
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biocatalysis making waves in organic chemistry
Ulf
Hanefeld
 *,
Frank
Hollmann
* and
Caroline E.
Paul
*
*,
Frank
Hollmann
* and
Caroline E.
Paul
*
Biocatalysis, Department of Biotechnology, Delft University of Technology, Van der Maasweg 9, The Netherlands. E-mail: u.hanefeld@tudelft.nl; f.hollmann@tudelft.nl; c.e.paul@tudelft.nl
First published on 20th December 2021
Abstract
Biocatalysis has an enormous impact on chemical synthesis. The waves in which biocatalysis has developed, and in doing so changed our perception of what organic chemistry is, were reviewed 20 and 10 years ago. Here we review the consequences of these waves of development. Nowadays, hydrolases are widely used on an industrial scale for the benign synthesis of commodity and bulk chemicals and are fully developed. In addition, further enzyme classes are gaining ever increasing interest. Particularly, enzymes catalysing selective C–C-bond formation reactions and enzymes catalysing selective oxidation and reduction reactions are solving long-standing synthetic challenges in organic chemistry. Combined efforts from molecular biology, systems biology, organic chemistry and chemical engineering will establish a whole new toolbox for chemistry. Recent developments are critically reviewed.
 Ulf Hanefeld | Ulf Hanefeld received his PhD from the Georg-August-Universität zu Göttingen, having performed the research both with Prof. H. Laatsch (Göttingen) and Prof. H. G. Floss (Seattle). After postdoctoral years with Prof. C. W. Rees (Imperial College London), Prof. J. Staunton (Cambridge) and Prof. J. J. Heijnen and Dr A. J. J. Straathof (TU Delft), he received a fellowship from the Royal Netherlands Academy of Arts and Sciences (KNAW). He rose through the ranks at the Technische Universiteit Delft and his research in Delft focuses on enzymes, their immobilisation and application in organic synthesis. |
 Frank Hollmann | Frank Hollmann studied Chemistry at the University of Bonn (Germany). After his PhD with Andreas Schmid (ETHZ Zurich, Switzerland) and a postdoc with Manfred T. Reetz (Max-Planck-Institute for Coal Research, Germany), he worked for some years at Evonik Industries. In 2008 he joined the Biocatalysis Group at the Delft University of Technology where he now is Professor of Biocatalysis. |
 Caroline E. Paul | Caroline E. Paul obtained her Honours BSc and MSc in Biological Chemistry at the University of Toronto, and her PhD in 2013 in Bioorganic Chemistry at the University of Oviedo with Profs. V. Gotor-Fernández and I. Lavandera. She carried out postdoctoral work with a Marie Curie fellowship with Prof. F. Hollmann at TU Delft followed by a NWO VENI grant at Wageningen University on cofactors biomimetic for oxidoreductases. Since 2018 she is an Assistant Professor in Biocatalysis at TU Delft. |
1. Introduction
In 1837 Liebig and Wöhler described the action of an enzyme: the crude preparation of hydroxynitrile lyase from the almond Prunus amygdalus (PaHNL), which they called emulsin.1 This emulsin released HCN from a compound also isolated from these almonds, later identified as (R)-mandelonitrile. At that time Berzelius had just coined the term catalysis based on, among others, Döbereiner's experiments with platinum.2 Liebig and Wöhler then went on to disprove that special forces (vis vitalis) were necessary to prepare organic compounds and chemistry was set to continue as a single field of science. But when Fischer published his lock and key hypothesis,3 chemistry was already split into several fields: in 1907 Buchner received the Nobel prize in Chemistry for his work on cell free application of enzymes for fermentation. But the first enantioselective organic synthesis, the enantioselective addition of HCN to benzaldehyde by Rosenthaler, reverting Liebig and Wöhler degradation of (R)-mandelonitrile, was published in 1908 in a biochemistry journal (Scheme 1a).4 Today this is considered the first wave of biocatalysis.5 It did not, however, move enzymes centre-stage of Chemistry. This lack of interest in the possibility of enzymes for selective and in particular enantioselective synthesis became even more visible with the Nobel prize of 1931: the discovery by Warburg of redox enzymes, all of which are highly enantioselective, was the prize for Physiology or Medicine not for Chemistry. Equally the first industrial enantioselective synthesis, the preparation of (R)-phenylacetylcarbinol (PAC) for L-ephedrine production catalysed by yeast whole cells dates back to that time and finds little to no recognition in chemistry textbooks (Scheme 1b).6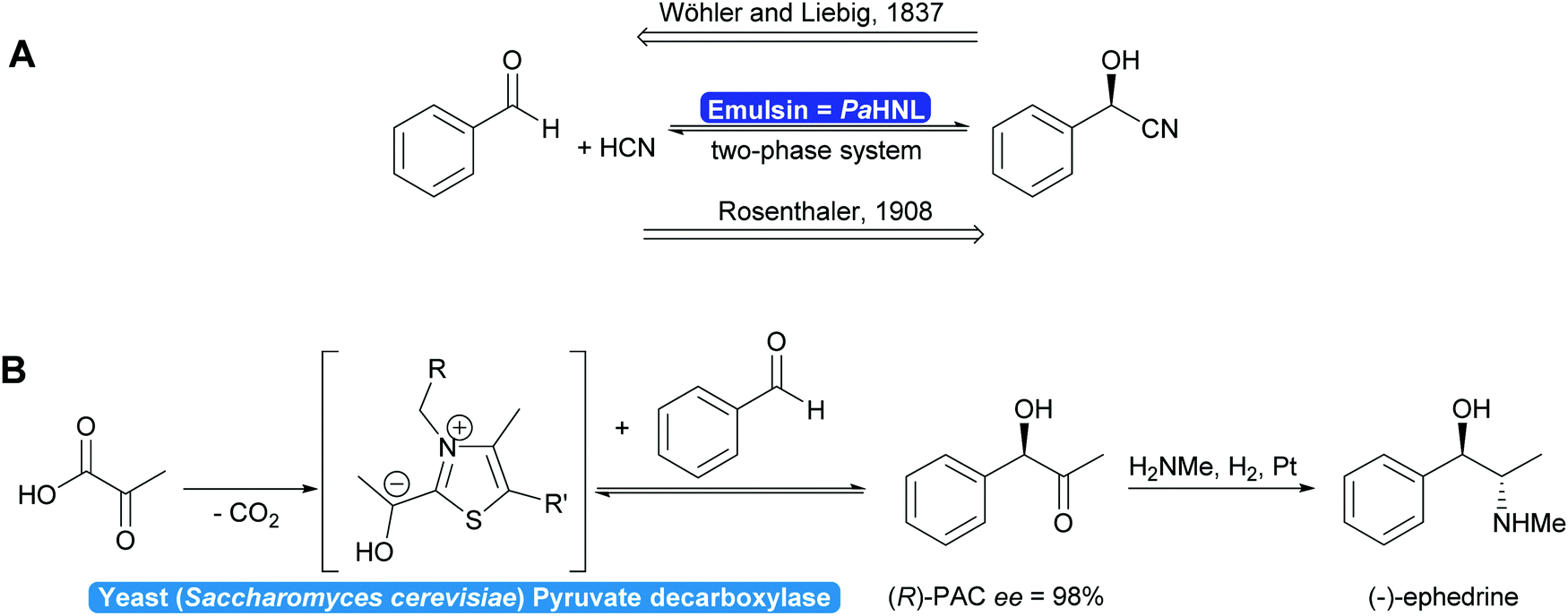 | ||
| Scheme 1 (A) In 1837 Wöhler and Liebig described the PaHNL-catalysed degradation of (R)-mandelonitrile. In 1908 Rosenthaler reversed this reaction in the synthesis direction, the first enantioselective reaction described. (B) Since the 1930s the industrial synthesis of L-ephedrine is based on the yeast-catalysed enantioselective synthesis of (R)-PAC. | ||
With the recognition of the need for selectivity in general and enantioselectivity in particular also came the recognition of enzymes as promising catalysts. The difference in perspective on selectivity is, however, still visible in the approach that the fields of catalysis take to describing their catalysts: while in most fields a catalyst that is broadly applicable, i.e. not very selective, is called versatile, in biocatalysis an enzyme that lacks selectivity is called promiscuous.7–9 This difference in terminology between the different fields of catalysis can lead to undesirable confusion.7
1.1 Enzymes: back to chemistry?
Two landmark reviews from 200310 and 20125 dispelled myths around biocatalysis and identified three waves in biocatalysis. The first wave started with the enantioselective synthesis of (R)-mandelonitrile and continued viaL-ephedrine synthesis towards the formation of selected drugs, such as penicillins all the way to the bulk production of fructose from glucose for the food production. All these processes rely on identifying suitable, naturally occurring enzymes without protein modifications. In all cases use was made of the excellent, if not occasionally too high, selectivity. The vast majority of these developments took place outside main stream chemistry, often in biochemical, food or pharmaceutical laboratories.11This was followed by a second and third wave of biocatalysis.5,10,11,14 In the second wave either well-thought through mutations were introduced or random mutagenesis coupled with massive screening yielded new enzymes that could answer the industrial needs. This included the required selectivity. The third wave combined the two approaches of enzyme improvement, reducing library sizes and allowing rapid screening. As a consequence, the adjustment of the properties of a given enzyme has become almost a standard laboratory technique. Broadening or narrowing the substrate scope, improving or inverting the regio- or enantioselectivity but also adjusting the optimal reaction conditions of enzymes are state-of-the-art nowadays. Furthermore, the reaction scope of biocatalysis is increasingly extended either by designing new biocatalysts or combinations of enzymes enabling transformations not thought possible a few years ago.15–18 As a consequence, the scope of biocatalytic bond formations today almost matches the scope of existing chemical technologies (Scheme 2).
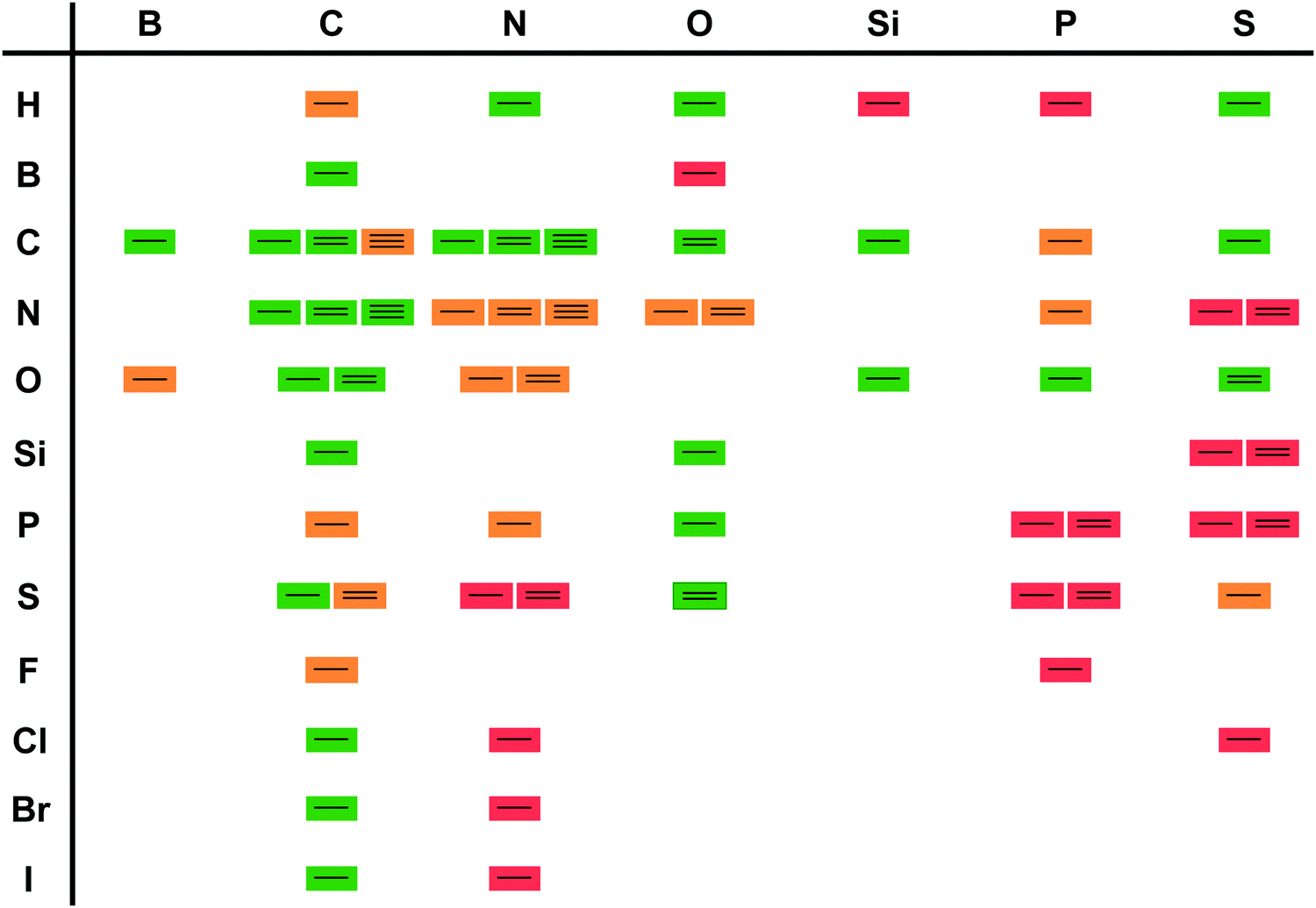 | ||
| Scheme 2 Synthetic transformations listed on https://www.organic-chemistry.org/synthesis/. Green boxes indicate that a biocatalytic equivalent is known; orange indicates that literature reports are available but that the reaction has not yet been applied synthetically. Red indicates a (so far) unknown biocatalytic bond formation. | ||
The results of this third wave now spreads to all fields of biocatalysis and is the focus of this review.
For example, enzymes involved in primary metabolism under natural conditions (intracellular environment) do not encounter high substrate or product loadings, nor are they typically confronted with drastic changes of conditions. All of this does however, occur in a batch reaction, in particular when working solvent-free.14,19 Here the second and third wave of biocatalysis help.5,10,11,14 The engineering of structural aspects helps to improve stability, in particular towards solvents20 and high concentrations of all reagents, substrates and products.21,22
An alternative method of enzyme stabilisation, their immobilisation, has been investigated with similar success and has been reviewed extensively. It is therefore not covered in this overview.23–29
2. Chemical transformations catalysed by biocatalysts
Based on the above written the reader might now expect that for the vast majority of chemical conversions a biocatalytic variant is available. This biocatalytic process should then have ideally several advantages over its uncatalysed chemical counterpart. While this status is not yet reached we here demonstrate the many waves that biocatalysis is by now making in organic synthesis. In many cases the biocatalytic methods complement the already available tools, enriching chemistry. We follow the sequence of the waves, C–C bond forming enzymes appeared early (Wöhler, Liebig, Rosenthaler),1,4,6 subsequently redox enzymes (Warburg) were intensely studied at a later stage.31,32 We combine this sequence with the disconnection approach common in organic chemistry.332.1 C–C-Bond forming reactions
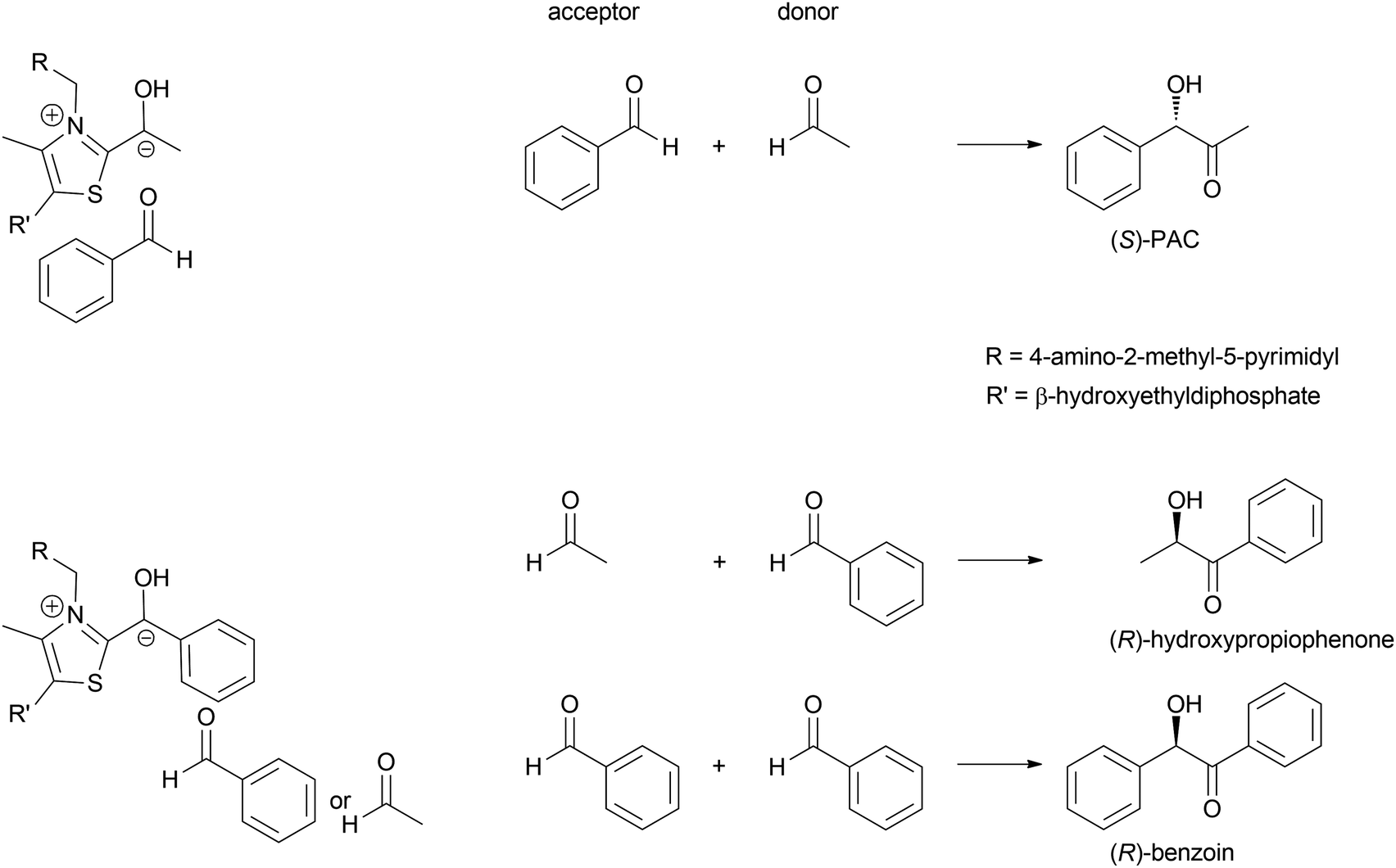
The formation of C–C bonds is essential for any organic synthesis to establish the carbon framework of the desired compound. This is in particular the case in total synthesis of natural compounds and pharmaceutical chemistry. Here the superior selectivity of enzymes is the basis of many successes. However, this selectivity to date also limits the broad application of enzymes, as there is no single enzyme that catalyses Friedel–Crafts chemistry in general, as AlCl3 does.6,7,15,34,35Up to the present time the largest part of the enzyme catalysed C–C bond formations are based on the nucleophilic attack on an aldehyde. This yields 1,2 disconnections in the case of cyanide or the umpolung of a second aldehyde or 1,3 disconnections when an enol(ate) is used as nucleophile.6,33,35,36 The aldehyde can be replaced by a ketone, though then the reactions often have lower yields as the reaction equilibria are less favourable. Essentially six nucleophiles (in biochemistry called donor) are today broadly utilised in combination with countless aldehydes or ketones (in biochemistry called acceptor) (Scheme 3). Additionally the aldehyde based biocatalytic equivalent of a Pictet–Spengler reaction is already well-known.37
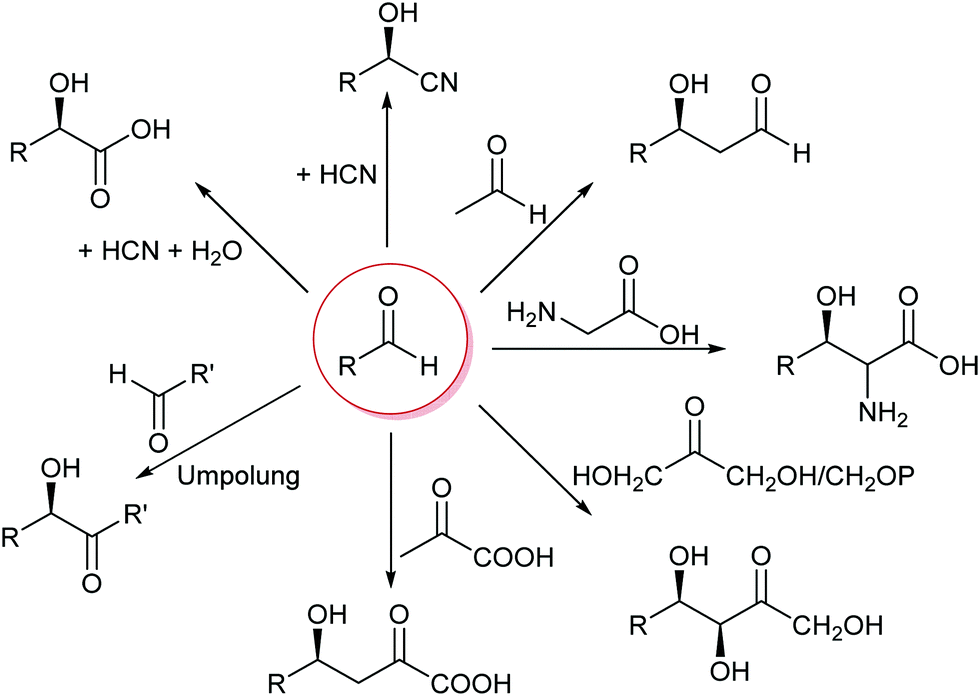 | ||
| Scheme 3 The majority of the today well-established biocatalytic C–C bond formations are based on the attack of a nucleophile on an aldehyde or ketone. | ||
2.1.1.1 HNL-Catalysed reactions. HNLs catalyse in nature the decomposition of cyanohydrins to release HCN as a defence mechanism.38,40,41 This catalytic activity is a prime example of convergent evolution. It evolved in many very different organisms, plants and animals utilising very different protein folds and thus catalytic mechanisms. Consequently a wealth of enzymes, both R- and S-selective is available. These enzymes work with sometimes very different mechanisms, from Lewis metal-catalysed to classical acid base catalysis, and have been further developed by enzyme engineering.40,41,44,45 Independent of the mechanism, the enzymes are typically utilised in two phase systems at low pH to suppress the undesired background reaction and ensuring high substrate loadings.38,45 Both industrially and for laboratory purposes the biocatalytic cyanohydrin synthesis is today the standard method.38,40,41,46 In addition to cyanide, nitromethane can also be employed as nucleophile (donor). This unnatural reaction is possible as both nucleophiles have similar pKa values and size and thus both fit into the active site. The synthesis then is a biocatalytic Henry reaction (Scheme 4).40,41,47,48
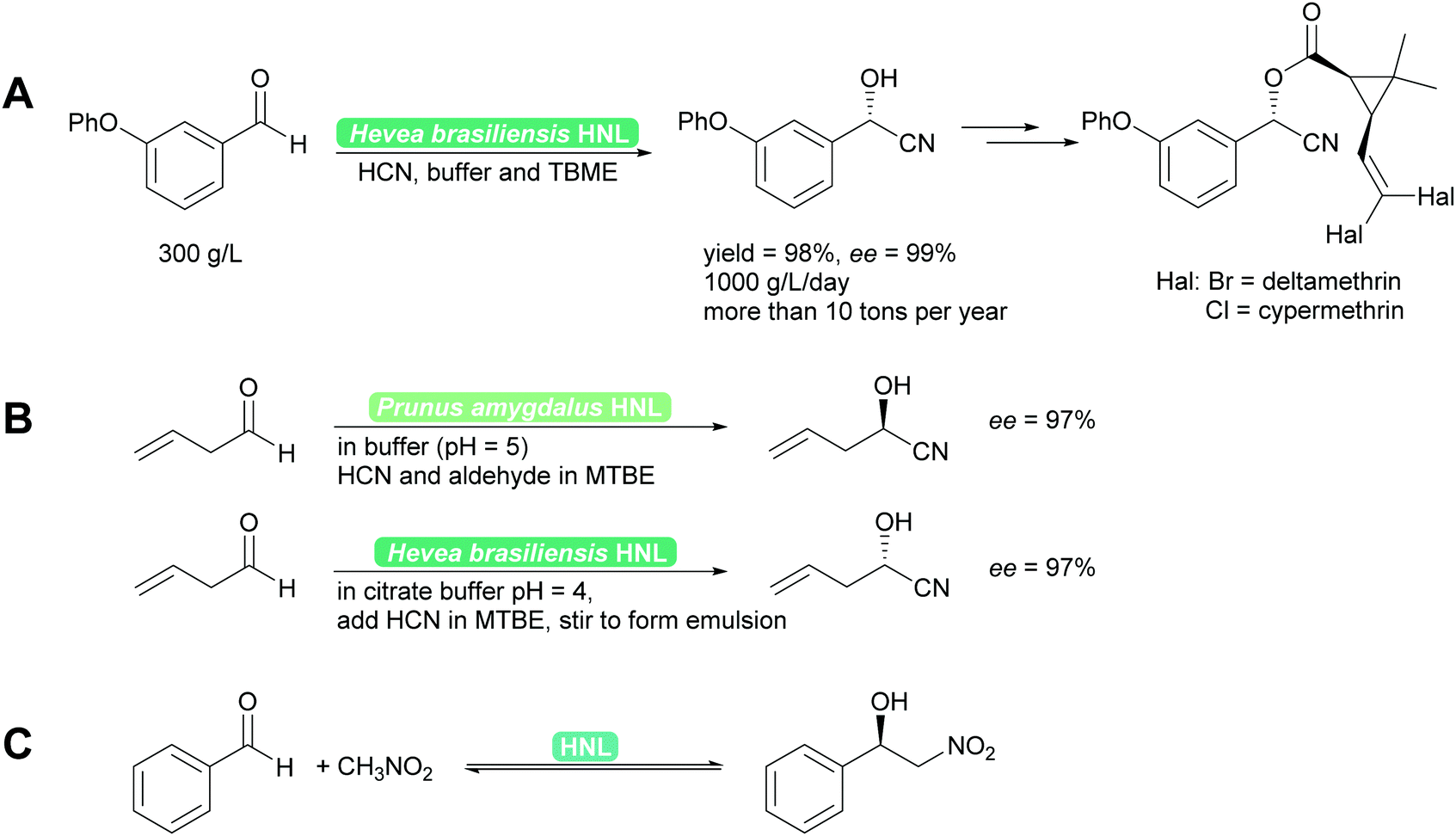 | ||
| Scheme 4 (A) Industrially HNLs are employed in biphasic systems. (B) For synthetic purposes they can be employed in organic solvents or biphasic systems. Both R- and S-cyanohydrins synthesis is straightforward49,50 and (C) equally the enantioselective Henry reaction can be performed. | ||
Recent developments have focussed on even more enzymes from unusual sources such as millipedes44,51–53 and bacteria,54–56 the exploration of evolutionary ancestors57 and other methods to interconvert HNL activity with other enzyme activities, in particular hydrolase activity.58 Reaction engineering with a special focus of the application of HNLs in flow has recently led to promising results. Single phase organic solvents could be employed and both high space time yields (STY) of up to 1200 g L−1 h−1 and cascade reactions were reported.59,60
2.1.1.2 ThDP-Dependent umpolungs reactions. All umpolungs chemistry in nature is based on ThDP as a cofactor. In its unphosphorylated form it is known as vitamin B1 and chronic lack of it causes the disease beriberi. The protein structure of the apoenzyme that then together with the ThDP forms the holoenzyme imparts both activity and selectivity to the ThDP.6,35,42,43 Given its unusual chemistry ThDP will be briefly described here (Scheme 5). Within the holoenzyme the ThDP is folded such that the pyrimidine and the thiazolium rings have an angle of 90 degree to each other. The amino group of the aminopyrimidine ring is thus in close proximity of the hydrogen of the C2 of the thiazolium ring and can abstract it. This deprotonation is supported by amino acids from the protein scaffold.61–63 The resulting ylide can also be viewed as tautomeric carbene.64 The ylide is then ideally posed to attack a carbonyl compound, the electrophile or acceptor. This acceptor can be either an aldehyde (ketones are very rare due to the steric hindrance) or an α-ketoacid. In the case of the aldehyde the initially formed adduct undergoes the umpolung; the negative charge now resides on the carbon of the aldehyde carbonyl group. In the case of decarboxylases α-ketoacids are decarboxylated, again resulting in the umpolung of the carbonyl group. This reactive intermediate can be drawn in two tautomeric forms, the enamine and the carbanion. It attacks the second carbonyl compound, again in most cases an aldehyde as ketones are sterically very demanding. Subsequent to the formation of the new C–C bond the ThDP is again split off and the product is released. The reaction can equally well proceed in reverse. In the case of transketolase this reverse reaction is the natural reaction it catalyses.
