
New model for aspartic acid species in aqueous calcium carbonate growth environments: challenges and perspectives†
Alicia
Schuitemaker
 abc,
Katarzyna B.
Koziara
c,
Paolo
Raiteri
c,
Julian D.
Gale
c and
Raffaella
Demichelis
*c
abc,
Katarzyna B.
Koziara
c,
Paolo
Raiteri
c,
Julian D.
Gale
c and
Raffaella
Demichelis
*c
aAustralian Research Council Centre of Excellence in Exciton Science, School of Chemistry, University of Sydney, Sydney, New South Wales, Australia
bThe University of Sydney Nano Institute, University of Sydney, Sydney, New South Wales, Australia
cSchool of Molecular and Life Sciences, Curtin University, GPO Box U1987, 6845 Perth, Western Australia, Australia. E-mail: raffaella.demichelis@curtin.edu.au
First published on 29th December 2023
Abstract
The lack of experimental data on the dynamics of aspartic acid species in water for its range of protonation states and the details of their atomic-level interaction with aqueous calcium carbonate species is a driver for accurate force field development. A classical model that is consistent with the few pieces of experimental data available and with first principles calculations has been developed. The complex dynamics of the aspartate anions relevant to biomineralization and calcium carbonate crystal growth has been explored in water, providing a quantitative description of solvation structure and free energies, including conformational free energy profiles and pairing free energies. The model has been used to probe the structure and dynamics of aqueous calcium aspartate homo- and hetero-chiral clusters, confirming their unlikelihood due to weak and water–mediated interactions. This supports the hypothesis that the formation of such clusters, observed while growing vaterite in the presence of acidic chiral amino acids, is favoured by the presence of the crystal surface.
1 Introduction
The ability to model with high accuracy organic molecules of biological relevance in mineral growth environments has the potential to reveal the atomic-level intermolecular details that dominate mineral synthesis during biomineralization. While biomineralization is a complex phenomenon involving a variety of physico-chemical and mechanical processes, biomolecules are considered as the key players in growing, maintaining and repairing the hard tissues of living organisms. In particular, organisms belonging to all living kingdoms can synthesise minerals with specific sizes, compositions, textures and shapes; an ability that goes beyond our current knowledge in materials synthesis.1–4Calcium carbonate is arguably the most widespread and studied biomineral, and proteins, peptides and amino acids are the biomolecules that have received most attention in the context of calcium carbonate biomineralization.5–12 Numerous studies are available that demonstrate how these molecules affect the nucleation and growth of calcium carbonate, acting as crystal structure modifiers, inhibitors, or forming organic–inorganic composites. Recently, non-racemic mixtures of chiral acidic amino acids, such as aspartic and glutamic acids, were found to induce the formation of chiral toroids of vaterite through the formation of homochiral clusters on the mineral surface.13,14
While a certain level of knowledge exists on the macroscopic effects of proteins, peptides and amino acids on the nucleation and growth of calcium carbonate, the fundamental interactions that underpin the process are largely unexplored. The size and time scale of the dynamics of these molecules in aqueous environments has so far limited their theoretical investigation.4 Additionally, the majority of models available for biomolecular simulation15–17 have not been parameterised for this purpose, and therefore may not be directly transferable to investigations in mineral growth environments.
Recently, some of the present authors have started the development of accurate potential models that can bridge this gap. The dynamics of small organic molecules in aqueous environments and their interaction with both calcium carbonate aqueous species and calcite terraces and steps have been investigated with both polarisable and rigid-ion models.18,19 In particular, the simplest molecules with the most relevant functional groups for calcium carbonate biomineralization were considered, i.e. methylammonium, acetate, and the glycine zwitterion.
This work is focused on aspartic acid, its speciation in water and interaction with calcium carbonate aqueous species. While being a relatively simple and small additive, this molecule has two additional levels of complexity with respect to those that have been considered previously: (1) the presence of an additional carboxylate group, offering an additional binding site; (2) the flexibility to change conformation through the torsional angle defined by the four carbon atoms. A rigid-ion potential model presented in this paper has already been used to provide theoretical support to the formation of homochiral aspartic acid clusters as a result of the interaction between vaterite and aspartic acid solutions.14 However, the details of its development and its performance have yet to be examined in detail. Additionally, while numerous computational investigations on aspartic acid species exist, its complex conformational energy profile and speciation have never been thoroughly probed in aqueous environments relevant to biomineralization, while its interaction with calcium and carbonate ions in water have only been superficially discussed without considering the full complexity of this amino acid. Below we highlight the main works that have examined relevant systems to date.
Nada20 modelled the interaction of aspartic acid with the various surface features of calcite. The CHARMM2221 force field was used for aspartic acid, in conjunction with an earlier calcite model from Raiteri et al.22 and TIP3P water.23 This water model yields a self-diffusion coefficient that is twice the experimental value, meaning much faster dynamics occur than in real water, and a dielectric constant that is overestimated by more than 25%, meaning that charged species are over-stabilized with respect to complexes and the solvation structure is altered due to the discrepancy in the correlations in the orientations of water dipoles.24 Additionally, the conformational arrangement of aspartic acid species was not fully explored and their dynamics and thermodynamics in water was not investigated.
Stepić et al.25 performed umbrella sampling of aspartic acid species attachment to the surface of calcite. Here, a similar approach to model choice as in Nada20 was adopted, combining the AMBER15 force field with the aforementioned calcite model from Raiteri et al.22 and TIP3P water.23 Apart from being affected by the same inaccuracies as Nada,20 due to the water potential model, again the flexibility of aspartic acid species to change conformation is not fully explored, as the attachment has been investigated only through the distance between the centre of mass of the molecule and the surface as a collective variable. Additionally, Stepić et al.'s results suggest preferential attachment to calcite via the amino group, rather than the carboxylate, which is counter-intuitive, while strongest binding for the monomer occurs on the terraces, rather than at steps or kinks. In contrast, Njegić-Džakula et al.9 and Wierzbicki et al.5 experimentally found attachment of polyaspartic acid and aspartic acid, respectively, to calcium sites on the calcite surface through chain carboxylate groups. Montanari et al.12 showed that aspartic acid competes with carbonate ions to attach to the surface of calcite (hence suggesting preferential attachment to calcium sites). Additionally, Verch et al.26 classified polyaspartic acid as an additive that binds to calcium in solution, though it is suggested that carboxylate groups are not solely responsible for the binding.
Innocenti Malini et al.27 simulated the effect of a few different amino acids, including aspartic acid, on amorphous calcium carbonate. This model combines the ff12sc AMBER28 force field with a more recent version of Raiteri et al.'s29 model, using the SPC/Fw water model. This is an improvement with respect to earlier force fields, and provides a binding free energy of aspartic acid with calcium that is in much better agreement with the experimental values than a previous model by some of the present authors,30 using the same calcium carbonate and water parameterisations. However, the dynamics of aspartic acid species in water, the energy profile of their interaction with water and dissolved ions was not examined. Additionally, also in this case it was shown that aspartic acid binds to calcium carbonate preferentially via the protonated amino group, which acts as a hydrogen bond donor. This is in contrast with the aforementioned pieces of experimental evidence, which do not exclude this kind of interaction, but suggest binding at biological pH occurs primarily through carboxylate groups.5,9,12,26 A similar model, though based on the ft14sp28 parameterisation of AMBER, was used by Finney et al.31 to simulate the effect of several amino acids, including aspartic acid, on calcium carbonate speciation at different concentrations in aqueous solution.
Recently, Koskamp et al.32 calibrated a model for the fully deprotonated aspartate species (pH = 10) through combining parameters from the ff99SB version of AMBER33 (aspartic acid, originally developed in combination with TIP3P23 water) and from Raiteri et al.29 (calcium carbonate and SPC/Fw water24) using combination rules. While the parameters between calcium and the carboxylate oxygen in aspartate were successfully calibrated to fit the numerical value of the experimental calcium aspartate association free energy at that pH, the model was not reparametrised to predict other properties that affect ion pairing, including the hydration free energy, conformational free energy profile, and pair distribution function.
As mentioned above, the force field developed in this paper is a first step towards being able to capture the complexity of aspartic acid species in environments relevant to crystal growth and biomineralization. In particular: (a) one of the most accurate models for liquid water at room temperature is used; (b) the model includes two different aspartic acid protonation states that are relevant to the biomineralization and crystal growth fields; (c) hydration free energies, water structure of the solvation shell, and conformational free energy profile of aspartic acid species are in agreement with first principles data; (d) ion pairing free energy values with calcium are in line with experiment. Despite lacking explicit inclusion of polarisability, the careful thermodynamic calibration means that it represents an improvement with respect to other existing models and a starting point for future applications to aqueous organic-biomineral interfaces.
2 Computational methods
Two aspartic acid anions were considered in this study; the negative zwitterion, namely Asp ZW−, which is the predominant species at typical pH values for biomineralization, and the fully deprotonated anion, namely Asp2−, which is the predominant species in highly alkaline environments, such as in recent experiments showing the growth of chiral toroids of vaterite in the presence of aspartic acid.14All classical simulations were run using the LAMMPS34 code, with a time step of 1 fs and trajectories recorded every 1 ps. Cubic boxes containing ∼520 molecules (corresponding to a cell length of ∼24.860 Å) were used to analyse the conformational energy profile and hydration of the aspartate anions, and cubic boxes containing ∼4175 molecules (cell length ∼49.969 Å) were used to explore the pairing free energy with calcium and the formation of clusters.
In all simulations the temperature (300 K) was controlled using a chain of five Nosé–Hoover thermostats with a relaxation time of 0.1 ps. For simulations in the isobaric–isothermal (NPT) ensemble, the pressure (1 atm) was maintained using a chain of five Nosé–Hoover barostats with a relaxation time of 1 ps. A MTK barostat35 was additionally applied to reproduce the true volume probability distribution of the NPT ensemble.
