
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective crossed intramolecular [2+2] photocycloaddition reactions mediated by a chiral chelating Lewis acid†
Thomas
Rigotti
 ,
Daniel P.
Schwinger
,
Raphaela
Graßl
,
Christian
Jandl
and
Thorsten
Bach
*
,
Daniel P.
Schwinger
,
Raphaela
Graßl
,
Christian
Jandl
and
Thorsten
Bach
*
School of Natural Sciences, Department Chemie, Catalysis Research Center (CRC), Technische Universität München, 85747 Garching, Germany. E-mail: thorsten.bach@ch.tum.de; Fax: +49 89 28913315; Tel: +49 89 28913330
First published on 1st February 2022
Abstract
In intramolecular [2+2] photocycloaddition reactions, the two tethered olefins can approach each other in a straight or in a crossed fashion. Despite the fact that the latter reaction mode leads to intriguing, otherwise inaccessible bridged skeletons, there has so far not been any enantioselective variants thereof. This study concerned the crossed [2+2]-photocycloaddition of 2-(alkenyloxy)cyclohex-2-enones to bridged cyclobutanes. It was found that the reaction could be performed with high enantioselectivity (80–94% ee) under visible light conditions when employing a chiral rhodium Lewis acid as a catalyst (2 mol%).
Introduction
Intramolecular [2+2] photocycloaddition reactions have been found extremely useful for the construction of cyclobutane rings and they have seen a multitude of applications in the synthesis of natural products.1 In contrast to intermolecular reactions, they display a more reliable and predictable regioselectivity which is determined by the length of the tether linking the two olefinic reaction partners. Typically, the photoexcited olefin is embedded in a five- or six-membered ring to avoid the rapid decay of its excited state by double bond rotation.2 In this scenario, if the tether is linked to one of the olefinic carbon atoms of the chromophore (Scheme 1), product formation can occur either to a product in which this carbon atom and the internal carbon atom of the tethered alkene are connected in a 1,2-fashion (A, straight product) or in a 1,3-fashion (B, crossed product). It has been realized very early that a tether with three atoms leads preferentially to the straight product, establishing a five-membered ring in the course of the cycloaddition.3 This preference reflects the nature of the first step of the photocycloaddition which can be envisioned for triplet reactions as a radical type addition4 to the tethered double bond leading to a 1,4-diradical intermediate. If the tether contains only two atoms, four-membered ring formation is avoided and the reaction proceeds to crossed products B. Although the first step in this case is also a five-membered ring formation it occurs between atoms α/β′ or α′/β but not at α/α′. From a synthetic perspective, the crossed reaction pathway is particularly rewarding because it enables to establish a bond set which is very difficult to attain in a thermal reaction.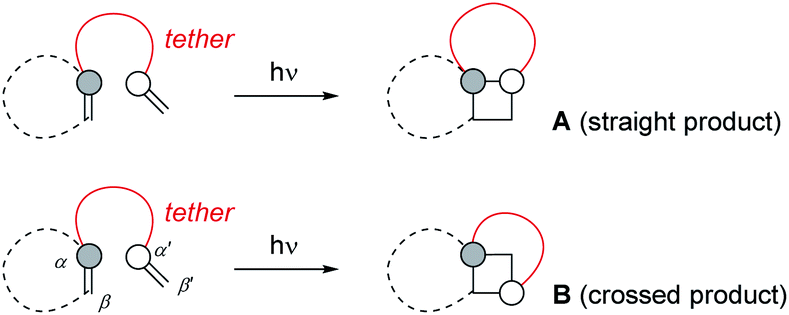 | ||
| Scheme 1 In intramolecular [2+2] photocycloaddition reactions, two regioisomeric products A and B are possible. The photoexcited olefin is often embedded in a ring to avoid competitive E/Z isomerization (dotted line). If the tether is attached to the reactive olefin components as shown, product A is called the straight and product B the crossed photocycloaddition product. | ||
To name a few notable examples, Hiemstra, van Maarseveen, and co-workers employed a crossed [2+2] photocycloaddition reaction to key intermediate 1 (Fig. 1) as pivotal step in the synthesis of the complex terpene natural product solanoeclepin A.5 The group of Kwon demonstrated that benzobicyclo[3.1.1]heptanones such as rac-2 can be cleanly obtained by a visible light-mediated, crossed [2+2] photocycloaddition employing an iridium complex as sensitizer.6 A photochemical access to strained, saturated bioisosteres of ortho-disubstituted benzenes, e.g. rac-3, was recently accomplished by Mykhailiuk and co-workers from 1,5-dienes.7
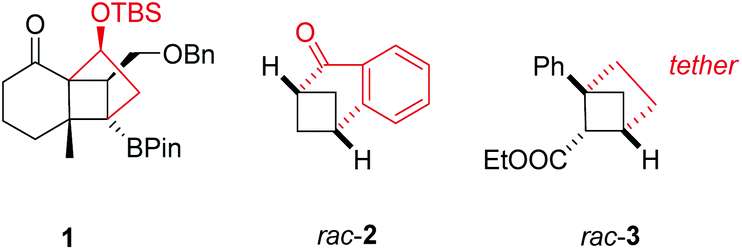 | ||
Fig. 1 Examples for intramolecular crossed [2+2] photocycloaddition products 1,5rac-2,6 and rac-3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 7 (Bn = benzyl, Pin = pinacolate, TBS = tert-butyldimethylsilyl). 7 (Bn = benzyl, Pin = pinacolate, TBS = tert-butyldimethylsilyl). | ||
It is very clear from these and many other studies,8 that crossed [2+2] photocycloadditions do not represent laboratory curiosities but rather play an important role in the toolbox of synthetic chemists. Despite their potential use and despite the fact that catalytic, enantioselective [2+2] photocycloaddition reactions have received increased attention in recent years,2,9 it is surprising that the enantioselective synthesis of crossed photocycloaddition products from achiral precursors has not been addressed.10 In this contribution, we present our results on the intramolecular, crossed [2+2] photocycloaddition of 2-(2′-alkenyloxy)cyclohex-2-enones which we found to proceed enantioselectively in the presence of a chelating Lewis acid.
