
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA carbonyl-rich covalent organic framework as a high-performance cathode material for aqueous rechargeable zinc-ion batteries†
Dingxuan
Ma
a,
Huimin
Zhao
a,
Fan
Cao
c,
Huihui
Zhao
a,
Jixin
Li
d,
Lei
Wang
 *b and
Kang
Liu
*a
*b and
Kang
Liu
*a
aCollege of Chemistry and Molecular Engineering, Key Laboratory of Optic-Electric Sensing and Analytical Chemistry for Life Science, MOE, Shandong Key Laboratory of Biochemical Analysis, Qingdao University of Science and Technology, Qingdao 266042, Shandong, P. R. China. E-mail: liukang82@126.com
bCollege of Environment and Safety Engineering, Qingdao University of Science and Technology, Qingdao 266042, Shandong, P. R. China. E-mail: inorchemwl@126.com
cSchool of Materials Science and Engineering, Shandong Jianzhu University, Jinan 250101, Shandong, P. R. China
dState Key Laboratory of Inorganic Synthesis and Preparative Chemistry, College of Chemistry, Jilin University, Changchun 130012, P. R. China
First published on 8th February 2022
Abstract
Aqueous rechargeable zinc-ion batteries (ZIBs) provide high theoretical capacity, operational safety, low-cost and environmental friendliness for large-scale energy storage and wearable electronic devices, but their future development is plagued by low capacity and poor cycle life due to the lack of suitable cathode materials. In this work, a covalent organic framework (Tp-PTO-COF) with multiple carbonyl active sites is synthesized and successfully introduced in aqueous rechargeable ZIBs for the first time. Tp-PTO-COF delivers high specific capacities of 301.4 and 192.8 mA h g−1 at current densities of 0.2 and 5 A g−1, respectively, along with long-term durability and flat charge–discharge plateaus. The remarkable electrochemical performance is attributed to the abundance of nucleophilic carbonyl active sites, well defined porous structure and inherent chemical stability of Tp-PTO-COF. Moreover, the structural evolution and Zn2+ ion intercalation mechanism are discussed and revealed by the experimental analysis and density functional theory calculations. These results highlight a new avenue to develop organic cathode materials for high performance and sustainable aqueous rechargeable ZIBs.
Introduction
To overcome the worldwide energy crisis, the utilization of green renewable energy sources such as solar, wind and tidal energy is highly desirable. Therefore, efficient energy storage devices play a crucial role in realizing the storage and release of renewable energy. Although widespread success has been achieved for rechargeable lithium-ion batteries (LIBs),1,2 increasing concerns about safety issues, high cost and constrained lithium resources strongly limit their applications.3 Compared to expensive and flammable LIBs, aqueous rechargeable zinc-ion batteries (ZIBs) are becoming an attractive alternative due to the natural abundance and high operational safety of zinc anodes.4–6 Furthermore, aqueous rechargeable ZIBs can theoretically achieve higher specific capacity and energy density, because of Zn2+ ions as multivalent charge carriers involved in the redox reaction and aqueous electrolytes offering much higher ionic conductivities compared to organic electrolytes. Overall, these economic and technical superiorities of aqueous ZIBs hold particular promise for their application in large-scale energy storage systems and even in wearable electronic devices. Nevertheless, one of the primary obstacles hindering the development of aqueous rechargeable ZIBs is the lack of suitable cathode materials which can reversibly host Zn2+ ions and maintain structural stability during the process of charge–discharge.7,8 As a result, great efforts have been devoted to building high-performance ZIBs using inorganic compound cathodes such as manganese-based oxides,9–12 vanadium-based oxides,13–15 spinel-type materials16–18 and Prussian blue analogues.19–21 However, these inorganic electrodes were plagued by structural and chemical instabilities, unsatisfactory capacity, significant capacity fading, and toxic elements in their components. Recently, various organic materials have been recognized as the next promising green electrodes because they offer the advantages of light weight, low-toxicity, inexpensiveness and sustainability.22–25 Organic compounds with carbonyl groups can store Zn2+ ions by coordination reactions, therefore, some carbonyl compounds have been explored as cathode materials in aqueous rechargeable ZIBs, and exhibited high specific capacity and electrochemical reversibility.26–30 Although these carbonyl compounds are almost insoluble in aqueous electrolytes, their discharge products always display high solubility, which inevitably limits their cycle stability and creates an urgent need for organic polymer electrode systems owing to the stability and structural tunability of organic polymer. However, the simple polymerization of carbonyl compounds often leads to reduced capacity and poor rate performance.31 Hence, both cycle stability and high capacity have to be further considered in the design of novel organic polymer cathode materials.Covalent organic frameworks (COFs),32–36 an emerging class of crystalline and porous polymeric materials, often possess high chemical stability, well defined porosity, and ability to predictably organize redox-active groups. The unique features of COFs endow them with great advantages and potential for application as electrode materials for rechargeable batteries.37–41 Unlike traditional amorphous organic polymers, COFs can achieve accurate