All multiple-walker36 well-tempered37 metadynamics38,39 simulations were run using the Plumed40 package. Gaussian functions with a width of 0.2 (radians or Å, depending on the collective variable being an angle or a distance, respectively) and 0.1 (dimensionless, coordination number) were deposited every 1 ps, with an initial height of kBT. Collective variables in all simulations used a bias factor of 5 to progressively reduce the heights of the Gaussian functions until convergence was achieved.
The collective variables used in this study, as defined in the Plumed manual40,41 and with atoms labelled as in Fig. 1, are listed below and their use is summarised in Table 1:
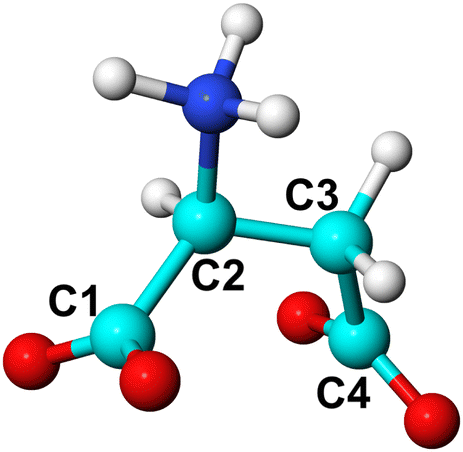 | ||
| Fig. 1 Ball and stick representation of L-Asp ZW−. C atoms (cyan) are labelled to define the C1–C2–C3–C4 torsional angle. O, H and N atoms are coloured in red, white and blue, respectively. The same labelling is used for L-Asp2−, with the amino group deprotonated, and for the D-enantiomers. | ||
| Asp | Ca + Asp | CaAsp + Asp | CaAsp2 + Ca | |
|---|---|---|---|---|
| d1 | ✓ | |||
| d2 | ✓ | |||
| d3 | ✓ | ✓ | ||
| d4 | ✓ | |||
| Torsional angle | ✓ | ✓ | ||
| Water coordination | ✓ | ✓ |
• Distance between Ca and C1 (d1).
• Distance between Ca and C4 (d2).
• Distance between Ca and the centre of mass (COM) of aspartic acid species (d3).
• Distance between Ca in water and Ca in cluster (d4).
• Torsional angle of aspartic acid species (C1–C2–C3–C4).
• Coordination of Ca2+ by water molecules (with d0 = 2.3 Å, r0 = 1.4 Å, n = 4, m = 10).
• Coordination of L-Asp ZW− by water molecules (with d0 = 3.1 Å, r0 = 1.4 Å, n = 4, m = 10).
Further details of the force field and the computational setup of the various types of simulations are reported in the following subsections and as ESI.†
2.1 Force field parameterisation
The potential model developed by Raiteri et al.29 for calcium carbonate, combined with the SPC/Fw water model by Wu et al.,24 was used. Although we have recently published an updated model for aqueous calcium carbonate simulation,42 the development of the organic parameters pre-dated this development. The force field for both aspartate anions was derived from GROMOS43 with intermolecular parameters obtained from the Automated Topology Builder (ATB) and Repository.44Due to the scarcity of experimental information regarding the structure of water around aspartic acid species, the interactions with water were fitted to match the pair distribution function obtained from ab initio molecular dynamics and refined against ab initio solvation free energies. Here, Lennard-Jones (LJ and LJ-AB) and Buckingham potentials were used. Charges were also fitted against quantum mechanical data.
The aspartate–aspartate interaction parameters were adopted from the GROMOS force field as obtained from the ATB and Repository without change. The interactions of aspartate with carbonate were derived from the methylammonium–carbonate interactions with the GROMOS force field as obtained from the ATB and Repository without change and from intermolecular carboxylate–carboxylate interactions of aqueous aspartate. The interactions between calcium and aspartate were derived from fitting of Buckingham potentials to gas phase quantum mechanical potential energy surface data, at the ωB97X-D3/ma-def2-QZVPP level of theory, for a Ca2+ ion interacting with acetate (to derive the Ca–carboxylate repulsion) and methyl amine (for the Ca–N repulsion). A short-range repulsive Lennard-Jones potential for the interaction between calcium and the carboxylate carbon was added to prevent unphysical configurations from occurring in instances where the ion was above the plane of this functional group, as opposed to in the natural bidentate binding structure. More details regarding the parameterisation of the force field for methylammonium and acetate are available in Schuitemaker et al.18 Additional details and the full set of parameters are reported as ESI.†
2.2 Hydration structure
The water structure around the aspartate anions was explored using unbiased classical and ab initio molecular dynamics simulations in an isochoric–isothermal (NVT) ensemble. Classical simulations were run for 10 ns, with the setup described above.Ab initio simulations were run using the CP2K45 program with the BLYP-D3 functional and a Gaussian planewave basis set. Core electrons and nuclei were described using GTH pseudopotentials46 with valence basis sets of TZ2P quality. The electron density was described with an auxiliary basis set of planewaves with a cutoff of 400 Ry, while the self-consistent field calculation was performed using the orbital transformation algorithm.47 Asp ZW− was placed in a cubic cell with length 13.774 Å containing 85 water molecules using an initial configuration derived from equilibration with classical molecular dynamics. Simulations were run for up to 50 ps in the NVT ensemble using a time step of 0.5 fs and the CSVR thermostat. A temperature of 330 K was used to compensate for the known systematic over-structuring of liquid water with GGA functionals,48 despite the inclusion of an empirical dispersion49 correction. Asp2− was treated with a similar procedure, except that the box length was 13.088 Å, containing 70 water molecules, and run for up to 32 ps.
2.3 Hydration free energies
Free energy perturbation50 was run in the NPT ensemble by progressively switching off the van der Waals’ and electrostatic interactions between the anions and water (20 equally spaced stages of 5 ns each per interaction, with 500 ps equilibration). To account for the partial screening of the 1–4 interactions leading to a non-zero internal energy, an additional gas-phase calculation was performed to remove the electrostatic interactions. The calculated hydration free energy values were then corrected for finite size effects.51Due to scarce experimental reference values, quantum mechanical calculations were performed to estimate the free energy of hydration. Three different approaches were used to examine the sensitivity of the computed value. In the first approach, the molecules were optimised using Density Functional Theory (DFT) with the M06 meta-GGA hybrid exchange–correlation functional,52 in conjunction with a 6-31+G** Gaussian basis set, with or without the SM8 implicit solvation model53 using QChem version 6.0.1.54 The second set of quantum mechanical calculations using implicit solvent were performed using ORCA55 with the SMD solvation model,56 either alone or with a first solvation shell of 7 explicit waters. For these calculations, the ωB97X-D3 functional and ma-def2-QZVPP basis set were used as they have been shown to yield good thermochemistry against benchmark datasets. In order to extract the hydration free energy for the case where explicit water is included, the cluster method was used with a water hexamer as the reference water cluster.57
2.4 Conformation of aspartate anions in water
In order to estimate the rotational barriers and preferred conformations of both enantiomers of Asp ZW− and Asp2− in water, a scan of the C1–C2–C3–C4 torsional angle (Fig. 1) was conducted in the NVT ensemble using metadynamics as described above. The torsional angle was the only collective variable and 24 individual walkers were run until convergence, resulting in a cumulative simulation time of 240 ns.2.5 Ion pairing and cluster formation free energies
Metadynamics, with the setup described above, was used to simulate the free energy profile of ion pairs and cluster aggregation in water. Subsets of 1 to 3 of the following collective variables were used; the calcium–water coordination, the C–C–C–C torsional angle of the anions, the distance between each carboxylate group with calcium ions (d1, d2) and the distance between COM of the aspartate anions and calcium (d3). For the formation of Ca2Asp2 clusters, the distance Ca–Ca (d4) was used as a collective variable. Details of the collective variables are reported in Table 1 and related discussion. Simulations were run for a total time of >1 μs for the ion pairs and >0.5 μs for the cluster, using 24 parallel walkers. All binding free energies reported in this paper include correction to the standard state.To reduce the collective variable space and speed up sampling, an upper wall at 20 Å was applied to distance collective variables with a spring constant of 0.1 eV Å−2. In cluster simulations, to keep the pre-formed cluster together, a second upper wall at 7.5 Å, again with a spring constant of 0.1 eV Å−2, was applied to the calcium-COM distance(s) that was (were) not being biased.
