
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA DNAzyme-augmented bioorthogonal catalysis system for synergistic cancer therapy†
Yawen
You
ab,
Hao
Liu
ab,
Jiawei
Zhu
ab,
Yibo
Wang
c,
Fang
Pu
 *ab,
Jinsong
Ren
*ab and
Xiaogang
Qu
*ab
*ab,
Jinsong
Ren
*ab and
Xiaogang
Qu
*ab
aState Key Laboratory of Rare Earth Resources Utilization, Laboratory of Chemical Biology, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, P. R. China. E-mail: pufang@ciac.ac.cn; jren@ciac.ac.cn; xqu@ciac.ac.cn
bUniversity of Science and Technology of China, Hefei, Anhui 230029, P. R. China
cLaboratory of Chemical Biology, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, P. R. China
First published on 10th June 2022
Abstract
As one of the representative bioorthogonal reactions, the copper-catalyzed click reaction provides a promising approach for in situ prodrug activation in cancer treatment. To solve the issue of inherent toxicity of Cu(I), biocompatible heterogeneous copper nanoparticles (CuNPs) were developed for the Cu-catalyzed azide–alkyne cycloaddition (CuAAC) reaction. However, the unsatisfactory catalytic activity and off-target effect still hindered their application in biological systems. Herein, we constructed a DNAzyme-augmented and targeted bioorthogonal catalyst for synergistic cancer therapy. The system could present specificity to cancer cells and promote the generation of Cu(I) via DNAzyme-induced value state conversion of DNA-templated ultrasmall CuNPs upon exposure to endogenous H2O2, thereby leading to high catalytic activity for in situ drug synthesis. Meanwhile, DNAzyme could produce radical species to damage cancer cells. The synergy of in situ drug synthesis and chemodynamic therapy exhibited excellent anti-cancer effects and minimal side effects. The study offers a simple and novel avenue to develop highly efficient and safe bioorthogonal catalysts for biological applications.
Introduction
As an alternative to conventional chemotherapy, prodrug approaches are considered a promising solution to the drug side effects caused by off-target effect. The CuAAC reaction as one of the representative bioorthogonal reactions,1–11 has now become a powerful tool for prodrug activation or drug synthesis in situ.12–16 However, the intrinsic toxicity of Cu(I) to organisms hampered its application in biological systems.17 To circumvent the limitation, biocompatible heterogeneous catalysts based on CuNPs were developed for the CuAAC reaction.18 Unfortunately, the inherent poor activity of Cu(0) makes these reactions often require a long time and the assistance of high temperature, microwave irradiation and additives.19,20 It has been reported that the surface of CuNPs was more reactive due to the unfilled valences of the surface atoms and a large surface-to-volume ratio.21 Therefore, promoting the conversion of surface Cu(0) to Cu(I) and regulating their size would accelerate the catalytic process.22 Recently, we designed biocompatible heterogeneous CuNPs for highly efficient CuAAC reaction, in which the valence state change of Cu was achieved by near-infrared light (NIR)-induced reactive oxygen species (ROS).23 However, the dependence of external excitation inevitably suffers from limited tissue-penetrating depth, and has adverse effects on the surrounding normal tissues. Therefore, the endogenous control of the valence state of CuNPs is highly desirable. In addition to unsatisfactory efficiency, the lack of targeting ability of these nanocatalysts may compromise the efficacy of the CuAAC reaction in specific regions and cause damage to healthy tissues. Although modification of targeting molecules, such as triphenylphosphonium,24 could endow nanocatalysts with increment of specific localization,25–28 it is still a challenge to develop highly efficient and precise targeted bioorthogonal catalysts for biological applications.Peroxidase-mimicking DNAzymes are single-stranded DNA molecules, which adopt a G-quadruplex structure and bind hemin molecules to exhibit high catalytic capability in the presence of H2O2.29–34 They have been widely explored in biology, sensing, and material sciences.35–37 Recently, it was reported that the peroxidase-mimicking DNAzyme could produce strong radical species to cut single-wall nanotubes (SWNTs) and degrade graphene oxide into small fragments.38,39 Inspired by the properties of DNAzyme, herein, we construct a DNAzyme-augmented bioorthogonal catalysis system for synergistic cancer therapy. Using the high local concentration of H2O2 in cancer cells, the DNAzyme could produce active radical species to facilitate the conversion of Cu(0) to Cu(I) on the surface of DNA-templated ultrasmall CuNPs, resulting in much enhanced bioorthogonal catalytic activity and enabling in situ prodrug activation. In addition, the aptamer AS1411/hemin-based DNAzyme exhibited cancer cell-targeting ability through specific recognition of nucleolin overexpressed on the surface of cancer cells. Meanwhile, the cancer-killing radical species produced by DNAzyme could offer synergistic chemodynamic effects.40–42 The combination of in situ drug synthesis with chemodynamic therapy successfully achieved highly specific and efficient therapeutic efficacy with minimal side effects.
Results and discussion
The design principle of the system is shown in Scheme 1. Featuring a G-quadruplex structure and specific recognition of nucleolin overexpressed by cancer cells,43 the aptamer AS1411 can form peroxidase-mimicking DNAzyme with cancer cell targeting ability.44–46 AS1411 is selected and linked with a poly-thymine (polyT) sequence, which serves as a template for CuNP formation.47,48 The obtained DNA sequence AS1411-linker-polyT was mixed with CuSO4 and sodium ascorbate to synthesize CuNPs (G-Cu), followed by incorporating hemin to form peroxidase-mimicking DNAzyme (D-Cu) (Fig. S1†). The combination of DNAzyme with CuNPs yielded a system of predesigned functionalities. The DNAzyme features triple functions: targeting cancer cells, endogenous control of the valence state of CuNPs, and production of cancer-killing radical species. | ||
| Scheme 1 Schematic illustration of a DNAzyme-augmented bioorthogonal catalytic system for synergistic cancer therapy. | ||
High-resolution transmission electron microscopy (TEM) images show that the as prepared DNAzyme-CuNPs had excellent monodispersity with an average size of 3.5 nm (Fig. 1a–c). D-Cu still maintained a stable particle size distribution in the cell culture medium (DMEM) with 10% fetal bovine serum (FBS), suggesting its stability under physiological conditions (Fig. S2†). Zeta potentials showed that G-Cu and D-Cu were negatively charged in H2O, PBS, and DMEM with 10% FBS (Fig. 1d). The inductively coupled plasma (ICP) data showed the accurate copper content of D-Cu (Table S2†). D-Cu showed fluorescence emissions at 600 nm (Fig. 1e). X-ray photoelectron spectroscopy (XPS) was conducted to examine the valence states of D-Cu. The spectrum of D-Cu exhibited two typical peaks at 932.6 eV (Cu 2p3/2) and 952.3 eV (Cu 2p1/2), corresponding to Cu(0) (Fig. 1f). These results indicated that the DNA-templated CuNPs were successfully formed.