Results and discussion
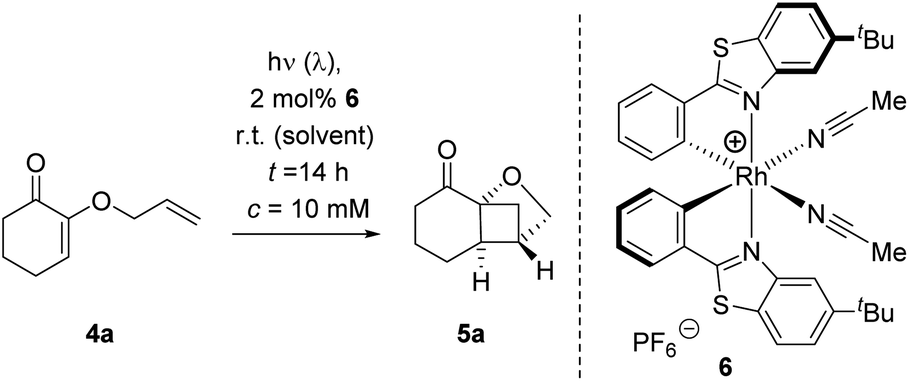
Recently, we revisited earlier work by Ikeda et al. who had reported the crossed [2+2] photocycloaddition of 2-(alkenyloxy)cyclohex-2-enones by excitation with a UV source (high-pressure mercury lamp, λ > 280 nm).11 We found the reaction to be possible upon visible light irradiation (λ = 420 nm) in the presence of thioxanthen-9-one (TXT) as the sensitizer and we studied the influence of substituents and of the enone ring size.12 The oxygen substituent attached to the carbon atom in α-position to the carbonyl group represents a remarkable and distinct feature of the substrates. Since the formation of five-membered chelates is well established with bidentate Lewis acids,13 we wondered whether it would be possible to employ a chiral Lewis acid for an enantioselective reaction of 2-(alkenyloxy)cyclohex-2-enones. Based on previous work with chiral Lewis acids as catalysts for photochemical reactions,14 we had reason to believe that a catalytic enantioselective reaction would be possible if the Lewis acid induced a long wavelength absorption that could be selectively addressed by a proper light source. As indicated above, the selection of possible Lewis acids is extremely wide and we screened several compound classes for this purpose. Bisoxazoline-based metal salts had been previously applied to a related chromophore15 but gave no enantioselectivity in the present case. Likewise, Lewis acids with a binol- or binap-derived ligand failed as did the monodentate boron catalysts we had successfully used for the reaction of enones.14 Eventually, we resorted to the chiral-at-metal Lewis catalysts which had been introduced by Meggers and co-workers and which have been shown to offer a broadly applicable bidentate binding.16 Specifically, we focussed on chiral rhodium complex 6 which can be readily prepared by a published procedure17 and which we had recently used in an intermolecular [2+2] photocycloaddition.18 2-(Allyloxy)cyclohex-2-enone (4a) served as the test substrate which undergoes a clean crossed [2+2] photocycloaddition at λ = 420 nm to product rac-5a under sensitizing conditions.12 If irradiated under the same conditions, there was no background reaction as verified in the present set of experiments by irradiation with a light emitting diode (LED) at λ = 425 nm. A first screen in different solvents (Table 1, entries 1–4) with a catalyst loading of 2 mol% gave a promising enantioselectivity (ee = enantiomeric excess) in dichloromethane (entry 4) at ambient temperature. However, the yield remained low despite the fact that the conversion was high after a reaction time of 16 hours. The major breakthrough was achieved when employing 1,2-dichloroethane (DCE) as the solvent which not only led to an increase in yield but also in enantioselectivity (entry 5). Subsequent modifications of the reaction conditions included the presence of air (entries 6 and 9), a longer reaction time (entry 7), a different excitation wavelength (entry 8), a higher catalyst loading (entry 10), a lower reaction temperature (entry 11), and a lower substrate concentration (entry 12). The only significant improvement was achieved when an LED was employed which emitted at a longer irradiation wavelength (entry 8). When the reaction was performed at λ = 437 nm instead of λ = 425 nm (entry 5) the yield increased by more than 20% (to 86%) without compromising the enantioselectivity (92% ee).|
|
|||||
|---|---|---|---|---|---|
| Entrya | Solvent | λ [nm] | Conv.b [%] | Yieldc [%] | % eed |
| a All reactions were performed under anaerobic conditions employing a light emitting diode (LED) in a previously described set-up.19 b The conversion (conv.) was determined by NMR analysis. c All yields refer to isolated material. d Enantiomeric excess (ee) as determined by GLC analysis on a chiral stationary phase. e The reaction was performed in the presence of air. f The reaction time was 24 hours. g The catalyst loading was 4 mol%. h The reaction was performed at 0 °C. i The substrate concentration was 4 mM. | |||||
| 1 | MeCN | 425 | 2 | — | — |
| 2 | PhMe | 425 | 11 | 11 | 15 |
| 3 | THF | 425 | 11 | 11 | 20 |
| 4 | CH2Cl2 | 425 | 85 | 22 | 87 |
| 5 | DCE | 425 | 83 | 61 | 91 |
| 6e | DCE | 425 | 91 | 68 | 87 |
| 7f | DCE | 425 | 95 | 86 | 85 |
| 8 | DCE | 437 | 93 | 86 | 92 |
| 9e | DCE | 437 | 73 | 55 | 90 |
| 10g | DCE | 437 | 79 | 64 | 94 |
| 11h | DCE | 437 | 89 | 78 | 87 |
| 12i | DCE | 437 | 72 | 70 | 87 |
Having established optimal conditions for the enantioselective crossed [2+2] photocycloaddition reaction, other possible substrates were tested. The compounds were readily available from the respective 1,2-diketones or α,β-epoxyketones (see the ESI† for details).12,20Scheme 2 summarizes the reactions which delivered the desired bridged tricyclic products 5. We found substitution at the internal carbon atom of the tethered olefin (R1) and at its terminal carbon atom (R2) compatible with the reaction conditions. Products 5b–5f were obtained in moderate yields but with consistently good to excellent enantioselectivity. Functional group compatibility with an aryl ring (products 5d, 5f), a bromine substituent (product 5f) and a methoxy group (product 5e) was demonstrated. Substitution at carbon atoms C5 and C6 within the cyclohex-2-enone ring was well tolerated (products 5g–5l) although the ee suffered in some cases slightly (80–90% ee). A salient feature of the reaction is the fact that its regioselectivity and diastereoselectivity are very high. Up to four stereogenic centres are created in a single reaction and all products were obtained free of any isomers.