assembly of the redox-active building blocks, which makes it reasonable to obtain COF electrodes with specific structure and redox properties. Besides, COFs provide continuous open channels due to various topological structures or the stacking of conjugated polymeric layers. Because of this porosity advantage, the Zn2+ ions are allowed to rapidly move in the COF electrode and reach the active sites. More importantly, the highly crystalline π-conjugated structure of COFs can provide a stable physical and chemical reaction environment, which is particularly beneficial to achieve stable operation at variable cathodic and anodic polarizations. However, the relevant research on COF-based aqueous rechargeable ZIBs is just in the beginning stage. Recently, Banerjee et al. proposed for the first time that a classical two-dimensional (2D) COF (HqTp) could be used as a cathode for aqueous rechargeable ZIBs.42 Unfortunately, HqTp delivered limited specific capacities of 276.0 mA h g−1 at 0.125 A g−1, and carbon nanofibers must be used as an electrical conductivity amplifier in the cathode. Furthermore, HqTp exhibited slopping charge–discharge curves without apparent plateaus, which was detrimental to achieving a stable voltage output. Although the state-of-the-art capacity performance of COF-based aqueous rechargeable ZIBs is far from expectation, these studies hinted at the promise of COF redox chemistry and structural stability.43
To achieve better electrochemical performance for COFs as cathodes of aqueous ZIBs, we report herein the synthesis and research of a 2D COF (Tp-PTO-COF) with dual active sites of adjacent carbonyl groups and β-keto carbonyl groups as nucleophilic centers to provide reversible and efficient Zn2+ ion storage during the charge–discharge process.44 Benefiting from the abundance of nucleophilic active sites, well-defined porous structure and inherent chemical stability, Tp-PTO-COF shows a remarkably high experimental capacity of 301.4 mA h g−1 at a current density of 0.2 A g−1. Impressively, even at a high current density of 5 A g−1, Tp-PTO-COF displays an impressive reversible capacity of 192.8 mA h g−1. Moreover, the structural evolution and Zn2+ ion intercalation mechanism in Tp-PTO-COF have been demonstrated by experimental analysis and density functional theory (DFT) calculations.
Results and discussion
Synthesis and characterization
Tp-PTO-COF was synthesized by condensing 1,3,5-triformylphloroglucinol (Tp) with 2,7-diaminopyrene-4,5,9,10-tetraone (DAPTO) via an acid-catalyzed solvothermal reaction (Fig. 1). The successful condensation and structural characterization of Tp-PTO-COF were confirmed by both Fourier transform infrared spectroscopy (FT-IR) and solid state 13C NMR spectroscopy. The FT-IR spectrum (Fig. S6†) exhibits two strong peaks at 1580 cm−1 and 1272 cm−1 for Tp-PTO-COF, which are typical characteristics of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond and C–N bond, indicating the formation of a β-ketoenamine-linked framework structure. The concomitant disappearing characteristic peaks of aldehydic CO (1640 cm−1) and diamine N–H (3349, 3436 cm−1) verify the complete consumption of the reactants. In addition, the characteristic peak of the β-keto CO stretch appears at 1616 cm−1, and the peak at 1672 cm−1 of Tp-PTO-COF is attributed to the carbonyl groups (CO) of 1,2-diketone units. Solid-state 13C NMR studies also indicate the local structures of Tp-PTO-COF (Fig. S7†). The appearance of the representative signal for the enamine carbon atom at 146.8 ppm validates the successful formation of the β-ketoenamine-linked framework. Furthermore, two well-resolved peaks are detected at 182.7 and 176.6 ppm indicating the two different carbonyl carbon atoms in Tp-PTO-COF. The structural stability of Tp-PTO-COF was evaluated by thermogravimetric analysis (TGA), which reveals its high thermal stability up to 400 °C (Fig. S8†). Furthermore, the permanent porosity of Tp-PTO-COF was assessed by N2 sorption-desorption measurements at 77 K. As indicated in Fig. S9,† the typical type-II sorption isotherms of Tp-PTO-COF reveal its mesoporous properties, and the surface area of Tp-PTO-COF was calculated to be 601 m2 g−1 on the basis of the Brunauer–Emmet and Teller (BET) model.
C bond and C–N bond, indicating the formation of a β-ketoenamine-linked framework structure. The concomitant disappearing characteristic peaks of aldehydic CO (1640 cm−1) and diamine N–H (3349, 3436 cm−1) verify the complete consumption of the reactants. In addition, the characteristic peak of the β-keto CO stretch appears at 1616 cm−1, and the peak at 1672 cm−1 of Tp-PTO-COF is attributed to the carbonyl groups (CO) of 1,2-diketone units. Solid-state 13C NMR studies also indicate the local structures of Tp-PTO-COF (Fig. S7†). The appearance of the representative signal for the enamine carbon atom at 146.8 ppm validates the successful formation of the β-ketoenamine-linked framework. Furthermore, two well-resolved peaks are detected at 182.7 and 176.6 ppm indicating the two different carbonyl carbon atoms in Tp-PTO-COF. The structural stability of Tp-PTO-COF was evaluated by thermogravimetric analysis (TGA), which reveals its high thermal stability up to 400 °C (Fig. S8†). Furthermore, the permanent porosity of Tp-PTO-COF was assessed by N2 sorption-desorption measurements at 77 K. As indicated in Fig. S9,† the typical type-II sorption isotherms of Tp-PTO-COF reveal its mesoporous properties, and the surface area of Tp-PTO-COF was calculated to be 601 m2 g−1 on the basis of the Brunauer–Emmet and Teller (BET) model.
