
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIs Cu instability during the CO2 reduction reaction governed by the applied potential or the local CO concentration?†
Patrick
Wilde‡
 a,
Peter B.
O'Mara‡
b,
João R. C.
Junqueira
a,
Tsvetan
Tarnev
a,
Tania M.
Benedetti
b,
Corina
Andronescu
c,
Yen-Ting
Chen
d,
Richard D.
Tilley
*be,
Wolfgang
Schuhmann
*a and
J. Justin
Gooding
*bf
a,
Peter B.
O'Mara‡
b,
João R. C.
Junqueira
a,
Tsvetan
Tarnev
a,
Tania M.
Benedetti
b,
Corina
Andronescu
c,
Yen-Ting
Chen
d,
Richard D.
Tilley
*be,
Wolfgang
Schuhmann
*a and
J. Justin
Gooding
*bf
aAnalytical Chemistry - Center for Electrochemical Sciences (CES), Faculty of Chemistry and Biochemistry, Ruhr-Universität Bochum, Universitätsstr. 150, D-44780 Bochum, Germany. E-mail: wolfgang.schuhmann@rub.de
bSchool of Chemistry, Australian Centre for NanoMedicine, University of New South Wales, Sydney 2052, Australia. E-mail: justin.gooding@unsw.edu.au; r.tilley@unsw.edu.au
cChemical Technology III, Faculty of Chemistry and CENIDE, Center for Nanointegration University Duisburg Essen, Carl-Benz-Str. 199, D-47057 Duisburg, Germany
dCenter for Solvation Science (ZEMOS), Ruhr-Universität Bochum, Universitätsstr. 150, D-44780 Bochum, Germany
eElectron Microscope Unit, Mark Wainwright Analytical Centre, University of New South Wales, Sydney 2052, Australia
fAustralian Research Council Centre of Excellence in Convergent Bio-Nano Science and Technology, University of New South Wales, Sydney 2052, Australia
First published on 27th January 2021
Abstract
Cu-based catalysts have shown structural instability during the electrochemical CO2 reduction reaction (CO2RR). However, studies on monometallic Cu catalysts do not allow a nuanced differentiation between the contribution of the applied potential and the local concentration of CO as the reaction intermediate since both are inevitably linked. We first use bimetallic Ag-core/porous Cu-shell nanoparticles, which utilise nanoconfinement to generate high local CO concentrations at the Ag core at potentials at which the Cu shell is still inactive for the CO2RR. Using operando liquid cell TEM in combination with ex situ TEM, we can unequivocally confirm that the local CO concentration is the main source for the Cu instability. The local CO concentration is then modulated by replacing the Ag-core with a Pd-core which further confirms the role of high local CO concentrations. Product quantification during CO2RR reveals an inherent trade-off between stability, selectivity and activity in both systems.
Introduction
The development of electrocatalysts involves consideration of the triumvirate of interlinked properties, namely activity, stability, and selectivity.1,2 The activity–stability relationship in particular has been a balance between competing properties. More active catalysts are invariably less stable and more stable catalysts are less active.3–5 For example, in many organometallic catalysts, a change in the oxidation state of a metal centre can mean the pendant ligands around the metal centre are less stably bonded and dissociate, causing loss of catalytic activity.6 The interest in developing catalysts that perform multiple catalytic steps in a cascade reaction is rapidly expanding.7–11 Such catalysts require the localisation of multiple different active sites in close proximity. By this approach, the local concentration of key intermediates can be increased to significantly enhance the follow up reaction. Thus a plethora of additional impactful parameters such as local pH value, local ion concentrations, wetting properties and chemical follow-up reactions are affected, which facilitate one over the many possible reaction pathways. However, the question of how local concentrations of intermediate products in confined reaction volumes influence the stability of the catalyst remains unanswered.We have become concerned about the impact of the concentration of intermediates on catalyst stability through the development of nanoparticle electrocatalysts for the CO2 reduction reaction (CO2RR). Cu is typically the electrocatalytic material of choice for the CO2RR due to its unique ability to perform C–C coupling reactions and produce valuable hydrocarbon (C≥2) products.12–15 It is generally accepted that the reaction proceeds via the reduction of CO2 to CO and then the surface combination of CO species to form the hydrocarbon products in a multi-step reaction.16 Improved catalytic performance and product selectivity has been achieved through nanostructuring of Cu catalysts allowing precise control over the exposure of specific surface facets,17,18 grain boundaries and defect structure19–21 as well as three dimensional morphology to locally and temporally trap intermediate species.22–24 Yet, the high overpotential required to form CO as the first intermediate product on Cu before higher hydrocarbons can be obtained, prevents the selection of specific products in follow-up reaction pathways. Instead, a complex mixture of products is obtained, which can even change its composition during the course of electrolysis.
