
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSmall luminescent silver clusters stabilized in porous crystalline solids
Naoya
Haraguchi
 ,
Taisei
Kurosaki
and
Sayaka
Uchida
*
,
Taisei
Kurosaki
and
Sayaka
Uchida
*
Department of Basic Science, School of Arts and Sciences, The University of Tokyo 3-8-1 Komaba, Meguro-ku, Tokyo 153-8902, Japan. E-mail: csayaka@g.ecc.u-tokyo.ac.jp
First published on 9th January 2024
Abstract
Subnanometric or small metal clusters (SMCs) have been extensively researched due to their unique electronic, optical, catalytic, and magnetic properties, which differ from those of bulk samples. Among the SMCs, silver (Ag) clusters have received significant interest due to their affordability and unique luminescent properties. Currently, two major approaches, gas-phase and liquid-phase synthesis, have been employed to obtain Ag clusters with precise control of size and structure. More recently, attention has been directed toward the utilization of porous crystalline solids such as metal–organic frameworks (MOFs), zeolites, and porous ionic crystals (PICs) to synthesize and stabilize Ag clusters. In this review, we aim to provide a comprehensive overview of the synthesis, structures, and luminescent properties of Ag clusters in porous crystalline solids.
1. Introduction
Subnanometric metal clusters, commonly referred to as small metal clusters (SMCs), have been intensively studied due to their unique electronic, optical, catalytic, or magnetic properties, stemming from their discrete electronic structure, distinctive atomic arrangements, and high surface-to-atom ratio, which are not observed in the corresponding bulk samples.1,2 Since the properties of SMCs are highly dependent on size (nuclearity or number of atoms), structure, and types of protecting ligands, it is important to control these parameters to fine-tune their properties. SMCs exhibit a molecule-like behavior due to their discrete electronic energy levels. Consequently, well-defined transitions with energies in the visible light region can occur, enabling strong fluorescence in some of these clusters. A landmark example was reported by Zheng, Dickson, and co-workers showing that water-soluble few-atom Au clusters encapsulated in poly(amidoamine) dendrimers exhibit size-dependent specific fluorescence (Au5 (UV), Au8 (blue), Au13 (green), Au23 (red)).3 Among the numerous reports on luminescent SMCs, considerable attention has been directed toward Ag clusters due to their relatively low price and distinctive color tone and luminescence properties different from their gold counterparts.4,5Meanwhile, gas-phase synthesis is one of the most commonly employed methods for synthesizing small Ag clusters.6 Within this approach, monodispersed small Ag clusters can be obtained through size-separation of clusters of various sizes generated by sputtering or vaporization. However, the yield of clusters is markedly low, and this method necessitates sophisticated equipment. Therefore, gas-phase synthesis is impractical for real-world applications of small Ag clusters. Another method involves the use of protecting ligands in the liquid phase.7 Ag ions are reduced by reducing reagents, and Ag clusters subsequently form, which are stabilized by the protecting ligands.
On the other hand, synthetic approaches have emerged to form and stabilize small Ag clusters in porous crystalline solids such as metal–organic frameworks (MOFs)8 and zeolites.4 The formation of small Ag clusters in the solid-phase offer the advantage of improved stability, resulting in a higher density or weight percentage of Ag clusters and an increase in luminescence intensity and efficiency. In addition, it is expected that the luminescence properties can be controlled by the solvent molecules adsorbed in the pores of crystalline solids (i.e., solvatochromism or vapochromism).9
Despite the large numbers of papers on small luminescent Ag clusters formed and stabilized in porous crystalline solids, to the best of our knowledge, there is no comprehensive review on this subject. The photophysical properties of Ag clusters, including the origin of photoluminescence (PL) in Ag clusters, particularly those formed in zeolites, have already been reported in papers and reviews.10,11 However, the synthetic methods employed for Ag clusters, which significantly influence their physical properties, have not yet been systematically categorized to date. We have recently reported the synthesis of luminescent small Ag clusters via electron transfer in redox-active porous ionic crystals.12–14 Therefore, the purpose of this perspective is to systematically categorize these composites and to provide an overview of the synthesis, structures, and luminescent properties of small Ag clusters formed and stabilized in porous crystalline solids.
2. Silver cluster-containing MOFs
2.1. Ag clusters as framework nodes of MOFs
MOFs are porous crystalline materials constructed by the assembly of metal ions/clusters and organic linkers through coordination bonds. Ag chalcogenide/chalcogenolate cluster-based MOF (SCC-MOF) was first reported by Huang, Zang, and co-workers in 20178 by using a luminescent SCC [Ag12(StBu)6(CF3COO)6(CH3CN)6]·CH3CN (Ag12). A SCC-MOF [(Ag12(StBu)8(CF3COO)4(bpy)4)]n (Ag12bpy) was obtained by replacing the CH3CN ligand in Ag12 with a bidentate 4,4′-bipyridine (bpy) linker. The Ag clusters in Ag12bpy were linked to each other by bpy. While Ag12 exhibited a three-layered structure of Ag5S2–Ag2S2–Ag5S2 (Fig. 1a), the middle layer changed from a square pyramidal Ag2S2 configuration to a tetrahedral Ag2S4 configuration by the formation of Ag12bpy (Fig. 1b). The stability of the Ag cluster remarkably increased by the ligand replacement, and the PL quantum yield (PLQY) improved from 0.2% to 12.1%. In the case of Ag12bpy, the luminescence originated from the triplet state, which rapidly turned off by O2. Additionally, Ag12bpy showed a bright green emission under vacuum, and multi-color switching of luminescence was realized by solvatochromism/vapochromism with volatile organic compounds (VOCs). The emission color under the VOCs red-shifted with the increase in their polarity. These results show that incorporating SCC into the MOF framework led to enhanced stability and acquisition of multi-color luminescent characteristics.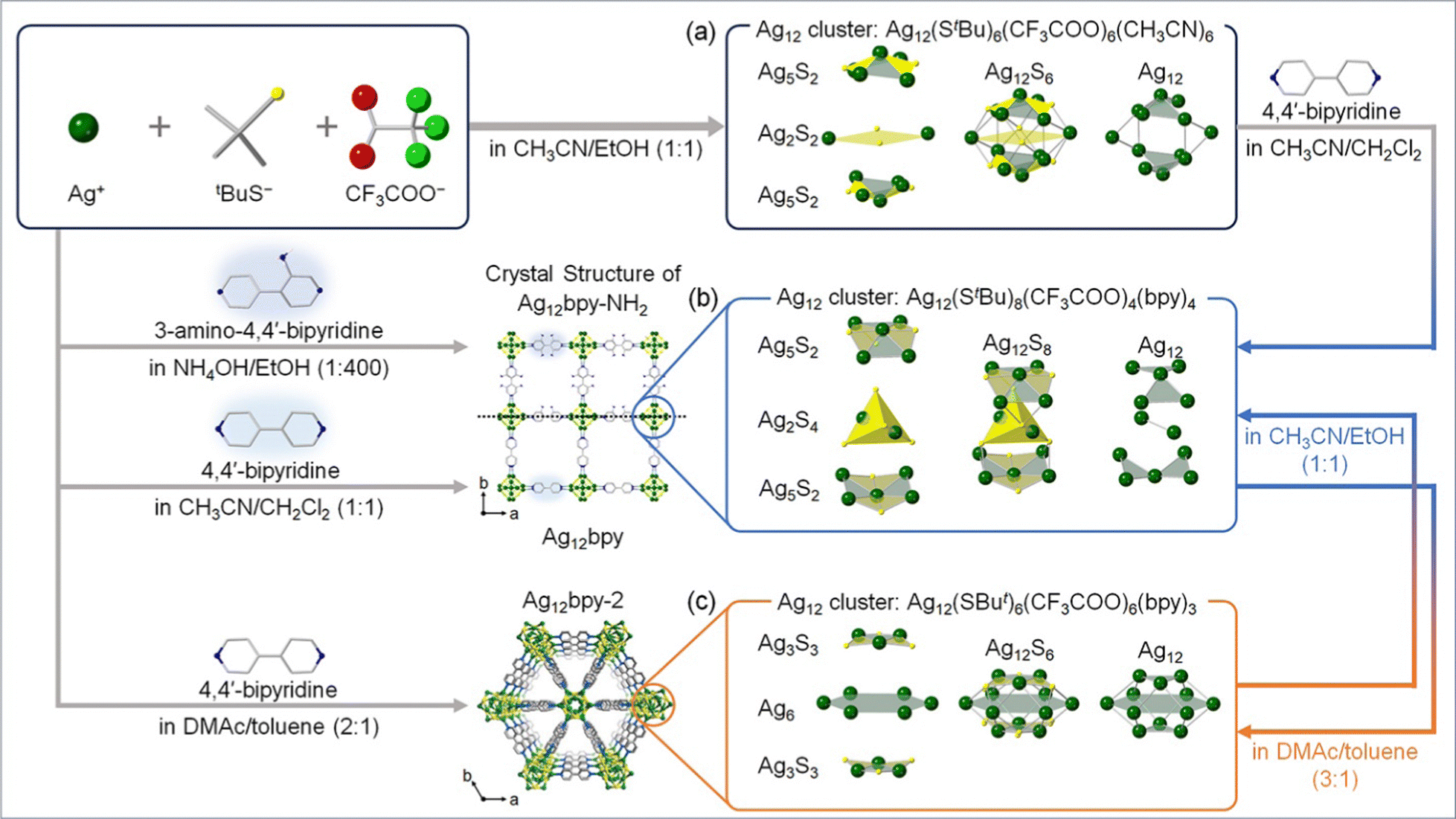 | ||
| Fig. 1 Design approach and schematic illustration of the syntheses and structures of Ag12-based SCC-MOFs. (a) Ag12, (b) Ag12bpy and Ag12bpy-NH2, and (c) Ag12bpy-2. | ||
After this study, Dong, Zang, and co-workers demonstrated that the luminescent properties of SCC-MOFs can be tuned by functional modification of the MOF linker. They reported the synthesis of [(Ag12(StBu)8(CF3COO)4(bpy-NH2)4)]n (Ag12bpy-NH2, Fig. 1b), which is isostructural with Ag12bpy,15 where the Ag12 cluster units were analogous by modifying the bpy linker with 3-amino-4,4′-bipyridine (bpy-NH2). It has been known that pristine bpy-NH2 in the solid state emits blue fluorescence.16Ag12bpy-NH2 exhibited dual emission of yellow phosphorescence (Ph) and blue fluorescence (Fl), while Ag12bpy emitted green Fl. As in the case of Ag12bpy, the Ph of Ag12bpy-NH2 is quenchable with O2, but the detection limit is much lower (32 Pa for Ag12bpy and 11.4 mPa for Ag12bpy-NH2). Additionally, the authors synthesized Ag12bpy-CH3, which is isostructural with Ag12bpy and Ag12bpy-NH2, but insensitive to oxygen. Therefore, bpy-NH2 and bpy-CH3 linkers contribute to increasing and decreasing, respectively, the sensitivity of Ph toward O2. Moreover, the O2-sensing pressure can be controlled by mixing and changing the ratio of bpy-NH2 and bpy-CH3 linkers in the MOF.
