
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective, radical-free activation of benzylic C–H bonds in methylarenes†
Antony P. Y.
Chan
a,
Martin
Jakoobi
 a,
Chenxu
Wang
a,
Yancong
Tian
a,
Nathan
Halcovitch
b,
Roman
Boulatov
a and
Alexey G.
Sergeev
*a
a,
Chenxu
Wang
a,
Yancong
Tian
a,
Nathan
Halcovitch
b,
Roman
Boulatov
a and
Alexey G.
Sergeev
*a
aDepartment of Chemistry, University of Liverpool, Crown Street, Liverpool, L69 7ZD, UK. E-mail: sergeev@liverpool.ac.uk
bDepartment of Chemistry, Lancaster University, Bailrigg, Lancaster, LA1 4YW, UK
First published on 13th July 2021
Abstract
We report rare examples of exclusive benzylic C–H oxidative addition in industrially important methylarenes using simple η4-arene iridium complexes. Mechanistic studies showed that coordinatively unsaturated η2-arene intermediates are responsible for the selective activation of benzylic, not aromatic C–H bonds and formation of stable benzyl complexes after trapping with a phosphine ligand.
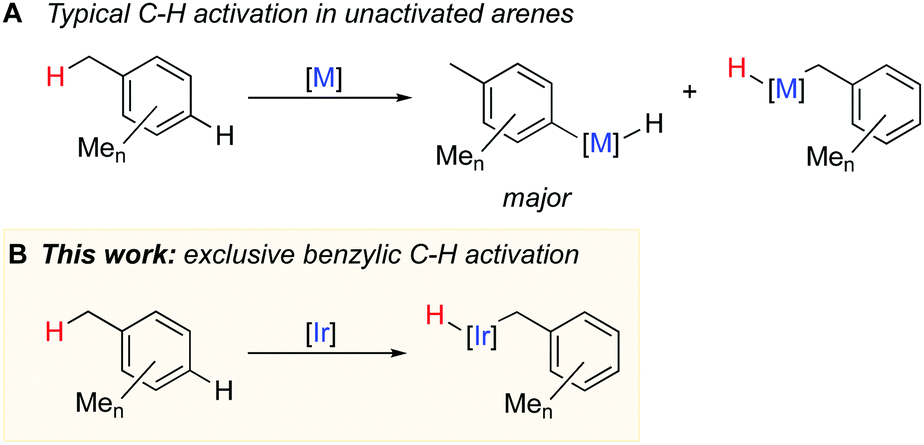
Syntheses of many fine and speciality chemicals rely on the C–H functionalisation of alkylarenes.1 However, such C–H functionalisation without directing groups occurs unselectively and yields mixtures of products. Substantial progress has recently been made in selective functionalisation of unactivated arene Csp2–H bonds using transition metal catalysts,2,3 but the selective activation of benzylic Csp3–H bonds remains challenging1,4 because the competing activation of the aromatic C–H bonds is generally thermodynamically and kinetically preferred (Fig. 1A).5–8 Known examples of catalytic functionalisation of benzylic C–H bonds typically involve radical intermediates and stoichiometric amounts of peroxides, or strong bases, which limit the synthetic utility of the methods.1,4 Catalytic activation of benzylic C–H bonds by metal complexes in the absence of radical or basic reagents occurs much less common and often suffers from selectivity issues.9–15
The key step for designing general and selective catalytic benzylic C–H functionalisation is selective activation, e.g. oxidative addition of a benzylic C–H bond in a range of unactivated alkylarenes. However, only few complexes mediate oxidative addition of benzylic C–H bonds and, in most cases, it is accompanied by oxidative addition of aromatic C–H bonds. Selective benzylic C–H oxidative addition occurs only with methylarenes with sterically hindered arene C–H bonds such as mesitylene (1,3,5-trimethylbenzene)16 or p-xylene;17 other arenes give mainly aromatic C–H activation products. Hence, the problem of highly selective benzylic C–H activation remains to be addressed.