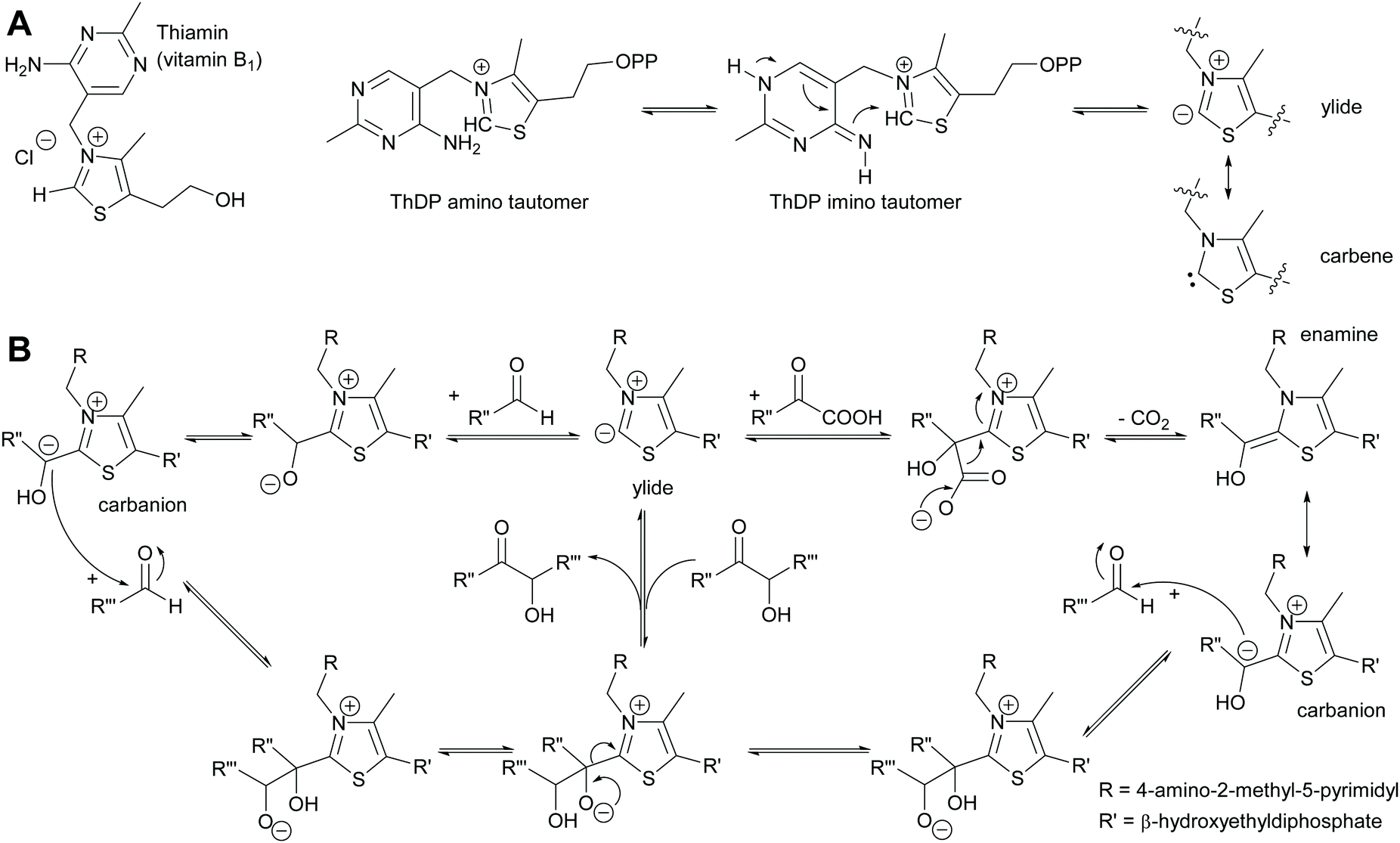 | ||
| Scheme 5 (A) Vitamin B1 in its phosphorylated form as ThDP is folded in the enzyme so that the two aromatic rings are at close to 90° to each other. The amino group can then deprotonate the thiazole ring. (B) The resulting ylide then attacks either an aldehyde or an α-ketoacid. This results in either case in the carbanion that as donor attacks another carbonyl compound. The new C–C bond is formed and the product released. In all cases the reaction is fully reversible and kinetically controlled. | ||
For a long time it was assumed that the forward reaction could be made irreversible by utilising α-ketoacids for the umpolung reaction. The release of gaseous CO2 was considered to thermodynamically make the revers reaction impossible.65,66 However, recently it has been demonstrated that this is a kinetic effect and that in both cases, with α-ketoacids and with aldehydes as donor molecule, the reaction is reversible.67
All ThDP-dependent enzymes are arrangements of proteins around the ThDP, typically with two ThDP cofactors between homodimeric proteins.42,43,61 It is these proteins that determine which molecule reacts with the ThDP, undergoes the umpolung and then acts as donor. This selectivity is typically high and often only relatively small molecules are accepted in the donor pocket. The second pocket enables the docking of the acceptor aldehyde and at the same time determines its orientation. This fixes the stereochemical outcome of the reaction. Elegant analysis of these binding pockets allowed the engineering thereof.68–72 As a result excellent control of the donor and acceptor molecule as well as the stereoselectivity was gained (Scheme 6). This work was further extended by engineering a decarboxylase in such a way that it could admit formaldehyde as acceptor, making it one of the few enzymes that can convert this toxic compound and at the same time enabling C1 chemistry also for the ThDP-dependent enzymes.73,74
 | ||
| Scheme 6 Based on the understanding of substrate docking in the active side both the donor pocket and the acceptor pocket can be modulated to accept the desired molecule. In this manner a toolbox of enzymes for all possible combinations could be established.68 | ||
Utilising the tools of the second and third wave of biocatalysis, ThDP dependent enzymes were also engineered to accept highly hydrophobic substrates and to enlarge the donor pocket. In this manner not only size but also other properties of the desired donors and acceptors have been taken into account.67,75–78 Additionally, sequence alignment and screening still yields other ThDP dependent enzymes that further expand the synthetic possibilities.79
The aldolases are grouped by the nucleophile they activate into four groups (Scheme 3).6,35,80 Overall three mechanisms are utilized. The acetaldehyde dependent aldolases utilises the enamine mechanism also known as class I and it is in its essence identical to the enamine chemistry in the organic chemistry textbook. A lysine reacts with the donor and then activates it as enamine which attacks the acceptor aldehyde. For the pyruvate and dihydroxyacetone phosphate (DHAP) dependent aldolases nature evolved two very different mechanisms. In higher plants and animals class I aldolases (enamine mechanism) are found. Class II aldolases are found in fungi and bacteria; here the mechanism is based on the Lewis acid activation of the donor and deprotonation thereof. For the glycine dependent aldolases a cofactor is required, pyridoxal phosphate (PLP). In its reduced, unphosphorylated form it is also known as vitamin B6, lack of which causes skin disorders.
2.1.2.1. Acetaldehyde-dependent aldolases. The only enzyme known to utilise acetaldehyde as donor is 2-deoxy-D-ribose-5-phosphate aldolase (DERA).86 It catalyses the reversible aldol reaction yielding 2-deoxyribose-5-phosphate (DR5P) from acetaldehyde and glyceraldehyde-3-phosphate (G3P). DR5P is stable, as it forms a hemiacetal, shifting the equilibrium to the aldol side. This hemiacetal formation is an element of reaction engineering that is utilised regularly in DERA-catalysed reactions and in aldolase-catalysed reaction in general.87,88 As the DERA-catalysed reaction is common in nature the number of DERA's is almost unlimited. The application of DERA struggles with one problem. The sensitivity of most DERA's to high acetaldehyde concentrations as acetaldehyde and in particular the side product of acetaldehyde self-condensation, crotonaldehyde, deactivate the enzyme.86,89 This has been addressed via screening and enzyme engineering while at the same time expanding the substrate scope.89–93 Although this improved the enzyme stability, additional reaction engineering, i.e. slow dosing of the acetaldehyde remains necessary. Immobilisation has also given promising results, including applications in flow.94,95
DERA is employed industrially.96–98 For several years already the synthesis of the statin side chain is based on an intriguing double aldol reaction. As the product of the first aldol reaction again yields an aldehyde this new aldehyde can also function as acceptor. After the second aldol reaction the product forms a stable hemiacetal, thus removing the product from the equilibrium. Recently this has been coupled to an ADH-catalysed oxidation yielding the desired lactone (Scheme 7).99,100 A more elaborate industrial cascade involving DERA is discussed below.
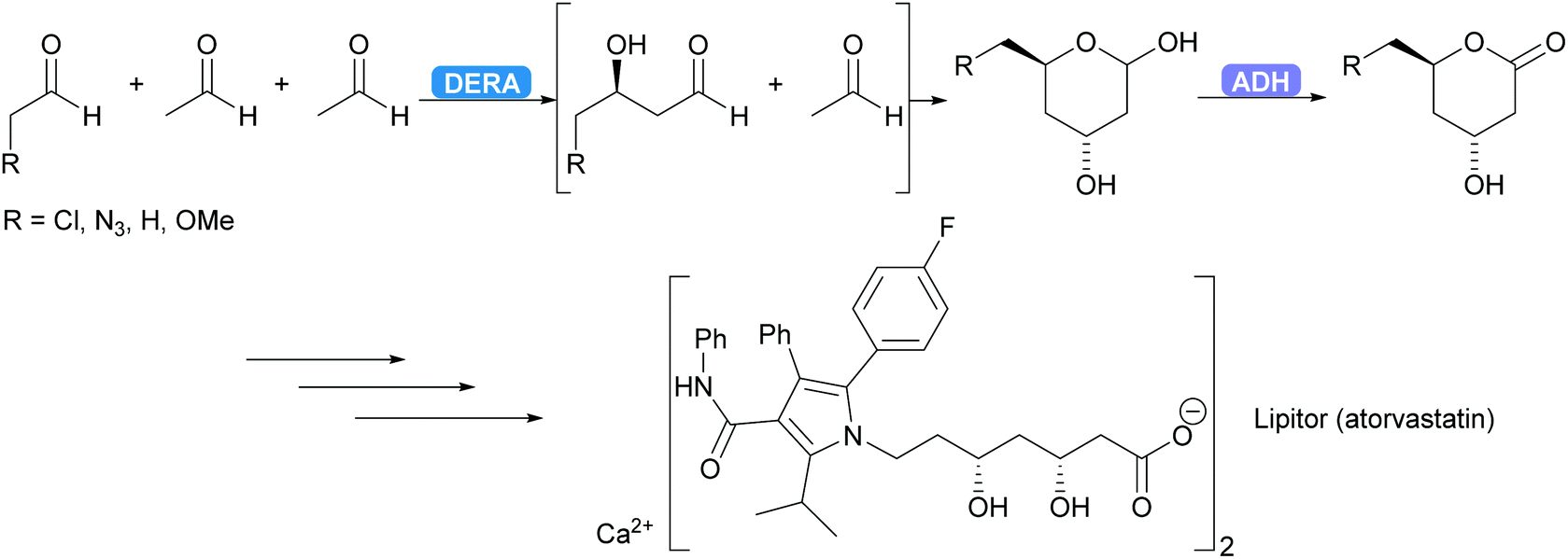 | ||
| Scheme 7 DERA catalyses a cascade of two aldol reactions. The stable hemiacetal is then oxidised by an ADH to the desired lactone, a key intermediate for the statin side chain. | ||
2.1.2.2. Pyruvate dependent aldolases. Pyruvate dependent aldolases play a central role in the synthesis of ulosonic acids and sialic acids which has recently been reviewed35,39,80,81,101,102 and a longstanding industrial example illustrates the power of the approach (Scheme 8), dating back to the first wave of biocatalysis.103–106 Class II pyruvate dependent aldolases are part of the lignin and polycyclic aromatic compound degrading enzymes. As degrading enzymes they are more flexible for their substrates and therefore in the focus of attention.107–109 In recent years enzyme engineering allowed to modify the active site of these class II aldolases to extend their scope.110–112 In particular a number of pyruvate nucleophile analogues was introduced. This not only increases the number of donors but it also means that the reaction now establishes two stereocentres. Similar results were obtained with screening efforts and today the pyruvate dependent aldolases accept a range of donors and first steps toward understanding of this relaxed selectivity were made.113–115 Interestingly also the stereoselectivity of the reaction seems to be somewhat relaxed as is the case with a number of glycine dependent aldolases.83,84 The earlier already observed thermodynamic stability of the product plays an important role in determining the diastereomeric outcome of the reaction here, too (Scheme 8). While the E/Z control over the enolate seems perfect, the attack of the acceptor is prone to lower selectivity.85,110
 | ||
| Scheme 8 (A) Already for more than 20 years pyruvate dependent aldolases are employed industrially to prepare N-acetylneuraminic acid on a ton scale. (B) in recent years the scope of pyruvate dependent aldolases has been extended and they now activate also pyruvate derivatives and establish two stereocentres in one step. The stereoselectivity is, however, often determined by the product stability. | ||
Another special type of pyruvate dependent enzymes have in recent years attracted attention. The hydratase–aldolase produce conjugated systems and therefore catalyse an aldol condensation rather than an aldol reaction. Isolated from organisms that degrade polycyclic compounds, these enzymes catalyse in the synthesis reaction the addition of pyruvate to aromatic aldehydes with subsequent elimination of water. Remarkably these enzymes have also been engineered for the synthesis of quinolones (Scheme 9).116,117
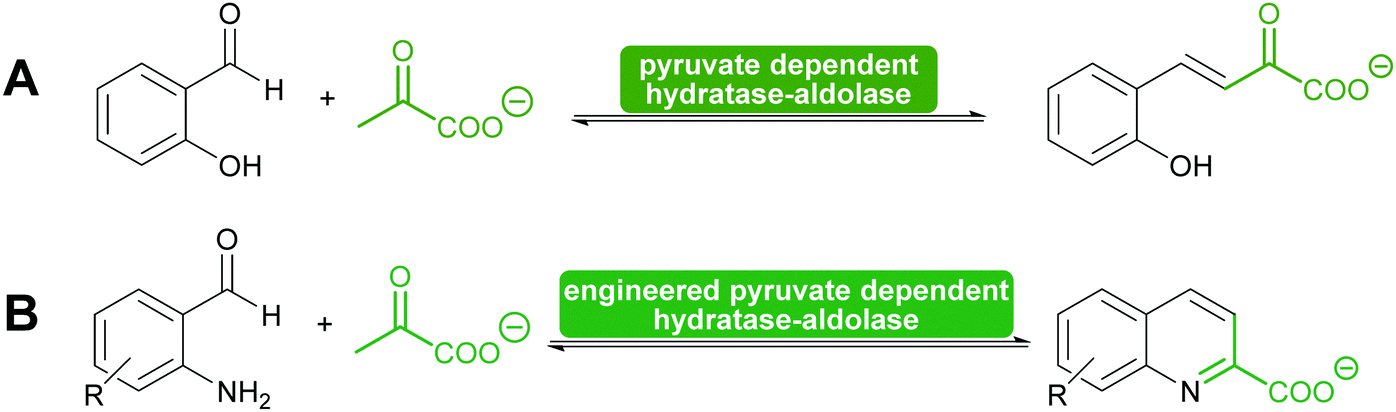 | ||
| Scheme 9 (A) Hydratase–aldolases are pyruvate dependent and degrade polycycling compound. (B) Engineering of a hydratase–aldolase enabled the synthesis of quinolones. The unstable amino group of the benzaldehydes was released just before the enzyme-catalysed reaction. | ||
2.1.2.3. DHA(P) dependent aldolases. The chemistry of the DAHP dependent aldolases is well explored.6,35,39,80,81 These powerful enzymes establish two stereocentres in a single reaction step. They are versatile catalysts for the synthesis of many sugars and their derivatives73,80,118–120 and also have been utilised for the preparation of many amino sugars.121 Overall four stereoisomers can be synthesised with these enzymes. For the syn-stereoisomer as well as for one of the anti-products the DHAP dependent aldolases are highly stereoselective. However, for the anti-product with the stereochemistry of tagatose, the corresponding tagatose 1,6-diphosphate aldolase (TDP A), displays low diastereoselectivity (Scheme 10). This is possibly due to a thermodynamically controlled product distribution in this case (see also Scheme 8B) as the observed lack of selectivity is diastereoselectivity and not enantioselectivity.85 While this lack of selectivity is typically considered a disadvantage, it recently has been utilised to prepare rare sugars such as L-psicose and L-sorbose.122
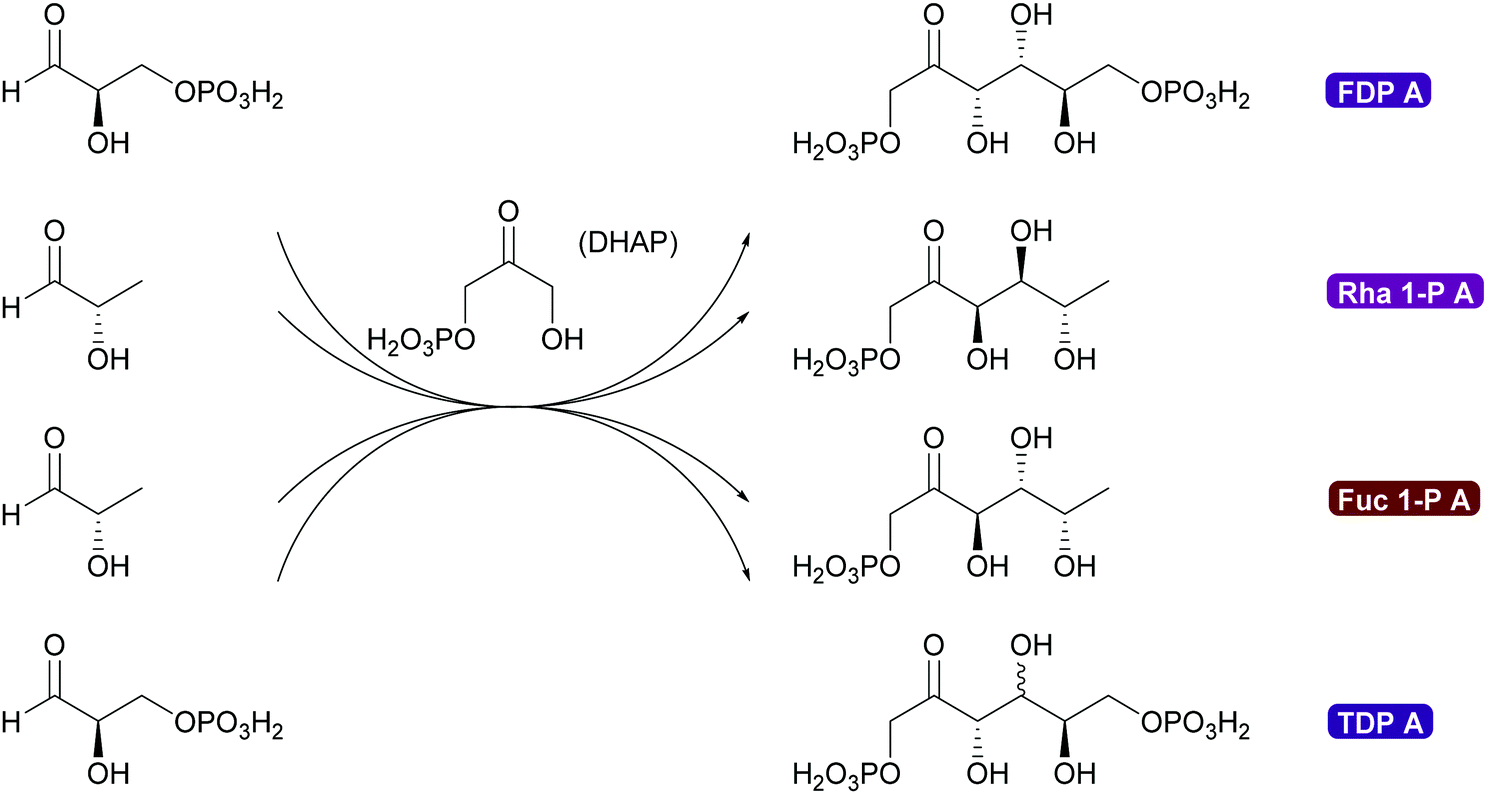 | ||
| Scheme 10 DHAP dependent aldolases establish two stereocentres in a single step. These enantioselective aldolases add to and complement chemical aldol technology. Formation of syn-products is catalysed with excellent enantio- and diastereoselectivity by rhamnulose 1-phosphate aldolase (Rha 1-PA) and fructose 1,6-diphosphate aldolase (FDP A). anti-Products are formed with excellent enantio- and diastereoselectivity catalysed by L-fuculose 1-phosphate aldolase (Fuc 1-PA), while tagatose 1,6-diphosphate (TDP A) aldolases displays lower diastereoselectivity. | ||
The DHAP aldolases are a versatile tool for the synthesis of sugars and sugar derivatives but their application is hampered by the low stability and high price of DHAP. To solve this problem two very different approaches were taken. In a classical approach a cascade of enzyme reactions was developed to generate DHAP in situ, starting from readily available glycerol. While first cascades required pH shifts this has by now been optimised and can even be applied in flow.80,120,123,124 The enzymatic DHAP generation has also been demonstrated to work well with any of the DHAP dependent aldolases. However elegant these approaches might be, the best answer to the problem is the direct application of dihydroxyacetone (DHA) instead of DHAP.