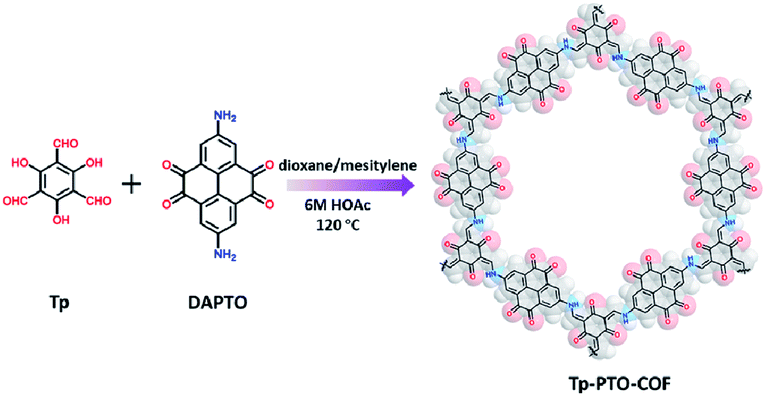 | ||
| Fig. 1 Schematic presentation of the synthesis process for Tp-PTO-COF. | ||
The crystalline structure feature of Tp-PTO-COF was analyzed by powder X-ray diffraction (PXRD) measurement in conjunction with structural simulation. As observed in Fig. 2a, Tp-PTO-COF exhibits well defined peaks, and the experimental pattern matches well with the simulated pattern of the eclipsed AA stacking model. The experimental PXRD pattern displays an apparent peak located at 3.4°, corresponding to the (100) reflection plane, and the hexagonal pore size of Tp-PTO-COF is calculated to be ∼25.7 Å, which is in accordance with the simulated result (Fig. 2b). In addition, the presence of a broad peak around 27° can be ascribed to the π–π stacking construction, revealing the existence of a multilayered COF structure. A full profile Pawley refinement was also performed for the experimentally obtained PXRD patterns based on the simulated AA stacking model, which has led to satisfactorily low residual values and acceptable profile differences (Fig. S13†). These obvious PXRD characteristic peaks in conjunction with structural simulation indicate a high crystallinity and typical 2D layered hexagonal network structure of the as-prepared Tp-PTO-COF.
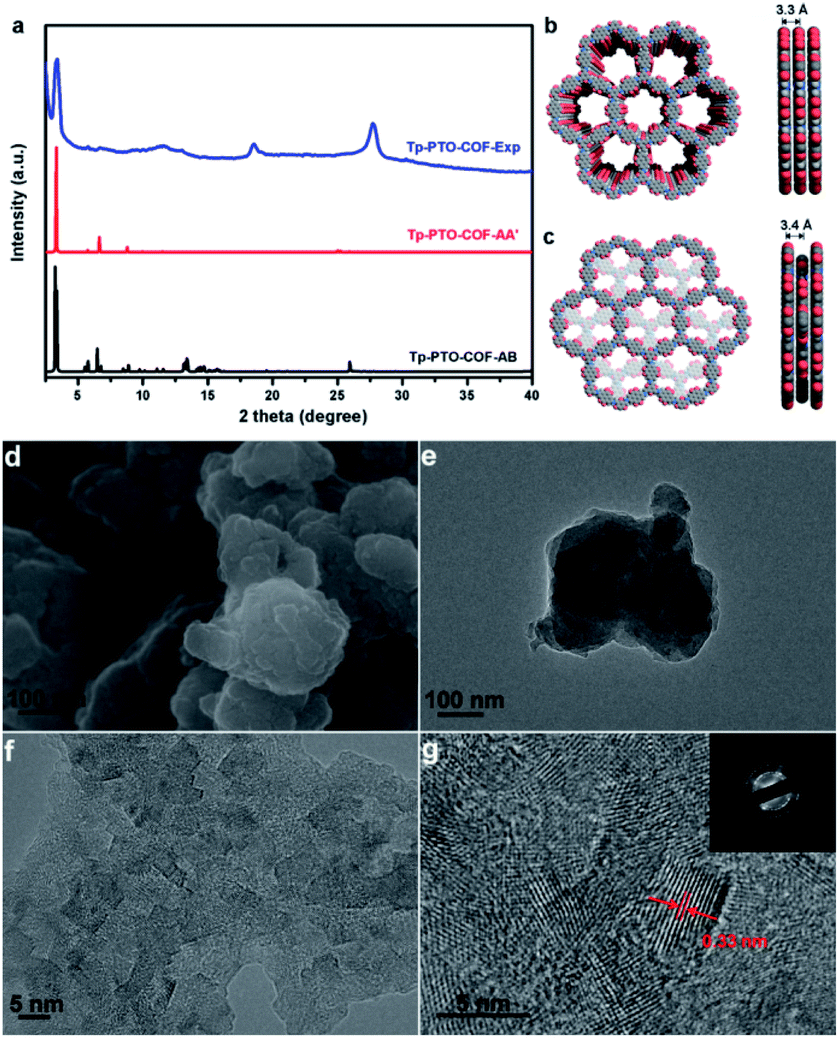 | ||
| Fig. 2 (a) Comparison of the PXRD patterns among the as-synthesized structure (blue), the simulated eclipsed AA structure (red), and the simulated staggered AB structure (black); (b) simulated eclipsed AA structure of Tp-PTO-COF; (c) simulated staggered AB structure of Tp-PTO-COF; (d) SEM image of Tp-PTO-COF; (e) TEM image of Tp-PTO-COF; (f and g) HRTEM images of Tp-PTO-COF. The inset is the corresponding SAED image. | ||
The morphology of Tp-PTO-COF was examined by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). The as-synthesized COF exhibits a 2D lamellar stacking morphology, and the boundaries of the TEM image show the layered-sheet arrangement of Tp-PTO-COF (Fig. 2d and e). In addition, an interplanar distance of approximately 0.33 nm can be clearly observed from the high-resolution TEM (HRTEM) images of Tp-PTO-COF (Fig. 2f and g), which is in accordance with the π–π stacking distance calculated based on the PXRD data and structural simulation. The HRTEM image and diffraction rings obtained from selected-area electron diffraction (SAED) analysis further confirm the intrinsic crystal structure of Tp-PTO-COF.