Here we report the first general metal system that mediates exclusive benzylic C–H activation in nine unactivated arenes bearing from one to five methyl groups in the aromatic ring (Fig. 1B). Our kinetic and computational studies suggest that the key to the unique selectivity is generation of coordinatively unsaturated η2-arene intermediate, which preferentially activates benzylic, not aromatic C–H bonds in methyl arenes.
 | ||
| Fig. 1 Metal-mediated C–H activation (oxidative addition) by metal complexes. | ||
During our investigation of arene C–C cleavage in rare Cp*Ir(η4-arene) complexes,18,19 we found that thermolysis of the mesitylene complex in the presence of PMe3 does not lead to C–C cleavage, but cleanly affords a product of benzylic C–H activation, Cp*Ir(CH2C6H3Me2-3,5)(PMe3).20 This selective benzylic C–H activation, however, could be explained by the steric hindrance of aromatic C–H bonds. To test if benzylic C–H activation would occur in the least sterically hindered methylarene, toluene, we heated the iridium complex Cp*Ir(η4-toluene) (1) in the presence of 4 equiv. of PMe3 in n-hexane at 100 °C for 1 h. Remarkably, all three isomers of the starting complex smoothly converted into a single benzylic C–H activation product (2) in a 97% isolated yield (Fig. 2A). We believe this is the first example of exclusive benzylic C–H oxidative addition in an unhindered methylarene. Nearly quantitative yields of 2 were also obtained upon thermolysis of 1 in the presence of 4 equiv. of more sterically hindered phosphines such as PPh3 and PCy3 (Fig. 2A).21 PMe3 was further used as the phosphine choice because the corresponding product 2 can be more conveniently isolated by the simple removal of the excess of the volatile phosphine in vacuo.
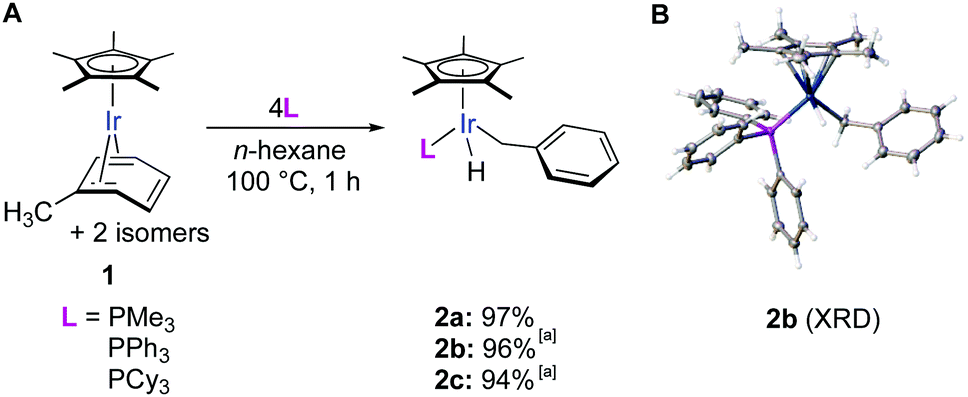 | ||
| Fig. 2 (A) Iridium-mediated selective benzylic C–H activation of toluene upon thermolysis of 1 in the presence of phosphines. (B) Crystal structure of 2b with displacement ellipsoids drawn at 50% probability level except for H atoms. Hydride ligand was modelled for clarity. [a] 1H NMR yield. | ||
The structure of 2b was confirmed by X-ray diffraction (Fig. 2A) and the hydride ligand, which was not explicitly detected by XRD, was observed in the 1H NMR spectrum as a doublet at −16.4 ppm.
