
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePutative reaction mechanism of nitrogenase with a half-dissociated S2B ligand†
Hao
Jiang
 and
Ulf
Ryde
*
and
Ulf
Ryde
*
Department of Computational Chemistry, Lund University, Chemical Centre, P. O. Box 124, SE-221 00 Lund, Sweden. E-mail: Ulf.Ryde@compchem.lu.se; Tel: +46-46 2224502
First published on 20th June 2024
Abstract
We have studied whether dissociation of the S2B sulfide ligand from one of its two coordinating Fe ions may affect the later parts of the reaction mechanism of nitrogenase. Such dissociation has been shown to be favourable for the E2–E4 states in the reaction mechanism, but previous studies have assumed that S2B either remains bridging or has fully dissociated from the active-site FeMo cluster. We employ combined quantum mechanical and molecular mechanical (QM/MM) calculations with two density-functional theory methods, r2SCAN and TPSSh. To make dissociation of S2B possible, we have added a proton to this group throughout the reaction. We study the reaction starting from the E4 state with N2H2 bound to the cluster. Our results indicate that half-dissociation of S2B is unfavourable in most steps of the reaction mechanism. We observe favourable half-dissociation of S2B only when NH or NH2 is bound to the cluster, bridging Fe2 and Fe6. However, the former state is most likely not involved in the reaction mechanism and the latter state is only an intermittent intermediate of the E7 state. Therefore, half-dissociation of S2B seems to play only a minor role in the later parts of the reaction mechanism of nitrogenase. Our suggested mechanism with a protonated S2B is alternating (the two N atoms of the substrate is protonated in an alternating manner) and the substrate prefers to bind to Fe2, in contrast to the preferred binding to Fe6 observed when S2B is unprotonated and bridging Fe2 and Fe6.
Introduction
Nitrogenase (EC 1.18/19.6.1) is the only enzyme that can cleave the triple bond in N2, by converting it to ammonia.1–4 Nitrogenase is produced by a few groups of bacteria but several higher plants live in symbiosis with such bacteria, e.g. legumes, rice and alder. Crystallographic studies have shown that the most common and active form of nitrogenase is a homodimer of heterodimers.5–9 It contains two unusual iron–sulfur clusters, the P-cluster (Fe8S7Cys6), which is used for electron transfer, and the FeMo cluster, which is the catalytic centre. The latter consists of a MoFe8S9C(homocitrate) cluster, which is connected to the protein by one His ligand to Mo and a Cys ligand to a Fe ion at the other end of the cluster (cf.Fig. 1a). There also exist alternative isoenzymes in which the Mo ion is replaced by V or Fe, but they have a lower activity.10 During catalysis, electrons are delivered to nitrogenase by the Fe protein, which contains a Fe4S4 cluster.3,4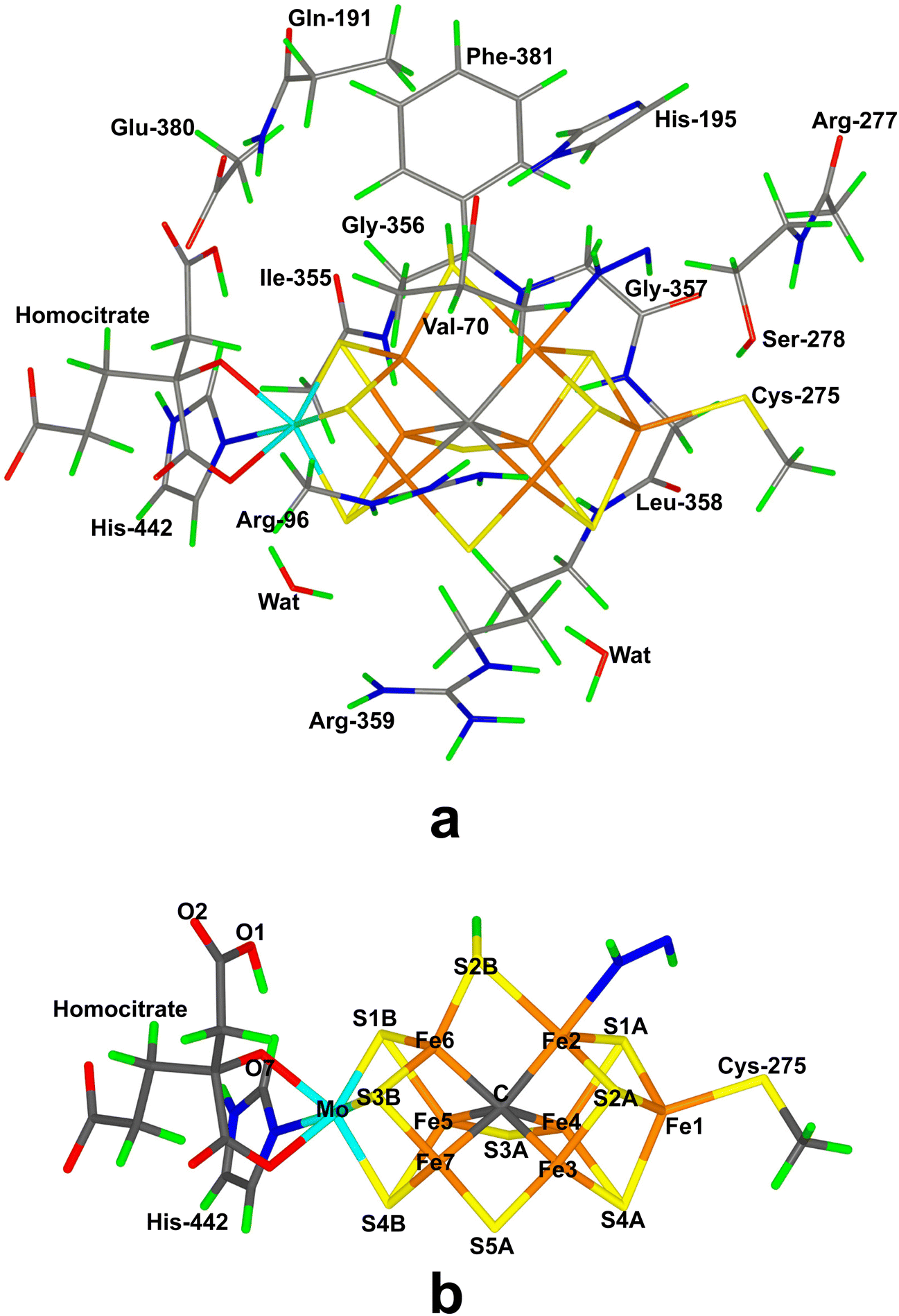 | ||
| Fig. 1 The FeMo cluster in nitrogenase (trans(2;1A)-S2B(26;3) structure). (a) Illustrates the QM system used in all calculations, as well as the names of the nearby residues, (b) shows only the FeMo cluster with atom names indicated. | ||
Nitrogenase catalyses the reaction3,4
| N2 + 8e− + 8H+ + 16ATP → 2NH3 + H2 + 16ADP + 16Pi | (1) |
Thus, eight electrons and protons are needed to form two molecules of NH3 from N2. Consequently, the reaction mechanism is typically described by nine states, E0–E8, differing in the number of added electrons and protons.11 Despite intensive biochemical, kinetic, spectroscopic and computational investigations,1–9,12,13 many details of the nitrogenase reaction mechanism are still controversial, partly because of technical problems to isolate pure samples of the various reaction intermediates En. It is known that N2 binds mainly to the E4 state and that N2 binding is coupled to the compulsory formation of H2, in a reductive elimination reaction.14–18 E4 has been shown to contain two hydride ions bridging two pairs of Fe ions,15,16,18 but the detailed structure of the E4 state is highly controversial.19–28
The later part of the reaction mechanism, i.e. after N2 binding has also been much discussed.3 In particular, it has been debated whether nitrogenase follows a distal or an alternating mechanism, i.e. whether the protons are first added to one N atom, so that NH3 dissociates already in the E5 state, before the second N atom is protonated, or whether protons are added alternatively to the two N atoms, so that HNNH and H2NNH2 are intermediates and the first NH3 molecule does not dissociate until the E7 state. A distal mechanism is supported by inorganic nitrogenase model complexes,29–33 but also by some computational and crystallographic studies of nitrogenase.25,34 An alternating mechanism is supported by the fact that H2NNH2 is a substrate of nitrogenase and can also be released from the enzyme by acid or base treatment during turnover.1,3,35,36 It has also been shown that N2, N2H2, CH3N2H and N2H4 all react via a common intermediate.37 Several computational studies have suggested reaction mechanisms that are alternating, at least during the end of the reaction (i.e. involving H2NNH2 as an intermediate).13,38,39
Our group has presented several studies of the later part of the nitrogenase reaction mechanism. We have performed an exhaustive search of possible binding sites and binding modes of N2H2 to the FeMo cluster in the E4 state.40 The study showed that the most favourable binding sites were Fe2 and Fe6 (atom numbers are shown in Fig. 1b). trans-HNNH bound to Fe2 was most stable, but structures with the same molecule bound to Fe6, cis-HNNH bound to Fe2, or HNNH2 bound to Fe6 (with the additional proton abstracted from homocitrate) were all competitive within 12 kJ mol−1. With a somewhat larger quantum-mechanical (QM) model, including also Val-70 and Phe-381, which may restrict the binding of the substrate, the preferences change somewhat in favour of trans-HNNH or HNNH2 binding to Fe6.39 That study also considered the complete reaction mechanism and suggested an alternating mechanism and that the homocitrate ligand may provide a proton buffer that may stabilise intermediates like H2NNH2 and NH3 already at the E5 and E7 states, respectively. Based on suggestions from crystal structures of inhibited nitrogenase, we also investigated the corresponding reactions when S2B has dissociated from the FeMo cluster, providing a natural binding site for the substrate.38 The results suggested that N2H2 binds as NNH2 bridging Fe2 and Fe6 (i.e. an intermediate connected to a distal mechanism), but that the mechanism the becomes alternating, with H2NNH2 bound at the E6 state and NH3 formation in the E7 state. Both mechanism seemed to be equally feasible, but a study of proton-transfer reactions in the cluster indicated that the proton transfer to the substrate is easier if S2B remains bound.41
Several recent studies have suggested that S2B may dissociate only from one of the two Fe ions, forming unhooked or half-dissociated structures.21,42–44 In fact, such structures seem to be among the most likely candidates for the E2–E4 states of Mo-nitrogenase.22,44,45 The question then naturally arises whether such structures are competitive also for the later part of the nitrogenase reaction, i.e. after binding of N2. The aim of the present study is to investigate this possibility using combined QM and molecular mechanics (QM/MM) calculations.