Electrochemical characterization
Such an ordered porous structure and high-density carbonyl centers are particularly favorable in Zn2+ ion storage electrodes due to their ability to offer ion diffusion channels and facilitate Zn2+ ion storage via reversible coordination/uncoordination. In order to evaluate the potential of Tp-PTO-COF as a cathode material for aqueous rechargeable ZIBs, the electrochemical performance was initially evaluated on a typical coin-type cell (Fig. 3a), which was assembled with Zn foil as the counter electrode and a glass fiber membrane adsorbed electrolyte (2 M ZnSO4) as the separator. Fig. 3b shows the cyclic voltammetry (CV) curve of Tp-PTO-COF at a scan rate of 0.5 mV s−1 in the potential range of 0.4–1.5 V vs. Zn2+/Zn. Two pairs of redox peaks around 0.72/1.01 V and 0.86/1.13 V (denoted as R1/O1 and R2/O2) can be observed, which correspond to a multistep Zn2+ ion intercalation process followed by the electrochemical reduction of Tp-PTO-COF. Furthermore, CV curves at different scan rates have also been recorded (Fig. S14†). There is a linear relationship between its peak current and the square root of the scan rate (Fig. S15†), indicating that the redox-reaction kinetics are controlled by the Zn2+ ion diffusion process. Furthermore, the Zn2+ ion diffusion coefficient of the Tp-PTO-COF electrode was studied using the galvanostatic intermittent titration technique (GITT) method. As shown in Fig. S16,† the diffusion coefficient of Zn2+ ions in the Tp-PTO-COF cathode is around 10−11 cm2 s−1. The fast kinetic performance is further proven by electrochemical impedance spectroscopy (EIS) measurements, which reflect a charge-transfer resistance (Rct) value of 29.08 Ω (Fig. S17 and Table S2†).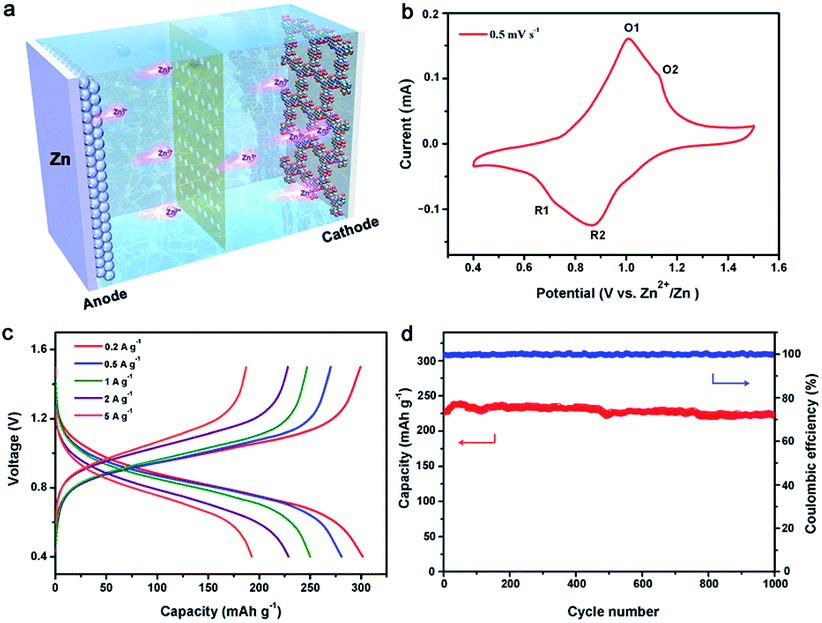 | ||
| Fig. 3 (a) Illustration of the aqueous Zn/Tp-PTO-COF battery; (b) CV curve at a scan rate of 0.5 mV s−1; (c) galvanostatic charge–discharge curves of the aqueous Zn/Tp-PTO-COF battery at various current densities; (d) cycle stability at a current density of 2 A g−1. | ||
The energy-storage properties of Tp-PTO-COF can be observed in the galvanostatic charge–discharge (GCD) profiles at different current densities in Fig. 3c, where the achieved current densities and capacities are calculated based on the mass of Tp-PTO-COF in the cathode. The Tp-PTO-COF cathode exhibits a remarkably high specific capacity of 301.4 mA h g−1 at a current density of 0.2 A g−1 and maintains specific capacities of 281.3, 250.1, and 228.6 mA h g−1 at current densities of 0.5, 1, and 2 A g−1 respectively. Remarkably, even at extremely high current densities of 5 and 10 A g−1, it also delivers capacities of 192.8 and 140.07 mA h g−1, much higher than that of many reported COF cathodes and inorganic or organic electrode materials (Fig. S18 and Table S3†). In addition, we further evaluated the rate performance of batteries with different Tp-PTO-COF loading masses (Fig. S19†). By comparing the results, similar capacity performances were found. It is worth mentioning that Tp-PTO-COF not only exhibits significantly better specific capacity performance, but also has a wide and stable discharging plateau contributing to the stable energy output. The durability and long-term cycling stability of Tp-PTO-COF are evaluated at a current density of 2 A g−1 (Fig. 3d). It delivers a capacity of 218.5 mA h g−1 over 1000 cycles with 95% retention of its initial capacity. And the coulombic efficiency