The luminescent properties of SCC-MOFs can be tuned by altering the geometric/electronic structure of the Ag cluster. For instance, Huang, Zang, and co-workers reported that isomerization of the Ag cluster occurs when dissolving Ag12bpy in N,N-dimethylacetamide (DMAc)/toluene (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), resulting in the formation of Ag12bpy-2 (Fig. 1c).16 This transformation changes the Ag cluster from a three-layered structure to a compressed empty cuboctahedron. Notably, Ag12bpy and Ag12bpy-2 exhibited distinct luminescence, with a single green emission for Ag12bpy and a dual blue-red emission for Ag12bpy-2. Subsequently, the authors conducted DFT calculations to gain insight into the structure–luminescence relationship. The results indicated that for Ag12bpy, degenerated LUMO–LUMO+7, characterized by π* orbitals of the bpy ligands, appeared. In contrast, for Ag12bpy-2 with a compressed cuboctahedron, Ag cluster-centered LUMO as well as LUMO+1–+6 with close energies distributed on the bpy ligands appeared. Combining these findings with time-dependent DFT calculations, the dual phosphorescence of Ag12bpy-2 with respective maxima at 463 nm and 620 nm can be deduced to originate from two separate excited states arising mainly from Ag cluster-centered transitions and intraligand transitions, respectively.
:1), resulting in the formation of Ag12bpy-2 (Fig. 1c).16 This transformation changes the Ag cluster from a three-layered structure to a compressed empty cuboctahedron. Notably, Ag12bpy and Ag12bpy-2 exhibited distinct luminescence, with a single green emission for Ag12bpy and a dual blue-red emission for Ag12bpy-2. Subsequently, the authors conducted DFT calculations to gain insight into the structure–luminescence relationship. The results indicated that for Ag12bpy, degenerated LUMO–LUMO+7, characterized by π* orbitals of the bpy ligands, appeared. In contrast, for Ag12bpy-2 with a compressed cuboctahedron, Ag cluster-centered LUMO as well as LUMO+1–+6 with close energies distributed on the bpy ligands appeared. Combining these findings with time-dependent DFT calculations, the dual phosphorescence of Ag12bpy-2 with respective maxima at 463 nm and 620 nm can be deduced to originate from two separate excited states arising mainly from Ag cluster-centered transitions and intraligand transitions, respectively.
2.2. Ag clusters formed inside the pores of MOFs
Houk, Allendorf, and co-workers reported the formation of Ag clusters inside the pores of three water-stable MOFs, Cu(BTC), MOF-508, and MIL-68(In), by infiltrating them in an aqueous-ethanolic solution of AgNO3, where ethanol could reduce Ag+ to Ag0in situ.17 EPR spectroscopy indicated that the Ag clusters formed in MOF-508 and MIL68(In) were linear [Ag3]0, which were particularly abundant in MOF-508. As shown in Fig. 2, Tiburcio, Pardo, and co-workers reported the synthesis of ligand-free [Ag2]0 clusters in the pores of a MOF with the formula of Ni2{Ni4[Cu2(Me3mpba)2]3}·54H2O [1] (Me3mpba4− = N,N′-2,4,6-trimethyl-1,3-phenylenebis(oxamate)),18 and Ag2@MOF was obtained by a two-step post synthetic process as specified below. In the first step, Ag-exchanged MOF [2] was obtained by immersing 1 in an aqueous solution of AgNO3. In the second step, Ag2 containing MOF [3] was obtained by reducing 2 with NaBH4. The crystal structure of 3 was successfully solved, and ligand-free [Ag2]0 clusters were located in the pores of 3. Compound 3 showed high catalytic activity in the Bunchner ring expansion reaction between toluene and ethyl diazoacetate, while 1 and 2 without the Ag cluster showed much lower activities. Still, the number of papers reporting the formation of small Ag clusters inside the pores of MOFs, especially mentioning their sizes, is limited. This limitation arises because MOFs are generally unstable under irradiation by an electron beam,17 and it is difficult to observe the clusters by electron microscopes due to degradation of MOFs and sintering of clusters.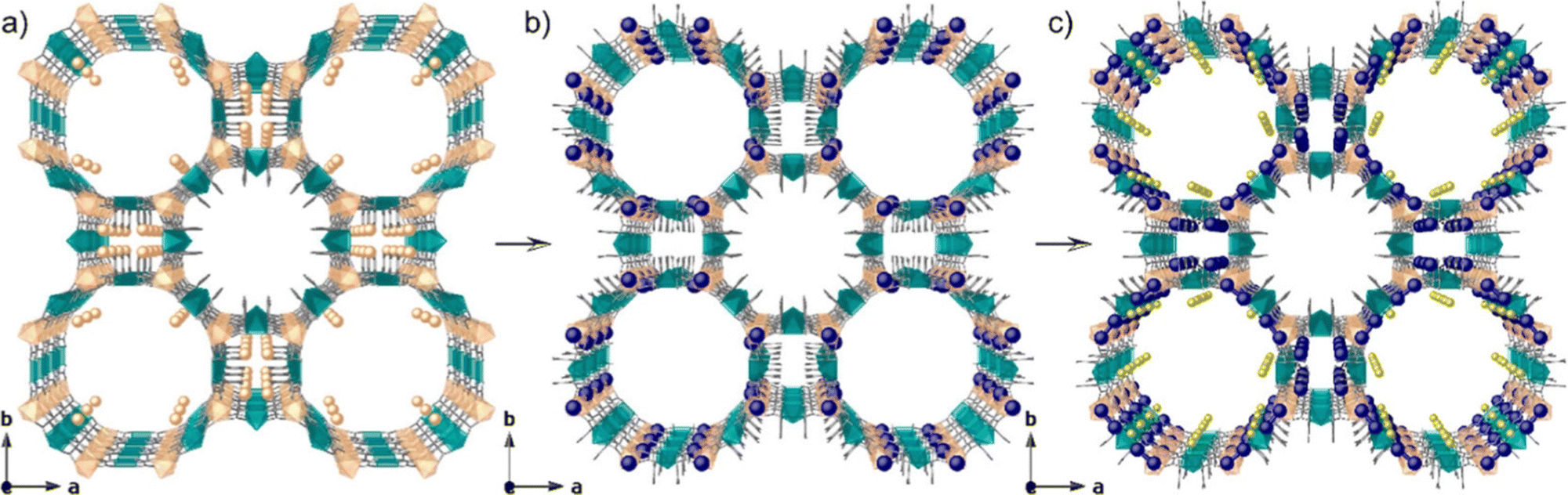 | ||
| Fig. 2 Design approach showing the crystal structures of (a) 1, (b) 2, and (c) 3, showing the two-step post-synthetic process of the formation of Ag2@MOF. Cu and Ni atoms from the network are represented by cyan and orange polyhedra, respectively, whereas organic ligands are depicted as gray sticks. Orange, yellow, and blue spheres represent Ni, Na, and Ag atoms, respectively. Reprinted with permission from Tiburcio et al. Copyright 2022 American Chemical Society. | ||
3. Silver clusters confined in zeolites
3.1. Silver-exchanged zeolites (Ag-zeolites)
Zeolites are porous crystalline aluminosilicates composed of covalently bonded tetrahedral TO4 (T = Si, Al) units. The framework of zeolites, composed of TO4 connections, can be of various structure types, and faujasite (FAU) and Linde type A (LTA) are well-known representative framework structures (Fig. 3).19 Extra-framework cations (e.g., Li+, Na+, K+, NH4+, Ca2+) reside within the pores of zeolites owing to the negatively charged nature of zeolite frameworks, depending on the Si/Al ratio. These extra-framework cations can be exchanged in solution with other metal cations. In particular, Ag-exchanged zeolites (Ag-zeolites) have been intensively studied since Ralek and co-workers first reported the unique color change of Ag-zeolites depending on the degree of hydration and Ag loading.20 One of the first studies on the PL of Ag-zeolites was reported by Ozin and co-workers,21 where blue and green emissions were observed in Ag-exchanged FAU with different Ag loadings. Later, Calzaferrin and co-workers reported the formation of luminescent quantum-sized Ag sulfide clusters in LTA.22 | ||
| Fig. 3 Framework structure of (a) FAU and (b) LTA.19 The Si–O–Si(Al) bonds are depicted as orange sticks. | ||
The unique luminescence properties of Ag-zeolites arise from small Ag clusters in zeolite scaffolds, which are activated with external stimuli. Cluster formation is initiated by reducing Ag+ to Ag0 in zeolite frameworks via loss of oxygen atoms from the zeolite framework (reaction (1))4,23 and/or dehydration (reaction (2))24,25
| 2Ag+ + ZO2− → 1/2O2 + 2Ag0 + Z | (1) |
| Ag+ + H2O → 2H+ + 2Ag0 + 1/2O2 | (2) |
Monoatomic Ag0, which form viareaction (1) and/or (2), subsequently self-assemblies with Ag+, and small Ag clusters form in the zeolite cages. It has been widely recognized that zeolites are appropriate scaffolds for small Ag clusters because the Ag clusters can be stabilized in the zeolite frameworks via electrostatic interaction or coordination bond with the oxygen atoms, as well as the confinement effect. Meanwhile, to proceed reaction (1) and/or (2), external stimuli such as thermal treatment and light irradiation are needed, which will be introduced in the next section.