Methods
The protein
The calculations were based on the 1.0 Å crystal structure of Mo nitrogenase from Azotobacter vinelandii (PDB code 3U7Q).5 The setup of the protein is identical to that of our previous studies.20,22,46–48 The entire heterotetramer was included in the calculations and the QM calculations were concentrated on the FeMo clusters in the C subunit because there is a buried imidazole molecule rather close to the active site (∼11 Å) in the A subunit. The two P clusters and the FeMo cluster in subunit A were modelled by molecular mechanics (MM) in the fully reduced and resting states, respectively, using a QM charge model.47 The protonation states of all residues were the same as before,47 and the homocitrate ligand was modelled in the singly protonated state with a proton shared between the hydroxyl group (O7 that coordinates to Mo) and the O1 carboxylate atom.47,49 The protein was solvated in a sphere with a radius of 65 Å around the geometrical centre of the protein. Cl− and Na+ ions were added to yield an ionic strength of 0.2 M.50 The final system contained 133![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 915 atoms. For the protein, we used the Amber ff14SB force field51 and water molecules were described by the TIP3P model.52 The metal sites were treated by a non-bonded model53 and charges were obtained with the restrained electrostatic potential method.54
915 atoms. For the protein, we used the Amber ff14SB force field51 and water molecules were described by the TIP3P model.52 The metal sites were treated by a non-bonded model53 and charges were obtained with the restrained electrostatic potential method.54
In the QM calculations, the FeMo cluster was modelled by MoFe7S9C(homocitrate)(CH3S)(imidazole), where the two last groups are models of Cys-275 and His-442. In addition, all groups that form hydrogen bonds to the FeMo cluster were also included, viz. Arg-96, Gln-191 and His-195 (sidechains), Ser-278 and Arg-359 (both backbone and sidechain, including the CA and C and O atoms from Arg-277), Gly-356, Gly-357 and Leu-358 (backbone, including the CA and C and O atoms from Ile-355), as well as two water molecules. Finally, the sidechain of Glu-380 was included because it forms hydrogen bonds to Gln191 and His-442, as well as the sidechains of Val-70 and Phe-381 because they are close to S2B, Fe2 and Fe6, i.e. the expected reactive site. The QM system contained 192–197 atoms depending on the En state and the bound substrate. It is shown in Fig. 1a. The net charge of QM region was −3e.
QM calculations
All QM calculations were performed with the Turbomole software (versions 7.5 and 7.6).55 All structures were studied with the r2SCAN56 density functional theory (DFT) method. To investigate the functional dependence, most structures were also studied with the TPSSh57 functional. r2SCAN is a meta generalised gradient approximation functional, whereas TPSSh is a hybrid functional with 10% Hartree–Fock exchange. Both functionals have been shown to give accurate structures of nitrogenase models.58 All calculations involved the def2-SV(P) basis set.59 The calculations were sped up by expanding the Coulomb interactions in an auxiliary basis set, the resolution-of-identity (RI) approximation.60,61 Empirical dispersion corrections were included with the DFT-D4 approach,62 as implemented in Turbomole.In this investigation we study the later part of the reaction mechanism of nitrogenase, starting after the binding and protonation of the substrate to N2H2 in the E4 state. Like in our previous two studies,38,39 we do this to avoid the problem that the nature of the E4 state is highly controversial, with a very large number of possible locations of the added four H atoms (protons or hydride ions), and extremely large discrepancy in the prediction of various DFT methods.13,20–23,46 The current consensus is that N2 binds to the FeMo cluster together with reductive elimination of H2.4 The remaining two protons are then used to protonate N2 to N2H2. As a consequence, the N2H2-bound E4 state is in the same formal oxidation state as the resting E0 state, viz. the MoIIIFeII3FeIII4 oxidation state.49,63,64
The aim of the present study is to investigate whether dissociation of S2B from either Fe2 or Fe6 may facilitate the conversion of N2H2 to two NH3 molecules. Previous studies have shown that such half-dissociation of S2B is energetically favourable only if S2B is protonated.21,43,44 Therefore, we added an extra proton to S2B in all the current models. Consequently, the charge of the models is one step less negative than previously studied models.
The electronic structure in all QM calculations was obtained with the broken-symmetry (BS) approach:65 Each of the seven Fe ions was modelled in the high-spin state, with either a surplus of α (four Fe ions) or β (three Fe ions) spin. Such a state can be selected in 35 different ways.65,66 The various BS states were obtained either by swapping the coordinates of the Fe ions67 or with the fragment approach by Szilagyi and Winslow.68 The BS states are named by listing the numbers of the three Fe ions with minority spin, e.g. BS-235. At each En-level, we first optimised all possible structures with one BS state (typically BS-235 or BS-147). For the most stable structure, a full investigation of all 35 BS was performed and if this was a different state, the best structures are reoptimised with that state. A similar procedure was performed with the spin state, which is also unknown for these intermediates.
QM/MM calculations
QM/MM calculations were performed with the ComQum software.69,70 In this approach, the protein and solvent are split into two subsystems: system 1 (the QM region) was relaxed by QM methods. System 2 was kept fixed at the original coordinates (equilibrated crystal structure), to avoid the risk that different calculations end up in different local minima.In the QM calculations, system 1 was represented by a wavefunction, whereas all the other atoms were represented by an array of partial point charges, one for each atom, taken from the MM setup (electrostatic embedding). Thereby, the polarisation of the QM system by the surroundings is included in a self-consistent manner. When there is a bond between systems 1 and 2 (a junction), the hydrogen link-atom approach was employed: the QM system was capped with hydrogen atoms, the positions of which are linearly related to the corresponding carbon atoms (carbon link atoms, CL) in the full system.69,71 All atoms were included in the point-charge model, except the CL atoms.72 ComQum employs a subtractive scheme with van der Waals link-atom corrections.73 No cut-off is used for the QM and QM–MM interactions. The geometry optimisations were continued until the energy change between two iterations was less than 2.6 J mol−1 (10−6 a.u.) and the maximum norm of the Cartesian gradients was below 10−3 a.u.
In most structures where S2B binds to both Fe2 and Fe6, one Fe–S distance is ∼2.3 Å and the other 0.1–0.3 Å longer. Thereby, S2B is in between the two Fe ions and both Fe–Fe–S2B angles are less than 90°. When S2B dissociates from one of the Fe ions, the substrate typically bridges Fe2 and Fe6 (with one or two N atoms) and one Fe–S2B distance increases to >3.4 Å and one Fe–Fe–S2B angle is larger than 90°. However, in some cases, the substrate binds only to one Fe ion and S2B to the other ion (often, but not always, receiving a hydrogen bond from the substrate). In those cases, it is less obvious whether S2B has dissociated or not. We used a cutoff of Fe–S2B > 2.9 Å to define a dissociated structure (no structure has a Fe–S2B bond length between 2.81 and 3.04 Å; likewise, there is a gap between 0.52 and 0.86 Å for the absolute difference between the two Fe–S2B distances). Mayer bond orders74 (calculated with the Multiwfn package75) for the Fe–S interactions are correlated to the Fe–S distances (correlation coefficient −0.87) and show similar trends; a cutoff at 0.2 can be used to define bonds.
Results and discussion
In this investigation, we have studied possible paths for the second half of the mechanism of nitrogenase, allowing the S2B ligand to dissociate from one Fe ion. The results are compared to our previous studies where S2B was either binding to both Fe2 and Fe6,39 or was completely dissociated.38 As in the previous studies, we start from a E4 state, where H2 has dissociated, N2 is bound and is protonated to N2H2. This is done to avoid the severe problems that different DFT method give widely different predictions regarding the most stable protonation states of E4 and the strength of the binding of N2.22,46For each new En state, we add one electron and a proton to the FeMo cluster. The electrons are provided by the Fe protein via the P-cluster.2,3 Protons come ultimately from water solvent and two possible proton channels have been suggested, ending either at His-195 or at a water molecule close to S3B, S4B and S5A.76–79 It has suggested that His-195 can only provide a single proton, because rotation of the imidazole group is restricted in the protein.76,80 Several groups have studied proton transfers within the FeMo cluster and have shown that the individual steps in general are facile,76,78,81 although sometimes certain protonation states may act as thermodynamic sinks making the net barriers somewhat high.41 However, Siegbahn has suggested that the barriers are strongly lowered by the employment of surrounding water molecules.28,82 Therefore, we have not explicitly studied proton transfers within the FeMo cluster in this study. Instead we concentrate on determining the thermodynamically most stable structures at each En level and the cleavage of the N–N bond. We discuss the various En states in separate sections.
E4 state
We started with the E4 state with N2H2 bound. We tested four different isomers of N2H2, viz. NNH2, cis-HNNH, trans-HNNH and HNNH2 (in the latter case, the third proton was taken from homocitrate, which is nearby when the ligand binds to Fe6). We studied binding of N2H2 only to Fe2 and Fe6, because these two Fe ions have been pointed out by experimental studies3,83,84 and it has also been shown to be the preferred binding sites by a systematic DFT search.40 We studied both end-on and side-on binding, to one Fe ion as well as to both Fe ions. If relevant, we allowed the non-coordinating N atom or the protons to point in different directions. We use the following nomenclature: the four isomers are called NNH2, cis, trans or HNNH2, respectively, which is followed by the binding site in brackets and indicating only the number of the Fe atom. 2 and 6 means that it binds to only Fe2 or Fe6, whereas 26 means that it bridges Fe2 and Fe6. Two numbers with a comma between means that both N atoms bind to Fe, e.g. (26,2), indicating that one N atom bridges Fe2 and Fe6, whereas the other binds only to Fe2. A number or an atom after a semicolon indicates the direction of the non-coordinating atoms, where 3 and 5 indicate S3A and S5A, respectively.In addition, we considered structures with the S2B ligand bridging Fe2 and Fe6 or binding only to one of the two Fe ions. In variance to the previous study,39 we assumed that S2B is singly protonated, allowing it to dissociate from either Fe2 or Fe6. When it still bound to both ions, we tried to force it to dissociate from one of the Fe ions. Moreover, we also studied cases where the proton on S2B pointed in different directions, typically towards S3A or S5A. The nomenclature is S2B with the coordinating Fe ions in brackets (2, 6, or 26) and with the direction indicated after a semicolon with numbers 3 or 5, or an atom.
We tested 61 different structures and managed to obtain 53 of them. They are listed in Table 1. It can be seen that the most stable structure is trans(2;1A)-S2B(26;3), i.e. with trans-HNNH binding to Fe2 with the non-bonding NH group pointing towards S1A (with a S–H hydrogen-bonding distance of 2.2 Å), and with S2B coordinating to both Fe2 and Fe6, with the proton pointing towards S3A. It is most stable in the quartet BS-235 state and it is shown in Fig. 2. Cleaving the Fe2–S2B bond is strongly unfavourable, so the best structure does not have any half-dissociated S2B group. Moving the proton on S2B to the opposite side, trans(2;1A)-S2B(26;5) (also in Fig. 2), changes the energy by only 3–4 kJ mol−1. The corresponding structures with HNNH binding instead to Fe6 are only 6 kJ mol−1 less stable with the r2SCAN functional, but 12–13 kJ mol−1 with TPSSh (trans(6;3B)-S2B(26;3) and trans(6;3B)-S2B(26;5) in Fig. 2). In this case, the non-coordinating N atom of HNNH forms a hydrogen bond to S3B (2.6 Å). On the other hand, structures with HNNH pointing in the opposite direction are appreciably less stable, by 34–44 kJ mol−1 when HNNH binds to Fe2 and 17–27 kJ mol−1 when it binds to Fe6. The non-coordinating N atom still forms hydrogen bonds to the cluster (2.4 Å to S2A or 2.5 Å to S1B), but Val-70 and Ser-278 are strongly restricting its movement.
