
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Impact of subtle change in branched amino acid on the assembly and properties of perylene bisimides hydrogels†
Jacquelyn G.
Egan
 a,
Glen
Brodie
a,
Daniel
McDowall
a,
Andrew J.
Smith
b,
Charlotte J. C.
Edwards-Gayle
b and
Emily R.
Draper
*a
a,
Glen
Brodie
a,
Daniel
McDowall
a,
Andrew J.
Smith
b,
Charlotte J. C.
Edwards-Gayle
b and
Emily R.
Draper
*a
aSchool of Chemistry, University of Glasgow, Glasgow, G12 8QQ, UK. E-mail: Emily.Draper@glasgow.ac.uk
bDiamond Light Source Ltd, Harwell Science and Innovation Campus, Didcot, OX11 0QX, UK
First published on 8th July 2021
Abstract
We investigate how apparent slight changes to the chemical structure of amino acid-functionalised perylene bisimides (PBIs) affect the self-assembled aggregates formed and their resulting physical and optical properties. PBIs functionalised with L-valine (PBI-V), L-leucine (PBI-L) and L-isoleucine (PBI-I) are investigated due to their similarly branched structure and their assemblies in water were studied using UV-vis absorption spectroscopy, cyclic voltammetry (CV), small angle X-ray scattering (SAXS) and viscosity at different pHs. It was seen that each PBI behaved differently. Each of the PBIs were then used to prepare hydrogels, and their properties again assessed, with PBI-I forming different hydrogels than the other PBIs. By understanding how slight changes in chemical structure can affect bulk properties we become a step closer to designing gels with specific physical and electrical properties.
Introduction
There has been an increased interest in the use of organic semi-conductors (OSCs) as precious metal supplies deplete. OSCs can be used in organic field transistors (OFETs), organic light emitting diodes (OLEDs) or organic photovoltaics (OPVs). There are many examples of OSCs, including single crystals,1–3 polymers,4–7 single molecules,8–10 and low molecular weight gelators (LMWGs).11,12 Of those listed, LMWGs stand out as their self-assembly can be changed using different stimulus to fine tune their properties.11,13,14 They undergo a hierarchy of self-assembly in which 1D structures form then further assemble to eventually entangle into a 3D network. With many of these systems, slight changes in chemical structure, assembly conditions and kinetics can result in materials with very different morphological, optical, and mechanical properties, for example.15 Understanding and controlling what affects the self-assembly process will allow for improved design of materials and even self-assembly prediction.The use of perylene bisimides (PBIs) OSCs as LWMGs has shown promise.16–20 Their highly conjugated π-system allows them to stack into two types of aggregates, generally H-type and J-type.21,22 They have also shown n-type semiconductor behavior and have good photoconductivity, due to their high light absorptivity and ability to readily form a radical anion making them interesting materials for OPVs.23–25 Properties of the PBIs such as solubility,26 electrical,27,28 and colour,29 can be modified through chemical structure changes, whilst self-assembly of PBIs can be modified using environmental stimulus such as solvent,30 concentration31 or temperature.17 The structures of PBIs can be modified at three main positions, the bay, ortho and imide position.32–34 Of the three positions, the imide position is easiest to modify through a common one pot synthesis, without the need for organic solvents or catalysts.
PBIs that have been functionalised at the imide position with amino acids have been used as LMWGs.35–38 They have the advantage of being soluble in water, form 1D structures at high pH, and can gel at low pH.36 We have previously shown that uniform hydrogels using amino-PBIs can be made using a glucono-δ-lactone (GdL) acid trigger.35 These hydrogels were then irradiated to form a highly stable conductive radical anion whilst still retaining their structure as hydrogels.36 Furthermore, we showed that by changing the amino acid used for functionalisation we could vary the amount of radical produced. We deduced this was due to different assemblies being formed rather than the electronic properties of the molecules themselves. Understanding these changes in self-assembly and the changes in properties is the key to design better OSCs and functional organic materials in general.
The impact of changes in molecular structure to the self-assembly properties of PBIs has been investigated.22,39 The self-assembly of a PBI functionalized with (D/L) phenylalanine isomers was investigated and found that they self-assembled into supramolecular helices via intermolecular hydrogen bonding rather than π–π stacking.40 The different chirality also led to the assembly of clockwise or anticlockwise helices depending on the isomers used. Others have seen two PBI derivatives modified with different side chains at the imide nitrogen has a major impact on aggregates formed.41 While one structure adopted both H and J aggregations, the other only adopted H aggregation. These two PBIs also saw an increase in conductivity for nanobelts formed in methanol compared to nanoleaves formed in hexane. By studying the factors that can impact the self-assembly we can use these to tune and understand the properties for better functional material design.