of the battery is maintained around 100%, demonstrating durable cycle stability of the Tp-PTO-COF cathode. SEM is further conducted to monitor the morphological evolution and structural stability of the Tp-PTO-COF cathode during the charge–discharge process. As shown in Fig. S22–S26,† there are no obvious morphological changes or by-products on the electrode,10,45,46 confirming the stability of Tp-PTO-COF. Besides, to verify whether the ordered porous structure and inherent chemical stability of Tp-PTO-COF are conducive to the storage of Zn2+ ions, we synthesized a similar amorphous organic material, Tp-PTO-POF, as the cathode for comparison. The durability and long-term cycling stability of Tp-PTO-POF are evaluated at a current density of 2 A g−1. It delivers a capacity of about 220 mA h g−1 over 45 cycles; then the performance decreases precipitously (Fig. S20†). This result is in sharp contrast to the performance of Tp-PTO-COF, which further confirmed the advantage of selecting the COF material as an electrode. All these results show that the Zn/Tp-PTO-COF battery has an excellent energy-storage performance, which can meet the requirement of high capacity as well as realize stable charge–discharge.
Charge–discharge mechanism research
To gain further insight into the charge–discharge process, a series of elemental and structural characteristics have been studied. X-ray photoelectron spectroscopy (XPS) measurement was used to investigate the reversible process of Zn2+ ion insertion/extraction on the Tp-PTO-COF cathode. As shown in Fig. 4a and S21,† no Zn element signal was detected from the pristine cathode. During the discharge process, the zinc anode is oxidized to Zn2+ ions, which pass across the electrolyte and coordinate with carbonyl groups of Tp-PTO-COF. Meanwhile, XPS analysis also indicated the appearance of Zn in the divalent state in the Tp-PTO-COF cathode. After charging to 1.5 V, the intensity of the XPS peaks of Zn2+ ions disappeared due to the removal of Zn2+ ions from the cathode. Furthermore, a reversible process of Zn appearance/disappearance can also be observed in the TEM mapping images of the Tp-PTO-COF cathode at pristine, fully discharged and fully charged states (Fig. 4b). The active sites for Zn2+ ion storage in Tp-PTO-COF were further detected by FT-IR (Fig. 4c). The stretching vibration of carbonyl groups is at about 1610–1680 cm−1. After discharging to 0.4 V, the intensity of the carbonyl vibration peak is evidently weakened, indicating the coordination reaction between Zn2+ ions and carbonyl groups. Subsequently, the peak intensity increases back to the pristine state after the charging process, demonstrating the removal of Zn2+ ions from the Tp-PTO-COF cathode.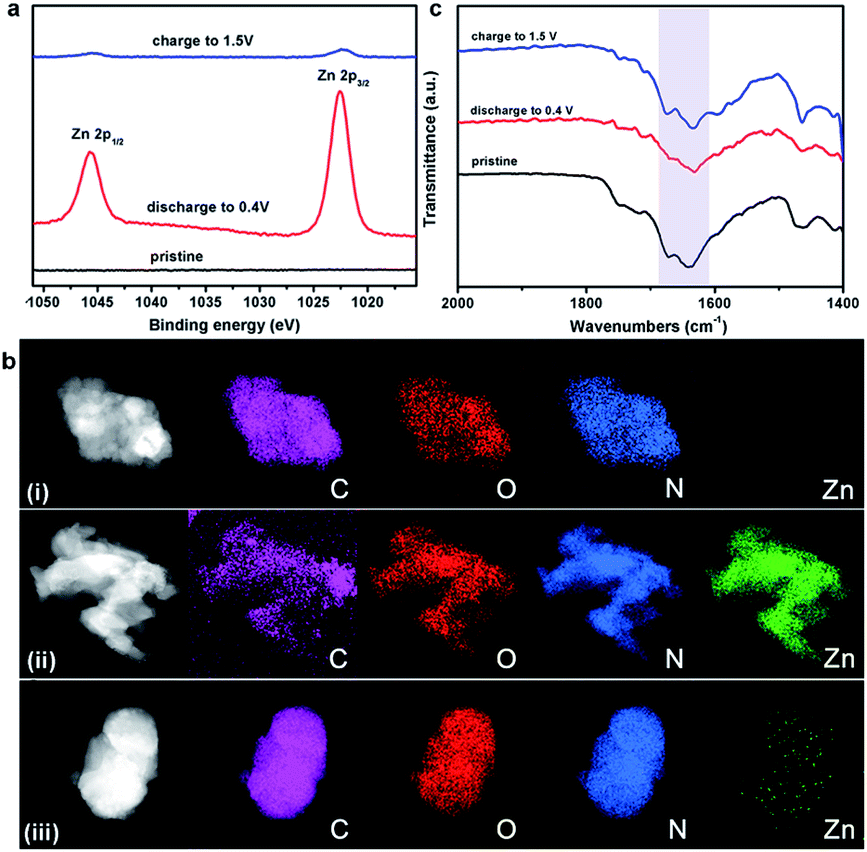 | ||
| Fig. 4 (a) Zn 2p XPS spectra of the Tp-PTO-COF electrode at pristine, fully discharged and fully charged states; (b) TEM mapping images of C, O, N, and Zn elemental distributions on the Tp-PTO-COF electrode at (i) pristine, (ii) fully discharged, and (iii) fully charged states; (c) FT-IR spectra of the Tp-PTO-COF electrode at pristine, fully discharged, and fully charged states. | ||