3 Results and discussion
3.1 Hydration structure and free energy
Interatomic interactions between the aspartate anions and water were parametrised to reproduce pair distribution functions obtained from ab initio molecular dynamics, as well as the ab initio hydration free energy. Fig. 2 shows that the structure of water around aspartic acid functional groups is reproduced with very similar accuracy by the force field and ab initio methods. In particular, very good agreement is observed for the position of the first and the second peaks in all the plots.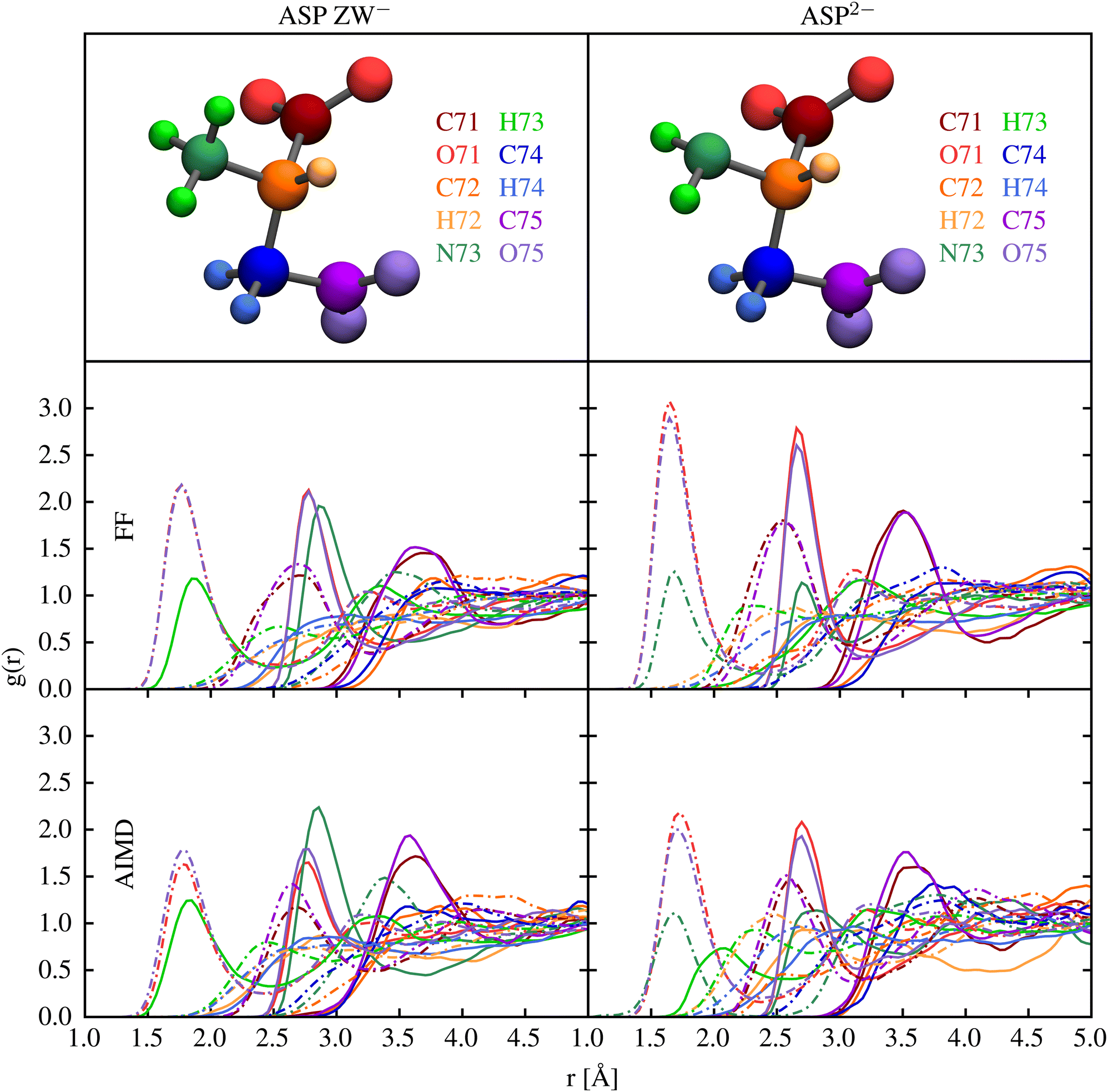 | ||
| Fig. 2 Pair distribution functions, g(r), between the atoms of the aspartic acid species and the hydrogen (dash-dotted lines) and oxygen (solid lines) of water calculated with our force field (FF) and ab initio molecular dynamics (AIMD). Plots are coloured according to the atoms represented in the top figures. | ||
Table 2 summarises the results of calculations of the hydration free energies for the aspartate species. Unfortunately, there are no experimental data to compare against, while the quantum mechanical results show some degree of variation depending on the treatment of solvation. Arguably QM2 is likely to be the most accurate value as it uses the highest level of theory, largest basis set and a shell of explicit water. A complication in comparing the force field hydration free energies to those computed quantum mechanically is that the aspartate zwitterion is unstable in the gas phase due to the proton on the amino group migrating to the carboxylate group. As a result, there is a large relaxation contribution in the quantum mechanical energy that cannot be accounted for by an unreactive force field where bonding is fixed. One solution to this is to focus on the free energy difference between the gas phase and solvated state at the optimised geometry for the aqueous state. Based on this approach, the hydration free energies for method QM1 (labelled as QM1* in the table) would be −356.2 and −945.0 kJ mol−1 for the two species. This substantially reduces the discrepancy between the force field and quantum mechanical data. Attempts to further reduce the difference between the force field and quantum-mechanical hydration free energies were unsuccessful, in that any improvement came at the expense of the reproduction of the pair distribution functions. Use of polarisable force fields in future work may be able to simultaneous describe the hydration thermodynamics and structure.
| FF | QM1* | QM1 | QM2 | QM3 | |
|---|---|---|---|---|---|
| Asp ZW− | −430.6 | −356.2 | −291.5 | −294.9 | −291.1 |
| Asp2− | −1018.1 | −945.0 | −879.1 | −846.8 | −839.4 |
3.2 Conformational energy profiles of aqueous aspartate anions
Comitani et al.58 provided an in-depth overview of the conformational profile of aspartic acid and its neutral zwitterion in the gas phase and in water, respectively, using classical molecular dynamics and ab initio methods. The CHARMM3659 parametrisation was used for aspartic acid species in conjunction with the TIP3P23 water model. While we argued in Section 1 that such models suffer from a number of limitations in the context of biomineralization, this study provides a comparison of the main findings with results obtained with other classical force fields, DFT and second order Møller-Plesset perturbation theory (MP2), highlighting aspects on which there is a very good qualitative agreement between all methods. Therefore, this paper will be used as the main reference in this discussion.Comitani et al.58 identified the full set of torsional angles for the conformers of aspartic acid and of its neutral zwitterion. They highlighted that the majority of states are separated by rotational barriers that are fully accessible by unbiased molecular dynamics calculations, the exception being the C1–C2–C3–C4 torsional angle (named β in the original paper and labelled as C4–C3–C2–C1 with respect to Fig. 1) and the two C–C–O–H torsional angles (δ and ζ). For clarity of comparison with Comitani et al.,58 in the remainder of this paper we will refer to the C1–C2–C3–C4 torsional angle as −β. Both species considered in this paper, i.e. the negative zwitterion Asp ZW− and Asp2−, have both carboxylic acid groups deprotonated, meaning that only −β is relevant to the conformational analysis.
The free energy profile along −β for both species, in their D- and L-enantiomers, is shown in Fig. 3. As expected, Asp ZW− and Asp2− have profiles that are nearly indistinguishable and the enantiomers have mirror-image profiles. Three minima are present at around 60, −60 and ±160 degrees, as also found in Comitani et al.58 (where the last minimum is shifted toward 170 degrees). In particular, L-enantiomers favour the conformation at −β = +60° and the second most stable conformation is at −β = +160°, whereas the D-enantiomers have their absolute free energy minimum at −β = −60° and the second lowest energy minimum at −β = −160°. To confirm that our simulations are fully capturing the dynamics of the molecule, an additional scan of the L-Asp ZW− torsional angle was run for a total of 120 ns using the water coordination number as a second collective variable. The 1-dimensional free energy profiles of the −β torsional angle, projected from the resulting 2-dimensional free energy surface, fully overlap with those obtained above.
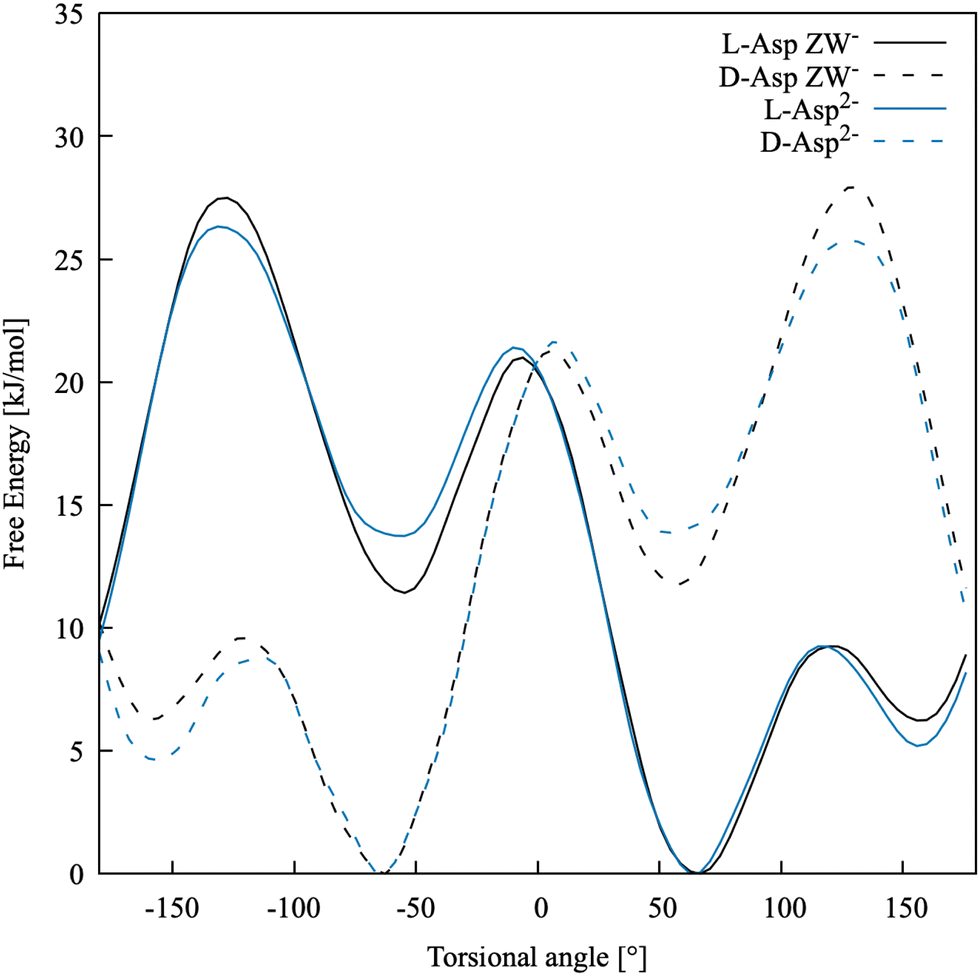 | ||
| Fig. 3 Free energy profiles for the torsional angle of the two enantiomers of Asp ZW− and Asp2− in water. | ||
Table 3 shows the relative free energies of the L-Asp minimum energy conformers. The results of Comitani et al.58 for the zwitterion (L-ZW), calculated with their force field, hybrid DFT (B3LYP) and MP2, are compared with those obtained in this work for the negative zwitterion (L-ZW−). Here, Comitani et al. calculated the energy for all L-ZW minima resulting from the rotation around both β and δ torsional angles, combined. However, δ cannot be defined in ZW− because both carboxylic acid groups are deprotonated. Therefore, there are only three possible minima in L-ZW−, resulting from the rotation around β, and their energy has been compared to the equivalent L-ZW conformer with δ in its most energetically favourable conformation. DFT, MP2 and our force field predict the same stability order for the three conformational minima, with −β = 60° and −β = −60° being the most and least stable ones, respectively. Results obtained with our force field are in reasonable quantitative agreement with MP2 calculations. The force field by Comitani et al.58 predicts the conformation at −β = 170° as the most stable one and the conformation at −β = 60° as slightly more stable than that at −β = −60°.