Herein we are investigating how seemingly small changes to the molecular structure of amino acid functionalised PBIs impacts the self-assembly and bulk properties. Specifically, we are looking at three very chemically and structurally similar imide functionalised perylene bisimides. We have used three branched amino acids, L-valine, L-leucine and L-isoleucine, which we call PBI-V, PBI-L, and PBI-I, respectively (Fig. 1). We look at the differences in the self-assembly process and resulting properties of aqueous solutions and gels at various pHs and concentrations using UV-vis absorption spectroscopy, cyclic voltammetry (CV), small angle X-ray scattering (SAXS), rheology and 1H-NMR.
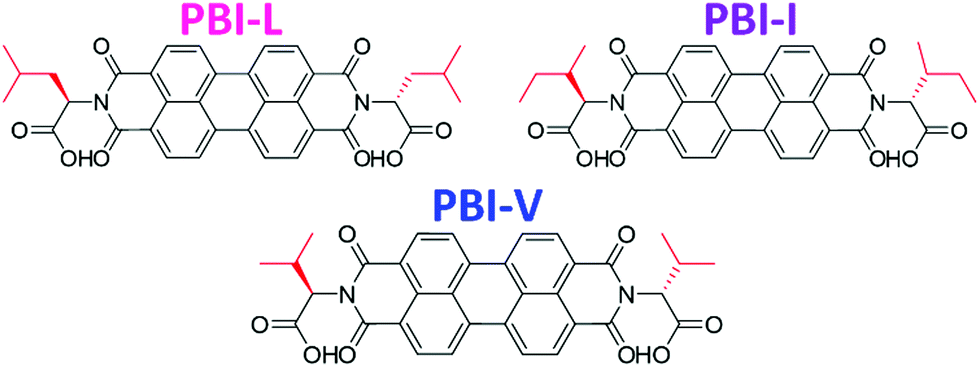 | ||
| Fig. 1 Structures of the PBIs used in this study with the different branched groups highlighted in red. | ||
Results and discussion
All PBIs were synthesized using a previously published method. We have previously reported PBI-L and PBI-V35,36,42,43 as have other groups.44–49PBI-I has not been synthesised in our group and all characterisation data is in the ESI† Section 3.1, (Fig. S1–S4).Each of the PBIs were prepared in water at a concentration of 10 mg mL−1 with 1 equivalent of 0.1 M NaOH. The assembly of the PBIs in solution at different pHs and concentrations were then investigated using viscosity, UV-vis absorption spectroscopy, small angle X-ray scattering (SAXS) and cyclic voltammetry (CV). Hydrogels of each of the PBIs were then formed using a change in pH. Again, the assembly, optical, electrical, and mechanical properties were compared between each other.
Firstly, the aggregation in water at pH 7 was examined by UV-vis absorption spectroscopy, then the PBIs ability to form the radical anion with UV light. The absorption for before and after irradiation was measured for each solution. The normalised spectra are shown in Fig. 2. Each of the three PBIs have similar peak positions at 510 and 545 nm with a ratio of 0.85 for the S0–S1 peaks, summarised in Table S2 (ESI†), suggesting that they have similar H-aggregated molecular packing. PBI-V however, has a slightly more pronounced shoulder at 480 nm, suggesting a slight difference in packing. All three PBIs show the characteristic three peaks relating to the radical anion formation at 725, 815 and 980 nm. PBI-V forms the most amount of radical after 5 minutes of irradiation which agrees with data previously collected on these systems at 5 mg mL−1.42
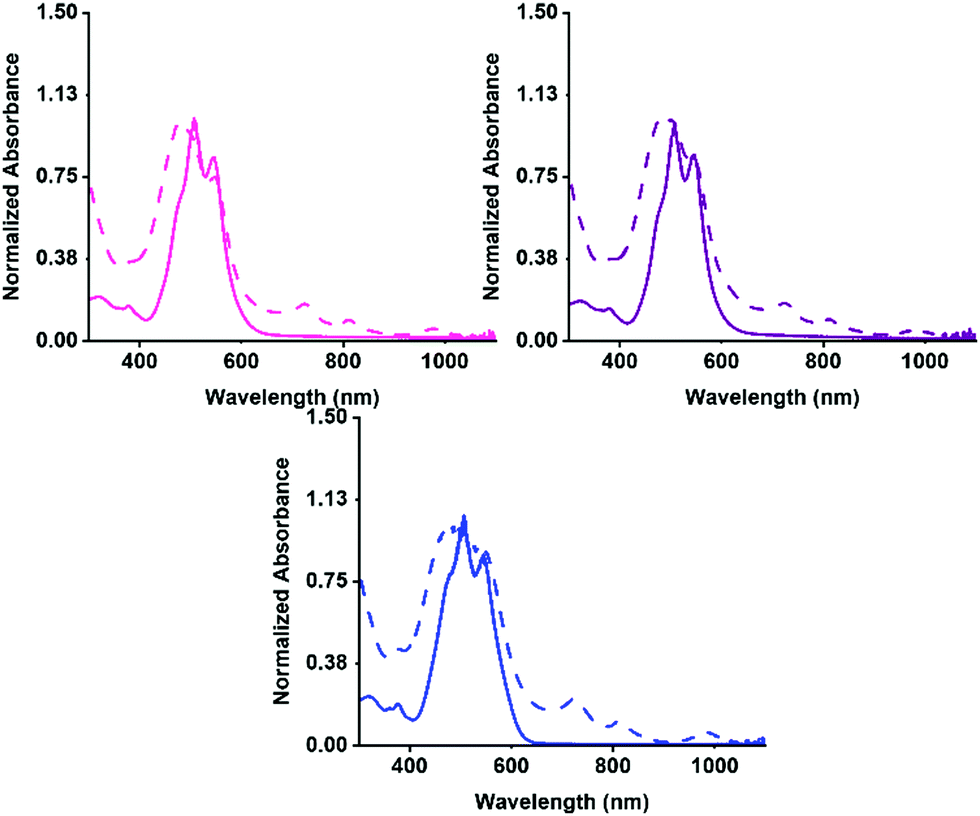 | ||
| Fig. 2 UV-vis absorption spectra of PBI-L (solid pink), PBI-I (solid purple), PBI-V (solid blue) as at pH 7 and 10 mg mL−1 solutions. UV-Vis absorption spectra of after being irradiated for 5 minutes using a 365 nm LED light for PBI-L (dashed pink), PBI-I (dashed purple), PBI-V (dashed blue). | ||