Given the above issues, detailed theoretical calculations with DFT were applied to further identify the Zn2+ ion storage sites during the charge–discharge process and the structure evolution of Tp-PTO-COF after Zn2+ ion uptake. The molecular electrostatic potential (MESP) regions near the carbonyl groups show a negative value (Fig. 5a), indicating strong chemical affinity for cation uptake. Especially, the carbonyl group of PTO has the local minima value of −19.66 kcal mol−1 (Fig. S27†), which indicates the strongest chemical affinity for Zn2+ ion uptake and tend to be the prime storage site. More specifically, Tp-PTO-COF may bind different numbers of Zn2+ ions with several possible modes, in consequence, further theoretical calculations based on the bilayer model fragment compound (MO-Tp-PTO) were conducted to deduce the structure of Tp-PTO-COF after Zn2+ ion uptake. The binding energies (BE) of MO-Tp-PTO after the redox reaction of Zn2+ ion uptake were calculated at the B3LYP/6-31+G* level in three possible model compounds.47,48 The optimized structures of Znx@MO-Tp-PTO (x = 4, 7, 10) and their total energies, defined by the sum of electronic and zero-point energies, are shown in Fig. 5b and Table S4,† respectively. It was found that, in all cases, a negative BE value was achieved after Zn2+ ion uptake, indicating efficient interaction between the carbonyl redox-active groups and Zn. Especially, the Zn7@MO-Tp-PTO model has the most negative BE (−320.03 kcal mol−1), representing a highly stable configuration (Fig. S28–S30†). Thus, Zn7@MO-Tp-PTO is considered as the most likely configuration after the discharge process. According to these DFT theoretical calculation results and some previous reports,25–27,42 we propose a conceivable electrochemical redox mechanism of Tp-PTO-COF during the reversible process of Zn2+ ion insertion/extraction: Tp-PTO-COF stores charge via an ‘ion-coordination’ process where each Zn2+ cation coordinates to two negatively charged oxygen atoms upon electrochemical reduction of the carbonyl redox-active groups, and uncoordinate reversibly during the charge process as shown in Fig. S31,† which corresponds to a theoretical capacity of 406 mA h g−1.
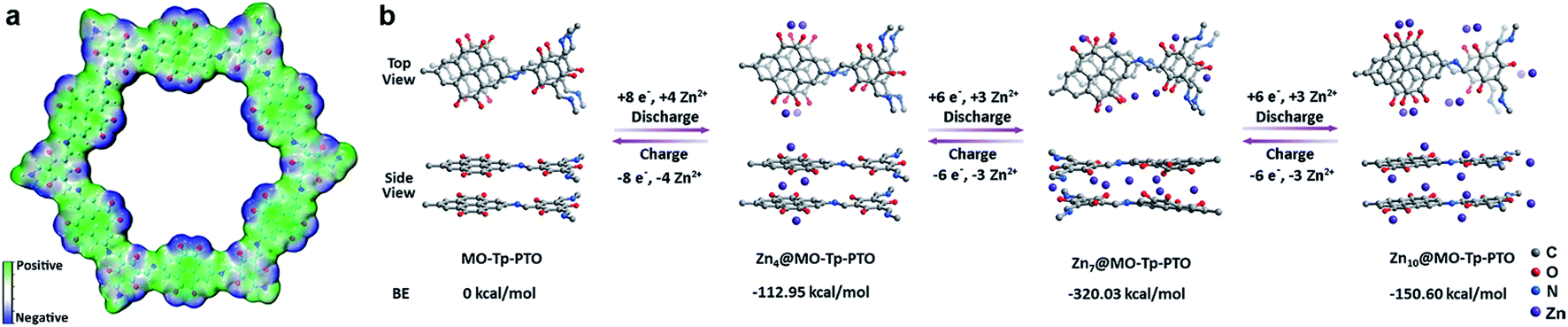 | ||
| Fig. 5 (a) Electronegativity simulation of Tp-PTO-COF; (b) simulation of binding energies with different numbers of Zn2+ ions in MO-Tp-PTO. | ||
Conclusions
In summary, Tp-PTO-COF with dual carbonyl active sites was proposed as a novel cathode material and successfully applied in aqueous rechargeable ZIBs. Tp-PTO-COF possesses abundant carbonyl nucleophilic centers, an ordered porous structure and inherent chemical stability, which are beneficial for Zn2+ ion storage and diffusion. The aqueous rechargeable ZIBs assembled with Tp-PTO-COF electrodes display superior capacity output, flat charge–discharge plateaus and excellent cycle stability. In addition, the Zn2+ ion intercalation mechanism has been studied through experimental analysis and theoretical simulations. The encouraging performance opens new possibilities for the design of advanced COF cathodes for high performance and sustainable aqueous rechargeable ZIBs.Data availability
All experimental and computational data are available in the ESI.†Author contributions
D. Ma and K. Liu designed the study. D. Ma, H. Zhao, H. Zhao and J. Li performed the experiments and analyzed the data. F. Cao and J. Li contributed to the theoretical calculations of the work. D. Ma, K. Liu and L. Wang prepared the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21601103 and 21701097) and the Natural Science Foundation of Shandong Province, China (No. ZR2020QB035).Notes and references
- F. Wu, J. Maier and Y. Yu, Chem. Soc. Rev., 2020, 49, 1569–1614 RSC.