Building on the successful result with toluene (Fig. 2), we explored the scope of benzylic C–H activation in a range of methylarenes bearing from two to five methyl groups (Fig. 3). The thermolysis of the corresponding η4-arene complexes in n-hexane at 100 °C for 1–48 h gave C–H oxidative addition products 3–10 in high yields (84–93%). As shown in Fig. 3, the reactivity of the coordinated arenes strongly depends on the accessibility of methyl groups: arenes with unhindered methyl groups fully reacted within 1 h (2a, 4 and 5), with methyl groups having one or two neighbouring substituents − 2 h (3, 8 and 9) and 3 h (7), respectively. Ultimately, the pentamethylbenzene complex [Cp*Ir (η4-C6Me5H)] was consumed within 48 h (10), while its fully substituted congener was the least reactive and reacted to only 45% conversion after 168 h.22
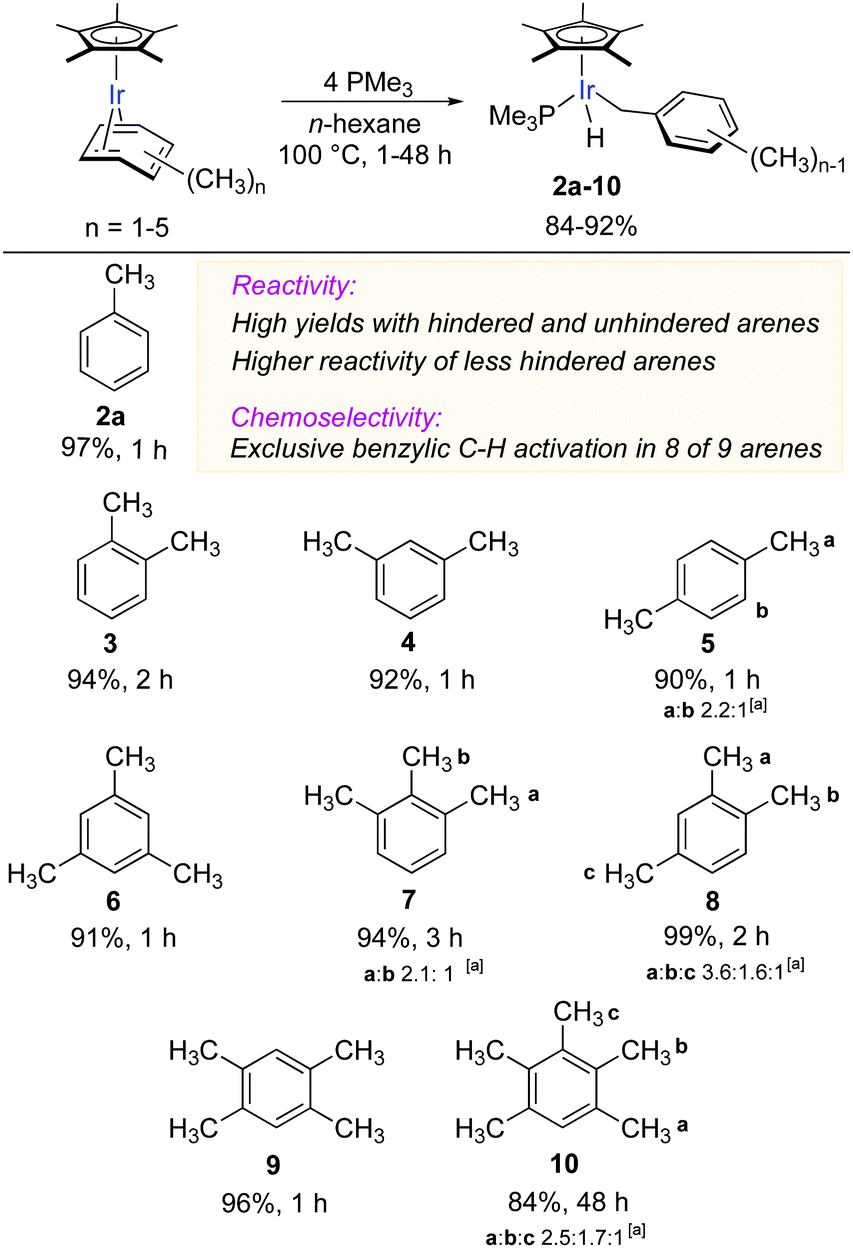 | ||
| Fig. 3 Scope of iridium-mediated benzylic C–H activation in coordinated methyl arenes. All yields reported are isolated yields. [a] Ratio of regioisomers. | ||
Remarkably, the C–H activation occurred with high chemoselectivity, affording exclusively benzylic complexes in eight examples out of nine (Fig. 3). The only exception was the activation of p-xylene (5), which gave a 69![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :31 ratio of benzylic to aromatic C–H oxidative addition products.23 To the best of our knowledge, this is the most general and selective system for oxidative addition of benzylic C–H bonds in alkyl arenes.