To overcome the limitation of the high overpotential required for the first reaction step causing a complex mixture of products, bimetallic systems were designed to provide at least two different active sites. The goal of the bimetallic approach for CO2RR is to enable the spillover of CO as an intermediate product from a first active site to Cu as the second active centre, where it is converted to the final product. In this scenario, the first active centre, e.g. Ag, acts as an efficient source of CO through electrochemical reduction of CO2 to CO at lower overpotentials than on Cu.25–31 With such a strategy in mind, we have previously designed a catalytic nanoparticle based on a solid Ag-core surrounded by a Cu2O-derived nanoporous Cu-shell.32 Nanoconfinement in the nanoporous Cu-shell facilitates increased C–C coupling reaction rates through CO, which is produced by the Ag, accumulates within the porous Cu-shell and leads to very high concentrations of CO within the nanoporous Cu-shell. These very high local CO concentrations provided a previously unprecedented selectivity for C3 products over C2 hydrocarbons at very low overpotentials.32 Uniquely, the specific architectural design of this core–shell system allows subtle modulation of the local CO concentration via the applied potential. That is, the solid Ag-core/nanoporous Cu-shell experimental system provides access to obtaining high local CO concentrations and enables the investigation of the impact of this intermediate on the structural stability of bimetallic systems for CO2RR.
The purpose of this paper is to explore how the stability of Ag-core/nanoporous Cu-shell nanoparticles is influenced by the applied potential and the amount of CO that is locally generated. Thus, this work is intended to provide insight into the stability of Cu-based mono- and bimetallic CO2RR catalysts during operation in general. Recent developments in our group in operando electrochemistry coupled liquid cell transmission electron microscopy (ELC-TEM) enable operando investigation of the electrocatalyst as the CO2RR proceeds. Solid Ag-core/porous Cu-shell nanoparticles were precisely deposited from suspension onto the ELC-TEM working electrode. For that purpose, a suspension filled nanometre-sized quartz capillary was brought onto the working electrode using approach techniques from scanning electrochemical cell microscopy (SECCM). Thus, puncturing of the fragile glassy carbon electrode/Si3N4 window or short-circuiting between the electrodes of the chip is prevented (see ESI page S4 for further information†).33
Recent studies of monometallic Cu systems have uncovered structural reorganisation and degradation under CO2RR conditions. However, these separate studies have suggested different factors as the main driving force for the processes (Fig. 1A). Both, applied potential and the presence of adsorbates, specifically CO, were suggested as possible causes for the observed restructuring.34,35 Understanding the main contributor to the catalyst degradation and its underlying mechanism is crucial to the design of stable Cu electrocatalysts. Such knowledge is especially important for next generation bimetallic CuX catalyst systems, where the superior performance during CO2RR is based on the enhanced initial formation of CO. The mechanism behind the structural evolution of Cu-based catalysts has remained elusive through a lack of operando microscopy investigations into the stability of CO2RR catalysts.36 Previously, one study has described the structural evolution of Cu-cubes under CO2RR conditions using ELC-TEM. However, the main driving force was not further investigated.37
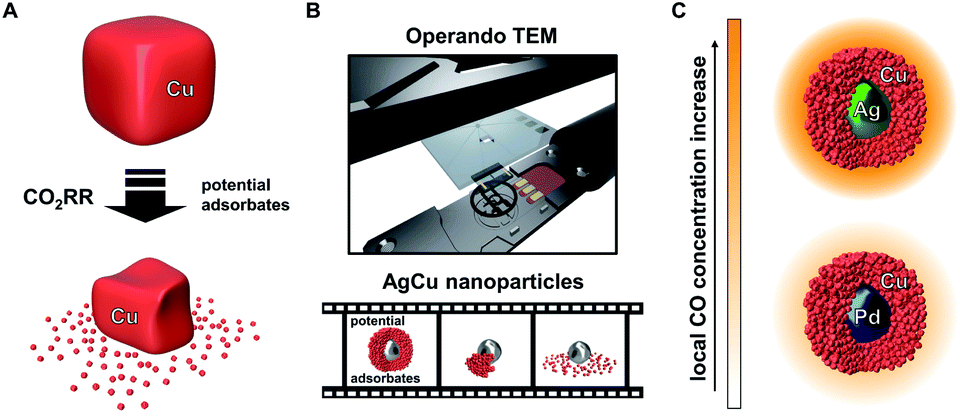 | ||
| Fig. 1 Structural evolution of Cu-only particles reported during CO2RR (A). ELC-TEM is employed to investigate the equivalent process on Ag-core/porous Cu-shell nanoparticles and the course of the reaction is visualised in an operando movie (B). Changing the core metal of the solid core/porous shell particle provides an opportunity to tune the local CO concentration (C). | ||
The ELC-TEM experiments we perform (Fig. 1B), coupled with ex situ TEM analysis, identify CO as the main contributing source of the instability of the electrocatalytic nanoparticles through the dissolution of the porous Cu-shell. The insights from the study suggest that the high catalytic activity of the Ag for CO2RR under formation of CO is therefore the source of the catalyst instability; thus showing that such core–shell nanoparticles and presumably other bimetallic catalyst systems for CO2RR suffer from the common challenge of higher activity meaning lower stability. We show this is the case by designing a similar bimetallic catalyst system by replacing Ag by Pd. Solid Pd-core/porous Cu-shell nanoparticles exhibit a lower rate of CO formation, however, the local CO concentration within the nanoconfined porous Cu is still sufficiently high to yield hydrocarbon products at low overpotential with substantially increased stability and higher selectivity (Fig. 1C).