The UV-vis absorption spectra of the PBIs at different concentrations (Fig. S8–S16, ESI†) was collected to determine their molar absorptivity and displayed on the figures. PBI-V was shown to have the highest absorptivity, which agrees with it producing the most amount of radical anion when irradiated with UV light. Next, the absorption spectra of PBIs at different pHs were compared (Fig. S17–S19, ESI†). All these systems become more aggregated at lower pH due to a change in solubility.50 At pH 5, PBI-L and PBI-V begin to change their aggregation, whilst PBI-I changed aggregation at pH 6. This suggests that the PBIs have different pKas as they are starting to assemble differently or form different aggregates to each other.
As demonstrated from the data above, pH influences the self-assembly of the PBIs, most notably for PBI-I, so this was investigated further. The apparent pKa of each PBI was measured through a titration with a 0.1M HCl solution (Fig. S20 and S21, ESI†). Apparent pKa titrations showed PBI-I had an apparent pKa of 6.3 whilst PBI-L and PBI-V had apparent pKa of 6.0 and 5.9 respectively. The difference in the pKa values explains the differences in assembly at different pHs. It seems that PBI-I forms different assemblies to PBI-L and PBI-V despite the similarity in chemical structure due to it having a higher pKa. This has been seen with dipeptide gelators where the amino acid sequence was reversed and led dramatically different behaviors in the two structures.51
To determine the differences in the resulting aggregates, the dynamic viscosity of the PBI solutions at different concentrations and different pHs were measured (Fig. S23–S28, ESI†). The viscosity for the PBIs at different concentrations all showed shear thinning, suggesting the presence of worm-like micelles, or fibrous type structures which we have previously reported.35,37,42,52,53 The viscosity of the PBI solutions at different pHs showed that as the pH is lowered the solutions become more viscous, which is expected as they begin to form longer, thicker fibers which can entangle. It was seen at pH 5 PBI-I formed the most viscous solution. Again, this indicates the formation of different aggregates, which could be different in morphology, length, or abundance than for PBI-V and PBI-L.
We next used SAXS to examine solutions at different pHs. Full fitting parameters for the PBIs are found in Section 2.4 of the ESI† (Fig. S29–S31). At pH 7 PBI-L fit a flexible elliptical cylinder combined with a sphere model indicating that there was a mixture of assembled structures present. Whilst PBI-I and PBI-V fit flexible cylinder and power law model. The SAXS data agrees with the absorption and viscosity data, that they all have fibrous structures present and form different types of aggregates to each other.