Genetic modification of L-rhamnulose-1-phosphate aldolase (Rha 1-PA) in the initial high throughput approach were outside the DHAP binding pocket.125 A second, more focussed approach, lead to modification of the DHAP binding pocket, making it more suitable for DHA.126,127 An even greater step forward was the results of a screening effort. Fructose-6-phosphate aldolase (FSA) does not require DHAP but DHA.128,129 This enzyme has completely replaced FDP A. Today modified Rha 1PA and FSA are utilised for countless syntheses of sugar derivatives and aliphatic alkaloids (Scheme 11).73,80,120,121 The donor in these enzyme variants is not limited to DHA, also other aliphatic and even cyclic ketones can be employed. The acceptor displays an even wider substrate range, including aromatic aldehydes.130–135
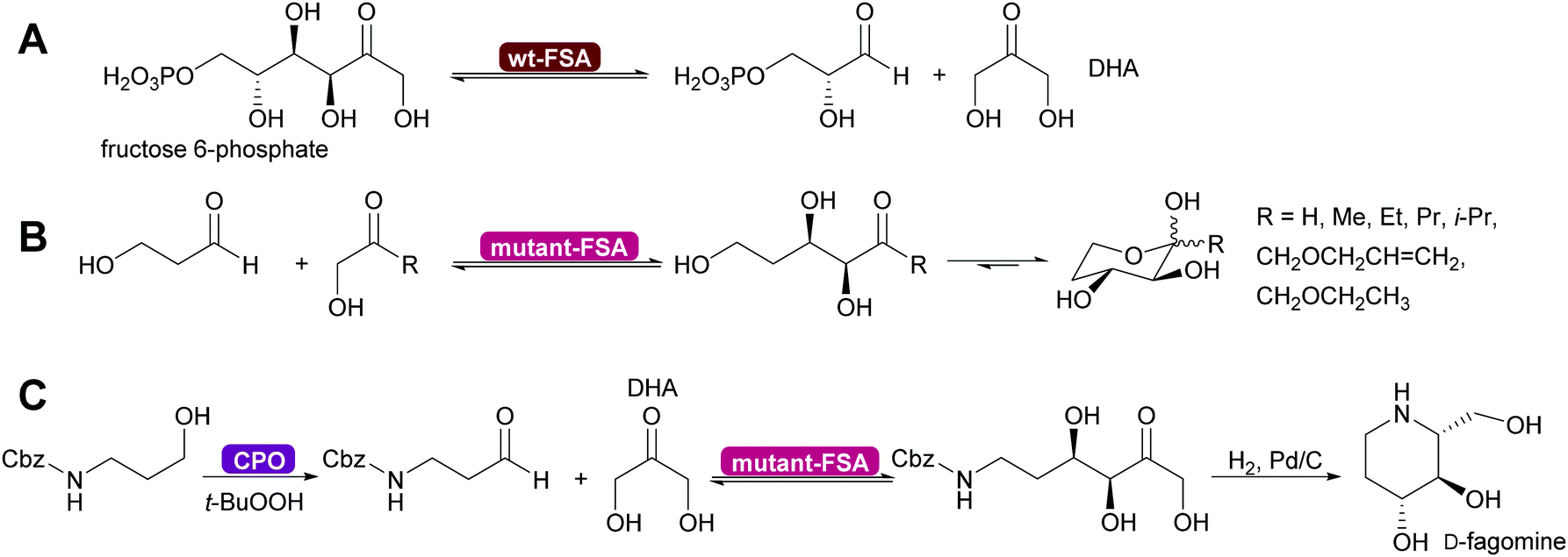 | ||
| Scheme 11 (A) Wild type D-fructose-6-phosphate aldolase (wt-FSA) does not require a phosphorylated DHAP but accepts DHA instead. (B) Mutations expanded the scope on the side of the donor molecule significantly, including even cyclic ketones. (C) The acceptor aldehyde can be varied extensively. Chloroperoxidase (CPO) catalysed in situ generation of sensitive aldehydes that then are converted into a precursor of D-fagomine.136 | ||
2.1.2.4. Glycine dependent aldolases. The glycine dependent aldolases require PLP as cofactor.6,35,39,80,137–139 This is essential for the activation of the donor. The amino group of the glycine is covalently bound to the cofactor and then attacks the acceptor. These enzymes convert a broad range of acceptors, inducing aromatic aldehydes and formaldehyde.73 Again the number of donors is very limited, next to glycine a small number of other amino acids with sterically not very demanding side chains can be utilised. The best studied enzymes are the threonine aldolases (TA). It was demonstrated that both the D- and the LTA are often hampered by low diastereoselectivity.83,84 While stereocontrol over the α-C is excellent the β-stereocentre is often badly defined. As explained above the more stable threo- or syn-products are the products under thermodynamic control, even if the enzyme has trans-selectivity. This problem has been approached by reaction engineering, i.e. utilising kinetic control, or by protein engineering.137,138,140 The equally glycine dependent aldolase serine hydroxymethyltransferase (SHMT) often complement the TA and together these enzymes enable new approaches to amino sugars, amino acids and pharmaceutically relevant 1,2 amino alcohols. In particular combinations with other aldolases allow for highly stereoselective syntheses (Scheme 12).141–143
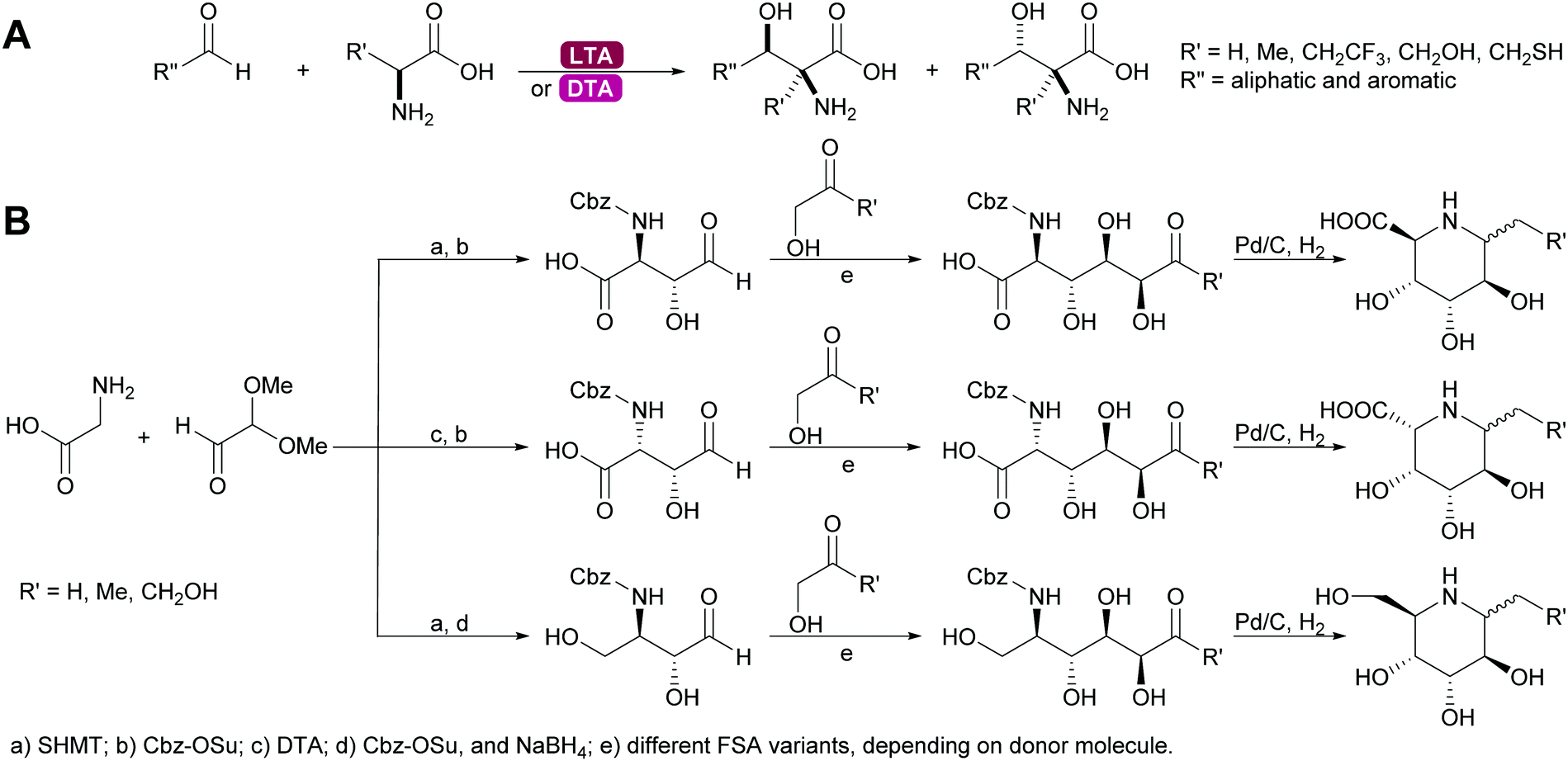 | ||
| Scheme 12 (A) Threonine aldolases catalyse the formation of a wide range of amino alcohols. (B) Glycine dependent aldolases in combination with other aldolases enable the synthesis of many alkaloids. | ||
2.1.2.5. Pictet–Spenglerases. This group of enzymes is part of the many enzymes that are involved in alkaloid biosynthesis which have been extensively reviewed. The chemistry is based on imine formation and subsequent Mannich chemistry in the case of the aliphatic substrates and Pictet–Spengler cyclisation in the case of aromatic substrates.37,144–146 The aliphatic substrates are often synthesised with aldolases (see above). For the aromatic alkaloids here phenethylamine or its derivatives react with the suitable aldehyde, forming the imine. The Pictet–Spenglerase then catalyses the cyclisation leading to tetrahydroisoquinolines. The best studied enzyme is the norcoclaurine synthase (NCS). It has been carefully evolved to accept many different substrates and was recently utilised for the formation spiro tetrahydroisoquinoline alkaloids (Scheme 13).147,148
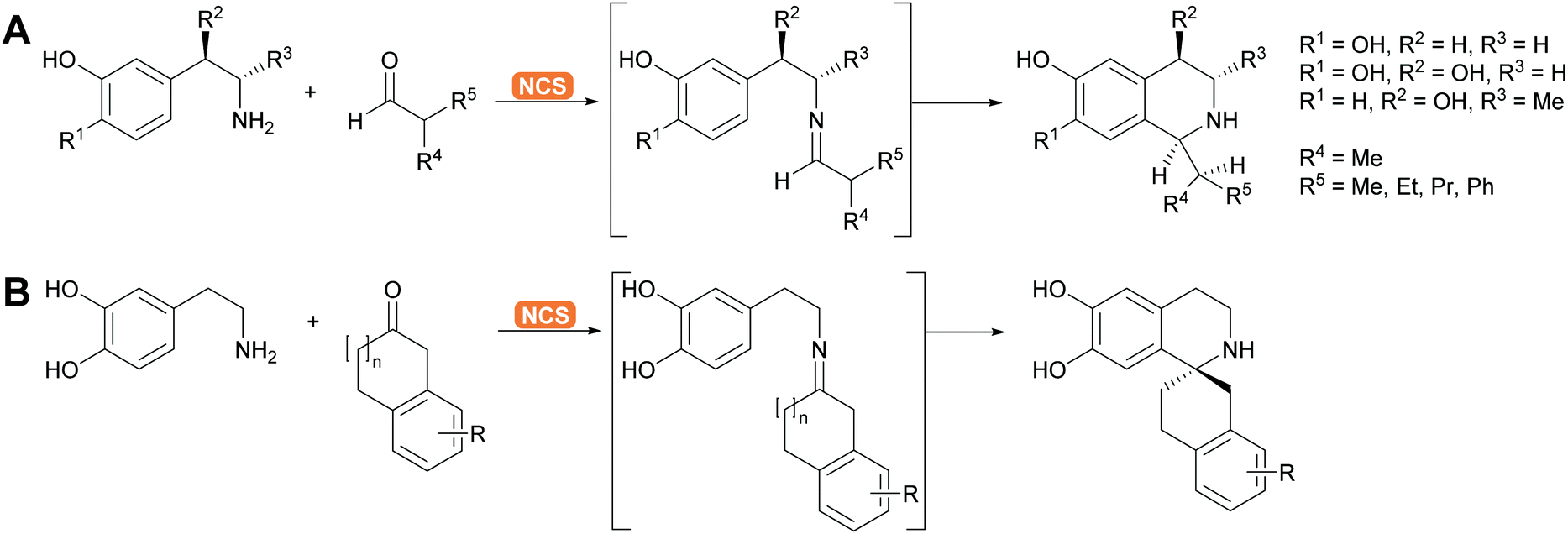 | ||
| Scheme 13 (A) Engineered NCS enables the enantioselective Pictet–Spengler cyclisation of chiral dopamine derivatives. (B) Equally spiro compounds are accessible via engineered NCS, catalysing in this case ring closure starting from a ketone rather than an aldehyde. | ||
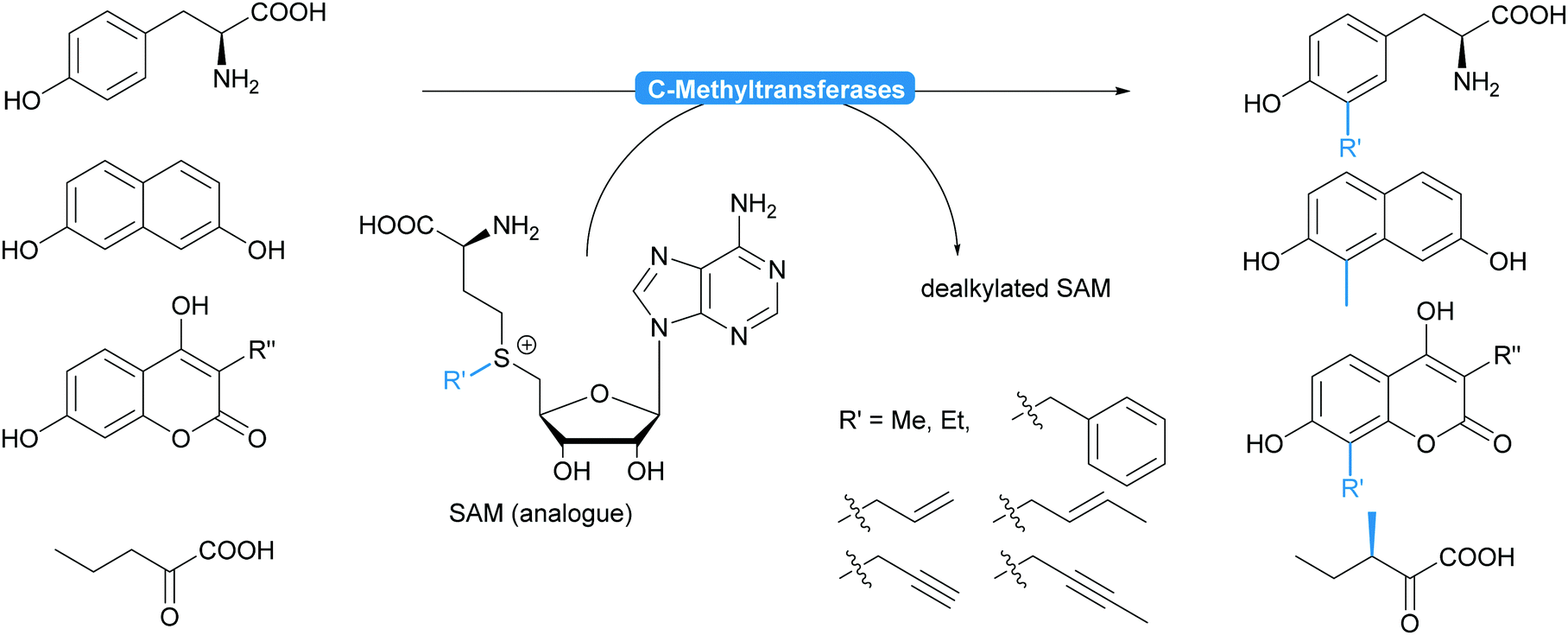 | ||
| Scheme 14 Methyl transferases chemoselectively alkylate electron rich aromatic compounds and enantioselectively ketoacids. While very selective for C–C bond formations, they accept a range of other alkyl groups in addition to the methyl group. | ||
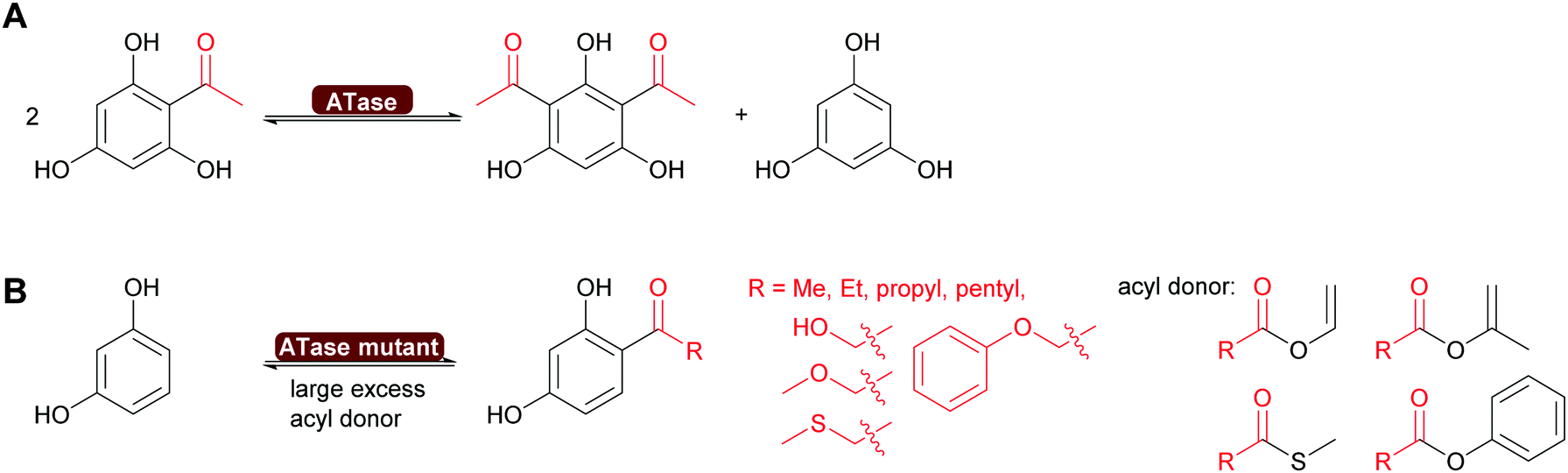 | ||
| Scheme 15 (A) ATase catalyses the disproportionation of a singly acylated phenol. (B) Engineered ATase can introduce a range of different acyl groups, starting with activated acyl donors in excess. | ||
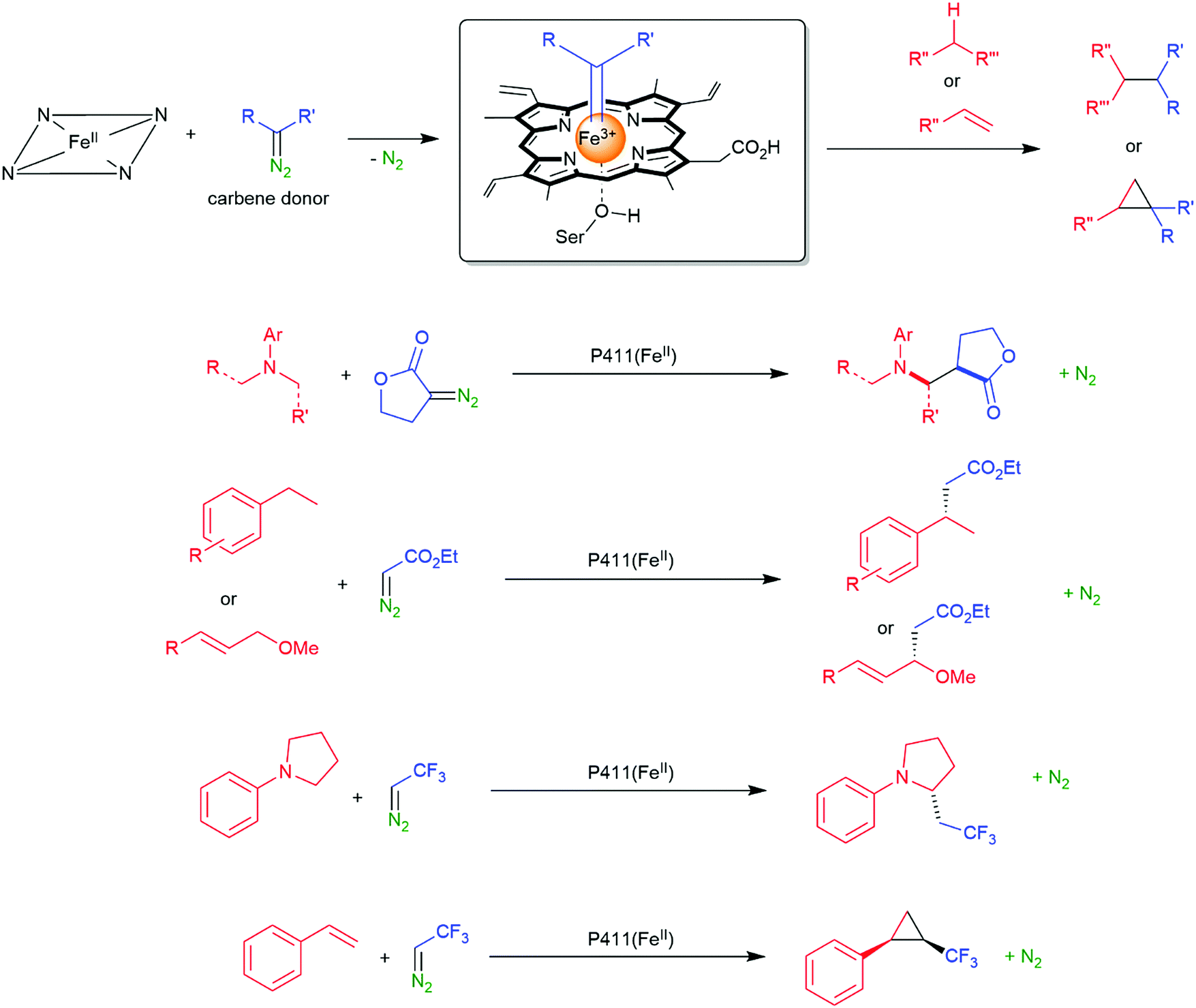 | ||
| Scheme 16 Direct C–H activation using P411 enzymes forming Fe carbene species.161,162,164–170 | ||
2.2 Selective functionalisation of C–H bonds (C–H to C–X)
Selective functionalisation, especially oxyfunctionalisation of non-activated C–H bonds is a showpiece of biocatalysis. Chemical catalysts exhibiting sufficient oxidation power to activate C–H bonds are recurrently plagued by selectivity issues whereas oxidoreductases often exhibit excellent regio- and stereoselectivity.The synthetic breadth of enzymatic oxyfunctionalisation chemistry has been covered by various review articles.31,171–176
2.2.1.1. Aromatic hydroxylation reactions. A range of flavoprotein monooxygenases (FPMOs) are available for the selective ortho- or para-hydroxylation of activated arenes such as phenols (Scheme 17).
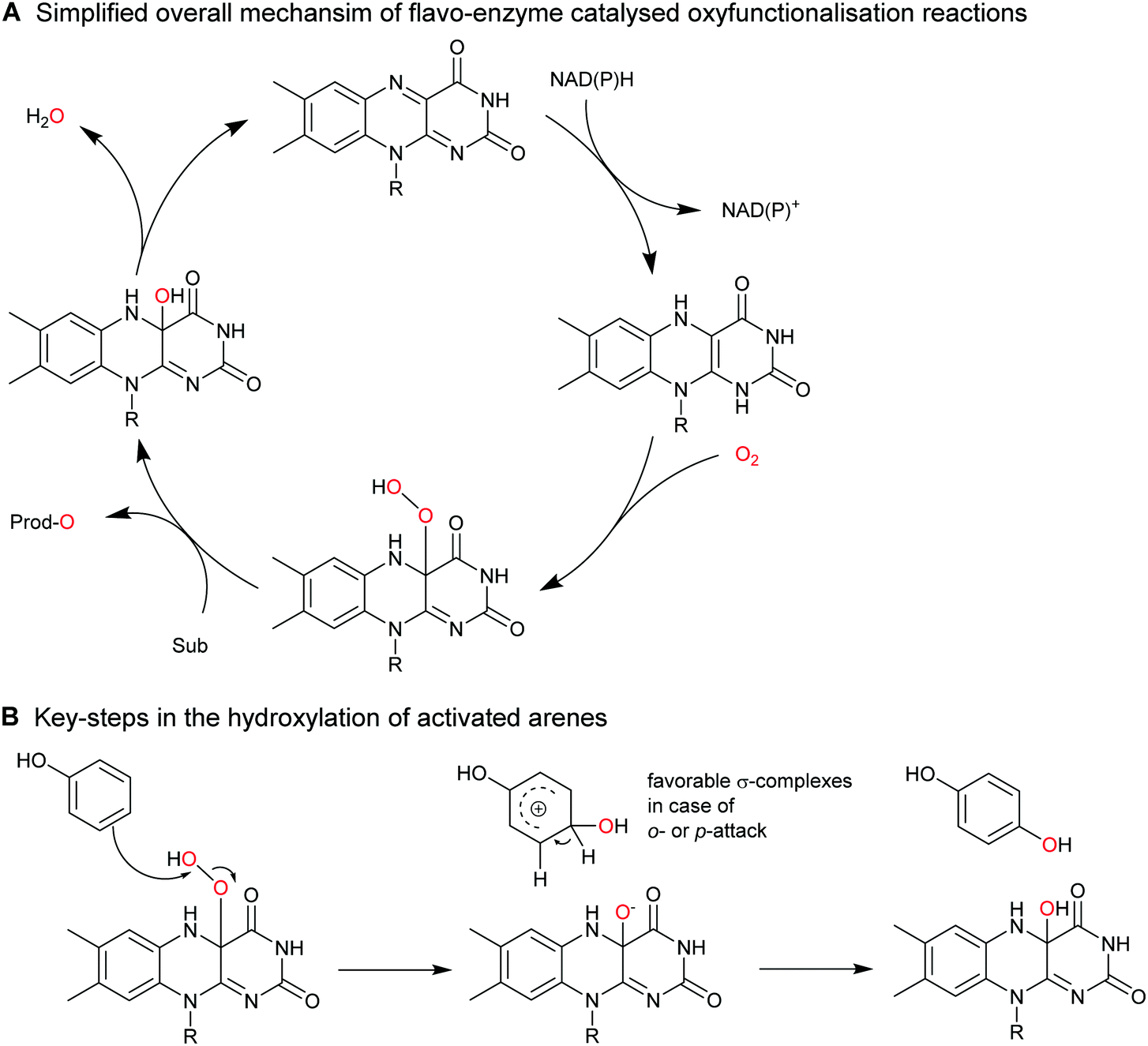 | ||
| Scheme 17 Aromatic hydroxylation of activated arenes (e.g. phenols) by FPMOs. (A) the general catalytic cycle of FPMOs comprises NAD(P)H-dependent reduction of the enzyme-bound flavin prosthetic group followed by (multistep, the individual intermediates are not shown) activation of O2 as flavin-4a-hydroperoxoflavin, which performs the oxyfunctionalisation reaction; the catalytic cycle is closed after water elimination. (B) The reactive 4a-hydroperoxoflavin is capable of electrophilic attack at electron-rich arenes such as phenols. | ||
Though, in principle a broad range of enzymes is available,171 only few transformations have found preparative applications such as the ortho-hydroxylation of ortho′-substituted phenols catalysed by the 2-hydroxy biphenyl-3-monooxygenase from Pseudomonas azelaica (HbpA).177,178 The wild-type enzyme shows high preference for 2-hydroxy biphenyl, which can be relaxed by enzyme engineering.179–181
Less activated arenes such as benzene are not converted by the above-mentioned biocatalysts but by some higher redox-potential heme- and non-heme iron monooxygenases.176 The catalytic mechanism of these hydroxylation reactions differs somewhat from the mechanism of FPMOs involving an intermediate arene epoxide, which spontaneously rearranges into the final phenol product (Scheme 18).
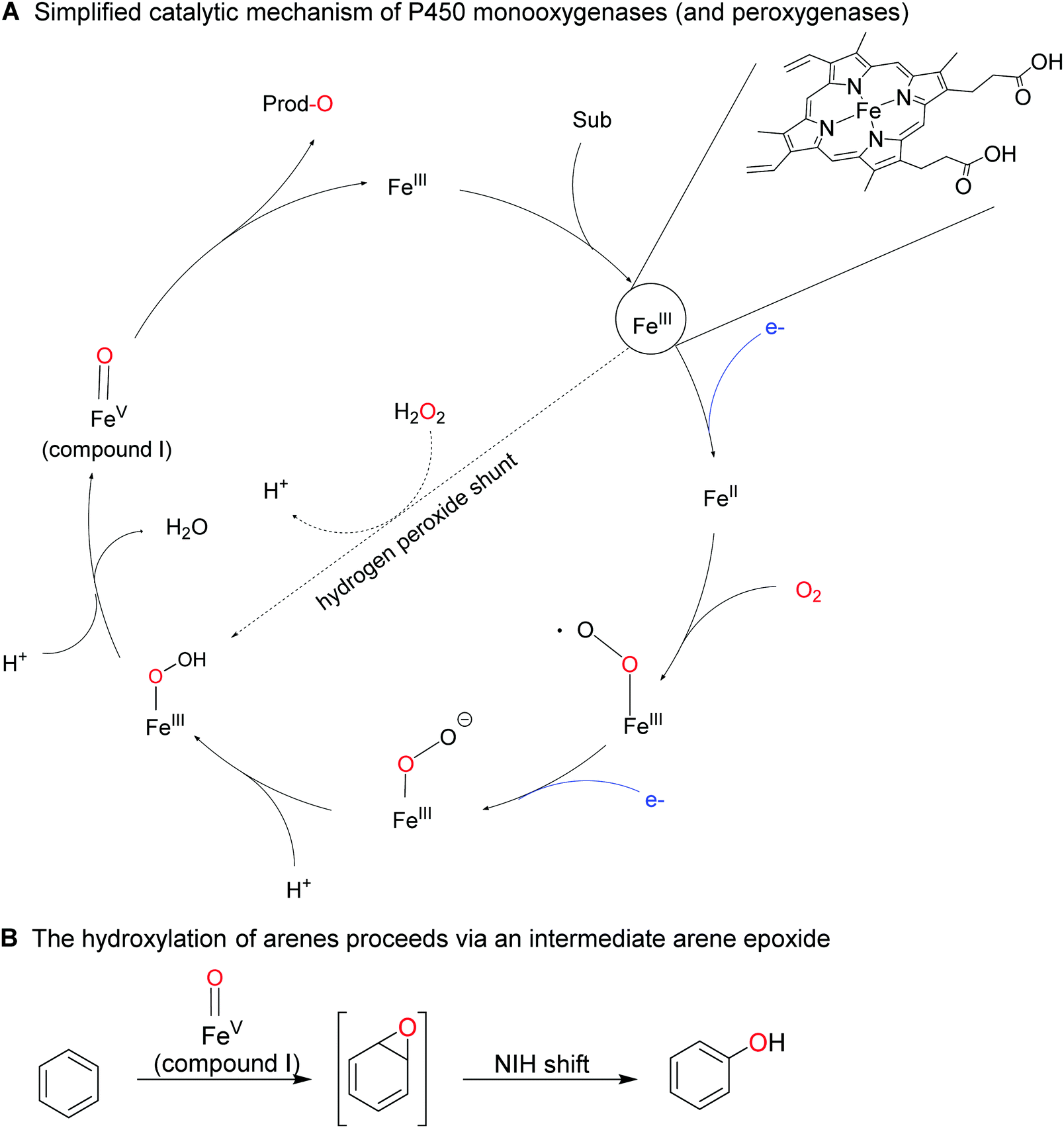 | ||
| Scheme 18 Simplified mechanism of P450 monooxygenase-catalysed activation of molecular oxygen resulting in the oxygenating species ‘compound I’ (formally an Fe(V) oxo species (a)). The reductive activation of molecular oxygen comprises sequential single electron reduction of the enzyme-bound iron and water elimination of the Fe-bound O2-species. In the presence of (already reduced) H2O2, compound I can also be formed directly from the resting Fe(III) state via the hydrogen peroxide shunt pathway. B: Aromatic compounds are converted into the corresponding phenols by a sequence of compound I-mediated epoxidation of the aromatic ring followed by spontaneous re-arrangement into the final product (NIH shift). | ||
P450 monooxygenases such as the enzyme from Bacillus megaterium (P450BM3) have been studied extensively for the ring-hydroxylation of benzene and its derivatives.182–187 Especially in case of alkyl substituted arenes the regioselectivity can be rather poor, yielding complex mixtures of ring- and side-chain hydroxylated products. Enzyme engineering however can alleviate this issue.185
An interesting enzymatic cascade utilising an engineered P450BM3 in combination with arbutin synthase was reported recently by Reetz and co-workers (Scheme 19).184 The semi-rationally designed P450BM3 variant enabled selective para-dihydroxylation of benzene (devoid of over-oxidation products commonly observed in chemical processes). Coexpression with arbutin synthase yielded an E. coli catalyst transforming benzene into arbutin with 60% isolated yield.
 | ||
| Scheme 19 Arbutin synthesis using an engineered P450BM3 variant to transform benzene into catechol. | ||
Next to the monooxygenases, also peroxygenases are worth mentioning as catalysts for the hydroxylation of arenes.188–191 Issues associated with the peroxidase activity of peroxygenases resulting in undesired radical polymerisation of the phenol products can be circumvented by the addition of antioxidants or using engineered peroxygenases.192,193
Next to the above-mentioned oxygenases, also a range of dehydrogenases are known (and used industrially) for the ortho-hydroxylation of pyridines (Scheme 20).194–196 The O-atom introduced does not stem from molecular oxygen (as to be expected from a monooxygenase-catalysed hydroxylation) but from water.197 The mechanism of this reaction is yet to be elucidated.
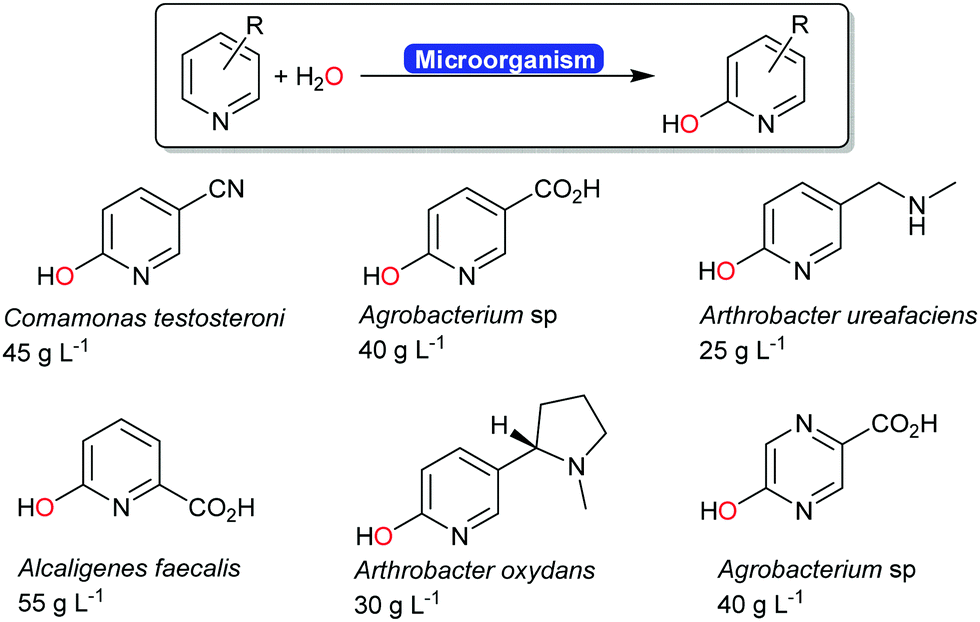 | ||
| Scheme 20 Industrial examples of ortho-hydroxylating transformations of pyridine derivates.194 The carboxylate groups shown in blue were obtained from the biotransformation of the corresponding nitrile. | ||
Finally, also aromatic nitration is worth being mentioned here. Even though this activity is rarely found in nature,198 the P450 monooxygenase from Streptomyces scabies (P450 TxtE) catalyses the nitration of tryptophan.199–202 In a directed evolution approach, Arnold and co-workers were even able to modulate the regioselectivity of the nitration reaction (Scheme 21).200
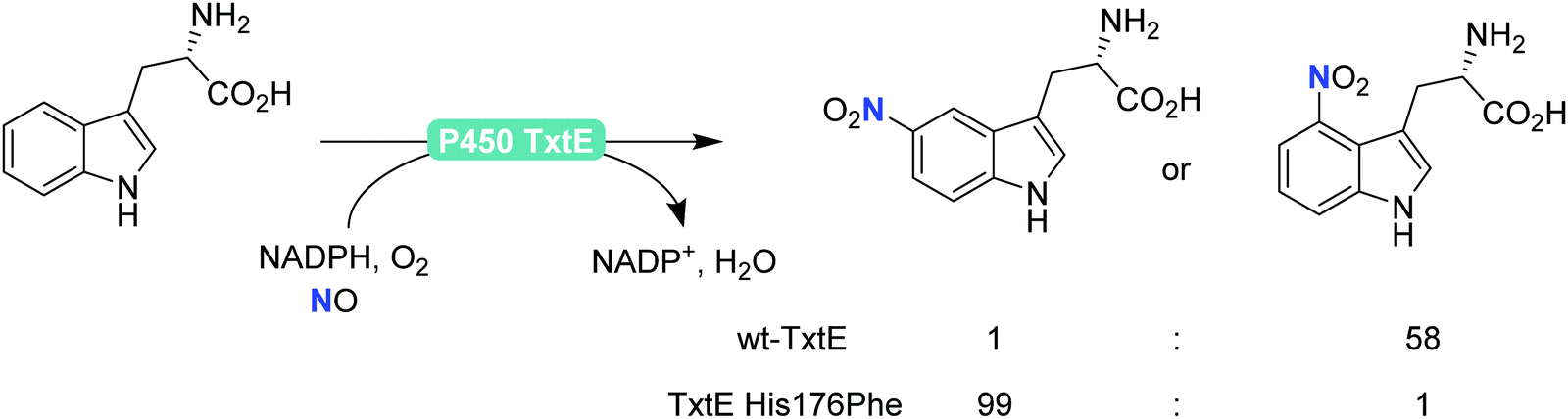 | ||
| Scheme 21 Regioselective nitration of tryptophan with the P450 monooxygenase from Streptomyces scabies (P450 TxtE) and one of its mutants. | ||
Though the catalytic activity of the enzyme still needs improvement, this method bears considerable synthetic potential.
2.2.1.2. Aromaticity breaking oxidation reactions. The majority of biocatalytic transformations of aromatic compounds yield aromatic products. Comparably few aromaticity-breaking transformations are known so far. The most prominent example for this is the cis-dihydroxylation of arenes using dioxygenases (Scheme 22). Particularly Hudlicky and co-workers have extensively explored the synthetic possibilities that dioxygenases offer for the organic chemist203–205 A range of (recombinant) dioxygenases are nowadays available for the regio- and stereoselective cis-hydroxylation of a broad range of arenes. As chemical alternatives exhibiting comparable selectivity patterns are rare, these catalysts have been recognised early on by preparative organic chemists in natural product synthesis (Scheme 22).