| L-ZW58 | L-ZW− | |||
|---|---|---|---|---|
| −β | FF | B3LYP | MP2 | FF |
| +60 | 1.8 | 0.0 | 0.0 | 0.0 |
| −60 | 2.0 | 8.6 | 10.8 | 11.4 |
| +170/+160 | 0.0 | 0.9 | 3.9 | 6.2 |
Activation barriers between the three minima are between 3 and 9 kT. A qualitative comparison with the force field results of Comitani et al.58 is reported in Table 4 and shows that with both models the transitions between the conformation at −β = 60° and those at −β = 160° (L-ZW−) and 170° (L-ZW) are the ones with the lowest barrier, whereas the other transitions are more energetically expensive. The main question here is whether classical dynamics simulations are able to access these barriers and to fully explore the conformational space of this molecule. Therefore, we ran 5 ns of unbiased molecular dynamics simulation with our force field model starting from the most stable conformation of L-Asp ZW− at about −β = 60°. Here, the conformation at −β = 160° was visited a few times whereas the conformation at −β = −60° was visited only once. This means that the β torsional angle should be used as a collective variable in free energy calculations to fully capture the dynamics and energy profile of processes involving aspartic acid species. Accelerating the conformational dynamics might be crucial when exploring the binding with other species and mineral surfaces, as the most stable conformation of the free solvated molecule might not necessarily correspond to the most favourable conformation of the same molecule when bound.
| −β | L-ZW58 | L-ZW− |
|---|---|---|
| +60 → +160 (L-ZW−) or +170 (L-ZW) | 18.8–25.1 | 9.3 |
| +60 → −60 | 37.6–44.0 | 21.0 |
| +160 (L-ZW−) or +170 (L-ZW) → −60 | 37.6–44.0 | 21.3 |
3.3 Ion pairing free energies
This section presents a detailed analysis of the ion pairing dynamics and free energy of Ca2+L-Asp ZW−. The choices associated with using different sets of collective variables and the implications of such choices are discussed for this specific case. The same tests are repeated and commented on for Ca2+L-Asp2−. The most convenient set of collective variables is then used to investigate the ion pairing free energy profile and dynamics between calcium and the D-aspartic acid species.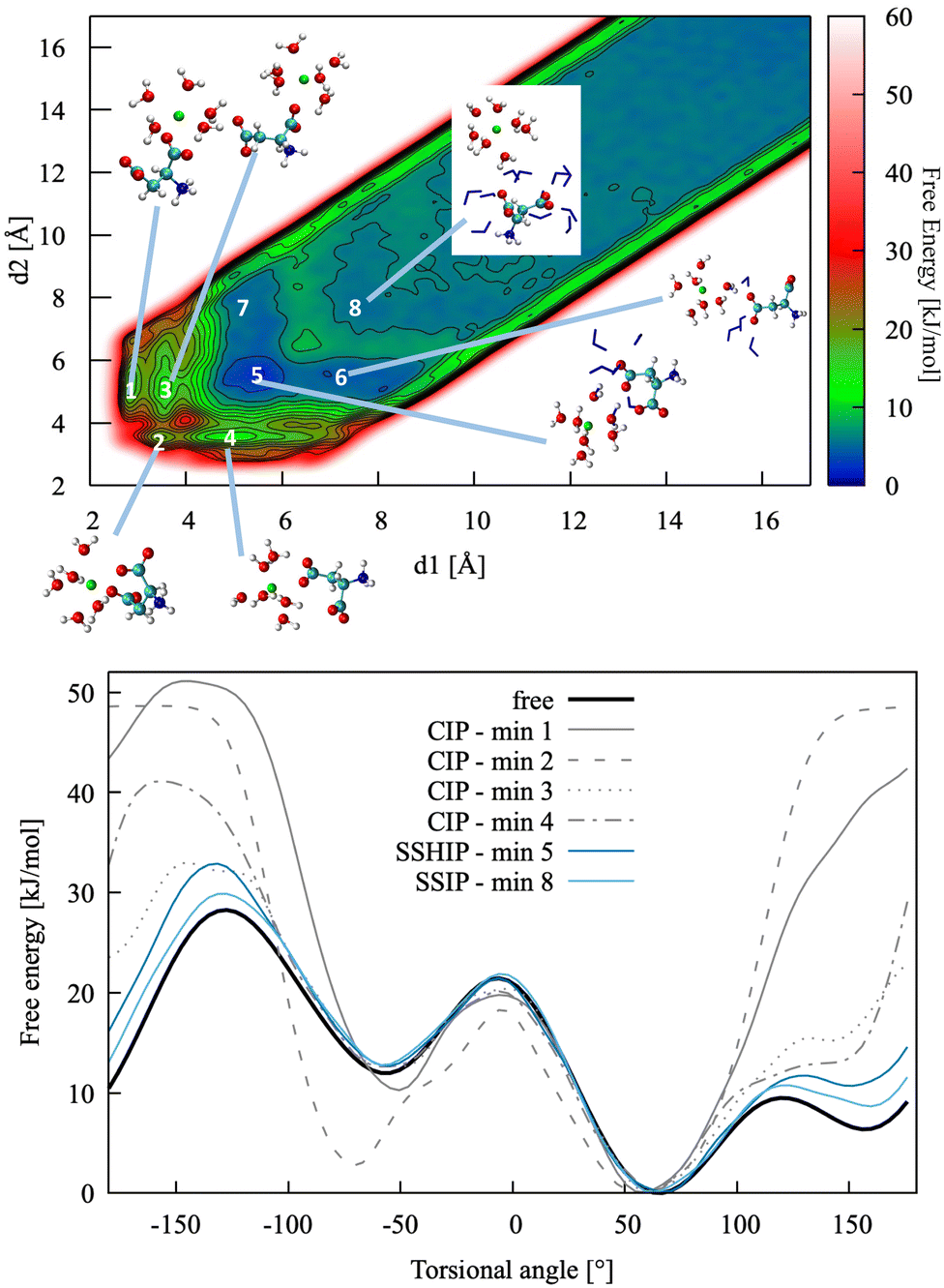 | ||
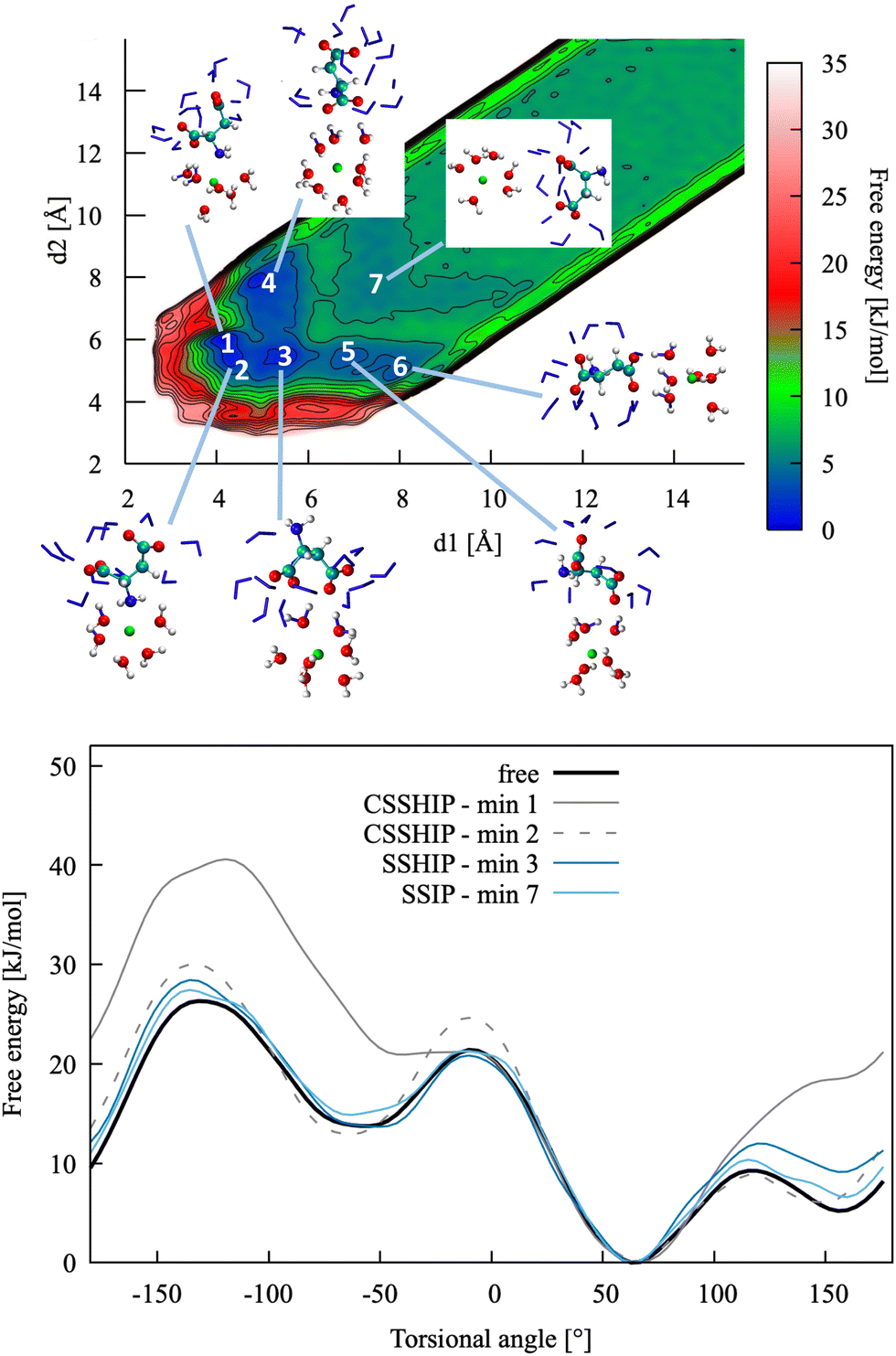
| Fig. 4 Top: Pairing free energy of Ca2+L-Asp ZW− projected along two Ca–C distances, d1 and d2 (defined in Fig. 5). Minima correspond to contact ion pairs (CIP, 1 to 4), solvent-shared ion pairs (SSHIP, 5 to 7) and solvent-separated ion pairs (SSIP, 8). Examples of configurations for each minimum are provided, with Asp atoms coloured as in Fig. 1, Ca atoms coloured in green, water molecules belonging to the calcium solvation sphere coloured in white (H) and red (O), and water molecules belonging to the solvation sphere of the COO− closest to calcium represented as blue sticks. Bottom: Rotational free energy around the −β torsional angle of L-Asp ZW−, free and bound with Ca2+. | ||