Scattering at different pHs were also measured to see the impact of pH on the structures. At higher pH, the scattering fit best to a spherical model combined with a power law. All three PBIs could be modelled using a sphere model, therefore it is expected that there are spherical micelles at this pH. PBI-V had the largest sphere radius of 3.83 nm compared to the other two PBIs, meaning it formed larger spheres at this pH. Generally, at lower pH, a flexible elliptical cylinder models combined with either a power law or a sphere model could fit all solutions, again confirming that the structures form fibre like structures upon assembly. No suitable model was found for PBI-L at pH 5 and pH 6, or PBI-I at pH 5, this could be attributed to a transition of structures around their pKas, making it difficult to fit to a specific model or combination of models.
To determine whether this difference in aggregation affected the reduction potentials of the assembled materials, the reduction potentials of the different PBIs in solution were collected using CV (Fig. S32–S34, ESI†). The reduction potentials are shown in Table S10 (ESI†). The reduction potentials for PBI-L and PBI-V are slightly more similar to each other than for PBI-I. The different chemical structure is not impacting the HOMO and LUMO here, but instead morphology is playing a role, especially in mobility of the structures leading to different reduction potentials. At pH 7 the peak positions for the reduction potentials are similar for all three PBIs, which is not surprising as each of the PBIs were quite structurally similar at this pH. The CVs of the PBIs at a range of pH from 5 to 9 were also measured (Fig. S35–S37, ESI†). The different reductions potentials at different pH are compiled into Table S11 (ESI†) in the SI. It was seen that as the pH changes there is an increase in electron affinity. This change in reduction potentials with a change in pH suggest that the different mobilities of the assemblies is having an impact on these properties. This again highlights the importance in the aggregated species present, but also gives us the opportunity to finely tune these properties for specific applications.
The PBIs were used to make hydrogels using a glucono-δ-lactone (GdL) acid trigger, and their mechanical bulk properties were studied using rheology, shown in Fig. 3(a), Table 1 and Fig. S39 (ESI†). All gels broke at low strain and were independent of frequency. PBI-I had the lowest yield point and flow point but was the stiffest of all the gels formed. All the data suggests that not only are there different aggregates forming, but also that this results in a different network that directly impacts the mechanical properties.
 | ||
| Fig. 3 (a) Strain sweep data for hydrogels of PBI-L (pink), PBI-I (purple) and PBI-V (blue). In all cases, the storage modulus (G′) is represented by the closed symbols and the loss modulus (G′′) is represented by open symbols. Measurements were performed in duplicated and errors were calculated from the standard deviation of 3 repeat measurements. UV-Vis absorption spectra of hydrogels made using (b) PBI-L (pink) and after being irradiated (dashed pink), (c) PBI-I (purple) and after being irradiated (dashed purple), (d) PBI-V (blue) and after being irradiated (dashed blue). | ||
| PBI | L | I | V |
|---|---|---|---|
| G′ (Pa) | 1350 ± 90 | 3930 ± 370 | 1820 ± 30 |
| G′′ (Pa) | 130 ± 20 | 520 ± 270 | 120 ± 20 |
| Yield point (%) | 10 | 5 | 12 |
| Flow point (%) | 320 | 60 | 200 |
Tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ (G′′/G′) δ (G′′/G′) |
0.11 | 0.13 | 0.07 |
The UV-vis absorption spectra for before and after irradiation were then measured for the hydrogels. The normalised spectra are shown in Fig. 3(b andc). There is a change in the intensity ratio for 500 nm and 550 nm compared to the solution spectra, which was expected due to the increased aggregation during the gelation process. The three PBIs no longer have the same intensity ratio as was seen for the solution, summarized in Table S13 (ESI†). The PBI-I hydrogel has the largest difference between intensity ratio for 500 nm and 550 nm. PBI-I looks much different to the absorbance for PBI-L and PBI-V hydrogels. This difference in absorption is likely because of the network and aggregates (as seen in the rheology data) impacting the absorbance. It was seen that solutions of PBI-V formed the most amount of radical anion, and here again it is seen that hydrogels of PBI-V form the most amount of radical anion. Again, this difference in radical formation has been seen in previous work,42 indicating that both the molecular packing and the networks formed through gelation are impacting the ability to form the radical anion.