Results and discussion
To deepen the understanding of mechanisms providing stability of bimetallic CO2 reduction electrocatalysts, and thus extend the discussion beyond activity and selectivity, ELC-TEM was used to visualise the morphological changes of Ag-core/Cu-shell nanoparticles under operando conditions inside the TEM chamber (movies M1 and M2 in the electronic ESI†). In the ELC-TEM cell, CO2RR was performed potentiostatically at a nominal working electrode potential of −2.0 V vs. Pt (−1.12 V vs. RHE) whilst the current response and a video of the evolving TEM images were recorded (for further information on ELC-TEM see ESI Fig. S1 and S2†). The major structural change occurs at 110 s with the structural collapse of one of the nanoparticles (Fig. 2A yellow circle and Fig. 2B). The porous Cu-shell starts to disintegrate adjacent to a small gap in the shell. This process unfolds evenly around the particle until the Cu-shell remains as a branch opposite to the initial gap. After 150 s the silver core begins to show signs of deformation and eventually after 170 s shrinkage of the core occurs. No further changes are visible after 170 s since the observed area became blocked by a gas bubble and thus was no longer in contact with the electrolyte. This leads to the question, what happens to the dissolved Cu? | ||
| Fig. 2 Screenshots from the operando movie recorded during ELC-TEM of at indicated seconds after potential step to −2.0 V vs. Pt (A). Coloured geometric shapes refer to processes described in the main text. Close inspection of the process in the yellow circle (B). Schematic representation of the process (I–IV). | ||
The fate of the dissolved Cu can be seen in the emergence of a low-contrast solid structure that grows from 110 s until 150 s (Fig. 2A red rectangle). Combined with the low contrast of the material in the dark-field STEM image, in situ EDX analysis of the electrode surface (see ESI Fig. S3†) reveals that the structure is made from Cu. The growth shows that irregular Cu structures are formed directly on already existing structures. Furthermore, another Cu structure can be identified (Fig. 2A green hexagon, 110–170 s), which appears to be a well-defined crystal growth from a seed.
The ELC-TEM ‘stress-test’ clearly demonstrates that there is a dynamic evolution of the catalyst structure during electrocatalysis. To better interpret these dynamic processes, and to relate these changes to specific reaction conditions, a series of ex situ experiments was performed to probe the influence of applied potential, time, CO concentration and CO2RR rate on the stability of the nanoparticles (Fig. 3 and ESI Fig. S4†).
 | ||
| Fig. 3 Ex situ EDX elemental maps of AgCu core–shell nanoparticles after electrocatalysis with different conditions: after 1 h CO2RR at −500 mV (A), −600 mV (B), −700 mV (C) and −800 mV (D) vs. RHE; in their pristine state (E), after 1 h at −800 mV vs. RHE in N2-saturated electrolyte (F) and after CO reduction reaction at −800 mV vs. RHE in 0.1 mM CO (G) and 1 mM CO (H). The Ag is depicted in green and Cu in red. Scale bar equals 250 nm. | ||
Varying the applied potential has a significant effect on the morphological evolution of the Ag-core/porous Cu-shell nanoparticles under exposure to CO2RR conditions in 0.1 M KHCO3 for 1 h (Fig. 3A–D). Whereas the porous shell structure, which consists of distinguishable Cu particulates (∼10–15 nm diameter, ESI Fig. S5†), is maintained at −500 mV vs. RHE, it begins to undergo structural changes with increasing overpotential. At −600 mV vs. RHE individual particulates cannot be differentiated anymore (Fig. 3B). This is due to the formation of a foam-like structure, which consists of considerably smaller Cu nanoparticles (∼2–4 nm diameter, ESI Fig. S6†) but still preserves the integral solid core/porous shell architecture. Similar nanoparticles have been observed during the degradation of a monometallic Cu system during CO2RR, suggesting a similar decomposition mechanism.34 The sizes of the particulates that form the shell and of the nanoparticles, which evolve during CO2RR, differ significantly from each other. This observation indicates that the evolving nanoparticles cannot be merely the result of a physical disintegration of the shell of the particle. Further increase of the overpotential leads to the complete loss of the core–shell structure at −800 mV vs. RHE (Fig. 3D). Cu and Ag phases become spatially segregated in the form of clustered Ag particles and highly ordered, mostly single crystalline Cu structures (ESI Fig. S7A–C†). The latter are identified as Cu2O (ESI Fig. S7D–F†), which is initially surprising considering that the Pourbaix diagram predicts either CuH or metallic Cu under these conditions.34 A possible explanation is that either there is a locally high pH value within the nanoconfined volume during reaction or rapid re-oxidation of the nanoparticles occurs when they are exposed to atmospheric conditions at open circuit potential, prior to transfer into the vacuum for ex situ TEM characterisation.35,38,39