3.2. Synthetic approaches of small silver clusters in zeolites
Ag-zeolites can be activated by various methods to obtain Ag clusters, and thermal treatment is one of the most commonly used methods.26–35 Upon thermal treatment, Ag-zeolites are heated gradually to prevent structure damage, and heating is kept for about a day after reaching the target temperature to produce Ag0 by reaction (1) and/or (2). For example, Ag4 clusters were formed in K-exchanged LTA (K-LTA) by heating partially Ag-exchanged K-LTA, which was pre-treated at 100 °C and 150 °C followed by heating to 450 °C and keeping overnight.31 In addition, the atmosphere for thermal treatment may also affect the properties of Ag clusters. In particular, applying high vacuum shows activating effect viareaction (2), resulting in the formation of small Ag clusters with luminescent properties.5,36Ag-zeolites can also be activated by irradiation with UV light,37,38 electron beam,39 X-ray,40–42 or γ-ray.43 Michalik and Kevan reported the formation of small Ag clusters in partially Ag-exchanged Na-LTA by γ-ray irradiation at 77 K, and the existence of linear [Ag3]0 and [Ag6]+ was suggested by electron spin resonance (ESR) measurement.43 By activation with UV light, Ag clusters can be formed in irradiated spots with high space resolution. This method enables well-localized formation of luminescent small Ag clusters and could be applied to optical encoding.38 Sasaki and Suzuki observed the formation and growth of Ag species in a zeolite scaffold from clusters of approximately 1 nm in size to much larger nanoparticles with electron beam irradiation of Ag-LTA and Ag-FAU.39 The distribution of Ag clusters corresponded to the pore arrangement in the zeolite crystal, which means that the Ag clusters are formed in the pores of zeolites. Importantly, electron beam irradiation can initiate large cluster formation and overcome the energy barriers to particle growth, while thermal treatment may not. Fenwick and co-workers monitored the growth of Ag species in Ag-FAUX zeolite with X-ray irradiation and found that the growth dynamics obey the Hill–Langmuir equation.42
Chemical reduction is another activation method of Ag-zeolites.44–46 For example, Shimizu and co-workers reported the formation of [Ag4]2+ in MFI zeolite, which formed independently of the degree of Ag-exchange, by the reduction of Ag-MFI with H2 at 573 K.45 The Ag clusters served as catalytically active sites for the reduction of NO by hydrocarbons (HC-SCR).47
3.3. The effect of parameters on the properties of silver cluster in zeolites
The main parameters to control the properties of small Ag clusters are as follows: (1) the topology of zeolite frameworks (LTA, FAU, MFI, etc.), (2) the charge of zeolite (Si/Al ratio), (3) Ag loading (degree of Ag-exchange), (4) the types or amounts of cations contained in the parent zeolite, (5) the degree of hydration, and (6) the activation method. Hereafter, we will introduce how these parameters affect the properties of small Ag clusters in zeolites by giving some representative examples.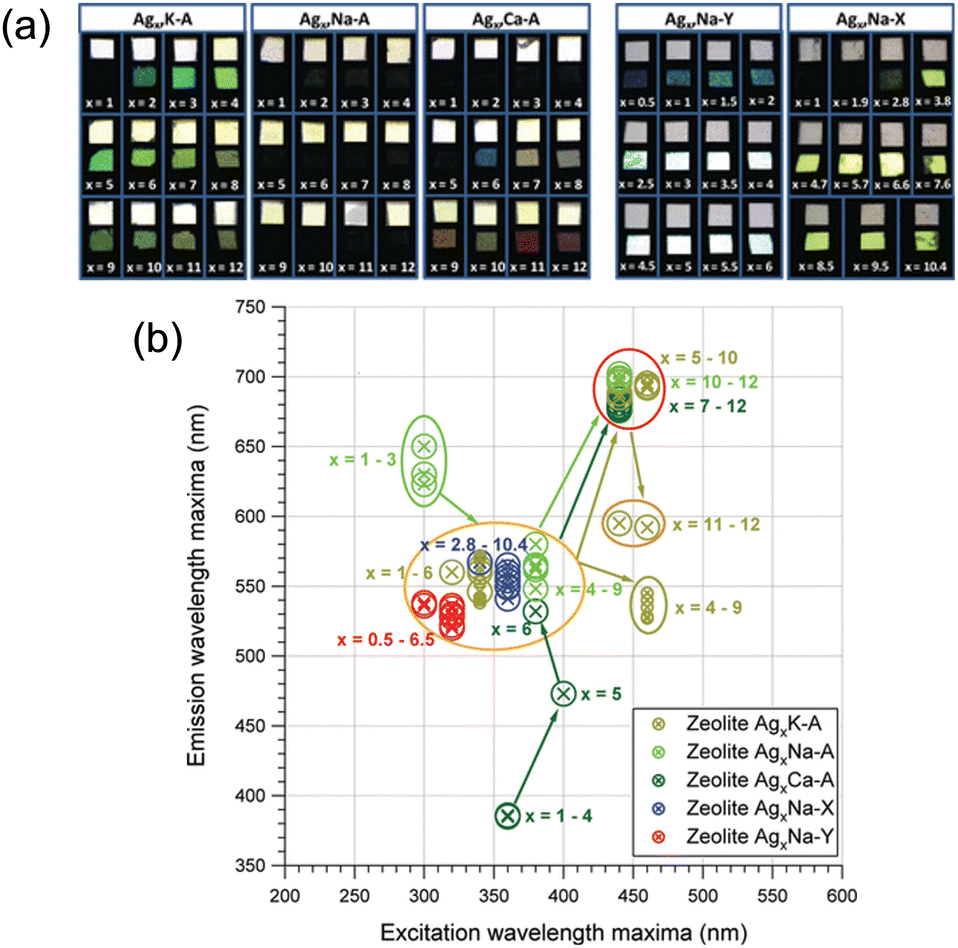 | ||
| Fig. 4 (a) Photographs of Ag-exchanged zeolites after heat treatment. For each Ag loading, the upper picture represents the sample under room light illumination, while the lower picture is taken under UV illumination (360 nm). (b) Position of the most pronounced luminescence bands of the heat-treated Ag-exchanged zeolites. For each set of samples, the global trends upon increasing the Ag loading are highlighted by the colored arrows. Reprinted with permission from De Cremer et al. Copyright 2009 American Chemical Society. | ||
Fenwick, Roeffaers, and co-workers investigated the effect of Ag loadings on the PLQY of Agx-FAUY (x = 0–6.5).31 The PLQY increased with the decrease in Ag loadings and reached almost 100% at x = 0.5. To understand this phenomenon, extended X-ray absorption fine structure (EXAFS) spectroscopy was carried out, which revealed that the size of clusters and the proportion of Ag atoms involved in cluster formation remained almost constant (67% and 62% for Ag0.5-FAUY and Ag6.5-FAUY, respectively), indicating that these two factors are not the main parameters for the high PLQY of FAUY. On the other hand, the Debye–Waller factor associated with the Ag–Ag shells of Ag0.5-FAUY (0.026) was much smaller than that of Ag6.5-FAUY (0.044), indicating that Ag4 in Ag0.5-FAUY are significantly more ordered than the Ag4 in Ag6.5-FAUY. Therefore, the high PLQY of FAUY at low Ag loadings originates from the ordered structure of Ag clusters. It is noteworthy to comment that PLQY can be optimized to nearly 100% in FAUY, while FAUX, with the same topology but higher extra-framework cation (Na+) content, showed very low PLQY. A detailed analysis of the EXAFS signal of Ag1-FAUX revealed a sodium shell around the Ag clusters, which is not detected in FAUY. This fact suggests that Na+, which is more concentrated and mobile in FAUX, plays a key role. Consequently, Na+ can move inside the sodalite cages, interfering with Ag cluster formation and affecting their electronic/geometric structure.
To summarize, these results indicate that the luminescent properties of small Ag clusters are highly dependent on the topology of zeolite frameworks, the charge of zeolite (Si/Al ratio), and Ag loading.