We used SAXS again to examine the difference in structures in the hydrogels. Full fitting parameters for the PBIs are found in Section 2.4 of the ESI† (Fig. S40–S42). All three PBIs as hydrogels fit a flexible elliptical cylinder model. The Kuhn length of each PBI hydrogel was longer than the Kuhn length in solution. This is expected because as the worm-like micelles start to form the network fibres, they become less flexible. PBI-I hydrogels saw the greatest increase to its Kuhn length from 7.55 nm to 30.6 nm, suggesting that it is the least flexible. This could also account for it having the stiffest network. The cylinder radius size was also larger for the hydrogels of all three PBIs compared to in solution. PBI-I saw a difference in cylinder radius size from 1.31 nm to 3.36 nm as a hydrogel compared to the solution. This can be attributed to the 1D fibres laterally associating to form thicker fibres, which again has been seen in our previous work.35,37,42,52,53
The CVs of the hydrogels were again measured to determine whether this difference in aggregation and network affected the reduction potentials (Fig. S43–S48, ESI†). The differences in the CV were greater than for the assemblies in solution. The reduction potential is −2.5 V for the PBI-L hydrogel and −2.8 V for the PBI-V hydrogel. However, the reduction potential for the hydrogel of PBI-I is not clearly defined, which could be due to the assemblies of the hydrogel, or the network restricting diffusion.
To investigate the difference in assembly between PBI-I and the other two PBIs further, the kinetics of the gel formation was monitored using rheology, 1H-NMR, and pH change. From the 1H-NMR spectra the percent assembled could be calculated as well as the time frames of assembly. Rheology shows when structures start to form and when they are a gel, all dictated by the change in pH over time. All three sets of kinetic experiments were overlaid and are shown in Fig. 4 and Section 3.3 of the ESI† (Fig. S49–S54). The rheology data shows that for PBI-I it begins to form solid like materials much sooner than the other two PBIs, agreeing with the other data collected, as it has the highest pKa. The 1H NMR data suggests that there is little to no structures initially, but they rapidly start to assemble as the pH drops to the pKa, forming worm-like micelles. This was seen previously with L-alanine functionalised PBI.52 However, after the pH drops below the pKa, PBI-I behaves like a gel. For PBI-V and PBI-L, by the time the first measurement is taken, already 40% of the material is assembled, suggesting assembled structures are present before gelation which we have seen previously in the SAXS, viscosity, and UV-vis absorption data. This is due to the pH already being below the pKa of each of the gelators at this time point. We then see G′ increase after around an hour which then plateaus as the gel is formed and the percentage assembly reaches nearly 100% as the gel has formed. The 1H NMR for PBI-L and PBI-V also shows a two-stage assembly process that is not seen in the rheology between 20 and 30 minutes, indicating that perhaps the structures in solution are elongating or thickening to entangle and then we are able to measure a G′ value. These kinetics data show a completely different behaviour for PBI-I compared to PBI-V and PBI-L. PBI-I forms different aggregates to that of the other two and therefore has different assembly kinetics resulting in a different gel network be formed.
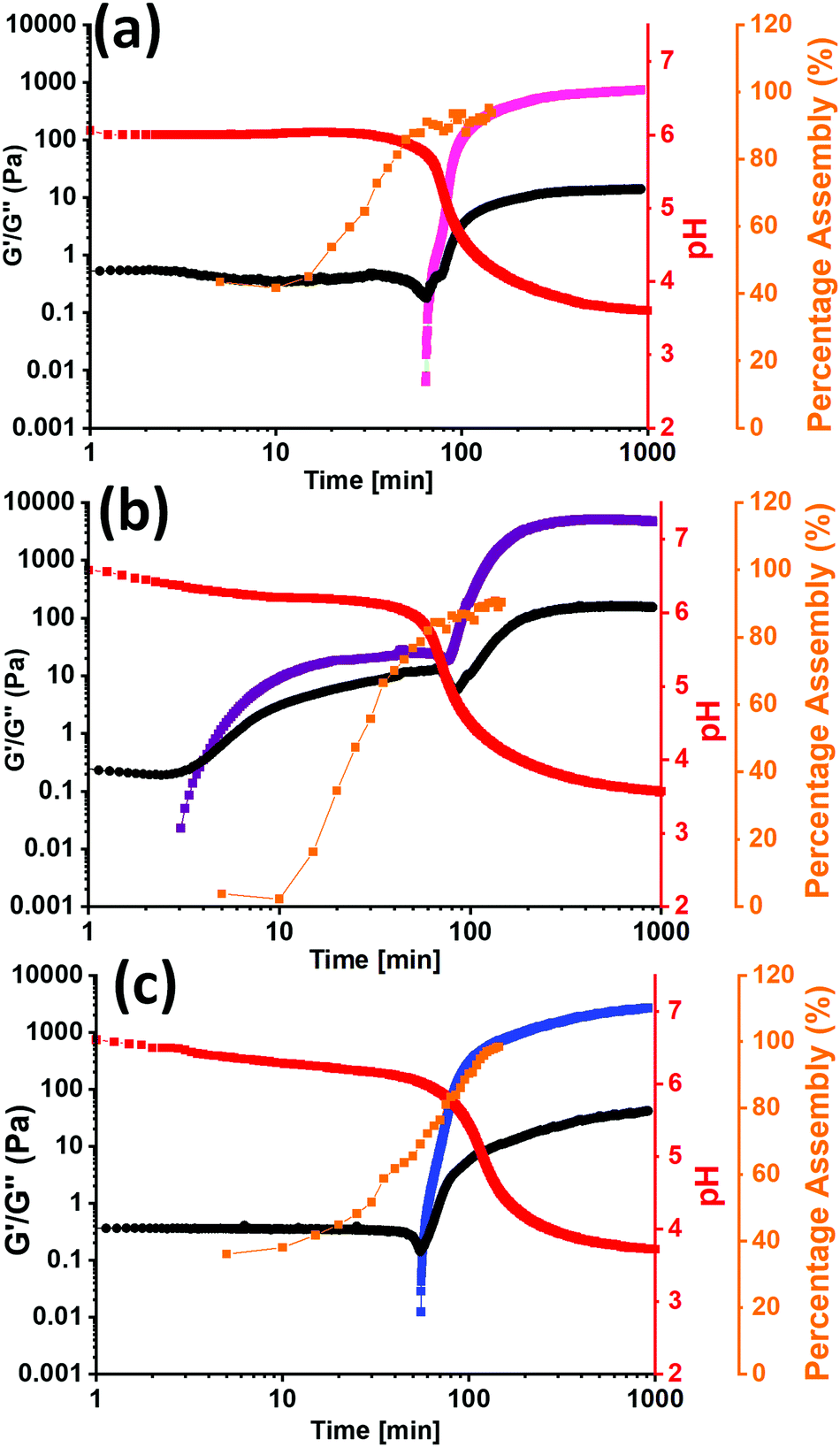 | ||