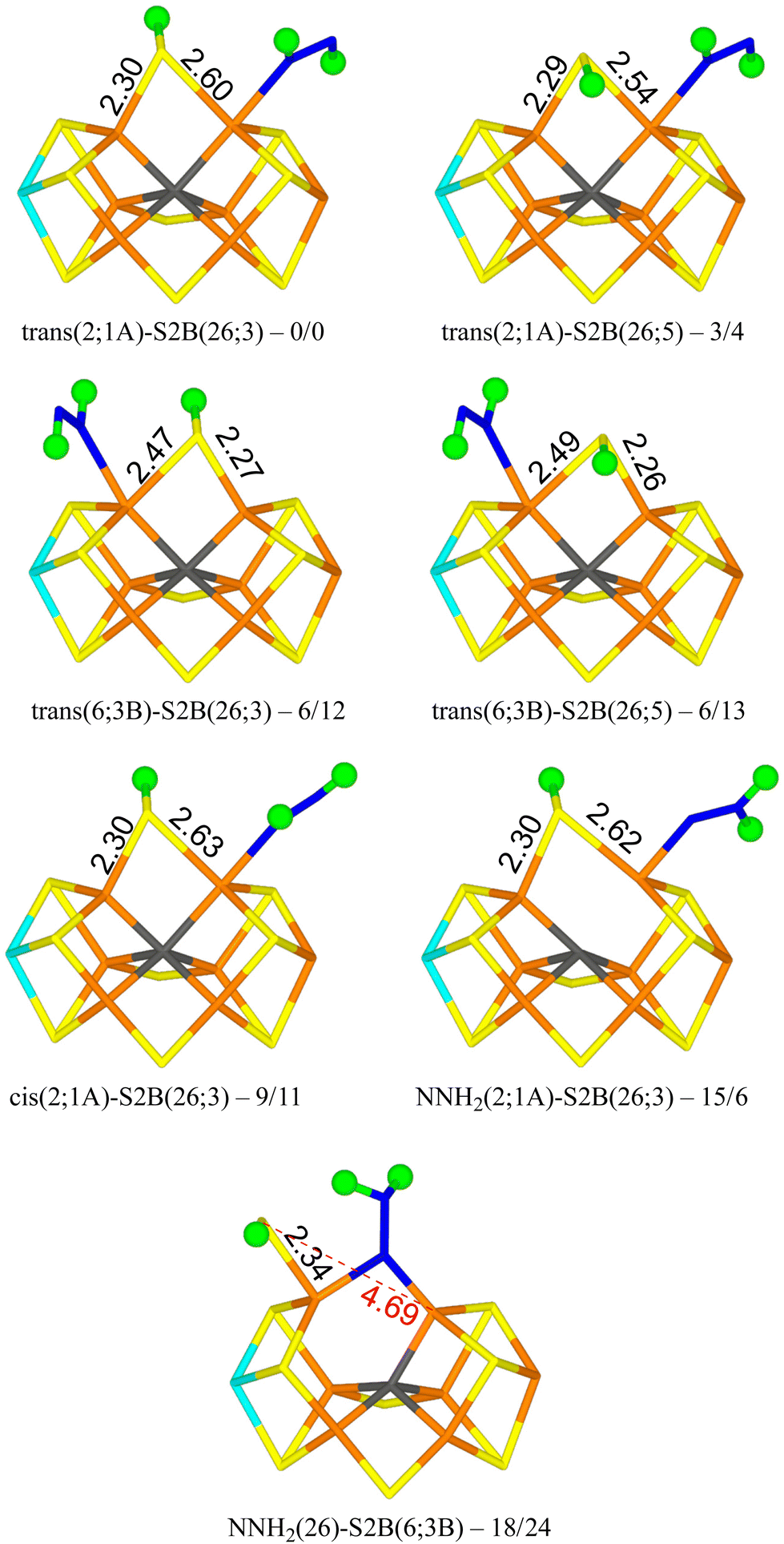 | ||
| Fig. 2 The best E4 structures with relative energies in kJ mol−1 indicated (r2SCAN/TPSSh). | ||
| Structure | r2SCAN | TPSSh |
|---|---|---|
| a Quartet BS-147 state. b Doublet BS-147 state. | ||
| HNNH2(6;HCA)-S2B(26;3) | 45 | 39 |
| HNNH2(6;HCA)-S2B(26;5) | 38 | 57 |
| NNH2(26)-S2B(2;Fe1) | 21 | 31 |
| NNH2(26)-S2B(2;5) | 34 | 39 |
| NNH2(26)-S2B(6;3B) | 18 | 24 |
| NNH2(26)-S2B(6;1B) | 18 | 40 |
| NNH2(2;1A)-S2B(26;3) | 15 | 6 |
| NNH2(2;1A)-S2B(26;5) | 25 | 33 |
| NNH2(2;2A)-S2B(6;3) | 60 | 57 |
| NNH2(2;2A)-S2B(26;5) | 76 | 71 |
| NNH2(6;1B)-S2B(2;Fe1) | 80 | 96 |
| NNH2(6;2B)-S2B(2;4A) | 91 | |
| NNH2(6;HCA)-S2B(26,3) | 56 | 57 |
| NNH2(6;3)-S2B(26;5) | 66 | 74 |
| cis(26;3)-S2B(2;Fe1) | 114 | 111 |
| cis(26;3)-S2B(2;5) | 129 | 139 |
| cis(26,2;3)-S2B(6;N) | 41 | 45 |
| cis(26,2;5)-S2B(6;Mo) | 34a | 52 |
| cis(26,2;5)-S2B(6;3) | 27a | 45 |
| cis(26,6;3)-S2B(2;Fe1) | 88 | 90 |
| cis(26,6;3)-S2B(2;5) | 102 | 104 |
| cis(26,6;5)-S2B(2;Fe1) | 71 | 73 |
| cis(26,6;5)-S2B(2;3) | 90 | 95 |
| cis(26,6;5)-S2B(2;5) | 78 | 81 |
| cis(2,6)-S2B(2;Fe1) | 103b | 108 |
| cis(2,6)-S2B(2;5) | 84a | 109 |
| cis(2,6)-S2B(6;Mo) | 92a | 112 |
| cis(2,2)-S2B(6;3B) | 81 | 99 |
| cis(2,2)-S2B(6;3) | 76 | 94 |
| cis(2;1A)-S2B(26;3) | 9 | 11 |
| cis(2;1A)-S2B(26;5) | 14 | 16 |
| cis(2;2A)-S2B(26;3) | 61 | 64 |
| cis(2;2A)-S2B(26;5) | 60 | 52 |
| cis(6,6;5)-S2B(2;2A) | 130 | |
| cis(6,6)-S2B(2;Fe1) | 130 | |
| cis(6;1B)-S2B(26;3) | 29 | 34 |
| cis(6;1B)-S2B(26;5) | 30 | 37 |
| cis(6;3B)-S2B(26;3) | 40 | 47 |
| cis(6;3B)-S2B(26;5) | 39 | 46 |
| trans(26;5)-S2B(2;Cys) | 83b | 86 |
| trans(26;5)-S2B(2;N) | 94 | 99 |
| trans(26,2;5)-S2B(6;3B) | 41 | 35 |
| trans(26,2;5)-S2B(6;N) | 33 | 28 |
| trans(26;3)-S2B(2;Cys) | 47 | 57 |
| trans(2;1A)-S2B(26;3) | 0 | 0 |
| trans(2;1A)-S2B(26;5) | 3 | 4 |
| trans(2;2A)-S2B(6;3) | 34 | 39 |
| trans(2;2A)-S2B(26;5) | 41 | 44 |
| trans(6;1B)-S2B(26;3) | 17 | 22 |
| trans(6;1B)-S2B(26;5) | 23 | 27 |
| trans(6;3B)-S2B(26;3) | 6 | 12 |
| trans(6;3B)-S2B(26;5) | 6 | 13 |
Structures with cis-HNNH binding to Fe2 (e.g. cis(2;1A)-S2B(26;3) in Fig. 2) are also competitive, 9–16 kJ mol−1 less stable than the best one, and still with S2B binding to both Fe2 and Fe6. The non-bonding N atom is still pointing towards S1A, but no hydrogen bond can form for the cis-isomer of the ligand. The opposite orientation of HNNH is much less stable, owing to steric interactions with Val-70 and Ser-278. Likewise, cis-HNNH binding to Fe6 is quite unfavourable, 29–46 kJ mol−1 less stable than the best structure.
A structure with NNH2 binding to Fe2 is also competitive, NNH2(2;1A)-S2B(26,3) in Fig. 2, especially with TPSSh, being 6–15 kJ mol−1 less stable than the best structure. The non-bonding N atom forms a hydrogen bond to S1A (2.3 Å) and S2B binds to both Fe2 and Fe6. Structures with the substrate pointing instead towards S2A (2.3 Å hydrogen-bonding distance) is ∼50 kJ mol−1 less stable.
The best structure with a half-dissociated S2B ligand is NNH2(26)-S2B(6;3B) in Fig. 2 with r2SCAN, i.e. with NNH2 bridging Fe2 and Fe6, and with one of the H atoms forming a hydrogen bond to S2B (2.14 Å H–S distance). S2B is dissociated from Fe2, but binds to Fe6. Its proton is pointing towards S3B (2.9 Å distance). The structure with the proton on S2B pointing in the opposite direction (towards S3B) is essentially degenerate, 18 kJ mol−1 less stable than the best trans(2;1A)-S2B(26;3) structure. With TPSSh, the latter structure is most stable by 16 kJ mol−1, 24 kJ mol−1 less stable than the best structure.
These results are quite similar to what has been observed in our previous studies: trans-HNNH binding to either Fe2 and Fe6 is nearly degenerate when S2B remains bound and bridging NNH2 most stable without S2B.38–40 The largest difference is that the HNNH2 structures are not competitive, being 38–57 kJ mol−1 less stable than the best structures. The reason may be the extra proton on S2B, which makes the hydrogen bond between the substrate and S2B less favourable (2.42 Å, compared to 2.24 Å in our previous study40) and also orients the substrate so that the hydrogen bond to homocitrate is worse (1.97 Å, compared to 1.69 Å (ref. 40)). However, the most important conclusion is that there is no advantage of S2B dissociation in the N2H2-bond E4 state.