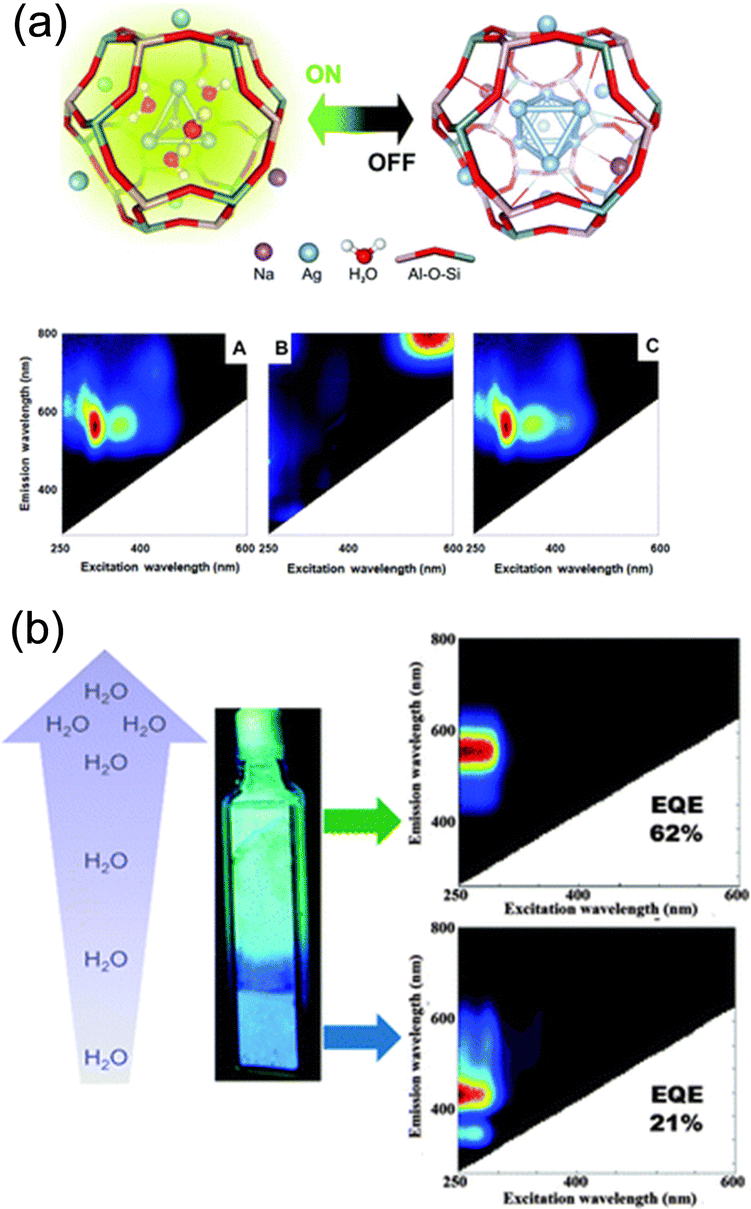 | ||
| Fig. 5 (a) (top) Schematic representation of the reversible opto-structural switching of Ag clusters confined in partially Ag-exchanged Na-LTA. (bottom) 2D excitation–emission plots of the sample in the (left) hydrated, (middle) dehydrated, and (right) rehydrated state. Reprinted with permission from Aghakhani et al. Copyright 2018 Royal Society of Chemistry. (b) Water dependence of the emission color observed in Ag1-LTA(Li). In fully hydrated samples, a green emission was found, whereas in partially hydrated samples blue emission was recorded. The water gradient observed in the cuvette was generated by leaving the materials exposed to air under ambient conditions. Reprinted with permission from Coutino-Gonzalez et al. Copyright 2015 Royal Society of Chemistry. | ||
Coutino-Gonzalez, Sels, and co-workers investigated the luminescence properties of Agx-LTA(Li) (x = 1–12), which is the Ag-exchanged form of Li4Na8-LTA after calcination at 450 °C.30 As in the case of the composites reported by De Cremer and co-workers,27 the samples showed green-yellow emissions at low Ag loadings (x < 6) and red emissions at high Ag loadings under hydrated conditions. In particular, at low Ag loadings (x = 1–4), an intense blue emission was observed in the partially hydrated state, while green/yellow emission was observed in the fully hydrated state (Fig. 5b). EXAFS analyses suggested that the blue and green/yellow emissions originate from Ag3 and Ag4, respectively.
Hoshino and co-workers investigated the PL properties of “air-packed samples” which is an Ag12-LTA heated at 500 °C for 24 h under air for dehydration and packed in a glass cell with air at various temperatures, followed by cooling to room temperature to control the degree of hydration.26 The samples packed at 100 °C exhibited an emission band, which was not observed for those packed at higher temperatures. In situ Ag K-edge EXAFS was measured to investigate the origin of the emission band, which revealed the formation of [Ag5]m+ (m ≧ 3) for the samples air packed below 100 °C, since dark Ag6 clusters were unstable in hydrated Ag12-LTA.
All these results show that the cluster sizes (e.g., Ag3, Ag4, Ag5, and Ag6) and the corresponding luminescence properties depend on the hydration level of zeolites.
The size change of Ag clusters with hydration/dehydration, as mentioned above,32 can also be considered as an example affected by the mobility of cations. Notably, the Ag clusters formed in calcined NaAg3-LTA exist as Ag4 (a mixture of 80% of Ag4(H2O)4 and 20% of Ag4(Oframework)2) and can be readily dehydrated to form dark [Ag6]2+. On the other hand, it is more difficult to dehydrate Ag4 in calcined KAg3-LTA (a mixture of 60% [Ag4(H2O)2]2+ and 40% of [Ag4(H2O)4]2+), resulting in the formation of emissive Ag4 with high stability. The authors mentioned that this phenomenon may be due to the larger size and thus lower mobility of K+ than Na+, leading to preserve the coordination water molecules. We note that the origin of the PL of Ag4 has been evaluated with time-dependent DFT calculations. Ag4 exhibits a closed-shell electronic configuration, in which the Ag 4d shell is completely filled, and the two remaining 5s electrons are delocalized over the cluster. Upon excitation, one electron of the s-type HOMO is promoted to the p-type LUMO and relaxes through enhanced intersystem crossing into long-lived triplet states.33
The counter cations of zeolites also affect the geometry of Ag clusters confined in the zeolite cages. As shown in Fig. 6, Baekelant, Steele, and co-workers investigated the luminescence properties of calcined Ag1LixNa(11−x)-LTA, prepared by introducing Ag+ into Li-exchanged LixNa(12−x)-LTA.48 They reported that the emission color of calcined Ag1LixNa(11−x)-LTA continuously changes from red to blue with increased Li loading. X-ray-excited optical luminescence (XEOL) combined with EXAFS revealed that the bond length between the Ag of Ag4 cluster and the O atoms of zeolite framework reduces with the increase in Li loading, causing the continuous emission color change.
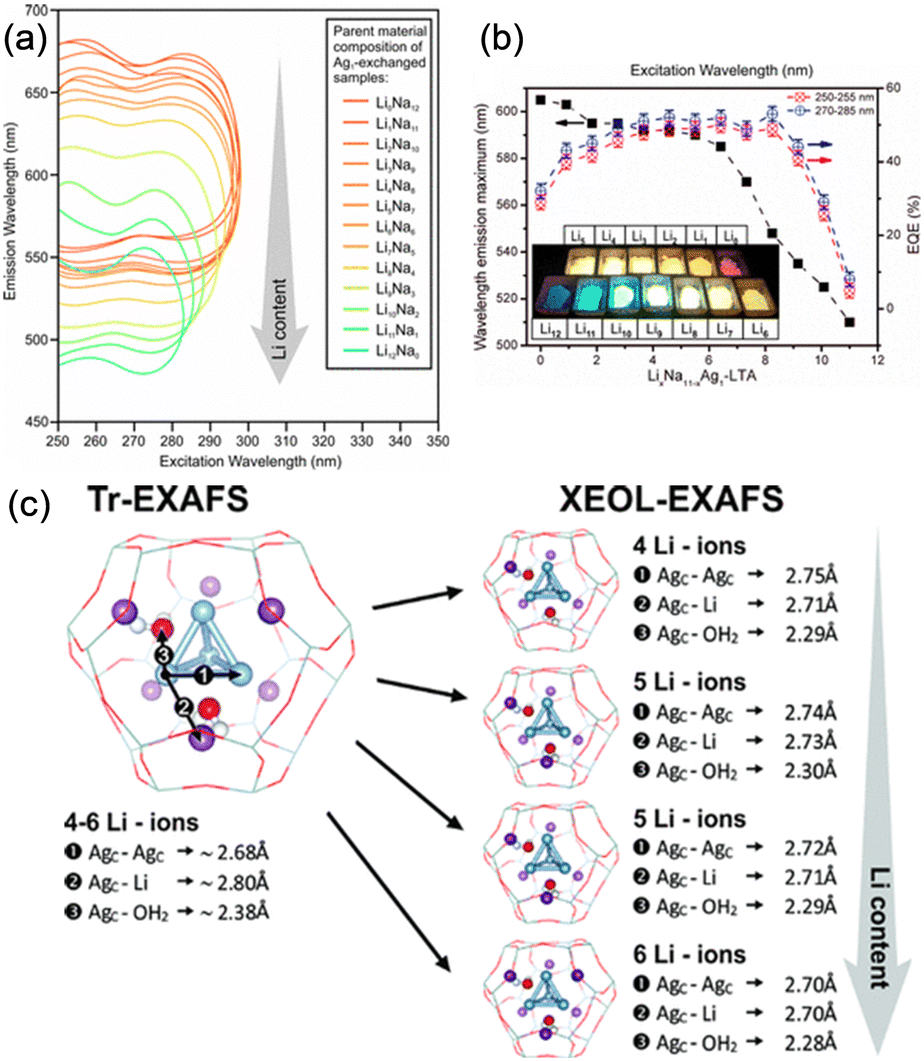 | ||
| Fig. 6 (a) 2D emission and excitation profiles of the different hydrated Ag-exchanged LTA. (b) Maximal emission wavelength (excitation at 270–285 nm) and external quantum efficiency of the Ag1LixNa(11−x)-LTA depending on the Li content. (c) Schematic representation of the changes observed for the ground and excited states of the tetrahedral Ag clusters as determined by Tr- and XEOL-EXAFS, respectively. Reprinted with permission from Baekelant et al. Copyright 2018 American Chemical Society. | ||
4. Ag clusters confined in porous ionic crystals (Ag-PICs)
4.1. Porous ionic crystals