| Fig. 4 Development of G′ (pink, purple, blue data) and G′′ (black data) over time during the gelation of (a) PBI-L (pink), (b) PBI-I (purple) and (c) PBI-V (blue) at a strain of 0.5% and a frequency of 10 rad s−1, compared to the change in pH (red data) over time and the change in percent assembled (orange data) over time calculated from the change in 1H-NMR. | ||
Conclusions
We introduced simple changes to the structure of amino acid functionalised PBIs to see how they impacted self-assembly and other properties. The UV-vis absorption spectroscopy and viscosity of the solutions at different pHs showed that PBI-I was behaving differently compared to PBI-V and PBI-L despite the similarity in chemical structure. This was due to PBI-I having the highest apparent pKa and so assembling at higher pH values. It was seen that in the UV-vis absorption of the solution that PBI-V formed the most radical anion and dianion. The SAXS showed that in solution there was a structural transition period at for PBI-L between pH 5 and pH 6 and for PBI-I at pH 5, again matching up with the pKa value of the PBIs. Hydrogels formed from PBI-I were stiffer than PBI-L and PBI-V. PBI-V hydrogels formed the most radical when irradiated at 365 nm. SAXS of the hydrogels showed that upon forming the gel network the fibres present at higher pHs became longer, thicker, and stiffer, with PBI-I being the most inflexible. The CVs of all the PBIs showed little difference in reduction potentials, which we attribute to the different structures having different mobilities and therefore diffusion in solution. Kinetics studies of the gel formation showed a completely different assembly for PBI-I compared to PBI-V and PBI-L, which explains why the bulk properties of PBI-I were different compared to PBI-V and PBI-L.This work really highlights how seemingly subtle changes in chemical structure can have a major outcome on assembly of the materials and the bulk properties of the materials produced. It is not correct to assume a slight change in chemical structure does not affect all properties of the material. This brings us a little closer to understanding how the chemical structure directly impacts assembly, and step closer to being able to design these materials with specific properties.
Author contributions
The synthesis and characterization of materials was carried out by JGE and GB. All experimental data was collected by JGE, with the exception of ESI-TOF MS, TGA and DSC (carried out by Uni. Of Glasgow technicians). ERD and JGE conceived the experiments and wrote the paper. ERD acquired the funding for the work. SAXS data was collected by AJS and CJCEG at Diamond Light Source and interpreted by JGE, DM and ERD. All authors contributed to the editing and final drafts of the paper.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
ERD and JGE thanks the University of Glasgow for a bank studentship. ERD thanks the Leverhulme Trust for an Early Career Fellowship (ECF-2017-223), the EPSRC for a New Investigator Award (EP/S032673/1). ERD and GB thank the University of Glasgow for an EPSRC Impact Acceleration Award. We thank Diamond for the award of beamtime on B21, reference number SM27906-1 and SM27520-1. This work benefitted from SasView software, originally developed by the DANSE project under NSF award DMR-0520547. We would like to thank Dr Tom Welsh for help with the reply to referees whilst ERD was on maternity leave. DM would like to thank the Leverhulme Trust for funding (RPG-2018-013).Notes and references
- S. Liu, W. M. Wang, S. C. B. Mannsfeld, J. Locklin, P. Erk, M. Gomez, F. Richter and Z. Bao, Langmuir, 2007, 23, 7428–7432 CrossRef CAS PubMed
.
