DOI:
10.1039/D3NH00061C
(Communication)
Nanoscale Horiz., 2023,
8, 1062-1072
Engineering magnetotactic bacteria MVs to synergize chemotherapy, ferroptosis and immunotherapy for augmented antitumor therapy†
Received
18th February 2023
, Accepted 22nd May 2023
First published on 12th June 2023
Abstract
One main obstacle to targeted cancer therapies is the immunosuppressive tumor microenvironment, which can facilitate tumor growth and induce resistance to antitumor treatments. Recent studies have indicated that treatment combined with immunotherapy often yields a better prognosis than monotherapy. Bacterial membrane vesicles (MVs), nanostructures released from the membrane of bacteria, can be used as natural nanocarriers for drug delivery and stimulate an immune response because of their immunogenicity. Inspired by the development of synergistic therapeutic strategies, we herein propose a novel nanovaccine-based platform to achieve chemotherapy, ferroptosis therapy, and immunotherapy simultaneously. By simply culturing magnetotactic bacteria in the medium with doxorubicin (DOX) and then extracting specialized MVs (BMVs), BMV@DOX, which are membrane vesicles containing iron ions and DOX, were obtained. We confirmed that in BMV@DOX, the BMV component can stimulate the innate immune system, DOX acts as the chemotherapeutic agent and iron ions will induce ferroptosis. Furthermore, BMV@DOX vesicles modified with DSPE-PEG-cRGD peptides (T-BMV@DOX) have minimized systemic toxicity and increased tumor-specificity. We demonstrated that the smart MVs-based nanovaccine system not only showed superior performance in the treatment of 4T1 breast cancer but also effectively restrained the growth of drug-resistant MCF-7/ADR tumors in mice. Moreover, the nanovaccine could abrogate in vivo lung metastasis of tumor cells in a 4T1-Luc cell induced-lung breast cancer metastasis model. Collectively, the MVs-based nanoplatform offers an alternative promise for surmounting the limitations of monotherapy and may deserve further study for application in synergistic cancer therapy.
New concepts
Among the emerging biomaterials for biomedical applications, there is an ongoing interest in the development of membrane vesicles (MVs) in tumor therapy due to several specific properties. In this research, we have utilized a special strain of Gram-negative bacteria, Magnetospirillum gryphiswaldense (MSR-1), which is nonpathogenic and possesses the ability to accumulate high concentrations of ferrous iron. Although the applications of such magnetotactic bacteria are extensively studied, limited work is available on the applied research of their derived MVs. Ferroptosis is a newly discovered cell death mechanism that is different from apoptosis and thus can be integrated with immunotherapy to overcome the drawbacks of conventional treatment modalities. With this, here we propose an eco-friendly strategy to obtain a magnetotactic bacteria MVs-based nanovaccine in which iron and antitumor drug DOX was encased in the MVs. We demonstrated that tumor cells were susceptible to ferroptosis and hence the proliferation could be inhibited not only for sensitive tumor cells but also for drug-resistant types after being challenged with such hybrid nanovaccine. Together with chemotherapy, the integration of immunotherapy with ferroptosis in drug-loaded MVs nanoplatform may surmount the challenges of monotherapy and augment the antitumor efficacy.
|
1. Introduction
Characterized by rapid growth, metastasis, and high mortality, cancer is one type of malignant disease that poses a great threat to human health.1 To combat cancer over the past decades, multiple therapeutic strategies have been developed for the treatment of tumors. Among them, chemotherapy, a traditional method, is one of the most universally used modalities in clinical trials, which works by inducing apoptotic cell death following exposure to chemotherapeutic drugs.2,3 Nevertheless, due to the risks of adverse side effects in patients and drug resistance in tumors, treatment by chemotherapy is frequently impeded. With an increased understanding of the immune system, cancer immunotherapy has emerged and sparked an intense interest in the development of immuno-based vaccines, including Alemtuzumab for chronic lymphocytic leukemia,4 and Imiquimod for basal cell carcinoma,5Dostarlimab for endometrial cancer,6 and so on. Despite the remarkable advances in recent years, the success rate and efficacy of most single immunotherapy modalities is usually hindered by the tumor complexity and heterogeneity, in particular the immunosuppression of the tumor microenvironment.7 Combined modality treatment, in which patients can receive immunotherapy after exhausting the chemotherapy or radiotherapy, for example, is a recently common therapy mode for solid tumors.8,9 Nevertheless, each modality was separated and operated independently in the same cycle of treatment. To overcome the drawbacks and boost the therapeutic efficacy of cancer, an emerging strategy involving the development of drugs that can combine immunotherapy with other conventional therapy modalities in one platform, has been explored and reported to be more effective in certain cases.10,11
Bacteria membrane vesicles (MVs), mainly produced and secreted by Gram-negative bacteria, are natural nanovesicles of 20–400 nm diameters with an outer leaflet of lipopolysaccharide (LPS) and wrapped with genetic substances, proteins, virulence factors, and transporters.12,13 There are multiple models for the formation mechanism of MVs, and most of them are generated by membrane blebbing when cell envelope disturbances occur during bacterial growth.13 Since the 1990s, Beveridge's team have conducted a series of research studies on the outer membrane vesicles of Pseudomonas aeruginosa and found that they have effective microbicidal activity and possess the potential to be drug nanocarriers.14–16 Furthermore, with the abundant presence of pathogen-associated molecular patterns, MVs are found to exhibit the ability to activate the innate immune system, thus offering an option for the development of an immune-based nanovaccine platform with the integration of other antitumor therapy modalities, which would overcome the challenges of tumor-induced suppression and drug-resistance, and thus amplify the anti-tumor effectiveness.17–22 Ferroptosis, a new type of non-apoptotic regulated cell death first discovered by Brent Stockwell in 2012, resulted from the process of iron-dependent lipid peroxidation.23 Interestingly, there have been studies implicating that activation of the ferroptosis pathway was able to not only enhance the chemosensitivity of drugs, especially in drug-resistant cancer cells, but also evoke immunogenic cell death (ICD) and magnify the immune response.24–27 The immune response, in turn, would participate in the ferroptosis pathway.28 Hence, the integration of iron-dependent ferroptosis with the immunomodulatory strategy would induce a novel synergistic way to treat tumors that are not sensitive to traditional apoptosis-induced treatments.
Herein, we proposed for the first time to simply construct a versatile nanoplatform based on Magnetospirillum gryphiswaldense MSR-1 bacteria membrane vesicles (BMVs). As one of the magnetotactic bacterium (MTB) strains, MSR-1 bacteria are nonpathogenic Gram-negative bacteria with the ability to absorb iron ions from the environment through active transportation and form biogenic magnetite.29–31 MSR-1 bacteria have been studied a lot because they are environmentally and biologically safe, and easy to isolate and culture in the lab. Although the applications of MSR-1 bacteria and magnetosomes are extensively studied, limited work is available on the applied research of membrane vesicles derived from MSR-1. In our previous work, we successfully constructed doxorubicin (DOX)-internalized bacteria swimmers by incubating bacteria with DOX in the growth medium. Considering the immunogenicity, the drug-internalized swimmers were administrated by direct peritumor injection and the desired therapeutic effect was achieved.32 Inspired by the fact that antibiotics could trigger vesicle formation,33,34 we studied the secreted fraction of the bacteria, successfully extracted membrane vesicles, and confirmed that DOX and ion irons were enclosed in the BMVs. The associations between the immune response and ferroptosis as mentioned above motivated us to develop the bacteria membrane vesicle-based nanovaccine that synergizes multiple treatment modalities. As shown in the conceptual diagram, DOX, a potent anthracycline type of chemotherapy aiming to treat a wide range of solid tumors, was selected for the construction of the MVs-based cancer therapy system. As a type of anthracycline antibiotic, DOX is a good iron chelator and the chelate formation can be used in drug delivery systems.35 By incubating MTB with DOX in the growth medium, the complexation between ferrous iron and DOX in the vesicles was obtained and DOX-Fe chelate-loaded MVs (BMV@DOX) were isolated. Furthermore, to endow the vesicles with tumor specificity and alleviate the systemic toxicity, BMV@DOX was functionalized with DSPE-PEG5000-cRGD peptides to form a T-BMV@DOX nanovaccine with tumor-targeting capacity (Scheme 1a). Beyond offering the intravenous administration, the T-BMV@DOX was able to actively target tumor tissue of interest, then exerted synergistic antitumor effects. To be specific, the immunostimulatory components from T-BMV@DOX would stimulate the dendritic cell (DC) maturation, which would subsequently activate effector T cells and switch on the antitumor immune responses to kill tumor cells. During the process of proliferation, T cells also produced interferon-gamma (IFN-γ), the upregulation of IFN-γ tended to trigger a burst of oxidative activity in System Xc− (a key regulatory target of ferroptosis) and the lipid peroxidation was enhanced, thus arousing immuno-induced ferroptosis. At the same time, DOX was expected to release and trigger apoptotic cell death by inhibiting the replication of DNA. Besides, the overloading of ferrous ions directly induced the occurrence of ferroptosis, accompanied by the surge of reactive oxygen species (ROS). Together with DOX-induced chemotherapy, the ROS-based ferroptosis mutually promoted ICD by upregulating the cellular expression of CRT, in turn promoting the anticancer immune process (Scheme 1b). To the best of our knowledge, few have reported the utilization of MTB-derived MVs as a tumor-targeting nanocarrier for immuno-based synergistic cancer therapy applications.
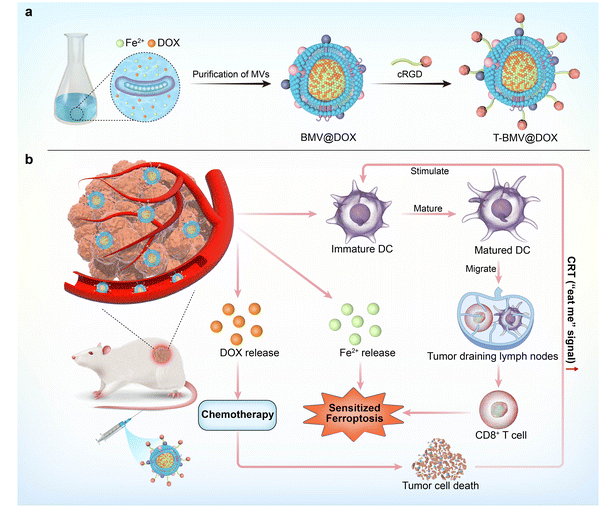 |
| | Scheme 1 Schematic illustration of the magnetotactic bacteria MVs-based nanoplatform. (a) The production of magnetotactic bacteria MVs. (b) Proposed mechanism for enhanced antitumor responses of MVs by synergizing chemotherapy, ferroptosis, and immunosuppression after intravenous injection into tumor-bearing mice. | |
2. Results
2.1. Characterization of BMV@DOX
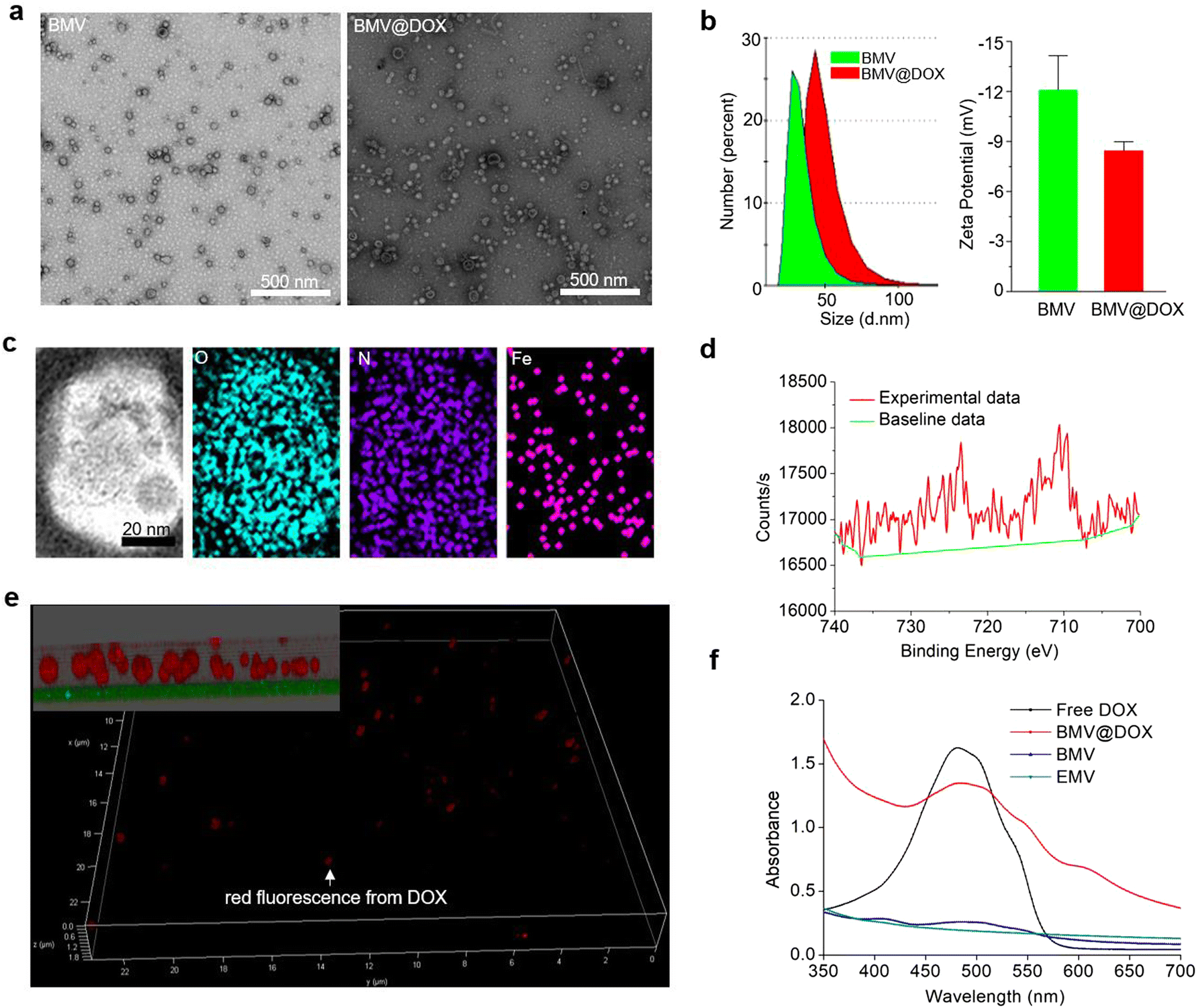
We isolated extracellular membrane vesicles (MVs) from the magnetotactic bacteria, Magnetospirillum gryphiswaldense (MSR-1) with the detailed procedure provided in the experimental section. Membrane vesicles extracted from MSR-1 bacteria growing in the original ferrous acetate-supplied LAY medium36 were named BMVs. For the preparation of drug-loaded MVs, as described in the experimental section, DOX at a final concentration of 15 μg mL−1 was added to the medium 24 h after the inoculation of MSR-1. After continuing the culture for another 48 h, the extracellular membrane vesicles with DOX-loading were extracted with the same method and named BMV@DOX. As shown in the transmission electron microscopy images (Fig. 1a), MVs derived from MSR-1 bacteria showed spherical structures with a size of around 20–50 nm in diameter, which was similar to other membrane vesicles derived from Gram-negative bacteria that were reported in previous studies.37 The dynamic light scattering (DLS) revealed that the hydrodynamic particle size distribution of BMVs centered at 20 nm, whereas an increased size was observed in BMV@DOX. Additionally, the average zeta potential values of BMV and MV@DOX were found to be −12.07 mV and −8.44 mV (Fig. 1b), which is reasonable because of the contribution of the positively charged DOX to BMV@DOX. The increased size and zeta potential of MVs should be attributed to the encapsulation of DOX inside the vesicles.
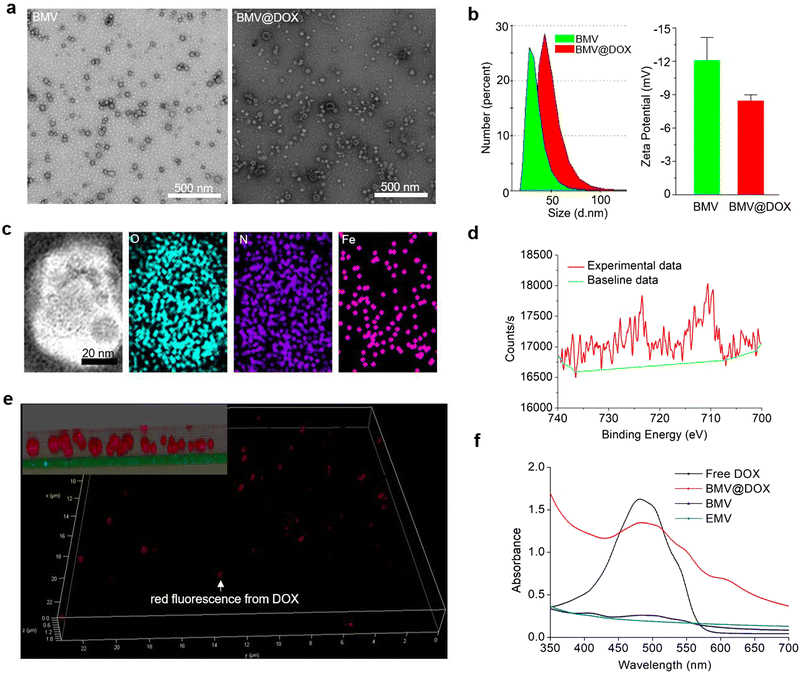 |
| | Fig. 1 The characterization of drug-loaded MVs. (a) The transmission electron microscopy images of MVs with and without DOX loading. Scale bar: 500 nm. (b) DLS measurements of size (left image) and zeta potential (right image) of MVs. (c) Scanning transmission electron microscopy image and corresponding elemental mapping of BMV@DOX. Scale bar: 20 nm. (d) Fe2p X-ray photoelectron spectroscopy spectra of BMV@DOX. (e) Super-resolution STED microscopy images of BMV@DOX. Red fluorescence: DOX. Inset in the top left corner: magnified z-stacked image. (f) The comparison of UV absorption for free DOX, BMV@DOX, BMV, and EMV. | |
Next, we analyzed the composition of MVs using the elemental mapping performed by an energy dispersive spectrometer. Fig. 1c reveals a massive distribution of oxygen and nitrogen elements, probably due to a large number of proteins in both BMVs and BMV@DOX. Notably, a significant amount of iron signal was also detected in the vesicles, confirming the existence of iron element in BMV@DOX. X-ray photoelectron spectroscopy was used to investigate the valence state of iron. As revealed in Fig. 1d, the absorption peak near 709.5 eV suggested the prescence of positive divalent iron ions. Additionally, an MR imaging study was performed by scanning the BMV@DOX with concentration gradients using a T2-weighted MR imaging system. The transverse relaxivity, the slope of transverse relaxation rate versus the BMV@DOX concentration (quantified as DOX), was 113 μg μL−1 s−1 (Fig. S1, ESI†), indicating enhanced contrast in T2-weighted MRI imaging. The presence of Fe-DOX inside the MVs may be the substance to prompt the MR imaging capacity of BMV@DOX.38,39
To further confirm the internalization of DOX to MVs, super-resolution STED confocal microscopy was used to examine BMV@DOX samples. Fig. 1e illustrates the distribution of the fluorescence signal of DOX in a 3-dimensional mode. As can be seen in the zoomed-in view (inset in the top left corner), a high intensity of DOX signals was observed in the whole spherical vesicles, implying that the DOX was equally distributed inside the MVs. Meanwhile, empty membrane vesicles (referred to as EMV), which were vesicles without iron ions or drugs, were extracted from bacteria and prepared as the control for UV-vis absorption analysis. Additionally, BMVs, which contain iron ions but no DOX, and free DOX as the positive control were also prepared for comparison. As shown in Fig. 1f, the UV-vis spectrum showed a characteristic absorption peak around 480 nm for both free DOX and BMV@DOX, while no obvious featured peak was observed in the EMV and BMV samples. Combining the above results, we have successfully obtained BMV@DOX with both DOX and iron ions enclosed in the membrane vesicles. With the assistance of Nanosight technology, per milligram of protein the BMV@DOX suspensions were determined to be composed of 1.39 × 1011 vesicle particles, in which 0.45 mg DOX and 0.21 mg iron were contained.
2.2. Cellular uptake
First, the primary cytometric analysis confirmed the cellular uptake of BMV@DOX following a 24 h incubation (Fig. S2, ESI†). To enhance the tumor-targeting efficiency of vesicles, the DSPE-PEG5000-cRGD peptide, a diblock polymer (DSPE-PEG5000) linked to the tumor-targeting peptide (cRGD) was used to modify MVs by physical coextrusion. Specifically, the BMV@DOX was functionalized with DSPE-PEG5000-cRGD, resulting in T-BMV@DOX vesicles. Such peptide functionalization was employed to impart better properties to the MVs. The formation of a polyethylene glycol (PEG) shell on the surface of the MVs was to increase the blood circulation time and overcome the systemic inflammatory responses. Once administered systemically, the T-BMV@DOX is expected to be enriched in the tumor tissue via active targeting and activate the innate immune system. Firstly, we compared the cellular uptake of BMV@DOX and T-BMV@DOX by first labeling MVs with Cy5 via the amidation reaction as described in the experimental section. Then, BMV@DOX-Cy5 and T-BMV@DOX-Cy5 were incubated with 4T1 cells for different time lengths and imaged by confocal laser scanning microscopy (CLSM). The acquired CLSM images in Fig. 2a demonstrated that cells incubated with T-BMV@DOX-Cy5 have significantly stronger Cy5 and DOX signals than those treated with BMV@DOX-Cy5 after 3 h and 5 h incubations, suggesting that T-BMV@DOX vesicles can be more efficiently internalized into cells than the unmodified vesicles.
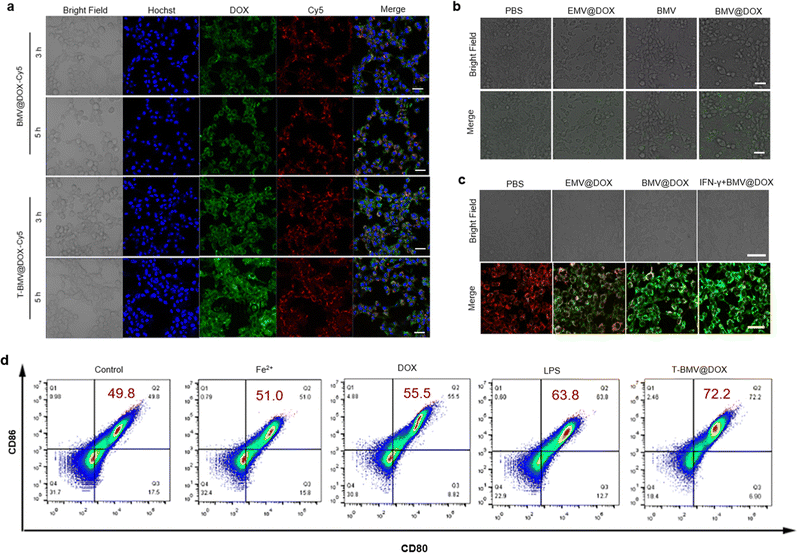 |
| | Fig. 2
In vitro cell experiment results of the MVs. (a) The uptake of BMV@DOX-Cy5 and T-BMV@DOX-Cy5 by 4T1 cells in vitro. Scale bars: 40 μm. (b) CLSM images of ROS levels in 4T1 cells with different treatments. Scale bars: 40 μm. (c) CLSM imaging of mitochondrial membrane potential in 4T1 cells treated with various samples. Scale bars: 40 μm. (d) Flow cytometry detection of maturation of DC cells after co-stimulation with different samples. The percentages of CD80 and CD86 double-positive cells are displayed in red color on the top right quadrant of each graph. | |
2.3. Verification of the ferroptosis procedure
As a special type of programmed cell death, ferroptosis is characterized by the accumulation of ROS and lipid peroxidation.40 When ROS react with cellular lipids, the formation of lipid hydroperoxides can be induced. The 2,7-dichlorodihydrofluorescein diacetate (DCFH-DA) method is commonly used to detect the levels of intracellular ROS in the cells from various treatment groups. Briefly, the probe can diffuse into cells, be hydrolyzed into DCFH by cytoplasmic esterase, and further oxidized to DCF by ROS with the emission of green fluorescence. As presented in the CLSM images (Fig. 2b), a significant amount of DCF signals were observed in BMV@DOX-treated cells, while the fluorescence in EMV@DOX and BMV was weaker, indicating that the iron ions and DOX derived from the MVs induce ROS generation, consistent with the “ROS and iron hypothesis”.41,42
Furthermore, ROS accumulation can also cause mitochondria membrane oxidation, leading to the hyperpolarization of mitochondrial membrane potential (MMP). Therefore, ferroptosis can be assessed using a JC-1 probe, which can evaluate the MMP levels, with a red fluorescence signal indicating healthy mitochondria, and green fluorescence indicating impaired mitochondrial function.43 As shown in Fig. 2c, the MMP levels of cells in the BMV@DOX were highly negative compared with that in the PBS and EMV@DOX treated groups, with the EMV@DOX showing less impaired mitochondria, and PBS-treated groups being normal. These results are consistent with the DCFH-DA assay. Furthermore, Kim et al. have discovered that the injection of immunogenic outer membrane vesicles could promote IFN-γ production in tumor microenvironments.25 Additionally, a study by Zou et al. has found that IFN-γ, a major effector secreted by natural killer cells (NK) cells, can sensitize and promote ferroptosis in tumor cells.28 Therefore, IFN-γ may function as a hub between immune signaling and ferroptosis pathways.44 To test this hypothesis, in vitro experiments involving IFN-γ-induced ferroptosis were conducted. In this part of the experiment, purified IFN-γ was purchased and used to perform the simulations of cytokines released by effector cells. As revealed in Fig. 2c, treatment with IFN-γ promoted the hyperpolarization of MMP levels in 4T1 cells. Taken together, BMV@DOX vesicles can induce ferroptosis by the loaded DOX and iron ions; in addition, it is feasible that MVs can sensitize immune-based ferroptosis in the tumor environment through the activation of the IFN-γ signaling pathway.
2.4.
In vitro DC mutation
DCs are crucial antigen-presenting cells derived from bone marrow. Bacterial membrane vesicles have previously been known to stimulate DC maturation due to the presence of LPS.45 To examine whether BMVs trigger the maturation of DCs, a flow cytometry study was performed to analyze the DC markers of cells exposed to T-BMV@DOX, DOX, Fe2+, LPS, and PBS for 24 h, whereas LPS and PBS served as the positive and negative controls. Co-stimulatory molecules CD80 and CD86 are known markers for mature DC cells, which are upregulated during the maturation process. As shown in Fig. 2d, the percentage of CD80/CD86 double-positive cells was shown in the upper right quadrant. Notably, both the Fe2+ and DOX treatment moderately promote the maturation of DC cells compared with that of the PBS group, while the T-BMV@DOX treated sample has more double-positive cells than LPS (72.2% and 63.8%, respectively), suggesting that the effect of T-BMV@DOX in DC maturation may be better than the positive control LPS.
2.5.
In vivo tumor targeting assessment
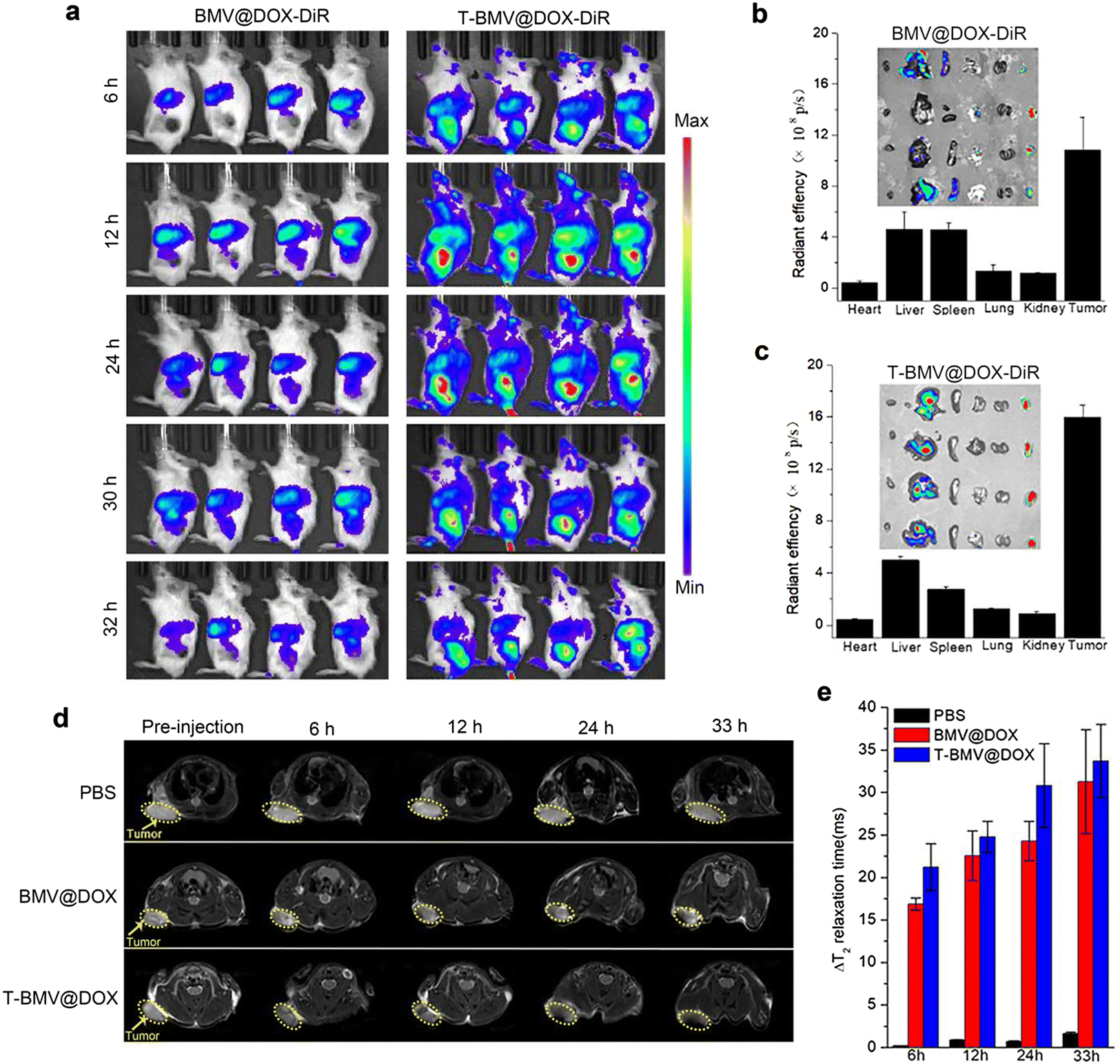
The above study has verified the targeting efficacy of DSPE-PEG-cRGD modified T-BMV@DOX and its synergistic effects in vitro, including ferroptosis and immune stimulation. We next employed in vivo imaging experiments to investigate the tumor-targeting ability of T-BMV@DOX in mice. As a fluorescent dye featuring low autofluorescence and high tissue penetration, DiR is appropriate for imaging in living animals. Hence, the in vivo biodistribution was analyzed by first labeling BMV@DOX and T-BMV@DOX with DiR dye and then injecting them into the 4T1 tumor-bearing mice intravenously. At 6 h post-injection, significant fluorescence was observed in the tumor site of the T-BMV@DOX-DiR treated mice, with the highest peak at approximately 12 h and lasting up to 30 h after injection. The fluorescence was not obvious in the tumors for mice injected with BMV@DOX. The results revealed that owing to the deposition of tumor-targeting cRGD peptide and PEG shell on the surface of the vesicles, T-BMV@DOX exhibited prominently improved tumor targeting ability as well as long circulation in mice bodies as compared to BMV@DOX vesicles (Fig. 3a). After dissection, isolated tumors and organs of mice in the two groups were harvested, and their quantitative distribution was consistent with corresponding ex vivo fluorescence images (Fig. 3b and c). In addition, in vivo T2-weighted MR imaging was also performed to evaluate the targeting behavior of T-BMV@DOX. As the transverse plane images presented (Fig. 3d), in comparison with the PBS and BMV@DOX treated groups, darker signals were observed in the specific tumor sites of mice receiving T-BMV@DOX injection. Quantification of the T2 relaxation time from the tumor sites at the indicated time points suggested that T-BMV@DOX vesicles exhibited a better T2-weighted MRI effect (Fig. 3e). Following the imaging experiments, the tumor tissues from mice in each group were embedded and sectioned for staining purposes. The Prussian blue staining and fluorescence imaging results confirmed the enrichment of iron and DOX in the tumors of T-BMV@DOX treated mice (Fig. S3, ESI†). Collectively, the accumulation capability in the tumor tissue of MVs in T-BMV@DOX treated mice was better than that in the BMV@DOX treated group.
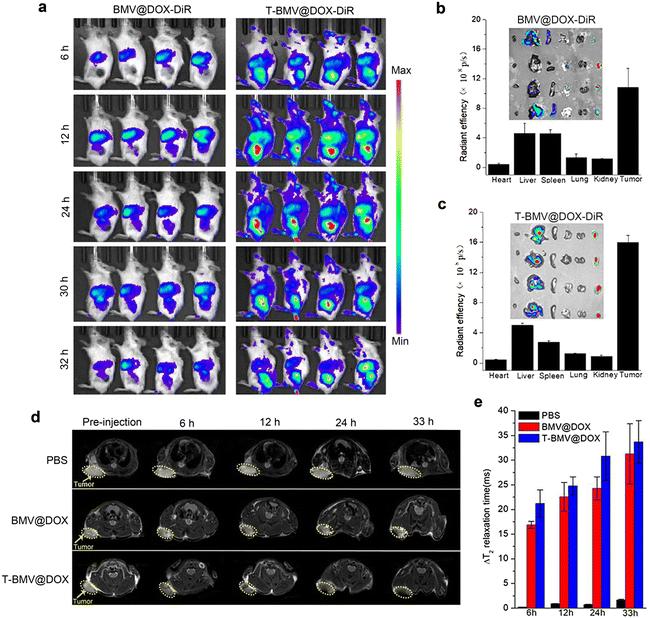 |
| | Fig. 3
In vivo tumor targeting assessment of MVs. (a) Whole-body fluorescence images for the mice injected with BMV@DOX-DiR and T-BMV@DOX-DiR (n = 4). (b) Ex vivo fluorescence intensity and images of isolated organs in BMV@DOX-DiR and (c) T-BMV@DOX-DiR treated mice. (d) T2-weighted MR images with the tumor sites circled by yellow dashed lines and (e) quantification of changes in T2 relaxation time in the tumors of PBS, BMV@DOX, and T-BMV@DOX treated mice (n = 4). | |
2.6.
In vivo immune response assessment
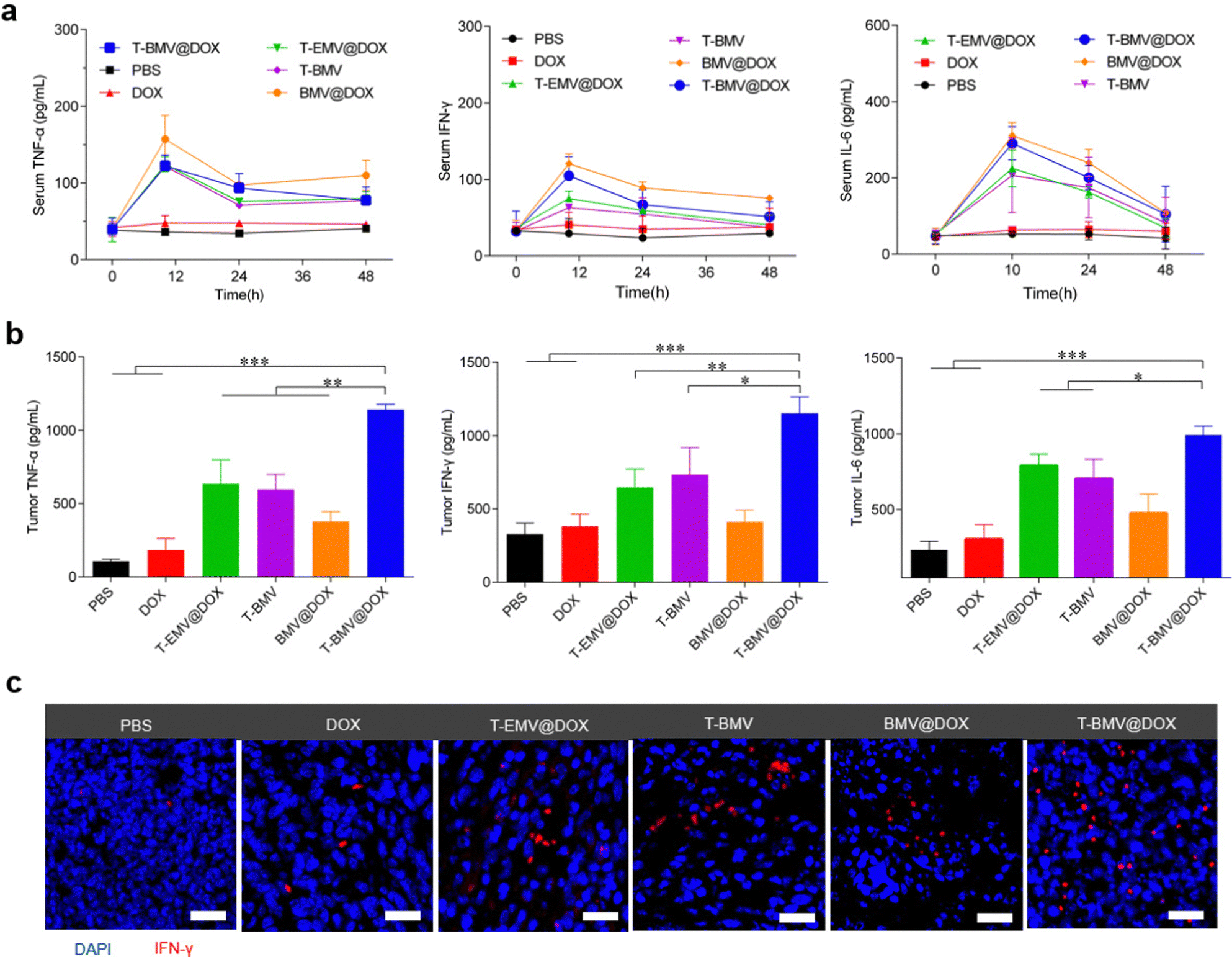
Before the anti-tumor study, firstly, an assessment of the levels of inflammatory factors in the 4T1 tumor-bearing BALB/c mice administrated with PBS, DOX, T-EMV@DOX, T-BMV, BMV@DOX, and T-BMV@DOX was performed. After receiving a designated dose of membrane vesicles (1 mg DOX kg−1) via tail vein injection as described in the experimental section, the serum levels of inflammatory factors including TNF-α, IFN-γ and IL-6 fluctuated significantly during the first 24 h and then returned to normal after 48 h (Fig. 4a), suggesting a relatively mild systemic inflammatory response, which may be attributed to the PEG modification. In terms of the tumor sites, strong levels of cytokine secretion were observed in the T-BMV@DOX group (Fig. 4b), which may boost antitumor immunity. The results confirmed that obvious local immune responses occurred in the MVs-treated tumor samples, with T-BMV@DOX treated tumors stronger due to the more effective targeting ability resulting in a higher amount of MVs accumulation. Meanwhile, DOX itself barely induced detectable TNF-α, IFN-γ and IL-6 expression in the tumor. The immunofluorescence image of tumor sections was consistent with the above observation (Fig. 4c).
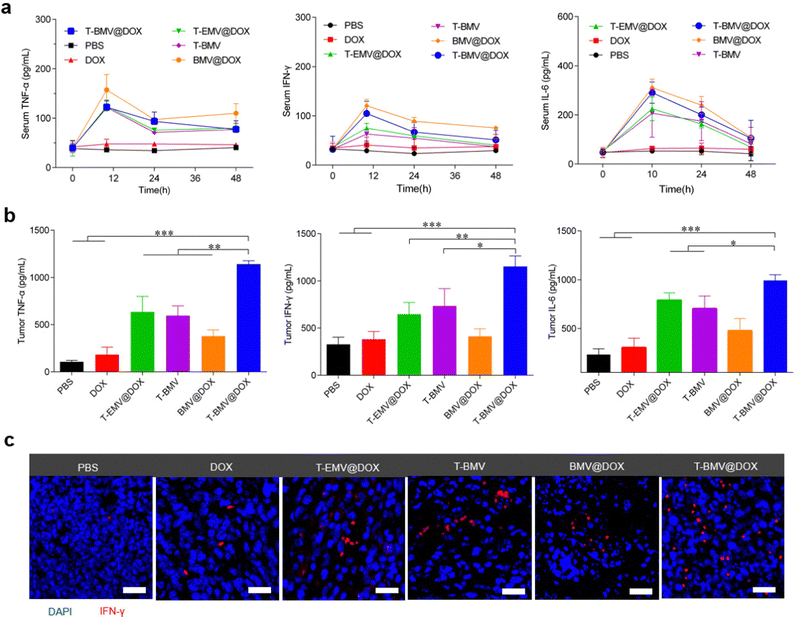 |
| | Fig. 4
In vivo immune responses of mice with the treatments of MVs. (a) Inflammatory factor levels of serum and (b) tumor region of mice treated with PBS, DOX, T-EMV@DOX, T-BMV, BMV@DOX, and T-BMV@DOX (n = 3). (c) Immunofluorescence images of tumor tissue sections of the mice with different treatments. DAPI is shown in blue and IFN-γ is shown in red. Scale bars: 20 μm. | |
2.7. Therapeutic effect on the 4T1 tumor model
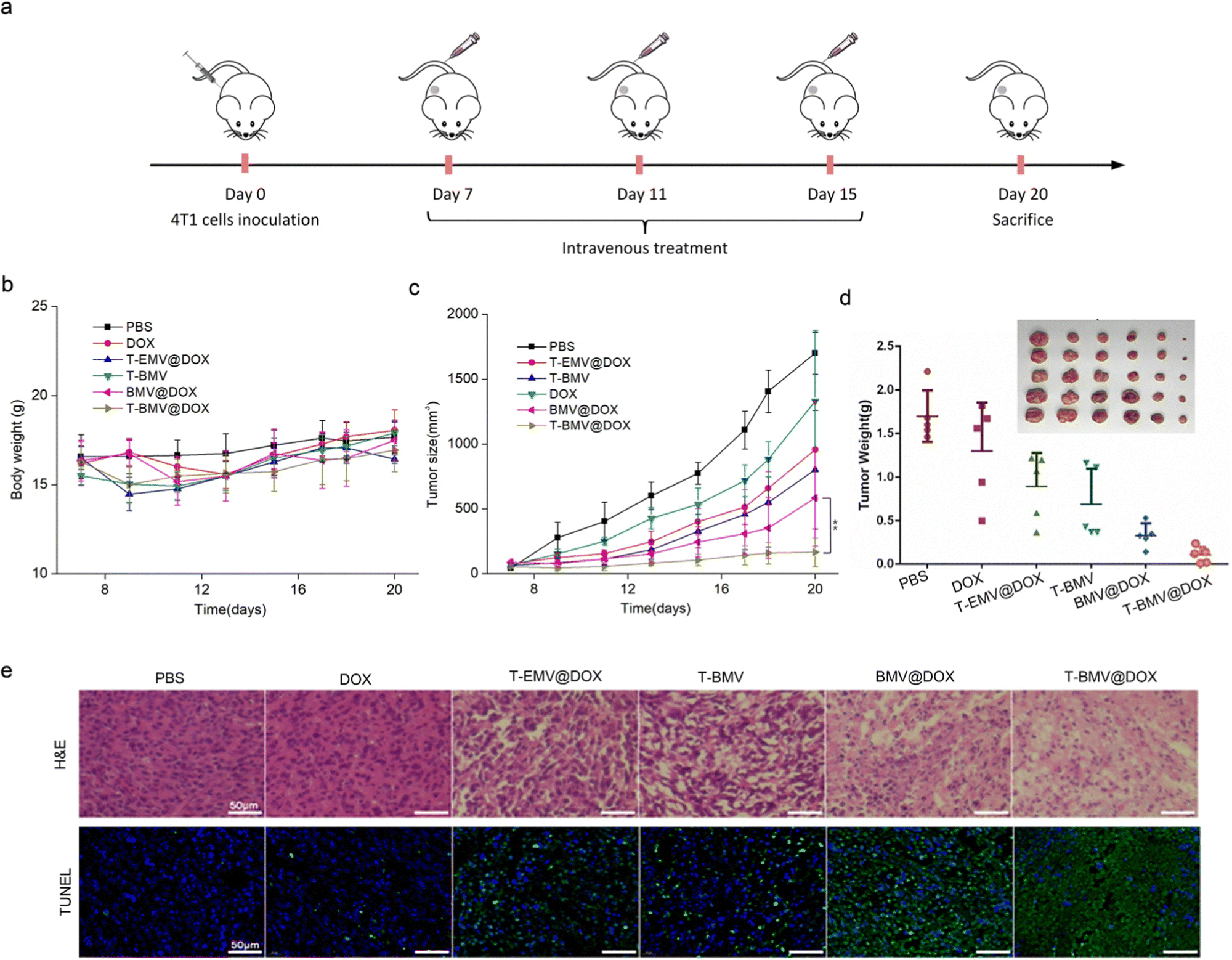
In the anti-tumor study performed using 4T1 tumor-bearing BALB/c mice model, doses of samples were injected intravenously and administered at 3-day intervals (Fig. 5a). Throughout the treatment, no significant loss of the body weight of mice was observed in various groups (Fig. 5b), whereas the tumor volumes in T-BMV@DOX treated mice were significantly smaller than those in other groups (Fig. 5c), including the control group of PBS, DOX, T-EMV@DOX (MVs without iron ions), T-BMV (MVs without DOX), and BMV@DOX (MVs without targeting peptides). Taken together, the results proved the in vivo augmented antitumor therapy due to the synergized effect of active targeting, chemotherapy, ferroptosis, and immunotherapy. At the end of the experiment, the tumor tissues in each group were removed, photographed, and weighed. The tumor weights were consistent with their measured sizes (Fig. 5d). Hematoxylin and eosin (H&E) and terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) staining of tumor sections revealed that the mice with T-BMV@DOX administration induced the highest levels of necrosis and apoptosis of tumor cells when compared with other groups (Fig. 5e). In addition, no apparent pathological damage and abnormality was found in the main organs in each group (Fig. S4, ESI†), indicating good biocompatibility of all of the MV samples.
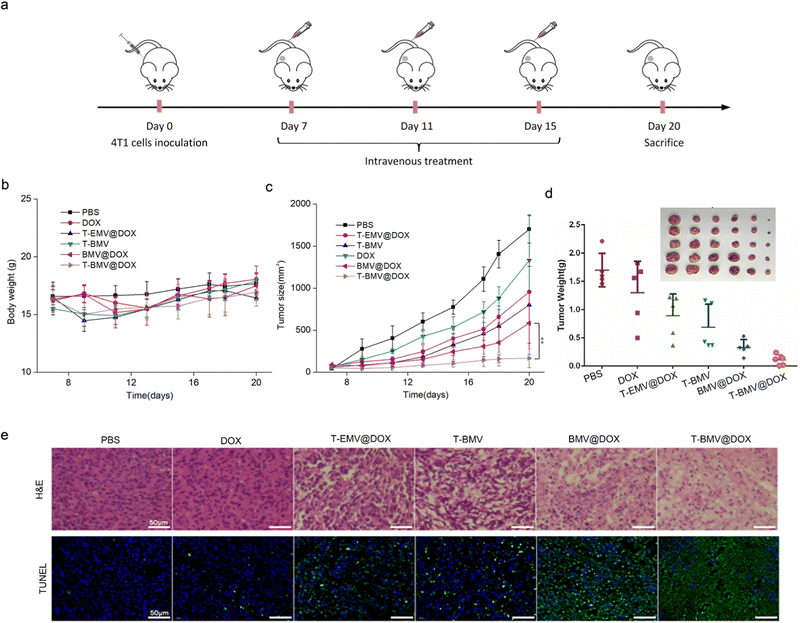 |
| | Fig. 5 Therapeutic study of MVs in 4T1 tumor-bearing mice. (a) The treatment flowchart of MVs against 4T1 breast cancer. (b) Body weight and (c) tumor size change during the treatment (n = 5). (d) Tumor weights of mice in each group after the treatment (n = 5). (e) The H&E and TUNEL images of the isolated tumors from mice after therapy. Scale bars: 50 μm. | |
Moreover, to investigate the potency of drug-loaded MVs in overcoming drug resistance to traditional chemotherapy, we studied the therapeutic effect of the MVs in drug-resistant cell model using the DOX-resistant breast cancer cells MCF-7/ADR. The in vitro cell toxicity assay demonstrated that MCF-7/ADR cells remained insensitive to DOX at the concentration range below 0.5 μg mL−1. In contrast, BMV@DOX and T-BMV@DOX displayed significantly higher killing potency while T-BMV@DOX was the best (Fig. S5, ESI†), which again may be due to ferroptosis plus immunogenic effects of the MVs derived from MTB. Afterward, the MCF-7/ADR tumor-bearing female BALB/c nude mice were administrated with PBS, DOX, and T-BMV@DOX intravenously by tail vein injection. The procedure was repeated every 3 days and lasted for 2 weeks. The representative images of tumors from mice in each group showed that tumor sizes from mice treated with T-BMV@DOX were consistently smaller than those treated with PBS and DOX. The superior anti-tumor efficacy of T-BMV@DOX in the drug-resistance model (Fig. S6 and S7, ESI†) should be attributed to both the tumor-targeting ability of T-BMV@DOX, ferroptosis, and immune stimulation. Nude mice were used in this experiment because it was hard to make MCF-7 tumor-bearing mice model using regular BALB/c mice. BALB/c nude mice show the robust activity of NK cells, B cells, and non-specific T cells, which compensates for thymus-dependent deficiencies in T lymphocyte function.46,47 They may work through non-specific immune mechanisms to resist tumor development. To confirm this, we performed immunohistochemistry and immunofluorescence staining of tumors. High-mobility group box 1 (HMGB1) is a protein that binds to DNA and initiates the transcription of various inflammatory markers, as well as plays a key role in immunity responses by interacting with the Toll-like receptor 4.48 Together with calreticulin (CRT), a distinct biomarker of immunogenic cell death, exposure to HMGB1 could activate DCs and further assist NK cells in eliminating tumors.49Via the immunohistochemistry and immunofluorescence staining results of tumor sections of MCF-7/ADR tumor-bearing mice, HMGB1 translocation (from the nucleus to the cytosol) and obvious CRT fluorescent signals were visualized in the T-BMV@DOX treated group (Fig. S8, ESI†). The above results demonstrated that T-BMV@DOX can overcome drug resistance of MCF-7/ADR tumors and immune activation plays an important role in the therapy.
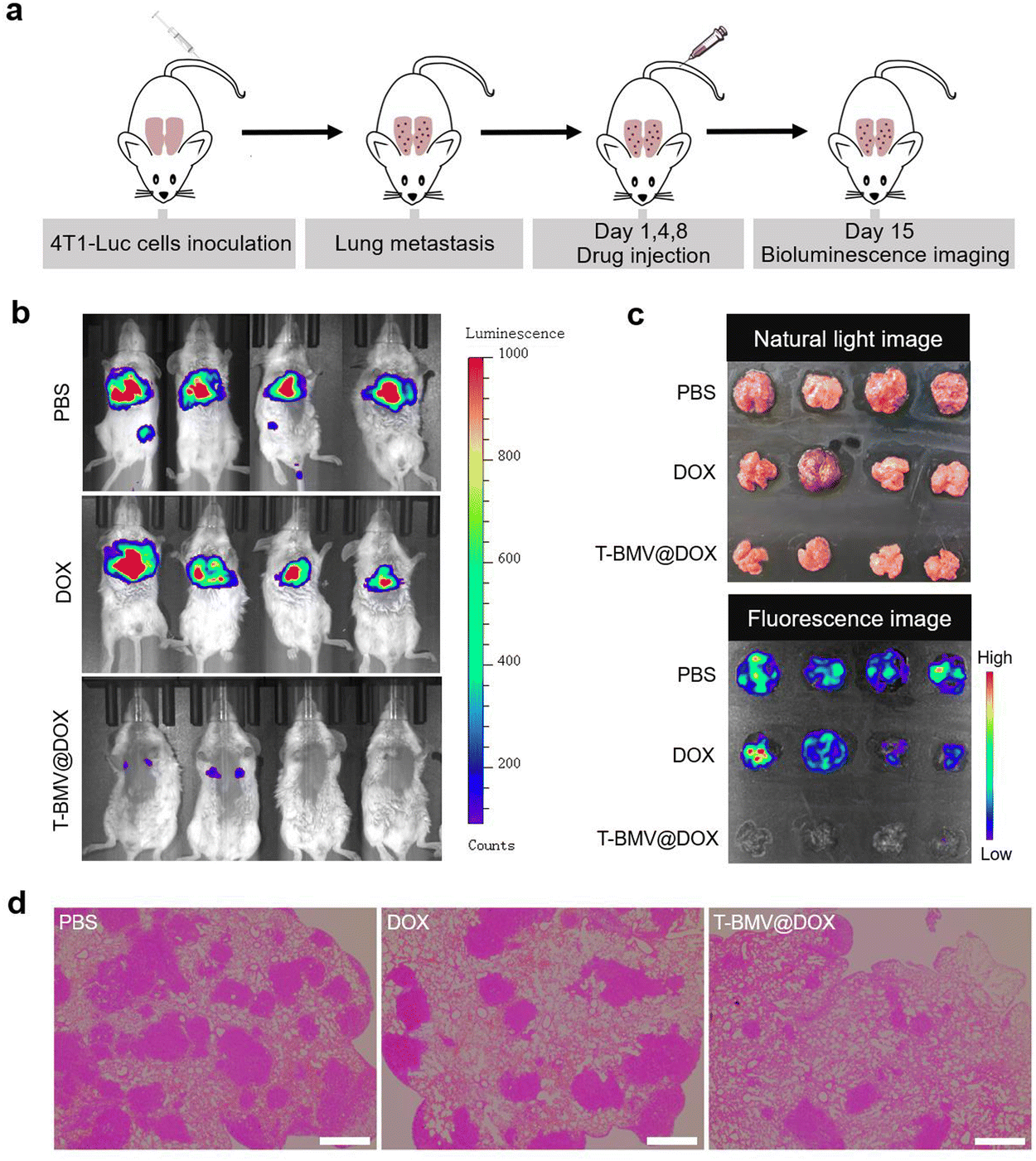
2.8. Anti-metastasis study
The above experiments have demonstrated the anti-tumor ability of T-BMV@DOX against 4T1 tumor-bearing mice; here we want to examine whether it has anti-metastasis efficacy. As demonstrated in Fig. 6a, a lung breast cancer metastasis model was constructed by injecting 4T1-Luc cells (4T1 cells with stable luciferase expression) intravenously into healthy mice as reported.50 Twenty-four hours after cell injection, the first T-BMV@DOX dosing was performed. After consecutive administration for 2 weeks, a minimal fluorescent signal of 4T1 tumor cells was observed in the lungs of T-BMV@DOX-treated mice in vivo, whereas the PBS- and DOX-treated groups exhibited remarkable tumor fluorescence signals (Fig. 6b), suggesting metastasis in PBS- and DOX-treated mice, but not in T-BMV@DOX treated ones. In addition, the lung tissues from 4T1-Luc tumor bearing mice with different treatments were dissected and photographed. Their corresponding fluorescence strengths were consistent with the observed fluorescence images (Fig. 6c). H&E staining showed notable metastatic nodules in the excised lungs of the PBS- and DOX-treated groups, while in the T-BMV@DOX treated group, lung metastasis was hardly observed (Fig. 6d). Based on the above study, it was reasonable to conclude that the injection of T-BMV@DOX may potently inhibit metastasis in the breast cancer metastasis model.
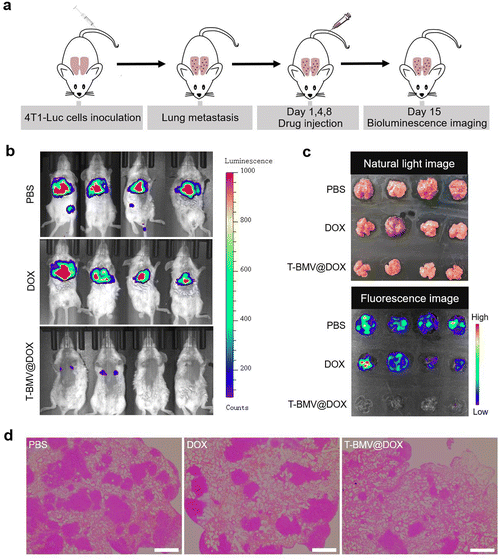 |
| | Fig. 6 Anti-metastasis study of T-BMV@DOX in the cancer metastasis model. (a) Schematic illustration of the construction of the 4T1-Luc cell induced-lung breast cancer metastasis model. (b) In vivo fluorescence images of mice received 3 different treatments: (1) i.v. injection of PBS, (2) i.v. injection of DOX solution, (3) i.v. injection of T-BMV@DOX (n = 4). (c) Photographs captured under natural light (top) and fluorescence images (bottom) of isolated ex vivo lung tissues in different groups of mice treated with PBS, DOX, and T-BMV@DOX, respectively. (d) Representative images of H&E staining of dissected lung tissues in each group. Scale bars: 200 μm. | |
3. Conclusion
In summary, we here reported a multifunctional nanoplatform that synergizes chemotherapy, ferroptosis, and immune-mediated antitumor therapy. By encapsulating an antitumor drug (DOX) and iron ions into MVs produced from magnetotactic bacteria MSR-1 via the culturing medium, the drug-loaded BMV@DOX vesicles can be extracted and used integrally, without breaking and reforming the MVs. A series of experiments have been performed to characterize and confirm the presence of DOX as well as iron in BMV@DOX. Besides, the aqueous solution of drug-loaded MVs demonstrated an impressive magnetic resonance T2 relaxivity. For further improvement, the BMV@DOX vesicles were functionalized with a tumor-targeting DSPE-PEG-cRGD peptide and became hybrid nanovaccine T-BMV@DOX. In vitro cellular assays suggested that, in addition to the effect of the chemotherapeutic drug, T-BMV@DOX could also activate the ferroptosis pathway. Once administered systemically, the resultant T-BMV@DOX nanovaccine, as expected, could target the tumor region in mice via active targeting and exert a synergistic action to enhance therapeutic efficacy in tumor ablation, even in the drug-resistance model. On the murine breast cancer model of lung metastasis, the efficacy of T-BMV@DOX on anti-tumor metastasis was further confirmed. Given the merits of the facile construction, MRI capabilities, and effective tumor-targeting abilities, our study suggested that the MSR-1 bacteria MVs-based T-BMV@DOX nanovaccine with the integration of chemotherapy, immunotherapy, and ferroptosis effect presented potential application prospects in cancer diagnosis and treatment.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
This study is supported by the National Key Research and Development Program (No. 2022YFA1203200) and the Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDB36000000).
References
- F. Bray, A. Jemal, N. Grey, J. Ferlay and D. Forman, Lancet Oncol., 2012, 13(8), 790–801 CrossRef PubMed.
- M. Tolomeo and D. Simoni, Curr. Med. Chem.: Anti-Cancer Agents, 2002, 2(3), 387–401 CrossRef CAS PubMed.
- J. Song, Z. Qu, X. Guo, Q. Zhao, X. Zhao, L. Gao, K. Sun, F. Shen, M. Wu and L. Wei, Autophagy, 2009, 5(8), 1131–1144 CrossRef CAS PubMed.
- T. Robak, Expert Rev. Anticancer Ther., 2008, 8(7), 1033–1051 CrossRef CAS PubMed.
- A. H. Sabri, Z. Cater, P. Gurnani, J. Ogilvie, J. Segal, D. J. Scurr and M. Marlow, Int. J. Pharm., 2020, 589, 119808 CrossRef CAS PubMed.
- A. Redondo, A. Gallego and M. Mendiola, Expert Rev. Clin. Pharmacol., 2022, 15(1), 1–9 CrossRef CAS PubMed.
- N. P. Restifo, M. J. Smyth and A. Snyder, Nat. Rev. Cancer, 2016, 16(2), 121–126 CrossRef CAS PubMed.
- D. Hoelzer, J. Walewski, H. Döhner, A. Viardot, W. Hiddemann, K. Spiekermann, H. Serve, U. Dührsen, A. Hüttmann, E. Thiel, J. Dengler, M. Kneba, M. Schaich, I. G. Schmidt-Wolf, J. Beck, B. Hertenstein, A. Reichle, K. Domanska-Czyz, R. Fietkau, H. A. Horst, H. Rieder, S. Schwartz, T. Burmeister and N. Gökbuget, Blood, 2014, 124(26), 3870–3879 CrossRef CAS PubMed.
- G. Y. Chen, G. L. Jiang, L. J. Wang, H. Qian, X. L. Fu, H. Yang, K. L. Wu and S. Zhao, Int. J. Radiat. Oncol., Biol., Phys., 2005, 61(1), 70–75 CrossRef CAS PubMed.
- J. Galon and D. Bruni, Nat. Rev. Drug. Discovery, 2019, 18(3), 197–218 CrossRef CAS PubMed.
- W. Yue, L. Chen, L. Yu, B. Zhou, H. Yin, W. Ren, C. Liu, L. Guo, Y. Zhang, L. Sun, K. Zhang, H. Xu and Y. Chen, Nat. Commun., 2019, 10(1), 2025 CrossRef PubMed.
- C. Schwechheimer and M. J. Kuehn, Nat. Rev. Microbiol., 2015, 13(10), 605–619 CrossRef CAS PubMed.
- M. Toyofuku, N. Nomura and L. Eberl, Nat. Rev. Microbiol., 2019, 17(1), 13–24 CrossRef CAS PubMed.
- J. L. Kadurugamuwa and T. J. Beveridge, J. Bacteriol., 1996, 178(10), 2767–2774 CrossRef CAS PubMed.
- J. L. Kadurugamuwa and T. J. Beveridge, J. Bacteriol., 1995, 177(14), 3998–4008 CrossRef CAS PubMed.
- Z. Li, A. J. Clarke and T. J. Beveridge, J. Bacteriol., 1996, 178(9), 2479–2488 CrossRef CAS PubMed.
- L. H. Peng, M. Z. Wang, Y. Chu, L. Zhang, J. Niu, H. T. Shao, T. J. Yuan, Z. H. Jiang, J. Q. Gao and X. H. Ning, Sci. Adv., 2020, 6(27), eaba2735 CrossRef CAS PubMed.
- K. Cheng, R. Zhao, Y. Li, Y. Qi, Y. Wang, Y. Zhang, H. Qin, Y. Qin, L. Chen, C. Li, J. Liang, Y. Li, J. Xu, X. Han, G. J. Anderson, J. Shi, L. Ren, X. Zhao and G. Nie, Nat. Commun., 2021, 12(1), 2041 CrossRef CAS PubMed.
- S. Qing, C. Lyu, L. Zhu, C. Pan, S. Wang, F. Li, J. Wang, H. Yue, X. Gao, R. Jia, W. Wei and G. Ma, Adv. Mater., 2020, 32(47), e2002085 CrossRef PubMed.
- M. Li, S. Li, H. Zhou, X. Tang, Y. Wu, W. Jiang, Z. Tian, X. Zhou, X. Yang and Y. Wang, Nat. Commun., 2020, 11(1), 1126 CrossRef CAS PubMed.
- Y. Li, R. Zhao, K. Cheng, K. Zhang, Y. Wang, Y. Zhang, Y. Li, G. Liu, J. Xu, J. Xu, G. J. Anderson, J. Shi, L. Ren, X. Zhao and G. Nie, ACS Nano, 2020, 14(12), 16698–16711 CrossRef CAS PubMed.
- Q. Chen, H. Bai, W. Wu, G. Huang, Y. Li, M. Wu, G. Tang and Y. Ping, Nano Lett., 2020, 20(1), 11–21 CrossRef CAS PubMed.
- S. J. Dixon, K. M. Lemberg, M. R. Lamprecht, R. Skouta, E. M. Zaitsev, C. E. Gleason, D. N. Patel, A. J. Bauer, A. M. Cantley, W. S. Yang, B. Morrison III and B. R. Stockwell, Cell, 2012, 149(5), 1060–1072 CrossRef CAS PubMed.
- J. P. Friedmann Angeli, D. V. Krysko and M. Conrad, Nat. Rev. Cancer, 2019, 19(7), 405–414 CrossRef CAS PubMed.
- O. Y. Kim, H. T. Park, N. T. H. Dinh, S. J. Choi, J. Lee, J. H. Kim, S. W. Lee and Y. S. Gho, Nat. Commun., 2017, 8(1), 626 CrossRef PubMed.
- Y. Zhang, L. Liu, L. Jin, X. Yi, E. Dang, Y. Yang, C. Li and T. Gao, J. Invest. Dermatol., 2014, 134(1), 183–191 CrossRef CAS PubMed.
- F. Zhou, B. Feng, H. Yu, D. Wang, T. Wang, Y. Ma, S. Wang and Y. Li, Adv. Mater., 2019, 31(14), e1805888 CrossRef PubMed.
- W. Wang, M. Green, J. E. Choi, M. Gijón, P. D. Kennedy, J. K. Johnson, P. Liao, X. Lang, I. Kryczek, A. Sell, H. Xia, J. Zhou, G. Li, J. Li, W. Li, S. Wei, L. Vatan, H. Zhang, W. Szeliga, W. Gu, R. Liu, T. S. Lawrence, C. Lamb, Y. Tanno, M. Cieslik, E. Stone, G. Georgiou, T. A. Chan, A. Chinnaiyan and W. Zou, Nature, 2019, 569(7755), 270–274 CrossRef CAS PubMed.
- R. Uebe and D. Schuler, Nat. Rev. Microbiol., 2016, 14(10), 621–637 CrossRef CAS PubMed.
- C. Rong, Y. Huang, W. Zhang, W. Jiang, Y. Li and J. Li, Res. Microbiol., 2008, 159(7–8), 530–536 CrossRef CAS PubMed.
- M. Amor, A. Ceballos, J. Wan, C. P. Simon, A. T. Aron, C. J. Chang, F. Hellman and A. Komeili, Appl. Environ. Microbiol., 2020, 86(22), e01278-20 CrossRef PubMed.
- Z. Xiang, G. Jiang, D. Fan, J. Tian, Z. Hu and Q. Fang, Nanoscale, 2020, 12(25), 13513–13522 RSC.
- N. Orench-Rivera and M. J. Kuehn, Cell. Microbiol., 2016, 18(11), 1525–1536 CrossRef CAS PubMed.
- A. Kulp and M. J. Kuehn, Annu. Rev. Microbiol., 2010, 64, 163–184 CrossRef CAS PubMed.
- M. M. Joseph, G. Aswathy, T. K. Manojkumar and T. T. Sreelekha, Int. J. Biol. Macromol., 2016, 92, 20–29 CrossRef CAS PubMed.
- Z. Xiang, X. Yang, J. Xu, W. Lai, Z. Wang, Z. Hu, J. Tian, L. Geng and Q. Fang, Biomaterials, 2017, 115, 53–64 CrossRef CAS PubMed.
- N. Zlatkov, A. Nadeem, B. E. Uhlin and S. N. Wai, FEMS Microbiol. Rev., 2021, 45(2), fuaa047 CrossRef CAS PubMed.
- Y. Wu, X. Song, W. Xu, K. Y. Sun, Z. Wang, Z. Lv, Y. Wang, Y. Wang, W. Zhong, J. Wei, H. L. Cai and X. Wu, Small, 2021, 17(33), e2101705 CrossRef PubMed.
- S. J. Dorazio and J. R. Morrow, Inorg. Chem., 2012, 51(14), 7448–7450 CrossRef CAS PubMed.
- C. Liang, X. Zhang, M. Yang and X. Dong, Adv. Mater., 2019, 31(51), e1904197 CrossRef PubMed.
- J. M. Berthiaume and K. B. Wallace, Cell Biol. Toxicol., 2007, 23(1), 15–25 CrossRef CAS PubMed.
- Y. Ichikawa, M. Ghanefar, M. Bayeva, R. Wu, A. Khechaduri, S. V. Naga Prasad, R. K. Mutharasan, T. J. Naik and H. Ardehali, J. Clin. Invest., 2014, 124(2), 617–630 CrossRef CAS PubMed.
- S. T. Smiley, M. Reers, C. Mottola-Hartshorn, M. Lin, A. Chen, T. W. Smith, G. D. Steele Jr and L. B. Chen, Proc. Natl. Acad. Sci. U. S. A., 1991, 88(9), 3671–3675 CrossRef CAS PubMed.
- M. Zhang, X. Qin, Z. Zhao, Q. Du, Q. Li, Y. Jiang and Y. Luan, Nanoscale Horiz., 2022, 7(2), 198–210 RSC.
- T. Uto, Y. Nishi, M. Toyama, K. Yoshinaga and M. Baba, Int. Immunopharmacol., 2011, 11(11), 1932–1938 CrossRef CAS PubMed.
- W. Budzynski and C. Radzikowski, Immunopharmacol. Immunotoxicol., 1994, 16(3), 319–346 CrossRef CAS PubMed.
- N. Hanna, T. W. Davis and I. J. Fidler, Int. J. Cancer, 1982, 30(3), 371–376 CrossRef CAS PubMed.
- J. S. Park, F. Gamboni-Robertson, Q. He, D. Svetkauskaite, J. Y. Kim, D. Strassheim, J. W. Sohn, S. Yamada, I. Maruyama, A. Banerjee, A. Ishizaka and E. Abraham, Am.
J. Physiol.: Cell Physiol., 2006, 290(3), C917–C924 CrossRef CAS PubMed.
- L. Apetoh, F. Ghiringhelli, A. Tesniere, M. Obeid, C. Ortiz, A. Criollo, G. Mignot, M. C. Maiuri, E. Ullrich, P. Saulnier, H. Yang, S. Amigorena, B. Ryffel, F. J. Barrat, P. Saftig, F. Levi, R. Lidereau, C. Nogues, J. P. Mira, A. Chompret, V. Joulin, F. Clavel-Chapelon, J. Bourhis, F. André, S. Delaloge, T. Tursz, G. Kroemer and L. Zitvogel, Nat. Med., 2007, 13(9), 1050–1059 CrossRef CAS PubMed.
- Z. Gong, M. Chen, Q. Ren, X. Yue and Z. Dai, Signal Transduction Targeted Ther., 2020, 5(1), 12 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *abc
*abc