In this section, we will introduce synthetic approaches of small Ag clusters utilizing polyoxometalate-based porous crystalline solids. Polyoxometalates (POMs) are nanosized anionic metal-oxygen clusters, and Keggin ([XM12O40]n−) and Dawson ([X2M18O62]n−) types are the widely used representative POMs. In recent years, POMs have been utilized as building blocks in combination with molecular macrocation to synthesize porous ionic crystals (PICs).49,50 Generally, POMs have a high negative charge, and the framework structure of PICs composed of POMs and macrocations are negatively charged: the negative charge of the framework is compensated by exchangeable cations such as K+, Cs+, and NH4+, resulting in no net charge on the materials as a whole.51 PICs possess ion-exchange properties, and Ag-exchanged PICs have been reported.52 Additionally, PICs can serve as electron reservoirs since POMs show reversible redox-active properties based on the changes in the valence of composing metal ions.53–55 Based on these observations, we have recently proposed to utilize PICs as scaffolds to form small Ag clusters based on their ion exchange and redox-active properties.12–14 For instance, a PIC scaffold with Dawson-type POM contains closed pores of approximately 1.4 × 2.2 nm2 in size, 55 sufficiently spacious to incorporate small Ag clusters, considering the van der Waals radius of Ag atoms (0.34 nm) (Fig. 7). As shown schematically in Fig. 8, upon soaking the reduced PICs into an aqueous solution of AgNO3, the introduction and reduction of Ag proceed simultaneously, forming Ag clusters in the pores of PICs. Since the reduction of Ag+ proceeds by the electron transfer from the reduced POM to Ag+, this synthetic approach does not need any external stimuli or reducing reagents, which makes the operation simple. Therefore, this synthetic approach can be considered as a novel method to synthesize small Ag clusters.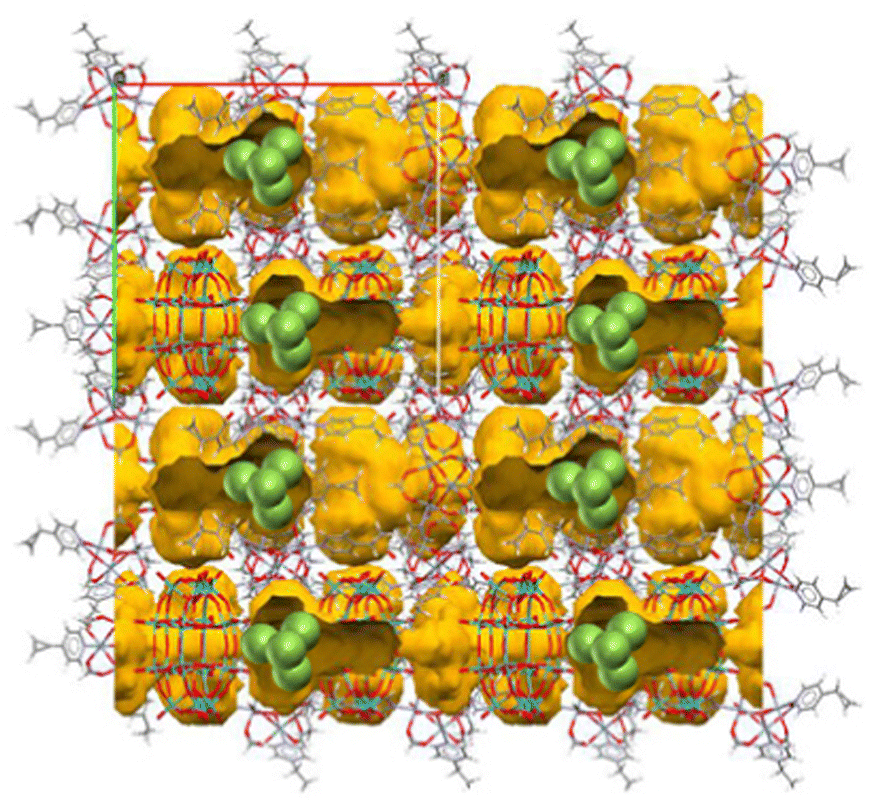 | ||
| Fig. 7 A PIC scaffold with Dawson-type POM.55 Yellow part indicates the closed pores of approximately 1.4 × 2.2 nm2 in size. The pore size is sufficiently large to accommodate a Ag4 cluster (green spheres indicate the Ag toms). | ||
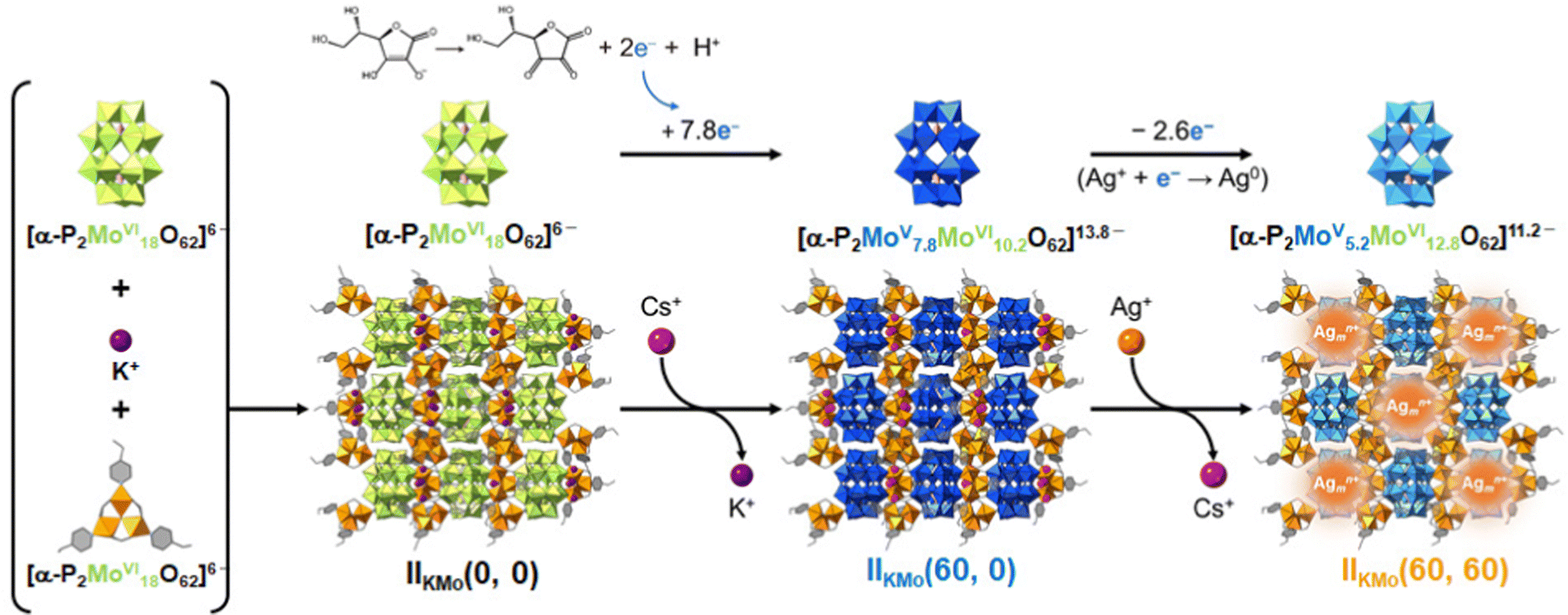 | ||
| Fig. 8 Schematic representation of the Ag cluster formation in a PIC scaffold. Relationship among as-synthesized PIC (IIKMo(0, 0)), reduction-induced ion-exchange of K+ with Cs+ (IIKMo(60, 0)), and Ag incorporation followed by Ag cluster formation (IIKMo(60, 60)). IIKMo(t1, t2) represents the PIC, which has been reduced and ion-exchanged with Cs+ for t1 min followed by the immersion in aqueous solution of AgNO3 for t2 min. Note that the number of transferred electrons with decimal points (7.8e− and 2.6e−) indicates bulk averages. Reprinted with permission from Haraguchi et al. Copyright 2023 Wiley-VCH. | ||
4.2. Formation and stabilization of small Ag clusters in PICs
Cs+ and electrons can be stored cooperatively in the PIC comprising [PMoVI12O40]3− as follows, upon dispersing the PIC in water and adding CsCl and ascorbic acid as a reducing reagent (red stands for the reduced state)53| (mepyH)[Cr3O(OOCH)6(mepy)3]2[PMoVI12O40] [I] + e− + Cs+ → Cs2[Cr3O(OOCH)6(mepy)3]2[PMoVI11MoVO40] [I-red-Cs] (mepy = 4-methylpyridine) | (3) |
Upon immersing I-red-Cs, which stores Cs+ and electron viareaction (3), in an aqueous solution containing AgNO3, ion-exchange between Cs+ and Ag+ and subsequent formation of Ag0 by electron-transfer from the POM to Ag+ occurs, followed by the self-assembly of the Ag species into small Ag clusters (reaction (4), ox stands for the oxidized state).12
| Cs2[Cr3O(OOCH)6(mepy)3]2[PMoVI11MoVO40] [I-red-Cs] + 2Ag+ → Ag0AgI[Cr3O(OOCH)6(mepy)3]2[PMoVI12O40] + 2Cs+ [I-ox-Ag] | (4) |
On the other hand, simple ion-exchange (IE) between Cs+ and Ag+ occurs by immersing I-ox-Cs, which is obtained by oxidizing I-red-Cs with an aqueous solution of chlorine (reaction (5), ox stands for the oxidizing reagent), in an aqueous solution containing AgNO3 (reaction (6)).
| Cs2[Cr3O(OOCH)6(mepy)3]2[PMoVI11MoVO40] [I-red-Cs] + ox → Cs[Cr3O(OOCH)6(mepy)3]2[PMoVI12O40] [I-ox-Cs] + e− + Cs+ | (5) |
| Cs[Cr3O(OOCH)6(mepy)3]2[PMoVI12O40] [I-ox-Cs] + Ag+ → AgI[Cr3O(OOCH)6(mepy)3]2[PMoVI12O40] + Cs+ [I-ox-AgIE] | (6) |
The reduction-induced ion-exchange (Fig. 9a, reaction (4)) occurs much faster than the simple ion-exchange (Fig. 9b, reaction (6)). I-ox-Ag is emissive while I-ox-AgIE is completely dark (Fig. 9c and d), suggesting the formation of small Ag clusters in I-ox-Ag. This difference clearly indicates that the formation of small Ag clusters was triggered by electron-transfer from the reduced POM to Ag+ and the subsequent formation of Ag0. Since a bright green emission is observed for [Ag4]2+ in zeolites,33 formation of [Ag4]2+ is suggested in I-ox-Ag. This study demonstrates that PICs are promising candidates as scaffolds for the formation of small Ag clusters.
 | ||
| Fig. 9 Time course of the amounts of Cs or Ag in (a) reduction-induced ion-exchange and (b) simple ion-exchange reactions. (c) PL spectrum of I-ox-Ag with excitation at 405 nm and (d) time course of PL images. Reprinted with permission from Uchida et al. Copyright 2019 Royal Society of Chemistry. | ||
Because the activation sites for the commercial Ag/Al2O3 ethylene and propylene epoxidation catalyst are Agn (n = 2–4) clusters, we aimed to utilize the porosity and [Ag4]2+ in I-ox-Ag for the selective adsorption of unsaturated hydrocarbons. Gas adsorption studies have found that [Ag4]2+ exhibits high affinity toward acetylene and ethylene,12 suggesting the potential of Ag cluster containing PICs as adsorbents and solid catalysts.