In this specific case, the conformational free energy profile changes only for contact ion pairs, which are energetically disfavoured. To assess the effect of including −β as a collective variable on the overall accuracy of the pairing free energy, the latter was also computed using only d1 and d2 as collective variables and the results compared with those discussed above. Fig. 5 shows the monodimensional free energy profiles projected along the d1 and d2 distances, obtained with and without using −β as a collective variable. Here, minima are the result of mixed states: from left to right, the four minima in d1 correspond to state 1, states 2 + 3, states 4 + 5 + 7 and states 6 + 8 of Fig. 4, respectively; the three minima in d2 correspond to states 2 + 4, states 1 + 3 + 5 + 6, and states 7 + 8 of Fig. 4, respectively. The four plots overlap nearly exactly in the region above 4 Å due to the solvent-shared states being significantly more stable. Differences between the d1 and d2 profiles, and between the plots obtained with and without biasing −β are observed in the contact ion pair region only.
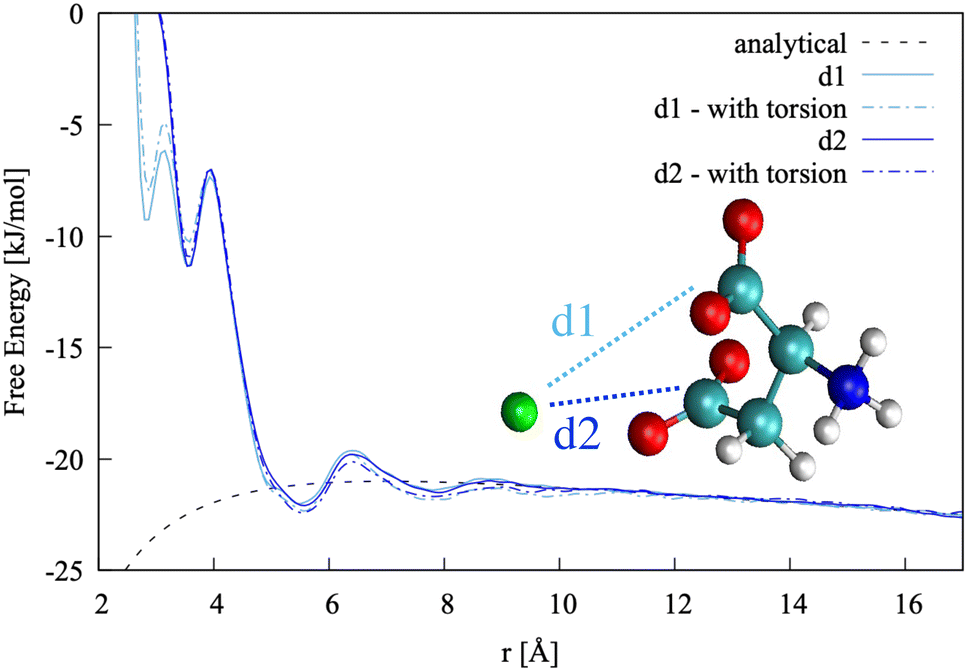 | ||
| Fig. 5 Pairing free energy of Ca2+L-Asp ZW− projected along the two Ca–C distances and obtained with and without sampling along the −β torsional angle collective variable. | ||
The pairing free energy can be extracted from our data using the Fuoss60,61 equation and it can be used as a quantitative measure of the differences observed above. Results of the integration along the two reaction coordinates are very similar, with ΔGd1 = −4.27 kJ mol−1 and ΔGd1 = −4.42 kJ mol−1 when rotation around −β is sampled. These values become −4.17 and −4.10 kJ mol−1, respectively, when the −β torsional angle is not considered as a collective variable, confirming the accuracy of the latter approach for this specific case.
An alternative to using the distance between calcium and each carboxylate group, resulting in having to bias two collective variables, is to use the distance between calcium and COM of the molecule, d3. Having to explore only one collective variable would significantly reduce the computational demand, something that becomes crucial when dealing with clusters and more complex systems, at the cost of losing information regarding the atomic contribution of each separate functional group and of the overall dynamics and geometry of the ion pair. This approach would also allow for a more direct comparison with experimental measures.
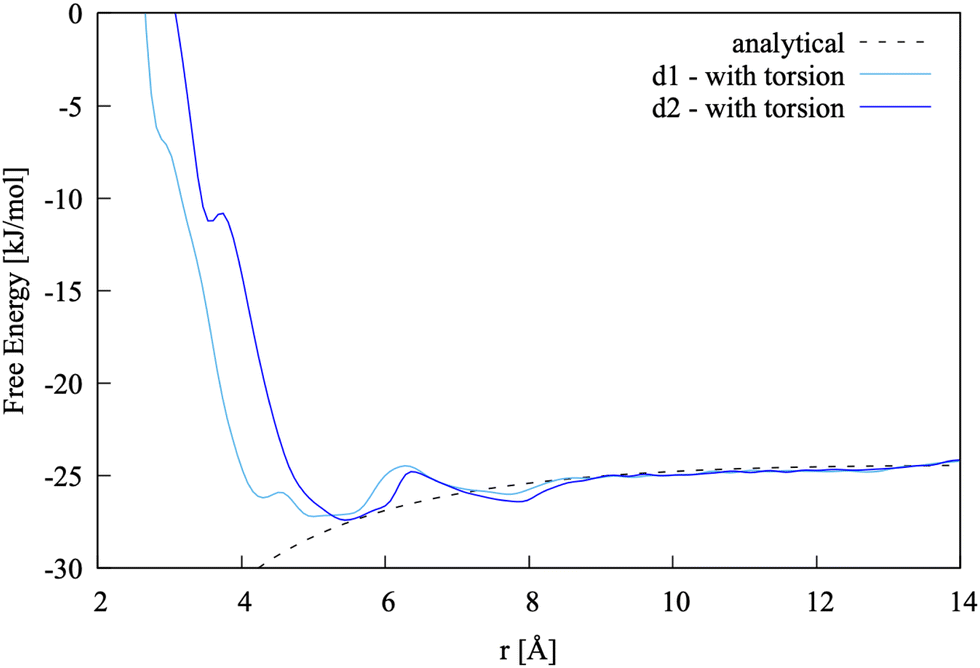
The pairing free energy between Ca2+ and L-Asp ZW− was computed using d3 as a collective variable. Direct comparison of the resulting free energy profile with those obtained using d1 and d2 is reported as ESI† and shows very good agreement, with minima at a slightly higher distance (around 0.5 Å more than for d1 and d2) due to reference to the COM instead of to the carbon atoms in the COO− groups. The pairing free energy value is ΔGd3 = −5.0 kJ mol−1.
In order to ensure that d3 can fully capture the dynamics and energetics of the ion pair, a simulation was run using the coordination of calcium with water as an additional collective variable. The resulting 1-D profile along d3 is reported as ESI,† showing the binding energy is slightly more negative (by 0.5 kJ mol−1) when the water coordination number around calcium is considered.
