing facilities in the High Performance Computing Centres of Nanjing University and Collaborative Innovation Centre of Advanced Microstructures and Collaborative Innovation Center of Solid-State Lighting and Energy-Saving Electronics.Notes and references
- P. J. Stang and F. Diederich, Modern Acetylene Chemistry, Wiley-VCH, Weinheim, 1995 Search PubMed.
- Y. Liu, J. W. Y. Lam and B. Z. Tang, Nat. Sci. Rev., 2015, 2, 493–509 CrossRef CAS.
- J. C. Jewett and C. R. Bertozzi, Chem. Soc. Rev., 2010, 39, 1272–1279 CAS.
- P. Thirumurugan, D. Matasiuk and K. Jozwiak, Chem. Rev., 2013, 113, 4905–4979 CrossRef CAS PubMed.
- K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 16, 4467–4470 CrossRef.
- (a) M. Eckhardt and G. C. Fu, J. Am. Chem. Soc., 2003, 125, 13642–13643 CrossRef CAS PubMed; (b) W. Shi, Y. Luo, X. Luo, L. Chao, H. Zhang, J. Wang and A. Lei, J. Am. Chem. Soc., 2008, 130, 14713–14720 CrossRef CAS PubMed; (c) L. Su, J. Dong, L. Liu, M. Sun, R. Qiu, Y. Zhou and S.-F. Yin, J. Am. Chem. Soc., 2016, 138, 12348–12351 CrossRef CAS PubMed; (d) X.-Y. Cui, Y. Ge, S. M. Tan, H. Jiang, D. Tan, Y. Lu, R. Lee and C.-H. Tan, J. Am. Chem. Soc., 2018, 140, 8448–8455 CrossRef CAS PubMed; (e) X.-Y. Dong, Y.-F. Zhang, C.-L. Ma, Q.-S. Gu, F.-L. Wang, Z.-L. Li, S.-P. Jiang and X.-Y. Liu, Nat. Chem., 2019, 11, 1158–1166 CrossRef CAS PubMed.
- (a) R. Chinchilla and C. Najera, Chem. Rev., 2007, 107, 874–922 CrossRef CAS PubMed; (b) R. Chinchilla and C. Najera, Chem. Soc. Rev., 2011, 40, 5084–5121 RSC.
- A. Hossain, A. Bhattacharyya and O. Reiser, Science, 2019, 364, eaav9713 CrossRef PubMed.
- (a) X.-Y. Yu, Q.-Q. Zhao, J. Chen, J.-R. Chen and W.-J. Xiao, Angew. Chem., Int. Ed., 2018, 57, 15505–15509 ( Angew. Chem. , 2018 , 130 , 15731–15735 ) CrossRef CAS PubMed; (b) M. Claros, F. Ungeheuer, F. Franco, V. Martin-Diaconescu, A. Casitas and J. Lloret-Fillol, Angew. Chem., Int. Ed., 2019, 58, 4869–4874 ( Angew. Chem. , 2019 , 131 , 4923–4928 ) CrossRef CAS PubMed; (c) S. Guo, D. I. AbuSalim and S. P. Cook, Angew. Chem., Int. Ed., 2019, 58, 11704–11708 ( Angew. Chem. , 2019 , 131 , 11830–11834 ) CrossRef CAS PubMed; (d) Q.-Y. Meng, X.-W. Gao, T. Lei, Z. Liu, F. Zhan, Z.-J. Li, J.-J. Zhong, H. Xiao, K. Feng, B. Chen, Y. Tao, C.-H. Tung and L.-Z. Wu, Sci. Adv., 2017, 3, e1700666 CrossRef PubMed; (e) Z. Liu, H. Chen, Y. Lv, X. Tan, H. Shen, H.-Z. Yu and C. Li, J. Am. Chem. Soc., 2018, 140, 6169–6175 CrossRef CAS PubMed; (f) Y. Li, K. Zhou, Z. Wen, S. Cao, X. Shen, M. Lei and L. Gong, J. Am. Chem. Soc., 2018, 140, 15850–15858 CrossRef CAS PubMed.