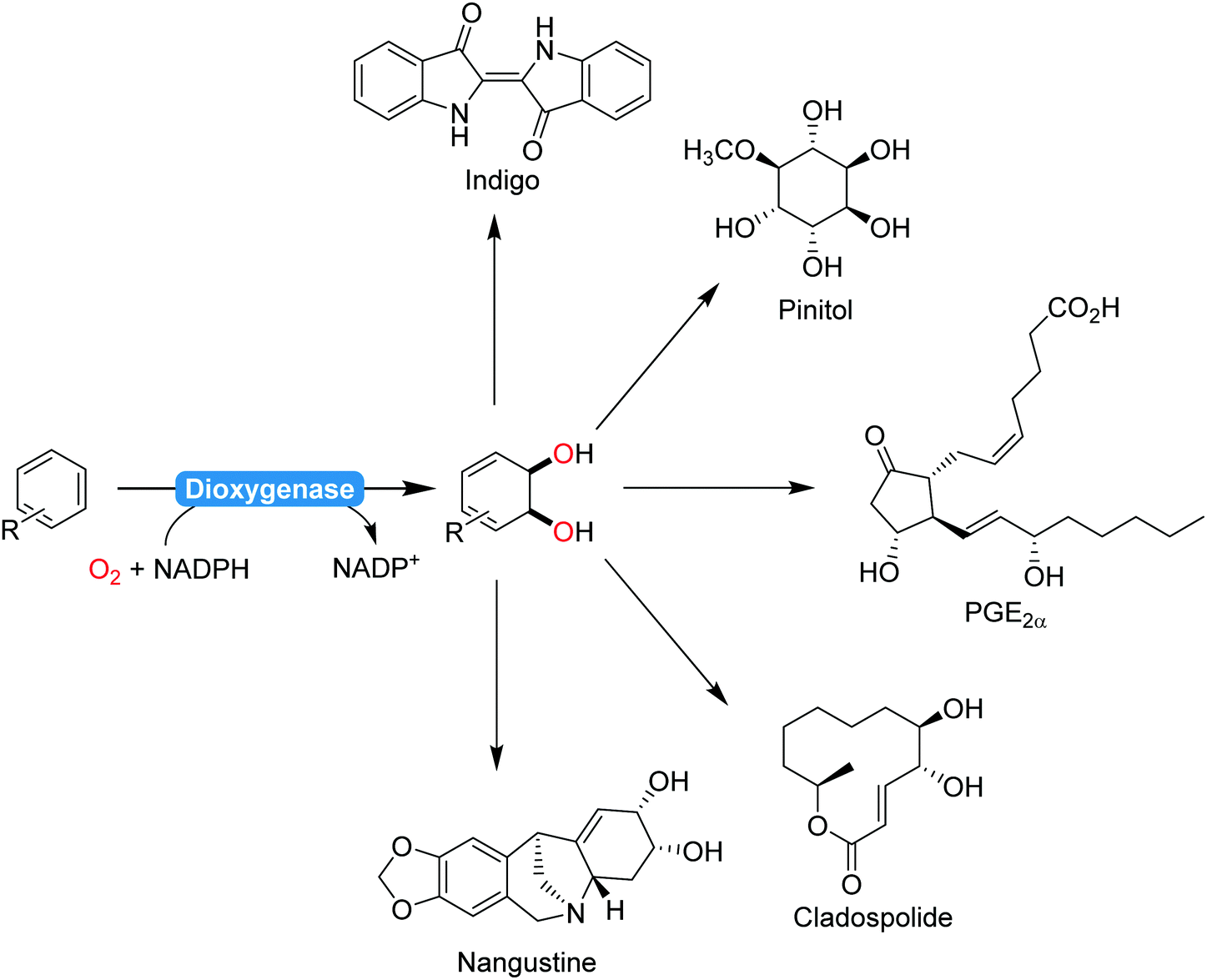 | ||
| Scheme 22 Selection (incomplete) of natural products synthesised from dioxygenase products:206 indigo,207 pinitol,208 prostaglandins,209 cladospolide,210 or nangistine.211 | ||
Aromatic hydroxylation reactions using heme-dependent enzymes proceed via an unstable arene oxide intermediate, which spontaneously rearranges into the corresponding phenol product (Scheme 18).191,212 This intermediate, however, can also be trapped via nucleophilic attack yielding trans-disubstituted cyclohexadienes (Scheme 23).213
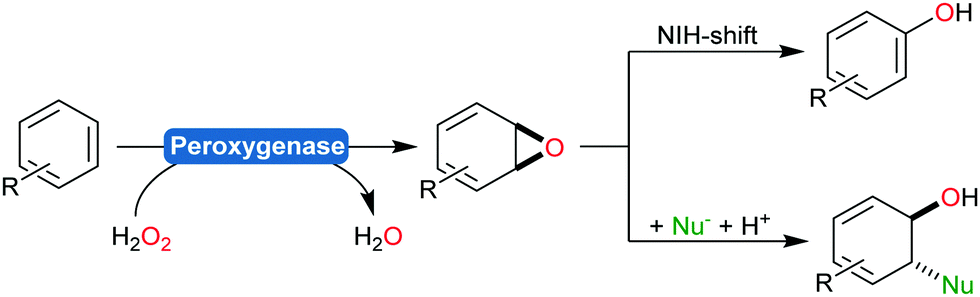 | ||
| Scheme 23 Peroxygenase-catalysed epoxidation of aromatic compounds followed by either a spontaneous rearrangement reaction (NIH shift) yielding phenols or by reacting the intermediate with nucleophiles yielding trans-disubstituted cyclohexadienes. | ||
Very recently, some new flavin-dependent monooxygenases have been discovered that catalyse the aromaticity-breaking hydroxylation of resorcinol derivates.214 So far the enzymes TropB, SorbC and AzaH with complementary site- and stereoselectivities have been identified from biosynthetic pathways. Like other flavin-dependent monooxygenases, these enzymes utilise 4a-hydroperoxoflavin as oxidation agent and direct it to the ortho- or para-position of a pre-existing OH substituent. Their selectivity, however, prefers attack at substituted ring-C-atoms. The resulting σ-complex therefore cannot collapse via simple deprotonation and re-aromatisation but rather by ortho-quinol formation (Scheme 24).
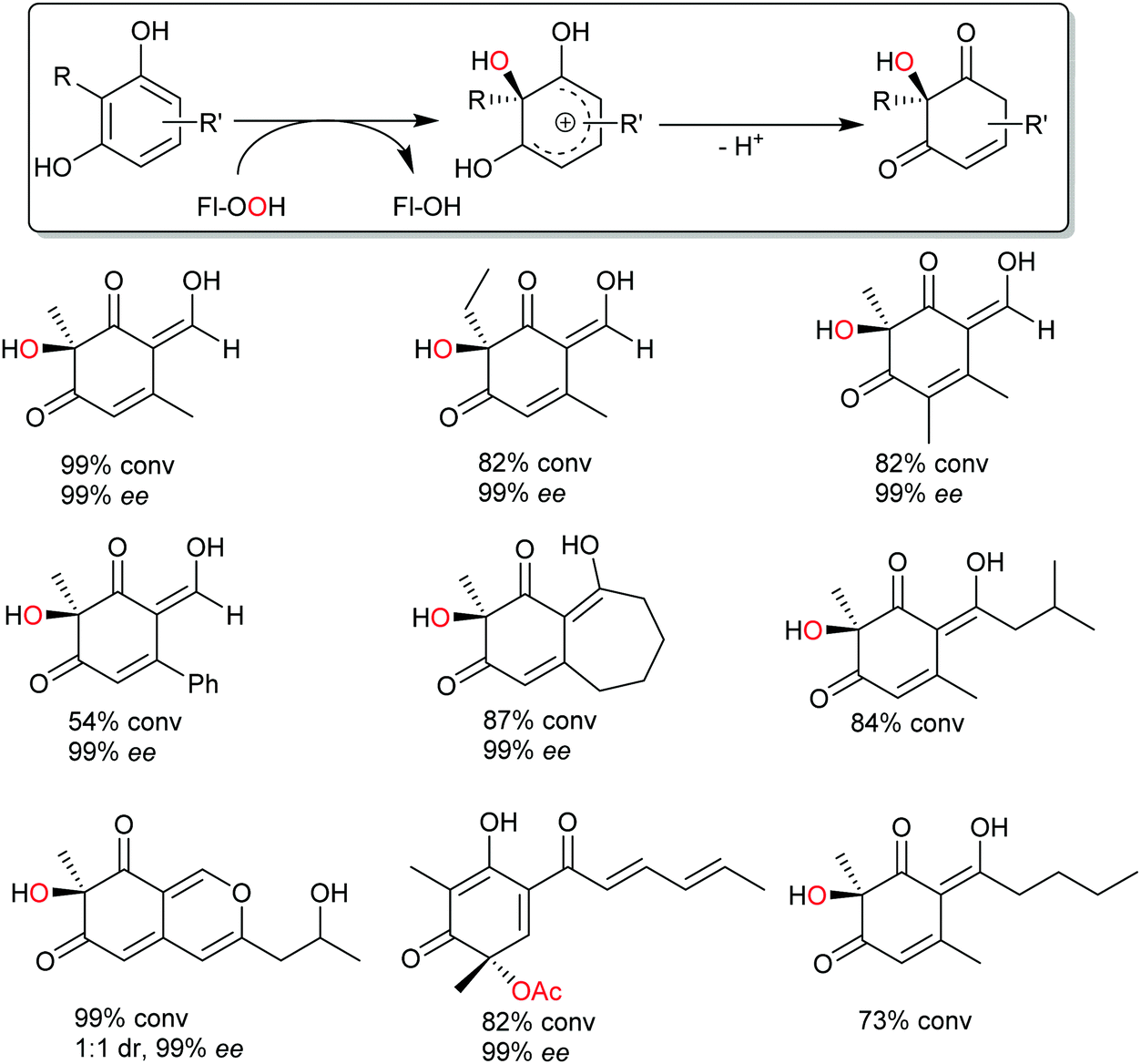 | ||
| Scheme 24 Aromaticity-breaking hydroxylation of substituted phenols. | ||
So far, few preparative examples using this promising reaction have been reported but we are confident that further mechanistic understanding and broadening of the substrate scope will result in a range of useful catalysts/transformations for the organic chemist.
2.2.1.3 Hydroxylation of sp3 C–H bonds. The inertia of sp3-hybridised C–H bonds (even in ‘activated’ benzylic or allylic position) requires highly potent O-transfer agents, which are rarely selective. P450 monooxygenases combine both properties by placing the highly active oxyferryl (FeIV
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O+) species (in situ generated from its resting FeIII state) in the well-defined framework of the enzyme active site. The same also controls binding of the substrate and thereby its positioning towards the oxidising agent. This way, though various positions within the starting material could react, only few of them get close enough to the oxiferryl species to react. Fatty acids for example can be hydroxylated selectively in α-,215 β-,216 or ω-position,217–220 depending on the enzyme used. More recently, ‘in-chain’ hydroxylating P450 monooxygenases have also been reported,221,222 giving rise to the assumption that soon a set of P450 monooxygenases with tailored selectivity to selectively transform a given C–H-bond within an alkyl chain will be available.
O+) species (in situ generated from its resting FeIII state) in the well-defined framework of the enzyme active site. The same also controls binding of the substrate and thereby its positioning towards the oxidising agent. This way, though various positions within the starting material could react, only few of them get close enough to the oxiferryl species to react. Fatty acids for example can be hydroxylated selectively in α-,215 β-,216 or ω-position,217–220 depending on the enzyme used. More recently, ‘in-chain’ hydroxylating P450 monooxygenases have also been reported,221,222 giving rise to the assumption that soon a set of P450 monooxygenases with tailored selectivity to selectively transform a given C–H-bond within an alkyl chain will be available.
The potentially enormous versatility of (non-)heme monooxygenases for the selective oxyfunctionalisation often is challenged by either too high or too low selectivity. As a rule of thumb, monooxygenases involved in specific biosynthetic pathways such as secondary metabolites tend to be highly substrate specific. Already small modifications within the structure of the starting material compared to the natural substrate can lead to complete inactivity of a given enzyme. Enzymes playing a role in detoxification/degradation tend to be rather unselective, converting a broad range of starting materials albeit with often low selectivity.
Enzyme engineering has proven being a powerful tool to address those selectivity issues (Scheme 25).
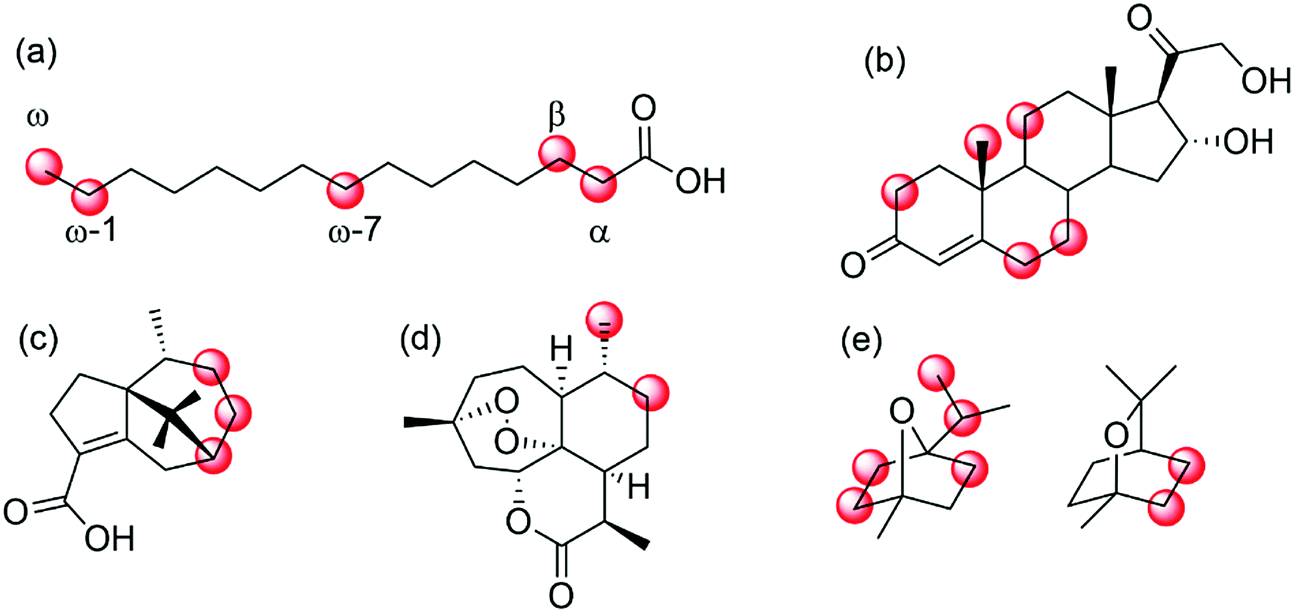 | ||
| Scheme 25 Selected examples of regio- and enantioselective hydroxylation of diverse hydrocarbons by means of (engineered) P450 monooxygenases. (a) regioselective hydroxylation of fatty acids in α-,215 β-,216 ω-7,221,222 ω- and ω-1-position;217–220 (b) hydroxylation of 11-deoxycortisol;223,224 (c) P450BM3 mutants for selective hydroxylation of cyperenoic acid;225 (d) P450 BM3 mutants for the selective hydroxylation of artemisinin,226 or (d) hydroxylation of 1,4- and 1,8-cineole.227 | ||
2.2.2.1 Amination of C–H bonds. So far, no natural enzyme is known to catalyse the direct amination of C–H-bonds. Nevertheless, this reaction can be realised in a cascade approach comprising C–H-bond hydroxylation followed by oxidation of the resulting alcohol and reductive amination of the carbonyl intermediate as exemplified by Turner and co-workers.228,229
The previously mentioned P450 monooxygenases also here opened up new possibilities.161,162 The functionalisation of C–H-bonds enabled the direct amination starting again with the appropriate reactive nitrogen species (Scheme 26).
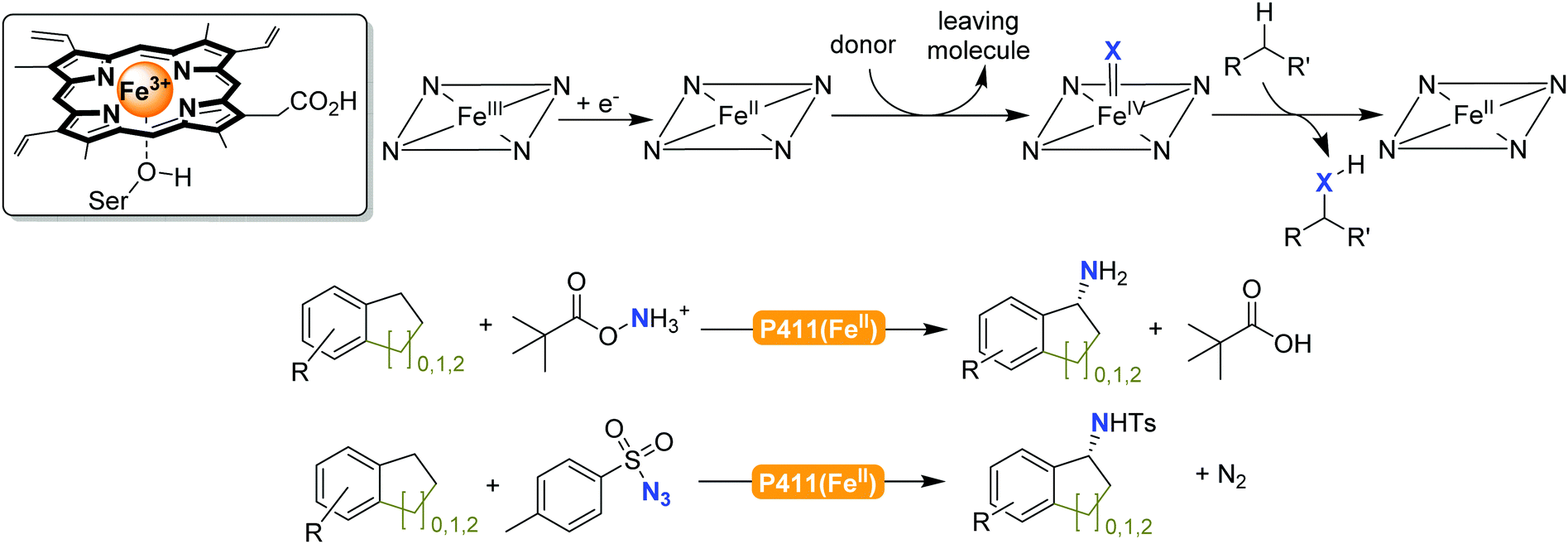 | ||
| Scheme 26 Direct C–H activation using P411 enzymes forming FeIV nitrene species.230,231 | ||
2.2.2.2 Halogenation of C–H-bonds. In recent years, biocatalytic methods introducing halide atoms into (non)activated C–H bonds have been receiving an increasing interest from the academic community.232,233 Today, a range of highly selective halogenases (halogenating monooxygenases) and haloperoxidases are available. Scheme 27 gives a representative overview over the biocatalytic halogenation reactions available today.
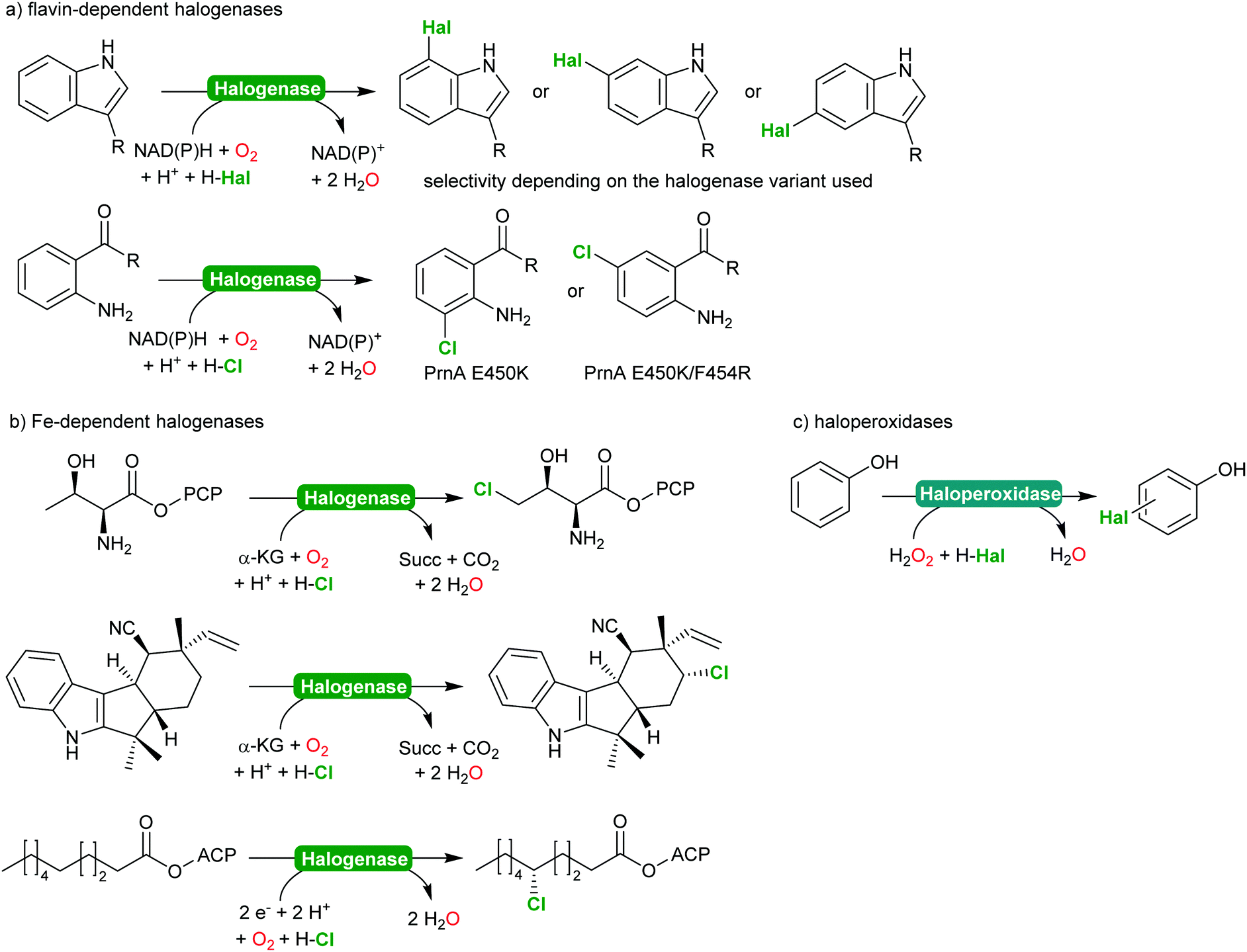 | ||
| Scheme 27 Selection of enzymatic halogenation reactions. | ||
From a synthetic perspective, halogenases and haloperoxidases can be distinguished. Halogenases excel by their often superb regio- and stereoselectivity as exemplified in Scheme 27. For example, for the halogenation of substituted aromatics, o-, m- and p-selective enzymes are available either from natural or from man-made diversity;232–238 impressively demonstrating how chemical properties of the starting materials (such as the o- and p-directing influence of electron donating substituents) can be over ridden by enzyme selectivity. In addition to the flavin-dependent halogenases, Fe-dependent halogenases are also increasingly moving into the focus of the research community.232,234 These enzymes exhibit an even stronger halogenating capacity and are capable of selective halogenation of even non-activated sp3-C–H-bonds.
This enormous potential, however, is impaired by the comparably low activity. In case of the flavin-dependent halogenases, catalytic activities in the range of several turnovers per minute are the rule rather than the exception.239 To some extend this can be explained by the molecular architecture of these enzymes comprising an NAD(P)H:flavin oxidoreductase and a halogenase subunit, functionally connected via the diffusible reduced flavin (FADH2).240,241 This makes flavin-dependent halogenases particularly susceptible to spontaneous oxidative uncoupling. On the other hand, this also enables simplifying the complex electron supply chain by other chemical reductants.240,242,243 Despite the rather low activity, Sewald and co-workers have established the gram-scale halogenation of tryptophan235 also an indigo precursor244 or as starting material for Pd-catalysed cross coupling reactions.245,246
In contrast, haloperoxidases excel by their superb catalytic activity reaching hundreds of turnovers per second even upon prolonged reaction times. Haloperoxidases, however, catalyse the H2O2-driven oxidation of halides (Cl−, Br− and I−) into the corresponding hypohalites,247–249 which then react spontaneously (non-enzymatically) with the starting material. As a consequence, the selectivity of haloperoxidase-catalysed halogenation reactions of activated aromatics such as phenols,247,250 or pyrroles,251–253 is dictated by the chemical reactivity of the starting material. Nevertheless, haloperoxidases have been demonstrated to be useful catalysts to initiate hypohalite-initiated reactions such as (aza)-Achmatowicz reactions,254–256 halolactonisation reactions,257–259 or oxidative decarboxylation of amino acids.260–262
2.3 Functional group interconversions
The interconversion of functional groups is at the heart of the disconnection approach and retrosynthesis. It allows to utilise synthons that are at first glance very different from the desired product. Nature struggles with similar synthetic problems as chemists do in the laboratory (this should in particular be true in natural product synthesis) and thus a multitude of enzymes is available that catalyses selective interconversions.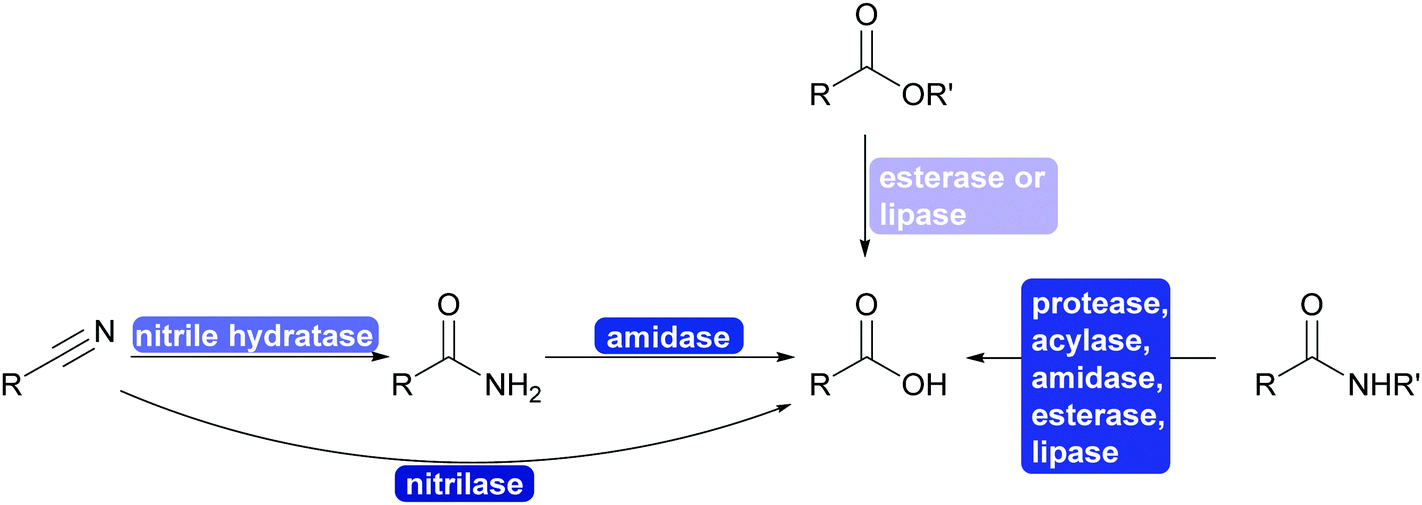 | ||
| Scheme 28 Enzyme catalysed functional group interconversion and protection group chemistry of nitriles, esters, amides and acids. All of the enzymes can be enantioselective and the reactions can be utilised for kinetic resolutions. | ||
Three recent developments will however be highlighted that added greatly to scope of the chemistry: ester and amide synthesis is water, PET hydrolysis and nitrile formation.
2.3.1.1. Synthesis of esters and amides in water. A major breakthrough in the application of hydrolases is the introduction of the acyltransferases and similar enzymes.277 These enzymes are closely related to lipases and esterases,278 however, they catalyse the transfer of an acyl group under aqueous conditions.279,280 This phenomenon had already been observed for a number of lipases such as CALA and CpLip2 but the scope was still rather limited.280,281 Strictly speaking this is a transesterification but since straightforward starting molecules as ethyl acetate of vinyl acetate are used in combination with alcohols of choice, it is viewed as ester synthesis.7,8 In particular the acyltransferase from Mycobacterium smegmatis (MsAcT) led to a burst of new applications.282 This octameric enzyme, structurally closely related to α,β-hydrolases like the esterases, was first described in 2007 to show unusual transesterification activity that allowed the preparation of esters, starting from ethyl acetate and an alcohol. It was demonstrated that the reactions was independent of whether ethyl acetate or water was the solvent. Additionally MsAcT displayed perhydrolase activity, i.e. the property to convert esters like ethyl acetate in the presence of hydrogen peroxide into their peracids (Scheme 29).
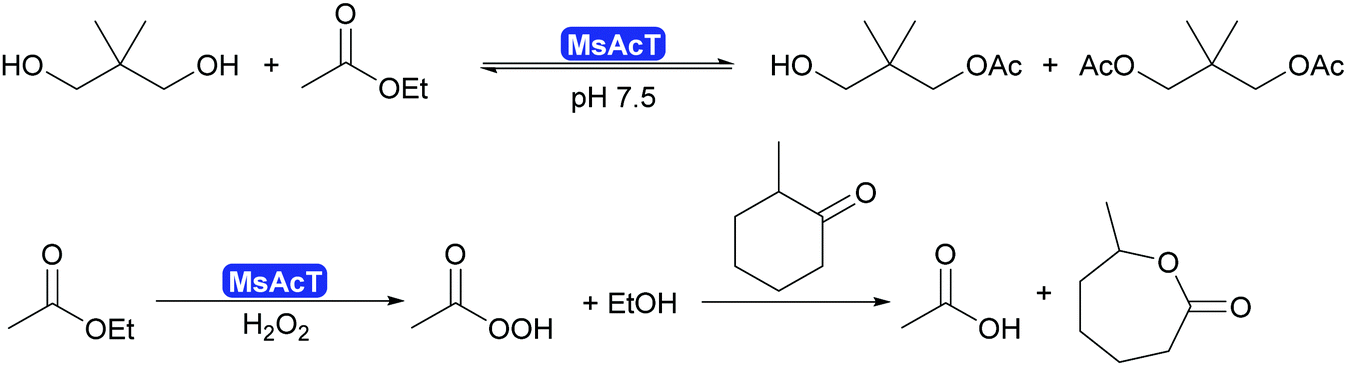 | ||
| Scheme 29 MsAcT catalyses the transesterification of primary alcohols with ethyl acetate in water as solvent. It also displays perhydrolase activity, generating peracid from esters. They can subsequently enable Baeyer–Villiger reactions. The BV reaction is not enzyme catalysed. | ||
Initially it was the latter activity that was utilised. MsAcT was immobilised and included in paint.283 This paint displayed a strongly disinfecting property and could be applied in rooms that require high hygiene standards. Furthermore, it was demonstrated that the peracids generated in the presence of 60% aqueous peroxide could be used in Baeyer–Villiger-oxidations.284 However, as the enzyme is not catalysing the oxidation reaction, no stereoselectivity was observed (Scheme 29).