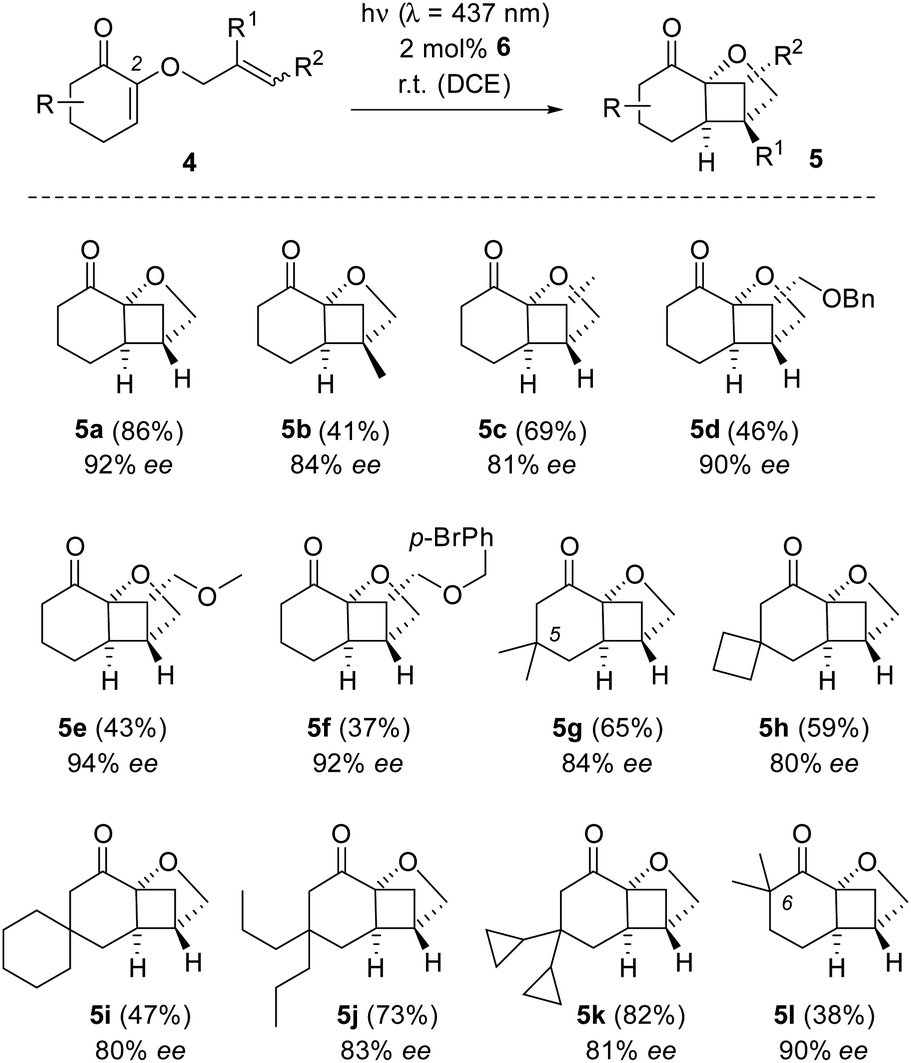 | ||
| Scheme 2 Intramolecular crossed [2+2] photocycloaddition of various substituted 2-(2′-alkenyloxy)cyclohex-2-enones 4 to bridged tricyclic products 5. Yields refer to isolated products. The ee was determined by GLC or HPLC analysis on a chiral stationary phase. | ||
Limitations of the reactions are intrinsic to the photocycloaddition itself but not to the catalytic conditions. In previous work,12 we had already seen that cyclopent-2-enones were not suitable for the reaction nor was it possible to obtain cyclobutanes from substrates with a two-fold substitution at the terminal carbon atom of the tethered olefin. A cyclohex-2-enone with a two-fold substitution at carbon atom C4 had in the racemic series already given a poor yield (38%) and failed to give any products under the catalytic conditions.
To establish the absolute configuration of the products, various derivatization methods were tried because none of the products, not even bromophenyl-substituted product 5f, gave suitable crystals for X-ray crystallographic analysis. Eventually, the classical way via the respective hydrazone was viable and compound 7 was obtained from product 5a in 73% yield upon treatment with para-nitrophenylsulfonylhydrazide in CH2Cl2 solution. The crystal structure (Fig. 2) not only facilitated the assignment of the absolute configuration but also confirmed the Lewis basicity of the ether oxygen atom.21 Indeed, product 7 was formed exclusively as (E)-isomer presumably because a hydrogen bond between the hydrazone NH and the oxygen atom stabilizes this diastereoisomer. Regarding the absolute configuration, the products are apparently formed as a result of an attack at the cyclohexenone double bond from the top face which in turn is the Si face relative to carbon atom C2.
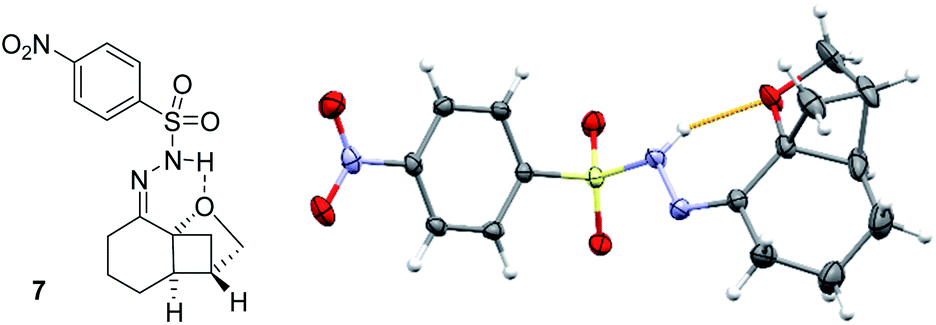 | ||
| Fig. 2 Molecular structure of hydrazone 7 in the solid state with ellipsoids set at the 50% probability level (one out of two independent molecules in the asymmetric unit). The structure displays an intramolecular hydrogen bond to the oxygen atom indicating its intrinsic basicity. The structure also served to establish the absolute configuration by anomalous dispersion. | ||
The experimental observation, whether the double bond configuration of an olefinic reaction partner is retained or not, has an implication for mechanistic analysis. Singlet reactions are concerted and occur stereospecifically, e.g. an (E)-configuration of the dienophile in the Diels–Alder reaction translates into a trans-configuration in the respective cyclohexene product. If the relative configuration is not retained, the reaction is not concerted but an intermediate must be involved in which the steric information is erased. Photocycloaddition reactions on the triplet hypersurface are known to proceed non-stereospecifically.22 In the current case, this issue was addressed by subjecting the (E)- and (Z)-isomer of substrate 4m in separate experiments to the conditions of the enantioselective photoreaction (Scheme 3). In both experiments, a single diastereoisomer was isolated to which the depicted relative configuration was assigned based on NOESY data. The assignment of the absolute configuration rests on analogy to product 5a. The reactions proceeded stereoconvergently to the formal cis-product, irrespective of whether the (E)- or (Z)-isomer (E)-4m or (Z)-4m was used as the starting material.