The main conclusions of this analysis are listed below and apply to ion pairs and other water–mediated interactions, while noting that differences of 0.5 kJ mol−1 are within the error of the method, but would need to be reassessed in systems dominated by direct–contact interactions:
• Calcium ions bind to L-Asp ZW− weakly in water, and the most stable state is solvent-shared.
• The pairing free energy values obtained with and without biasing the −β torsional angle are of comparable accuracy due to the solvent-shared nature of the ion pair.
• The pairing free energy value obtained via biasing the two individual distances between the carboxylate groups and calcium differ by about 0.5 kJ mol−1 from that obtained by using the distance between the COM of the anion and calcium as a sole collective variable, which is within the accuracy of the method.
• The pairing free energy values obtained with and without biasing the water coordination of calcium also differ by 0.5 kJ mol−1.
 | ||
| Fig. 6 Top: Pairing free energy of Ca2+L-Asp2− projected along two Ca–C distances, d1 and d2 (defined in Fig. 5). Minima correspond to contact (N–Ca) plus solvent-shared (C–Ca) ion pairs (CSSHIP, 1 and 2), solvent-shared ion pairs (SSHIP, 3 to 6) and solvent separated ion pairs (SSIP, 7). Examples of configurations for each minimum are provided, with atoms coloured as in Fig. 4. Bottom: Rotational free energy profile around the −β torsional angle of L-Asp2−, free and bound with Ca2+. | ||
 | ||
| Fig. 7 Pairing free energy of Ca2+L-Asp2− projected along two Ca–C distance and obtained with sampling along the −β torsional angle collective variable. | ||
To further validate this finding, we performed DFT calculations with ORCA using the ωB97X-V level of theory and the triple-ζ basis set, both in the gas phase and with implicit solvent. As a starting configuration, we chose a snapshot extracted from the metadynamics trajectory where the Ca ion is close to the nitrogen and removed all the water molecules that did not belong to the calcium first hydration shell. We then performed a geometry optimisation of the structure first in vacuum and then with the implicit solvent. During the geometry optimisation, the aspartate undergoes a significant conformational rearrangement but the N–Ca distance remains around 2.4 Å. Although these calculations do not provide an indication of what the binding energy of the complex is, they do show that Ca is not immediately repelled by the amino group and suggest that Ca–N is not an unfavourable interaction.
This is also in agreement with the findings of Tang and Skibsted,62 though also in their case calculations were not run in explicit solvent and no conformational analysis was performed.
The main conclusions of the analysis of Ca2+L-Asp2− association are listed below. Apart from a few features, they are overall similar to those reported for the zwitterion and show that ion association can be simulated using one collective variable only, i.e. the distance between the COM of the anion and calcium, d3:
• The pairing free energy between calcium and L-Asp2− is stronger than that between calcium and L-Asp ZW− due to a higher charge and a contact interaction between the amino group and calcium.
• The interactions between calcium and the carboxylate groups are still weak and solvent shared, with contact interactions being highly disfavoured.
• The most stable conformation of L-Asp2− is the same in all the main minimum energy ion pair configurations.
• The pairing free energy values obtained via biasing the two individual distances between the carboxylate groups and calcium are −10.9 (d1) and −10.5 (d2) kJ mol−1, therefore the difference with the value obtained by using the distance between COM and calcium (d3, −10.7 kJ mol−1) as a sole collective variable is largely within the accuracy of the method.
| Asp ZW− | ΔG | CV |
|---|---|---|
| L− + Ca2+ | −5.0 | d3 |
| D− + Ca2+ | −5.5 | d3 |
| CaL+ + L− | −2.3 | d3 |
| CaL+ + D− | −3.0 | d3 |
| CaL2 + Ca2+ | −4.8 | d4 |
| CaLD + Ca2+ | −4.6 | d4 |
| Asp2− | ΔG | |
|---|---|---|
| L2− + Ca2+ | −10.7 | d3 |
| D2− + Ca2+ | −11.6 | d3 |
| CaL + L2− | −2.9 | d3 |
| CaL + D2− | −3.0 | d3 |
| CaL22− + Ca2+ | −11.3 | d4 |
| CaLD2− + Ca2+ | −11.3 | d4 |
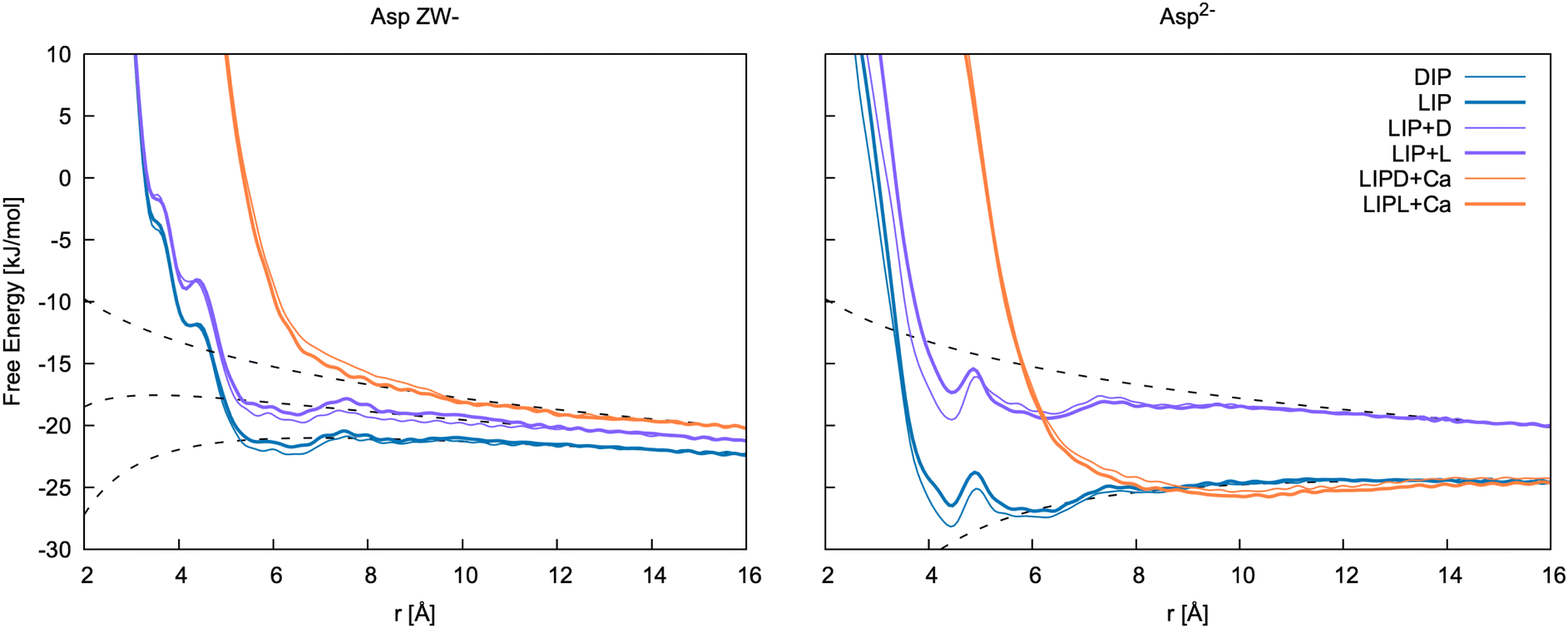 | ||
| Fig. 8 Free energy profiles for the formation of ion pairs and clusters of Asp ZW− and Asp2− with Ca2+. DIP and LIP: ion pair between calcium and D-/L-aspartate; LIP + D and LIP + L: association between LIP and L-/D-aspartate; LIPD/L + Ca: association between LIP + D/L cluster and calcium. r is d3 for LIP, DIP, LIP + L and LIP + D, and the distance between Ca2+ in the cluster and Ca2+ ions for LIPD + Ca and LIPL + Ca. The binding free energies determined from these 1D profiles are included in Table 5. | ||
Of the few experimental data available in the literature, Tang and Skibsted62 provide the latest and most complete dataset of association constants between aspartic acid and calcium at various pH and at different concentrations. Here, the pairing free energy measured between pH = 6 and pH = 8 (where Asp ZW− is the dominant species, pKa1 = 1.88 and pKa2 = 3.6563) at 298 K ranges from −5.61 to −6.43 kJ mol−1, which is in very good agreement with our calculations for the Ca2+–Asp ZW− ion pair. The pairing free energy measured at pH = 10 (where Asp2− is the dominant species, pKb = 9.6063) is −9.78 ± 0.05 kJ mol−1, which is also in good agreement with our calculations for the Ca2+–Asp2− ion pair. The calculated values show a slight overbinding (pairing free energy of −10.7 and −11.6 kJ mol−1 for the L- and D-enantiomers, respectively), but the difference between the calculated and the experimental values is close to the calculation error bar.
Tang and Skibsted62 also provide an estimation of the average binding at neutral or acidic pH (Kzwitter) and at alkaline pH (Kbase) obtained through fitting their data collected at various pH values. The pairing free energy in this case is −5.68 and −10.31 kJ mol−1 for the zwitterion and the fully deprotonated aspartate, respectively. These values are, again, fully in line with our results.
A few other studies are available in the literature that provide experimental calcium aspartate binding free energies, most of which are in overall good agreement with our and Tang and Skibsted's62 values. Blaquiere and Berthong64 provide similar values for the binding free energies of both species with calcium; −6.52 ± 0.95 kJ mol−1 (Asp2−) and −6.78 ± 0.18 kJ mol−1 (Asp ZW−). To obtain these values, we have assumed the same notation as in Covington et al.65 was used in this paper. As only values for βMLH are reported, we used the value of βHL from Covington et al.65 to estimate the Ca–Asp ZW− binding free energy. Later, Maeda et al.66 provided values of −11.42 and −6.97 kJ mol−1 for the binding free energy of calcium with Asp2− and Asp ZW−, respectively. Similar values, though slightly more binding for Asp ZW−, were measured by Covington et al.65 using two different electrodes; −10.68 ± 1.21 and −11.89 ± 0.29 kJ mol−1 for Ca–Asp2−, and −8.67 ± 1.72 and −9.88 ± 0.80 kJ mol−1 for Ca–Asp ZW−.