:31 ratio of benzylic to aromatic C–H oxidative addition products.23 To the best of our knowledge, this is the most general and selective system for oxidative addition of benzylic C–H bonds in alkyl arenes.
Another remarkable aspect of this benzylic C–H activation is that the regioselectivity does not appear to depend on steric effects. In case of 1,2,3-trimethylbenzene, 7, (Fig. 3), the regioselectivity for activation of either of the two less hindered CH3 groups vs. the more hindered internal CH3 equals 2.1:1, which after statistical correction for the number of methyl groups gives nearly equal regioselectivity of 1.1:1. Similarly, in pentamethylbenzene all three types of methyl groups have similar reactivity as evidenced by the statistically adjusted regioselectivity of 1.3:0.9:1 (10). More surprisingly, in 1,2,4-trimethylbenzene either of the more hindered CH3 groups is greater than two times more reactive than the sterically unhindered CH3 group.24 This regioselectivity contrasts to known C–H activations in alkylarenes, where the least sterically hindered aromatic C–H bonds are cleaved preferentially.
To get insight into the mechanism, we first conducted kinetic studies using thermolysis of mesitylene complex Cp*Ir(η4-mesitylene) (11) and PMe3 as a model reaction (Fig. 4A). The reason for this choice is that 11 and product 6 exist as single isomers, which simplifies monitoring and interpretation of the data. The thermolysis was conducted in cyclohexane-d12 at 75 °C and monitored by 1H NMR. We measured the initial rates of the reaction by varying the concentration of PMe3 and 11 independently. The rate of the reaction was first order in [11] and zero-order in [PMe3] (Fig. S3 and S5, ESI†). Analogous tentative data were obtained for PPh3 and PCy3. Note that although the variation of the concentration of these bulky phosphines did not change the rate of the reaction, the use of 1 equiv. instead of 4 equiv. of the phosphine relative to 11 led to the formation of the C–C cleavage side products.25
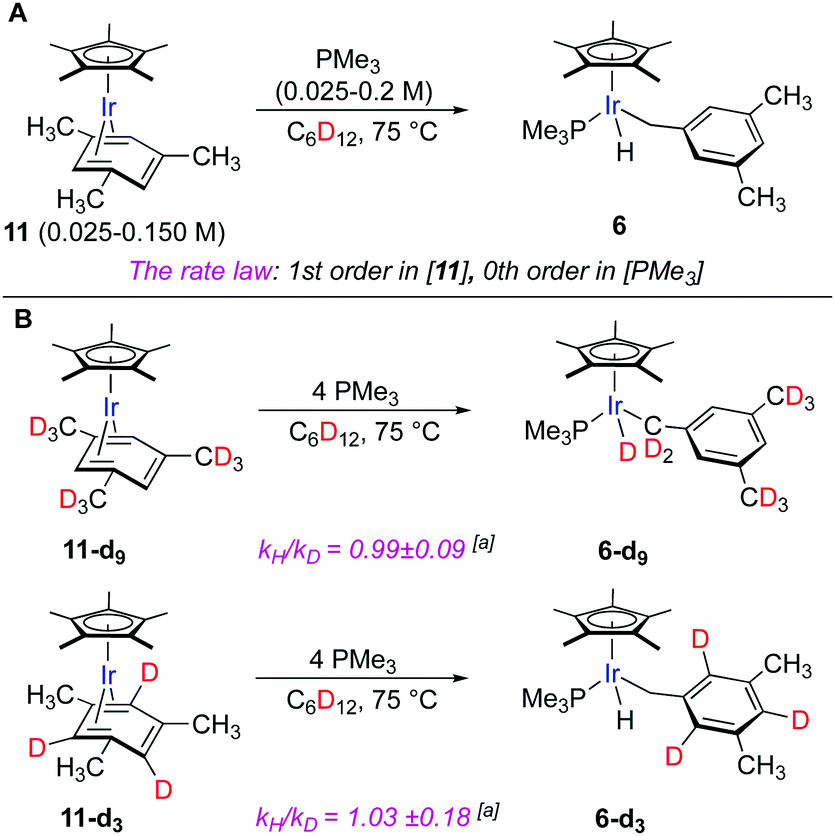 | ||
| Fig. 4 (A) The model reaction to determine reaction order in Cp*Ir(η4-mesitylene) (11) and PMe3. (B) Thermolysis of 11-d9 and 11-d3: kinetic isotope effects and the lack of H/D scrambling. [a] KIEs were calculated from the initial rates for separate reactions of the deuterated and non-deuterated species. | ||