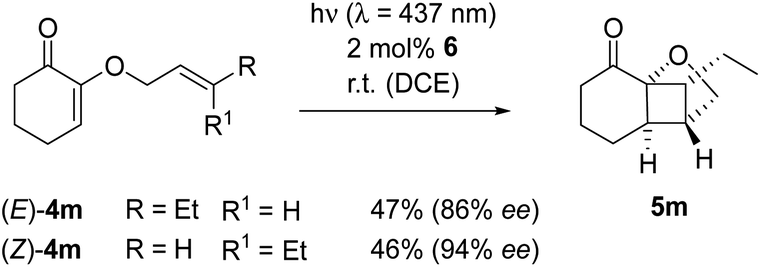 | ||
| Scheme 3 The Lewis acid-catalyzed, intramolecular [2+2] photocycloaddition of (E)- and (Z)-substituted 2-(2′-butenyloxy)cyclohex-2-enones (4m) proceeds stereoconvergently to a single product 5m. | ||
The observed enantioselectivity for the reactions 4 → 5 indicates that there is a binding of the substrate to the catalyst during or even before the photochemical event (vide infra). UV/vis spectra were recorded to possibly identify a change of absorbance upon substrate coordination. Enone 4a is transparent at a wavelength λ > 400 nm. Catalyst rac-6 is coloured and its most red-shifted absorption stretches into the visible region. An equimolar mixture of both components (4a/rac-6) showed only a minor change as compared to the spectrum of the catalyst itself (Fig. 3). A bathochromic shift of the longest wavelength absorption is notable but small. Compared to previous photocycloaddition reactions catalysed by compound 6,16a there was little change in the absorbance nor a pronounced bathochromic absorption shift.
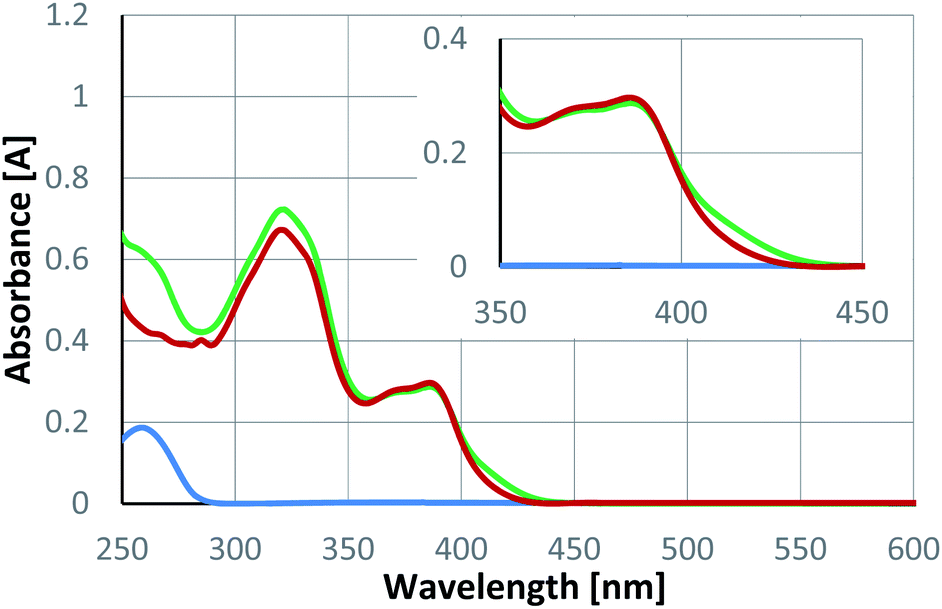 | ||
| Fig. 3 UV/vis spectra of substrate 4a (blue), racemic Lewis acid rac-6 (red) and an equimolar mixture (green) of substrate 4a and rac-6 (c = 0.5 mM in CH2Cl2). The wavelength region from 350–450 nm is enlarged in the inset. | ||
The UV/vis data indicated that the association between catalyst and substrate was weak. NMR titration studies remained unsuccessful but it was tried to evaluate the efficiency of the catalyst from the quantum yield of the reaction 4a → 5a (Scheme 4). The intrinsic quantum yield of the reaction was accessed by direct excitation at λ = 368 nm. Although the nπ* band of the cyclohex-2-enone is symmetry-forbidden, it leads to rapid population of the reactive T1 state which has ππ* character.23 The fact that the quantum yield (0.321 ± 0.034) is smaller than 1 can be accounted for by other decay pathways but also by an initial cyclization to an unproductive 1,4-diradical (vide infra).12 More or less the same value (0.316 ± 0.011) was obtained if a sensitizer24 was used (λ = 382 nm) suggesting that sensitization leads with high quantum yield to the reactive T1 state. In order to allow for a comparison with the conditions of the Rh-catalysed reaction the quantum yield measurements were performed at λ = 424 nm in DCE as the solvent with a lower sensitizer loading. The value (0.299 ± 0.055) was – within the error margin – identical to the value recorded at λ = 382 nm in CH2Cl2 at a higher catalyst concentration. In all cases, racemic product (rac-5a) was obtained.