4.3. Analysis of the state of silver clusters by combining multiple measurements
In our previous study,12 the assignment of Ag species ([Ag4]2+) was made based on the emission color (green) and composition. Therefore, further investigation is necessary to obtain detailed information on the states of Ag clusters in PICs. Hereafter, we will introduce a combination of techniques including PL, XPS, and XAFS measurements to investigate the state of Ag species in PICs. We utilized K3[Cr3O(OOCH)6(etpy)3]3[P2WVI18O62] [II] (etpy = 4-ethylpyridine) as a PIC scaffold to generate small Ag clusters via the following reactions.13| K3[Cr3O(OOCH)6(etpy)3]3[P2WVI18O62] [II] + 5e− + 5H+ + 3Cs+ → Cs3H5[Cr3O(OOCH)6(etpy)3]3[P2WVI13WV5O62] [II-red-Cs] + 3K+ | (7) |
| Cs3H5[Cr3O(OOCH)6(etpy)3]3[P2WVI13WV5O62] [II-red-Cs] + 3Ag+ → Ag3H4.5[Cr3O(OOCH)6(etpy)3]3[P2WVI15WV3O62] [II-ox-Ag] + 3Cs++ 0.5H+ | (8) |
Compound II-ox-Ag showed green emission similar to that of our previous work (I-ox-Ag),12 suggesting the formation of Ag4n+ (Fig. 10a and b). Then, we determined the oxidation state of Ag in II-ox-Ag by XPS. The narrow-scan XPS spectra of II-ox-Ag (Fig. 10c and d) suggest that 2 electrons were transferred from the reduced PIC to Ag+viareaction (8), and Ag0 and Ag+ existed in the ratio of 1:1. To gain information on the average size of the Ag cluster, XAFS measurement was carried out. The FT-EXAFS analysis revealed that the Ag atoms in II-ox-Ag have an average coordination number (CNAg–Ag) of 4.0, much smaller than CNAg–Ag of the bulk sample (CNAg–Ag = 12.6 obtained for Ag foil). This result indicates the formation of cluster-sized Ag species in II-ox-Ag. The Debye–Waller factor was small, suggesting that the Ag cluster has a symmetric geometry. Based on these results, the Ag species in II-ox-Ag could be reasonably assigned to [Ag4]2+.56,57 Besides, Ag–O was not clearly observed, indicating that the PIC contains ligand-free small Ag clusters, highlighting the uniqueness of this system.
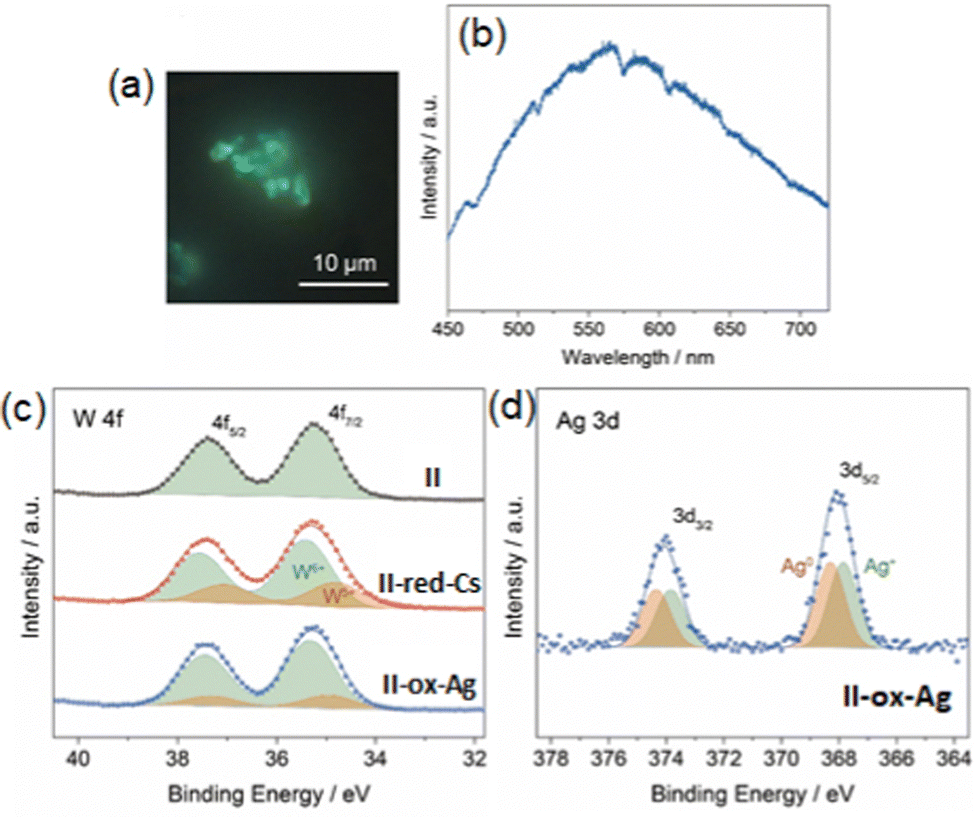 | ||
| Fig. 10 PL (a) image and (b) spectrum of II-ox-Ag with excitation at 405 nm. Narrow scan XPS spectra (c) W 4f and (d) Ag 3d of with deconvolution. Reprinted with permission from Haraguchi et al. Copyright 2021 Wiley-VCH. | ||
4.4. Size-controlled synthesis of small Ag clusters in PICs with sole parameters
As summarized above, while Ag clusters of various sizes can be formed and stabilized in Ag-zeolite composites, which can be considered as analogues of Ag-PICs, the size of Ag clusters is only empirically controlled by parameters such as topology of zeolite structures, Ag loadings, type/amount of counter cations, temperature, heat treatment, and other activation methods. Therefore, our next objective was to strategically control the size of Ag clusters with parameters such as crystal structures, redox properties, reaction times, and counter cations of the PICs. Then, we employed isostructural redox-active PICs A3[Cr3O(OOCH)6(etpy)3]3[P2MVI18O62] (A = K or NH4; M = Mo or W) as scaffolds to systematically investigate the parameters for controlling the Ag cluster size in PICs.14 Hereafter, these PICs will be denoted as IIAM(t1, t2). In this abbreviation, A and M show the counter cation (K+ or NH4+) and metal elements contained in POMs (Mo or W), respectively, and t1 and t2 show the reaction times for reducing the PIC and Ag introduction, respectively.Firstly, we synthesized small Ag clusters in a similar manner to our previous study (reaction (9) and (10)).13 We note that the time for reduction (t1) is a variable, which can control the degree of reduction (i.e., the number of stored electrons in PICs), to investigate the effect of electron transfer from PICs to Ag+ on cluster formation.
| K3[Cr3O(OOCH)6(etpy)3]3[P2MoVI18O62] + xCs+ + yH+ + 7.8e− → Cs3.3H7.5[Cr3O(OOCH)6(etpy)3]3[P2MoV7.8MoVI10.2O62] (t1 = 60 min) | (9) |
| Cs3.3H7.5[Cr3O(OOCH)6(etpy)3]3[P2MoV7.8MoVI10.2O62] + zAg+ → Ag3.9H6.9[Cr3O(OOCH)6(etpy)3]3[P2MoV5.2MoVI12.8O62] + 3.3Cs+ (t2 = 60 min) | (10) |
Then, we synthesized samples with long or short t1 for the three isostructural PICs and characterized these samples to investigate the parameters influencing the cluster size. In Fig. 11, the PL spectra of samples with long t1 (Fig. 11a–c, IIKMo(60, 60), IIKW(60, 60), and IINH4Mo(120, 60)) and short t1 (Fig. 11d–f, IIKMo(1, 60), IIKW(1, 60), and IINH4Mo(5, 60)) are shown. By comparing samples with long and short t1 for each PIC, we observed that the maximum emission wavelength (λmax) is red-shifted with an increase in t1. It is well known that λmax of small Ag clusters generally red-shifts with increasing size.58 Therefore, these results indicate that the cluster size formed in samples with longer t1 is larger than those with shorter t1 for all three PIC scaffolds (IIKMo, IIKW, and IINH4Mo).