The observed preferential activation of weaker benzylic C–H bonds may suggest the involvement of radical pathways.4 One of the common tests for the participation of the radical mechanisms is the addition of radical traps such as TEMPO that inhibits radical reactions.26 We probed this scenario by conducting thermolysis of 11 in the presence of 1 equiv. of TEMPO at 100 °C for 1 h. Full consumption of 1 was observed and the expected hydride product 6 was obtained in a 14% yield, which suggests that the involvement of radical intermediates is unlikely. The lower yield of 6 in the presence of the radical trap as compared to that in its absence (91%, Fig. 3) might be explained by the TEMPO-induced decomposition of 6 under reaction conditions as confirmed by control experiments.27,28 The lack of involvement of radicals in the benzylic C–H oxidative addition also agrees with the absence of observable incorporation of deuterium from the solvent upon thermolysis of 6 in cyclohexane-d12 (Fig. 4A).
To confirm that the hydride ligand in 6 results from the C–H oxidative addition of the methyl groups of the arene ligand, we heated the model complex 11-d9 having fully deuterated methyl groups on the aryl ligand (Fig. 4B). This led to the exclusive formation of the deuteride product 6-d9. Heating of the model complex 11-d3 bearing deuterium labels on the three aromatic positions predictably gave hydride complex 6-d3. No observable H/D scrambling was detected in either case. Kinetic isotope effect measurements conducted in separate experiments with 11-d3 and 11-d9 complexes and the nondeuterated 11 resulted in negligible KIEs (kH/kD = 1.03 ± 0.18 and 0.99 ± 0.09), which suggests that the rate-determining step does not involve benzylic or aromatic C–H bond cleavage (Fig. S7 and S9, ESI†).
To identify the mechanism that agrees with the observed experimental data and explains the unusual chemoselectivity, we computed a range of energy pathways for benzylic and aromatic C–H activation in 6. We conducted all geometry optimizations, reaction path calculations and calculations of thermodynamic corrections with the B3LYP functional and a mixed basis set of LANL2DZ for Ir and 6-31G(d) for all other atoms, which gave excellent results for the calculations of C–C oxidative addition in η4-arene iridium complexes.20
The DFT computations indicated that the benzylic C–H activation 11 → 6 (Fig. 5, green path) has indeed the lowest energy as compared to the aromatic C–H activation 11→ 17 (Fig. 5, red and blue paths). The benzylic C–H oxidative addition 11 → 6 involves the initial rate-determining η4 to η2 arene ring slippage in the starting complex over the barrier of 24.4 kcal mol−1. The resulting η2-arene intermediate 12 undergoes facile C–H activation of a methyl group to form an η3-benzyl complex 13 that is subsequently trapped by PMe3 to yield the isolated benzylic complex 6. The benzylic C–H activation preferentially occurs via the phosphine-free η2-complex 12, rather than via phosphine ligated complex 14 because the latter is much less kinetically accessible from the starting complex 11 either directly or via12 (Fig. 5).