E5 state
Next, we added one proton and one electron to consider the E5 state. We studied four variants of the substrate, HNNH2, NNH3, H2NNH2 (hydrazine) and HNNH3. In the latter two cases, the substrate has abstracted a proton from homocitrate. As for E4, we studied both end-on and side-on binding, to Fe2, Fe6 or both. Likewise, we considered structures with S2B bound to only Fe2 or Fe6, or to both. The structures are named as in the previous section and the results are listed in Table 2.| Structure | r2SCAN | TPSSh |
|---|---|---|
| a BS-247 state. b BS-147 state. | ||
| H2NNH2(6;1B)-S2B(26;3) | 11 | 29 |
| H2NNH2(6;1B)-S2B(26;5) | 11 | 24 |
| H2NNH2(6;2B)-S2B(26;3) | 9 | 20 |
| H2NNH2(6;2B)-S2B(26;5) | 14 | 32 |
| H2NNH(26,6)-S2B(2;Cys) | 90 | 82 |
| H2NNH(26,6)-S2B(2;2A) | 96 | 91 |
| H2NNH(2,6)-S2B(6;3B) | 113 | 120 |
| H2NNH(2,6)-S2B(6;1B) | 117 | |
| H2NNH(2;2B)-S2B(6;3B) | 79 | 93 |
| H2NNH(2,2)-S2B(6;1B) | 40 | 51 |
| HNNH2(6,2)-S2B(2;Cys) | 109 | 112 |
| HNNH2(6,2)-S2B(2;N) | 121 | 114 |
| HNNH2(2,2)-S2B(6;3B) | 47 | 42 |
| HNNH2(2,2)-S2B(6;1B) | 37 | 49 |
| HNNH2(2;2A)-S2B(6;3B) | 36 | 43 |
| HNNH2(2;2A)-S2B(6;1B) | 28 | 35 |
| HNNH2(2;Cys)-S2B(6;1B) | 9a | 15b |
| HNNH2(2;1A)-S2B(26;3) | 0 | 0 |
| HNNH2(2;1A)-S2B(26;5) | 7 | 8 |
| HNNH2(2;2A)-S2B(6;1B) | 45 | |
| HNNH2(2;2A)-S2B(26;5) | 67 | 68 |
| HNNH2(6,6)-S2B(2;Cys) | 139 | 146 |
| HNNH2(6;2B)-S2B(26;3) | 47 | 53 |
| HNNH2(6;2B)-S2B(26;5) | 32 | 55 |
| HNNH3(6;1B)-S2B(26;3) | 82 | 93 |
| HNNH3(6;1B)-S2B(26;5) | 76 | 88 |
| HNNH3(6;3B)-S2B(26;3) | 80 | 80 |
| HNNH3(6;3B)-S2B(26;5) | 67 | 75 |
| NNH3(26)-S2B(2;1A) | 140 | 139 |
| NNH3(26)-S2B(2;2A) | 131 | 134 |
| NNH3(26)-S2B(6;3B) | 59 | 79 |
| NNH3(26)-S2B(6;1B) | 53 | 76 |
| NNH3(2;1A)-S2B(26;3) | 137 | 131 |
| NNH3(2)-S2B(26;5) | 143 | 137 |
| NNH3(6;2B)-S2B(2;2A) | 218 | 243 |
| NNH3(6;1B)-S2B(26;3) | 197 | 214 |
| NNH3(6;1B)-S2B(26;5) | 186 | 197 |
| NNH3(6;3B)-S2B(26;3) | 180 | 190 |
| NNH3(6;3B)-S2B(26;5) | 185 | 191 |
We tried to optimise 45 structures and 39 of them were obtained. The best (HNNH2(2;1A)-S2B(26;3)) has HNNH2 bound to Fe2, with the non-coordinating NH2 group pointing towards S1A, forming a hydrogen bond with a H–S1A distance of 2.22 Å. The structure is most stable in the quintet BS-235 state and is shown in Fig. 3. The other H atom of the NH2 group points towards the backbone NH group of Ser-278 in a perpendicular manner (2.18 Å H–N distance). S2B binds both Fe2 and Fe6, with the proton on the S3A side. Dissociating S2B from Fe2 is strongly unfavourable. Moving the proton to the other side gives a structure that is 7–8 kJ mol−1 less stable (HNNH2(2;1A)-S2B(26;5) in Fig. 3). On the other hand, rotating the substrate around the Fe2–N bond so that the NH2 group instead forms a hydrogen bond to S2A (2.37 Å), gives a structure that is 45 kJ mol−1 less stable and S2B dissociates from Fe2. Likewise, structures with HNNH2 binding to Fe6 are quite unfavourable (by 32–55 kJ mol−1).
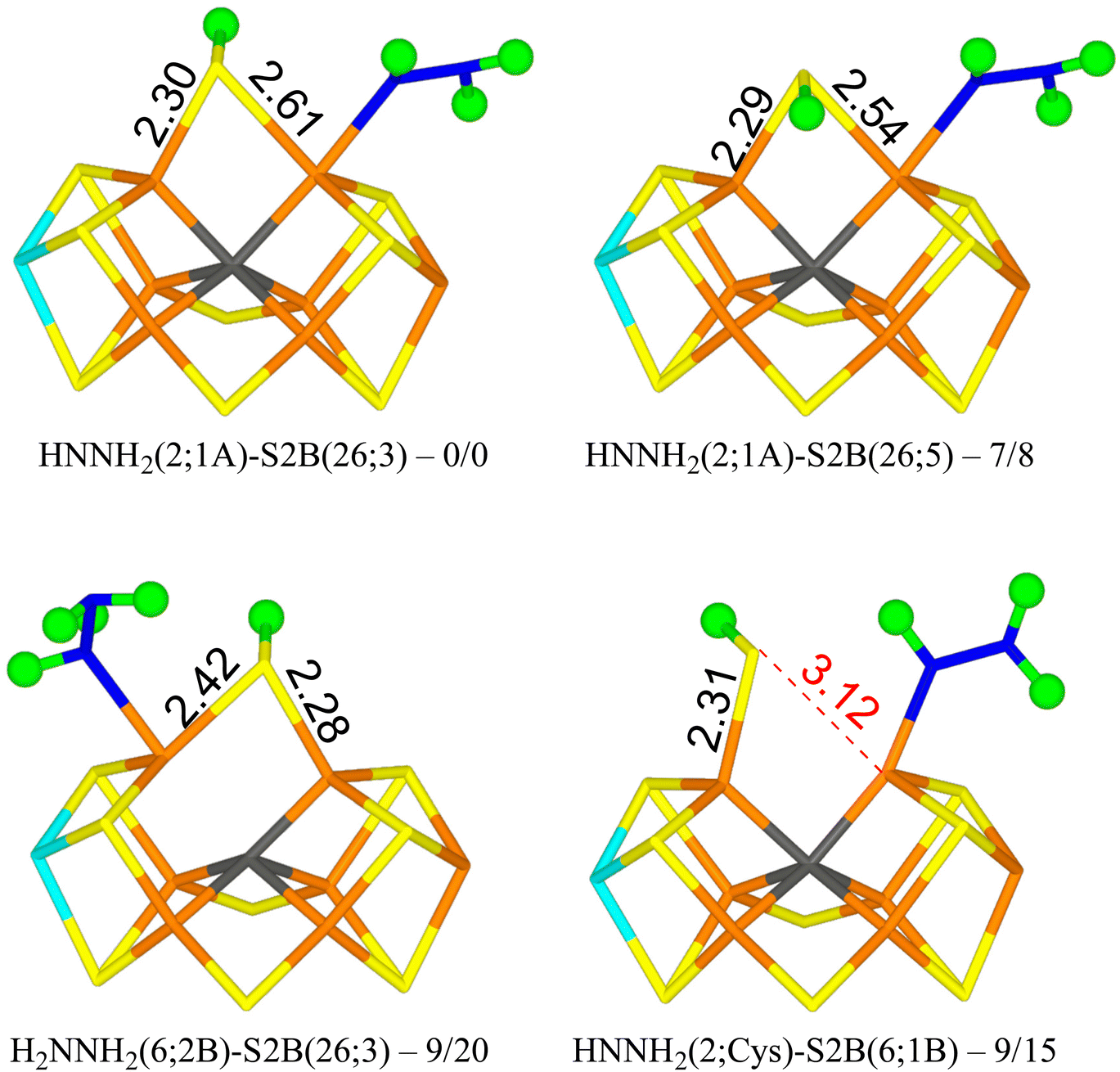 | ||
| Fig. 3 The best E5 structures with relative energies in kJ mol−1 indicated (r2SCAN/TPSSh). | ||
With r2SCAN, structures with H2NNH2 bound to Fe6 are competitive, only 8–14 kJ mol−1 less stable than HNNH2(2;1A)-S2B(26;3). The best is H2NNH2(6;2B)-S2B(26;3) in Fig. 3, in which one proton on the binding NH2 group points to the alcohol oxygen of homocitrate (2.11 Å), whereas the two protons of the non-bonding NH2 group point towards S2B (2.36 Å) and an acetate oxygen of homocitrate (3.13 Å) but with far from optimal hydrogen-bond geometries (explaining why the various conformations have similar energies). With TPSSh, these structures are 20–32 kJ mol−1 less stable than the best structure.
The best structure with S2B half-dissociated is HNNH2(2;Cys)-S2B(6;1B) in Fig. 3, i.e. with HNNH2 binding end-on to Fe2, the non-bonding NH2 group forming a hydrogen bond to SG of Cys-275 (2.77 Å) and the proton of the bonding NH group pointing towards S2B (2.55 Å). S2B binds only to Fe6 with the proton pointing towards S1B. It is only 9–15 kJ mol−1 less stable than the best structure.
Structures with HNNH3 binding to Fe6 are quite unfavourable, 67–93 kJ mol−1 less stable than the best structure. They all have S2B binding to both Fe2 and Fe6. Structures with NNH3 are strongly unfavourable by 131–243 kJ mol−1. The only exception are two structures with the unprotonated N atom bridging Fe2 and Fe6, S2B binding only to Fe6 and the proton on S2B pointing either two S1B or S3B. They are 53–59 (r2SCAN) or 76–79 kJ mol−1 less stable than the best structure.
E6 state
We next studied the E6 state by adding another electron and proton to the E5 state. We considered three variants of the substrate: H2NNH2, HNNH3 or H2NNH3 (in the latter case with one proton abstracted from homocitrate). In total, 26 structures were tested and 24 of these were obtained. They are described in Table 3.| Structure | r2SCAN | TPSSh |
|---|---|---|
| a BS-147 state. | ||
| H2NNH2(2,6)-S2B(2;Cys) | 133a | 131 |
| H2NNH2(2,6)-S2B(6;3B) | 161 | 148 |
| H2NNH2(2,6)-S2B(6;1B) | 173 | 160 |
| H2NNH2(2;1A)-S2B(6;3B) | 118 | 107 |
| H2NNH2(2;1A)-S2B(26;3) | 3 | −1 |
| H2NNH2(2;1A)-S2B(26;5) | 17 | 9 |
| H2NNH2(2;2A)-S2B(26;3) | 33 | 27 |
| H2NNH2(2;2A)-S2B(26;5) | 25 | 20 |
| H2NNH2(6;HCA)-S2B(2;2A) | 47 | 52 |
| H2NNH2(6;1B)-S2B(26;3) | 13 | 13 |
| H2NNH2(6;1B)-S2B(26;5) | 11 | 11 |
| H2NNH2(6;3B)-S2B(26;3) | 0 | 0 |
| H2NNH2(6;3B)-S2B(26;5) | 1 | 2 |
| H2NNH3(6;3B)-S2B(26;3) | 24 | 21 |
| H2NNH3(6;3B)-S2B(26;5) | 25 | 23 |
| HNNH3(2;1A)-S2B(26;3) | 63 | 58 |
| HNNH3(2;1A)-S2B(26;5) | 71 | 67 |
| HNNH3(2;2A)-S2B(26;3) | 113 | 103 |
| HNNH3(2;2A)-S2B(26;5) | 118 | 108 |
| HNNH3(6;1B)-S2B(26;3) | 95 | 97 |
| HNNH3(6;1B)-S2B(26;5) | 97 | 100 |
| HNNH3(6;3B)-S2B(26;3) | 102 | 107 |
| HNNH3(6;3B)-S2B(26;5) | 103 | 101 |
| NH3 + NH2(6)-S2B(26,3) | 85 | 93 |
| NH3 + NH(26)-S2B(6,3) | 49 | −10 |
The best structure with r2SCAN, H2NNH2(6;3B)-S2B(26;3) in Fig. 4, has H2NNH2 bound end-on to Fe2. The two H atoms of the non-coordinating NH2 group point towards S2B and S3B with distances of 2.53 and 2.83 Å, respectively. One of the H atoms of the coordinating NH2 group points towards the alcohol and acetate O atoms of homocitrate with distances of 2.28 and 2.27 Å, but the two O atoms are also involved in an internal hydrogen bond (1.42 Å) and a hydrogen bond to the sidechain NH2 group of Gln-191. S2B bridges Fe2 and Fe6, and the proton points towards S3A. The structure is most stable in the quartet BS-235 state. The structure with the proton on S2B pointing in the opposite direction (H2NNH2(6;3B)-S2B(26;5) in Fig. 4) is only 1–2 kJ mol−1 less stable.