Further ex situ measurements were performed at (i) open circuit potential, (ii) at −600 mV for different durations and (iii) under the exclusion of CO2. The results (ESI Fig. S8†) show that structural rearrangements of the nanoparticles were not observed (i) in the mere presence of the electrolyte at OCP (ESI Fig. S8A†), (ii) through the prolongation of the reaction time under CO2RR conditions (ESI Fig. S8B–D†), and (iii) under the application of potential without the presence of CO2 (ESI Fig. S8E and F†). Previously, the CO adsorbate, the key intermediate product of the CO2RR, was proposed as a major influencing factor on the stability of monometallic Cu systems.34,35 Control experiments in 0.1 M KHCO3 with different CO concentrations were performed to assess the influence of the local CO concentration on the structural evolution of the catalyst. TEM analysis reveals that there is a correlation between the degree of restructuring and the CO concentration combined with the applied potential. At −800 mV vs. RHE, a concentration of 0.1 mM CO does not initiate the previously observed decomposition of the porous Cu-shell (Fig. 3G). In contrast, a ten-fold increase to 1 mM CO induces the loss of the core–shell architecture and the formation of nanoscopic Cu particles surrounding the Ag-cores (Fig. 3H). At −600 mV vs. RHE, both 1 mM and 0.1 mM CO invoke little change in structure (ESI, Fig. S8G and H†). The latter result emphasises the importance of both the amount of CO and the magnitude of the applied potential. Interestingly, the disintegration observed under CO reduction conditions at −800 mV vs. RHE results in a significantly different structure compared to the one observed after 1 h at the same potential under CO2RR conditions (compare Fig. 3D with Fig. 3H). This indicates that the local confinement of CO in the nanoporous Cu-shell has a significant effect on the chemical environment around the nanoparticle in comparison to when it is freely available from the bulk of the electrolyte. Based on the observation that the size of the Cu nanoparticles that were observed surrounding the Ag cores after CO2RR at −600 mV vs. RHE decreases gradually with increasing distance (ESI, Fig. S6†) from the Ag-cores – the source of CO – supports a degradation mechanism40 where: (i) primarily formed CO binds to Cu sites, (ii) weakens its binding strength to adjacent Cu atoms, (iii) removes Cu atoms from the under-coordinated sites of the particulates in the shell, which spread unidirectionally around the particulate of origin, and (iv) eventually aggregate to clusters or single crystals, if they are not subjected to further degradation (ESI Fig. S9†). Larger polycrystalline nanoparticles (ESI Fig. S10†) can be explained through the coalescence of multiple smaller nanoparticles.34
It is the combination of the operando ELC-TEM with results from ex situ analysis, which ultimately unlocks mechanistic insights into the structural evolution of the bimetallic core–shell particles that would otherwise remain elusive. The results indicate that the structural stability of Cu systems for the CO2RR heavily depends on the local concentration of the CO intermediate around the Cu catalyst, which is predominantly influenced by the catalytic CO2 conversion rate of the CO source (e.g. Ag for bimetallic CuX or Cu for pure Cu catalysts). This conclusion is reflected in the structural evolution of the core–shell nanoparticle observed in the video (Fig. 2B), which reveals that in this specific case the shell degradation originates from a pre-existing gap in the otherwise intact shell. The locally facilitated access of CO2 to the Ag-core is expected to lead to a locally enhanced CO formation rate and hence increased local CO concentration. The latter then accelerates an asymmetric reconstruction process (Fig. 2B(I–IV)), which causes the rapid collapse of the entire nanoparticle.