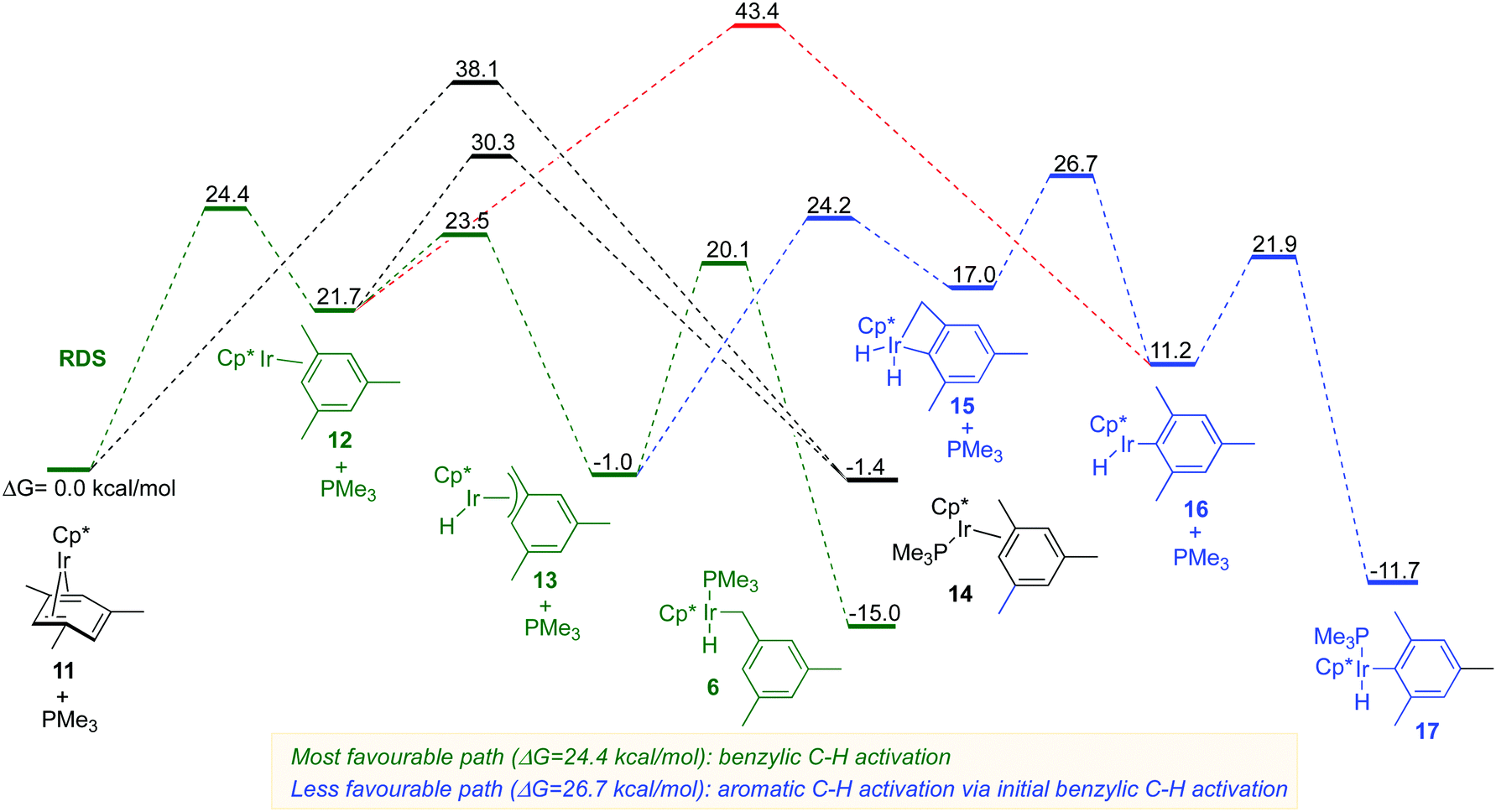 | ||
| Fig. 5 The calculated mechanisms for benzylic and aromatic C–H activation in 11 in the presence of added PMe3 ligand. Energies at M06-L/(6-311+G*+LANL2TZ/cpcm = hexane)//B3LYP/(6-31G*+LANL2DZ) and thermodynamic corrections at 50 °C. | ||
All paths for aromatic C–H activation 11 → 17 also involve the η2-arene complex 12 as an essential intermediate. The direct aromatic C–H oxidative addition 12 → 16 requires a 20.1 kcal mol−1 higher barrier than the corresponding benzylic C–H activation 12 → 13 (Fig. 5; compare red and green paths). A much more favourable path to 17 involves the initial benzylic C–H activation 12 → 13 followed by the aromatic C–H activation yielding the dihydride intermediate 15. The dihydride 15 reforms the benzylic C–H bond and the resulting coordinatively unsaturated aryl hydride 16 picks up PMe3 to give the final product 17. This lowest energy aromatic C–H oxidative addition still has a 6.6 kcal mol−1 higher barrier than the benzylic C–H activation path (Fig. 5, compare 13 → 16vs.13 → 6).