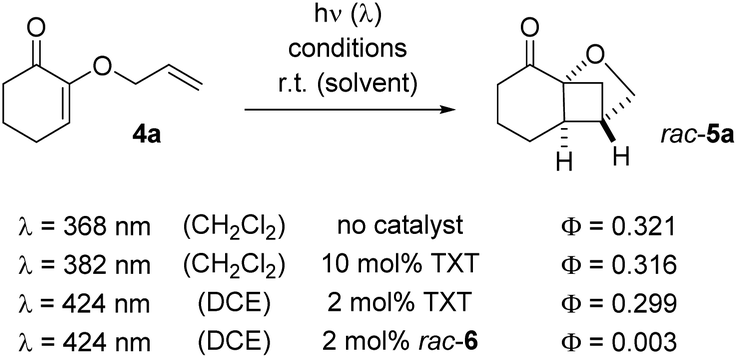 | ||
| Scheme 4 Quantum yields as determined for the reaction 4a → rac-5a under different conditions: while 9-thioxanthenone (TXT) as sensitizer delivers identical quantum yields as the uncatalyzed reaction, the catalysis by Lewis acid rac-6 is inefficient. | ||
In comparison to the sensitized conditions, the reaction with chiral catalyst rac-6 was inefficient. The quantum yield was almost a factor 100 lower than the intrinsic quantum yield of the reaction. Although several deactivation pathways may contribute to the loss in efficiency, the most likely explanation for the low quantum yield appears to be a decreased population of the reactive triplet state. The hypothesis is in accord with recent studies by Baik and co-workers25 who collected evidence that rhodium catalyst 6 – as opposed to the related iridium catalyst – does not operate by sensitization but rather by activating the substrate directly within a catalyst–substrate complex. Mechanistically, a picture emerges which includes a productive cycle involving the complex between the catalyst and the substrate, and an unproductive cycle for the catalyst itself (Scheme 5). If a substrate, such as 4a, successfully displaces the acetonitrile ligands from complex 6, an intermediate 8 is formed in which chirality can be transferred from the catalyst to the substrate. The importance of this ligand exchange is apparent from the result obtained in acetonitrile as the solvent (Table 1, entry 1). No reaction occurred because there was no ligand exchange and light absorption led only to an energy dissipation within the unproductive cycle. In complex 8, however, which may benefit from an irradiation at long wavelength (Fig. 3; Table 1, entry 5 vs. entry 8: λ = 425 nm vs. λ = 437 nm), excitation of the substrate within the complex is possible. The stereoconvergent reaction course (Scheme 3) indicates that the ensuing photocycloaddition occurs on the triplet manifold which in turn means that intersystem crossing (ISC) precedes the first carbon–carbon bond forming step.
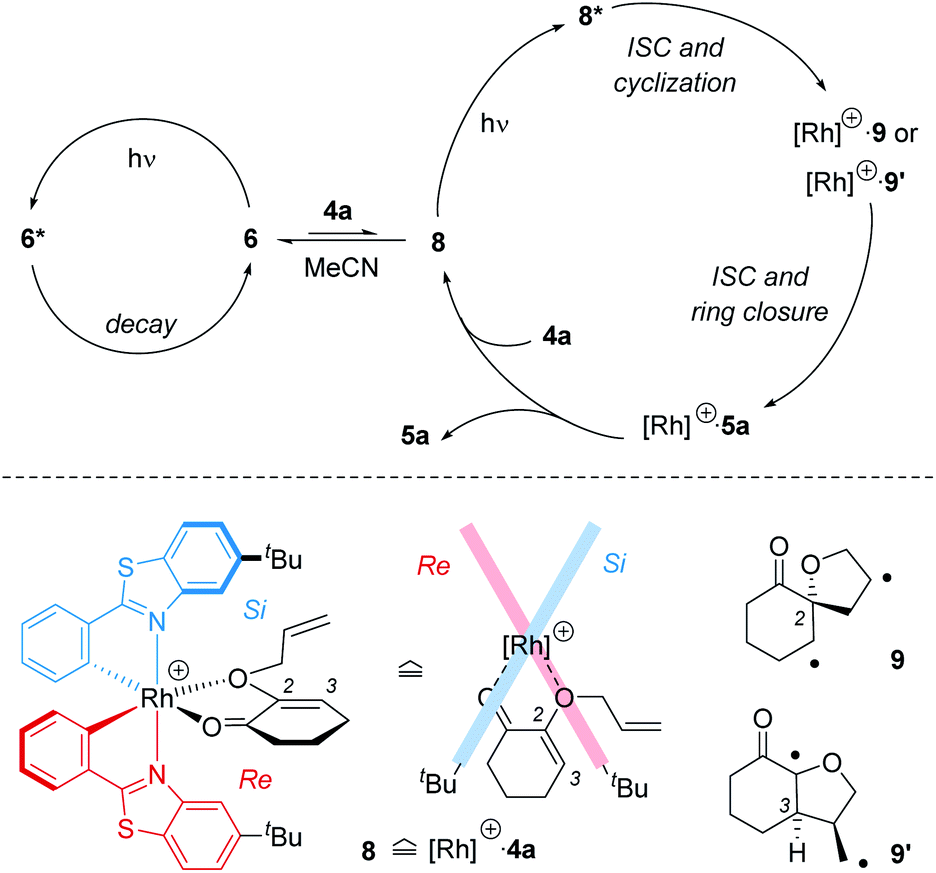 | ||
| Scheme 5 Mechanistic picture of the crossed intramolecular [2+2] photocycloaddition catalysed by Rh complex 6: (top) most photons are absorbed by complex 6 but not by complex 8 between cationic rhodium fragment [Rh]+ and substrate 4a. If excited, complex 8* reacts via 1,4-diradical intermediates 9 or 9′ to product 5a. (Bottom) simplified structure of complex 8 from the side and from the top illustrating that access onto the enone double bond from the Si face (relative to C2) is preferred. The first C–C bond forming step establishes the absolute configuration in 9 or 9′. | ||
Irrespective of which bond formation occurs first, the absolute configuration is established with the first carbon–carbon bond. The C2-symmetric rhodium complex shields the Re face (bottom face) of the olefinic double bond. Accidently, the top face is the Si face for both prostereogenic centres C2 and C3. Attack at C2 leads to 1,4-diradical 9 while attack at C3 produces the regioisomeric radical 9′.26 As previously mentioned there is a diastereomeric form of radical 9′ in which the hydrogen atom at C3 and the adjacent methylene group are cis- but not trans-oriented. This diastereoisomer is not productive but will cleave to the starting material after ISC or lead to side products. In general, any constitutional or configurational 1,4-diradical isomers which can for steric reasons not lead to bond formation can decay to the ground state by retrocleavage. The observed quantum yields for the uncatalysed process (Scheme 4) indicate that only one out of three photons induces product formation. If possible on steric grounds, the reaction is completed by the second carbon–carbon bond forming event which can take place within the complex as depicted. Release of product 5a is required before a new substrate molecule can enter the catalytic cycle. The fact that the reactions did not go to completion under any of the reaction conditions (Table 1) hints at a non-negligible product inhibition.
In a final set of experiments, we performed some consecutive reactions with the enantioenriched cyclobutanes 5 obtained by the crossed [2+2] photocycloaddition (Scheme 6). The reduction with diisobutylaluminium hydride (Dibal-H)11b exemplifies the high diastereoselectivity that can be achieved within the conformationally restricted tricyclic skeleton. Alcohols 10a and 10b were isolated as single diastereoisomers since the hydride nucleophile approaches the cyclohexanone in its locked chair conformation exclusively in an equatorial fashion.