- Y. Zhou, T. Lei, L. Wang, J. Pei, Y. Cao and J. Wang, Adv. Mater., 2010, 22, 1484–1487 CrossRef CAS PubMed
- Y. Kim, B. Yoo, J. E. Anthony and S. K. Park, Adv. Mater., 2012, 497–502 CrossRef
- J. A. Merlo and C. D. Frisbie, J. Polym. Sci., Part B: Polym. Phys., 2003, 41, 2674–2680 CrossRef CAS
- J. A. Merlo and C. D. Frisbie, J. Phys. Chem. B, 2004, 108, 19169–19179 CrossRef CAS
- X. Guo, M. Baumgarten and K. Müllen, Prog. Polym. Sci., 2013, 38, 1832–1908 CrossRef CAS
- I. Osaka, T. Abe, M. Shimawaki, T. Koganezawa and K. Takimiya, ACS Macro Lett., 2012, 1–4 Search PubMed
- K. Asadi, Y. Wu, F. Gholamrezaie, P. Rudolf and P. W. M. Blom, Adv. Mater., 2009, 21, 4109–4114 CrossRef CAS
- Y. Shirota and H. Kageyama, Chem. Rev., 2007, 107, 953–1010 CrossRef CAS PubMed
- T. Izawa, E. Miyazaki and K. Takimiya, Adv. Mater., 2008, 20, 3388–3392 CrossRef CAS
- E. R. Draper and D. J. Adams, Chem. Commun., 2016, 52, 8196–8206 RSC
- S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973–2129 CrossRef CAS PubMed
- E. R. Draper and D. J. Adams, Langmuir, 2019, 35, 6506–6521 CAS
- X. Yang, G. Zhang and D. Zhang, J. Mater. Chem., 2012, 22, 38–50 RSC
- J. Raeburn, A. Z. Cardoso and D. J. Adams, Chem. Soc. Rev., 2013, 42, 5143–5156 RSC
- E. Krieg, E. Shirman, H. Weissman, E. Shimoni, S. G. Wolf, I. Pinkas and B. Rybtchinski, J. Am. Chem. Soc., 2009, 131, 14365–14373 CrossRef CAS PubMed
- V. Grande, B. Soberats, S. Herbst, V. Stepanenko and F. Würthner, Chem. Sci., 2018, 9, 6904–6911 RSC
- P. Bairi, B. Roy and A. K. Nandi, RSC Adv., 2012, 2, 264–272 RSC
- X. Ren, W. Yu, Z. Zhang, N. Xia, G. Fu, X. Lu and W. Wang, Colloids Surf., A, 2011, 375, 156–162 CrossRef CAS
- X. Lin, H. Kurata, D. D. Prabhu, M. Yamauchi, T. Ohba and S. Yagai, Chem. Commun., 2017, 53, 168–171 RSC
- F. Würthner, Chem. Commun., 2004, 1564–1579 RSC
- F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962–1052 CrossRef PubMed
- J. E. Anthony, Chem. Mater., 2011, 23, 583–590 CrossRef CAS
- C. Li and H. Wonneberger, Adv. Mater., 2012, 24, 613–636 CrossRef CAS PubMed
- E. Kozma and M. Catellani, Dyes Pigm., 2013, 98, 160–179 CrossRef CAS
- Z. Chen, V. Stepanenko, V. Dehm, P. Prins, L. D. A. Siebbeles, J. Seibt, P. Marquetand, V. Engel and F. Würthner, Chem. – Eur. J., 2007, 13, 436–449 CrossRef CAS PubMed
- E. Zhou, J. Cong, Q. Wei, K. Tajima, C. Yang and K. Hashimoto, Angew. Chem., 2011, 123, 2851–2855 CrossRef
- E. Kozma, D. Kotowski, F. Bertini, S. Luzzati and M. Catellani, Polymer, 2010, 51, 2264–2270 CrossRef CAS
- M. P. Aldred, G. F. Zhang, C. Li, G. Chen, T. Chen and M. Q. Zhu, J. Mater. Chem. C, 2013, 1, 6709–6718 RSC
- Z. Chen, B. Fimmel and F. Würthner, Org. Biomol. Chem., 2012, 10, 5845–5855 RSC
- F. D’Anna, S. Marullo, G. Lazzara, P. Vitale and R. Noto, Chem. – Eur. J., 2015, 14780–14790 CrossRef PubMed