- A. Manthiram, Nat. Commun., 2020, 11, 1550 CrossRef CAS PubMed.
- K. Liu, Y. Liu, D. Lin, A. Pei and Y. Cui, Sci. Adv., 2018, 4, 9820 CrossRef PubMed.
- H. Dong, J. Li, J. Guo, F. Lai, F. Zhao, Y. Jiao, D. Brett, T. Liu, G. He and I. P. Parkin, Adv. Mater., 2021, 33, 2007548 CrossRef CAS PubMed.
- F. Wan, X. Zhou, Y. Lu, Z. Niu and J. Chen, ACS Energy Lett., 2020, 5, 3569–3590 CrossRef CAS.
- X. Jia, C. Liu, Z. G. Neale, J. Yang and G. Cao, Chem. Rev., 2020, 120, 7795–7866 CrossRef CAS PubMed.
- D. Selvakumaran, A. Pan, S. Liang and G. Cao, J. Mater. Chem. A, 2019, 7, 18209–18236 RSC.
- B. Yong, D. Ma, Y. Wang, H. Mi, C. He and P. Zhang, Adv. Energy Mater., 2020, 10, 2002354 CrossRef CAS.
- N. Zhang, F. Cheng, J. Liu, L. Wang, X. Long, X. Liu, F. Li and J. Chen, Nat. Commun., 2017, 8, 405 CrossRef PubMed.
- J. Huang, Z. Wang, M. Hou, X. Dong, Y. Liu, Y. Wang and Y. Xia, Nat. Commun., 2018, 9, 2906 CrossRef PubMed.
- A. Huang, W. Zhou, A. Wang, M. Chen, Q. Tian and J. Chen, J. Energy Chem., 2021, 54, 475–481 CrossRef.
- V. Mathew, B. Sambandam, S. Kim, S. Kim, S. Park, S. Lee, M. H. Alfaruqi, V. Soundharrajan, S. Islam, D. Y. Putro, J.-Y. Hwang, Y.-K. Sun and J. Kim, ACS Energy Lett., 2020, 5, 2376–2400 CrossRef CAS.
- Q. Pang, C. Sun, Y. Yu, K. Zhao, Z. Zhang, P. M. Voyles, G. Chen, Y. Wei and X. Wang, Adv. Energy Mater., 2018, 8, 1800144 CrossRef.
- J. Ding, H. Gao, D. Ji, K. Zhao, S. Wang and F. Cheng, J. Mater. Chem. A, 2021, 9, 5258–5275 RSC.
- J. Guo, J. Ming, Y. Lei, W. Zhang, C. Xia, Y. Cui and H. N. Alshareef, ACS Energy Lett., 2019, 4, 2776–2781 CrossRef CAS.
- N. Zhang, F. Cheng, Y. Liu, Q. Zhao, K. Lei, C. Chen, X. Liu and J. Chen, J. Am. Chem. Soc., 2016, 138, 12894–12901 CrossRef CAS PubMed.
- M. Shi, B. Wang, Y. Shen, J. Jiang, W. Zhu, Y. Su, M. Narayanasamy, S. Angaiah, C. Yan and Q. Peng, Chem. Eng. J., 2020, 399, 125627 CrossRef CAS.
- Y. Lu, J. Wang, S. Zeng, L. Zhou, W. Xu, D. Zheng, J. Liu, Y. Zeng and X. Lu, J. Mater. Chem. A, 2019, 7, 21678–21683 RSC.
- L. Zhang, L. Chen, X. Zhou and Z. Liu, Adv. Energy Mater., 2014, 5, 1400930 CrossRef.
- G. Zampardi and F. L. Mantia, Curr. Opin. Electrochem., 2020, 21, 84–92 CrossRef CAS.
- G. Du and H. Pang, Energy Storage Mater., 2021, 36, 387–408 CrossRef.
- K. Qin, J. Huang, K. Holguin and C. Luo, Energy Environ. Sci., 2020, 13, 3950–3992 RSC.