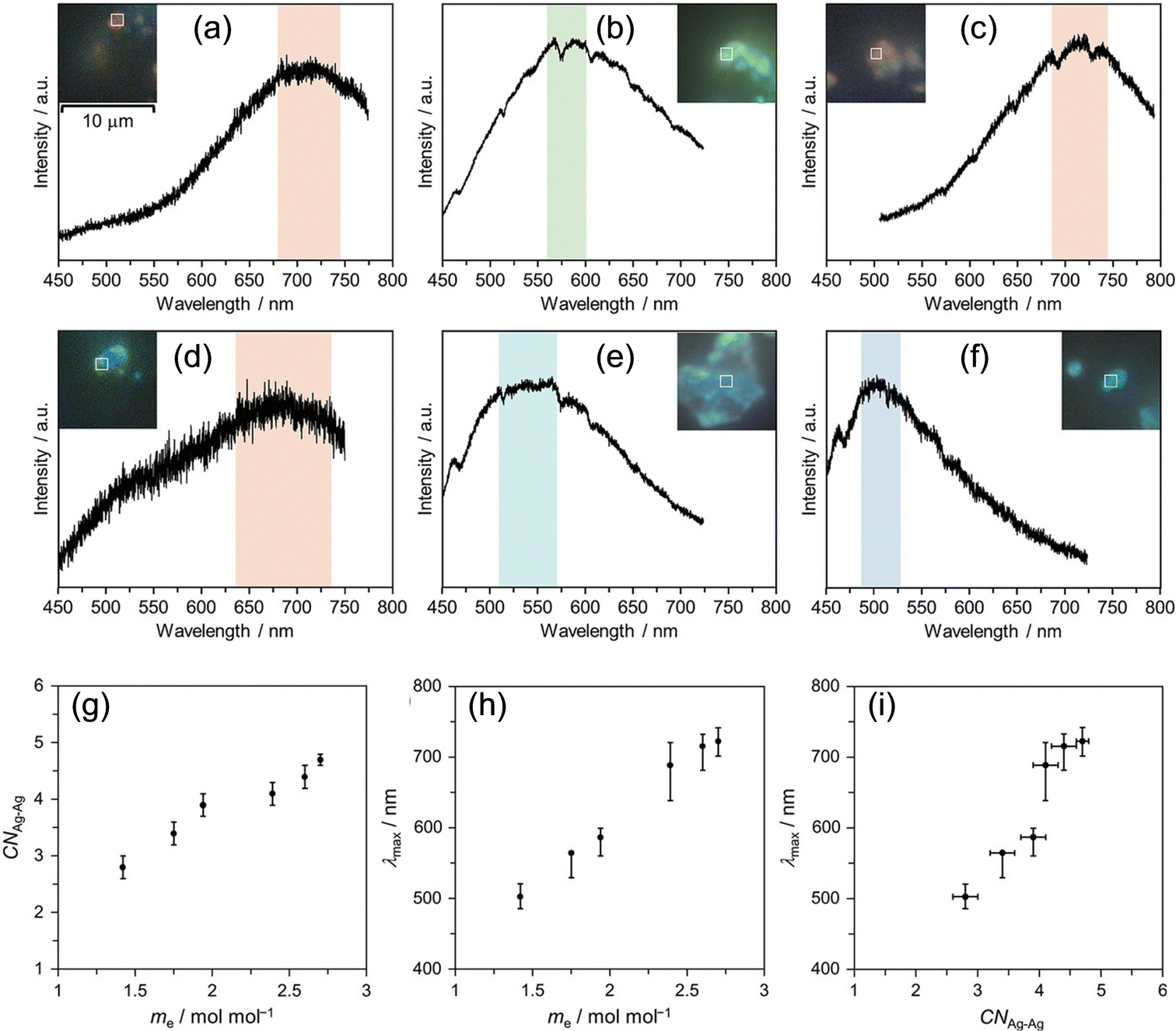 | ||
| Fig. 11 PL images and spectra of (a) IIKMo(60, 60), (b) IIKW(60, 60), (c) IINH4Mo(120, 60), (d) IIKMo(1, 60), (e) IIKW(1, 60), and (f) IINH4Mo(5, 60). Each spectrum is the sum of PL emitted from the area indicated by the white open square. (g) CNAg–Agvs. me (number of transferred electron), (h) λmaxvs. me, and (i) λmaxvs. CNAg–Ag. The error bar of λmax is estimated by extracting the top 10% intensity range from the smoothed PL spectrum drawn by a moving average of five measurement points. Reprinted with permission from Haraguchi et al. Copyright 2023 Wiley-VCH. | ||
These findings indicate that the degree of electron transfer from PICs to Ag+ influences the size of Ag clusters formed in PICs. To clarify this trend, we conducted XAFS and XPS to investigate the correlation between cluster size and degree of electron transfer. Fig. 11g–i show the trends of CNAg–Ag and λmax as a function of the average amounts of electrons transferred from POM to Ag+ in the PICs. These figures clearly illustrate that the size and emission color of the Ag clusters correlate with the degree of electron transfer. The combination of elemental analysis (Ag loading), XPS (Ag 3d, W 4f, and Mo 3d regions to obtain the degree of electron transfer from POM to Ag and charge balance in the chemical formula), and XAFS (CN of Ag clusters) measurements reveals that the blue, green, and red emissions observed in IINH4Mo(5, 60), IIKW(60, 60), and IIKMo(60, 60)/IINH4Mo(120, 60), respectively, can be attributed to [Ag3]+, [Ag4]2+, and [Ag6]2+, respectively. Therefore, it is shown that Ag cluster size can be controlled with a sole parameter, the degree of electron transfer, in the Ag-PICs system.
5. Summary and outlook
In this perspective, we provided an overview and categorized small Ag clusters formed and stabilized in porous crystalline solids, namely MOFs, zeolites, and PICs. Future challenges in utilizing MOFs include the synthesis of ligand-free metal clusters, which could result in physical properties different from those of ligand-protected ones. In fact, ligand-free Au25 cluster was successfully synthesized in the pores of a MOF (UiO-66) through in situ self-assembly of Zr6Ox clusters, Au25(Capt)18 (Capt = captopril), and benzene-1,4-dicarboxylate under ambient conditions followed by heating under vacuum.59 The ligand-free Au25 cluster exhibited much superior stability and catalytic activity compared with the pristine clusters. This method could also be applied to stabilize ligand-free luminescent Ag clusters in MOFs. Regarding the zeolite scaffold, numerous studies mention the structure-function (luminescence) relationship, which will facilitate the development of rational design rules for luminescent base metal60 and metal alloy clusters in zeolites.The PIC scaffold, a relatively new member of the porous crystalline solids family, utilizes the redox property of POMs. The Ag cluster size is controlled by the degree of electron transfer from the POM to Ag+. No external stimuli or reducing reagents are required, simplifying the Ag cluster synthesis method. In particular, the ligand-free Ag clusters formed in PIC scaffolds exhibit remarkable stability in the presence of air, maintaining their emission color even after one year.14 However, the size distribution of these clusters is relatively larger than those formed in zeolite, and achieving greater homogeneity in size remains a challenge. Our future challenge is to synthesize monodisperse Ag clusters by investigating the kinetics of Ag cluster formation, considering factors such as the choice of synthetic solvents, reaction temperature, Ag content, and the method of reduction, whether by reagents, electrochemical, or photochemical methods.
Author contributions
All authors contributed to collecting information and writing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the JSPS Grants-in-Aid for Scientific Research from MEXT of Japan (JP23K17952, JP22H04914, JP21K18975, and JP20H02750), the International Network on Polyoxometalate Science at Hiroshima University, JSPS Core-to-Core program, and the joint research program No. R03003 and R04020 of Molecular Photoscience Research Center, Kobe University. N. H. wishes to acknowledge support from JSPS research fellowship for Young Scientist (JP22J23130).Notes and references
- K. Yonesato, D. Yanai, S. Yamazoe, D. Yokogawa, T. Kikuchi, K. Yamaguchi and K. Suzuki, Nat. Chem., 2023, 15, 940–947 CrossRef CAS PubMed.
- Y. Chai, W. Shang, W. Li, G. Wu, W. Dai, N. Guan and L. Li, Adv. Sci., 2019, 6, 1900299 CrossRef PubMed.
- J. Zheng, C. Zhang and R. M. Dickson, Phys. Rev. Lett., 2004, 93, 077402 CrossRef PubMed.
- T. Sun and K. Seff, Chem. Rev., 1994, 94, 857–870 CrossRef CAS.
- R. Seifert, A. Kunzmann and G. Calzaferri, Angew. Chem., Int. Ed., 1998, 37, 1521–1524 CrossRef.
- K. LaiHing, P. Y. Cheng and M. A. Duncan, Z. Phys. D, 1989, 13, 161–169 CrossRef CAS.
- Z. Wu, E. Lanni, W. Chen, M. E. Bier, D. Ly and R. Jin, J. Am. Chem. Soc., 2009, 131, 16672–16674 CrossRef CAS.
- R. W. Huang, Y. S. Wei, X. Y. Dong, X. H. Wu, C. X. Du, S. Q. Zang and T. C. W. Mak, Nat. Chem., 2017, 9, 689–697 CrossRef CAS PubMed.
- X.-Y. Dong, H.-L. Huang, J.-Y. Wang, H.-Y. Li and S.-Q. Zang, Chem. Mater., 2018, 30, 2160–2167 CrossRef CAS.
- E. Coutiño-Gonzalez, W. Baskelant, J. A. Steele, C. W. Kim, M. B. J. Roeffaers and J. Hofkens, Acc. Chem. Res., 2017, 50, 2353–2361 CrossRef PubMed.
- L. Sun, M. Keshavarz, G. Romolini, B. Dieu, J. Hofkens, F. de Jong, E. Fron, M. B. J. Roeffaers and M. Van der Auweraer, Chem. Sci., 2022, 13, 11560–11569 RSC.
- S. Uchida, T. Okunaga, Y. Harada, S. Magira, Y. Noda, T. Mizuno and T. Tachikawa, Nanoscale, 2019, 11, 5460–5466 RSC.
- N. Haraguchi, T. Okunaga, Y. Shimoyama, N. Ogiwara, S. Kikkawa, S. Yamazoe, M. Inada, T. Tachikawa and S. Uchida, Eur. J. Inorg. Chem., 2021, 1531–1535 CrossRef CAS.
- N. Haraguchi, N. Ogiwara, Y. Kumabe, S. Kikkawa, S. Yamazoe, T. Tachikawa and S. Uchida, Small, 2023, 19, e2300743 CrossRef PubMed.
- X. Y. Dong, Y. Si, J. S. Yang, C. Zhang, Z. Han, P. Luo, Z. Y. Wang, S. Q. Zang and T. C. W. Mak, Nat. Commun., 2020, 11, 3678 CrossRef CAS PubMed.
- R. W. Huang, X. Y. Dong, B. J. Yan, X. S. Du, D. H. Wei, S. Q. Zang and T. C. W. Mak, Angew. Chem., Int. Ed., 2018, 57, 8560–8566 CrossRef CAS.