- Z. B. Hill, D. B. Rodovsky, J. M. Leger and G. P. Bartholomew, Chem. Commun., 2008, 6594–6596 RSC
- L. Rocard, A. Goujon and P. Hudhomme, Molecules, 2020, 25, 1402 CrossRef CAS PubMed
- A. Nowak-Król and F. Würthner, Org. Chem. Front., 2019, 6, 1272–1318 RSC
- E. R. Draper, J. J. Walsh, T. O. McDonald, M. A. Zwijnenburg, P. J. Cameron, A. J. Cowan and D. J. Adams, J. Mater. Chem. C, 2014, 2, 5570–5575 RSC
- E. R. Draper, R. Schweins, R. Akhtar, P. Groves, V. Chechik, M. A. Zwijnenburg and D. J. Adams, Chem. Mater., 2016, 28, 6336–6341 CrossRef CAS
- J. J. Walsh, J. R. Lee, E. R. Draper, S. M. King, F. Jäckel, M. A. Zwijnenburg, D. J. Adams and A. J. Cowan, J. Phys. Chem. C, 2016, 120, 18479–18486 CrossRef CAS
- E. R. Draper, O. O. Mykhaylyk and D. J. Adams, Chem. Commun., 2016, 52, 6934–6937 RSC
- Z. Guo, X. Zhang, Y. Wang and Z. Li, Langmuir, 2019, 35, 342–358 CrossRef CAS PubMed
- X. Lu, Z. Guo, C. Sun, H. Tian and W. Zhu, J. Phys. Chem. B, 2011, 37, 10871–10876 CrossRef PubMed
- Y. Chen, Y. Feng, J. Gao and M. Bouvet, J. Colloid Interface Sci., 2012, 368, 387–394 CrossRef CAS PubMed
- E. R. Draper, L. J. Archibald, M. C. Nolan, R. Schweins, M. A. Zwijnenburg, S. Sproules and D. J. Adams, Chem. – Eur. J., 2018, 24, 4006–4010 CrossRef CAS PubMed
- D. McDowall, B. J. Greeves, R. Clowes, K. McAulay, A. M. Fuentes-Caparrós, L. Thomson, N. Khunti, N. Cowieson, M. C. Nolan, M. Wallace, A. I. Cooper, E. R. Draper, A. J. Cowan and D. J. Adams, Adv. Energy Mater., 2020, 2002469, 1–10 Search PubMed
- N. B. Bowden, K. A. Willets, W. E. Moerner and R. M. Waymouth, Macromolecules, 2002, 35, 8122–8125 CrossRef CAS
- Y. Xu, S. Leng, C. Xue, R. Sun, J. Pan, J. Ford and S. Jin, Angew. Chem., Int. Ed., 2007, 46, 3896–3899 CrossRef CAS PubMed
- M. J. Farooqi, M. A. Penick, J. Burch, G. R. Negrete and L. Brancaleon, Spectrochim. Acta, Part A, 2016, 153, 124–131 CrossRef CAS PubMed
- S. A. Boer, C. S. Hawes and D. R. Turner, Chem. Commun., 2014, 50, 1125–1127 RSC
- C. Xue, M. Chen and S. Jin, Polymer, 2008, 49, 5314–5321 CrossRef CAS
- R. F. Araújo, C. J. R. Silva, M. C. Paiva, M. M. Franco and M. F. Proença, RSC Adv., 2013, 3, 24535–24542 RSC
- A. Datar, K. Balakrishnan and L. Zang, Chem. Commun., 2013, 49, 6894–6896 RSC
- L. Chen, S. Revel, K. Morris, L. C. Serpell and D. J. Adams, Langmuir, 2010, 26, 13466–13471 CrossRef CAS PubMed
- E. R. Draper, J. R. Lee, M. Wallace, F. Jäckel, A. J. Cowan and D. J. Adams, Chem. Sci., 2016, 7, 6499–6505 RSC
- M. C. Nolan, J. J. Walsh, L. L. E. Mears, E. R. Draper, M. Wallace, M. Barrow, B. Dietrich, S. M. King, A. J. Cowan and D. J. Adams, J. Mater. Chem. A, 2017, 5, 7555–7563 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Experimental protocols and supplementary characterizations. See DOI: 10.1039/d1ma00470k |
| This journal is © The Royal Society of Chemistry 2021 |