The key breakthrough was the controlled synthesis of esters in water. After a preliminary study which indicated both activity and enantioselectivity, it was shown that MsAcT can be used in water to perform a successful transesterification. MsAcT displays a very high preference of the formation of esters from activated acids and alcohols, in particular primary ones (Scheme 30). In contrast, lipases and esterases hydrolyse the activated acid straightaway. This special property of MsAcT opens up new possibilities. Remarkably not just ethyl acetate can be utilised as activated acid but a range of vinyl esters can be used, too. This underlines the high selectivity of MsAcT for ester synthesis rather than hydrolysis.285–287
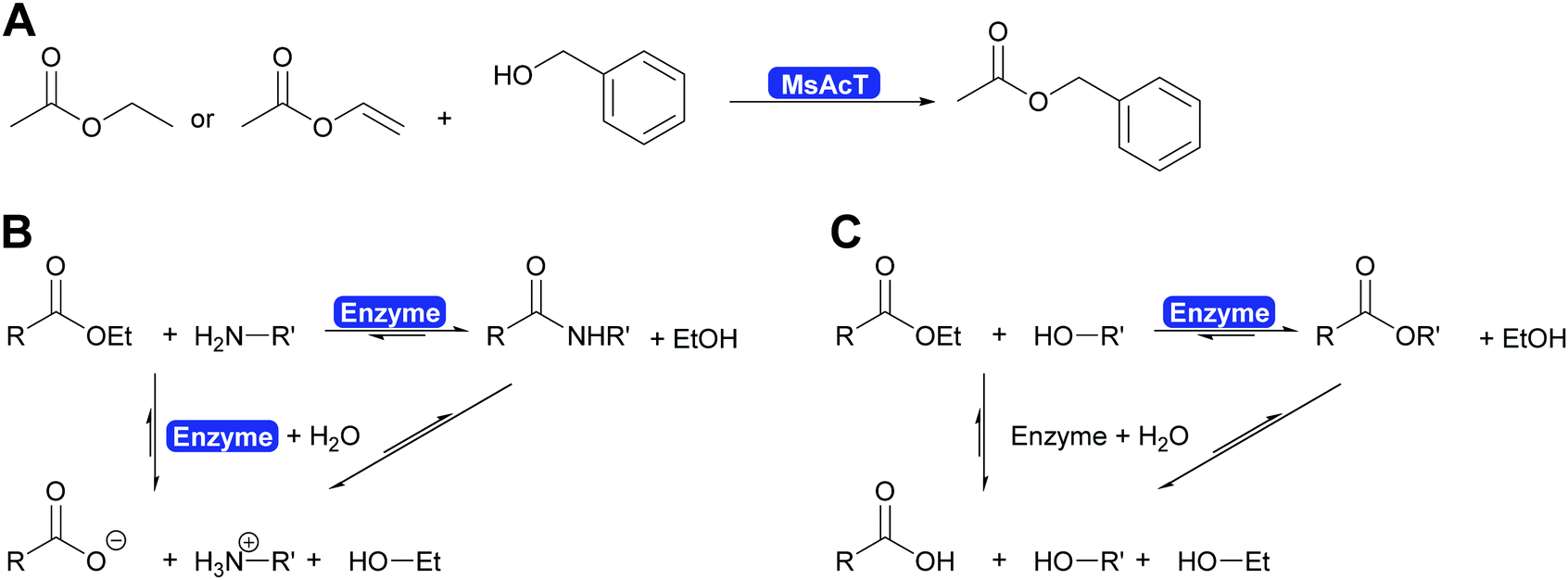 | ||
| Scheme 30 (A) Synthesis of esters in water catalysed by MsAcT. Either ethyl acetate or more reactive acid derivatives such as vinyl acetate can be utilised. (B) The formation of an amide in water is based on the rapid conversion of an activated acid, here an ester, with an amine to an amide. The competing hydrolysis of the starting material and of the product are relatively slow and thus high yields of amide can be achieved. (C) MsAcT can utilise esters as activated acids and catalyses their transesterification to the desired product. Again the competing hydrolysis of the starting material of the desired product is slow and thus high yields of ester can be obtained even though the solvent is water. | ||
Careful investigation of the reaction revealed an underlying principle that was already known from the industrial synthesis of penicillins but was to date perceived to be limited to amide synthesis (Scheme 30). Both, in the penicillin acylase catalysed penicillin synthesis and the MsAcT-catalysed ester formation an activated acid will react with a nucleophile more rapidly than with water. This leads to product formation, in the case of penicillin acylase the amide bond, in the case of MsAcT an ester. Hydrolysis of the activated acid and the product is significantly slower than product formation but good control of the reaction kinetics is essential. Otherwise full hydrolysis will still occur.82,280,288 Soon it was demonstrated that MsAcT can also be employed to synthesise amides in water, broadening its applicability.289,290 In the latter case it follows the kinetic model described earlier for the penicillin synthesis.
MsAcT displayed good activity in the ester synthesis, both in batch and in flow chemistry.285,291–293 Engineering extended the substrate scope and improved as well as inverted the enantioselectivity. Today a wide range of primary and secondary alcohols can be esterified, the latter enantioselectively both to R or S ester.285–287 As with other hydrolases, tertiary alcohols and their esters remain a bottleneck. The search for homologous enzymes with similar characteristics has expanded the range of enzymes available for this type of chemistry. Carboxylesterases of family VIII as well as an alcohol acyl transferase from yeast show great promise.294–296 A particularly noteworthy approach is the application of CAR for the synthesis of esters in water.297 While up to now limited to primary alcohols, it shows that the catalytic machinery or several enzymes might be suitable for ester synthesis in water.
The results described so far are not limited to batch. It was shown that MsAcT also works well for the synthesis of esters and amides in water or in biphasic mixture of water and acyl donor in flow, and this chemistry was broadly explored (Scheme 31).298–300
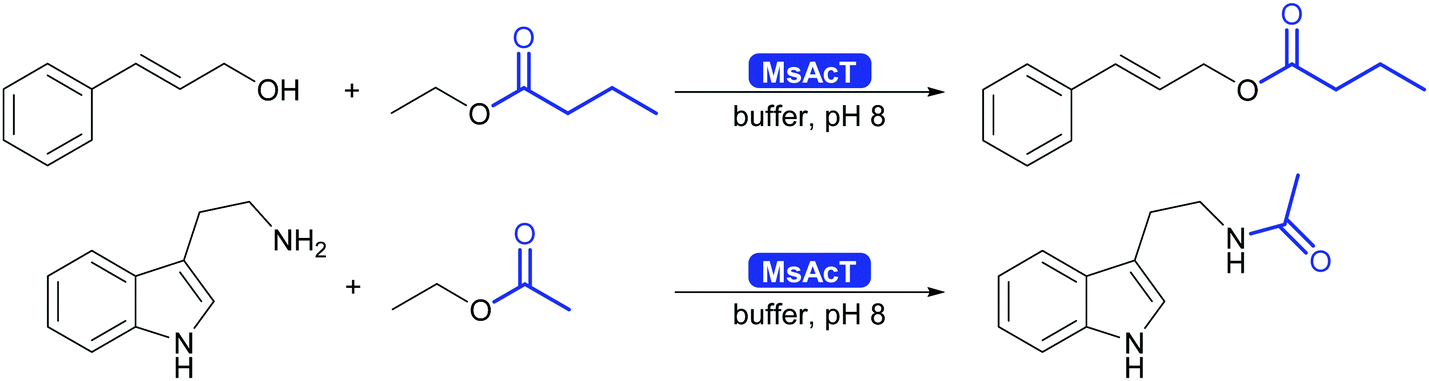 | ||
| Scheme 31 MsAcT catalyses the formation of esters and amides in water. | ||
A single mutation of the nucleophile in the catalytic triad of MsAcT revealed even more possibilities for the application of this intriguing enzyme. By changing the catalytic triade from the standard Ser-His-Asp to Cys-His-Asp the hydroxy group of serine was replaced by the thiol of cysteine. This enabled the synthesis of thioesters, up to then essentially impossible with hydrolases. A wide range of thiol and thiophenol can now be converted. Particularly intriguing was that even CoA could be converted to acetyl-CoA. This was possible although the reaction times were long. However, the Ser11Cys MsAcT did not hydrolyse the product, allowing these long reaction times. The enzyme in this case utilised a different tunnel to the active side, allowing the large substrate to enter. With the Ser11Cys mutant it was also possible to convert amines into amides. Particularly interesting is the conversion of secondary amines to tertiary amides. This challenge had earlier not been met (Scheme 32). Thus, by a single mutation two major challenges for Green Chemistry have been solved, the straightforward and clean syntheses of thioesters and tertiary amides.301
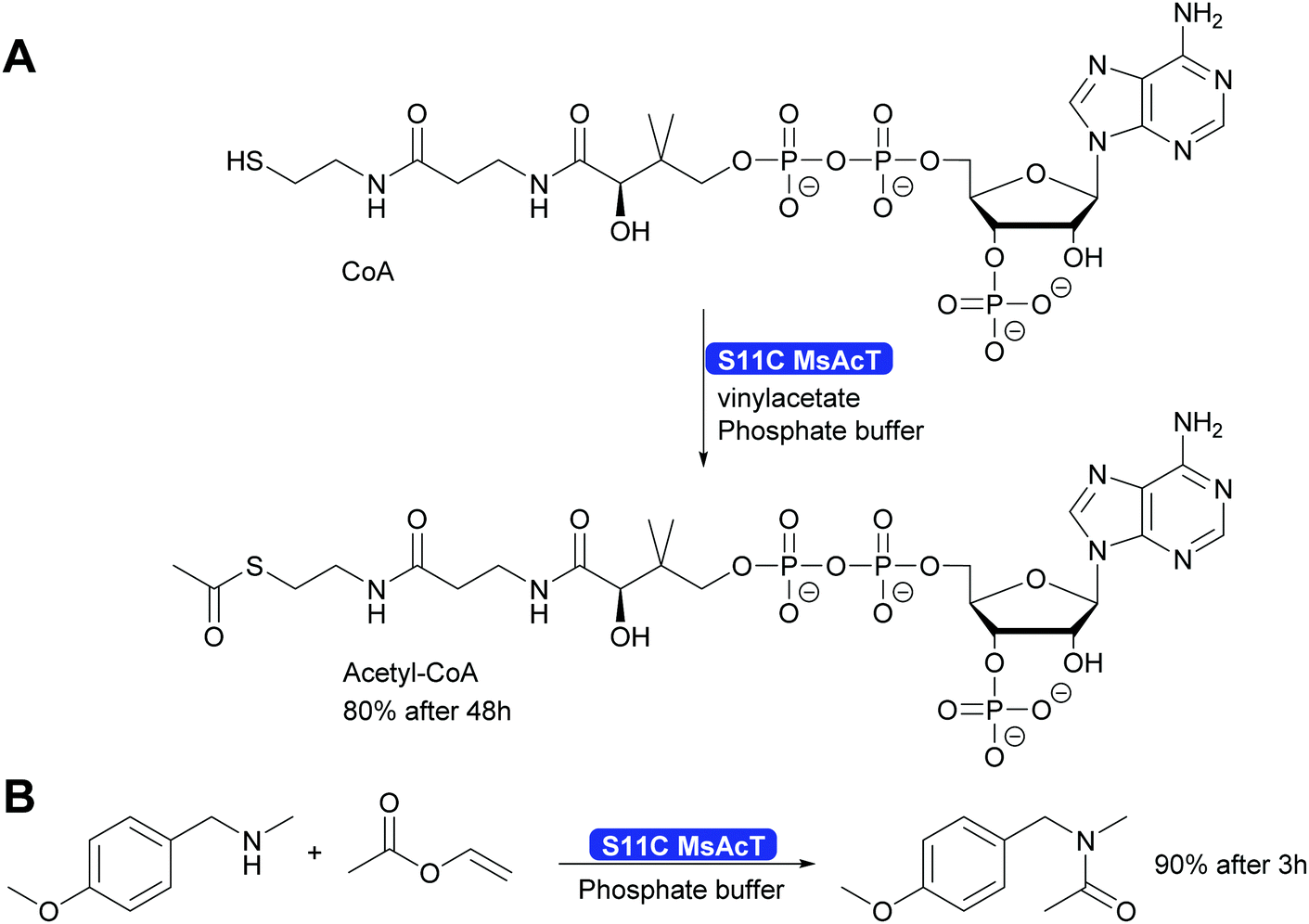 | ||
| Scheme 32 The mutation of the catalytic Ser11 to Cys enables the synthesis of thioesters, including acetyl-Co-A as well as the synthesis of tertiary amides. | ||
An alternative methodology to synthesise amides in aqueous media has recently been introduced by Flitsch and co-workers.302 Based on a careful analysis of the catalytic mechanism of carboxylic acid reductases (CARs, vide infra) the authors hypothesised that CARs, next to the ATP-and NADPH-driven reduction of carboxylic acids, should also be able to catalyse the amidation of enzyme-bound and ATP-activated carboxylic acids (Scheme 33). A broad range of carboxylic acids and amine nucleophiles could be converted in high yields using this creative exploitation of mechanistic understanding!
 | ||
| Scheme 33 Turning a carboxylic acid reductase into a carboxylic acid amidase. | ||
2.3.1.2. Hydrolysis of PET. The application of esterases, lipases and amidases in polymer chemistry was for a long time focussed on the synthesis of polymers. The hydrolysis of polyesters or polyamides was much less investigated.12 This is actually surprising as the hydrolysis of esters and amides in water should be straightforward. However, the hydrophobic surface properties and the low accessibility of the functional groups of the polymers essentially prevented this. While it is highly important that polymers are not biodegraded when they are applied in building materials and electrical appliances the ever increasing amount of plastic waste does need to be degraded. Indeed, plastics that are intended for short term usage should be designed in a biodegradable manner. Here splitting into the starting materials for recycling as truly new plastic would be of particular interest. This should in particular be possible for plastics with a hydrolysable ester or amide group.303
A major step toward polymer degradation and recycling was made when a Polyethylene Terephthalate (PET) degrading bacterium was identified.304 This, together with earlier work on polymer degrading enzymes, gave a new boost to recycling of PET to the monomers. The natural answer to the difficult accessibility of the functional groups of the polymer was to evolve a swallow active site open along the surface. This type of active site already existed in cutinases, enzymes that hydrolyse waxes on leaves. Both the naturally evolved PETase and enzymes that were evolved in the laboratory proved interesting starting points for PET recycling. Very recently an enzyme was reported that was successfully evolved for this purpose. This mutated cutinase is highly suitable for the rapid hydrolysis of amorphous PET at relatively high temperatures (72 °C). It is important that the PET is amorphous in structure as crystalline PET is much more difficult to hydrolyse. At these relatively high temperatures the cutinase mutant rapidly hydrolyses post-consumer waste PET to approx. 90%, releasing terephthalic acid (Scheme 34). The high rate of hydrolysis is essential as PET otherwise crystallises under the reaction conditions. The obtained terephthalic acid was utilised to again prepare virgin PET of the same quality as before. While this does introduce two additional steps into the recycling it also means that always pristine polymer is (re-)generated. If the polymer is recycled as such the quality quickly degrades and down-cycling rather than re-cycling takes place.305,306
 | ||
| Scheme 34 A cutinase mutant evolved in the laboratory for the hydrolysis of PET can hydrolyse approx. 90% of post-consumer waste PET at elevated temperature. | ||
2.3.1.3. Cyanide-free nitrile synthesis. The selective hydrolysis of nitriles to amides or acids is well-established industrial practice. The enzymes utilised for this, nitrilases and nitrile hydratases cannot be used in reverse. However, more recently a new type of enzyme was described that can convert aldoximes into nitriles. These aldoxime dehydratases (Oxd) contain a heme iron centre to which the aldoxime coordinates with its nitrogen atom. Subsequent elimination of the hydroxy group, induced by the catalytic cascade of Arg His Ser and enabled by the oxidation of Fe(II) to Fe(IV) is followed by the release of carbonyl hydrogen as proton with simultaneous reduction of the Fe back to Fe(II) and release of the nitrile from the active site.307,308
The Oxd can be employed in whole cells with the separately prepared aldoxime or the aldoxime can be generated in situ and then directly converted to the desired nitrile (Scheme 35). Oxd have been demonstrated to be enantioselective and they distinguish between E and Z aldoximes. Their application for the preparation of dinitriles, relevant for the polymer industry, has been demonstrated. Whole cell preparations have successfully been utilised in flow, demonstrating the versatility of these enzymes.308–310
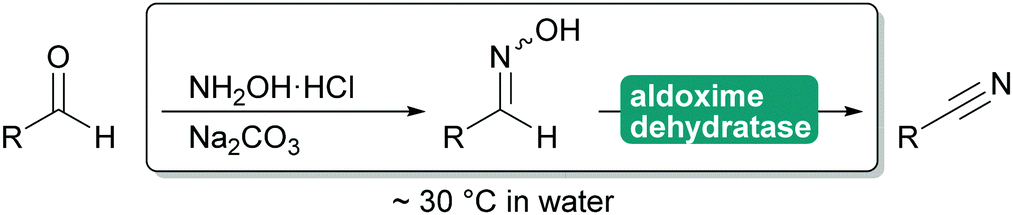 | ||
| Scheme 35 Application of Oxd to prepare nitriles from aldehydes. The initial preparation of the aldoxime is not catalysed but can be performed in situ. | ||
2.3.2.1 Alcohol oxidation to aldehydes. Enzyme-catalysed alcohol oxidation to aldehydes, relevant compounds for the flavour and fragrance industry, can be carried out by the classic nicotinamide-dependent alcohol dehydrogenases (ADHs).31 However, recent renewed interest combined with extensive mining of enzymes led to the discovery and applications of interesting flavin-dependent (FAD-AlcOx) and copper-radical alcohol oxidases (CRO-AlcOx).311,312 A major attraction of AlcOx is their lack of dependence on the NAD(P) cofactor, simply using molecular oxygen as a terminal electron acceptor. These AlcOx enzymes allow access to terpenes and derivatives used in the fragrance industry.311 Over-oxidation to the carboxylic acid remains minor and catalase is used to remove the formed hydrogen peroxide in solution (Scheme 36).
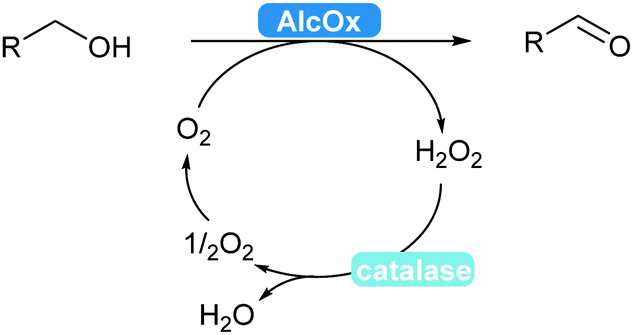 | ||
| Scheme 36 AlcOx-Catalysed oxidation of alcohols to aldehydes. | ||
2.3.2.3 C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/i_char_e001.gif) C epoxidation.
Asymmetric epoxidation of alkenes provides access to valuable chiral epoxide building blocks. Established with P450-BM3, epoxidation of styrenes and terpenoids occurs with high chemo-, regio-, and stereoselectivity.313 In addition, unspecific peroxygenases (UPOs) and flavoprotein styrene monooxygenases (SMOs)171 allow for complementary substrate scope towards steroid and aromatic products (Scheme 37). In particular, a new clade of SMO that catalyse the epoxidation of alkenes now complements the already extensive (S)-selective SMO family to give access to the corresponding (R)-chiral epoxides.314
C epoxidation.
Asymmetric epoxidation of alkenes provides access to valuable chiral epoxide building blocks. Established with P450-BM3, epoxidation of styrenes and terpenoids occurs with high chemo-, regio-, and stereoselectivity.313 In addition, unspecific peroxygenases (UPOs) and flavoprotein styrene monooxygenases (SMOs)171 allow for complementary substrate scope towards steroid and aromatic products (Scheme 37). In particular, a new clade of SMO that catalyse the epoxidation of alkenes now complements the already extensive (S)-selective SMO family to give access to the corresponding (R)-chiral epoxides.314
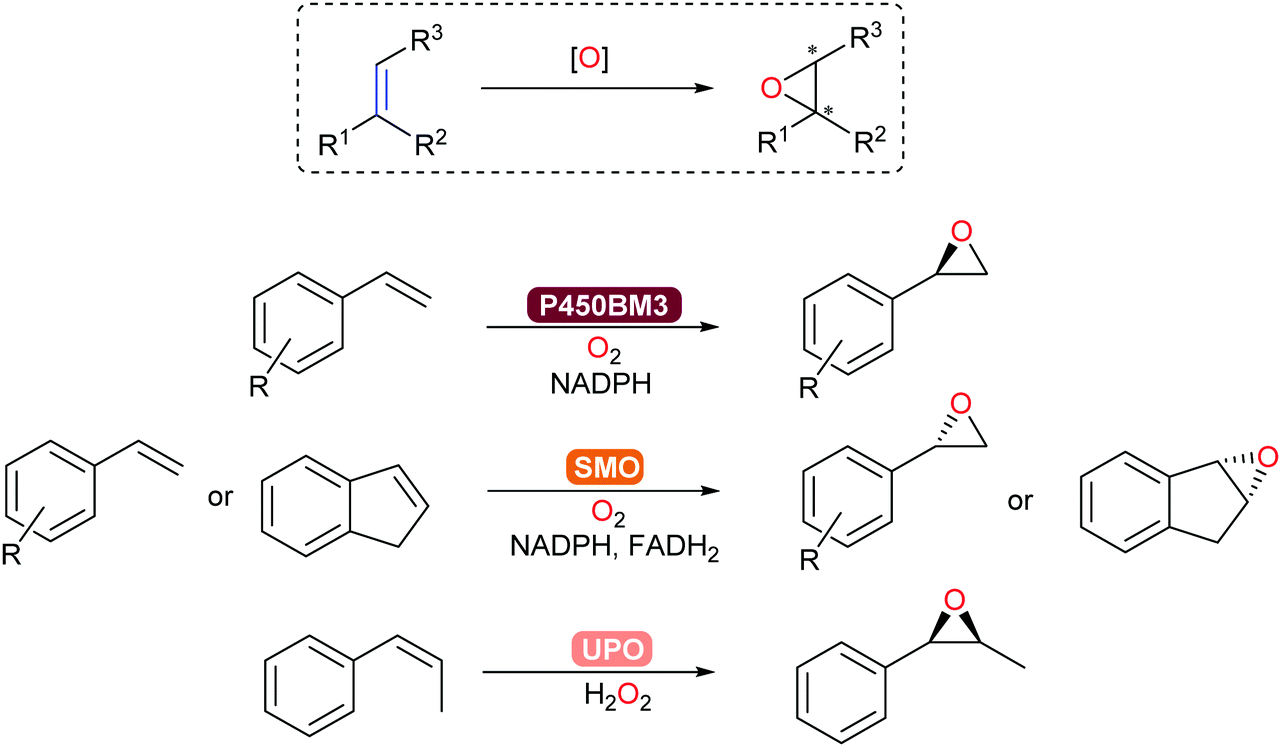 | ||
| Scheme 37 Asymmetric epoxidation of alkenes can be catalysed by P450-BM3, SMO or UPO. | ||
Preparative-scale (up to the kg-range) whole-cell-catalysed epoxidations with SMOs have been demonstrated by Schmid and co-workers.315,316
2.3.2.4 Oxidation of sulfur. Flavoprotein monooxygenases (FPMOs) have been recently reviewed to catalyse a wide variety of reactions ranging from electrophilic oxygenation, electrophilic aromatic substitution, nucleophilic oxygenation, radical mechanisms to miscellaneous reactions.171 Asymmetric sulfoxidation using an engineered flavoprotein cyclohexane monooxygenase CHMO was demonstrated for the production of the proton-pump inhibitor (S)-omeprazole (esomeprazole) with the higher enantiomeric excess (>99%) than its current chemical catalytic counterpart (Scheme 38).317
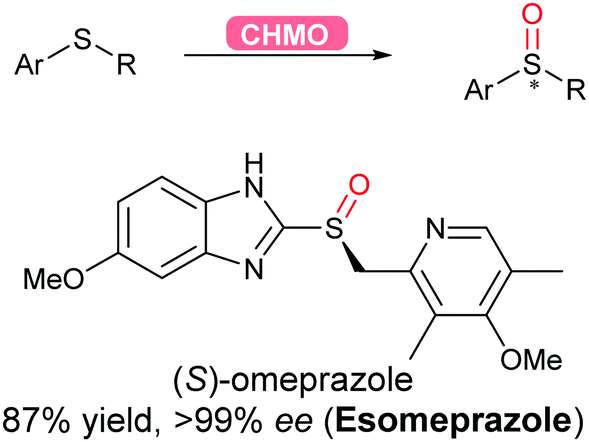 | ||
| Scheme 38 CHMO-Catalysed asymmetric sulfoxidation to afford esomeprazole in high yield and enantiomeric excess. | ||
Asymmetric sulfoxidation can also be catalysed by flavoprotein SMOs mentioned above and allow for complementary selectivity and substrate scope.318,319
2.3.3.1. Reduction of ketones and aldehydes to alcohol. The most widely applied oxidoreductase enzymes for reduction in organic chemistry are the ketoreductases (KREDs), which catalyse the reversible asymmetric hydrogenation of ketones to the corresponding alcohol (Scheme 39).32 The name is usually interchanged between the typical zinc-dependent medium-chain dehydrogenase family, represented by the horse liver ADH,320,321 and short-chain dehydrogenase ADHs such as from Lactobacillus kefir.322,323 Long-chain dehydrogenases and aldo-ketoreductases remain underused in biocatalytic applications.322 The dependence of KREDs on stoichiometric amounts of NADH or phosphorylated form NADPH as a reductant can be overcome with efficient and established in situ recycling systems (Scheme 39). Most common systems either use the same KRED and a sacrificial co-substrate (e.g. isopropanol) in large excess, or couple another oxidoreductase enzyme such as glucose dehydrogenase (GDH), formate dehydrogenase (FDH) or phosphite dehydrogenase (PDH) with their corresponding substrate (glucose, carbonate or phosphite).154,324
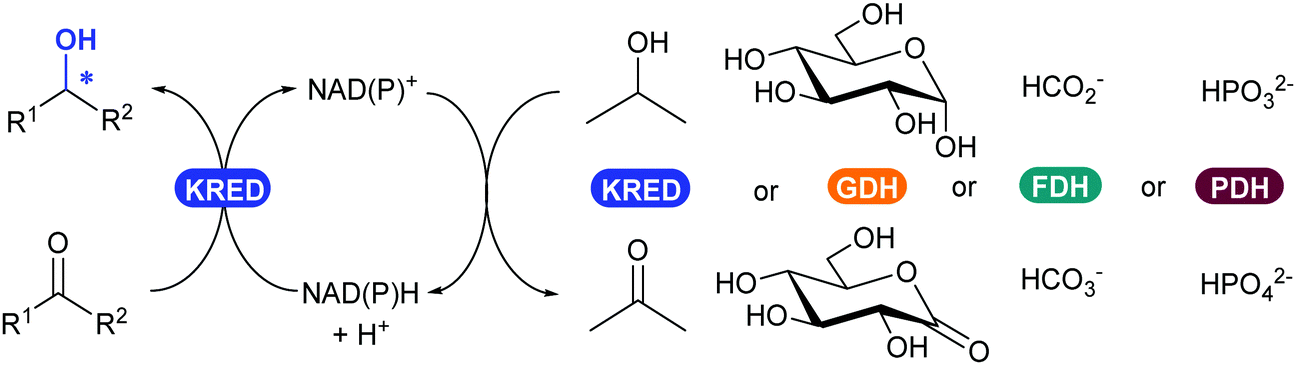 | ||
| Scheme 39 KRED-Catalysed reduction of an aldehyde or ketone to alcohol (left), with available NAD(P)H enzymatic cofactor recycling systems (right). | ||
The asymmetric reduction of prochiral ketones using ADHs has evolved to a mainstream robust and reliable technology with examples reaching a variety of chemical motifs for the chemical industry.325 ADHs have been implemented for the asymmetric reduction of prochiral ketones in the production of blockbuster pharmaceutical drugs such as Lipitor (atorvastatin), affording high standards for enantiopurity.326,327 Several stereoselective ADH-catalysed ketone reductions have led to the production of chiral intermediates and pharmaceutical products such as montelukast,327 emend,328 sulopenem,329 at industrial scale (Scheme 40).