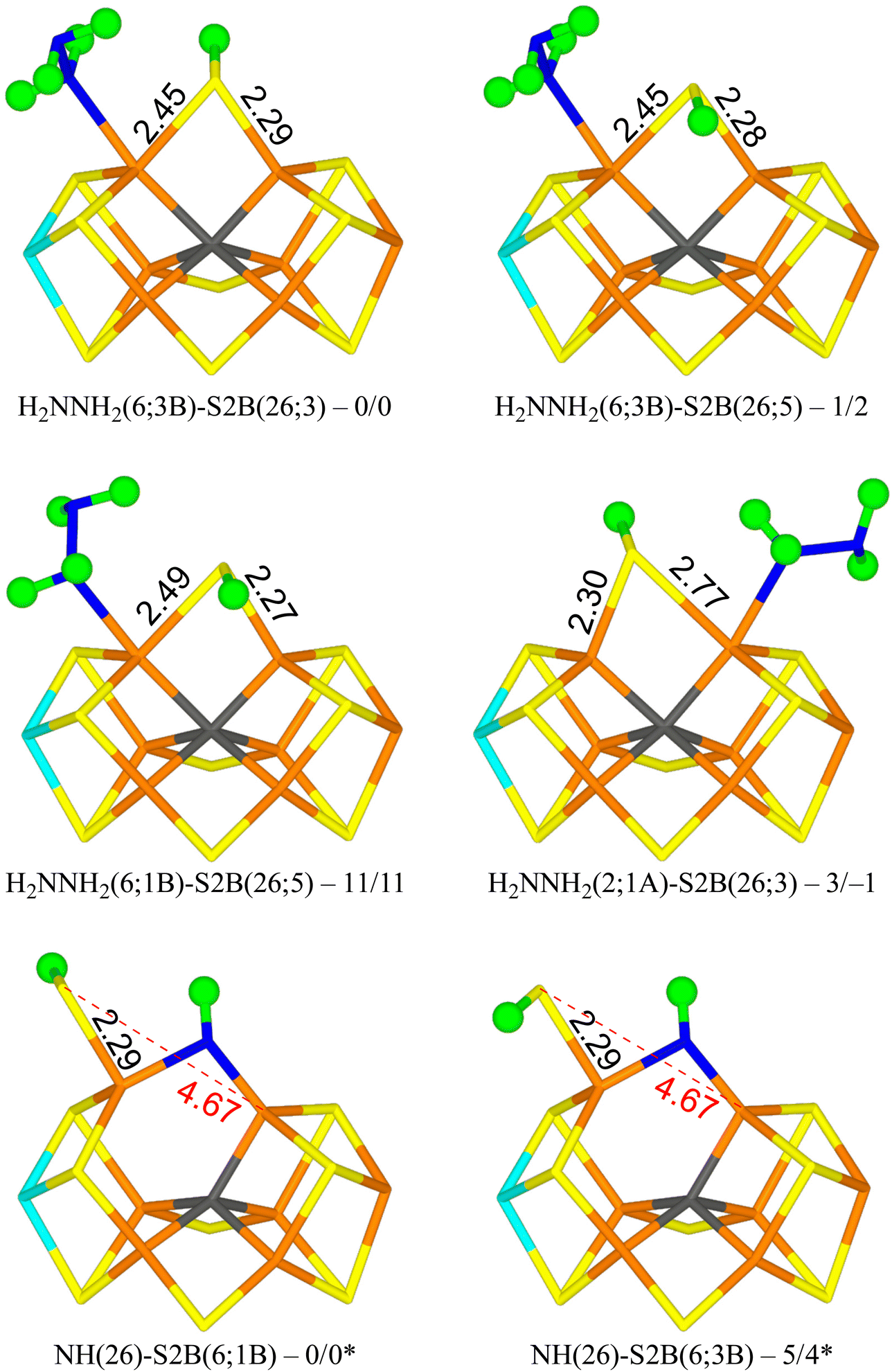 | ||
| Fig. 4 The best E6 structures with relative energies in kJ mol−1 indicated (r2SCAN/TPSSh; * indicates a distinct reference state). | ||
A structure with the non-bonding NH2 group of the substrate pointing in the opposite direction (H2NNH2(6;1B); with the H atoms pointing towards S2B and S1B with distances of 2.48 and 2.97 Å) is 11–13 kJ mol−1 less stable (e.g. H2NNH2(6;1B)-S2B(26;5) in Fig. 4). Still a third conformation, with the non-bonding NH2 group pointing towards homocitrate, which leads to dissociation of S2B from Fe6 (H2NNH2(6;HCA)-S2B(2;2A)), is 47–52 kJ mol−1 less stable than the best structure and is the most stable structure with a half-dissociated S2B.
Structures with H2NNH2 binding to Fe2 are strongly competitive. The best is H2NNH2(2;1A)-S2B(26;3) in Fig. 4, i.e. with one H atom of the non-coordinating NH2 group forming a hydrogen bond to S1A at a distance of 2.32 Å. Both the H atoms of the coordinating NH2 group point towards S2B, but with poor hydrogen-bond geometries and distances of 2.61 and 3.00 Å. S2B binds to both Fe2 and Fe6, but with quite different distances of 2.77 and 2.30 Å. This structure is only 3 kJ mol−1 less stable than the best structure. With TPSSh, it is actually 1 kJ mol−1 better. In both cases, it is most stable in the quartet BS-235 state. This probably the most relevant isomer for the E6 state, considering that the best structures of the E4 and E5 states had the substrate bound to Fe2. The corresponding S2B(26;3) structure is 10-14 kJ mol−1 less stable. Several other conformations of the substrate and the proton on S2B were also tested and they are 4–30 kJ mol−1 less stable. We also tested some structures with H2NNH2 bridging Fe2 and Fe6 and with S2B binding to only one of the Fe ions. However, they were all unfavourable by 131–173 kJ mol−1.
Two structures with H2NNH3 bound to Fe6 were optimised. They were 21–25 kJ mol−1 less stable than the best one, indicating that proton transfer from homocitrate is slightly uphill.
Eight structures with HNNH3 binding to either Fe2 or Fe6 were also tested, but they were all unfavourable by 58–118 kJ mol−1. This shows that hydrazine is the most stable isomer of the substrate at the E6 state. Again, structures with S2B half-dissociated seem to be of minor relevance.
We have tried to cleave the N–N bond in the two best structures (H2NNH2(2;1A)-S2B(26;3) and H2NNH2(2;1A)-S2B(26;3)). However, the reaction is quite prohibitive with barriers of 90–128 kJ mol−1 and the product is higher in energy than the starting structures.
For completeness, we also evaluated E6 structures after dissociation of NH3, i.e. structures with only NH bound to the FeMo cluster. Two structures with NH2 bound to Fe6, after abstraction of the proton from homocitrate were also considered. The results are gathered in Table 4.
| Structure | r2SCAN | TPSSh |
|---|---|---|
| a BS-147 state. | ||
| NH2(6)-S2B(26;3) | 86 | 79 |
| NH2(6)-S2B(26;5) | 87a | 82 |
| NH(26)-S2B(2;Cys) | 40 | 37 |
| NH(26)-S2B(2;5A) | 44 | 41 |
| NH(26)-S2B(6;3B) | 5 | 4 |
| NH(26)-S2B(6;1B) | 0 | 0 |
| NH(2;1A)-S2B(26;3) | 120 | 99 |
| NH(2;1A)-S2B(26;5) | 127 | 108 |
| NH(2)-S2B(6;HCA) | 160 | 136 |
| NH(6;1B)-S2B(2;Cys) | 205 | 201 |
| NH(6;3A)-S2B(2;Cys) | 192 | 189 |
| NH(6;3A)-S2B(26;3) | 130a | 130 |
| NH(6;3A)-S2B(26;5) | 142a | 130 |
The most stable state has NH bridging the Fe2 and Fe6 ions, and S2B binding only to Fe6 with the proton pointing towards S1B (NH(26)-S2B(6;1B) in Fig. 4). A structure with the proton pointing instead towards S3B is only 4–5 kJ mol−1 less stable (NH(26)-S2B(6;3B) in Fig. 4). Structures with S2B binding only to Fe2 are 37–44 kJ mol−1 less stable, whereas structures with NH binding only to Fe2 or Fe6 are 120–205 kJ mol−1 less stable. The two structures with NH2 binding to Fe6 are 79–91 kJ mol−1 less stable. Thus, structures with S2B half-dissociated are important when NH binds to the cluster. However, compared to the structures with an intact N–N bond, the HN structures are 31–46 kJ mol−1 less stable (adding the energy of an isolated NH3 molecule in a water-like continuum solvent).