Since structural stability under operando conditions is essential, the ability to tune the CO formation rate in bimetallic systems through smart catalyst design is required to achieve the optimum balance between activity, selectivity and stability. In the specific case of the solid Ag-core/porous Cu-shell nanoparticles, the preservation of the structural integrity of the Cu-shell around the Ag-core is the basis for a cascade mechanism for increased activity and selectivity for C3 products. Against this background, the replacement of the CO source (Ag) with a different catalyst (X) that exhibits a lower CO formation rate could improve the structural stability, with a trade-off in activity and possibly the selectivity of the cascade reaction products. Pd can act as a viable substitute since it exhibits a considerably lower CO2 to CO conversion rate in comparison to Ag (Fig. 1C).41 Furthermore, replacement of the core metal in this bimetallic system ultimately helps to differentiate between the effects of applied potential and local CO concentration on the stability of Cu during CO2RR. In case of a monometallic Cu-catalyst, more cathodic potentials inevitably lead to an increased CO formation rate. By comparing AgCu and PdCu nanoparticles, however, the CO formation rate can be tuned even though the same potential is applied. Nanoparticles with solid Pd-cores and nanoporous Cu-shells were synthesised and tested for their structural stability (Fig. 4A and ESI Fig. S11†). TEM analysis after CO2RR revealed, that the PdCu nanoparticles maintain their solid core/porous shell architecture even at −800 mV vs. RHE, where AgCu particles had previously suffered complete loss of their initial structure (compare Fig. 3D and E to 4A and B). It is this observation, which demonstrates that the application of a certain potential alone is not responsible for the instability of Cu during CO2RR. Instead, it is the combination of the applied potential with the local CO concentration, which determines how stable Cu is under reaction conditions. The influence of a different strain effect for the AgCu particles compared with PdCu particles was discounted since the Cu shell thickness of more than 30 nm is beyond the size regime where strain is active.42–44
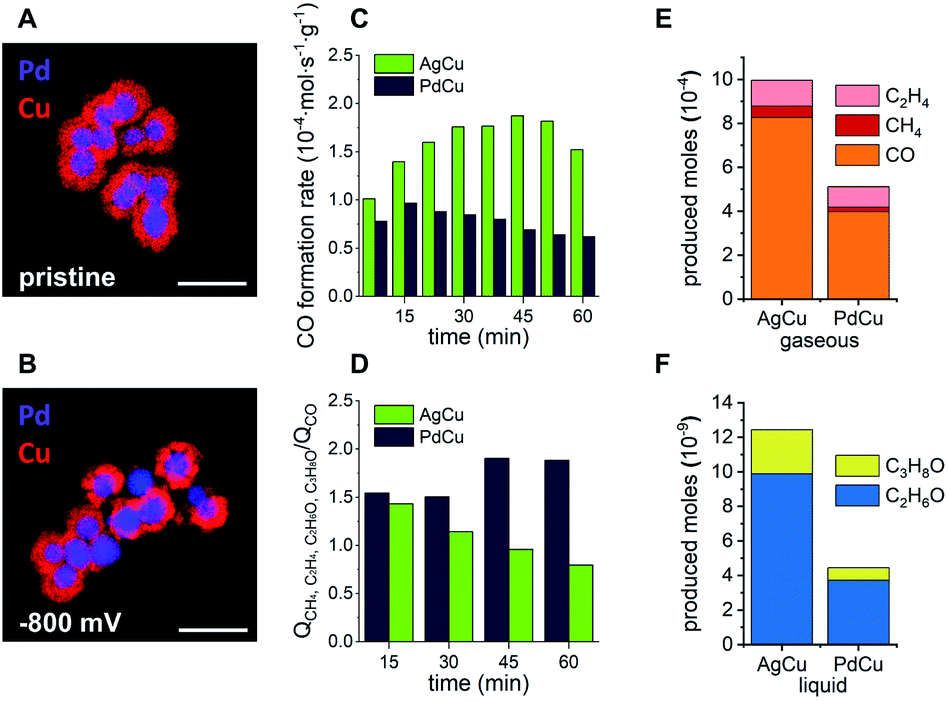 | ||
| Fig. 4 Ex situ EDX elemental maps of PdCu solid core/porous shell particles in their pristine state (A) and after CO2RR for 1 h at −800 mV vs. RHE (B). Scale bars equal 250 nm. Comparison of CO2RR performance at −800 mV vs. RHE over time of AgCu (green) and PdCu (blue) in terms of CO formation rate (C) and the ratio of charge that was consumed to form cascade products (CH4, C2H4, C2H6O and C3H8O) over charge that was consumed to form CO (D). Produced moles of gaseous products (E) and liquid products (F) for AgCu and PdCu particles after 60 min. Values are the average of 2 independent measurements. | ||
An increased stability of PdCu under similar operating conditions to where the AgCu was unstable (compare Fig. 4B and 3D) satisfies one aspect of the triumvirate balancing act, which leaves the question as to how the selectivity and activity are affected. Looking at the activity for the CO formation first, the product distributions of both catalytic systems were investigated under CO2RR conditions at −800 mV vs. RHE for 1 h (ESI Fig. S12†). The AgCu system exhibits a steady increase of CO formation rate per gram of electrocatalyst from 1.01 × 10−4 mol s−1 g−1 after 7.5 min to 1.87 × 10−4 mol s−1 g−1 after 45 min before it drops rapidly to 1.52 × 10−4 mol s−1 g−1 after 60 min (Fig. 4C). In direct comparison, the PdCu system shows a lower overall CO formation rate, which is expected given the lower activity of Pd for the formation of CO. The CO formation rate on Pd initially increases between 7.5 min and 15 min from 0.78 × 10−4 mol s−1 g−1 to 0.97 × 10−4 mol s−1 g−1, followed by a steady decrease to 0.62 × 10−4 mol s−1 g−1 after 60 min (Fig. 4C).