- (a) S. E. Creutz, K. J. Lotito, G. C. Fu and J. C. Peters, Science, 2012, 338, 647–651 CrossRef CAS PubMed; (b) Q. M. Kainz, C. D. Matier, A. Bartoszewicz, S. L. Zultanski, J. C. Peters and G. C. Fu, Science, 2016, 351, 681–684 CrossRef CAS PubMed; (c) W. Zhao, R. P. Wurz, J. C. Peters and G. C. Fu, J. Am. Chem. Soc., 2017, 139, 12153–12156 CrossRef CAS PubMed; (d) C. D. Matier, J. Schwaben, J. C. Peters and G. C. Fu, J. Am. Chem. Soc., 2017, 139, 17707–17710 CrossRef CAS PubMed; (e) J. M. Ahn, J. C. Peters and G. C. Fu, J. Am. Chem. Soc., 2017, 139, 18101–18106 CrossRef CAS PubMed.
- (a) D. B. Bagal, G. Kachkovskyi, M. Knorn, T. Rawner, B. M. Bhanage and O. Reiser, Angew. Chem., Int. Ed., 2015, 54, 6999–7002 ( Angew. Chem. , 2015 , 127 , 7105–7108 ) CrossRef CAS PubMed; (b) A. Hossain, A. Vidyasagar, C. Eichinger, C. Lankes, J. Phan, J. Rehbein and O. Reiser, Angew. Chem., Int. Ed., 2018, 57, 8288–8292 ( Angew. Chem. , 2018 , 130 , 8420–8424 ) CrossRef CAS PubMed.
- (a) A. Sagadevan and K. C. Hwang, Adv. Synth. Catal., 2012, 354, 3421–3427 CrossRef CAS; (b) A. Sagadevan, A. Ragupathi and K. C. Hwang, Angew. Chem., Int. Ed., 2015, 54, 13896–13901 ( Angew. Chem. , 2015 , 127 , 14102–14107 ) CrossRef CAS PubMed; (c) A. Sagadevan, V. P. Charpe, A. Ragupathi and K. C. Hwang, J. Am. Chem. Soc., 2017, 139, 2896–2899 CrossRef CAS PubMed; (d) A. Sagadevan, A. Ragupathi, C.-C. Lin, J. R. Hwu and K. C. Hwang, Green Chem., 2015, 17, 1113–1119 RSC; (e) V. P. Charpe, A. A. Hande, A. Sagadevan and K. C. Hwang, Green Chem., 2018, 20, 4859–4864 RSC.
- A. Hazra, M. T. Lee, J. F. Chiu and G. Lalic, Angew. Chem., Int. Ed., 2018, 57, 5492–5496 ( Angew. Chem. , 2018 , 130 , 5590–5594 ) CrossRef CAS PubMed.
- (a) Q.-Q. Zhou, W. Guo, W. Ding, X. Wu, X. Chen, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 11196–11199 ( Angew. Chem. , 2015 , 127 , 11348–11351 ) CrossRef CAS PubMed; (b) F. Le Vaillant, T. Courant and J. Waser, Angew. Chem., Int. Ed., 2015, 54, 11200–11204 ( Angew. Chem. , 2015 , 127 , 11352–11356 ) CrossRef CAS PubMed; (c) H.-P. Bi, L. Zhao, Y.-M. Liang and C.-J. Li, Angew. Chem., Int. Ed., 2009, 48, 792–795 ( Angew. Chem. , 2009 , 121 , 806–809 ) CrossRef CAS PubMed; (d) C. Zhang and D. Seidel, J. Am. Chem. Soc., 2010, 132, 1798–1799 CrossRef CAS PubMed; (e) Y.-S. Feng, Z.-Q. Xu, L. Mao, F.-F. Zhang and H.-J. Xu, Org. Lett., 2013, 15, 1472–1475 CrossRef CAS PubMed; (f) C. Ye, Y. Li and H. Bao, Adv. Synth. Catal., 2017, 359, 3720–3724 CrossRef CAS; (g) C. Yang, J.-D. Yang, Y.-H. Li, X. Li and J.-P. Cheng, J. Org. Chem., 2016, 81, 12357–12363 CrossRef CAS PubMed.