The results of DFT calculations indicate that the generation of a phosphine-free Cp*Ir(η2-arene) intermediate is the key to the exclusive regioselectivity of benzylic C–H activation in η4-arene iridium complexes (Fig. S12A, ESI†). That is, the Cp*Ir fragment selectively activates benzylic C–H bonds in the η2-methyl arene ligand and then picks up a phosphine ligand to afford the final product (Fig. S12A, ESI†). The reactivity of this Cp*Ir fragment is orthogonal to the reactivity of the corresponding well-known phosphine-ligated species, Cp*Ir(PMe3), which preferentially activates aromatic C–H bonds as reported by Jones29 and Bergman (Fig. S12B, ESI†).30
These Cp*Ir(η2-arene) intermediates have unique reactivity not only in the benzylic C–H activation. As we showed previously, when these species are generated in the absence of an added ligand, they promote oxidative addition of the aromatic C–C bonds.7,8 The starting Cp*Ir(η4-arene) complexes serve as excellent precursors for the generation of these highly reactive intermediates.
In summary, we reported an unprecedented highly selective oxidative addition of benzylic C–H bonds in a range of methylarenes, which occurs in simple Cp*Ir(η4-arene) complexes. Experimental and computational data suggest that the process occurs via the rate-determining formation of highly reactive η2-arene iridium species that undergo selective oxidative addition of benzylic C–H bonds. The unique reactivity of the η2-arene iridium species in benzylic C–H activation provides an exciting starting point for the design of novel highly selective functionalisations, which are currently investigated in our laboratory.
We thank the Leverhulme Trust, UK (Grant RPG-2018-406), EPSRC Early Career Fellowship (EP/L000075/1 to R. B.) and Petroleum Research Fund (58885-ND7 to R. B.) for financial support. Computations reported here were performed in the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number ACI-1548562, with computational resources provided by the SDSC under allocation TG-CHE140039. Analytical services were provided by University of Liverpool.
Conflicts of interest
There are no conflicts to declare.Notes and references
- R. Vanjari and K. N. Singh, Chem. Soc. Rev., 2015, 44, 8062–8096 RSC.
- P. Wedi and M. van Gemmeren, Angew. Chem., Int. Ed., 2018, 57, 13016–13027 CrossRef CAS PubMed.
- N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 10236–10254 CrossRef CAS PubMed.
- R. Yazaki and T. Ohshima, Tetrahedron Lett., 2019, 60, 151225 CrossRef.
- W. D. Jones and F. J. Feher, J. Am. Chem. Soc., 1982, 104, 4240–4242 CrossRef CAS.
- P. E. M. Siegbahn, J. Phys. Chem., 1995, 99, 12723–12729 CrossRef CAS.
- W. D. Jones and E. T. Hessell, J. Am. Chem. Soc., 1993, 115, 554–562 CrossRef CAS.
- E. Clot, C. Mégret, O. Eisenstein and R. N. Perutz, J. Am. Chem. Soc., 2006, 128, 8350–8357 CrossRef CAS PubMed.