E7 state
Next, we added an electron and a proton to reach state E7. We studied first the H2NNH3 form of the substrate. The results are gathered in Table 5. The most stable structure has H2NNH3 bound end-on to Fe2 with the non-bonded NH3 group forming a hydrogen bond to S1A (1.92 Å) and with another of the H atoms pointing transversely to the backbone N atom of Ser-278 (1.90 Å). S2B bridges Fe2 and Fe6 with the proton on the S3A side (H2NNH3(2;1A)-S2B(26;3) in Fig. 5). It is most stable in the triplet BS-147 state. A structure with the S2B proton on the other side is 10 kJ mol−1 less stable. Structures with the non-bonding NH3 group pointing in the other direction, forming hydrogen bonds to S2A and S2B (2.13 and 2.16 Å) and with one of the NH2 protons forming a hydrogen bond to SG of Cys-275 (2.68 Å) are 26–43 kJ mol−1 less stable.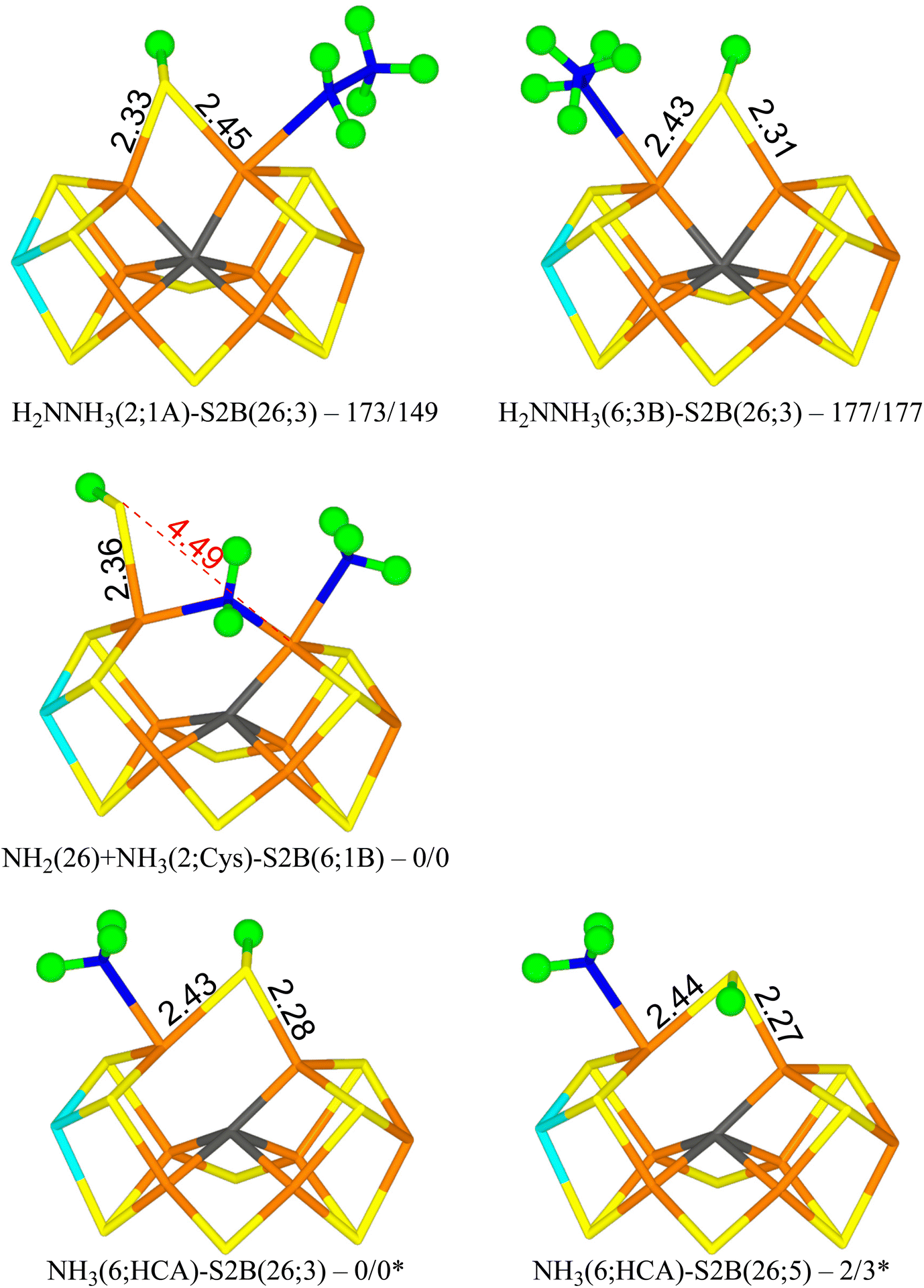 | ||
| Fig. 5 The E7 structures with relative energies in kJ mol−1 indicated (r2SCAN/TPSSh; * indicates another reference state). | ||
| Structure | r2SCAN | TPSSh |
|---|---|---|
| a BS-235 state. b BS-346 state. c BS-156 state. | ||
| H2NNH3(2;1A)-S2B(26;3) | 173 | 149 |
| H2NNH3(2;1A)-S2B(26;5) | 183 | 158 |
| H2NNH3(2;2A)-S2B(26;3) | 199 | 177 |
| H2NNH3(2;2A)-S2B(26;5) | 214 | 192 |
| H2NNH3(6;1B-diss)-S2B(26;3) | 168 | 154 |
| H2NNH3(6;1B-diss)-S2B(26;5) | 160a | 158 |
| H2NNH3(6;3B)-S2B(26;3) | 177a | 177 |
| H2NNH3(6;3B)-S2B(26;5) | 178 | 183 |
| NH2(2;1A) + NH3(26)-S2B(6;HCA) | 83b | 71 |
| NH2(26) + NH3(6;HCA)-S2B(2;Cys) | 23b | 14 |
| NH2(26) + NH3(6;HCA)-S2B(2;N) | 23b | 24 |
| NH2(26) + NH3(2;Cys)-S2B(6;1B) | 0c | 0c |
| NH2(26) + NH3(diss)-S2B(6;1B) | 29b | |
| NH2(2;NH3) + NH3(diss)-S2B(6;HCA) | 58c | 44b |
Structures with H2NNH3 bound end-on to Fe6 are only 4–5 kJ mol−1 less stable with r2SCAN, but 28–34 kJ mol−1 less stable with TPSSh. In the best structure (H2NNH3(6;3B)-S2B(26;3) in Fig. 5), the non-bonding NH3 group forms hydrogen bonds to S3B and S2B (2.21 and 2.27 Å). S2B still bridges Fe2 and Fe6, with the proton on the S3A side. If the NH3 group is moved to the other side, the ligand actually dissociates, giving a structure that is actually 9 kJ mol−1 more stable than the best state with r2SCAN but 6 kJ mol−1 less stable with TPSSh.
From the H2NNH3(2;1A)-S2B(26;3) structure, the N–N bond can easily be cleaved with an activation energy of only 41–50 kJ mol−1. Therefore, we studied several product (NH2 + NH3) structures. In the best structure (NH2(26) + NH3(2;1A)-S2B(6;1B) in Fig. 5), NH2 bridges Fe2 and Fe6, whereas NH3 binds to Fe2, forming a hydrogen bond to S1A (2.67 Å). S2B binds only to Fe6, with the proton pointing towards S1B (3.29 Å). It is most stable in the triplet BS-156 state. This structure is 149–173 kJ mol−1 more stable than the best H2NNH3 structure. Structures with NH3 instead binding to Fe6 is only 14–23 kJ mol−1 less stable.
NH3 can dissociate from the FeMo cluster with an activation energy of only 32 kJ mol−1. Therefore, we also studied structures without NH3 (i.e. dissociated and excluded from the calculations). The results are shown in Table 6. The most stable structure has NH3 bound to Fe6, i.e. with a proton abstracted from homocitrate (NH3(6;HCA)-S2B(26;3) in Fig. 5). One of the protons forms a hydrogen bond to the alcohol O atom of homocitrate (2.23 Å), whereas another is directed towards S2B (2.81 Å). The latter atom bridges Fe2 and Fe6 with the proton on the S3A side. It is most stable in the quintet BS-235 state. A structure with the S2B proton on the other side is 2–3 kJ mol−1 less stable (NH3(6;HCA)-S2B(26;5) in Fig. 5).
| Structure | r2SCAN | TPSSh |
|---|---|---|
| a BS-147 state. | ||
| NH2(26)-S2B(2;Cys) | 47 | 35 |
| NH2(26)-S2B(2;N) | 54 | 42 |
| NH2(26)-S2B(6;3B) | 24 | 26 |
| NH2(26)-S2B(6;1B) | 26 | 19 |
| NH2(2)-S2B(26;3) | 78a | 66 |
| NH2(2)-S2B(26;5) | 75a | 66 |
| NH2(2;2A)-S2B(6;3B) | 217 | |
| NH2(6;1B)-S2B(26;3) | 77 | 79 |
| NH2(6;1B)-S2B(26;5) | 76 | 80 |
| NH3(6;HCA)-S2B(26;3) | 0 | 0 |
| NH3(6;HCA)-S2B(26;5) | 2 | 3 |
Structures with NH2 bridging Fe2 and Fe6 are at least 19–24 kJ mol−1 less stable. They have S2B binding to only one Fe ion, preferably Fe6. Structures with NH2 binding to either Fe2 or Fe6 (with S2B bridging Fe2 and Fe6) are at least 66–79 or 76–79 kJ mol−1 less stable than the NH3(6;HCA)-S2B(26;3) structure.
E8 state
Finally, we studied the E8 state with NH3 bound to the FeMo cluster. The results are collected in Table 7. Four structures are essentially degenerate (within 5 kJ mol−1). Two have NH3 bound to Fe6 with one of the protons forming a hydrogen bond to the acetate group of homocitrate (1.87–1.90 Å) and S2B bridging Fe2 and Fe6 with the proton either on the S3A or S5A side (NH3(6)-S2B(26;3) and NH3(6)-S2B(26;5) in Fig. 6). The other two have NH3 bound to Fe2 with the three protons approximately in the directions of SG of Cys-275, S1A and S2A (all distances are 3.0–3.2 Å) and S2B bridging Fe2 and Fe6 with the proton either on the S3A or S5A side (NH3(2)-S2B(26;3) and NH3(2)-S2B(26;5) in Fig. 6). Structures with S2B binding only to Fe6 and NH3 to Fe2 are 16–26 kJ mol−1 less stable. Structures with NH4, where the proton has been abstracted from homocitrate, are 11–13 (r2SCAN) or 21–22 kJ mol−1 (TPSSh) less stable and in these NH4 has dissociated from the cluster (and S2B bridges Fe2 and Fe6). All structures are most stable in the quartet BS-235 state.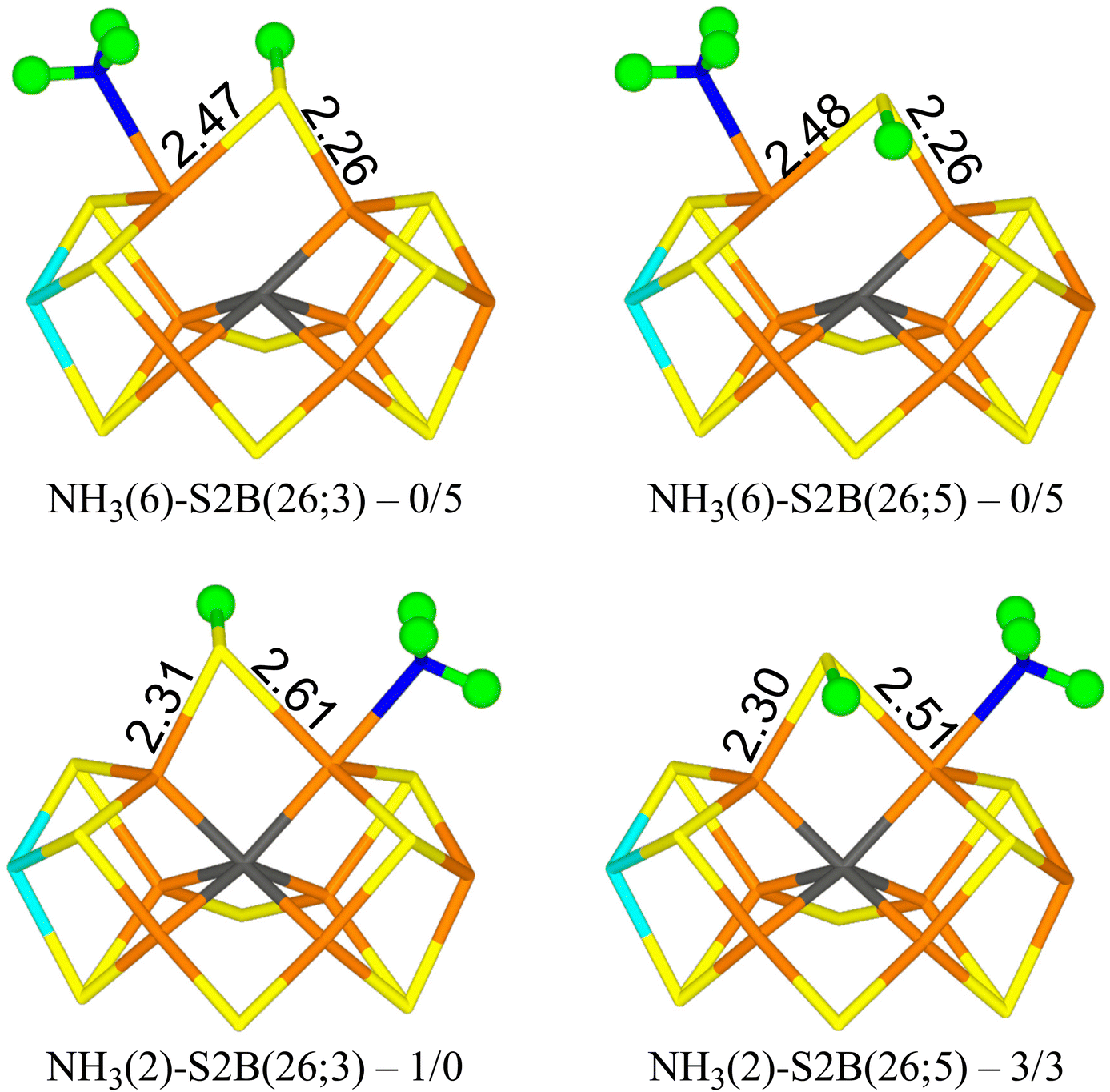 | ||
| Fig. 6 The best E8 structures with relative energies in kJ mol−1 indicated (r2SCAN/TPSSh). | ||
| Structure | r2SCAN | TPSSh |
|---|---|---|