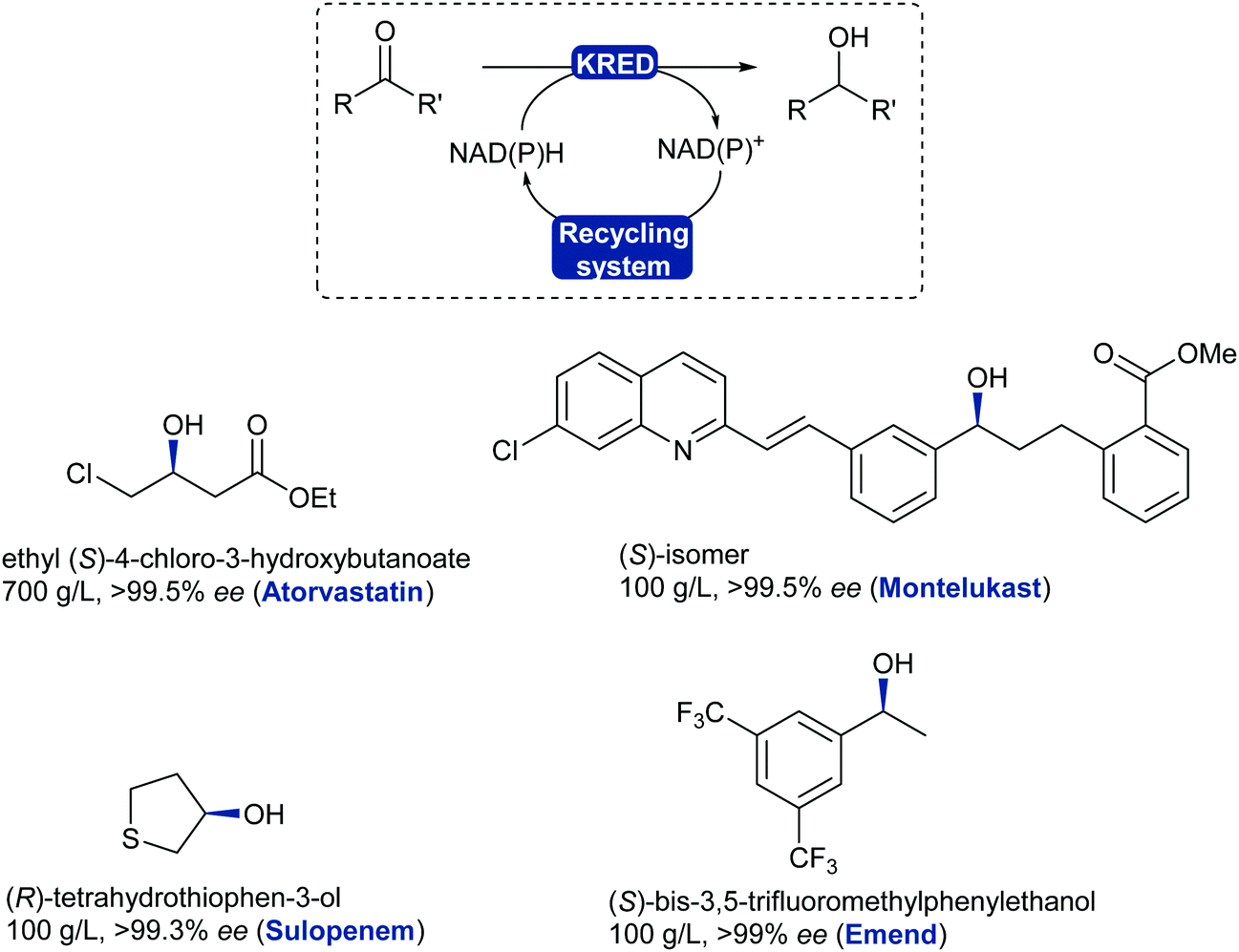 | ||
| Scheme 40 KRED/ADH-Catalysed reduction of ketones or aldehydes. Examples of chiral alcohols produced on large scale with high enantiopurity; drug name in brackets. | ||
KRED enzymes are now commonly used for the reduction of aromatic, aliphatic and cyclic ketones to their corresponding chiral alcohol with exquisite selectivity. These enzymes generally display a wide substrate scope and high selectivity towards the desired enantiomer thanks to available stereocomplementary KREDs, which also allows for deracemisation and stereoinversion processes.330,331 The challenging reduction of bulky substituted ketones has now been addressed as more enzymes are being discovered and evolved to tolerate these substrates such as the montelukast example. KREDs can also be employed to reduce aldehydes and obtain chiral substituted alcohols in a dynamic kinetic resolution system.332,333
2.3.3.2. Reductive amination and transamination of aldehydes and ketones. The reductive amination of ketones leading to chiral amines can now be achieved by different family of enzymes developed in the 2010's. Thus, imine reductases (IREDs),334–336 amine dehydrogenases (AmDHs)337–341 and reductive aminases (RedAms)342,343 catalyse the formation of the imine and its subsequent reduction (Scheme 41).344,345 These reductive enzymes, together with amino acid dehydrogenases (AaDHs) that transform keto acids to amino acids, and PLP-dependent transaminases (TAs) that catalyse transamination, give access to primary, secondary and tertiary amines and have been applied on gram to kilogram scale. Transaminases in particular have been used in many processes already, for example in the synthesis of sitagliptin.346,347
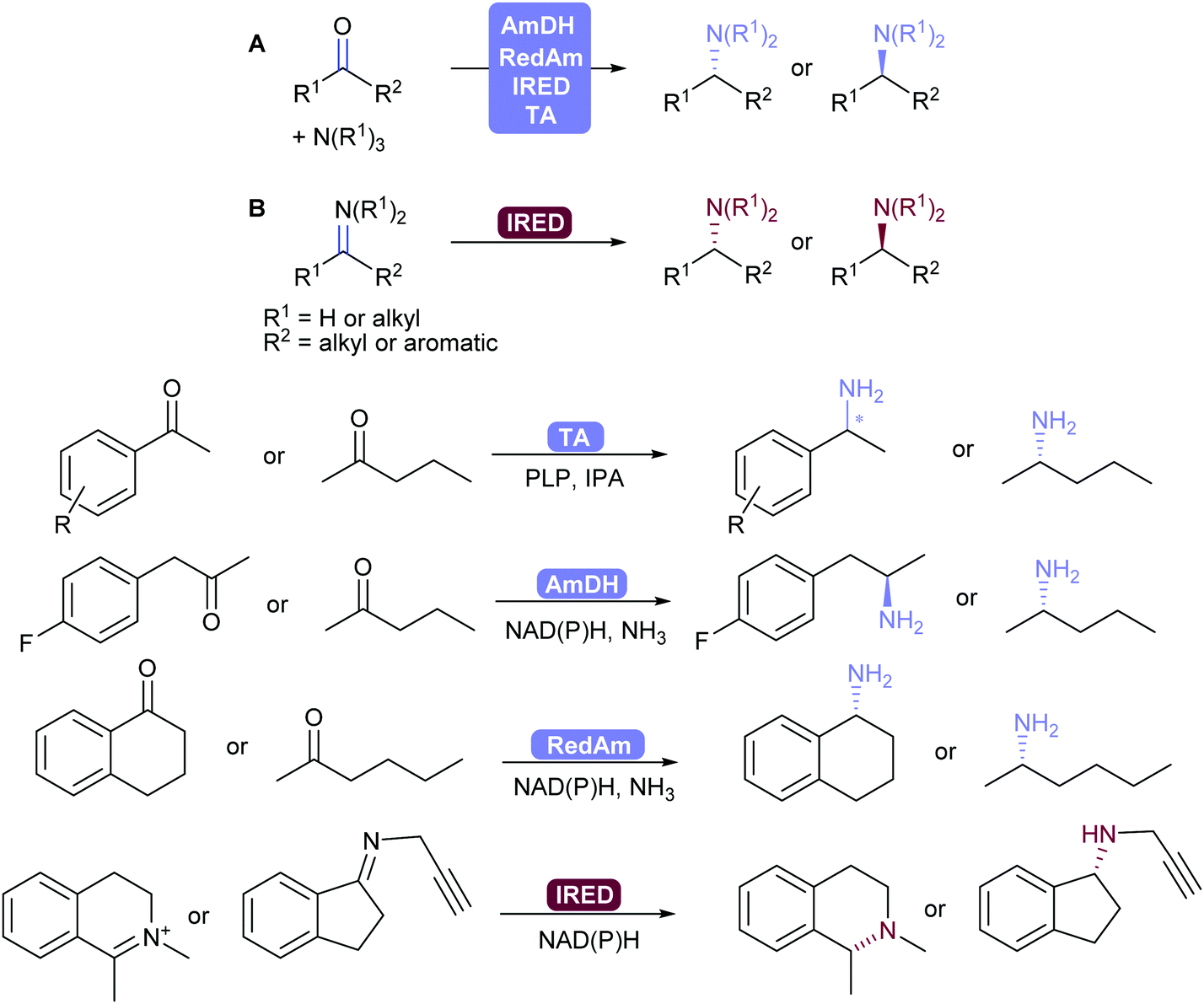 | ||
| Scheme 41 (A) AmDH-, RedAm-, IRED-Catalysed reductive amination and TA-catalysed transamination with ammonia or alkyl amines. (B) IRED-catalysed reduction of imines. Selected examples of chiral amine products. | ||
IREDs also have the ability to catalyse the reduction of (usually cyclic) imines formed in situ to the corresponding amine. Turner and co-workers recently explored the structure–activity relationship of IREDs, which seem to be highly dependent on the substrate thus does not allow to predict the outcome based on an IRED and substrate pair.348
In order to shift the equilibrium to the desired amine product, AmDHs, IREDs and TAs require an excess of amine donor to reach full conversion, whereas the recently discovered RedAms do not, and can generate secondary and tertiary amines with stoichiometric carbonyl and amine substrates.342,345,349
Seminal work by Turner and co-workers showed that chiral amines can also be obtained using flavin-dependent monoamine oxidases (MAOs) in a chemo-enzymatic process, by oxidising amines to imines with concomitant non-selective chemical reduction of the imine.350
2.3.3.3. Carboxylic acid reduction to aldehyde. The chemical reduction of carboxylic acids to aldehydes is not trivial, usually requiring a strong reductant, and the desired aldehyde tends to be further reduced to the alcohol. The discovery of carboxylic acid reductases (CARs) has allowed the reduction of mostly aromatic carboxylic acids and fatty acids under mild reaction conditions through activation of the carboxylate with ATP, forming a thioester further reduced by NADPH.351,352
The attractiveness of these enzymes for wider applications in vitro remains limited due to their requirement for ATP, hence their use in microbial cells. Nevertheless, CAR-catalysed reduction of carboxylic acids are relevant to obtain volatile aldehyde products for the flavour and fragrance industry, or intermediate building blocks in cascade reactions (Scheme 42).351,352 Examples include the production of amines, diamines, aldol compounds and vanillin.351 Recently CARs were even used for amide bond formation using amines bypassing the NADPH reduction step (see above),302,353 thus expanding the utility of these enzymes on carboxylic acid functional group interconversion.
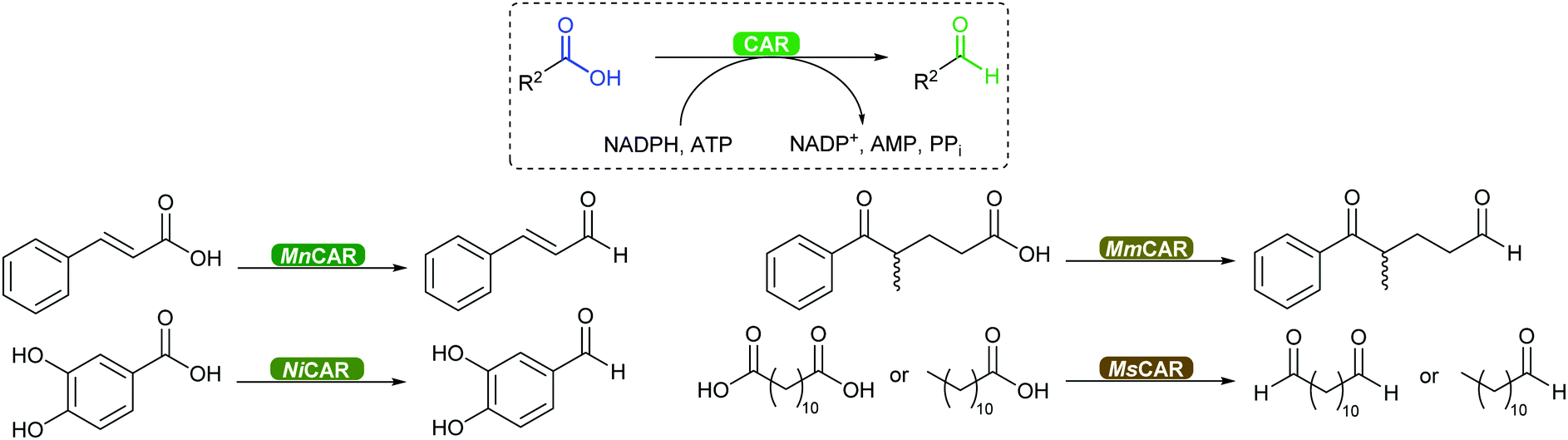 | ||
| Scheme 42 CAR-catalysed reduction of aromatic and aliphatic carboxylic acids to aldehydes further used as building blocks. MnCAR: from M. neoaurum; NiCAR: N. iowensis; MmCAR: from M. marinum; MsCAR: from M. smegmatis. | ||
2.3.3.4 Asymmetric C
C reduction.
The enzymatic asymmetric hydrogenation catalysed by flavin-dependent Old Yellow Enzyme ene reductases (EREDs), first identified by Warburg in 1932, has clearly been in the lead in the past decade. In particular, OYEs were proven to catalyse highly regio-, chemo- and stereoselective reduction of activated alkenes in a trans-addition fashion.354,355 The alkenes substrates typically require an electron withdrawing group such as a carbonyl, nitro, or nitrile (Scheme 43).
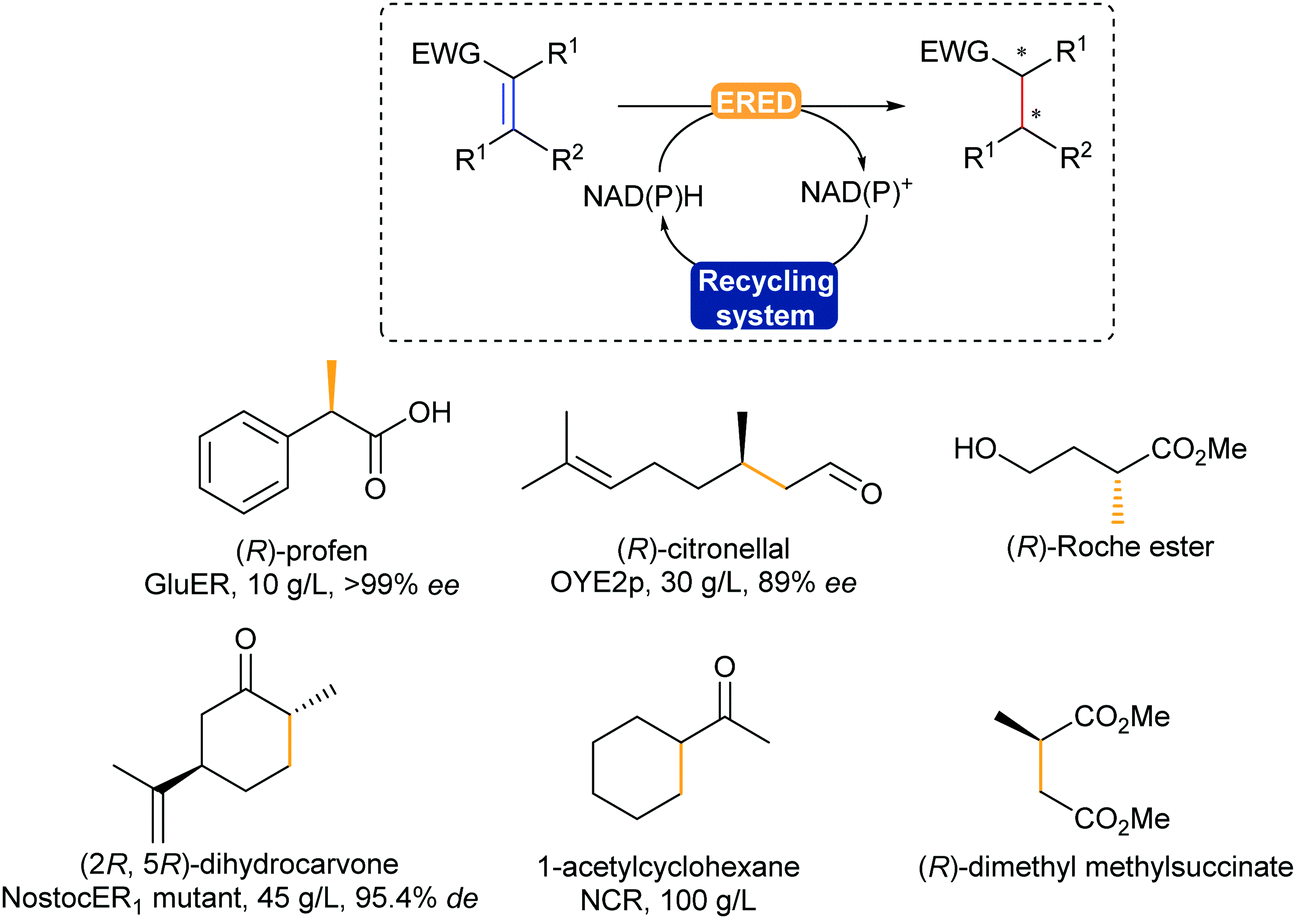 | ||
| Scheme 43 ERED-Catalysed asymmetric hydrogenation of activated CC bonds to the corresponding (chiral) alkane building block or final product. EWG = electron withdrawing group: aldehyde, ketone, carboxylic acid, ester, nitrile, nitro. | ||
EREDs are promising catalysts, allowing for hydrogenation reactions without the use of a palladium catalyst or hydrogen gas, under mild reaction conditions in water. This does bring along the challenge of substrate solubility, which has been addressed using biphasic systems and co-solvents.356 In addition, these enzymes display cofactor promiscuity due to their prosthetic flavin cofactor laying in a TIM-barrel motif, allowing access to not only both natural NAD(P)H, but also artificial analogues,357,358 and electron mediators.359 Various chiral building blocks and products have already been scaled-up, such as (R)-profens, (R)-citronellal, a valuable intermediate for the synthesis of menthol, and dihydrocarvone (Scheme 43).32
OYEs can also catalyse the reduction of alkynes in a selective fashion, obtaining the trans-alkene.360 If the reaction is left longer then full reduction to the alkane is observed. The additional use of light with these enzymes has recently unfolded different types of activities such as dehalogenation, and intra- and intermolecular carbon–carbon bond formation.361,362
2.4 Multi-enzyme cascades – a few examples highlighting the potential
Multi-enzyme cascades are the essence of life. All metabolic routes are such cascades, circular in the case of the citric acid cycle and the many cycles known from the huge posters that decorate many (bio)chemists offices and is today an online tool (https://web.expasy.org/pathways/). In all cases the cascades keep concentrations of all intermediates low, thus avoiding inhibition of the enzymes. In particular the cascades that lead to secondary metabolites have attracted much attention as they often have interesting biological activities. These cascades were therefore from the very beginning of biosynthetic studies a focus of attentions. A first milestone was the synthesis of the corrin scaffold in 1994. Starting from 5-aminolevulinic acid, utilizing 12 biosynthetic enzymes 17 steps were catalysed, creating 9 stereocentres and with an overall yield of 20% hydrogenobyrinic acid was formed (Scheme 44). This is a key intermediate for vitamin B12. It can be converted into the vitamin with just two further steps. All the elegance of the earlier chemical synthesis fades besides this achievement.363,364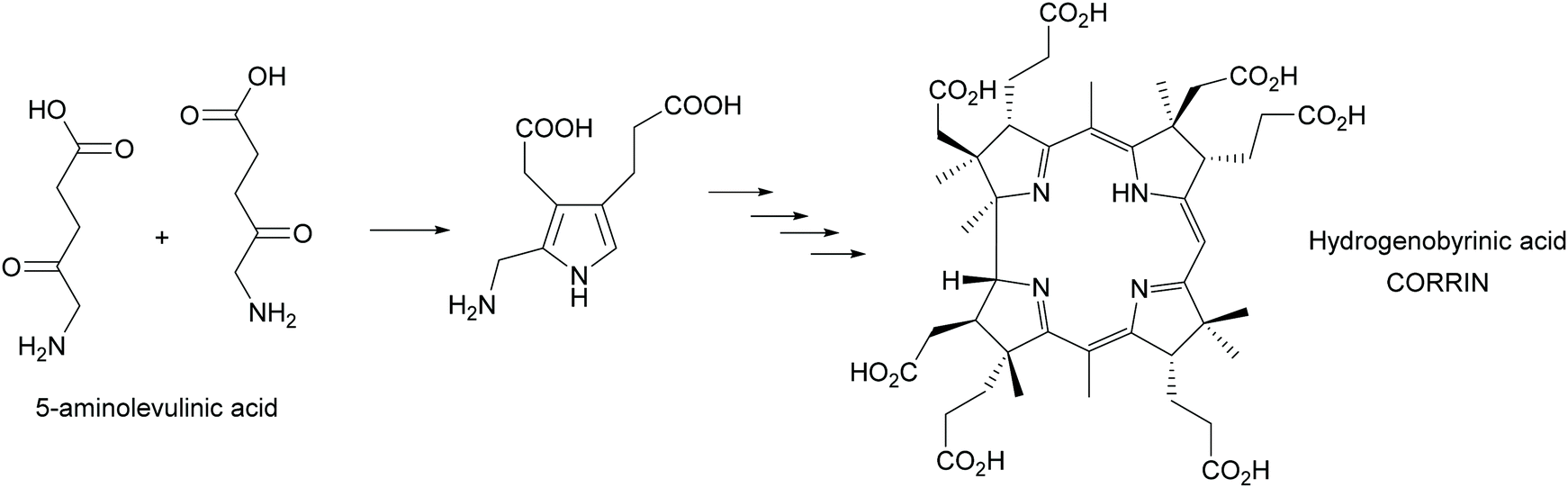 | ||
| Scheme 44 In vitro biosynthesis of the corrin ring of vitamin B12. In a glass flask 12 enzymes perform the 17 step biosynthesis with excellent yield. | ||
Ever since then cascades of enzyme-catalysed reactions have attracted attention.365–369 Several arguments are in favour of this approach. Cascade reactions are much closer to the natural conditions under which enzymes operate, relatively low substrate and product concentrations throughout the cascade occur, as only the starting material and the final product will be present in high concentrations. Furthermore the large number of steps allows to plan the synthesis in such a manner that the last step also constitutes a thermodynamically favourable reaction, shifting the overall equilibrium towards the product side. This is the case in the hydrogenobyrinic acid synthesis and the example below. Additionally, cofactor recycling is much easier in cascades, in particular if they involve oxidation and reduction steps that can be balanced.
Recent reviews compiled the combination of multiple enzymes in sequence, enabling access to complex functionalised molecules starting from basic starting materials.370,371 An example of a multi-enzymatic cascade was demonstrated by Merck for the production of the drug islatravir (Scheme 45).21 The shift of the equilibrium towards the product was achieved by converting the phosphate released in the PNP catalysed step in an irreversible additional, removal step.
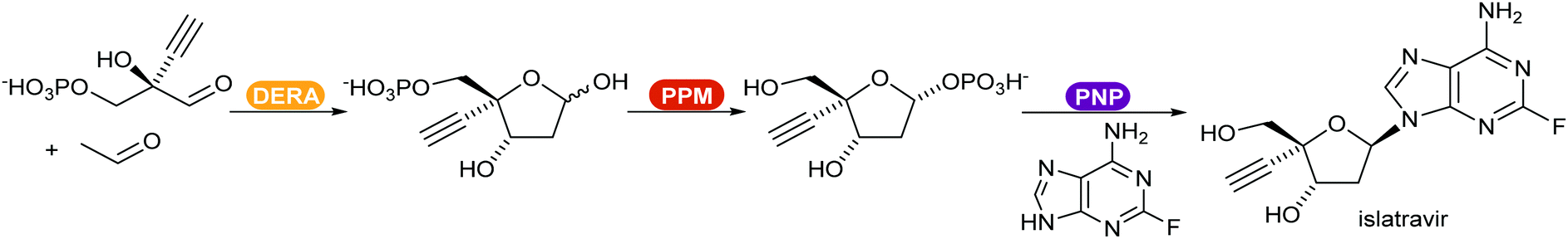 | ||
| Scheme 45 Enzyme-catalysed synthetic route for the production of the drug islatravir developed by Merck. DERA = deoxyribose-phosphate aldolase; PPM = phosphopentomutase; PNP = purine nucleoside phosphorylase. | ||
3. Conclusions
Closing in on almost two centuries since the discovery of emulsin, biocatalysis has increasing impact the field of organic synthesis by providing alternative methods for the synthesis of many classes of compounds with unrivalled selectivity. The first wave of biocatalysis was rather limited in the number of reactions that it enabled. Often only a single enzyme was available for the desired reaction. This landscape has completely changed. The second and third waves ensured that today biocatalysis is based on an understanding of the enzymes, including their scaffolds, which allows rapid identification of countless variations of a specific enzymatic activity, making a catalyst available for essentially all substrates with the functional group the synthetic chemist wishes to modify. More importantly, new reactions can be catalysed that were beforehand considered outside the scope of enzymes. This, in combination with an improved understanding of kinetics and thermodynamics governing the reactions, today allow the fast, efficient and clean synthesis of very complex structures under very mild conditions.New types of biocatalytic reactions remain to be unravelled with already known or soon to be discovered enzymes that would allow access to further classes of molecules. With this, biocatalysis is part of the many waves of change in chemistry.
Author contributions
All authors equally contributed to the conceptualization, visualization, writing of the original draft and in the final review and editing.Conflicts of interest
There are no conflicts of interest to declare.References
- F. Wöhler and J. Liebig, Ann. Pharm., 1837, 22, 1–24 CrossRef.
- P. M. D. Collins, Platinum Met. Rev., 1986, 30, 141–146 CAS.
- E. Fischer, Ber. Dtsch. Chem. Ges., 1894, 27, 2985–2993 CrossRef CAS.
- L. Rosenthaler, Biochem. Z., 1908, 14, 238–253 CAS.
- U. T. Bornscheuer, G. W. Huisman, R. J. Kazlauskas, S. Lutz, J. C. Moore and K. Robins, Nature, 2012, 485, 185–194 CrossRef CAS PubMed.
- J. Sukumaran and U. Hanefeld, Chem. Soc. Rev., 2005, 34, 530–542 RSC.
- U. Hanefeld and L. Lefferts, Catalysis: An Integrated Textbook for Students, Wiley-VCH, 2017 Search PubMed.
- K. Faber, Biotransformations in Organic Chemistry, Springer Verlag, 2018 Search PubMed.
- K. Hult and P. Berglund, Trends Biotechnol., 2007, 25, 231–238 CrossRef CAS PubMed.
- H. E. Schoemaker, D. Mink and M. G. Wubbolts, Science, 2003, 299, 1694–1697 CrossRef CAS PubMed.
- A. Schmid, J. S. Dordick, B. Hauer, A. Kiener, M. Wubbolts and B. Witholt, Nature, 2001, 409, 258–268 CrossRef CAS PubMed.
- E. M. M. Abdelraheem, H. Busch, U. Hanefeld and F. Tonin, React. Chem. Eng., 2019, 4, 1878–1894 RSC.
- M. A. Findeis and G. M. Whitesides, J. Org. Chem., 1987, 52, 2838–2848 CrossRef CAS.
- A. M. Klibanov, Nature, 2001, 409, 241–246 CrossRef CAS PubMed.
- L. E. Zetzsche and A. R. H. Narayan, Nat. Rev. Chem., 2020, 4, 334–346 CrossRef CAS PubMed.
- C. K. Winkler, J. H. Schrittwieser and W. Kroutil, ACS Cent. Sci, 2021, 7, 55–71 CrossRef CAS PubMed.
- E. Romero, B. S. Jones, B. N. Hogg, A. Rué Casamajo, M. A. Hayes, S. L. Flitsch, N. J. Turner and C. Schnepel, Angew. Chem., Int. Ed., 2021, 60, 16824–16855 CrossRef CAS PubMed.
- F. H. Arnold, Angew. Chem., Int. Ed., 2019, 58, 14420–14426 CrossRef CAS PubMed.
- M. M. C. H. van Schie, J. D. Spöring, M. Bocola, P. Domínguez de María and D. Rother, Green Chem., 2021, 23, 3191–3206 RSC.
- U. Markel, L. L. Zhu, V. J. Frauenkron-Machedjou, J. Zhao, M. Bocola, M. D. Davari, K. E. Jaeger and U. Schwaneberg, Catalysts, 2017, 7, 142 CrossRef.
- M. A. Huffman, A. Fryszkowska, O. Alvizo, M. Borra-Garske, K. R. Campos, K. A. Canada, P. N. Devine, D. Duan, J. H. Forstater, S. T. Grosser, H. M. Halsey, G. J. Hughes, J. Jo, L. A. Joyce, J. N. Kolev, J. Liang, K. M. Maloney, B. F. Mann, N. M. Marshall, M. McLaughlin, J. C. Moore, G. S. Murphy, C. C. Nawrat, J. Nazor, S. Novick, N. R. Patel, A. Rodriguez-Granillo, S. A. Robaire, E. C. Sherer, M. D. Truppo, A. M. Whittaker, D. Verma, L. Xiao, Y. Xu and H. Yang, Science, 2019, 366, 1255–1259 CrossRef CAS PubMed.
- X. Q. Meng, L. Yang, Y. Liu, H. L. Wang, Y. L. Shen and D. Z. Wei, ChemCatChem, 2021, 13, 3340–3349 CrossRef CAS.
- U. Hanefeld, L. Gardossi and E. Magner, Chem. Soc. Rev., 2009, 38, 453–468 RSC.
- R. A. Sheldon and S. van Pelt, Chem. Soc. Rev., 2013, 42, 6223–6235 RSC.
- A. Basso and S. Serban, Mol. Catal., 2019, 479, 35–54 Search PubMed.
- P. De Santis, L.-E. Meyer and S. Kara, React. Chem. Eng., 2020, 5, 2155–2184 RSC.
- V. D. Jäger, R. Lamm, K. Küsters, G. Ölcücü, M. Oldiges, K. E. Jaeger, J. Büchs and U. Krauss, Appl. Microbiol. Biotechnol., 2020, 104, 7313–7329 CrossRef PubMed.
- W. B. Liang, P. Wied, F. Carraro, C. J. Sumby, B. Nidetzky, C. K. Tsung, P. Falcaro and C. J. Doonan, Chem. Rev., 2021, 121, 1077–1129 CrossRef CAS PubMed.
- D. Videira-Quintela, O. Martin and G. Montalvo, Microchem. J., 2021, 167, 106318 CrossRef CAS.
- C. Ortiz, M. L. Ferreira, O. Barbosa, J. C. S. dos Santos, R. C. Rodrigues, A. Berenguer-Murcia, L. E. Briand and R. Fernandez-Lafuente, Catal. Sci. Technol., 2019, 9, 2380–2420 RSC.
- J. J. Dong, E. Fernández-Fueyo, F. Hollmann, C. E. Paul, M. Pesic, S. Schmidt, Y. H. Wang, S. Younes and W. Y. Zhang, Angew. Chem., Int. Ed., 2018, 57, 9238–9261 CrossRef CAS PubMed.
- F. Hollmann, D. J. Opperman and C. E. Paul, Angew. Chem., Int. Ed., 2021, 60, 5644–5665 CrossRef CAS PubMed.
- R. O. M. A. de Souza, L. S. M. Miranda and U. T. Bornscheuer, Chem. – Eur. J., 2017, 23, 12040–12063 CrossRef CAS PubMed.
- K. Fesko and M. Gruber-Khadjawi, ChemCatChem, 2013, 5, 1248–1272 CrossRef CAS.
- M. Brovetto, D. Gamenara, P. S. Mendez and G. A. Seoane, Chem. Rev., 2011, 111, 4346–4403 CrossRef CAS PubMed.
- D. Rother and S. Malzacher, Nat. Catal, 2021, 4, 92–93 CrossRef CAS.
- R. Roddan, J. M. Ward, N. H. Keep and H. C. Hailes, Curr. Opin. Chem. Biol., 2020, 55, 69–76 CrossRef CAS PubMed.
- P. Bracco, H. Busch, J. von Langermann and U. Hanefeld, Org. Biomol. Chem., 2016, 14, 6375–6389 RSC.
- M. Liu, D. Wei, Z. X. Wen and J. B. Wang, Front. Bioeng. Biotechnol., 2021, 9, 653682 CrossRef PubMed.
- M. Winkler, A. Glieder and K. Steiner, in Comprehensive Chirality, ed. E. M. Carreira and H. Yamamoto, Elsevier, Amsterdam, 2012, pp. 350–371 Search PubMed.
- R. Wiedner, H. Schwab and K. Steiner, Green Biocatalysis, 2016, pp. 603–628 Search PubMed.
- H. C. Hailes, D. Rother, M. Muller, R. Westphal, J. M. Ward, J. Pleiss, C. Vogel and M. Pohl, FEBS J., 2013, 280, 6374–6394 CrossRef CAS PubMed.
- M. Pohl, C. Wechsler and M. Meller, in Science of Synthesis Biocatalysis in Organic Synthesis 2, ed. K. Faber, W.-D. Fessner and N. J. Turner, Georg Thieme Verlag, Stuttgart, 2014, pp. 93–127 Search PubMed.
- F. Motojima, A. Izumi, A. Nuylert, Z. Y. Zhai, M. Dadashipour, S. Shichida, T. Yamaguchi, S. Nakano and Y. Asano, FEBS J., 2021, 288, 1679–1695 CrossRef CAS PubMed.
- U. Hanefeld, Chem. Soc. Rev., 2013, 42, 6308–6321 RSC.
- J. Holt and U. Hanefeld, Curr. Org. Synth., 2009, 6, 15–37 CrossRef CAS.
- T. Purkarthofer, K. Gruber, M. Gruber-Khadjawi, K. Waich, W. Skranc, D. Mink and H. Griengl, Angew. Chem., Int. Ed., 2006, 45, 3454–3456 CrossRef CAS PubMed.
- D. H. S. Rao, A. Chatterjee and S. K. Padhi, Org. Biomol. Chem., 2021, 19, 322–337 RSC.
- A. M. C. H. van den Nieuwendijk, A. B. T. Ghisaidoobe, H. S. Overkleeft, J. Brussee and A. van der Gen, Tetrahedron, 2004, 60, 10385–10396 CrossRef CAS.
- D. J. Vugts, L. Veum, K. al-Mafraji, R. Lemmens, R. F. Schmitz, F. J. J. de Kanter, M. B. Groen, U. Hanefeld and R. V. A. Orru, Eur. J. Org. Chem., 2006, 1672–1677 CrossRef CAS.
- A. Nuylert, M. Nakabayashi, T. Yamaguchi and Y. Asano, Acs Omega, 2020, 5, 27896–27908 CrossRef CAS PubMed.
- M. Dadashipour, Y. Ishida, K. Yamamoto and Y. Asano, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 10605–10610 CrossRef CAS PubMed.
- Y. Kuwahara, T. Yamaguchi, Y. Ichiki, T. Tanabe and Y. Asano, Sci. Nat., 2017, 104, 19 CrossRef PubMed.
- I. Hajnal, A. Lyskowski, U. Hanefeld, K. Gruber, H. Schwab and K. Steiner, FEBS J., 2013, 280, 5815–5828 CrossRef CAS PubMed.