Combined with the insights from the video, the significant increase in CO formation rate of the AgCu system can be explained through the breaking up of the Cu-shell. The shell breakup then provides better access for CO2 to the more active sites at the Ag-core as the Cu-shell is removed, hence the rate of conversion of the CO2 to CO increases. The decay occurring during the last 15 min of the measurement could be explained by a loss of Ag activity due to the structural degradation of the Ag-cores (Fig. 2 blue rhomboid).40 The PdCu system experiences less change in the CO formation rate. Considering the structural integrity of the PdCu catalyst at the investigated potential, −800 mV vs. RHE, the gradual decrease of CO formation rate could be explained by an activation of the Cu-shell through reduction of Cu2O for the conversion of CO to higher hydrocarbons (Fig. 4D).45
We next turn our attention to the selectivity. The selectivity of these bimetallic systems, which employ a cascade mechanism that utilises nanoconfined CO adjacent to where it is produced, can be defined as the ratio between CO that escapes the system without any further reaction and the cascade products, which are formed through re-adsorption of the intermediate CO and reaction on Cu to higher hydrocarbons. These products include CH4, C2H4, C2H6O and C3H8O. To express this ratio, the charge, which is involved in the formation of the escaping CO, is compared to the total charge, which is involved in the formation of cascade products. The decrease of this ratio from 1.43 to 0.79 in the case of AgCu quantifies the loss of its capability to perform cascade reactions due to the degradation of the core–shell structure. In comparison, the PdCu system experiences an increasing efficiency for cascade reactions as indicated by an increase in this ratio from 1.54 to 1.89.
The observations underline that the enhanced stability of the PdCu catalyst results in an enhanced and prolonged selectivity for higher hydrocarbons. The nature of the three-way balancing act becomes apparent when the activity of both systems is compared. Despite the loss of its nanostructure and the consequential lack of stability and selectivity, the AgCu catalyst shows higher activity over a period of 60 min for the formation of all cascade reaction products (Fig. 4E and F). Propanol, ethanol and methane are produced in approximately double the quantity on AgCu compared to PdCu, and ethylene production is enhanced on AgCu by a factor of 1.18. At −800 mV vs. RHE, AgCu possesses the highest activity for these cascade reaction products but the structure degrades due to an increased local concentration of CO. Decreasing the local concentration of CO by replacing the metal core with Pd sees lower activity at −800 mV vs. RHE but greater stability and selectivity. In this way we have shown that activity can be traded for increased selectivity and stability.
Conclusions
The results of the presented work demonstrate how a detailed study of stability using operando ELC-TEM combined with TEM analysis of ex situ experiments enable us to understand the origin of catalyst degradation and unlock a deeper discussion of the trade-off between activity, selectivity and stability. In this case, the combination of the applied potential with high local concentrations of the CO intermediate, which are often the goal of bimetallic CO2RR catalysts, can adversely affect the structural stability of Cu whilst simultaneously enhancing the production of higher order molecules. Also, other fields of catalysis are recommended to use operando techniques to better understand structural stability to unlock similar nuanced discussions of activity, selectivity and stability.Conflicts of interest
There are no conflicts to declare.Acknowledgements
W. S. is grateful for financial support by the Deutsche Forschungsgemeinschaft under Germany's Excellence Strategy – EXC 2033 – 390677874 – RESOLV and the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (CasCat; grant agreement No. 833408). This work was supported by the DAAD in the framework of the PPP project 57446293 as well as the Ruhr University Research School PLUS through a research stay of P. W. at UNSW. This research was financially supported by the Australian Research Council of Centre of Excellence in Convergent Bio-Nano Science and Technology (CE140100036), the ARC Laureate Fellowship (FL150100060) and the Discovery Project (DP190102659). The Mark Wainwright Analytical Centre (MWAC) at UNSW. This work used the facilities supported by Microscopy Australia at the Electron Microscope Unit at UNSW. Wei Xia is acknowledged for his contribution to the CO measurements. P. B. O'M. acknowledges the Australian Government Research Training Program Scholarship for financial support. P. W. is grateful to the Association of the Chemical Industry (VCI) for funding of the PhD fellowship.Notes and references
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, 1–12 CrossRef
.