| a Converged to another structure. | ||
| NH3(2)-S2B(26;3) | 0.6 | 0 |
| NH3(2)-S2B(26;5) | 3 | 3 |
| NH3(2)-S2B(6;3B) | 26 | |
| NH3(2)-S2B(6;1B) | 16 | |
| NH3(6)-S2B(26;3) | 0.0 | 5 |
| NH3(6)-S2B(26;5) | 0.1 | 5 |
| NH4(diss)-S2B(26;3) | 13 | 22 |
| NH4(diss)-S2B(26;5) | 11 | 21 |
Conclusions
We have investigated whether structures with S2B dissociated from either Fe2 or Fe6 may be involved in the second half of the reaction mechanism of nitrogenase. As mentioned in the Introduction, we have previously studied this part of the reaction mechanism assuming either that S2B binds both to Fe2 and Fe6 or that it is fully dissociated from the FeMo cluster.38,39 However, recent studies have indicated that for the E2–E4 states, structures with S2B dissociated from either Fe2 or Fe6 are more stable than structures with a bridging S2B.22,44,45 Therefore, it is of great interest to know how this finding affects the second half of the reaction mechanism. To make such a half-dissociation possible, we have added one extra proton on S2B, compared to the previous studies. Moreover, we employ two DFT functionals, r2SCAN and TPSSh, which in previous studies have supported and favoured half-dissociation of S2B,22,44,45 but also giving accurate results for the FeMo cluster of nitrogenase.58Interestingly, we see little advantage of half-dissociation of S2B. For the E4 and E5 states, such structures are at least 16–24 and 9–15 kJ mol−1 less stable than the best structures with a bridging S2B, respectively. For the E6 state, structures with a half-dissociated S2B are disfavoured by 47–52 kJ mol−1. However, with NH3 dissociated, the best E6 structure has S2B bound only to Fe6, because the NH ligand takes the Fe2–Fe6 bridging position. On the other hand, our results indicate that such structures are not involved in the mechanism. The situation is similar for the E7 state: With an intact N–N bond, only structures with a bridging S2B ligand is found, whereas after cleavage of N–N bond, NH2 prefers to bridge between Fe2 and Fe6, forcing S2B to dissociate from Fe2 in the most stable state. However, the substrate may also extract a proton from homocitrate, giving NH3, which prefers to bind to Fe6 and then S2B goes back to a bridging position. In the E8 state, NH3 may bind either to Fe2 or Fe6, but S2B prefers a bridging position by at least 16 kJ mol−1.
Fig. 7 shows our suggested reaction mechanism for nitrogenase with an extra proton on S2B. It contains only a single half-dissociated structure, NH2(26) + NH3(2;1A)-S2B(6;1B), and only intermittently during the E7 state. Therefore, we conclude that half-dissociation of S2B needs to be considered in the reaction mechanism of nitrogenase but seems to be of minor importance during the second half-reaction. The reason for this may be that there are no states with a bridging hydride ion in the second half-reaction.
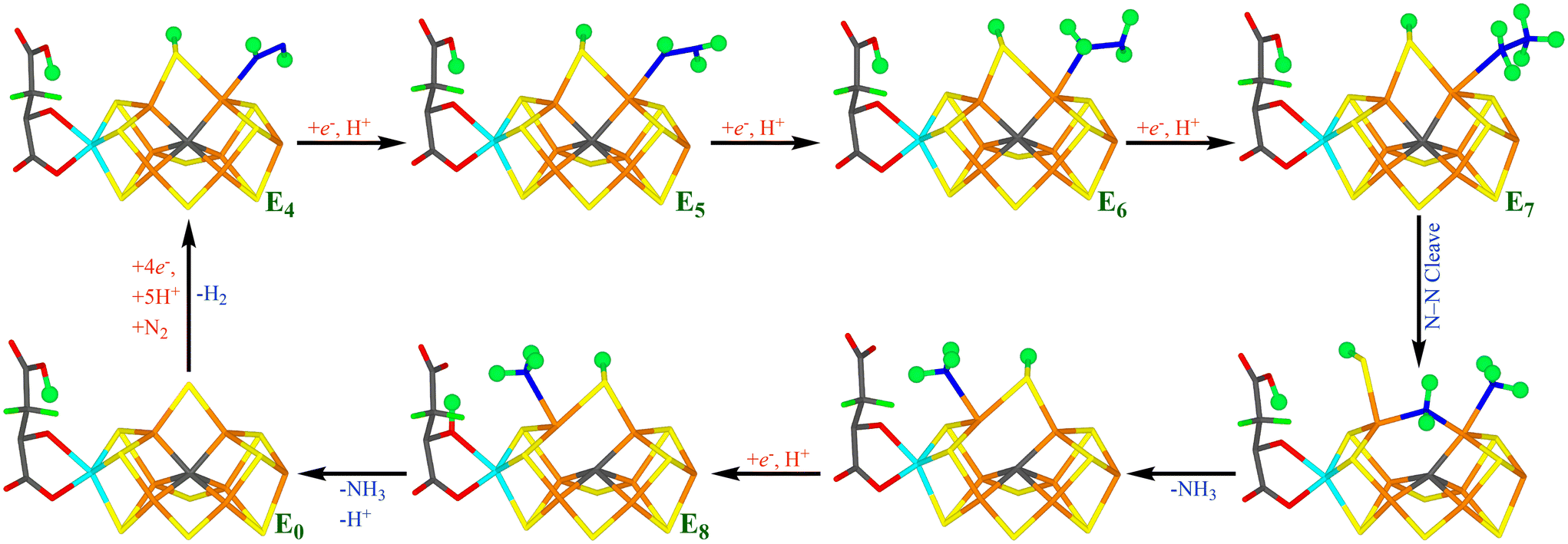 | ||
| Fig. 7 Suggested reaction mechanism for nitrogenase, assuming that the S2B ligand is protonated. | ||
In this study we have compared the results of two DFT functionals, r2SCAN and TPSSh. The two functionals give similar results for both structures and relative energies. For example, mean absolute deviation of the relative energies calculated with the two methods in Tables 1–7 are 5–10 kJ mol−1 and for the most stable structures in Fig. 2–6, the maximum difference in the relative energies is 1–11 kJ mol−1. The only exception is when different BS states are involved; then the difference can increase up to 25 kJ mol−1 (for example H2NNH3(2;1A)-S2B(26;3) in Fig. 5). Thus, the two functionals give quite similar results although one of them is a meta generalised gradient approximation functional, whereas TPSSh is a hybrid functional with 10% Hartree–Fock exchange. The two functionals were selected because they give accurate structures of nitrogenase models58 but they also give a stronger preference of half-dissociated structures, e.g. compared to pure generalised gradient approximation functionals like TPSS.45
Finally, we note that the suggested reaction mechanism is alternating, i.e. the protons are added alternating to the two N atoms of the substrate, so that HNNH and H2NNH2 are intermediates in the mechanism and that NH3 does not dissociate until the E7 state. This is in agreement with our previously suggested mechanism with S2B bridging or dissociated from the cluster.38,39 On the other hand, the substrate binds preferably to Fe2, which is in contrast to the previously suggested mechanism with a bridging S2B,39 where the substrate bound preferably to Fe6. The difference seems to come from the protonation of S2B. The protonation state of S2B remains to be settled, but the previous mechanism with unprotonated S2B has the advantage that it employs homocitrate as a proton buffer, which explains why this group is needed for the nitrogenase reaction.4,85
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This investigation has been supported by grants from the Swedish Research Council (project 2022-04978) and from China Scholarship Council. The computations were enabled by resources provided by LUNARC, the Centre for Scientific and Technical Computing at Lund University, and by the National Academic Infrastructure for Supercomputing in Sweden (NAISS) at NSC at Linköping University (Tetralith) and PDC at KTH Royal Institute of Technology (Dardel), partially funded by the Swedish Research Council through grant agreements no. 2022-06725 and 2018-05973.References
- B. K. Burgess and D. J. Lowe, Chem. Rev., 1996, 96, 2983–3012 CrossRef CAS PubMed
.