- J. Huang, X. Dong, Z. Guo and Y. Wang, Angew. Chem., Int. Ed., 2020, 59, 18322–18333 CrossRef CAS PubMed.
- Z. Tie and Z. Niu, Angew. Chem., Int. Ed., 2020, 59, 21293–21303 CrossRef CAS PubMed.
- Y. Liang, Y. Jing, S. Gheytani, K.-Y. Lee, P. Liu, A. Facchetti and Y. Yao, Nat. Mater., 2017, 16, 841 CrossRef CAS PubMed.
- Z. Guo, Y. Ma, X. Dong, J. Huang, Y. Wang and Y. Xia, Angew. Chem., Int. Ed., 2018, 57, 11737–11741 CrossRef CAS PubMed.
- Q. Zhao, W. Huang, Z. Luo, L. Liu, Y. Lu, Y. Li, L. Li, J. Hu, H. Ma and J. Chen, Sci. Adv., 2018, 4, 1761 CrossRef PubMed.
- K. W. Nam, H. Kim, Y. Beldjoudi, T. Kwon, D. J. Kim and J. F. Stoddart, J. Am. Chem. Soc., 2020, 142, 2541–2548 CrossRef CAS PubMed.
- Y. Wang, C. Wang, Z. Ni, Y. Gu, B. Wang, Z. Guo, Z. Wang, D. Bin, J. Ma and Y. Wang, Adv. Mater., 2020, 32, 2000338 CrossRef CAS PubMed.
- D. Kundu, P. Oberholzer, C. Glaros, A. Bouzid, E. Tervoort, A. Pasquarello and M. Niederberger, Chem. Mater., 2018, 30, 3874–3881 CrossRef CAS.
- X. Yue, H. Liu and P. Liu, Chem. Commun., 2019, 55, 1647–1650 RSC.
- H. L. Nguyen, Chem. Sci., 2021, 12, 8632–8647 RSC.
- K. Geng, T. He, R. Liu, S. Dalapati, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Chem. Rev., 2020, 120, 8814–8933 CrossRef CAS PubMed.
- Y. Yusran, Q. Fang and V. Valtchev, Adv. Mater., 2020, 32, 2002038 CrossRef CAS PubMed.
- X. Guan, F. Chen, Q. Fang and S. Qiu, Chem. Soc. Rev., 2020, 49, 1357–1384 RSC.
- J. Li, X. Jing, Q. Li, S. Li, X. Gao, X. Feng and B. Wang, Chem. Soc. Rev., 2020, 49, 3565–3604 RSC.
- R. Shi, L. Liu, Y. Lu, C. Wang, Y. Li, L. Li, Z. Yan and J. Chen, Nat. Commun., 2020, 11, 178 CrossRef CAS PubMed.
- Z. Lei, Q. Yang, Y. Xu, S. Guo, W. Sun, H. Liu, L.-P. Lv, Y. Zhang and Y. Wang, Nat. Commun., 2018, 9, 576 CrossRef PubMed.
- A. Schneemann, R. Dong, F. Schwotzer, H. Zhong, I. Senkovska, X. Feng and S. Kaskel, Chem. Sci., 2021, 12, 1600–1619 RSC.
- S.-Y. Li, W.-H. Li, X.-L. Wu, Y. Tian, J. Yue and G. Zhu, Chem. Sci., 2019, 10, 7695–7701 RSC.
- D.-G. Wang, T. Qiu, W. Guo, Z. Liang, H. Tabassum, D. Xia and R. Zou, Energy Environ. Sci., 2021, 14, 688–728 RSC.
- A. Khayum M, M. Ghosh, V. Vijayakumar, A. Halder, M. Nurhuda, S. Kumar, M. Addicoat, S. Kurungot and R. Banerjee, Chem. Sci., 2019, 10, 8889–8894 RSC.
- W. Wang, V. S Kale, Z. Cao, S. Kandambeth, W. Zhang, J. Ming, P. T. Parvatkar, E. Abou-Hamad, O. Shekhah, L. Cavallo, M. Eddaoudi and H. N. Alshareef, ACS Energy Lett., 2020, 5, 2256–2264 CrossRef CAS.
- M. Li, J. Liu, Y. Li, G. Xing, X. Yu, C. Peng and L. Chen, CCS Chem., 2020, 2, 696–706 Search PubMed.
- M. Na, Y. Oh and H. R. Byon, Chem. Mater., 2020, 32, 6990–6997 CrossRef CAS.
- Z. Tie, L. Liu, S. Deng, D. Zhao and Z. Niu, Angew. Chem., Int. Ed., 2020, 59, 4920–4924 CrossRef CAS PubMed.
- K. C. Kim, T. Liu, S. W. Lee and S. S. Jang, J. Am. Chem. Soc., 2016, 138, 2374–2382 CrossRef CAS PubMed.
- C. Peng, G.-H. Ning, J. Su, G. Zhong, W. Tang, B. Tian, C. Su, D. Yu, L. Zu, J. Yang, M.-F. Ng, Y.-S. Hu, Y. Yang, M. Armand and K. P. Loh, Nat. Energy, 2017, 2, 17074 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc06412f |
| This journal is © The Royal Society of Chemistry 2022 |