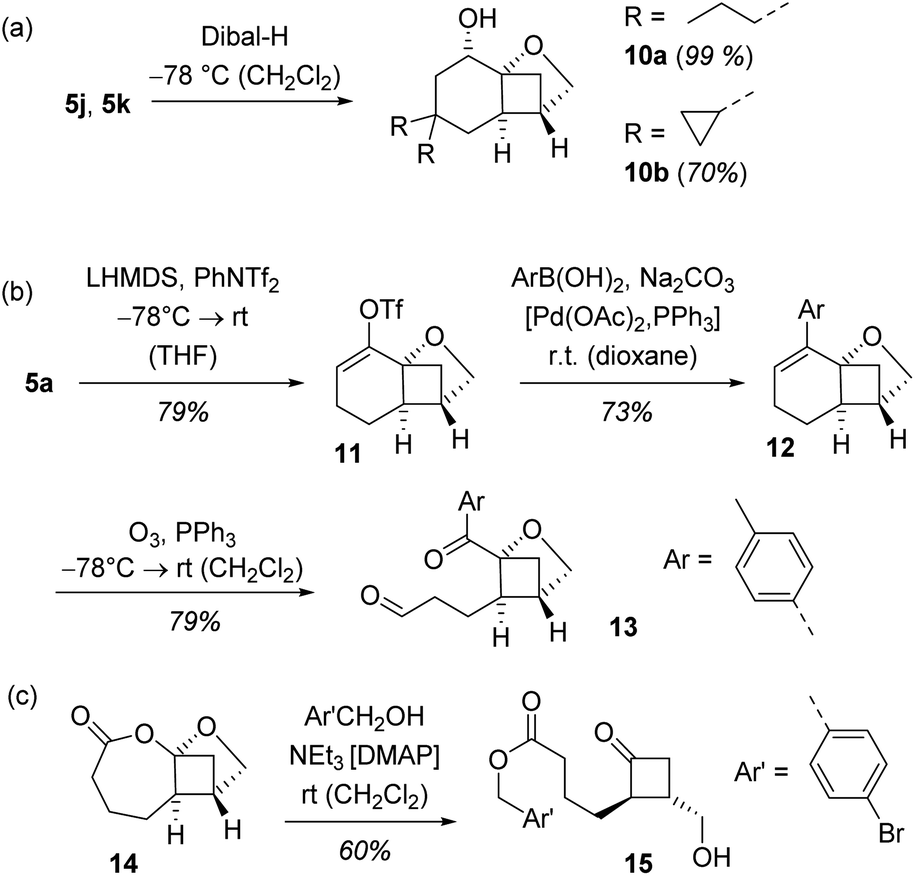 | ||
| Scheme 6 Consecutive reactions of tricyclic photocycloaddition products 5: (a) diastereoselective reduction to products 10, (b) opening of the six-membered ring by ozonolysis to product 13, and (c) formation of cyclobutanone 15. | ||
Conversion of ketone 5a to triflate 11 was achieved by treatment with lithium hexamethyldisilazide (LHMDS) and PhNTf2 (Tf = trifluoromethanesulfonyl).27 The triflate in turn was used as a coupling partner in a Suzuki cross-coupling.28 A representative aryl boronic acid served as nucleophilic reagent delivering trisubstituted olefin 12 as the only product and without interference with the adjacent stereogenic centre. When subjected to an ozonolysis29 the oxabicyclo[2.1.1]hexane core remained intact and the dicarbonyl compound 13 was isolated in high yield and with preserved enantiomeric purity (92% ee). Ikeda et al. had already shown that racemic photocycloaddition products analogous to rac-5 underwent a stereospecific Baeyer–Villiger oxidation.11 Under the same conditions, compound 14, was obtained in enantioenriched form and served as a useful precursor to 2,3-disubstituted cyclobutanones such as compound 15 (92% ee).
Conclusion
In summary, we have shown that crossed [2+2] photocycloaddition reactions of cycloalkenones can be performed enantioselectively in the presence of a chiral Lewis acid. In the present case, the additional oxygen substituent invited the use of a chelating, C2-symmetric Lewis acid. One key feature of the reaction is its high enantioselectivity which is testimony to the enantiodifferentiating power of the ligand framework around the stereogenic rhodium centre. A second key feature is the unique structure of the products which is difficult if not impossible to establish by thermal methods. The products display not only a bridged structure but also a tertiary oxygen substituted carbon atom in α-position to the carbonyl group. Future research efforts aim at possible applications of enantioselective, crossed [2+2] photocycloaddition reactions to the total synthesis of natural products.Data availability
Original NMR datasets (FIDs) are available at Open Science Framework at https://osf.io/73k6e.Author contributions
T. R., R. G., and T. B. developed the project. Funding was acquired by T. B., D. P. S. and T. R. T. R. designed and performed the synthetic experiments. C. J. performed the X-ray crystallographic analysis of compound 7. T. R., D. P. S., and R. G. planned and performed the quantum yield experiments. T. R., D. P. S., and R. G. generated and validated the data. T. B. administered the project and supervised the research. T. R., D. P. S., and T. B. wrote, reviewed, and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – TRR 325 (project B1) – 444632635, by the Studienstiftung des deutschen Volkes (PhD fellowship to D. P. S.), and by the TUM foundation (postdoctoral fellowship to T. R.) is gratefully acknowledged.Notes and references
- Reviews: (a) M. D. Kärkäs, J. A. Porco Jr and C. R. J. Stephenson, Chem. Rev., 2016, 116, 9683–9747 CrossRef PubMed; (b) T. Bach and J. P. Hehn, Angew. Chem., Int. Ed., 2011, 50, 1000–1045 CrossRef CAS PubMed; (c) N. Hoffmann, Chem. Rev., 2008, 108, 1052–1103 CrossRef CAS PubMed.
- Reviews: (a) S. Poplata, A. Tröster, Y.-Q. Zou and T. Bach, Chem. Rev., 2016, 116, 9748–9815 CrossRef CAS PubMed; (b) D. Becker and N. Haddad, Org. Photochem., 1989, 10, 1–162 CAS; (c) M. T. Crimmins, Chem. Rev., 1988, 88, 1453–1473 CrossRef CAS.
- (a) R. Srinivasan and K. H. Carlough, J. Am. Chem. Soc., 1967, 89, 4932–4936 CrossRef CAS; (b) R. S. H. Liu and G. S. Hammond, J. Am. Chem. Soc., 1967, 89, 4936–4944 CrossRef CAS; (c) D. J. Maradyn and A. C. Weedon, J. Am. Chem. Soc., 1995, 117, 5359–5360 CrossRef CAS.