3.4 Formation of Ca–Asp clusters in solution
The formation of homochiral clusters of aspartic acid species was identified as a main factor responsible for the formation of chiral toroids of vaterite by Jiang et al.13,14 While investigation of the structure and dynamics of such species at the calcium carbonate surface is beyond the scope of this paper, simulations were run to assess whether such clusters can form in solution, and whether there is any preference in forming homochiral aggregates in solution.Free energies of formation are reported in Table 5 and free energy profiles in Fig. 8. These data show weak binding, of the same magnitude of the ion pairs, with CO22−–Ca2+ interactions mediated by water in a solvent-shared mode and with weak contact between NH2–Ca2+ at high pH. There is no significant difference between homochiral and heterochiral interactions.
For comparison to the free energies determined in aqueous solution using the present force field, the relative energies of homo- vs. hetero-chiral clusters of aspartate with calcium were determined in the gas phase using quantum mechanical calculations at the ωB97X-D3/ma-def2-QZVPP level of theory, as used earlier for the hydration free energies. In this case, the homochiral cluster was found to be more stable by 93.1 kJ mol−1 for Asp ZW− and 26.3 kJ mol−1 for Asp2− based on the internal energy at the optimised structure. Inclusion of zero point energy and thermal corrections to room temperature increased the values by only 0.3 and 0.7 kJ mol−1, respectively. While this confirms the expected preference for homochiral complexes in the gas phase, it is harder to validate the lack of clear preference in solution. Inclusion of a continuum solvent model (SMD) is found to greatly reduce the preference for homochiral clusters (e.g. for Asp ZW− with Ca2+ the internal energy difference drops to 22.4 kJ mol−1), though a stability difference remains. This is likely to be as much a reflection on the approximate nature of continuum solvation as it is suggestive of a discrepancy with the force field model.
4 Conclusions
This paper presents an in-depth analysis of the dynamics of aspartic acid species and of their association with calcium ions in water at pH values relevant to biomineralization.A new classical model was developed that provides results consistent with first principles data, such as hydration structure and free energy, as well as conformational free energy differences. The model was then applied to the calculation of ion pairing free energy profiles, obtaining values that are in agreement with experiment. A weak and solvent-shared association was observed between aspartic acid species and calcium, resulting in a weak binding to form aqueous clusters, and in no difference in the formation free energy of homochiral vs. heterochiral clusters.
The simulations presented in this paper show that modelling molecules with conformational freedom in the context of biomineralization comes with additional levels of complexity, due to the fact that different conformations can be more or less stable depending on whether the molecule is free or bound. In fact, if the rotational free energy barrier is high, an appropriate bias needs to be added to accelerate the dynamics.
In the specific case of aspartic acid species, we found that some of the activation barriers between different conformations are not accessible with unbiased molecular dynamics. Additionally, the most stable conformation of bound (contact interaction) and solvent-shared or free aspartic acid species are different. While this turned out to be irrelevant for ion pairs, due to their solvent-shared nature, whether or not to bias the torsional angle would need to be reassessed in systems dominated by direct-contact interactions (e.g. mineral surfaces).
Author contributions
RD planned the research and wrote the manuscript, with inputs from all authors. AS and RD ran and analysed most calculations. JDG ran and analysed most first principles calculations. KBK, PR, JDG and RD developed the force field model.Conflicts of interest
There are no conflicts to declare.Acknowledgements
RD and JDG thank the Australian Research Council for support though a Discovery Project (DP160100677), Future and Laureate Fellowships (FT180100385, FL180100087). AS and RD thank Curtin University for supporting this project through a Science and Engineering Faculty Small Grant. The Pawsey Supercomputing Centre and the Australian National Computational Infrastructure are also acknowledged for the provision of computing time through the NCMAS and Pawsey Partners merit allocation schemes.References
- L. B. Gower, Chem. Rev., 2008, 108, 4551–4627 CrossRef CAS PubMed.
- S. Weiner and L. Addadi, Annu. Rev. Mater. Res., 2011, 41, 21–40 CrossRef CAS.
- F. C. Meldrum and H. Cölfen, Chem. Rev., 2008, 108, 4332–4432 CrossRef CAS PubMed.
- R. Demichelis, A. Schuitemaker, N. A. Garcia, K. B. Koziara, M. De La Pierre, P. Raiteri and J. D. Gale, Annu. Rev. Mater. Res., 2018, 48, 327–352 CrossRef CAS.
- A. Wierzbicki, C. S. Sikes, J. D. Madura and B. Drake, Calcif. Tissue Int., 1994, 54, 133–141 CrossRef CAS PubMed.
- C. A. Orme, A. Noy, A. Wierzbicki, M. T. McBride, M. Grantham, H. H. Teng, P. M. Dove and J. J. DeYoreo, Nature, 2001, 411, 775–779 CrossRef CAS PubMed.
- S. Elhadj, J. J. DeYoreo, J. R. Hoyer and P. M. Dove, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 19237–19242 CrossRef CAS PubMed.
- I. W. Kim, M. R. Darragh, C. Orme and J. S. Evans, Cryst. Growth Des., 2006, 6, 5–10 CrossRef CAS.
- B. Njegić-Džakula, L. Brečević, G. Falini and D. Kralj, Cryst. Growth Des., 2009, 9, 2425–2434 CrossRef.
- D. Tobler, J. R. Blanco, K. Dideriksen, K. Sand, N. Bovet, L. Benning and S. Stipp, Procedia Earth Planet. Sci., 2014, 10, 143–148 CrossRef CAS.
- Y.-Y. Kim, J. D. Carloni, B. Demarchi, D. Sparks, D. G. Reid, M. E. Kunitake, C. C. Tang, M. J. Duer, C. L. Freeman, B. Pokroy, K. Penkman, J. H. Harding, L. A. Estroff, S. P. Baker and F. C. Meldrum, Nat. Mater., 2016, 15, 903–910 CrossRef CAS PubMed.
- G. Montanari, L. Z. Lakshtanov, D. J. Tobler, K. Dideriksen, K. N. Dalby, N. Bovet and S. L. S. Stipp, Cryst. Growth Des., 2016, 16, 4813–4821 CrossRef CAS.
- W. Jiang, M. S. Pacella, D. Athanasiadou, V. Nelea, H. Vali, R. M. Hazen, J. J. Gray and M. D. McKee, Nat. Commun., 2017, 8, 15066 CrossRef CAS PubMed.
- W. Jiang, D. Athanasiadou, S. Zhang, R. Demichelis, K. B. Koziara, P. Raiteri, V. Nelea, W. Mi, J.-A. Ma, J. D. Gale and M. D. McKee, Nat. Commun., 2019, 10, 2318 CrossRef PubMed.
- D. Case, T. Cheatham III, T. Darden, H. Gohlke, R. Luo, K. Merz Jr., A. Onufriev, C. Simmerling, B. Wang and R. Woods, J. Comput. Chem., 2005, 26, 1668–1688 CrossRef CAS PubMed.
- B. R. Brooks, C. L. Brooks III, A. D. Mackerell, L. Nilsson, R. J. Petrella, Y. W. B. Roux, G. Archontis, C. Bartels, S. Boresch, A. Caflisch, L. Caves, Q. Cui, A. R. Dinner, M. Feig, S. Fischer, J. Gao, M. Hodoscek, W. Im, K. Kuczera, T. Lazaridis, J. Ma, V. Ovchinnikov, E. Paci, R. W. Pastor, C. B. Post, J. Z. Pu, M. Schaefer, B. Tidor, R. M. Venable, H. L. Woodcock, X. Wu, W. Yang, D. M. York and M. Karplus, J. Comput. Chem., 2009, 30, 1545–1615 CrossRef CAS PubMed.
- W. R. P. Scott, P. H. Hünenberger, I. G. Tironi, A. E. Mark, S. R. Billeter, J. Fennen, A. E. Torda, T. Huber, P. Krüger and W. F. van Gunsteren, J. Phys. Chem. A, 1999, 103, 3596–3607 CrossRef CAS.
- A. Schuitemaker, J. Aufort, K. K. Koziara, R. Demichelis, P. Raiteri and J. D. Gale, Phys. Chem. Chem. Phys., 2021, 23, 27253–27265 RSC.
- J. Aufort, A. Schuitemaker, R. Green, R. Demichelis, P. Raiteri and J. D. Gale, Cryst. Growth Des., 2022, 22, 1445–1458 CrossRef CAS.
- H. Nada, J. Phys. Chem. C, 2014, 118, 14335–14345 CrossRef CAS.
- A. D. MacKerell, D. Bashford, M. Bellott, R. L. Dunbrack, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiórkiewicz-Kuczera, D. Yin and M. Karplus, J. Phys. Chem. B, 1998, 102, 3586–3616 CrossRef CAS PubMed.
- P. Raiteri, J. D. Gale, D. Quigley and P. M. Rodger, J. Phys. Chem. C, 2010, 114, 5997–6010 CrossRef CAS.
- W. L. Jorgensen, J. Chandrasekhar and J. D. Madura, J. Chem. Phys., 1983, 79, 926 CrossRef CAS.
- Y. Wu, H. L. Tepper and G. A. Voth, J. Chem. Phys., 2006, 124, 024503 CrossRef PubMed.