- S. Shimada, A. S. Batsanov, J. A. K. Howard and T. B. Marder, Angew. Chem., Int. Ed., 2001, 40, 2168–2171 CrossRef CAS PubMed.
- T. Ishiyama, K. Ishida, J. Takagi and N. Miyaura, Chem. Lett., 2001, 1082–1083 CrossRef CAS.
- M. A. Larsen, C. V. Wilson and J. F. Hartwig, J. Am. Chem. Soc., 2015, 137, 8633–8643 CrossRef CAS PubMed.
- W. N. Palmer, J. V. Obligacion, I. Pappas and P. J. Chirik, J. Am. Chem. Soc., 2016, 138, 766–769 CrossRef CAS PubMed.
- C. M. Kelly, J. T. Fuller, C. M. Macaulay, R. McDonald, M. J. Ferguson, S. M. Bischof, O. L. Sydora, D. H. Ess, M. Stradiotto and L. Turculet, Angew. Chem., Int. Ed., 2017, 56, 6312–6316 CrossRef CAS PubMed.
- T. Furukawa, M. Tobisu and N. Chatani, Chem. Commun., 2015, 51, 6508–6511 RSC.
- K. Mertins, A. Zapf and M. Beller, J. Mol. Catal. A: Chem., 2004, 207, 21–25 CrossRef CAS.
- Y. Zhu, L. Fan, C.-H. Chen, S. R. Finnell, B. M. Foxman and O. V. Ozerov, Organometallics, 2007, 26, 6701–6703 CrossRef CAS.
- P. Burger and R. G. Bergman, J. Am. Chem. Soc., 1993, 115, 10462–10463 CrossRef CAS.
- M. Jakoobi, N. Halcovitch, G. F. S. Whitehead and A. G. Sergeev, Angew. Chem., Int. Ed., 2017, 56, 3266–3269 CrossRef CAS PubMed.
- M. Jakoobi, Y. Tian, R. Boulatov and A. G. Sergeev, J. Am. Chem. Soc., 2019, 141, 6048–6053 CrossRef CAS PubMed.
- Y. Tian, M. Jakoobi, R. Boulatov and A. G. Sergeev, Chem. Sci., 2021, 12, 3568–3579 RSC.
- Note that the reaction also proceeds smoothly with just one equivalent of PMe3 but C–C cleavage is not fully suppressed in the case of the larger phosphines.
- Heating Cp*Ir(η4-C6Me6) and PMe3 at 150 °C for 48 h led to the full conversion of the starting complex but gave a complex mixture of products. See the ESI†.
- This selectivity is reverse to that of the Cp*Ir(PMe3) species generated in situ from Cp*Ir(PMe3)H2, which reacts with p-xylene to afford the corresponding benzylic and aromatic C H activated products in a 29:71 ratio, respectively. See ref. 30.
- Although the origin of this selectivity has yet to be investigated, the selectivity results from kinetic, not thermodynamic control as evidenced by the DFT calculations of the relative stability of the corresponding products, which scale as 8c > 8a > 8b (Fig. S11).
- See the ESI†.
- K. Matyjaszewski, Macromolecules, 1998, 31, 4710–4717 CrossRef CAS PubMed.
- A. C. Albeniz, P. Espinet, R. Lopez-Fernandez and A. Sen, J. Am. Chem. Soc., 2002, 124, 11278–11279 CrossRef CAS PubMed.
- Heating 6 in the presence of 1 equiv. of TEMPO at 100 °C for 1 h led to 83% degradation of 6. Please see the ESI.† TEMPO-induced decomposition of related palladium benzyl hydride complexes is described in ref 27.
- W. D. Jones and F. J. Feher, J. Am. Chem. Soc., 1984, 106, 1650–1663 CrossRef CAS.
- A. H. Janowicz and R. G. Bergman, J. Am. Chem. Soc., 1983, 105, 3929–3939 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2090576 (2b). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cc03445f |
| This journal is © The Royal Society of Chemistry 2021 |