- (a) C. Walling and A. Cioffari, J. Am. Chem. Soc., 1972, 94, 6059–6064 CrossRef CAS; (b) J. E. Baldwin, J. Chem. Soc., Chem. Commun., 1976, 734–736 RSC; (c) A. L. J. Beckwith and C. H. Schiesser, Tetrahedron, 1985, 41, 3925–3941 CrossRef CAS.
- R. A. Kleinnijenhuis, B. J. J. Timmer, G. Lutteke, J. M. M. Smits, R. de Gelder, J. H. van Maarseveen and H. Hiemstra, Chem.–Eur. J., 2016, 22, 1266–1269 CrossRef CAS PubMed.
- J. Zhao, J. L. Brosmer, Q. Tang, Z. Yang, K. N. Houk, P. L. Diaconescu and O. Kwon, J. Am. Chem. Soc., 2017, 139, 9807–9810 CrossRef CAS PubMed.
- A. Denisenko, P. Garbuz, S. V. Shishkina, N. M. Voloshchuk and P. K. Mykhailiuk, Angew. Chem., Int. Ed., 2020, 59, 20515–20521 CrossRef CAS PubMed.
- For further, selected examples of crossed [2+2] photocycloadditions, see: (a) E. J. Corey, D. E. Cane and L. Libit, J. Am. Chem. Soc., 1971, 93, 7016–7021 CrossRef CAS; (b) M. Miyashita and A. Yoshikoshi, J. Am. Chem. Soc., 1974, 96, 1917–1925 CrossRef CAS; (c) Y. Tamura, H. Ishibashi, M. Hirai, Y. Kita and M. Ikeda, J. Org. Chem., 1975, 40, 2702–2710 CrossRef CAS; (d) S. Wolff and W. C. Agosta, J. Am. Chem. Soc., 1983, 105, 1292–1299 CrossRef CAS; (e) R. Hertel, J. Mattay and J. Runsink, J. Am. Chem. Soc., 1991, 113, 657–665 CrossRef CAS; (f) G. Pandey, S. Hajra, M. K. Ghorai and K. R. Kumar, J. Am. Chem. Soc., 1997, 119, 8777–8787 CrossRef CAS; (g) D. W. Piotrowski, Synlett, 1999, 1091–1093 CrossRef CAS; (h) T. Bach, H. Bergmann and K. Harms, Angew. Chem., Int. Ed., 2000, 39, 2302–2304 CrossRef CAS; (i) S. J. Bader and M. L. Snapper, J. Am. Chem. Soc., 2005, 127, 1201–1205 CrossRef CAS PubMed; (j) S. M. Ng, S. J. Bader and M. L. Snapper, J. Am. Chem. Soc., 2006, 128, 7315–7319 CrossRef CAS PubMed; (k) J. R. Ragains and J. D. Winkler, Org. Lett., 2006, 8, 4437–4440 CrossRef CAS PubMed; (l) A. Iyer, S. Jockusch and J. Sivaguru, J. Phys. Chem. A, 2014, 118, 10596–10602 CrossRef CAS PubMed; (m) J. M. Saya, K. Vos, R. A. Kleinnijenhuis, J. H. van Maarseveen, S. Ingemann and H. Hiemstra, Org. Lett., 2015, 17, 3892–3894 CrossRef CAS PubMed; (n) L. D. Elliott and K. I. Booker-Milburn, Org. Lett., 2019, 21, 1463–1466 CrossRef CAS PubMed; (o) S. Ahuja, A. Iyer, S. K. Kandappa and J. Sivaguru, J. Photochem. Photobiol., A, 2019, 382, 111883 CrossRef CAS.
- Reviews: (a) M. J. Genzink, J. B. Kidd, W. B. Swords and T. P. Yoon, Chem. Rev., 2022, 122, 1654–1716 CrossRef CAS PubMed; (b) J. Großkopf, T. Kratz, T. Rigotti and T. Bach, Chem. Rev., 2022, 122, 1626–1653 CrossRef PubMed; (c) W. Yao, E. A. Bazan-Bergamino and M.-Y. Ngai, ChemCatChem, 2022, 14, e202101292 CAS; (d) C. Prentice, J. Morrisson, A. D. Smith and E. Zysman-Colman, Beilstein J. Org. Chem., 2020, 16, 2363–2441 CrossRef PubMed; (e) T. Rigotti and J. Alemán, Chem. Commun., 2020, 56, 11169 RSC; (f) M. Silvi and P. Melchiorre, Nature, 2018, 554, 41–49 CrossRef CAS PubMed; (g) R. Brimioulle, D. Lenhart, M. M. Maturi and T. Bach, Angew. Chem., Int. Ed., 2015, 54, 3872–3890 CrossRef CAS PubMed.
- To the best of our knowledge, a by-product found in a straight [2+2] photocycloaddition and a single, low-yielding example of a crossed seven-membered ring containing cyclobutane represent the only previous catalytic examples: (a) C. Müller, A. Bauer and T. Bach, Angew. Chem., Int. Ed., 2009, 48, 6640–6642 CrossRef PubMed; (b) A. M. Martínez-Gualda, P. Domingo-Legarda, T. Rigotti, S. Díaz-Tendero, A. Fraile and J. Alemán, Chem. Commun., 2021, 57, 3046–3049 RSC.
- (a) M. Ikeda, M. Takahashi, K. Ohno, Y. Tamura and M. Kido, Chem. Pharm. Bull., 1982, 30, 2269–2271 CrossRef CAS; (b) M. Ikeda, M. Takahashi, T. Uchino, K. Ohno, Y. Tamura and M. Kido, J. Org. Chem., 1983, 48, 4241–4247 CrossRef CAS.
- R. Graßl, C. Jandl and T. Bach, J. Org. Chem., 2020, 85, 11426–11439 CrossRef PubMed.
- (a) G. Desimoni, G. Faita and K. A. Jørgensen, Chem. Rev., 2006, 106, 3561–3651 CrossRef CAS PubMed; (b) Catalytic Asymmetric Synthesis, ed. I. Ojima, Wiley, Hoboken, 3rd edn, 2010 Search PubMed; (c) J. S. Johnson and D. A. Evans, Acc. Chem. Res., 2000, 33, 325–335 CrossRef CAS PubMed; (d) L. Zhang and E. Meggers, Acc. Chem. Res., 2017, 50, 320–330 CrossRef CAS PubMed; (e) D. Yang, L. Wang, D. Li and R. Wang, Chem, 2019, 5, 1108–1166 CrossRef CAS.