- A. S. Bandarenka, E. Ventosa, A. Maljusch, J. Masa and W. Schuhmann, Analyst, 2014, 139, 1274–1291 RSC
- C. Spöri, J. T. H. Kwan, A. Bonakdarpour, D. P. Wilkinson and P. Strasser, Angew. Chem., Int. Ed., 2017, 56, 5994–6021 CrossRef
- H.-S. Chen, T. M. Benedetti, V. R. Gonçales, N. M. Bedford, R. W. J. Scott, R. F. Webster, S. Cheong, J. J. Gooding and R. D. Tilley, J. Am. Chem. Soc., 2020, 142, 3231–3239 CrossRef CAS
- R. Narayanan and M. A. El-Sayed, J. Phys. Chem. B, 2005, 109, 12663–12676 CrossRef CAS
- R. H. Crabtree, Chem. Rev., 2015, 115, 127–150 CrossRef CAS
- M. Zhao, K. Deng, L. He, Y. Liu, G. Li, H. Zhao and Z. Tang, J. Am. Chem. Soc., 2014, 136, 1738–1741 CrossRef CAS
- F. Rudroff, M. D. Mihovilovic, H. Gröger, R. Snajdrova, H. Iding and U. T. Bornscheuer, Nat. Catal., 2018, 1, 12–22 CrossRef
- J. Muschiol, C. Peters, N. Oberleitner, M. D. Mihovilovic, U. T. Bornscheuer and F. Rudroff, Chem. Commun., 2015, 51, 5798–5811 RSC
- A. V. Biradar, V. S. Patil, P. Chandra, D. S. Doke and T. A. Asefa, Chem. Commun., 2015, 51, 8496–8499 RSC
- G. C. Y. Choo, H. Miyamura and S. Kobayashi, Chem. Sci., 2015, 6, 1719–1727 RSC
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC
- T.-T. Zhuang, Z.-Q. Liang, A. Seifitokaldani, Y. Li, P. de Luna, T. Burdyny, F. Che, F. Meng, Y. Min, R. Quintero-Bermudez, C. T. Dinh, Y. Pang, M. Zhong, B. Zhang, J. Li, P.-N. Chen, X.-L. Zheng, H. Liang, W.-N. Ge, B.-J. Ye, D. Sinton, S.-H. Yu and E. H. Sargent, Nat. Catal., 2018, 1, 421–428 CrossRef CAS
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS
- Y. Hori, K. Kikuchi, A. Murata and S. Suzuki, Chem. Lett., 1986, 15, 897–898 CrossRef
- R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. M. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS
- Y. Hori, I. Takahashi, O. Koga and N. Hoshi, J. Mol. Catal. A: Chem., 2003, 199, 39–47 CrossRef CAS
- A. Bagger, W. Ju, A. S. Varela, P. Strasser and J. Rossmeisl, ACS Catal., 2019, 9, 7894–7899 CrossRef CAS
- R. G. Mariano, K. McKelvey, H. S. White and M. W. Kanan, Science, 2017, 358, 1187–1192 CrossRef CAS
- Z. Dan, F. Qin, A. Makino, Y. Sugawara, I. Muto and N. Hara, J. Alloys Compd., 2014, 586, S134–S138 CrossRef CAS
- X. Feng, K. Jiang, S. Fan and M. W. Kanan, J. Am. Chem. Soc., 2015, 137, 4606–4609 CrossRef CAS
- K. D. Yang, W. R. Ko, J. H. Lee, S. J. Kim, H. Lee, M. H. Lee and K. T. Nam, Angew. Chem., Int. Ed., 2017, 56, 796–800 CrossRef CAS
- A. Dutta, M. Rahaman, N. C. Luedi, M. Mohos and P. Broekmann, ACS Catal., 2016, 6, 3804–3814 CrossRef CAS
- S. Sen, D. Liu and G. T. R. Palmore, ACS Catal., 2014, 4, 3091–3095 CrossRef CAS
- C. G. Morales-Guio, E. R. Cave, S. A. Nitopi, J. T. Feaster, L. Wang, K. P. Kuhl, A. Jackson, N. C. Johnson, D. N. Abram, T. Hatsukade, C. Hahn and T. F. Jaramillo, Nat. Catal., 2018, 1, 764–771 CrossRef CAS
- S. Lee, G. Park and J. Lee, ACS Catal., 2017, 7, 8594–8604 CrossRef CAS
- D. Gao, Y. Zhang, Z. Zhou, F. Cai, X. Zhao, W. Huang, Y. Li, J. Zhu, P. Liu, F. Yang, G. Wang and X. Bao, J. Am. Chem. Soc., 2017, 139, 5652–5655 CrossRef CAS
- J. Huang, M. Mensi, E. Oveisi, V. Mantella and R. Buonsanti, J. Am. Chem. Soc., 2019, 141, 2490–2499 CrossRef CAS