-
B. Schmid, H.-J. Chiu, V. Ramakrishnan, J. B. Howard and D. C. Rees, in Handbook of Metalloproteins, John Wiley & Sons, Ltd, 2006, pp. 1025–1036, DOI:10.1002/0470028637.met174
- B. M. Hoffman, D. Lukoyanov, Z.-Y. Yang, D. R. Dean and L. C. Seefeldt, Chem. Rev., 2014, 114, 4041–4062 CrossRef CAS PubMed
- L. C. Seefeldt, Z.-Y. Yang, D. A. Lukoyanov, D. F. Harris, D. R. Dean, S. Raugei and B. M. Hoffman, Chem. Rev., 2020, 120, 5082–5106 CrossRef CAS PubMed
- T. Spatzal, M. Aksoyoglu, L. Zhang, S. L. A. Andrade, E. Schleicher, S. Weber, D. C. Rees and O. Einsle, Science, 2011, 334, 940–940 CrossRef CAS PubMed
- J. Kim and D. C. Rees, Science, 1992, 257, 1677–1682 CrossRef CAS PubMed
- O. Einsle, F. A. Tezcan, S. L. A. Andrade, B. Schmid, M. Yoshida, J. B. Howard and D. C. Rees, Science, 2002, 297, 1696–1696 CrossRef CAS PubMed
- T. Spatzal, K. A. Perez, O. Einsle, J. B. Howard and D. C. Rees, Science, 2014, 345, 1620–1623 CrossRef CAS PubMed
- O. Einsle, J. Biol. Inorg. Chem., 2014, 19, 737–745 CrossRef CAS PubMed
- A. J. Jasniewski, C. C. Lee, M. W. Ribbe and Y. Hu, Chem. Rev., 2020, 120, 5107–5157 CrossRef CAS PubMed
-
R. N. F. Thorneley and D. J. Lowe, in Molybdenum Enzymes, ed. T. G. Spiro, Wiley, New York, 1985, pp. 221–284 Search PubMed
- C. Van Stappen, L. Decamps, G. E. Cutsail, R. Bjornsson, J. T. Henthorn, J. A. Birrell and S. DeBeer, Chem. Rev., 2020, 120, 5005–5081 CrossRef CAS PubMed
- I. Dance, ChemBioChem, 2020, 21, 1671–1709 CrossRef CAS PubMed
- Z. Y. Yang, N. Khadka, D. Lukoyanov, B. M. Hoffman, D. R. Dean and L. C. Seefeldt, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 16327–16332 CrossRef CAS PubMed
- D. Lukoyanov, N. Khadka, Z.-Y. Yang, D. R. Dean, L. C. Seefeldt and B. M. Hoffman, J. Am. Chem. Soc., 2016, 138, 10674–10683 CrossRef CAS PubMed
- V. Hoeke, L. Tociu, D. A. Case, L. C. Seefeldt, S. Raugei and B. M. Hoffman, J. Am. Chem. Soc., 2019, 141, 11984–11996 CrossRef CAS PubMed
- H. Yang, J. Rittle, A. R. Marts, J. C. Peters and B. M. Hoffman, Inorg. Chem., 2018, 57, 12323–12330 CrossRef CAS PubMed
- R. Y. Igarashi, M. Laryukhin, P. C. Dos Santos, H.-I. Lee, D. R. Dean, L. C. Seefeldt and B. M. Hoffman, J. Am. Chem. Soc., 2005, 127, 6231–6241 CrossRef CAS PubMed
- S. Raugei, L. C. Seefeldt and B. M. Hoffman, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 10521–10530 CrossRef PubMed
- L. Cao and U. Ryde, J. Chem. Theory Comput., 2020, 16, 1936–1952 CrossRef CAS PubMed
- A. T. Thorhallsson, B. Benediktsson and R. Bjornsson, Chem. Sci., 2019, 10, 11110–11124 RSC
- H. Jiang and U. Ryde, Dalton Trans., 2023, 52, 9104–9120 RSC
- P. E. M. Siegbahn, J. Am. Chem. Soc., 2016, 138, 10485–10495 CrossRef CAS PubMed
- P. E. M. Siegbahn, J. Comput. Chem., 2018, 39, 743–747 CrossRef CAS PubMed
- J. B. Varley, Y. Wang, K. Chan, F. Studt and J. K. Nørskov, Phys. Chem. Chem. Phys., 2015, 17, 29541–29547 RSC
- M. Rohde, D. Sippel, C. Trncik, S. L. A. Andrade and O. Einsle, Biochemistry, 2018, 57, 5497–5504 CrossRef CAS PubMed
- W.-L. Li, Y. Li, J. Li and T. Head-Gordon, Chem Catal., 2023, 3, 100662 CrossRef CAS
- P. E. M. Siegbahn, Phys. Chem. Chem. Phys., 2023, 25, 23602–23613 RSC
- J. Chatt, J. R. Dilworth and R. L. Richards, Chem. Rev., 1978, 78, 589–625 CrossRef CAS
- J. Chatt, Annu. Proc. Phytochem. Soc. Eur., 1980, 18, 1–18 CAS
- D. V. Yandulov and R. R. Schrock, Science, 2003, 301, 76–78 CrossRef CAS PubMed
- R. R. Schrock, Acc. Chem. Res., 2005, 38, 955–962 CrossRef CAS PubMed
- R. R. Schrock, Angew. Chem., Int. Ed., 2008, 47, 5512–5522 CrossRef CAS PubMed
- D. Sippel, M. Rohde, J. Netzer, C. Trncik, J. Gies, K. Grunau, I. Djurdjevic, L. Decamps, S. L. A. Andrade and O. Einsle, Science, 2018, 359, 1484–1489 CrossRef CAS PubMed
- R. N. F. Thornely, R. R. Eady and D. J. Lowe, Nature, 1978, 272, 557–558 CrossRef
- R. N. F. Thorneley and D. J. Lowe, Biochem. J., 1984, 224, 887–894 CrossRef CAS PubMed
- D. Lukoyanov, S. A. Dikanov, Z.-Y. Yang, B. M. Barney, R. I. Samoilova, K. V. Narasimhulu, D. R. Dean, L. C. Seefeldt and B. M. Hoffman, J. Am. Chem. Soc., 2011, 133, 11655–11664 CrossRef CAS PubMed
- L. Cao and U. Ryde, J. Catal., 2020, 391, 247–259 CrossRef CAS
- H. Jiang and U. Ryde, Chem. – Eur. J., 2022, 28, e202103933 CrossRef CAS PubMed
- L. Cao and U. Ryde, J. Biol. Inorg. Chem., 2020, 25, 521–540 CrossRef CAS PubMed
- H. Jiang, O. K. G. Svensson, L. Cao and U. Ryde, Angew. Chem., Int. Ed., 2022, 61, e202208544 CrossRef CAS PubMed
- P. P. Hallmen and J. Kästner, Z. Anorg. Allg. Chem., 2015, 641, 118–122 CrossRef CAS
- I. Dance, Dalton Trans., 2019, 48, 1251–1262 RSC
- A. T. Thorhallsson and R. Bjornsson, Chem. – Eur. J., 2021, 27, 16788–16800 CrossRef CAS PubMed
- H. Jiang, O. K. G. Svensson and U. Ryde, Inorg. Chem., 2022, 61, 18067–18076 CrossRef CAS PubMed
- L. Cao and U. Ryde, Phys. Chem. Chem. Phys., 2019, 21, 2480–2488 RSC
- L. Cao, O. Caldararu and U. Ryde, J. Phys. Chem. B, 2017, 121, 8242–8262 CrossRef CAS PubMed
- H. Jiang and U. Ryde, Phys. Chem. Chem. Phys., 2024, 26, 1364–1375 RSC
- R. Bjornsson, F. Neese and S. DeBeer, Inorg. Chem., 2017, 56, 1470–1477 CrossRef CAS PubMed
- B. M. Barney, J. McClead, D. Lukoyanov, M. Laryukhin, T.-c. Yang, D. R. Dean, B. M. Hoffman and L. C. Seefeldt, Biochemistry, 2007, 46, 6784–6794 CrossRef CAS PubMed
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS PubMed
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS
- L. Hu and U. Ryde, J. Chem. Theory Comput., 2011, 7, 2452–2463 CrossRef CAS PubMed
- C. I. Bayly, P. Cieplak, W. D. Cornell and P. A. Kollman, J. Phys. Chem., 1993, 97, 10269–10280 CrossRef CAS
- F. Furche, R. Ahlrichs, C. Hättig, W. Klopper, M. Sierka and F. Weigend, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 91–100 CAS
- J. W. Furness, A. D. Kaplan, J. Ning, J. P. Perdew and J. Sun, J. Phys. Chem. Lett., 2020, 11, 8208–8215 CrossRef CAS PubMed
- V. N. Staroverov, G. E. Scuseria, J. Tao and J. P. Perdew, J. Chem. Phys., 2003, 119, 12129–12137 CrossRef CAS
- B. Benediktsson and R. Bjornsson, J. Chem. Theory Comput., 2022, 18, 1437–1457 CrossRef CAS PubMed
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef
- K. Eichkorn, O. Treutler, H. Öhm, M. Häser and R. Ahlrichs, Chem. Phys. Lett., 1995, 240, 283–289 CrossRef CAS
- K. Eichkorn, F. Weigend, O. Treutler and R. Ahlrichs, Theor. Chem. Acc., 1997, 97, 119–124 Search PubMed
- E. Caldeweyher, S. Ehlert, A. Hansen, H. Neugebauer, S. Spicher, C. Bannwarth and S. Grimme, J. Chem. Phys., 2019, 150, 154122–154122 CrossRef PubMed
- T. Spatzal, J. Schlesier, E.-M. Burger, D. Sippel, L. Zhang, S. L. A. Andrade, D. C. Rees and O. Einsle, Nat. Commun., 2016, 7, 10902–10902 CrossRef CAS PubMed
- R. Bjornsson, F. A. Lima, T. Spatzal, T. Weyhermüller, P. Glatzel, E. Bill, O. Einsle, F. Neese and S. DeBeer, Chem. Sci., 2014, 5, 3096–3103 RSC
- T. Lovell, J. Li, T. Liu, D. A. Case and L. Noodleman, J. Am. Chem. Soc., 2001, 123, 12392–12410 CrossRef CAS PubMed
- L. Cao and U. Ryde, Int. J. Quantum Chem., 2018, 118, e25627 CrossRef
- C. Greco, P. Fantucci, U. Ryde and L. Di Gioia, Int. J. Quantum Chem., 2011, 111, 3949–3960 CrossRef CAS
- R. K. Szilagyi and M. A. Winslow, J. Comput. Chem., 2006, 27, 1385–1397 CrossRef CAS PubMed
- U. Ryde, J. Comput.-Aided Mol. Des., 1996, 10, 153–164 CrossRef CAS PubMed
- U. Ryde and M. H. M. Olsson, Int. J. Quantum Chem., 2001, 81, 335–347 CrossRef CAS
- N. Reuter, A. Dejaegere, B. Maigret and M. Karplus, J. Phys. Chem. A, 2000, 104, 1720–1735 CrossRef CAS
- L. Hu, P. Söderhjelm and U. Ryde, J. Chem. Theory Comput., 2011, 7, 761–777 CrossRef CAS PubMed
- L. Cao and U. Ryde, Front. Chem., 2018, 6, 89–89 CrossRef PubMed
- I. Mayer, Chem. Phys. Lett., 1983, 97, 270–274 CrossRef CAS
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed
- I. Dance, J. Am. Chem. Soc., 2005, 127, 10925–10942 CrossRef CAS PubMed
- I. Dance, Dalton Trans., 2012, 41, 7647–7659 RSC
- I. Dance, Inorg. Chem., 2013, 52, 13068–13077 CrossRef CAS PubMed
- I. Dance, Dalton Trans., 2015, 44, 18167–18186 RSC
- I. Dance, J. Inorg. Biochem., 2017, 169, 32–43 CrossRef CAS PubMed
- I. Dance, Biochemistry, 2006, 45, 6328–6340 CrossRef CAS PubMed
- P. E. M. Siegbahn, J. Phys. Chem. B, 2023, 127, 2156–2159 CrossRef CAS PubMed
- L. C. Seefeldt, I. G. Dance and D. R. Dean, Biochemistry, 2004, 43, 1401–1409 CrossRef CAS PubMed
- S. J. George, B. M. Barney, D. Mitra, R. Y. Igarashi, Y. Guo, D. R. Dean, S. P. Cramer and L. C. Seefeldt, J. Inorg. Biochem., 2012, 112, 85–92 CrossRef CAS PubMed
- J. Imperial, T. R. Hoover, M. S. Madden, P. W. Ludden and V. K. Shah, Biochemistry, 1989, 28, 7796–7799 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dt00937a |
| This journal is © The Royal Society of Chemistry 2024 |