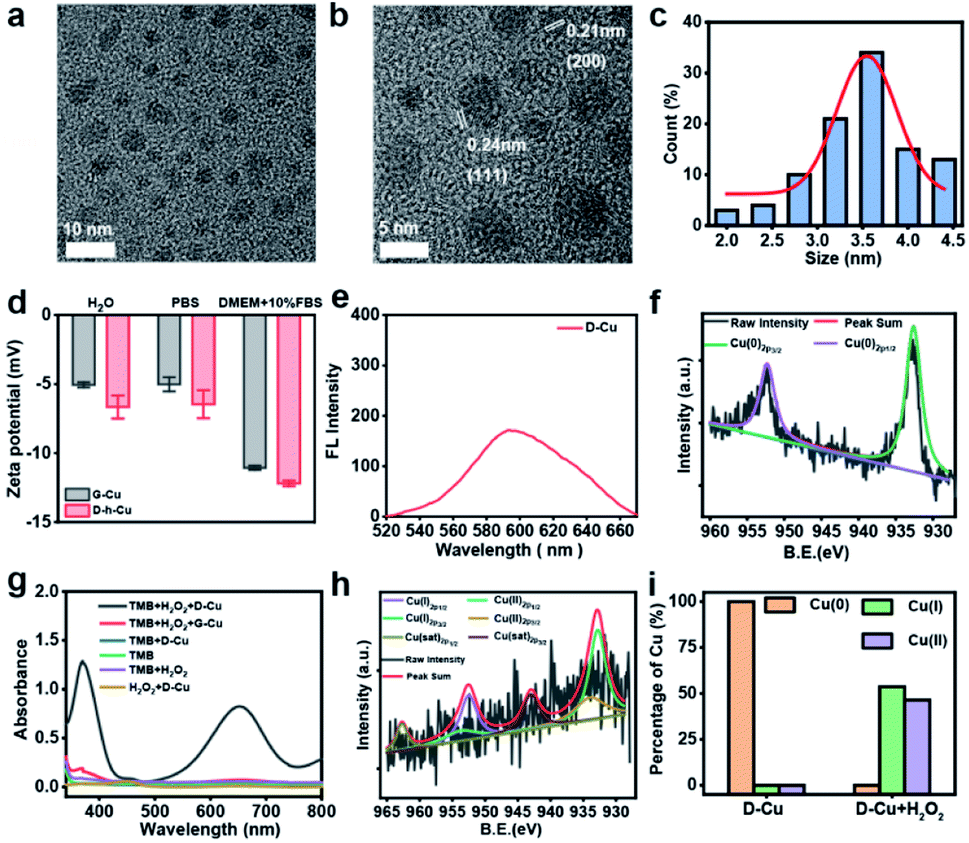 | ||
| Fig. 1 Characterization of the DNAzyme-Cu bioorthogonal catalyst. (a and b) TEM images of D-Cu. (c) Histogram of the size distribution of D-Cu (counted nanoparticles: 100). (d) Zeta-potentials of G-Cu and D-Cu in H2O, PBS and DMEM with 10% FBS. Data are presented as mean ± SD (n = 3). (e) Fluorescence spectrum of D-Cu (excited at 340 nm). (f) XPS spectra of D-Cu. (g) The peroxidase-like activity of D-Cu. (h) XPS spectrum of D-Cu with H2O2. (i) XPS analysis of D-Cu with and without H2O2. The y-axis represents the percentage of copper in different valence states. | ||
Subsequently, we investigated the peroxidase-mimicking activity of D-Cu via the 3,3′,5,5′-tetramethylbenzidine (TMB) oxidation reaction. D-Cu catalyzed TMB into oxTMB in the presence of H2O2, leading to a color change from colorless to blue and an increase in absorbance at 652 nm. In contrast, D-Cu or H2O2 alone did not produce a color change, suggesting the peroxidase-like activity of D-Cu (Fig. 1g). Furthermore, we systematically studied the catalytic activity of D-Cu under various reaction conditions, including pH, temperature, and different concentrations of D-Cu, H2O2, and TMB (Fig. S3–S7†). These results indicated that D-Cu could effectively transform H2O2 into ROS.
To confirm the value state conversion effect of DNAzyme, the catalytic performance of D-Cu in the CuAAC reaction in solution was examined. Pro-fluorophore 1 and 2, which were nonfluorescent, were chosen as models (Fig. 2a and S8†). As shown in Fig. S9,† the mixture of 1 and 2 had no fluorescence (λex = 340 nm; λem = 460 nm). Upon addition of D-Cu to 1 + 2 for 30 min, the sample emitted cyan-blue fluorescence, indicating that triazole 3 was produced. When D-Cu and H2O2 were added to 1 + 2 for 30 min, the product presented stronger fluorescence, implying higher catalytic efficiency (Fig. 2b). It could be attributed to the higher content of Cu(I) on the surface of D-Cu. The time-dependent fluorescence changes of products generated from 1 + 2, 1 + 2 + D-Cu, and 1 + 2 + D-Cu + H2O2 implied that the valence transition induced by DNAzyme led to the highest yield of the CuAAC reaction at the same time (Fig. 2c–e). The valence state of D-Cu after the addition of H2O2 was determined by XPS. As shown in Fig. 1h, two typical peaks at 932.8 eV (Cu 2p3/2) and 952.4 eV (Cu 2p1/2) were observed, implying that DNAzyme could promote the conversion of Cu(0) to Cu(I). The low redox potential of Cu(II)/Cu(I) makes Cu(I) easily oxidized to Cu(II). Therefore, the existence of Cu(II) is inevitable. It presented distinct peaks at 933.9 (Cu 2p3/2) and 953.7 eV (Cu 2p1/2), and satellite peaks at 943.0 eV (Cu 2p3/2) and 962.7 eV (Cu 2p1/2). The quantitative analysis of XPS data was performed to evaluate the value states of D-Cu in the absence and presence of H2O2. A large amount of Cu(0) existed in D-Cu. Upon addition of H2O2, Cu(0) was converted to Cu(I) and Cu(II), of which Cu(I) accounts for 53.6% (Fig. 1i). These results demonstrated that DNAzyme could facilitate the transformation of Cu(0) to Cu(I).
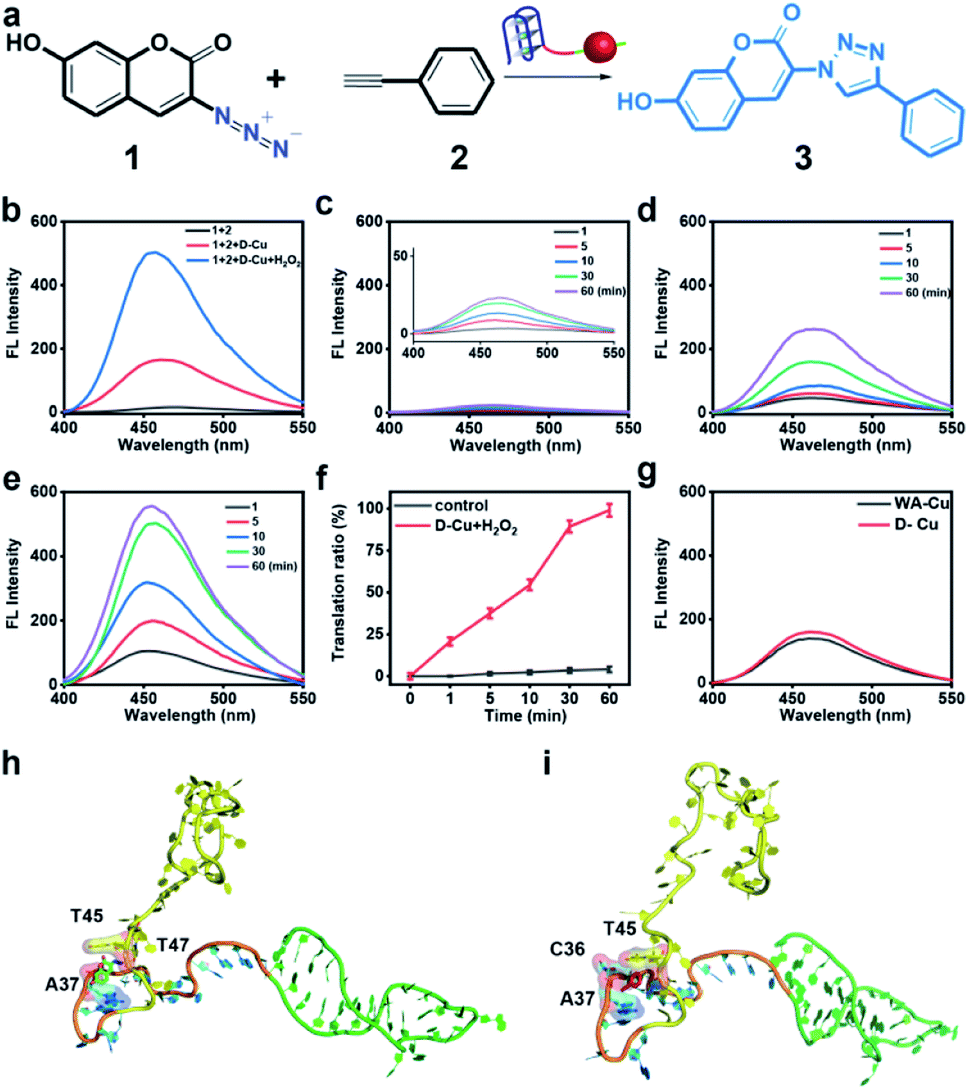 | ||
| Fig. 2 CuAAC reaction of D-Cu in solution. (a) Schematic illustration of the structures of 1, 2, and 3 and D-Cu-catalyzed CuAAC reaction. (b) Fluorescence spectra of the D-Cu-catalyzed CuAAC reaction in H2O. The spectra were recorded after adding D-Cu and D-Cu + H2O2 respectively to 1 + 2 for 30 min (equivalent amount of Cu 5 μM). (c) Fluorescence changes of the mixture of 1 and 2 at 1, 5, 10, 30, and 60 min, respectively. (d) Fluorescence spectra of 1 + 2 + D-Cu at 1, 5, 10, 30, and 60 min, respectively. (e) Fluorescence spectra of 1 + 2 + D-Cu + H2O2 at 1, 5, 10, 30, and 60 min, respectively. (f) Translation ratio of 3 upon addition of D-Cu and H2O2 with time. Data are presented as mean ± SD (n = 3). (g) Fluorescence spectra of the D-Cu- and WA-Cu-catalyzed CuAAC reactions in H2O. (h and i) Molecular docking for simulating the binding mode of 1 and 2 with D-Cu, respectively. | ||
Both fluorescence spectra and images (Fig. S10†) confirmed the superior catalytic activity of D-Cu in solution. Compared with the commonly used catalyst CuSO4/NaASC (Fig. S11†), the yield of triazole 3 increased by over 7 times (D-Cu) and 24 times (D-Cu with H2O2) within 30 minutes, respectively (Table S3†). The catalytic transformation performance of D-Cu was further measured by high-performance liquid chromatography (HPLC) at 0, 1, 10, 30 and 60 min (Fig. S12†). The transformation rates were consistent with the results of fluorescence analysis (Fig. 2f). D-Cu maintained catalytic activity under different physiological conditions (Fig. S13†), indicating its adaptability in biological systems. Furthermore, a nanocatalyst based on a DNA strand without the AS1411 aptamer (WA-Cu) was tested for comparison. The fluorescence spectra showed that the presence of aptamer did not affect the catalytic behavior in vitro (Fig. 2g).
The ultrasmall size of CuNPs and the valence state conversion of DNAzyme endowed D-Cu with high catalytic activity. In addition, we used molecular dynamics simulation (MDS) to study whether DNA had an additional contribution to catalytic activity.49 The DNA structures were first relaxed via MDS. The root-mean-square deviation (RMSD)50 values during the simulation suggested that the linker (15-base motif) and AS1411 could reach a stable state in 25 ns, while polyT kept free string style (Fig. S14†). The two precursors 1 and 2 were docked with the DNA structure in the last frame of simulations. The hydroxyl group of 1 could form two hydrogen bonds with A33 and C40 of the linker sequence. And the compound was stabilized through hydrophobic interactions between 1 and A37 and T45 and T47 (Fig. 2h). The hydrophobic interactions between 2 and C36, A37 and T45 of the linker could stabilize the complex (Fig. 2i). The results indicated that reaction substrates pulled close by DNA contributed to the high catalytic activity.
To investigate the targeting capability of D-Cu, a nucleolin-positive cell, HeLa (human cervical cancer cells) was used as a model and a normal cell, HEK-293 (human embryonic kidney cells) was used as a control. First, 3-(4,5-dimethyl-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was used to test the cytotoxicity of D-Cu. The results revealed that D-Cu was biocompatible for both HeLa and HEK-293 cells evenly at 50 μM (Fig. S15 and S16†). Then D-Cu and WA-Cu were incubated with HeLa and HEK-293 cells, respectively. Their cellular internalization was monitored using a confocal laser scanning microscope (CLSM) and quantified using inductively coupled plasma mass spectrometry (ICP-MS). The results indicated that D-Cu could be efficiently endocytosed by HeLa cells and reached the maximum within 4 hours, while a relatively low amount of WA-Cu was endocytosed even within 24 hours (Fig. S17–S19†). For HEK-293 cells, weak red fluorescence was observed within 24 hours, indicating that very little D-Cu was endocytosed (Fig. S20 and S21†). These results indicated that AS1411 could boost cellular internalization of D-Cu via receptor-mediated endocytosis.
After exploring the specific recognition capability of D-Cu, we evaluated its catalytic ability in the CuAAC reaction in HeLa and HEK-293 cells. The signals of cell membrane dye-DiO (green), D-Cu (red) and triazole 3 (blue) were monitored by CLSM. As shown in Fig. 3a, HeLa cells treated with precursors and D-Cu presented significant red and blue fluorescence enhancement compared with the cells treated with precursors only. The red and blue fluorescence was also much stronger than the cells treated with precursors and WA-Cu. HEK-293 cells treated with precursors and D-Cu showed weak red and blue fluorescence (Fig. 3b). These results indicated that a higher amount of CuNPs as a catalyst was accumulated in HeLa cells via the targeting effect of the aptamer, resulting in a higher transformation rate of precursors into triazole 3. The results of flow cytometry analysis were consistent with those of CLSM. As shown in Fig. 3c, HeLa cells treated with precursors and D-Cu presented about a 60-fold fluorescence enhancement compared with the cells treated with precursors only. However, only 1.6-fold fluorescence enhancement was shown in the HEK-293 cells under the same conditions (Fig. 3d). Furthermore, LC-MS analysis indicated that triazole 3 was synthesized in HeLa cells (Fig. S22†). The above results demonstrated that D-Cu could achieve DNAzyme-augmented and targeted CuAAC reactions in living cells.
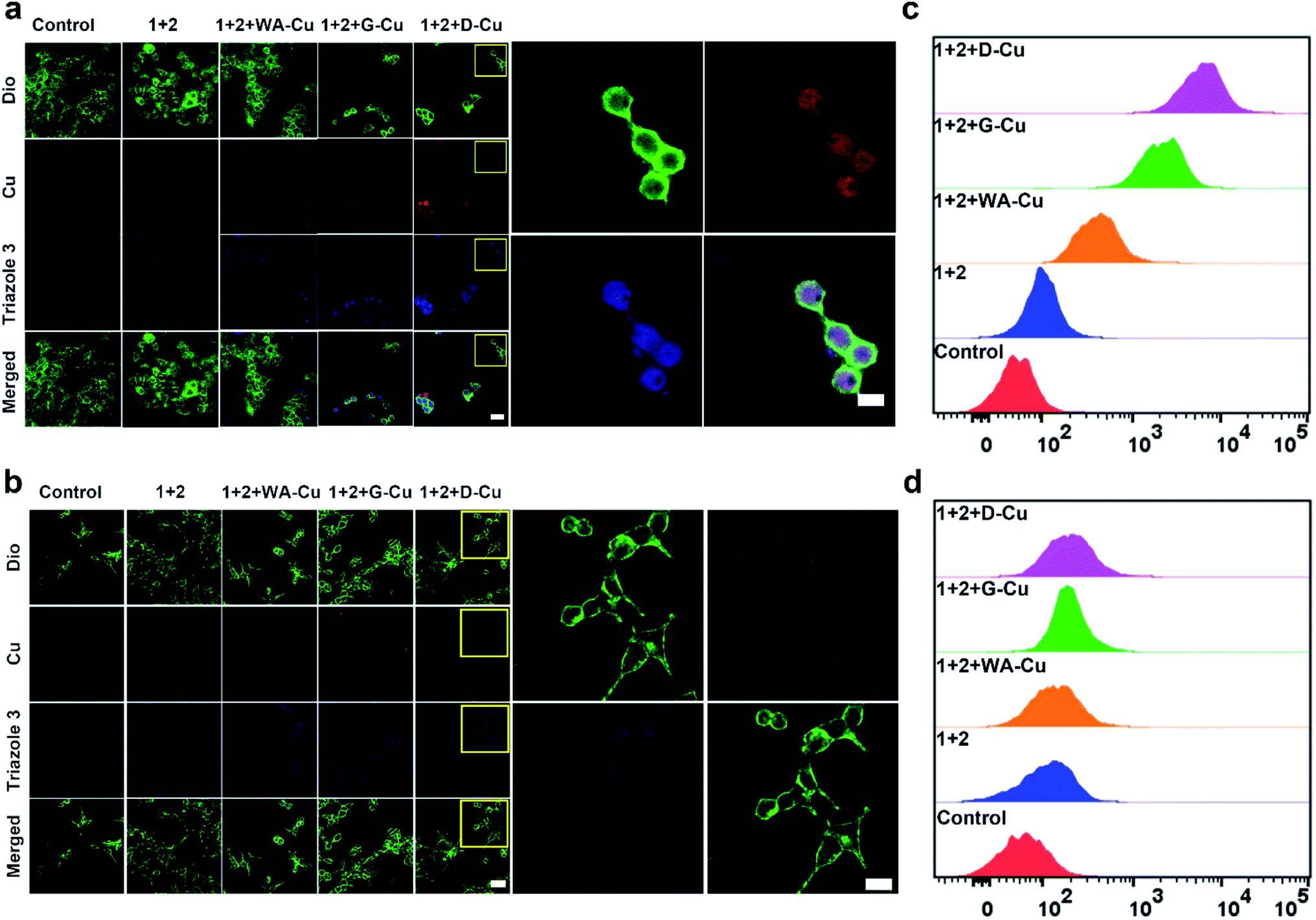 | ||
| Fig. 3 CuAAC reaction catalyzed by D-Cu in HeLa and HEK-293 cells. (a and b) Confocal microscopy images of HeLa and HEK-293 cells treated with different conditions. Scale bars: 50 μm in main images and 10 μm in high resolution images. (c and d) Flow cytometry assay of HeLa and HEK-293 cells treated with different conditions, respectively. | ||
Subsequently, we explored whether D-Cu could catalyze prodrugs 4 and 5 to produce the resveratrol (3,5,4′-trihydroxystilbene; Rsv) analog 6 (Fig. 4a), a model drug with antitumor activity. It is reported that the Rsv analog 6 can restrain tumor initiation, promotion, and progression via inhibiting the cyclooxygenase activity of COX-1.51,52 MTT assay showed that the prodrugs 4 and 5 had little cytotoxicity to HeLa and HEK-293 cells even at 50 μM (Fig. 4b and S23†). The half maximal inhibitory concentration (IC50) values of 4 and 5 against HeLa cells were 74.8, and 77.3 μM, respectively (Fig. S24†), indicating that the prodrugs were biocompatible at the concentration used in our experiment. After being treated with prodrugs 4 + 5 and G-Cu, the viability of HeLa cells decreased significantly compared with those treated with prodrugs 4 + 5. The viability further decreased when the cells were treated with prodrugs 4 + 5 and D-Cu (Fig. 4c). The IC50 value of prodrugs 4 + 5 and D-Cu against HeLa cells was 18.6 μM (Fig. S24†), indicating that the synthesized drug 6 had a strong cytotoxic effect. These results demonstrated that D-Cu could effectively catalyze the in situ synthesis of 6 to kill cancer cells. However, HEK-293 cells treated with prodrugs, prodrugs + G-Cu, and prodrugs + D-Cu showed a slight decrease in cell viability (Fig. 4d and S25†).
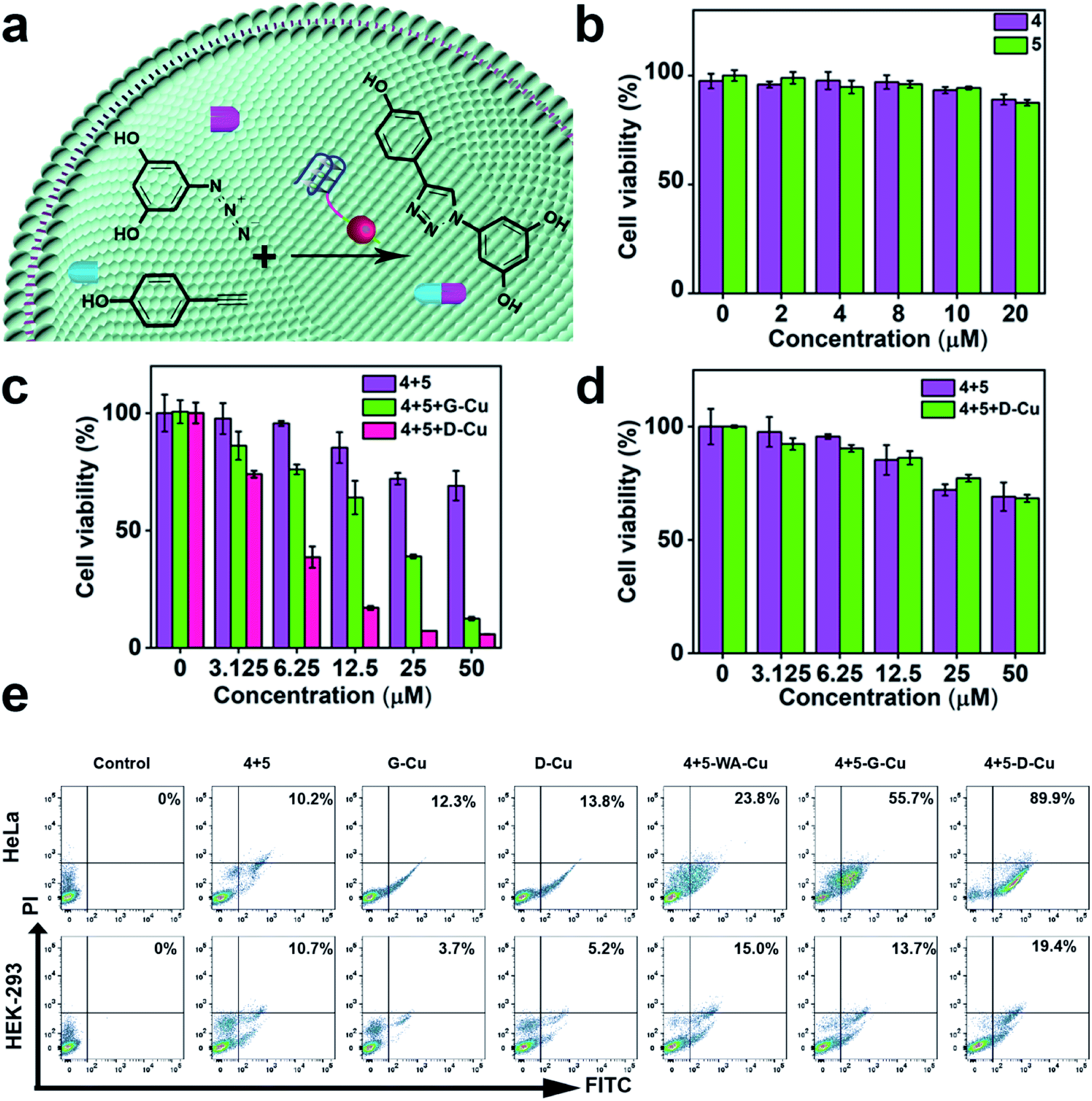 | ||
| Fig. 4 CuAAC reaction catalyzed by D-Cu in HeLa and HEK-293 cells for prodrug activation. (a) Schematic illustration of the CuAAC reaction catalyzed by D-Cu for intracellular prodrug activation. (b) Cytotoxicity of prodrugs on HeLa cells. (c) Cell viability of HeLa cells treated with 4 + 5, 4 + 5 + G-Cu and 4 + 5 + D-Cu, respectively. (d) Cell viability of HEK-293 cells treated with 4 + 5 and 4 + 5 + D-Cu, respectively. (e) Flow cytometry analysis of the apoptotic rates of HeLa and HEK-293 cells treated with different conditions, respectively. Data are presented as mean ± SD (n = 3). | ||
To further explore the self-augmented properties of D-Cu, we primarily detected the generation of ROS from the high local concentration of H2O2 in cancer cells using the DCFH (2,7-dichlorofluorescein) fluorescent probe. As shown in Fig. S26, D-Cu† could produce a large number of active radical species that boosted the transformation of Cu(0) into Cu(I). The cells were further examined using flow cytometry analysis by double staining. The apoptosis rate of HeLa cells was much higher than that of HEK-293 cells (Fig. 4e). The viability obviously decreased when the HeLa cells were treated with prodrugs and D-Cu. The results demonstrated that cancer cells could be killed by Rsv analogue 6 catalyzed by D-Cu and ROS produced by DNAzyme. Moreover, the synthesis of Rsv analogue 6 in cancer cells was confirmed by HPLC (Fig. S27†) and LC-MS analysis (Fig. S28†). The above research proved that D-Cu could promote the intracellular CuAAC reaction, enhancing anticancer efficacy in tumor cells and simultaneously minimizing the side-effects in normal cells.
We next monitored the catalytic performance of D-Cu in vivo. Caenorhabditis elegans (C. elegans) was used as a model to investigate the toxicology and phenotype alteration of D-Cu. After being treated with D-Cu, the N2 wild-type strain worms were totally paralyzed in 16 days, which was consistent with the control group (Fig. 5a and b), suggesting the biosafety of D-Cu in vivo.
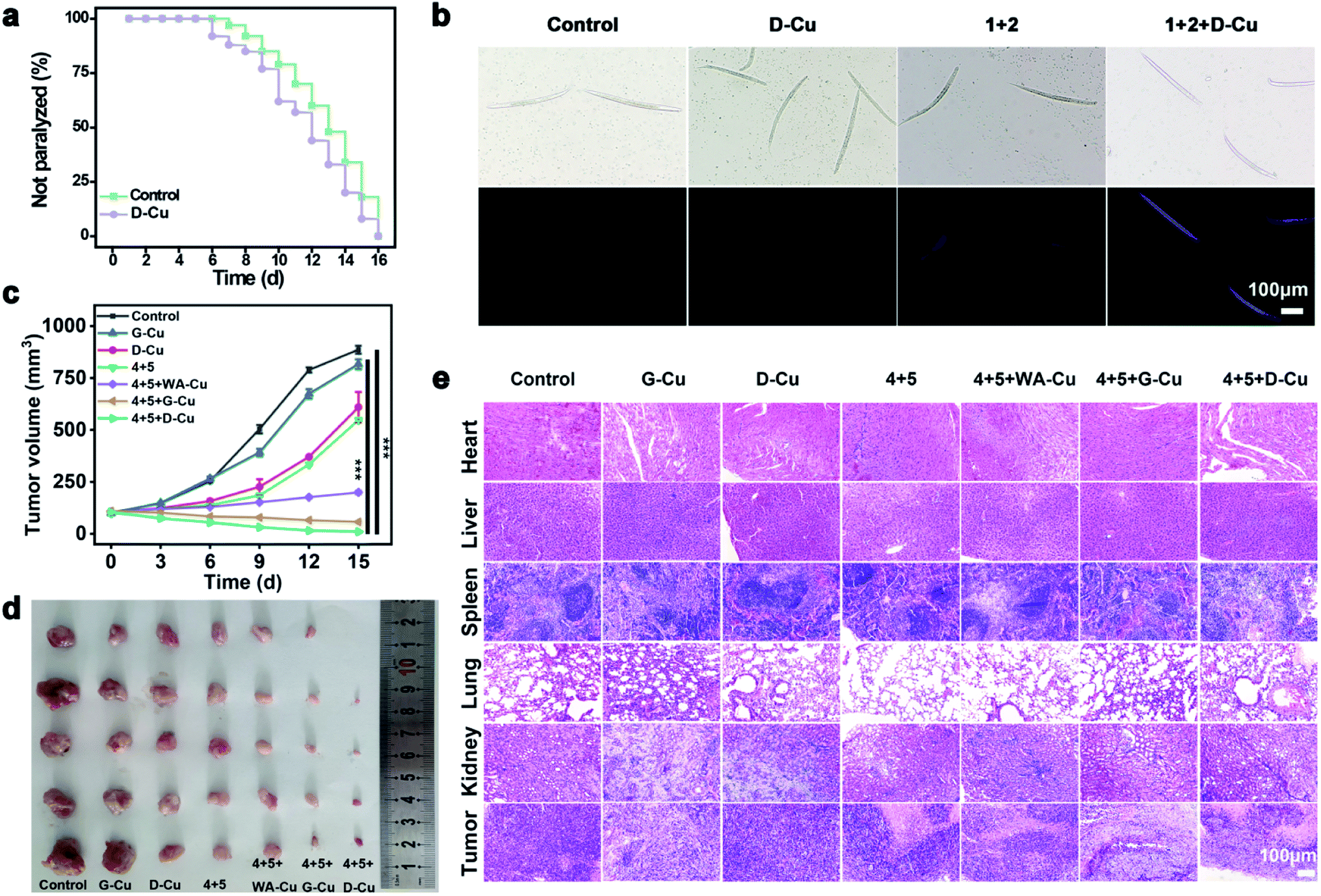 | ||
| Fig. 5 CuAAC reaction catalyzed by D-Cu in vivo. (a) Kaplan–Meier survival curves of worms treated with and without D-Cu. (b) Bright-field and fluorescence images (blue channel) of worms treated with different conditions. Scale bars: 100 μm. (c) Average tumor volume of the mice in different groups (n = 5). Student's t-test for significance analysis. P values: 0.00031 for HTANM treated with 4 + 5 + G-Cu vs. control; 0.000037 for HTANM treated with 4 + 5 + D-Cu vs. control (***P < 0.001) (d) excised tumors for different treatments on D14. (e) H&E-stained slices from different treatments. Scale bars: 100 μm. | ||
To systematically investigate the toxicology of D-Cu, hemolytic assay was performed. D-Cu showed little hemolysis even at a concentration of 50 μM (Fig. S29†). After that, the pharmacokinetics of D-Cu was investigated using HeLa tumor-bearing nude mice via ICP-MS analysis. The results showed that D-Cu had favorable accumulation and retention in the tumor region due to AS1411 with high specificity and affinity to nucleolin (Fig. S30†). Meanwhile, the growth of the mice was not affected (Fig. S31†). Furthermore, the results of hematological analysis, blood biochemical assay and hematoxylin and eosin (H&E) staining of major organs show that D-Cu was non-toxic, which favored its further biological applications (Fig. S32 and S33†). We further evaluated the therapy efficacy of D-Cu in vivo using the HeLa tumor-bearing athymic nude mouse model (HTANM). The HTANMmice were randomly divided into seven groups: (1) control; (2) G-Cu; (3) D-Cu; (4) 4 + 5; (5) 4 + 5 + WA-Cu; (6) 4 + 5 + G-Cu; and (7) 4 + 5 + D-Cu. The body weight (Fig. S34†) and tumor size (Fig. 5c) of mice in each group were monitored during the treatment. By comparing the excised tumor mass of each group, admirable antitumor efficacy was observed in group 7 (Fig. 5d). The H&E staining of tumor tissue revealed that the cancer cells in group 7 were extremely damaged. The apoptotic cells increased significantly, implying that the tumor was sufficiently suppressed (Fig. 5e). Taken together, based on targeting recognition and self-augmented capability, D-Cu could accumulate at the tumor site and produce ROS, which catalyzed drug synthesis in situ and directly kill cancer cells for synergistic cancer therapy.
Conclusions
In summary, a DNAzyme-augmented and targeted bioorthogonal catalyst was successfully designed and constructed for synergistic cancer therapy. Combining the peroxidase-mimicking activity of DNAzyme, specific recognition of the aptamer and DNA-templated CuNPs, the bioorthogonal catalysis system could be actively delivered to tumor cells and then produce ROS to convert the valence state of CuNPs for enhanced bioorthogonal drug synthesis and to kill cells directly. Compared with the common catalyst CuSO4/sodium ascorbate, the catalytic transformation of the system in the presence of H2O2 was increased by more than 20 times in 30 minutes. The system combined in situ drug synthesis with chemodynamic therapy, offering highly specific and efficient cancer therapy effects with minimal side effects. The study offers a simple and novel avenue to develop efficient bioorthogonal catalysts for biomedical applications.Data availability
Data are available in the ESI† online.Author contributions
Y. Y. designed and conceived the experiments, implemented the experiments, analysed the data and wrote the manuscript; H. L. and J. Z. implemented the experiments; Y. W. implemented the molecular dynamics simulation experiments; F. P., J. R. and X. Q. designed and conceived the experiments, supervised the study and revised the manuscript. All authors approved the final version.Conflicts of interest
All authors declare no competing interests.Acknowledgements
This work was supported by the National Key R&D Program of China (2021YFF1200701 and 2019YFA0709202), the National Nature Science Foundation of China (21871249, 21820102009, 91856205, and 21977091), the Key Program of Frontier of Sciences (CAS QYZDJ-SSW-SLH052), and the Jilin Province Science and Technology Development Plan Project (20200201241JC).Notes and references
- Z. Chen, H. Li, Y. Bian, Z. Wang, G. Chen, X. Zhang, Y. Miao, D. Wen, J. Wang, G. Wan, Y. Zeng, P. Abdou, J. Fang, S. Li, C. J. Sun and Z. Gu, Nat. Nanotechnol., 2021, 16, 933–941 CrossRef CAS PubMed
.
- C. Ritter, N. Nett, C. G. Acevedo-Rocha, R. Lonsdale, K. Kraling, F. Dempwolff, S. Hoebenreich, P. L. Graumann, M. T. Reetz and E. Meggers, Angew. Chem., Int. Ed., 2015, 54, 13440–13443 CrossRef CAS PubMed
- J. Clavadetscher, S. Hoffmann, A. Lilienkampf, L. Mackay, R. M. Yusop, S. A. Rider, J. J. Mullins and M. Bradley, Angew. Chem., Int. Ed., 2016, 55, 15662–15666 CrossRef CAS PubMed
- C. Wang, G. Jia, J. Zhou, Y. Li, Y. Liu, S. Lu and C. Li, Angew. Chem., Int. Ed., 2012, 51, 9352–9355 CrossRef CAS PubMed
- J. Geng, Y. Zhang, Q. Gao, K. Neumann, H. Dong, H. Porter, M. Potter, H. Ren, D. Argyle and M. Bradley, Nat. Chem., 2021, 13, 805–810 CrossRef CAS PubMed
- A. Unciti-Broceta, E. M. Johansson, R. M. Yusop, R. M. Sanchez-Martin and M. Bradley, Nat. Protoc., 2012, 7, 1207–1218 CrossRef CAS PubMed
- Y. Bai, X. Feng, H. Xing, Y. Xu, B. K. Kim, N. Baig, T. Zhou, A. A. Gewirth, Y. Lu, E. Oldfield and S. C. Zimmerman, J. Am. Chem. Soc., 2016, 138, 11077–11080 CrossRef CAS PubMed
- R. M. Yusop, A. Unciti-Broceta, E. M. Johansson, R. M. Sanchez-Martin and M. Bradley, Nat. Chem., 2011, 3, 239–243 CrossRef CAS PubMed
- C. Streu and E. Meggers, Angew. Chem., Int. Ed., 2006, 118, 5773–5776 CrossRef
- G. Y. Tonga, Y. Jeong, B. Duncan, T. Mizuhara, R. Mout, R. Das, S. T. Kim, Y. C. Yeh, B. Yan, S. Hou and V. M. Rotello, Nat. Chem., 2015, 7, 597–603 CrossRef CAS PubMed
- D. Wu, K. Yang, Z. Zhang, Y. Feng, L. Rao, X. Chen and G. Yu, Chem. Soc. Rev., 2022, 51, 1336–1376 RSC
- Q. Yao, F. Lin, X. Fan, Y. Wang, Y. Liu, Z. Liu, X. Jiang, P. R. Chen and Y. Gao, Nat. Commun., 2018, 9, 5032 CrossRef PubMed
- J. T. Weiss, J. C. Dawson, K. G. Macleod, W. Rybski, C. Fraser, C. Torres-Sánchez, E. E. Patton, M. Bradley, N. O. Carragher and A. Unciti-Broceta, Nat. Commun., 2014, 5, 3277 CrossRef PubMed
- D. J. Norman, A. Gambardella, A. R. Mount, A. F. Murray and M. Bradley, Angew. Chem., Int. Ed., 2019, 58, 14189–14192 CrossRef CAS PubMed
- Y. Dong, Y. Tu, K. Wang, C. Xu, Y. Yuan and J. Wang, Angew. Chem., Int. Ed., 2020, 59, 7168–7172 CrossRef CAS PubMed
- W. Wang, X. Zhang, R. Huang, C. M. Hirschbiegel, H. Wang, Y. Ding and V. M. Rotello, Adv. Drug Delivery Rev., 2021, 176, 113893 CrossRef CAS PubMed
- K. Liu, P. K. Lat, H. Z. Yu and D. Sen, Nucleic Acids Res., 2020, 48, 7356–7370 CAS
- J. Chen, J. Wang, Y. Bai, K. Li, E. S. Garcia, A. L. Ferguson and S. C. Zimmerman, J. Am. Chem. Soc., 2018, 140, 13695–13702 CrossRef CAS PubMed
- C. Vazquez-Vazquez, B. Vaz, V. Giannini, M. Perez-Lorenzo, R. A. Alvarez-Puebla and M. A. Correa-Duarte, J. Am. Chem. Soc., 2013, 135, 13616–13619 CrossRef CAS PubMed
- J. Lee, S. Dubbu, N. Kumari, A. Kumar, J. Lim, S. Kim and I. S. Lee, Nano Lett., 2020, 20, 6981–6988 CrossRef CAS PubMed
- M. R. Decan, S. Impellizzeri, M. L. Marin and J. C. Scaiano, Nat. Commun., 2014, 5, 4612 CrossRef CAS PubMed
- A. Sarkar, T. Mukherjee and S. Kapoor, J. Phys. Chem. C, 2008, 112, 3334–3340 CrossRef CAS
- Y. You, F. Cao, Y. Zhao, Q. Deng, Y. Sang, Y. Li, K. Dong, J. Ren and X. Qu, ACS Nano, 2020, 14, 4178–4187 CrossRef CAS PubMed
- F. Wang, Y. Zhang, Z. Liu, Z. Du, L. Zhang, J. Ren and X. Qu, Angew. Chem., Int. Ed., 2019, 58, 6987–6992 CrossRef CAS PubMed
- V. Sebastian, M. Sancho-Albero, M. Arruebo, A. M. Perez-Lopez, B. Rubio-Ruiz, P. Martin-Duque, A. Unciti-Broceta and J. Santamaria, Nat. Protoc., 2021, 16, 131–163 CrossRef CAS PubMed
- M. Sancho-Albero, B. Rubio-Ruiz, A. M. Perez-Lopez, V. Sebastian, P. Martin-Duque, M. Arruebo, J. Santamaria and A. Unciti-Broceta, Nat. Catal., 2019, 2, 864–872 CrossRef CAS PubMed
- W. Wang, K. Vellaisamy, G. Li, C. Wu, C. N. Ko, C. H. Leung and D. L. Ma, Anal. Chem., 2017, 89, 11679–11684 CrossRef CAS PubMed
- Y. You, Q. Deng, Y. Wang, Y. Sang, G. Li, F. Pu, J. Ren and X. Qu, Nat. Commun., 2022, 13, 1459 CrossRef CAS PubMed
- R. J. Lake, Z. Yang, J. Zhang and Y. Lu, Acc. Chem. Res., 2019, 52, 3275–3286 CrossRef CAS PubMed
- E. Golub, H. B. Albada, W. C. Liao, Y. Biniuri and I. Willner, J. Am. Chem. Soc., 2016, 138, 164–172 CrossRef CAS PubMed
- M. Vázquez-González, C. Wang and I. Willner, Nat. Catal., 2020, 3, 256–273 CrossRef
- Z. Zhang and J. Liu, Mater. Horiz., 2018, 5, 738–744 RSC
- Y. Cao, P. Ding, L. Yang, W. Li, Y. Luo, J. Wang and R. Pei, Chem. Sci., 2020, 11, 6896–6906 RSC
- S. Wang, Y. Chen, S. Wang, P. Li, C. A. Mirkin and O. K. Farha, J. Am. Chem. Soc., 2019, 141, 2215–2219 CrossRef CAS PubMed
- Z. Wang, J. Niu, C. Zhao, X. Wang, J. Ren and X. Qu, Angew. Chem., Int. Ed., 2021, 60, 12431–12437 CrossRef CAS PubMed
- P. Huang, D. Rochambeau, H. Sleiman and J. Liu, Angew. Chem., Int. Ed., 2020, 59, 3573–3577 CrossRef CAS PubMed
- B. Yu, W. Wang, W. Sun, C. Jiang and L. Lu, J. Am. Chem. Soc., 2021, 143, 8855–8865 CrossRef CAS PubMed
- H. Atsumi and A. M. Belcher, ACS Nano, 2018, 12, 7986–7995 CrossRef CAS PubMed
- H. Lee, J. Kim, J. Lee, H. Park, Y. Park, S. Jung, J. Lim, H. C. Choi and W. J. Kim, Biomaterials, 2020, 263, 120402 CrossRef CAS PubMed
- C. Liu, Y. Chen, J. Zhao, Y. Wang, Y. Shao, Z. Gu, L. Li and Y. Zhao, Angew. Chem., Int. Ed., 2021, 60, 14324–14328 CrossRef CAS PubMed
- Y. Sang, F. Cao, W. Li, L. Zhang, Y. You, Q. Deng, K. Dong, J. Ren and X. Qu, J. Am. Chem. Soc., 2020, 142, 5177–5183 CrossRef CAS PubMed
- B. Yang, Y. Chen and J. Shi, Adv. Mater., 2019, 31, 1901778 CrossRef PubMed
- A. F. Jou, Y. T. Chou, I. Willner and J. A. Ho, Angew. Chem., Int. Ed., 2021, 60, 21673–21678 CrossRef CAS PubMed
- W. Xuan, Y. Xia, T. Li, L. Wang, Y. Liu and W. Tan, J. Am. Chem. Soc., 2019, 142, 937–944 CrossRef PubMed
- Y. Yang, W. Zhu, L. Feng, Y. Chao, X. Yi, Z. Dong, K. Yang, W. Tan, Z. Liu and M. Chen, Nano Lett., 2018, 18, 6867–6875 CrossRef CAS PubMed
- W. Wei, X. He and N. Ma, Angew. Chem., Int. Ed., 2014, 53, 5573–5577 CrossRef CAS PubMed
- Q. Li, J. Zhao, L. Liu, S. Jonchhe, F. Rizzuto, S. Mandal, H. He, S. Wei, H. Sleiman, H. Mao and C. Mao, Nat. Mater., 2020, 19, 1012–1018 CrossRef CAS PubMed
- Z. Qing, X. He, D. He, K. Wang, F. Xu, T. Qing and X. Yang, Angew. Chem., Int. Ed., 2013, 52, 9719–9722 CrossRef CAS PubMed
- S. E. Feller, Y. Zhang, R. W. Pastor and B. R. Brooks, J. Chem. Phys., 1995, 103, 4613–4621 CrossRef CAS
- J. S. Reuter and D. H. Mathews, BMC Bioinf., 2010, 11, 129 CrossRef PubMed
- S. Fulda, Drug Discovery Today, 2010, 15, 757–765 CrossRef CAS PubMed
- J. A. Baur and D. A. Sinclair, Nat. Rev. Drug Discovery, 2006, 5, 493–506 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc02050e |
| This journal is © The Royal Society of Chemistry 2022 |