- C. W. Lee, K. D. Yang, D.-H. Nam, J. H. Jang, N. H. Cho, S. W. Im and K. T. Nam, Adv. Mater., 2018, 30, 1704717 CrossRef
- S. Ma, M. Sadakiyo, M. Heim, R. Luo, R. T. Haasch, J. I. Gold, M. Yamauchi and P. J. A. Kenis, J. Am. Chem. Soc., 2017, 139, 47–50 CrossRef CAS
- F. Li, Y. C. Li, Z. Wang, J. Li, D.-H. Nam, Y. Lum, M. Luo, X. Wang, A. Ozden, S.-F. Hung, B. Chen, Y. Wang, J. Wicks, Y. Xu, Y. Li, C. M. Gabardo, C.-T. Dinh, Y. Wang, T.-T. Zhuang, D. Sinton and E. H. Sargent, Nat. Catal., 2020, 3, 75–82 CrossRef CAS
- P. B. O'Mara, P. Wilde, T. M. Benedetti, C. Andronescu, S. Cheong, J. J. Gooding, R. D. Tilley and W. Schuhmann, J. Am. Chem. Soc., 2019, 141, 14093–14097 CrossRef
- T. Tarnev, S. Cychy, C. Andronescu, M. Muhler, W. Schuhmann and Y.-T. Chen, Angew. Chem., Int. Ed., 2020, 59, 5586–5590 CrossRef CAS
- J. Huang, N. Hörmann, E. Oveisi, A. Loiudice, G. L. de Gregorio, O. Andreussi, N. Marzari and R. Buonsanti, Nat. Commun., 2018, 9, 3117 CrossRef
- W. T. Osowiecki, J. J. Nussbaum, G. A. Kamat, G. Katsoukis, M. Ledendecker, H. Frei, A. T. Bell and A. P. Alivisatos, ACS Appl. Energy Mater., 2019, 2, 7744–7749 CrossRef CAS
- A. D. Handoko, F. Wei, Jenndy, B. S. Yeo and Z. W. Seh, Nat. Catal., 2018, 1, 922–934 CrossRef CAS
- R. M. Arán-Ais, R. Rizo, P. Grosse, G. Algara-Siller, K. Dembélé, M. Plodinec, T. Lunkenbein, S. W. Chee and B. R. Cuenya, Nat. Commun., 2020, 11, 3489 CrossRef
- H. Jung, S. Y. Lee, C. W. Lee, M. K. Cho, D. H. Won, C. Kim, H.-S. Oh, B. K. Min and Y. J. Hwang, J. Am. Chem. Soc., 2019, 141, 4624–4633 CrossRef CAS
- Y. Lum and J. W. Ager, Angew. Chem., Int. Ed., 2018, 57, 551–554 CrossRef CAS
- B. Eren, D. Zherebetskyy, L. L. Patera, C. H. Wu, H. Bluhm, C. Africh, L.-W. Wang, G. A. Somorjai and M. Salmeron, Science, 2016, 351, 475–478 CrossRef CAS
-
Y. Hori, in Modern Aspects of Electrochemistry 42, ed. C. G. Vayenas, R. E. White and M. E. Gamboa-Aldeco, Springer, Berlin, 2008, vol. 42, ch. 3, pp. 89–189 Search PubMed
- R. Reske, M. Duca, M. Oezaslan, K. J. P. Schouten, M. T. M. Koper and P. Strasser, J. Phys. Chem. Lett., 2013, 4, 2410–2413 CrossRef CAS
- A. Alinezhad, L. Gloag, T. M. Benedetti, S. Cheong, R. F. Webster, M. Roelsgaard, B. B. Iversen, W. Schuhmann, J. J. Gooding and R. D. Tilley, J. Am. Chem. Soc., 2019, 141, 16202–16207 CrossRef CAS
- Z. Xia and S. Guo, Chem. Soc. Rev., 2019, 48, 3265–3278 RSC
- L. Mandal, K. R. Yang, M. R. Motapothula, D. Ren, P. Lobaccaro, A. Patra, M. Sherburne, V. S. Batista, B. S. Yeo, J. W. Ager, J. Martin and T. Venkatesan, ACS Appl. Mater. Interfaces, 2018, 10, 8574–8584 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: For materials and methods section, further information on ELC-TEM, electron beam effect, ELC-TEM EDX spectra, size distributions of synthesised AgCu particles and Cu nanoparticles, high resolution TEM images of single crystalline phases, crystal lattice analysis, effect of additional parameters on structural stability, proposed degradation mechanism and chronoamperograms. See DOI: 10.1039/d0sc05990k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |