
Catecholase-catalyzed synthesis of wedelolactone, a natural coumestan and its analogs†
Anushree
Achari‡
a,
Sourav
Chatterjee‡
ab,
Sudip
Dey
a,
Tapas K.
Kundu
c and
Parasuraman
Jaisankar
 *a
*a
aLaboratory of Catalysis and Chemical Biology, Organic and Medicinal Chemistry Division, CSIR-Indian Institute of Chemical Biology, Kolkata-700032, India. E-mail: jaisankar@iicb.res.in
bDepartment of Chemistry, University of Minnesota, Minneapolis, MN55455, USA
cTranscription and Disease Laboratory, Molecular Biology and Genetics Unit, Jawaharlal Nehru Centre for Advanced Scientific Research, Jakkur, Bengaluru-560064, India
First published on 2nd December 2022
Abstract
Biocatalysis plays an important role in the synthesis of complex organic molecules. Wedelolactone, a natural coumestan, has been reported to have many bioactive properties. A novel and efficient enzyme obtained from sweet potato juice was used for condensation of 4-hydroxycoumarins with catechols to produce wedelolactone and its structurally diverse analogs in moderate to good yields under mild reaction conditions. Hence, this enzymatic approach creates an opportunity to access many coumestan-based compounds that are potential building blocks for the synthesis of pharmaceutically important molecules.
In the last two decades, applications of enzymes in biocatalytic transformations have increased significantly both in industry and academia. The urgency for greener reaction conditions with fewer synthetic steps, coupled with the importance of chirally pure compounds, especially in the pharmaceutical industry, has greatly provided an impetus for enzymatic catalysis. Many of the enzymatic reactions, unlike transition metal-catalyzed reactions, fulfill several criteria of green chemistry, e.g., use of aerial oxygen for some oxidative transformations wherein the waste product generated is water, which is non-toxic. Biocatalysis is now being merged with traditional syntheses as cascades to make them operable at industrial levels and to ensure sustainability in the production of natural products and many other pharmaceutically relevant compounds.1
Wedelolactone, a phytoconstituent of Eclipta spp., is one of the medicinal coumestans studied for its bioactive potential. It exhibits several pharmacological properties such as anti-cancer,2,3 anti-adipogenic,4 osteoprotective, and hepatoprotective activities.5,6 A limited number of total synthesis methods of wedelolactone have been reported that so far mostly comprise multiple synthetic steps involving harsh reaction conditions.7–9 A few enzymes such as laccase,10 peroxidase from solid onion waste,11 and tyrosinase from mushroom12 also showed catalytic activity in the synthesis of coumestan-based natural products. We have now found sweet potato to be a rich source of catecholase that could drive our model condensation reaction to deliver the natural coumestan wedelolactone (4a), even when the non-purified crude extract is tested.
Results
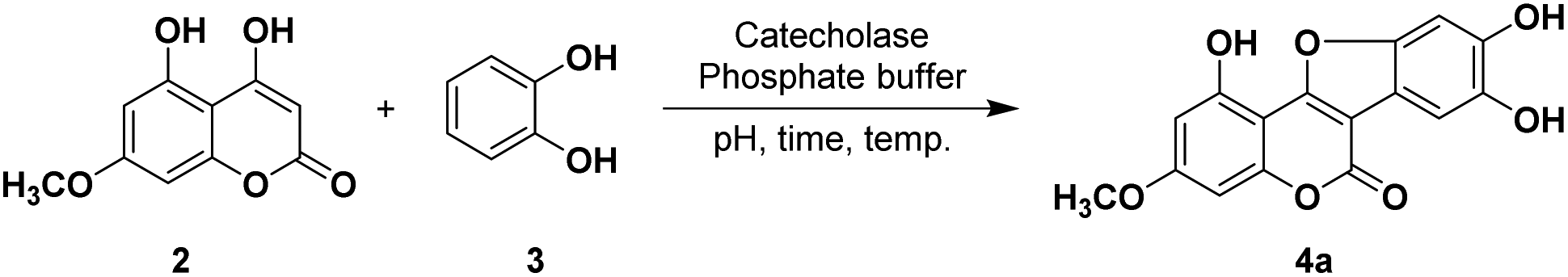
To optimize the total synthesis of wedelolactone (Scheme 1) and to further carry out its structural modification, a crude extract of sweet potato was first tested in our model reaction. Briefly, selective monomethylation of 2,4,6-trihydroxyacetophenone by dimethylsulfate yielded 2,6-dihydroxy-4-methoxyacetophenone (1) (see the ESI†), which was cyclized by heating with diethylcarbonate in the presence of sodium to afford 4,5-dihydroxy-7-methoxycoumarin (2). Oxidative cyclization of 2 with catechol (3) using various oxidizing agents was then attempted to isolate the desired product 4a. However, all the screenings attempted resulted in unsatisfactory reaction outcomes (see the ESI†). As the reaction is supposed to proceed through orthoquinone, a catechol oxidized product that subsequently undergoes dual cross-coupling with 4,5-dihydroxy-7-methoxycoumarin (2), we selected the catecholase enzyme that could oxidize a catechol moiety to orthoquinone en route to furnish the desired product wedelolactone. Herein, we report a biocatalytic, mild, scalable, oxidative cyclocondensation of catechol derivatives with hydroxy coumarins to afford the medicinal coumestan wedelolactone along with its analogs by making use of the catecholase enzyme from the common source sweet potato.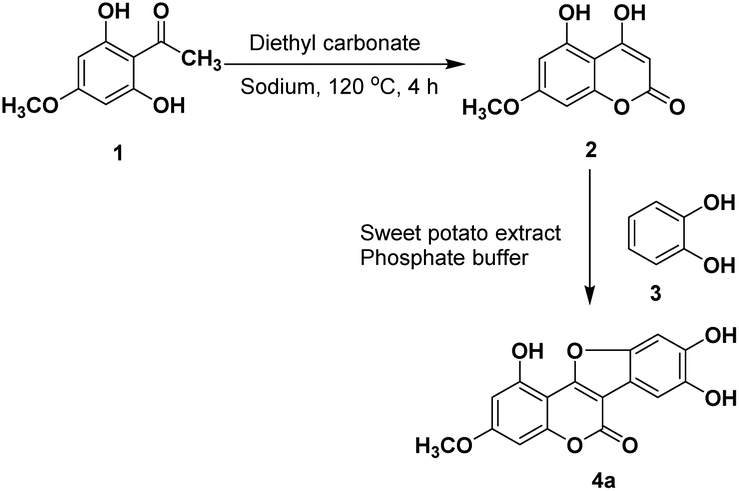 | ||
| Scheme 1 Representative synthesis of a coumestan using the sweet potato extract. | ||
Initially, the reaction was performed by adding the crude sweet potato extract (50 mL in phosphate buffer, pH 7.4) to a mixture of 4,5-dihydroxy-7-methoxycoumarin (2, 1 mmol) and catechol (3, 1.1 mmol) at 25 °C and stirring for 24 h. This yielded the corresponding coumestan (wedelolactone, 4a) in a good yield (55%, Table 1, entry 11). We observed that the enzyme is crucial for this oxidative cyclization and no product formation was observed in the absence of the enzyme (Table 1, entry 10). To improve the yield further, the sweet potato extract sourced catecholase was semi-purified for further use (see the ESI, Fig. S1†).13 On carrying out the reaction using 125 μM of the partially purified sample, the isolated yield went up to 66% (Table 1, entry 3). We, therefore, proceeded with varying the other reaction parameters (catalyst loading, temperature, time, and pH) to optimize the process (Table 1).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Cat. (μL) | Temp. (°C) | Time (h) | pH | Isolated yield (%) |
| a Reaction conditions: 2 (0.5 mmol), catechol 3 (0.55 mmol), catecholase of indicated conc. in 2.0 mL phosphate buffer of indicated pH. The resulting mixture was stirred at mentioned temperatures for the indicated time. | |||||
| 1 | 120 | 25 | 24 | 7.4 | 65 |
| 2 | 60 | 25 | 24 | 7.4 | 69 |
| 3 | 30 | 25 | 24 | 7.4 | 66 |
| 4 | 15 | 25 | 24 | 7.4 | 52 |
| 5 | 5 | 25 | 24 | 7.4 | 49 |
| 6 | 30 | 10 | 24 | 7.4 | 44 |
| 7 | 30 | 25 | 48 | 7.4 | 69 |
| 8 | 30 | 25 | 24 | 6.0 | 15 |
| 9 | 30 | 25 | 24 | 8.0 | 58 |
| 10 | 0 | 25 | 24 | 7.4 | No reaction |
| 11 | Crude extract | 25 | 24 | 7.4 | 55 |
It was observed that increasing the catalyst loading to 60 μL (Table 1, entry 2) or increasing the reaction time to 48 h (Table 1, entry 7) had no significant impact on the product yield. However, lowering the pH to 6.0 or increasing it to 8.0 had negative impacts on the yield. Hence, the use of 1 mmol of 4,5-dihydroxy-7-methoxycoumarin 2b and 1.1 mmol of catechol 3 in the presence of 125 μM (30 μL) of catecholase in phosphate buffer (pH 7.4) at 25 °C for 24 h was found to be the optimized reaction conditions. A number of analogs of wedelolactone were synthesized under these conditions (Fig. 1). The structures of all the compounds were determined using 1H, 13C NMR and ESI-MS techniques. It is pertinent to mention here that this synthetic approach permitted the total synthesis of wedelolactone (4a) along with a number of its analogs (4b to 4j, except 4i and 4j) in 35–70% yields (Fig. 1).
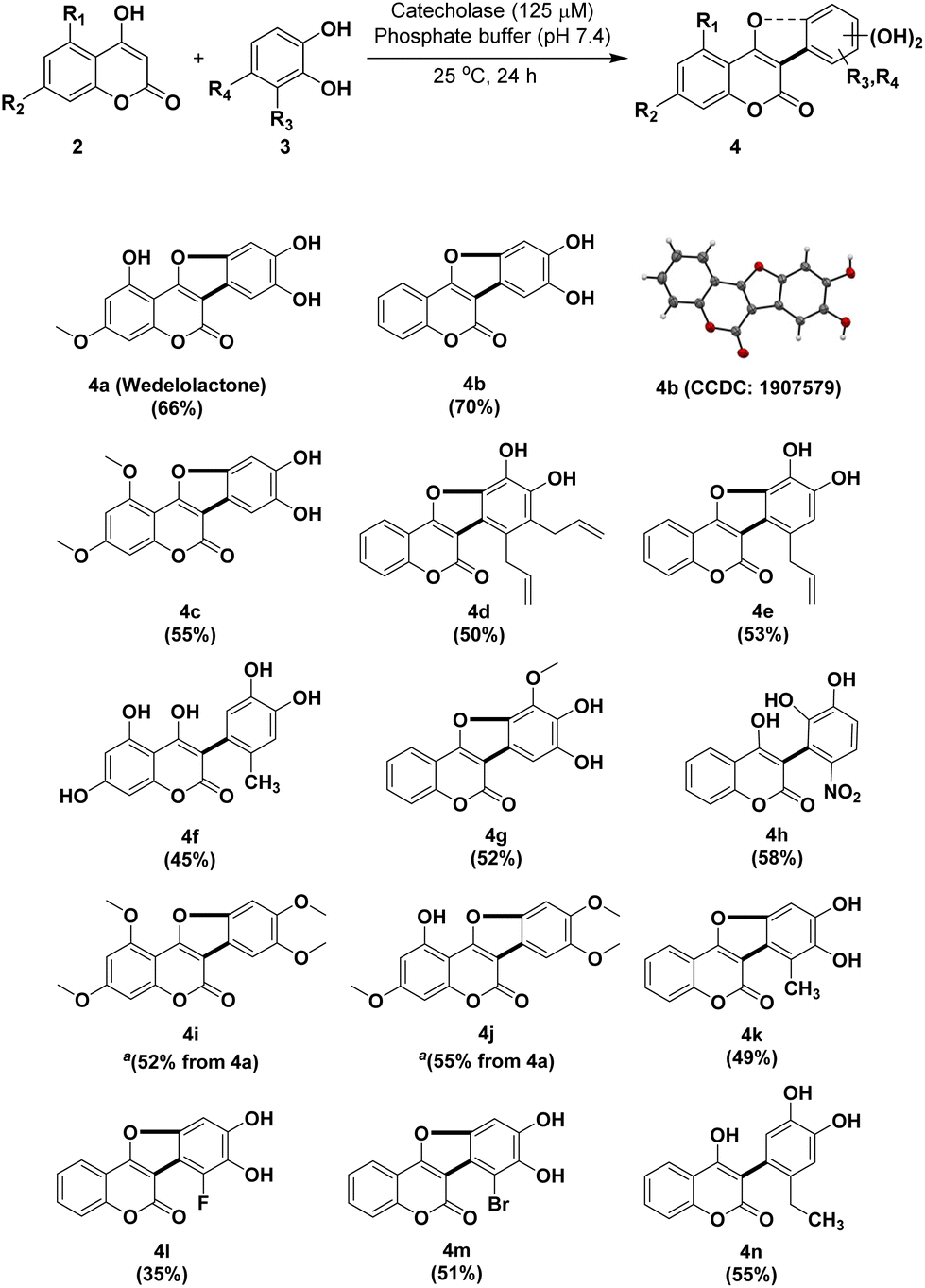 | ||
Fig. 1 Substrate scope of sweet potato enzyme-catalyzed reactions of 4-hydroxy coumarins and substituted catechols. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 4i and 4j were obtained from 4a through selective methylation. For the yields of 4i and 4j, refer to the selective methylation of 4a. Reaction conditions: 2 (0.5 mmol), 3 (0.55 mmol), catecholase (125 μM, 30 μL), phosphate buffer (2.0 mL, pH 7.4), rt, 24 h. 4i and 4j were obtained from 4a through selective methylation. For the yields of 4i and 4j, refer to the selective methylation of 4a. Reaction conditions: 2 (0.5 mmol), 3 (0.55 mmol), catecholase (125 μM, 30 μL), phosphate buffer (2.0 mL, pH 7.4), rt, 24 h. | ||
We investigated the influence of different coumarin and catechol substituents on the yields of the corresponding coumestan derivatives (Fig. 1) which ranged from moderate to good yields (45–70%). The effect of both electron-withdrawing and electron-donating groups on the ring-closing oxidative cyclization reaction was also investigated. Interestingly, when a substituent like CH3, CH2CH3, or NO2 was located at the 4-position of catechol, the hydroxyl group of the 4-hydroxy coumarin counterpart did not condense with the catechol ring to form the furan ring (Fig. 1, entries 4f, 4h, and 4n). The nitro group appears to have directed the incoming nucleophile to attack the α,β-unsaturated nitro unit, thereby foreclosing further cyclization. But the reasons for the non-reactivity of the methyl (4f) or ethyl substituents (4n), in sharp contrast to the 3-methyl, 3-allyl or 3,4-diallyl derivatives (4k, 4e and 4d), are not clear at present. The reaction worked with fluoro- (4l) or bromocatechols (4m), although the yields were on the lower side. Interestingly, when we used 4-chlorocatechol or 4-carboxycatechol with 4-hydroxy coumarin, it resulted in dechlorination and decarboxylation, respectively, and both afforded 4b as the product. However, no reactivity was observed when 3-cyano catechol and 3-carboxy catechol were tested for the reaction.