- R. Stepić, L. Jurković, K. Klementyeva, M. Ukrainczyk, M. Gredică, D. M. Smith, D. Kralj and A.-S. Smith, Cryst. Growth Des., 2020, 20, 2853–2859 CrossRef.
- A. Verch, D. Gebauer, M. Antonietti and H. Cölfen, Phys. Chem. Chem. Phys., 2011, 13, 16811–16820 RSC.
- R. Innocenti Malini, A. R. Finney, S. A. Hall, C. L. Freeman and J. H. Harding, Cryst. Growth Des., 2017, 17, 5811–5822 CrossRef CAS.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS PubMed.
- P. Raiteri, R. Demichelis and J. D. Gale, J. Phys. Chem. C, 2015, 119, 24447–24458 CrossRef CAS.
- P. Raiteri, R. Demichelis, J. D. Gale, M. Kellermeier, D. Gebauer, D. Quigley, L. Wright and T. R. Walsh, Faraday Discuss., 2012, 159, 61–85 RSC.
- A. R. Finney, R. Innocenti Malini, C. L. Freeman and J. H. Harding, Cryst. Growth Des., 2020, 20, 3077–3092 CrossRef CAS PubMed.
- J. A. Koskamp, S. E. Ruiz-Hernandez, N. H. deLeeuw and M. Wolthers, Phys. Chem. Chem. Phys., 2022, 25, 1220–1235 RSC.
- V. Hornak, R. Abel, A. Okur, B. Strockbine, A. Roitberg and C. Simmerling, Proteins, 2006, 65, 712–725 CrossRef CAS PubMed.
- S. Plimpton, J. Comput. Chem., 1995, 117, 1–19 CAS.
- G. J. Martyna, D. J. Tobias and M. L. Klein, J. Chem. Phys., 1994, 101, 4177–4189 CrossRef CAS.
- P. Raiteri, A. Laio, F. L. Gervasio, C. Micheletti and M. Parrinello, J. Phys. Chem. B, 2006, 110, 3533–3539 CrossRef CAS PubMed.
- A. Barducci, G. Bussi and M. Parrinello, Phys. Rev. Lett., 2008, 100, 20603 CrossRef PubMed.
- A. Laio and M. Parrinello, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12562–12566 CrossRef CAS PubMed.
- A. Laio and F. L. Gervasio, Rep. Prog. Phys., 2008, 71, 126601 CrossRef.
- G. A. Tribello, M. Bonomi, D. Branduardi, C. Camilloni and G. Bussi, Comput. Phys. Commun., 2014, 185, 604–613 CrossRef CAS.
- G. A. Tribello, M. Bonomi, D. Branduardi, C. Camilloni and G. Bussi, CV documentation, https://www.plumed.org/doc-v2.8/user-doc/html/_colvar.html.
- B. Armstrong, A. Silvestri, R. Demichelis, P. Raiteri and J. D. Gale, Philos. Trans. R. Soc., A, 2023, 381, 202220250 CrossRef PubMed.
- C. Oostenbrink, A. Villa, A. Mark and W. F. van Gunsteren, J. Comput. Chem., 2004, 25, 1656–1676 CrossRef CAS PubMed.
- A. Malde, L. Zuo, M. Breeze, M. Stroet, D. Poger, P. Nair, C. Oostenbrink and A. Mark, J. Chem. Theory Comput., 2011, 7, 4026–4037 CrossRef CAS PubMed.
- J. Hutter, M. Iannuzzi, F. Schiffmann and J. VandeVondele, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 15–25 CAS.
- S. Goedecker, M. Teter and J. Hutter, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 1703–1710 CrossRef CAS PubMed.
- J. VandeVondele and J. Hutter, J. Chem. Phys., 2003, 118, 4365–4369 CrossRef CAS.
- J. Wang, G. Román-Pérez, J. M. Soler, E. Artacho and M. V. Fernández-Serra, J. Chem. Phys., 2011, 134, 024516 CrossRef PubMed.
- R. A. DiStasio Jr., B. Santra, Z. Li, X. Wu and R. Car, J. Chem. Phys., 2014, 141, 084502 CrossRef PubMed.
- R. W. Zwanzig, J. Chem. Phys., 1954, 22, 1420–1426 CrossRef CAS.
- G. Hummer, L. R. Pratt and A. Garcia, J. Chem. Phys., 1997, 107, 9275–9277 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- A. V. Marenich, R. M. Olson, C. P. Kelly, C. J. Cramer and D. G. Truhlar, J. Chem. Theory Comput., 2007, 3, 2011–2033 CrossRef CAS PubMed.
- Y. Shao, Z. Gan, E. Epifanovsky, A. T. B. Gilbert, M. Wormit, J. Kussmann, A. W. Lange, A. Behn, J. Deng, X. Feng, D. Ghosh, M. G. P. R. Horn, L. D. Jacobson, I. Kaliman, R. Z. Khaliullin, T. Kús, A. Landau, J. Liu, E. I. Proynov, Y. M. Rhee, R. M. Richard, M. A. Rohrdanz, R. P. Steele, E. J. Sundstrom, H. Lee Woodcock III, P. M. Zimmerman, D. Zuev, B. Albrecht, E. Alguire, B. Austin, G. J. O. Beran, Y. A. Bernard, E. Berquist, K. Brandhorst, K. B. Bravaya, S. T. Brown, D. Casanova, C.-M. Chang, Y. Chen, S. H. Chien, K. D. Closser, D. L. Crittenden, M. Diedenhofen, R. A. D. Jr., H. Dop, A. D. Dutoi, R. G. Edgar, S. Fatehi, L. Fusti-Molnar, A. Ghysels, A. Golubeva-Zadorozhnaya, J. Gomes, M. W. D. Hanson-Heine, P. H. P. Harbach, A. W. Hauser, E. G. Hohenstein, Z. C. Holden, T.-C. Jagau, H. Ji, B. Kaduk, K. Khistyaev, J. Kim, J. Kim, R. A. King, P. Klunzinger, D. Kosenkov, T. Kowalczyk, C. M. Krauter, K. U. Lao, A. Laurent, K. V. Lawler, S. V. Levchenko, C. Y. Lin, F. Liu, E. Livshits, R. C. Lochan, A. Luenser, P. Manohar, S. F. Manzer, S.-P. Mao, N. Mardirossian, A. V. Marenich, S. A. Maurer, N. J. Mayhall, C. M. Oana, R. Olivares-Amaya, D. P. ONeill, J. A. Parkhill, T. M. Perrine, R. Peverati, P. A. Pieniazek, A. Prociuk, D. R. Rehn, E. Rosta, N. J. Russ, N. Sergueev, S. M. Sharada, S. Sharmaa, D. W. Small, A. Sodt, T. Stein, D. Stück, Y.-C. Su, A. J. W. Thom, T. Tsuchimochi, L. Vogt, O. Vydrov, T. Wang, M. A. Watson, J. Wenzel, A. White, C. F. Williams, V. Vanovschi, S. Yeganeh, S. R. Yost, Z.-Q. You, I. Y. Zhang, X. Zhang, Y. Zhou, B. R. Brooks, G. K. L. Chan, D. M. Chipman, C. J. Cramer, W. A. Goddard III, M. S. Gordon, W. J. Hehre, A. Klamt, H. F. Schaefer III, M. W. Schmidt, C. D. Sherrill, D. G. Truhlar, A. Warshel, X. Xua, A. Aspuru-Guzik, R. Baer, A. T. Bell, N. A. Besley, J.-D. Chai, A. Dreuw, B. D. Dunietz, T. R. Furlani, S. R. Gwaltney, C.-P. Hsu, Y. Jung, J. Kong, D. S. Lambrecht, W. Liang, C. Ochsenfeld, V. A. Rassolov, L. V. Slipchenko, J. E. Subotnik, T. V. Voorhis, J. M. Herbert, A. I. Krylov, P. M. W. Gill and M. Head-Gordon, Mol. Phys., 2015, 113, 184–215 CrossRef CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- V. Bryantsev, M. Diallo and W. A. Goddard III, J. Phys. Chem. B, 2008, 112, 9709–9719 CrossRef CAS PubMed.
- F. Comitani, K. Rossi, M. Ceriotti, M. E. Sanz and C. Molteni, J. Chem. Phys., 2017, 146, 145102 CrossRef PubMed.
- R. B. Best, J. S. X. Zhu, P. E. M. Lopes, J. Mittal, M. Feig and A. D. Mackerell, J. Chem. Theory Comput., 2012, 8, 3257–3273 CrossRef CAS PubMed.
- R. M. Fuoss and C. A. Krauss, J. Am. Chem. Soc., 1933, 55, 1019–1028 CrossRef CAS.
- J. Aufort, P. Raiteri and J. D. Gale, Computational Insights into Mg2+ Dehydration in the Presence of Carbonate, Acs Earth and Space Chemistry, 2022, 6, 733–745, DOI:10.1021/acsearthspacechem.1c00389.
- N. Tang and L. H. Skibsted, J. Agric. Food Chem., 2016, 64, 4376–4389 CrossRef CAS PubMed.
- D. Lide, Handbook of Chemistry and Physics, CRC Press, Boca Raton, FL, 72nd edn, 1991 Search PubMed.
- C. Blaquiere and G. Berthon, Inorg. Chim. Acta, 1987, 135, 179–189 CrossRef CAS.
- A. Covington and E. Danish, J. Solution Chem., 2009, 38, 1449–1462 CrossRef CAS.
- M. Maeda, K. Okada, Y. Tsukamoto, K. Wakabayashi and K. Ito, J. Chem. Soc., Dalton Trans., 1990, 2337–2339 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp04674e |
| This journal is © the Owner Societies 2024 |