- R. J. T. Houk, B. W. Jacobs, F. E. Gabaly, N. N. Chang, A. A. Talin, D. D. Graham, S. D. House, I. M. Robertson and M. D. Allendorf, Nano Lett., 2009, 9, 3413–3418 CrossRef CAS PubMed.
- E. Tiburcio, Y. Zheng, M. Mon, N. Martin, J. Ferrando Soria, D. Armentano, A. Leyva Perez and E. Pardo, Inorg. Chem., 2022, 61, 11796–11802 CrossRef CAS PubMed.
- Database of Zeolite Structures, https://www.iza-structure.org/databases/.
- M. Rálek, P. Jírů, O. Grubner and H. Beyer, Collect. Czech. Chem. Commun., 1962, 27, 142–146 CrossRef.
- G. A. Ozin, F. Hugues, S. M. Mattar and D. F. McIntosh, J. Phys. Chem., 1983, 87, 3445–3450 CrossRef CAS.
- D. Brühwiler, R. Seifert and G. Calzaferri, J. Phys. Chem. B, 1999, 103, 6397–6399 CrossRef.
- L. R. Gellens, J. V. Smith and J. J. Pluth, J. Am. Chem. Soc., 1983, 105, 51–55 CrossRef CAS.
- Y. Kim and K. Seff, J. Am. Chem. Soc., 1978, 100, 6989–6997 CrossRef CAS.
- Y. Kim and K. Seff, J. Am. Chem. Soc., 1978, 100, 3801–3805 CrossRef CAS.
- H. Hoshino, Y. Sannohe, Y. Suzuki, T. Azuhata, T. Miyanaga, K. Yaginuma, M. Itoh, T. Shigeno, Y. Osawa and Y. Kimura, J. Phys. Soc. Jpn., 2008, 77, 064712 CrossRef.
- G. De Cremer, E. Coutiño-Gonzalez, M. B. J. Roeffaers, B. Moens, J. Ollevier, M. Van der Auweraer, R. Schoonheydt, P. A. Jacobs, F. C. De Schryver, J. Hofkens, D. E. De Vos, B. F. Sels and T. Vosch, J. Am. Chem. Soc., 2009, 131, 3049–3056 CrossRef CAS.
- G. De Cremer, E. Coutino-Gonzalez, M. B. Roeffaers, D. E. De Vos, J. Hofkens, T. Vosch and B. F. Sels, ChemPhysChem, 2010, 11, 1627–1631 CrossRef CAS PubMed.
- A. Mayoral, T. Carey, P. A. Anderson, A. Lubk and I. Diaz, Angew. Chem., Int. Ed., 2011, 50, 11230–11233 CrossRef CAS PubMed.
- E. Coutino-Gonzalez, W. Baekelant, D. Grandjean, M. B. J. Roeffaers, E. Fron, M. S. Aghakhani, N. Bovet, M. Van der Auweraer, P. Lievens, T. Vosch, B. Sels and J. Hofkens, J. Mater. Chem. C, 2015, 3, 11857–11867 RSC.
- O. Fenwick, E. Coutino-Gonzalez, D. Grandjean, W. Baekelant, F. Richard, S. Bonacchi, D. De Vos, P. Lievens, M. Roeffaers, J. Hofkens and P. Samori, Nat. Mater., 2016, 15, 1017–1022 CrossRef CAS.
- S. Aghakhani, D. Grandjean, W. Baekelant, E. Coutino-Gonzalez, E. Fron, K. Kvashnina, M. B. J. Roeffaers, J. Hofkens, B. F. Sels and P. Lievens, Nanoscale, 2018, 10, 11467–11476 RSC.
- D. Grandjean, E. Coutiño-Gonzalez, N. T. Cuong, E. Fron, W. Baekelant, S. Aghakhani, P. Schlexer, F. D’Acapito, D. Banerjee, M. B. J. Roeffaers, M. T. Nguyen, J. Hofkens and P. Lievens, Science, 2018, 361, 686–690 CrossRef CAS PubMed.
- E. Fron, S. Aghakhani, W. Baekelant, D. Grandjean, E. Coutino-Gonzalez, M. Van der Auweraer, M. B. J. Roeffaers, P. Lievens and J. Hofkens, J. Phys. Chem. C, 2019, 123, 10630–10638 CrossRef CAS.
- E. Johan, Y. Kanda, N. Matsue, Y. Itagaki and H. Aono, J. Lumin., 2019, 213, 482–488 CrossRef CAS.
- T. Altantzis, E. Coutino-Gonzalez, W. Baekelant, G. T. Martinez, A. M. Abakumov, G. V. Tendeloo, M. B. Roeffaers, S. Bals and J. Hofkens, ACS Nano, 2016, 10, 7604–7611 CrossRef CAS PubMed.
- G. De Cremer, Y. Antoku, M. B. J. Roeffaers, M. Sliwa, J. Van Noyen, S. Smout, J. Hofkens, D. E. De Vos, B. F. Sels and T. Vosch, Angew. Chem., Int. Ed., 2008, 47, 2813–2816 CrossRef CAS PubMed.
- G. De Cremer, B. F. Sels, J.-I. Hotta, M. B. J. Roeffaers, E. Bartholomeeusen, E. Coutiño-Gonzalez, V. Valtchev, D. E. De Vos, T. Vosch and J. Hofkens, Adv. Mater., 2010, 22, 957–960 CrossRef CAS.
- Y. Sasaki and T. Suzuki, Mater. Trans., 2009, 50, 1050–1053 CrossRef CAS.
- E. Coutino-Gonzalez, D. Grandjean, M. Roeffaers, K. Kvashnina, E. Fron, B. Dieu, G. De Cremer, P. Lievens, B. Sels and J. Hofkens, Chem. Commun., 2014, 50, 1350–1352 RSC.
- K. Taiji, Y. Iso and T. Isobe, J. Lumin., 2018, 196, 214–220 CrossRef CAS.
- O. Fenwick, E. Coutino-Gonzalez, F. Richard, S. Bonacchi, W. Baekelant, D. de Vos, M. B. J. Roeffaers, J. Hofkens and P. Samori, Small, 2020, 16, e2002063 CrossRef PubMed.
- J. Michalik and L. Kevan, J. Am. Chem. Soc., 1986, 108, 4247–4253 CrossRef CAS.
- L. R. Gellens, W. J. Mortier and J. B. Uytterhoeven, Zeolites, 1981, 1, 85–90 CrossRef CAS.
- K.-i Shimizu, K. Sugino, K. Kato, S. Yokota, K. Okumura and A. Satsuma, J. Phys. Chem. C, 2007, 111, 1683–1688 CrossRef CAS.
- A. Baldansuren, H. Dilger, R.-A. Eichel, J. A. van Bokhoven and E. Roduner, J. Phys. Chem. C, 2009, 113, 19623–19632 CrossRef CAS.
- A. Satsuma, J. Shibata, K.-I. Shimizu and T. Hattori, Catal. Surv. Asia, 2005, 9, 75–85 CrossRef CAS.
- W. Baekelant, S. Aghakhani, E. Coutino-Gonzalez, K. Kennes, F. D'Acapito, D. Grandjean, M. Van der Auweraer, P. Lievens, M. B. J. Roeffaers, J. Hofkens and J. A. Steele, J. Phys. Chem. Lett., 2018, 9, 5344–5350 CrossRef CAS.
- S. Uchida, Chem. Sci., 2019, 10, 7670–7679 RSC.
- Y. Shimoyama and S. Uchida, Chem. Lett., 2021, 50, 21–30 CrossRef CAS.
- A. Lesbani, R. Kawamoto, S. Uchida and N. Mizuno, Inorg. Chem., 2008, 47, 3349–3357 CrossRef CAS PubMed.
- S. Uchida, R. Kawamoto, H. Tagami, Y. Nakagawa and N. Mizuno, J. Am. Chem. Soc., 2008, 130, 12370–12376 CrossRef CAS PubMed.
- R. Kawahara, S. Uchida and N. Mizuno, Chem. Mater., 2015, 27, 2092–2099 CrossRef CAS.
- S. Seino, R. Kawahara, Y. Ogasawara, N. Mizuno and S. Uchida, Angew. Chem., Int. Ed., 2016, 55, 3987–3991 CrossRef CAS PubMed.
- S. Hitose and S. Uchida, Inorg. Chem., 2018, 57, 4833–4836 CrossRef CAS PubMed.
- Ideally, a tetrahedral Ag4 cluster should have a CN of 3 and not 4. The assignment (Ag4) was made based on the following information: (i) many papers indicate that green emission is observed for Ag4 clusters, while larger clusters (e.g., Ag6) shows reddish emission, (ii) the bulk sample gave a CN of 12.6, which is larger than the ideal value of 12, and (iii) PXRD and XPS measurements suggest that a small amount of bulk Ag has formed on the surface of the particles which may contribute to increase the CN.
- It has been recognized that Ag-zeolite is sensitive to electron beam and X-ray irradiation. The CN obtained from EXAFS tends to be larger than the actual value because Ag clusters further sinters by electron beam and X-ray irradiation. Recently, efforts have been made to directly obtain structural information of luminescent species by using X-ray excited optical luminescence (XEOL)-XAFS33.
- A. Gonzalez-Rosell, C. Cerretani, P. Mastracco, T. Vosch and S. M. Copp, Nanoscale Adv., 2021, 3, 1230–1260 RSC.
- H. Wang, X. Liu, W. Yang, G. Mao, Z. Meng, Z. Wu and H. L. Jiang, J. Am. Chem. Soc., 2022, 144, 22008–22017 CrossRef CAS PubMed.
- W. Baekelant, S. Aghakhani, E. Coutino-Gonzalez, D. Grandjean, K. Kennes, D. Jonckheere, E. Fron, F. d’Acapito, A. Longo, P. Lievens, M. B. J. Roeffaers and J. Hofkens, J. Phys. Chem. C, 2018, 122, 13953–13961 CrossRef CAS.
| This journal is © the Owner Societies 2024 |