Conclusions
In conclusion, we have disclosed a unique and improved method for the synthesis of wedelolactone using catecholase for the first time. Besides the inherent advantages of the biocatalytic synthesis process, our unique method demonstrates the use of economical and abundantly available edible source sweet potato for conversion. Many hitherto unreported analogs of wedelolactone could be synthesized using this method. These compounds are yet to be tested for their effects on the biological system.Total synthesis of wedelolactone and its structural derivatives
:40) to give 4a as a pale brown solid.
Author contributions
Conceptualization, S. C. and P. J.; compound design, S. C., A. A., S. D., and P. J.; compound synthesis, A. A., S. C., and S. D.; crystallography, S. C. and A. A.; manuscript preparation, A. A., S. C., P. J., and T. K. K.Conflicts of interest
The authors confirm that there are no conflicts to declare.Acknowledgements
S. C. and A. A. thankfully acknowledge UGC, New Delhi, India and CSIR, New Delhi, India, respectively, for the award of Research Fellowships. This project (HCP-40) was funded by CSIR, New Delhi. The authors thank the Central Instrumentation Facility of CSIR-IICB for their constant support and help.References
- A. I. Benítez-Mateos and F. Paradisi, ChemSusChem, 2022, 15, e2021020 CrossRef PubMed.
- T. Kučírková, M. Stiborek, M. Dúcka, J. Navrátilová, J. BogdanovićPristov, A. Popović-Bijelić, S. Vojvodić, J. Preisler, V. Kanický, J. Šmarda, I. Spasojević and P. Beneš, Metallomics, 2018, 10, 1524 CrossRef PubMed.
- T. Nehybová, J. Šmarda, L. Daniel, M. Stiborek, V. Kanický, I. Spasojevič, J. Preisler, J. Damborský and P. Beneš, Int. J. Mol. Sci., 2017, 18, 729 CrossRef PubMed.
- S. Lim, H. J. Jang, E. H. Park, J. K. Kim, J. M. Kim, E. K. Kim, K. Yea, Y. H. Kim, W. Lee-Kwon, S. H. Ryu and P. G. Suh, J. Cell. Biochem., 2012, 113, 3436 CrossRef CAS PubMed.
- Y. Q. Liu, X. F. Han, J. X. Bo and H. P. Ma, Front. Pharmacol., 2016, 7, 375 CAS.
- Q. Luo, J. Ding, L. Zhu, F. Chen and L. Xu, Am. J. Chin. Med., 2018, 46, 819 CrossRef CAS PubMed.
- C. F. Chang, L. Y. Yang, S. W. Chang, Y. T. Fang and Y. J. Lee, Tetrahedron, 2008, 64, 3661 CrossRef CAS.
- H. Huang, J. Chen, J. Ren, C. Zhang and F. Ji, Molecules, 2019, 24, 4130 CrossRef CAS PubMed.
- C. C. Li, Z. X. Xie, Y. D. Zhang and J. H. Chen, J. Org. Chem., 2003, 68, 8500 CrossRef CAS PubMed.
- T. Qwebani-Ogunleye, N. I. Kolesnikova, P. Steenkamp, C. B. de Koning, D. Brady and K. W. Wellington, Bioorg. Med. Chem., 2017, 25, 1172 CrossRef CAS PubMed.
- S. Angeleska, P. Kefalas and A. Detsi, Tetrahedron Lett., 2013, 19, 2325 CrossRef.
- G. Pandey, C. Muralikrishna and U. T. Bhalerao, Tetrahedron, 1989, 45, 6867 CrossRef CAS.
- C. Eicken, F. Zippel, K. Büldt-Karentzopoulos and B. Krebs, FEBS Lett., 1998, 436, 293 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary data (spectroscopic data of compounds 4a–4l, copies of 1H and 13C NMR spectra, and crystallographic data for compound 4b). CCDC 1907579. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ob02081e |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2023 |