- F. Vertregt, G. Torrelo, S. Trunk, H. Wiltsche, W. R. Hagen, U. Hanefeld and K. Steiner, ACS Catal., 2016, 6, 5081–5085 CrossRef CAS.
- R. Wiedner, B. Kothbauer, T. Pavkov-Keller, M. Gruber-Khadjawi, K. Gruber, H. Schwab and K. Steiner, ChemCatChem, 2015, 7, 325–332 CrossRef CAS.
- B. J. Jones, R. L. Evans, N. J. Mylrea, D. Chaudhury, C. Luo, B. Guan, C. T. Pierce, W. R. Gordon, C. M. Wilmot and R. J. Kazlauskas, PLoS One, 2020, 15, e0235341 CrossRef CAS PubMed.
- S. K. Padhi, R. Fujii, G. A. Legatt, S. L. Fossum, R. Berchtold and R. J. Kazlauskas, Chem. Biol., 2010, 17, 863–871 CrossRef CAS PubMed.
- M. P. van der Helm, P. Bracco, H. Busch, K. Szymanska, A. B. Jarzebski and U. Hanefeld, Catal. Sci. Technol., 2019, 9, 1189–1200 RSC.
- A. Brahma, B. Musio, U. Ismayilova, N. Nikbin, S. B. Kamptmann, P. Siegert, G. E. Jeromin, S. V. Ley and M. Pohl, Synlett, 2016, 262–266 CAS.
- R. Kluger and K. Tittmann, Chem. Rev., 2008, 108, 1797–1833 CrossRef CAS PubMed.
- D. Meyer, P. Neumann, R. Ficner and K. Tittmann, Nat. Chem. Biol., 2013, 9, 488–U484 CrossRef CAS PubMed.
- S. Dai, L. M. Funk, F. R. von Pappenheim, V. Sautner, M. Paulikat, B. Schroder, J. Uranga, R. A. Mata and K. Tittmann, Nature, 2019, 573, 609–613 CrossRef CAS PubMed.
- M. Pareek, Y. Reddi and R. B. Sunoj, Chem. Sci., 2021, 12, 7973–7992 RSC.
- G. Goetz, P. Iwan, B. Hauer, M. Breuer and M. Pohl, Biotechnol. Bioeng., 2001, 74, 317–325 CrossRef CAS PubMed.
- Y. Kobori, D. C. Myles and G. M. Whitesides, J. Org. Chem., 1992, 57, 5899–5907 CrossRef CAS.
- S. R. Marsden, L. Gjonaj, S. J. Eustace and U. Hanefeld, ChemCatChem, 2017, 9, 1808–1814 CrossRef CAS PubMed.
- R. Westphal, C. Vogel, C. Schmitz, J. Pleiss, M. Muller, M. Pohl and D. Rother, Angew. Chem., Int. Ed., 2014, 53, 9376–9379 CrossRef CAS PubMed.
- L. Wang, W. Song, B. Wang, Y. Zhang, X. Xu, J. Wu, C. Gao, J. Liu, X. Chen, J. Chen and L. Liu, ACS Catal., 2021, 11, 2808–2818 CrossRef CAS.
- V. Erdmann, T. Sehl, I. Frindi-Wosch, R. C. Simon, W. Kroutil and D. Rother, ACS Catal., 2019, 9, 7380–7388 CrossRef CAS.
- Q. Liu, X. Xie, M. Tang, W. Tao, T. Shi, Y. Zhang, T. Huang, Y. Zhao, Z. Deng and S. Lin, ACS Catal., 2021, 11, 7477–7488 CrossRef CAS.
- X. Chen, Z. Wang, Y. Lou, Y. Peng, Q. Zhu, J. Xu and Q. Wu, Angew. Chem., Int. Ed., 2021, 60, 9326–9329 CrossRef CAS PubMed.
- S. Desnions, R. Faure and S. Bontemps, ACS Catal., 2019, 9, 9575–9588 CrossRef.
- M. Nattermann, S. Burgener, P. Pfister, A. Chou, L. Schulz, S. H. Lee, N. Paczia, J. Zarzycki, R. Gonzalez and T. J. Erb, ACS Catal., 2021, 11, 5396–5404 CrossRef CAS PubMed.
- J. L. Galman, D. Steadman, S. Bacon, P. Morris, M. E. B. Smith, J. M. Ward, P. A. Dalby and H. C. Hailes, Chem. Commun., 2010, 46, 7608–7610 RSC.
- A. Cazares, J. L. Galman, L. G. Crago, M. E. B. Smith, J. Strafford, L. Rios-Solis, G. J. Lye, P. A. Dalby and H. C. Hailes, Org. Biomol. Chem., 2010, 8, 1301–1309 RSC.
- H. Casajus, A. Lagarde, M. Leremboure, T. D. Miguel, L. Nauton, V. Thery, W. D. Fessner, N. Duguet, F. Charmantray and L. Hecquet, ChemCatChem, 2020, 12, 5772–5779 CrossRef CAS.
- T. Saravanan, S. Junker, M. Kickstein, S. Hein, M. K. Link, J. Ranglack, S. Witt, M. Lorilliere, L. Hecquet and W. D. Fessner, Angew. Chem., Int. Ed., 2017, 56, 5358–5362 CrossRef CAS PubMed.
- S. R. Marsden, D. G. G. McMillan and U. Hanefeld, Int. J. Mol. Sci., 2020, 21, 8641 CrossRef CAS PubMed.
- P. Clapés, Green Biocatalysis, 2016, pp. 267–306 Search PubMed.
- E. Busto, ChemCatChem, 2016, 8, 2589–2598 CrossRef CAS.
- S. R. Marsden, L. Mestrom, D. G. G. McMillan and U. Hanefeld, ChemCatChem, 2020, 12, 426–437 CrossRef CAS.
- K. Fesko, C. Reisinger, J. Steinreiber, H. Weber, M. Schurmann and H. Griengl, J. Mol. Catal. B: Enzym., 2008, 52–53, 19–26 CrossRef CAS.
- K. Shibata, K. Shingu, V. P. Vassilev, K. Nishide, T. Fujita, M. Node, T. Kajimoto and C. H. Wong, Tetrahedron Lett., 1996, 37, 2791–2794 CrossRef CAS.
- S. R. Marsden, L. Mestrom, H. J. Wijma, S. J. Noordam, D. G. G. McMillan and U. Hanefeld, ChemCatChem, 2021, 13, 2530–2536 CrossRef CAS.
- M. Haridas, E. M. M. Abdelraheem and U. Hanefeld, Appl. Microbiol. Biotechnol., 2018, 102, 9959–9971 CrossRef CAS PubMed.
- L. R. Chen, D. P. Dumas and C. H. Wong, J. Am. Chem. Soc., 1992, 114, 741–748 CrossRef CAS.
- H. J. M. Gijsen and C. H. Wong, J. Am. Chem. Soc., 1994, 116, 8422–8423 CrossRef CAS.
- M. Dick, R. Hartmann, O. H. Weiergraber, C. Bisterfeld, T. Classen, M. Schwarten, P. Neudecker, D. Willbold and J. Pietruszka, Chem. Sci., 2016, 7, 4492–4502 RSC.
- S. Voutilainen, M. Heinonen, M. Andberg, E. Jokinen, H. Maaheimo, J. Paakkonen, N. Hakulinen, J. Rouvinen, H. Lahdesmaki, S. Kaski, J. Rousu, M. Penttila and A. Koivula, Appl. Microbiol. Biotechnol., 2020, 104, 10515–10529 CrossRef CAS PubMed.
- M. Haridas, C. Bisterfeld, L. Chen, S. R. Marsden, F. Tonin, R. Medici, A. Iribarren, E. Lewkowicz, P. L. Hagedoorn, U. Hanefeld and E. Abdelraheem, Catalysts, 2020, 10, 883 CrossRef CAS.
- C. Bisterfeld, T. Classen, I. Kuberl, B. Henssen, A. Metz, H. Gohlke and J. Pietruszka, PLoS One, 2016, 11, e0156525 CrossRef PubMed.
- X. C. Jiao, J. Pan, G. C. Xu, X. D. Kong, Q. Chen, Z. J. Zhang and J. H. Xu, Catal. Sci. Technol., 2015, 5, 4048–4054 RSC.
- S. H. Zhang, J. Bramski, M. Tutus, J. Pietruszka, A. Boker and S. Reinicke, ACS Appl. Mater. Interfaces, 2019, 11, 34441–34453 CrossRef CAS PubMed.
- B. Grabner, Y. Pokhilchuk and H. Gruber-Woelfler, Catalysts, 2020, 10, 137 CrossRef CAS.
- P. Hoyos, V. Pace and A. R. Alcantara, Catalysts, 2019, 9, 260 CrossRef CAS.
- M. Schürmann, Industrial Enzyme Applications, 2019, pp. 385–403 Search PubMed.
- J. J. Liu, C. C. Hsu and C. H. Wong, Tetrahedron Lett., 2004, 45, 2439–2441 CrossRef CAS.
- A. Svarc, M. Fekete, K. Hernandez, P. Clapes, Z. F. Blazevic, A. Szekrenyi, D. Skendrovic, D. Vasic-Racki, S. J. Charnock and A. V. Presecki, Chem. Eng. Sci., 2021, 231, 116312 CrossRef CAS.
- A. Svarc, Z. F. Blazevic, D. Vasic-Racki, S. J. Charnock and A. V. Presecki, Chem. Eng. Res. Des., 2020, 164, 35–45 CrossRef CAS.
- K. Pradhan and S. S. Kulkarni, Eur. J. Org. Chem., 2020, 6819–6830 CrossRef CAS.
- C. C. Yu and S. G. Withers, Adv. Synth. Catal., 2015, 357, 1633–1654 CrossRef CAS.
- U. Kragl, D. Gygax, O. Ghisalba and C. Wandrey, Angew. Chem., Int. Ed. Engl., 1991, 30, 827–828 CrossRef.
- V. R. L. J. Bloemendal, S. J. Moons, J. J. A. Heming, M. Chayoua, O. Niesink, J. C. M. van Hest, T. J. Boltje and F. P. J. T. Rutjes, Adv. Synth. Catal., 2019, 361, 2443–2447 CrossRef CAS PubMed.
- M. T. de Martino, F. Tonin, V. R. L. J. Bloemendal, U. Hanefeld, F. P. J. T. Rutjes and J. C. M. van Hest, RSC Adv., 2021, 11, 21857–21861 RSC.
- M. Mahmoudian, D. Noble, C. S. Drake, R. F. Middleton, D. S. Montgomery, J. E. Piercey, D. Ramlakhan, M. Todd and M. J. Dawson, Enzyme Microb. Technol., 1997, 20, 393–400 CrossRef CAS PubMed.
- E. Masai, Y. Katayama and M. Fukuda, Biosci. Biotechnol. Biochem., 2007, 71, 1–15 CrossRef CAS PubMed.
- D. Rea, R. Hovington, J. F. Rakus, J. A. Gerlt, V. Fulop, T. D. H. Bugg and D. I. Roper, Biochemistry, 2008, 47, 9955–9965 CrossRef CAS PubMed.
- R. W. Eaton and P. J. Chapman, J. Bacteriol., 1992, 174, 7542–7554 CrossRef CAS PubMed.
- J. Fang, D. Hait, M. Head-Gordon and M. C. Y. Chang, Angew. Chem., Int. Ed., 2019, 58, 11841–11845 CrossRef CAS PubMed.
- K. Hernandez, J. Joglar, J. Bujons, T. Parella and P. Clapes, Angew. Chem., Int. Ed., 2018, 57, 3583–3587 CrossRef CAS PubMed.
- S. Bosch, E. Sanchez-Freire, M. L. del Pozo, M. Cesnik, J. Quesada, D. M. Mate, K. Hernandez, Y. Y. Qi, P. Clapes, D. Vasic-Racki, Z. F. Blazevic, J. Berenguer and A. Hidalgo, ACS Sustainable Chem. Eng., 2021, 9, 5430–5436 CrossRef CAS PubMed.
- V. de Berardinis, C. Guerard-Helaine, E. Darii, K. Bastard, V. Helaine, A. Mariage, J. L. Petit, N. Poupard, I. Sanchez-Moreno, M. Stam, T. Gefflaut, M. Salanoubat and M. Lemaire, Green Chem, 2017, 19, 519–526 RSC.
- V. Laurent, L. Gourbeyre, A. Uzel, V. Helaine, L. Nauton, M. Traikia, V. de Berardinis, M. Salanoubat, T. Gefflaut, M. Lemaire and C. Guerard-Helaine, ACS Catal., 2020, 10, 2538–2543 CrossRef CAS.
- S. R. Marsden, L. Mestrom, I. Bento, P. L. Hagedoorn, D. G. G. McMillan and U. Hanefeld, Adv. Synth. Catal., 2019, 361, 2649–2658 CrossRef CAS.
- T. Suzuki and N. Takizawa, Biosci. Biotechnol. Biochem., 2019, 83, 1884–1888 CrossRef CAS PubMed.
- D. J. Fansher, R. Granger, S. Kaur and D. R. J. Palmer, ACS Catal., 2021, 11, 6939–6943 CrossRef CAS.
- A. M. Li, L. Cai, Z. Chen, M. Y. Wang, N. Wang, H. Nakanishi, X. D. Gao and Z. J. Li, Carbohydr. Res., 2017, 452, 108–115 CrossRef CAS PubMed.
- V. Laurent, V. Helaine, C. Vergne-Vaxelaire, L. Nauton, M. Traikia, J. L. Petit, M. Salanoubat, V. de Berardinis, M. Lemaire and C. Guerard-Helaine, ACS Catal., 2019, 9, 9508–9512 CrossRef CAS.
- L. Wen, K. Huang, M. Wei, J. Meisner, Y. Liu, K. Garner, L. Zang, X. Wang, X. Li, J. Fang, H. Zhang and P. G. Wang, Angew. Chem., Int. Ed., 2015, 54, 12654–12658 CrossRef CAS PubMed.
- Z. Ferjancic and R. N. Saicic, Eur. J. Org. Chem., 2021, 3241–3250 CrossRef CAS.
- J. G. Yang, J. T. Li, Y. Men, Y. M. Zhu, Y. Zhang, Y. X. Sun and Y. H. Ma, Appl. Environ. Microbiol., 2015, 81, 4284–4294 CrossRef CAS PubMed.
- L. Babich, A. F. Hartog, L. J. C. van Hemert, F. P. J. T. Rutjes and R. Wever, ChemSusChem, 2012, 5, 2348–2353 CrossRef CAS PubMed.
- Z. J. Li, F. Li, L. Cai, Z. Chen, L. Qin and X. D. Gao, J. Agric. Food Chem., 2020, 68, 1347–1353 CrossRef CAS PubMed.
- M. Sugiyama, Z. Y. Hong, W. A. Greenberg and C. H. Wong, Bioorg. Med. Chem., 2007, 15, 5905–5911 CrossRef CAS PubMed.
- A. J. Rigual, J. Cantero, M. Risso, P. Rodriguez, S. Rodriguez, M. Paulino, D. Gamenara and N. Veiga, Mol. Catal., 2020, 495, 111131 CrossRef CAS.
- X. Garrabou, J. Joglar, T. Parella, R. Crehuet, J. Bujons and P. Clapés, Adv. Synth. Catal., 2011, 353, 89–99 CrossRef CAS.
- J. A. Castillo, J. Calveras, J. Casas, M. Mitjans, M. P. Vinardell, T. Parella, T. Inoue, G. A. Sprenger, J. Joglar and P. Clapés, Org. Lett., 2006, 8, 6067–6070 CrossRef PubMed.
- M. Sugiyama, Z. Hong, P. H. Liang, S. M. Dean, L. J. Whalen, W. A. Greenberg and C. H. Wong, J. Am. Chem. Soc., 2007, 129, 14811–14817 CrossRef CAS PubMed.
- X. Garrabou, J. Calveras, J. Joglar, T. Parella, J. Bujons and P. Clapés, Org. Biomol. Chem., 2011, 9, 8430–8436 RSC.
- A. L. Concia, L. Gomez, J. Bujons, T. Parella, C. Vilaplana, P. J. Cardona, J. Joglar and P. Clapés, Org. Biomol. Chem., 2013, 11, 2005–2021 RSC.
- M. Gutierrez, T. Parella, J. Joglar, J. Bujons and P. Clapés, Chem. Commun., 2011, 47, 5762–5764 RSC.
- R. Roldán, K. Hernández, J. Joglar, J. Bujons, T. Parella, W. D. Fessner and P. Clapés, Adv. Synth. Catal., 2019, 361, 2673–2687 CrossRef PubMed.
- D. Güclü, A. Szekrenyi, X. Garrabou, M. Kickstein, S. Junker, P. Clapés and W. D. Fessner, ACS Catal., 2016, 6, 1848–1852 CrossRef.
- M. Widersten, Enzyme Engineering and Evolution: Specific Enzyme Applications, 2020, vol. 644, pp. 149–167 Search PubMed.
- G. Masdeu, Z. F. Blazevic, S. Kralj, D. Makovec, J. Lopez-Santin and G. Alvaro, Chem. Eng. Sci., 2021, 239, 116602 CrossRef CAS.
- S. Wang and H. Deng, Appl. Microbiol. Biotechnol., 2021, 105, 3507–3520 CrossRef CAS PubMed.
- K. Fesko, Appl. Microbiol. Biotechnol., 2016, 100, 2579–2590 CrossRef CAS PubMed.
- S. E. Franz and J. D. Stewart, Adv. Appl. Microbiol., 2014, 88, 57–101 Search PubMed.
- Q. J. Chen, X. Chen, Y. F. Cui, J. Ren, W. Lu, J. H. Feng, Q. Q. Wu and D. M. Zhu, Catal. Sci. Technol., 2017, 7, 5964–5973 RSC.
- M. L. Gutierrez, X. Garrabou, E. Agosta, S. Servi, T. Parella, J. Joglar and P. Clapes, Chem. – Eur. J., 2008, 14, 4647–4656 CrossRef CAS PubMed.
- M. Pickl, R. Marin-Valls, J. Joglar, J. Bujons and P. Clapes, Adv. Synth. Catal., 2021, 363, 2866–2876 CrossRef CAS PubMed.
- A. Soler, X. Garrabou, K. Hernández, M. L. Gutiérrez, E. Busto, J. Bujons, T. Parella, J. Joglar and P. Clapés, Adv. Synth. Catal., 2014, 356, 3007–3024 CrossRef CAS.
- M. D. Patil, G. Grogan and H. Yun, ChemCatChem, 2018, 10, 4797–4818 CrossRef CAS.
- B. R. Lichman, Nat. Prod. Rep., 2021, 38, 103–129 RSC.
- C. S. Jamieson, J. Misa, Y. Tang and J. M. Billingsley, Chem. Soc. Rev., 2021, 50, 6950–7008 RSC.
- J. X. Zhao, D. Mendez-Sanchez, R. Roddan, J. M. Ward and H. C. Hailes, ACS Catal., 2021, 11, 131–138 CrossRef CAS.
- R. Roddan, G. Gygli, A. Sula, D. Mendez-Sanchez, J. Pleiss, J. M. Ward, N. H. Keep and H. C. Hailes, ACS Catal., 2019, 9, 9640–9649 CrossRef CAS.
- H. Stecher, M. Tengg, B. J. Ueberbacher, P. Remler, H. Schwab, H. Griengl and M. Gruber-Khadjawi, Angew. Chem., Int. Ed., 2009, 48, 9546–9548 CrossRef CAS PubMed.
- M. Tengg, H. Stecher, L. Offner, K. Plasch, F. Anderl, H. Weber, H. Schwab and M. Gruber-Khadjawi, ChemCatChem, 2016, 8, 1354–1360 CrossRef CAS.
- I. J. W. McKean, J. C. Sadler, A. Cuetos, A. Frese, L. D. Humphreys, G. Grogan, P. A. Hoskisson and G. A. Burley, Angew. Chem., Int. Ed., 2019, 58, 17583–17588 CrossRef CAS PubMed.
- J. C. Sadler, L. D. Humphreys, R. Snajdrova and G. A. Burley, ChemBioChem, 2017, 18, 992–995 CrossRef CAS PubMed.
- C. Sommer-Kamann, A. Fries, S. Mordhorst, J. N. Andexer and M. Müller, Angew. Chem., Int. Ed., 2017, 56, 4033–4036 CrossRef CAS PubMed.
- S. Mordhorst and J. N. Andexer, Nat. Prod. Rep., 2020, 37, 1316–1333 RSC.
- F. Michailidou and A. Rentmeister, Org. Biomol. Chem., 2021, 19, 3756–3762 RSC.
- N. G. Schmidt, T. Pavkov-Keller, N. Richter, B. Wiltschi, K. Gruber and W. Kroutil, Angew. Chem., Int. Ed., 2017, 56, 7615–7619 CrossRef CAS PubMed.
- A. Żądło-Dobrowolska, N. G. Schmidt and W. Kroutil, ChemCatChem, 2019, 11, 1064–1068 CrossRef PubMed.
- N. G. Schmidt and W. Kroutil, Eur. J. Org. Chem., 2017, 5865–5871 CrossRef CAS.
- X. Sheng, M. Kazemi, A. Żądło-Dobrowolska, W. Kroutil and F. Himo, ACS Catal., 2020, 10, 570–577 CrossRef CAS PubMed.
- A. Żądło-Dobrowolska, L. Hammerer, T. Paykov-Keller, K. Gruber and W. Kroutil, ACS Catal., 2020, 10, 1094–1101 CrossRef PubMed.
- P. S. Coelho, E. M. Brustad, A. Kannan and F. H. Arnold, Science, 2013, 339, 307–310 CrossRef CAS PubMed.
- P. S. Coelho, Z. J. Wang, M. E. Ener, S. A. Baril, A. Kannan, F. H. Arnold and E. M. Brustad, Nat. Chem. Biol., 2013, 9, 485–U433 CrossRef CAS PubMed.
- N. S. Sarai, B. J. Levin, J. M. Roberts, D. E. Katsoulis and F. H. Arnold, ACS Cent. Sci., 2021, 7, 944–953 CrossRef CAS PubMed.
- A. Z. Zhou, K. Chen and F. H. Arnold, ACS Catal., 2020, 10, 5393–5398 CrossRef CAS.
- B. J. Wittmann, A. M. Knight, J. L. Hofstra, S. E. Reisman, S. B. J. Kan and F. H. Arnold, ACS Catal., 2020, 10, 7112–7116 CrossRef CAS PubMed.
- O. F. Brandenberg, K. Chen and F. H. Arnold, J. Am. Chem. Soc., 2019, 141, 8989–8995 CrossRef CAS PubMed.
- O. F. Brandenberg, R. Fasan and F. H. Arnold, Curr. Opin. Biotechnol., 2017, 47, 102–111 CrossRef CAS PubMed.
- Z. J. Wang, H. Renata, N. E. Peck, C. C. Farwell, P. S. Coelho and F. H. Arnold, Angew. Chem., Int. Ed., 2014, 53, 6810–6813 CrossRef CAS PubMed.
- J. E. Zhang, X. Y. Huang, R. J. K. Zhang and F. H. Arnold, J. Am. Chem. Soc., 2019, 141, 9798–9802 CrossRef CAS PubMed.
- K. Chen and F. H. Arnold, J. Am. Chem. Soc., 2020, 142, 6891–6895 CrossRef CAS PubMed.
- C. E. Paul, D. Eggerichs, A. H. Westphal, D. Tischler and W. J. H. van Berkel, Biotechnol. Adv., 2021, 51, 107712 CrossRef CAS PubMed.
- Y. H. Wang, D. M. Lan, R. Durrani and F. Hollmann, Curr. Opin. Chem. Biol., 2017, 37, 1–9 CrossRef PubMed.
- V. B. Urlacher and M. Girhard, Trends Biotechnol., 2019, 37, 882–897 CrossRef CAS PubMed.
- M. K. Julsing, S. Cornelissen, B. Buhler and A. Schmid, Curr. Opin. Chem. Biol., 2008, 12, 177–186 CrossRef CAS PubMed.
- S. Chakrabarty, Y. Wang, J. C. Perkins and A. R. H. Narayan, Chem. Soc. Rev., 2020, 49, 8137–8155 RSC.
- L. M. Blank, B. E. Ebert, K. Buehler and B. Buhler, Antioxid. Redox Signaling, 2010, 13, 349–394 CrossRef CAS PubMed.
- W. A. Suske, W. J. H. van Berkel and H. P. E. Kohler, J. Biol. Chem., 1999, 274, 33355–33365 CrossRef CAS PubMed.
- W. A. Suske, M. Held, A. Schmid, T. Fleischmann, M. G. Wubbolts and H. P. E. Kohler, J. Biol. Chem., 1997, 272, 24257–24265 CrossRef CAS PubMed.
- A. Meyer, A. Schmid, M. Held, A. H. Westphal, M. Rothlisberger, H. P. E. Kohler, W. J. H. van Berkel and B. Witholt, J. Biol. Chem., 2002, 277, 5575–5582 CrossRef CAS PubMed.
- A. Meyer, M. Wursten, A. Schmid, H. P. E. Kohler and B. Witholt, J. Biol. Chem., 2002, 277, 34161–34167 CrossRef CAS PubMed.
- A. Meyer, M. Held, A. Schmid, H. P. E. Kohler and B. Witholt, Biotechnol. Bioeng., 2003, 81, 518–524 CrossRef CAS PubMed.
- O. Shoji, T. Kunimatsu, N. Kawakami and Y. Watanabe, Angew. Chem., Int. Ed., 2013, 52, 6606–6610 CrossRef CAS PubMed.
- S. Ariyasu, J. K. Stanfield, Y. Aiba and O. Shoji, Curr. Opin. Chem. Biol., 2020, 59, 155–163 CrossRef CAS PubMed.
- H. Y. Zhou, B. J. Wang, F. Wang, X. J. Yu, L. X. Ma, A. T. Li and M. T. Reetz, Angew. Chem., Int. Ed., 2019, 58, 764–768 CrossRef CAS PubMed.
- S. D. Munday, S. Dezvarei, I. C. K. Lau and S. G. Bell, ChemCatChem, 2017, 9, 2512–2522 CrossRef CAS.
- A. Dennig, A. M. Weingartner, T. Kardashliev, C. A. Muller, E. Tassano, M. Schurmann, A. J. Ruff and U. Schwaneberg, Chem. – Eur. J., 2017, 23, 17981–17991 CrossRef CAS PubMed.
- M. Karasawa, J. K. Stanfield, S. Yanagisawa, O. Shoji and Y. Watanabe, Angew. Chem., Int. Ed., 2018, 57, 12264–12269 CrossRef CAS PubMed.
- P. G. Gomez de Santos, M. Cañellas, F. Tieves, S. H. H. Younes, P. Molina-Espeja, M. Hofrichter, F. Hollmann, V. Guallar and M. Alcalde, ACS Catal., 2018, 8, 4789–4799 CrossRef CAS.
- E. Aranda, R. Ullrich and M. Hofrichter, Biodegradation, 2010, 21, 267–281 CrossRef CAS PubMed.
- M. Kinne, R. Ullrich, K. E. Hammel, K. Scheibner and M. Hofrichter, Tetrahedron Lett., 2008, 49, 5950–5953 CrossRef CAS.
- R. Ullrich and M. Hofrichter, Cell. Mol. Life Sci., 2007, 64, 271–293 CrossRef CAS PubMed.
- P. Molina-Espeja, J. Viña-Gonzalez, B. J. Gomez-Fernandez, J. Martin-Diaz, E. Garcia-Ruiz and M. Alcalde, Biotechnol. Adv., 2016, 34, 754–767 CrossRef PubMed.
- D. M. Mate, M. A. Palomino, P. Molina-Espeja, J. Martin-Diaz and M. Alcalde, Protein Eng. Des. Sel., 2017, 30, 191–198 CAS.
- M. Petersen and A. Kiener, Green Chem., 1999, 1, 99–106 RSC.
- A. Kiener, J. P. Roduit, A. Tschech, A. Tinschert and K. Heinzmann, Synlett, 1994, 814–816 CrossRef CAS.
- H. G. Kulla, Chimia, 1991, 45, 81–85 CAS.
- M. Nagel and J. R. Andreesen, Arch. Microbiol., 1990, 154, 605–613 CrossRef CAS.
- K. S. Ju and R. E. Parales, Microbiol. Mol. Biol. Rev., 2010, 74, 250–272 CrossRef CAS PubMed.
- M. R. Buddha, T. Tao, R. J. Parry and B. R. Crane, J. Biol. Chem., 2004, 279, 49567–49570 CrossRef CAS PubMed.
- S. C. Dodani, G. Kiss, J. K. B. Cahn, Y. Su, V. S. Pande and F. H. Arnold, Nat. Chem., 2016, 8, 419–425 CrossRef CAS PubMed.
- S. M. Barry, J. A. Kers, E. G. Johnson, L. J. Song, P. R. Aston, B. Patel, S. B. Krasnoff, B. R. Crane, D. M. Gibson, R. Loria and G. L. Challis, Nat. Chem. Biol., 2012, 8, 814–816 CrossRef CAS PubMed.
- S. Louka, S. M. Barry, D. J. Heyes, M. Q. E. Mubarak, H. S. Ali, L. M. Alkhalaf, A. W. Munro, N. S. Scrutton, G. L. Challis and S. P. de Visser, J. Am. Chem. Soc., 2020, 142, 15764–15779 CrossRef CAS PubMed.
- D. R. Boyd and T. D. H. Bugg, Org. Biomol. Chem., 2006, 4, 181–192 RSC.
- D. R. Boyd, N. D. Sharma and C. C. R. Allen, Curr. Opin. Biotechnol., 2001, 12, 564–573 CrossRef CAS PubMed.
- T. Hudlicky, D. Gonzalez and D. T. Gibson, Aldrichim. Acta, 1999, 32, 35–62 CAS.
- T. Hudlicky and J. W. Reed, Chem. Soc. Rev., 2009, 38, 3117–3132 RSC.
- B. D. Ensley and D. T. Gibson, J. Bacteriol., 1983, 155, 505–511 CrossRef CAS PubMed.
- S. V. Ley and F. Sternfeld, Tetrahedron, 1989, 45, 3463–3476 CrossRef CAS.
- T. Hudlicky, H. Luna, G. Barbieri and L. D. Kwart, J. Am. Chem. Soc., 1988, 110, 4735–4741 CrossRef CAS.
- M. G. Banwell, A. J. Edwards, D. W. Lupton and G. Whited, Aust. J. Chem., 2005, 58, 14–17 CrossRef CAS.
- O. J. Kokas, M. G. Banwell and A. C. Willis, Tetrahedron, 2008, 64, 6444–6451 CrossRef CAS.
- G. Guroff, J. W. Daly, D. M. Jerina, J. Renson, B. Witkop and S. Udenfriend, Science, 1967, 157, 1524–1530 CrossRef CAS PubMed.
- W. Y. Zhang, H. H. Li, S. H. H. Younes, P. G. de Santos, F. Tieves, G. Grogan, M. Pabst, M. Alcalde, A. C. Whitwood and F. Hollmann, ACS Catal., 2021, 11, 2644–2649 CrossRef CAS PubMed.