- Reviews: (a) D. P. Schwinger and T. Bach, Acc. Chem. Res., 2020, 53, 1933–1943 CrossRef CAS PubMed; (b) C. Brenninger, J. D. Jolliffe and T. Bach, Angew. Chem., Int. Ed., 2018, 57, 14338–14349 CrossRef CAS PubMed.
- V. Edtmüller, A. Pöthig and T. Bach, Tetrahedron, 2017, 73, 5038–5047 CrossRef.
- (a) X. Huang, T. R. Quinn, K. Harms, R. D. Webster, L. Zhang, O. Wiest and E. Meggers, J. Am. Chem. Soc., 2017, 139, 9120–9123 CrossRef CAS PubMed; (b) N. Hu, H. Jung, Y. Zheng, J. Lee, L. Zhang, Z. Ullah, X. Xie, K. Harms, M.-H. Baik and E. Meggers, Angew. Chem., Int. Ed., 2018, 57, 6242–6246 CrossRef CAS PubMed; (c) X. Huang and E. Meggers, Acc. Chem. Res., 2019, 52, 833–847 CrossRef CAS PubMed.
- J. Ma, X. Zhang, X. Huang, S. Luo and E. Meggers, Nat. Protoc., 2018, 13, 605–632 CrossRef CAS PubMed.
- N. Jeremias, L.-M. Mohr and T. Bach, Org. Lett., 2021, 22, 5674–5678 CrossRef PubMed.
- D. Lenhart, A. Bauer, A. Pöthig and T. Bach, Chem.–Eur. J., 2016, 22, 6519–6523 CrossRef CAS PubMed.
- (a) W. G. Dauben, A. A. Ponaras and A. Chollet, J. Org. Chem., 1980, 45, 4413–4417 CrossRef CAS; (b) K. Matoba, N. Karibe and T. Yamazaki, Chem. Pharm. Bull., 1982, 30, 3906–3911 CrossRef CAS; (c) J. R. Rodríguez, L. Castedo and J. L. Mascareñas, Chem.–Eur. J., 2002, 8, 2923–2930 CrossRef.
- Supplementary crystallographic data for 7 have been deposited with the Cambridge Crystallographic Data Centre (CCDC 2130504).†.
- (a) W. L. Dilling, T. E. Tabor, F. P. Boer and P. P. North, J. Am. Chem. Soc., 1970, 92, 1399–1400 CrossRef CAS; (b) N. P. Peet, R. L. Cargill and D. F. Bushey, J. Org. Chem., 1973, 38, 1218–1221 CrossRef CAS; (c) D. Becker, M. Nagler, S. Hirsh and J. Ramun, J. Chem. Soc., Chem. Commun., 1983, 371–373 RSC; (d) D. Becker, M. Nagler, Y. Sahali and N. Haddad, J. Org. Chem., 1991, 56, 4537–4543 CrossRef CAS; (e) J. F. D. Kelly, J. M. Kelly and T. B. H. McMurry, J. Chem. Soc., Perkin Trans. 2, 1999, 1933–1941 RSC; (f) R. Brimioulle, A. Bauer and T. Bach, J. Am. Chem. Soc., 2015, 137, 5170–5176 CrossRef CAS; (g) S. Poplata, A. Bauer, G. Storch and T. Bach, Chem.–Eur. J., 2019, 25, 8135–8148 CrossRef CAS PubMed.
- M. T. Peschel, P. Kabaciński, D. P. Schwinger, E. Thyrhaug, G. Cerullo, T. Bach, J. Hauer and R. de Vivie-Riedle, Angew. Chem., Int. Ed., 2021, 60, 10155–10163 CrossRef CAS PubMed.
- For recent work on the sensitization of enone [2+2] photocycloaddition reactions, see: (a) A. Clay, N. Vallavoju, R. Krishnan, A. Ugrinov and J. Sivaguru, J. Org. Chem., 2016, 81, 7191–7200 CrossRef CAS; (b) L. D. Elliott, M. Berry, B. Harji, D. Klauber, J. Leonard and K. I. Booker-Milburn, Org. Process Res. Dev., 2016, 20, 1806–1811 CrossRef CAS; (c) J. D. Williams, M. Nakano, R. Gérardy, J. A. Rincón, Ó. de Frutos, C. Mateos, J.-C. M. Monbaliu and C. O. Kappe, Org. Process Res. Dev., 2019, 23, 78–87 CrossRef CAS; (d) F. Strieth-Kalthoff, C. Henkel, M. Teders, A. Kahnt, W. Knolle, A. Gómez-Suárez, K. Dirian, W. Alex, K. Bergander, C. G. Daniliuc, B. Abel, D. M. Guldi and F. Glorius, Chem, 2019, 5, 2183–2194 CrossRef CAS; (e) F. Pecho, Y.-Q. Zou, J. Gramüller, T. Mori, S. M. Huber, A. Bauer, R. M. Gschwind and T. Bach, Chem.–Eur. J., 2020, 26, 5190–5194 CrossRef CAS PubMed.
- H. Jung, M. Hong, M. Marchini, M. Villa, P. S. Steinlandt, X. Huang, M. Hemming, E. Meggers, P. Ceroni, J. Park and M.-H. Baik, Chem. Sci., 2021, 12, 9673–9681 RSC.
- (a) D. I. Schuster, G. Lem and N. A. Kaprinidis, Chem. Rev., 1993, 93, 3–22 CrossRef CAS; (b) D. I. Schuster, Mechanistic Issues in [2+2] Photocycloadditions of Cyclic Enones to Alkenes, in CRC Handbook of Photochemistry and Photobiology, ed. W. M. Horspool and F. Lenci, CRC Press, Boca Raton, 2nd edn, 2004, pp. 72/1–72/24 Search PubMed.
- M. D. Lord, J. T. Negri and L. A. Paquette, J. Org. Chem., 1995, 60, 191–195 CrossRef CAS.
- M. Ichiki, H. Tanimoto, S. Miwa, R. Saito, T. Sato and N. Chida, Chem.–Eur. J., 2013, 19, 264–269 CrossRef CAS PubMed.
- E. V. Boltukhina, A. E. Sheshenev and I. M. Lyapkalo, Tetrahedron, 2011, 67, 5382–5388 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures and full characterization for all starting materials and products, mechanistic experiments, NMR spectra, GLC and HPLC traces. CCDC 2130504. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2sc00113f |
| This journal is © The Royal Society of Chemistry 2022 |