- (a) J. T. Edwards, R. R. Merchant, K. S. McClymont, K. W. Knouse, T. Qin, L. R. Malins, B. Vokits, S. A. Shaw, D.-H. Bao, F.-L. Wei, T. Zhou, M. D. Eastgate and P. S. Baran, Nature, 2017, 545, 213–218 CrossRef CAS PubMed; (b) T. Chen, L. M. Barton, Y. Lin, J. Tsien, D. Kossler, I. Bastida, S. Asai, C. Bi, J. S. Chen, M. Shan, H. Fang, F. G. Fang, H.-w Choi, L. Hawkins, T. Qin and P. S. Baran, Nature, 2018, 560, 350–354 CrossRef CAS PubMed; (c) A. Fawcett, J. Pradeilles, Y. Wang, T. Mutsuga, E. L. Myers and V. K. Aggarwal, Science, 2017, 357, 283–286 CrossRef CAS PubMed; (d) T. Qin, J. Cornella, C. Li, L. R. Malins, J. T. Edwards, S. Kawamura, B. D. Maxwell, M. D. Eastgate and P. S. Baran, Science, 2016, 352, 801–805 CrossRef CAS PubMed; (e) L. Huang, A. M. Olivares and D. J. Weix, Angew. Chem., Int. Ed., 2017, 56, 11901–11905 ( Angew. Chem. , 2017 , 129 , 12063–12067 ) CrossRef CAS PubMed; (f) J. M. Smith, T. Qin, R. R. Merchant, J. T. Edwards, L. R. Malins, Z. Liu, G. Che, Z. Shen, S. A. Shaw, M. D. Eastgate and P. S. Baran, Angew. Chem., Int. Ed., 2017, 56, 11906–11910 ( Angew. Chem. , 2017 , 129 , 12068–12072 ) CrossRef CAS PubMed; (g) S. Ni, A. F. Garrido-Castro, R. R. Merchant, J. N. de Gruyter, D. C. Schmitt, J. J. Mousseau, G. M. Gallego, S. Yang, M. R. Collins, J. X. Qiao, K.-S. Yeung, D. R. Langley, M. A. Poss, P. M. Scola, T. Qin and P. S. Baran, Angew. Chem., Int. Ed., 2018, 57, 14560–14565 ( Angew. Chem. , 2018 , 130 , 14768–14773 ) CrossRef CAS PubMed; (h) T.-G. Chen, H. Zhang, P. K. Mykhailiuk, R. R. Merchant, C. A. Smith, T. Qin and P. S. Baran, Angew. Chem., Int. Ed., 2019, 58, 2454–2458 ( Angew. Chem. , 2019 , 131 , 2476–2480 ) CrossRef CAS PubMed; (i) F. Sandfort, M. J. O'Neill, J. Cornella, L. Wimmer and P. S. Baran, Angew. Chem., Int. Ed., 2017, 56, 3319–3323 ( Angew. Chem. , 2017 , 129 , 3367–3371 ) CrossRef CAS PubMed; (j) J. Wang, T. Qin, T.-G. Chen, L. Wimmer, J. T. Edwards, J. Cornella, B. Vokits, S. A. Shaw and P. S. Baran, Angew. Chem., Int. Ed., 2016, 55, 9676–9679 ( Angew. Chem. , 2016 , 128 , 9828–9831 ) CrossRef CAS PubMed; (k) R. Mao, J. Balon and X. Hu, Angew. Chem., Int. Ed., 2018, 57, 13624–13628 ( Angew. Chem. , 2018 , 130 , 13812–13816 ) CrossRef CAS PubMed; (l) J. Cornella, J. T. Edwards, T. Qin, S. Kawamura, J. Wang, C.-M. Pan, R. Gianatassio, M. Schmidt, M. D. Eastgate and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 2174–2177 CAS; (m) K. M. M. Huihui, J. A. Caputo, Z. Melchor, A. M. Olivares, A. M. Spiewak, K. A. Johnson, T. A. DiBenedetto, S. Kim, L. K. G. Ackerman and D. J. Weix, J. Am. Chem. Soc., 2016, 138, 5016–5019 CrossRef CAS PubMed; (n) F. Toriyama, J. Cornella, L. Wimmer, T.-G. Chen, D. D. Dixon, G. Creech and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 11132–11135 CrossRef CAS PubMed; (o) L. Candish, M. Teders and F. Glorius, J. Am. Chem. Soc., 2017, 139, 7440–7443 CrossRef CAS PubMed; (p) C. Kingston, M. A. Wallace, A. J. Allentoff, J. N. deGruyter, J. S. Chen, S. X. Gong, S. Bonacorsi and P. S. Baran, J. Am. Chem. Soc., 2019, 141, 774–779 CrossRef CAS PubMed; (q) S. Ni, N. M. Padial, C. Kingston, J. C. Vantourout, D. C. Schmitt, J. T. Edwards, M. M. Kruszyk, R. R. Merchant, P. K. Mykhailiuk, B. B. Sanchez, S. Yang, M. A. Perry, G. M. Gallego, J. J. Mousseau, M. R. Collins, R. J. Cherney, P. S. Lebed, J. S. Chen, T. Qin and P. S. Baran, J. Am. Chem. Soc., 2019, 141, 6726–6739 CrossRef CAS PubMed; (r) H. Li, C. P. Breen, H. Seo, T. F. Jamison, Y.-Q. Fang and M. M. Bio, Org. Lett., 2018, 20, 1338–1341 CrossRef CAS PubMed; (s) H. Zhang, P. Zhang, M. Jiang, H. Yang and H. Fu, Org. Lett., 2017, 19, 1016–1019 CrossRef CAS PubMed; (t) J. Yang, J. Zhang, L. Qi, C. Hu and Y. Chen, Chem. Commun., 2015, 51, 5275–5278 RSC; (u) C. R. Jamison and L. E. Overman, Acc. Chem. Res., 2016, 49, 1578–1586 CrossRef CAS PubMed.
- For computational details, see the ESI.‡.
- P. Xiao, C. X. Li, W. H. Fang, G. Cui and W. Thiel, J. Am. Chem. Soc., 2018, 140, 15099–15113 CrossRef CAS PubMed.
- J. W. Hilborn and J. A. Pincock, J. Am. Chem. Soc., 1991, 113, 2683–2686 CrossRef CAS.
- (a) C. Le, T. Q. Chen, T. Liang, P. Zhang and D. W. C. MacMillan, Science, 2018, 360, 1010–1014 CrossRef CAS PubMed; (b) A. Tlahuext-Aca, L. Candish, R. A. Garza-Sanchez and F. Glorius, ACS Catal., 2018, 8, 1715–1719 CrossRef CAS; (c) Y. Ye and M. S. Sanford, J. Am. Chem. Soc., 2012, 134, 9034–9037 CrossRef CAS PubMed.
- F. D. Lu, D. Liu, L. Zhu, L. Q. Lu, Q. Yang, Q. Q. Zhou, Y. Wei, Y. Lan and W. J. Xiao, J. Am. Chem. Soc., 2019, 141, 6167–6172 CrossRef CAS PubMed.
- J. Jover, J. Chem., 2015, 2015, 1–8 CrossRef.
- (a) P. Leophairatana, S. Samanta, C. C. De Silva and J. T. Koberstein, J. Am. Chem. Soc., 2017, 139, 3756 CrossRef CAS PubMed; (b) D. K. Das, V. K. Kumar Pampana and K. C. Hwang, Chem. Sci., 2018, 9, 7318 RSC.
Footnotes |
| † Dedicated to the 100th anniversary of the School of Chemistry and Chemical Engineering, Nanjing University. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc02213f |
| This journal is © The Royal Society of Chemistry 2020 |