- S. A. Baker Dockrey, A. L. Lukowski, M. R. Becker and A. R. H. Narayan, Nat. Chem., 2018, 10, 119–125 CrossRef CAS PubMed.
- M. Girhard, S. Schuster, M. Dietrich, P. Durre and V. B. Urlacher, Biochem. Biophys. Res. Commun., 2007, 362, 114–119 CrossRef CAS PubMed.
- I. Matsunaga, A. Ueda, N. Fujiwara, T. Sumimoto and K. Ichihara, Lipids, 1999, 34, 841–846 CrossRef CAS PubMed.
- I. Funk, N. Rimmel, C. Schorsch, V. Sieber and J. Schmid, J. Ind. Microbiol. Biotechnol., 2017, 44, 1491–1502 CrossRef CAS PubMed.
- P. Durairaj, S. Malla, S. P. Nadarajan, P. G. Lee, E. Jung, H. H. Park, B. G. Kim and H. Yun, Microb. Cell Factories, 2015, 14, 45 CrossRef PubMed.
- P. Meinhold, M. W. Peters, A. Hartwick, A. R. Hernandez and F. H. Arnold, Adv. Synth. Catal., 2006, 348, 763–772 CrossRef CAS.
- C. Sathesh-Prabu and S. K. Lee, J. Agric. Food Chem., 2015, 63, 8199–8208 CrossRef CAS PubMed.
- M. J. Maseme, A. Pennec, J. van Marwijk, D. J. Opperman and M. S. Smit, Angew. Chem., Int. Ed., 2020, 59, 10359–10362 CrossRef CAS PubMed.
- J. Manning, M. Tavanti, J. L. Porter, N. Kress, S. P. De Visser, N. J. Turner and S. L. Flitsch, Angew. Chem., Int. Ed., 2019, 58, 5668–5671 CrossRef CAS PubMed.
- W. Lu, J. H. Feng, X. Chen, Y. J. Bao, Y. Wang, Q. Q. Wu, Y. H. Ma and D. M. Zhu, Appl. Environ. Microbiol., 2019, 85, e01182–01119 CAS.
- W. Lu, X. Chen, J. H. Feng, Y. J. Bao, Y. Wang, Q. Q. Wu and D. M. Zhu, Appl. Environ. Microbiol., 2018, 84, e00503–00518 CAS.
- Y. X. Li, B. Qin, X. Q. Li, J. Tang, Y. Chen, L. N. Zhou and S. You, ChemCatChem, 2018, 10, 559–565 CrossRef CAS.
- K. D. Zhang, B. M. Shafer, M. D. Demars, H. A. Stern and R. Fasan, J. Am. Chem. Soc., 2012, 134, 18695–18704 CrossRef CAS PubMed.
- J. H. Z. Lee, S. H. Wong, J. E. Stok, S. A. Bagster, J. Beckett, J. K. Clegg, A. J. Brock, J. J. De Voss and S. G. Bell, Arch. Biochem. Biophys., 2019, 663, 54–63 CrossRef CAS PubMed.
- M. Tavanti, J. Mangas-Sanchez, S. L. Montgomery, M. P. Thompson and N. J. Turner, Org. Biomol. Chem., 2017, 15, 9790–9793 RSC.
- H. Wang, Y. C. Zheng, F. F. Chen, J. H. Xu and H. L. Yu, ChemCatChem, 2020, 12, 2077–2082 CrossRef CAS.
- Z. J. Jia, S. L. Gao and F. H. Arnold, J. Am. Chem. Soc., 2020, 142, 10279–10283 CrossRef CAS PubMed.
- C. K. Prier, R. J. K. Zhang, A. R. Buller, S. Brinkmann-Chen and F. H. Arnold, Nat. Chem., 2017, 9, 629–634 CrossRef CAS PubMed.
- J. Latham, E. Brandenburger, S. A. Shepherd, B. R. K. Menon and J. Micklefield, Chem. Rev., 2018, 118, 232–269 CrossRef CAS PubMed.
- J. Büchler, A. Papadopoulou and R. Buller, Catalysts, 2019, 9, 1030 CrossRef.
- D. S. Gkotsi, J. Dhaliwal, M. M. W. McLachlan, K. R. Mulholand and R. J. M. Goss, Curr. Opin. Chem. Biol., 2018, 43, 119–126 CrossRef CAS PubMed.
- M. Frese, P. H. Guzowska, H. Voss and N. Sewald, ChemCatChem, 2014, 6, 1270–1276 CAS.
- J. T. Payne, M. C. Andorfer and J. C. Lewis, Angew. Chem., Int. Ed., 2013, 52, 5271–5274 CrossRef CAS PubMed.
- J. Zeng and J. X. Zhan, Biotechnol. Lett., 2011, 33, 1607–1613 CrossRef CAS PubMed.
- S. A. Shepherd, C. Karthikeyan, J. Latham, A. W. Struck, M. L. Thompson, B. R. K. Menon, M. Q. Styles, C. Levy, D. Leys and J. Micklefield, Chem. Sci., 2015, 6, 3454–3460 RSC.
- A. Phintha, K. Prakinee and P. Chaiyen, in The Enzymes, ed. P. Chaiyen and F. Tamanoi, Academic Press, 2020, vol. 47, ch. 11, pp. 327–364 Search PubMed.
- S. Unversucht, F. Hollmann, A. Schmid and K.-H. van Pée, Adv. Synth. Catal., 2005, 347, 1163–1167 CrossRef CAS.
- C. J. Dong, S. Flecks, S. Unversucht, C. Haupt, K.-H. van Pée and J. H. Naismith, Science, 2005, 309, 2216–2219 CrossRef CAS PubMed.
- M. Ismail, L. Schroeder, M. Frese, T. Kottke, F. Hollmann, C. E. Paul and N. Sewald, ACS Catal., 2019, 9, 1389–1395 CrossRef CAS PubMed.
- L. Schroeder, M. Frese, C. Müller, N. Sewald and T. Kottke, ChemCatChem, 2018, 10, 3336–3341 CrossRef CAS.
- C. Schnepel, V. I. Dodero and N. Sewald, Chem. – Eur. J., 2021, 27, 5404–5411 CrossRef CAS PubMed.
- C. Schnepel, H. Minges, M. Frese and N. Sewald, Angew. Chem., Int. Ed., 2016, 55, 14159–14163 CrossRef CAS PubMed.
- S. Dachwitz, D. H. Duwe, Y. H. Wang, H. Gruss, Y. Hannappel, T. Hellweg and N. Sewald, Chem. – Eur. J., 2020, 26, 16357–16364 CrossRef CAS PubMed.
- G. T. Höfler, A. But and F. Hollmann, Org. Biomol. Chem., 2019, 17, 9267–9274 RSC.
- R. Wever, R. Renirie and F. Hollmann, Vanadium Catalysis, The Royal Society of Chemistry, 2021, pp. 548–563 Search PubMed.
- R. Wever, B. E. Krenn and R. Renirie, in Methods Enzymol., ed. B. S. Moore, Academic Press, 2018, vol. 605, pp. 141–201 Search PubMed.
- E. Fernández-Fueyo, M. van Wingerden, R. Renirie, R. Wever, Y. Ni, D. Holtmann and F. Hollmann, ChemCatChem, 2015, 7, 4035–4038 CrossRef.
- V. Weichold, D. Milbredt and K. H. van Pee, Angew. Chem., Int. Ed., 2016, 55, 6374–6389 CrossRef CAS PubMed.
- D. Wischang and J. Hartung, Tetrahedron, 2011, 67, 4048–4054 CrossRef CAS.
- D. Wischang, J. Hartung, T. Hahn, R. Ulber, T. Stumpf and C. Fecher-Trost, Green Chem, 2011, 13, 102–108 RSC.
- D. Thiel, F. Blume, C. Jaeger and J. Deska, Eur. J. Org. Chem., 2018, 2717–2725 CrossRef CAS.
- D. Thiel, D. Doknic and J. Deska, Nat. Commun., 2014, 5, 5278 CrossRef CAS PubMed.
- E. Fernández-Fueyo, S. H. H. Younes, S. van Rootselaar, R. W. M. Aben, R. Renirie, R. Wever, D. Holtmann, F. P. J. T. Rutjes and F. Hollmann, ACS Catal., 2016, 6, 5904–5907 CrossRef.
- S. H. H. Younes, F. Tieves, D. M. Lan, Y. H. Wang, P. Süss, H. Brundiek, R. Wever and F. Hollmann, ChemSusChem, 2020, 13, 97–101 CrossRef CAS PubMed.
- G. T. Höfler, A. But, S. H. H. Younes, R. Wever, C. E. Paul, I. W. C. E. Arends and F. Hollmann, ACS Sustainable Chem. Eng., 2020, 8, 2602–2607 CrossRef PubMed.
- J. Naapuri, J. D. Rolfes, J. Keil, C. M. Sapu and J. Deska, Green Chem., 2017, 19, 447–452 RSC.
- X. M. Xu, A. But, R. Wever and F. Hollmann, ChemCatChem, 2020, 12, 2180–2183 CrossRef CAS.
- A. But, E. van der Wijst, J. Le Nôtre, R. Wever, J. P. M. Sanders, J. H. Bitter and E. L. Scott, Green Chem., 2017, 19, 5178–5186 RSC.
- A. But, J. Le Nôtre, E. L. Scott, R. Wever and J. P. M. Sanders, ChemSusChem, 2012, 5, 1199–1202 CrossRef CAS PubMed.
- G. Y. Li, M. Garcia-Borràs, M. J. L. J. Fürst, A. Ilie, M. W. Fraaije, K. N. Houk and M. T. Reetz, J. Am. Chem. Soc., 2018, 140, 10464–10472 CrossRef CAS PubMed.
- Z. G. Zhang, G. D. Roiban, J. P. Acevedo, I. Polyak and M. T. Reetz, Adv. Synth. Catal., 2013, 355, 99–106 CrossRef CAS.
- J. Rehdorf, M. D. Mihovilovic and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2010, 49, 4506–4508 CrossRef CAS PubMed.
- G. de Gonzalo, M. D. Mihovilovic and M. W. Fraaije, ChemBioChem, 2010, 11, 2208–2231 CrossRef CAS PubMed.
- S. Schmidt, C. Scherkus, J. Muschiol, U. Menyes, T. Winkler, W. Hummel, H. Gröger, A. Liese, H. G. Herz and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2015, 54, 2784–2787 CrossRef CAS PubMed.
- R. Karande, D. Salamanca, A. Schmid and K. Buehler, Biotechnol. Bioeng., 2018, 115, 312–320 CrossRef CAS PubMed.
- F. Hollmann, S. Kara, D. J. Opperman and Y. H. Wang, Chem. – Asian J., 2018, 13, 3601–3610 CrossRef CAS PubMed.
- A. Pennec, F. Hollmann, M. S. Smit and D. J. Opperman, ChemCatChem, 2015, 7, 236–239 CrossRef CAS.
- K. Chen, S. Q. Zhang, O. F. Brandenberg, X. Hong and F. H. Arnold, J. Am. Chem. Soc., 2018, 140, 16402–16407 CrossRef CAS PubMed.
- S. B. J. Kan, X. Y. Huang, Y. Gumulya, K. Chen and F. H. Arnold, Nature, 2017, 552, 132–136 CrossRef CAS PubMed.
- S. B. J. Kan, R. D. Lewis, K. Chen and F. H. Arnold, Science, 2016, 354, 1048–1051 CrossRef CAS PubMed.
- Z. Wu, S. B. J. Kan, R. D. Lewis, B. J. Wittmann and F. H. Arnold, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 8852–8858 CrossRef CAS PubMed.
- S. Bahr, S. Brinkmann-Chen, M. Garcia-Borras, J. M. Roberts, D. E. Katsoulis, K. N. Houk and F. H. Arnold, Angew. Chem., Int. Ed., 2020, 59, 15507–15511 CrossRef PubMed.
- C. C. Farwell, J. A. McIntosh, T. K. Hyster, Z. J. Wang and F. H. Arnold, J. Am. Chem. Soc., 2014, 136, 8766–8771 CrossRef CAS PubMed.
- C. Patinios, L. Lanza, I. Corino, M. C. R. Franssen, J. van der Oost, R. A. Weusthuis and S. W. M. Kengen, Front. Microbiol., 2020, 11, 579844 CrossRef PubMed.
- P. Bracco, N. van Midden, E. Arango, G. Torrelo, V. Ferrario, L. Gardossi and U. Hanefeld, Catalysts, 2020, 10, 308 CrossRef CAS.
- A. H. J. Deniau, M. Subileau and E. Dubreucq, ChemBioChem, 2018, 19, 1839–1844 CrossRef PubMed.
- M. Subileau, A. H. Jan, J. Drone, C. Rutyna, V. Perrier and E. Dubreucq, Catal. Sci. Technol., 2017, 7, 2566–2578 RSC.
- T. N. Nguyen, E. Dubreucq, V. Perrier, Q. H. Tran, C. Charpentier, C. Charnay, F. Terki, C. Jay-Allemand and L. P. R. Bidel, Food Chem., 2020, 318, 126482 CrossRef CAS PubMed.
- I. Mathews, M. Soltis, M. Saldajeno, G. Ganshaw, R. Sala, W. Weyler, M. A. Cervin, G. Whited and R. Bott, Biochemistry, 2007, 46, 8969–8979 CrossRef CAS PubMed.
- C. Z. Dinu, I. V. Borkar, S. S. Bale, A. S. Campbell, R. S. Kane and J. S. Dordick, J. Mol. Catal. B: Enzym., 2012, 75, 20–26 CrossRef CAS.
- A. Drozdz, U. Hanefeld, K. Szymanska, A. Jarzebski and A. Chrobok, Catal. Commun., 2016, 81, 37–40 CrossRef CAS.
- N. de Leeuw, G. Torrelo, C. Bisterfeld, V. Resch, L. Mestrom, E. Straulino, L. van der Weel and U. Hanefeld, Adv. Synth. Catal., 2018, 360, 242–249 CrossRef CAS.
- E. Jost, M. Kazemi, V. Mrkonjic, F. Himo, C. K. Winkler and W. Kroutil, ACS Catal., 2020, 10, 10500–10507 CrossRef CAS.
- S. P. Godehard, C. P. S. Badenhorst, H. Müller and U. T. Bornscheuer, ACS Catal., 2020, 10, 7552–7562 CrossRef CAS.
- L. Mestrom, J. G. R. Claessen and U. Hanefeld, ChemCatChem, 2019, 11, 2004–2010 CrossRef CAS.
- H. Land, P. Hendil-Forssell, M. Martinelle and P. Berglund, Catal. Sci. Technol., 2016, 6, 2897–2900 RSC.
- M. L. Contente, A. Pinto, F. Molinari and F. Paradisi, Adv. Synth. Catal., 2018, 360, 4814–4819 CrossRef CAS.
- M. L. Contente, L. Tamborini, F. Molinari and F. Paradisi, J. Flow Chem., 2020, 10, 235–240 CrossRef CAS.
- K. Szymanska, K. Odrozek, A. Zniszczol, G. Torrelo, V. Resch, U. Hanefeld and A. B. Jarzebski, Catal. Sci. Technol., 2016, 6, 4882–4888 RSC.
- M. Finnveden, S. Semlitsch, O. He and M. Martinelle, Catal. Sci. Technol., 2019, 9, 4920–4927 RSC.
- H. Müller, S. P. Godehard, G. J. Palm, L. Berndt, C. P. S. Badenhorst, A. K. Becker, M. Lammers and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2021, 60, 2013–2017 CrossRef PubMed.
- Z. Z. Yan, L. P. Ding, D. D. Zou, L. Y. Wang, Y. Z. Tan, S. T. Guo, Y. C. Zhang and Z. H. Xin, Arch. Microbiol., 2021, 203, 4113–4125 CrossRef CAS PubMed.
- H. Müller, A. K. Becker, G. J. Palm, L. Berndt, C. P. S. Badenhorst, S. P. Godehard, L. Reisky, M. Lammers and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2020, 59, 11607–11612 CrossRef PubMed.
- P. Pongpamorn, C. Kiattisewee, N. Kittipanukul, J. Jaroensuk, D. Trisrivirat, S. Maenpuen and P. Chaiyen, Angew. Chem., Int. Ed., 2021, 60, 5749–5753 CrossRef CAS PubMed.
- M. L. Contente, S. Farris, L. Tamborini, F. Molinari and F. Paradisi, Green Chem., 2019, 21, 3263–3266 RSC.
- I. C. Perdomo, S. Gianolio, A. Pinto, D. Romano, M. L. Contente, F. Paradisi and F. Molinari, J. Agric. Food Chem., 2019, 67, 6517–6522 CrossRef CAS PubMed.
- F. Annunziata, M. L. Contente, D. Betti, C. Pinna, F. Molinari, L. Tamborini and A. Pinto, Catalysts, 2020, 10, 939 CrossRef CAS.
- M. L. Contente, D. R. Padrosa, F. Molinari and F. Paradisi, Nat. Catal., 2020, 3, 1020–1026 CrossRef CAS.
- A. J. L. Wood, N. J. Weise, J. D. Frampton, M. S. Dunstan, M. A. Hollas, S. R. Derrington, R. C. Lloyd, D. Quaglia, F. Parmeggiani, D. Leys, N. J. Turner and S. L. Flitsch, Angew. Chem., Int. Ed., 2017, 56, 14498–14501 CrossRef CAS PubMed.
- A. N. Johnson, D. E. Barlow, A. L. Kelly, V. A. Varaljay, W. J. Crookes-Goodson and J. C. Biffinger, Polym. Int., 2021, 70, 977–983 CrossRef CAS.
- S. Yoshida, K. Hiraga, T. Takehana, I. Taniguchi, H. Yamaji, Y. Maeda, K. Toyohara, K. Miyamoto, Y. Kimura and K. Oda, Science, 2016, 351, 1196–1199 CrossRef CAS PubMed.
- M. O'Reilly and J. Stubbe, Biochemistry, 2020, 59, 2316–2318 CrossRef PubMed.
- V. Tournier, C. M. Topham, A. Gilles, B. David, C. Folgoas, E. Moya-Leclair, E. Kamionka, M. L. Desrousseaux, H. Texier, S. Gavalda, M. Cot, E. Guemard, M. Dalibey, J. Nomme, G. Cioci, S. Barbe, M. Chateau, I. Andre, S. Duquesne and A. Marty, Nature, 2020, 580, 216–219 CrossRef CAS PubMed.
- H. Sawai, H. Sugimoto, Y. Kato, Y. Asano, Y. Shiro and S. Aono, J. Biol. Chem., 2009, 284, 32089–32096 CrossRef CAS PubMed.
- A. Hinzmann, T. Betke, Y. Asano and H. Gröger, Chem. – Eur. J., 2021, 27, 5313–5321 CrossRef CAS PubMed.
- A. Hinzmann, M. Stricker and H. Gröger, ACS Sustainable Chem. Eng., 2020, 8, 17088–17096 CrossRef CAS.
- H. Gröger and Y. Asano, J. Org. Chem., 2020, 85, 6243–6251 CrossRef PubMed.
- D. Ribeaucourt, B. Bissaro, V. Guallar, M. Yemloul, M. Haon, S. Grisel, V. Alphand, H. Brumer, F. Lambert, J. G. Berrin and M. Lafond, ACS Sustainable Chem. Eng., 2021, 9, 4411–4421 CrossRef CAS.
- D. Ribeaucourt, B. Bissaro, F. Lambert, M. Lafond and J.-G. Berrin, Biotechnol. Adv., 2021, 107787 CrossRef PubMed.
- P. Le-Huu, T. Heidt, B. Claasen, S. Laschat and V. B. Urlacher, ACS Catal., 2015, 5, 1772–1780 CrossRef CAS.
- H. Xiao, S. Dong, Y. Liu, X. Q. Pei, H. Lin and Z. L. Wu, Catal. Sci. Technol., 2021, 11, 2195–2201 RSC.
- M. K. Julsing, D. Kuhn, A. Schmid and B. Buhler, Biotechnol. Bioeng., 2012, 109, 1109–1119 CrossRef CAS PubMed.
- D. Kuhn, M. A. Kholiq, E. Heinzle, B. Buhler and A. Schmid, Green Chem, 2010, 12, 815–827 RSC.
- Y. Zhang, Y. Q. Wu, N. Xu, Q. Zhao, H. L. Yu and J. H. Xu, ACS Sustainable Chem. Eng., 2019, 7, 7218–7226 CrossRef CAS.
- T. Heine, A. Scholtissek, S. Hofmann, R. Koch and D. Tischler, ChemCatChem, 2020, 12, 199–209 CrossRef CAS.
- D. Tischler, A. Kumpf, D. Eggerichs and T. Heine, in The Enzymes, ed. P. Chaiyen and F. Tamanoi, Academic Press, 2020, vol. 47, ch. 13, pp. 399–425 Search PubMed.
- B. V. Plapp, B. R. Savarimuthu, D. J. Ferraro, J. K. Rubach, E. N. Brown and S. Ramaswamy, Biochemistry, 2017, 56, 3632–3646 CrossRef CAS PubMed.
- B. V. Plapp, Arch. Biochem. Biophys., 2010, 493, 3–12 CrossRef CAS PubMed.
- J. H. An, Y. Nie and Y. Xu, Crit. Rev. Biotechnol., 2019, 39, 366–379 CrossRef CAS PubMed.
- A. Weckbecker and W. Hummel, Biocatal. Biotransform., 2006, 24, 380–389 CrossRef CAS.
- S. Kara, J. H. Schrittwieser, F. Hollmann and M. B. Ansorge-Schumacher, Appl. Microbiol. Biotechnol., 2014, 98, 1517–1529 CrossRef CAS PubMed.
- T. S. Moody and J. D. Rozzell, in Organic Synthesis Using Biocatalysis, ed. A. Goswami and J. D. Stewart, Academic Press, 2016, pp. 149–185 Search PubMed.
- W. Jiang and B. S. Fang, Appl. Biochem. Biotechnol., 2020, 192, 146–179 CrossRef CAS PubMed.
- S. K. Ma, J. Gruber, C. Davis, L. Newman, D. Gray, A. Wang, J. Grate, G. W. Huisman and R. A. Sheldon, Green Chem., 2010, 12, 81–86 RSC.
- D. Pollard, M. Truppo, J. Pollard, C. Y. Chen and J. Moore, Tetrahedron: Asymmetry, 2006, 17, 554–559 CrossRef CAS.
- J. Liang, E. Mundorff, R. Voladri, S. Jenne, L. Gilson, A. Conway, A. Krebber, J. Wong, G. Huisman, S. Truesdell and J. Lalonde, Org. Process Res. Dev., 2010, 14, 188–192 CrossRef CAS.
- C. V. Voss, C. C. Gruber and W. Kroutil, Synlett, 2010, 991–998 CAS.
- C. E. Paul, I. Lavandera, V. Gotor-Fernández, W. Kroutil and V. Gotor, ChemCatChem, 2013, 5, 3875–3881 CrossRef CAS.
- O. W. Gooding, R. Voladri, A. Bautista, T. Hopkins, G. Huisman, S. Jenne, S. Ma, E. C. Mundorff and M. M. Savile, Org. Process Res. Dev., 2010, 14, 119–126 CrossRef CAS.
- J. A. Friest, Y. Maezato, S. Broussy, P. Blum and D. B. Berkowitz, J. Am. Chem. Soc., 2010, 132, 5930–5931 CrossRef CAS PubMed.
- M. Hohne, Nat. Catal, 2019, 2, 841–842 CrossRef.
- S. C. Cosgrove, A. Brzezniak, S. P. France, J. I. Ramsden, J. Mangas-Sanchez, S. L. Montgomery, R. S. Heath and N. J. Turner, Enzymes Synth. Biol., 2018, 608, 131–149 CAS.
- J. Mangas-Sánchez, S. P. France, S. L. Montgomery, G. A. Aleku, H. Man, M. Sharma, J. I. Ramsden, G. Grogan and N. J. Turner, Curr. Opin. Chem. Biol., 2017, 37, 19–25 CrossRef PubMed.
- O. Mayol, K. Bastard, L. Beloti, A. Frese, J. P. Turkenburg, J. L. Petit, A. Mariage, A. Debard, V. Pellouin, A. Perret, V. de Berardinis, A. Zaparucha, G. Grogan and C. Vergne-Vaxelaire, Nat. Catal., 2019, 2, 324–333 CrossRef CAS.
- O. Mayol, S. David, E. Darii, A. Debard, A. Mariage, V. Pellouin, J. L. Petit, M. Salanoubat, V. de Berardinis, A. Zaparucha and C. Vergne-Vaxelaire, Catal. Sci. Technol., 2016, 6, 7421–7428 RSC.
- T. Knaus, W. Bohmer and F. G. Mutti, Green Chem., 2017, 19, 453–463 RSC.
- A. S. Bommarius, Nat. Catal., 2019, 2, 288–289 CrossRef CAS.
- M. J. Abrahamson, E. Vazquez-Figueroa, N. B. Woodall, J. C. Moore and A. S. Bommarius, Angew. Chem., Int. Ed., 2012, 51, 3969–3972 CrossRef CAS PubMed.
- G. A. Aleku, S. P. France, H. Man, J. Mangas-Sanchez, S. L. Montgomery, M. Sharma, F. Leipold, S. Hussain, G. Grogan and N. J. Turner, Nat. Chem., 2017, 9, 961–969 CrossRef CAS PubMed.
- M. Sharma, J. Mangas-Sanchez, S. P. France, G. A. Aleku, S. L. Montgomery, J. I. Ramsden, N. J. Turner and G. Grogan, ACS Catal., 2018, 8, 11534–11541 CrossRef CAS.
- L. Ducrot, M. Bennett, G. Grogan and C. Vergne-Vaxelaire, Adv. Synth. Catal., 2021, 363, 328–351 CrossRef CAS.
- S. P. France, R. M. Howard, J. Steflik, N. J. Weise, J. Mangas-Sanchez, S. L. Montgomery, R. Crook, R. Kumar and N. J. Turner, ChemCatChem, 2018, 10, 510–514 CrossRef CAS.
- G. H. Kim, H. Jeon, T. P. Khobragade, M. D. Patil, S. Sung, S. Yoon, Y. Won, I. S. Choi and H. Yun, Enzyme Microb. Technol., 2019, 120, 52–60 CrossRef CAS PubMed.
- C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis, J. C. Colbeck, A. Krebber, F. J. Fleitz, J. Brands, P. N. Devine, G. W. Huisman and G. J. Hughes, Science, 2010, 329, 305–309 CrossRef CAS PubMed.
- S. L. Montgomery, A. Pushpanath, R. S. Heath, J. R. Marshall, U. Klemstein, J. L. Galman, D. Woodlock, S. Bisagni, C. J. Taylor, J. Mangas-Sanchez, J. I. Ramsden, B. Dominguez and N. J. Turner, Sci. Adv., 2020, 6, eaay9320 CrossRef CAS PubMed.
- J. Mangas-Sanchez, M. Sharma, S. C. Cosgrove, J. I. Ramsden, J. R. Marshall, T. W. Thorpe, R. B. Palmer, G. Grogan and N. J. Turner, Chem. Sci., 2020, 11, 5052–5057 RSC.
- V. F. Batista, J. L. Galman, D. C. G. A. Pinto, A. M. S. Silva and N. J. Turner, ACS Catal., 2018, 8, 11889–11907 CrossRef CAS.
- S. R. Derrington, N. J. Turner and S. P. France, J. Biotechnol., 2019, 304, 78–88 CrossRef CAS PubMed.
- M. Winkler, Curr. Opin. Chem. Biol., 2018, 43, 23–29 CrossRef CAS PubMed.
- M. Lubberink, C. Schnepel, J. Citoler, S. R. Derrington, W. Finnigan, M. A. Hayes, N. J. Turner and S. L. Flitsch, ACS Catal., 2020, 10, 10005–10009 CrossRef CAS.
- C. K. Winkler, K. Faber and M. Hall, Curr. Opin. Chem. Biol., 2018, 43, 97–105 CrossRef CAS PubMed.
- H. S. Toogood and N. S. Scrutton, ACS Catal., 2018, 8, 3532–3549 CrossRef CAS PubMed.
- T. Ress, W. Hummel, S. P. Hanlon, H. Iding and H. Gröger, ChemCatChem, 2015, 7, 1302–1311 CrossRef CAS.
- A. Scholtissek, D. Tischler, A. H. Westphal, W. J. H. van Berkel and C. E. Paul, Catalysts, 2017, 7, 130 CrossRef.
- T. Knaus, C. E. Paul, C. W. Levy, S. de Vries, F. G. Mutti, F. Hollmann and N. S. Scrutton, J. Am. Chem. Soc., 2016, 138, 1033–1039 CrossRef CAS PubMed.
- H. S. Toogood, T. Knaus and N. S. Scrutton, ChemCatChem, 2014, 6, 951–954 CrossRef CAS.
- A. Muller, R. Sturmer, B. Hauer and B. Rosche, Angew. Chem., Int. Ed., 2007, 46, 3316–3318 CrossRef PubMed.
- B. A. Sandoval and T. K. Hyster, Curr. Opin. Chem. Biol., 2020, 55, 45–51 CrossRef CAS PubMed.
- X. Huang, B. Wang, Y. Wang, G. Jiang, J. Feng and H. Zhao, Nature, 2020, 584, 69–74 CrossRef CAS PubMed.
- C. A. Roessner, J. B. Spencer, N. J. Stolowich, J. Wang, G. Parmesh Nayar, P. J. Santander, C. Pichon, C. Min, M. T. Holderman and A. I. Scott, Chem. Biol., 1994, 1, 119–124 CrossRef CAS PubMed.
- A. I. Scott, Synlett, 1994, 871–883 CrossRef CAS.
- N. Losada-Garcia, Z. Cabrera, P. Urrutia, C. Garcia-Sanz, A. Andreu and J. M. Palomo, Catalysts, 2020, 10, 1258 CrossRef CAS.
- M. P. Thompson, I. Penafiel, S. C. Cosgrove and N. J. Turner, Org. Process Res. Dev., 2019, 23, 9–18 CrossRef CAS.
- M. Santi, L. Sancineto, V. Nascimento, J. B. Azeredo, E. V. M. Orozco, L. H. Andrade, H. Gröger and C. Santi, Int. J. Mol. Sci., 2021, 22, 990 CrossRef CAS PubMed.
- J. M. Woodley, Green Biocatalysis, 2016, pp. 503–518 Search PubMed.
- S. M. K. McKinnie, Z. D. Miles, P. A. Jordan, T. Awakawa, H. P. Pepper, L. A. M. Murray, J. H. George and B. S. Moore, J. Am. Chem. Soc., 2018, 140, 17840–17845 CrossRef CAS PubMed.
- Z. L. Wang, B. S. Sekar and Z. Li, Bioresour. Technol., 2021, 323, 124551 CrossRef CAS PubMed.
- J. H. Schrittwieser, S. Velikogne, M. Hall and W. Kroutil, Chem. Rev., 2018, 118, 270–348 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |