
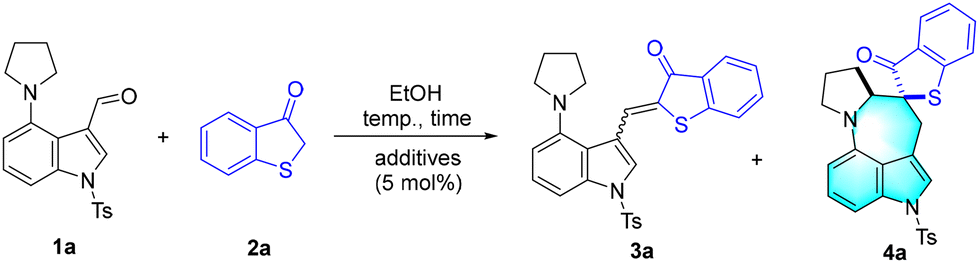
EtOH-mediated cascade C(sp3)–H alkylation via aromatization-driven [1,6]-hydride transfer: green and divergent synthesis of spirocyclic azepino[4,3,2-cd]indoles†
Yao-Bin
Shen‡
 *a,
Qian-Hao
Zhuang‡
b,
Xiao-Lin
Wang
b,
Xiao-De
An
b,
Bin
Qiu
b,
Tiesheng
Shi
a and
Jian
Xiao
*b
*a,
Qian-Hao
Zhuang‡
b,
Xiao-Lin
Wang
b,
Xiao-De
An
b,
Bin
Qiu
b,
Tiesheng
Shi
a and
Jian
Xiao
*b
aCollege of Chemistry, Chemical Engineering and Materials Science, Zaozhuang University, Zaozhuang, Shandong 277160, China. E-mail: shenyaobin2011@163.com
bCollege of Chemistry and Pharmaceutical Sciences, Qingdao Agricultural University, Qingdao, Shandong 266109, China. E-mail: chemjianxiao@163.com
First published on 5th November 2024
Abstract
The development of green and efficient methods for the construction of azepinoindole skeletons remains highly desirable yet challenging. Described herein are the EtOH-mediated cascade C(sp3)–H alkylation reactions of 4-dialkylamino-indole-3-carbaldehydes for green and divergent synthesis of spirocyclic azepino[4,3,2-cd]indole derivatives. This protocol proceeded through a cascade in situ assembly of pre-aromatics/aromatization-driven [1,6]-hydride transfer/cyclization sequence, which exhibited many advantages such as green bio-sourced EtOH as the reaction medium, metal-free and redox-neutral conditions, high step-/atom-economy, water as waste, high yields, excellent diastereoselectivities (up to >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr), a wide substrate scope, and diverse transformations.
:1 dr), a wide substrate scope, and diverse transformations.
Introduction
Nowadays, green synthetic chemistry has emerged as a prevailing research field, which advocates 12 principles of less hazardous synthesis, atom-/step-economy, benign solvents, waste prevention, and so on.1 The ‘Green ChemisTREE’,1c presented by Anastas in 2018, vividly showcased a wide range of research areas stemming from green chemistry. For example, non-metal catalysis and C–H bond functionalization align with the principle of less hazardous synthesis, while one-pot cascade reactions meet the principles of atom-/step-economy and waste prevention. The bio-sourced ethanol (EtOH) satisfies the principle of benign solvents.2 In view of the escalating concern about sustainability, the design of green synthetic methods, such as EtOH-mediated metal-free C–H functionalization cascade reactions, for the synthesis of high value-added molecules is undoubtedly appealing.Azepinoindoles, which combine the privileged structures of azepine and indole, are attractive targets in both synthetic chemistry and the medicinal industry.3,4 Among the azepinoindole family, azepino[4,3,2-cd]indole and its derivatives have garnered considerable attention owing to their potential as vasopressin V2 antagonists, RIPK1 inhibitors, and CGRP V2 receptor antagonists (Scheme 1A).5 Given their importance, efficient synthesis of such scaffolds is highly desirable; however, to our knowledge, only a limited number of methods have been documented to date (Scheme 1B).5–9 One feasible strategy relies on the construction of an indole nucleus, which involves a three-step synthetic procedure via tandem palladium catalysis (Scheme 1B, a).6 Another effective strategy depends on azepine formation through reactions of pre-functionalized 4-NO2-, 4-NH2-, and 4-NHR-indoles, including two-step AgOTf-catalyzed nucleophilic addition and Pd/C-catalyzed reductive cyclization (Scheme 1B, b),7 and TsOH-catalyzed or heat-promoted intramolecular amidation (Scheme 1B, c),5,8 Recently, in 2023, Guo and Xie's group disclosed an elegant nickel-catalyzed cascade [4 + 3] annulation reaction between N-alkyl-4-aminoindoles and β,γ-unsaturated α-ketoesters for the synthesis of chiral azepino[4,3,2-cd]indole derivatives (Scheme 1B, d).9 In spite of the merits of these finite but impressive works, the dependence on metal catalysis, tedious substrate pre-synthesis, multi-step procedures, and toxic reaction media may contradict the 12 principles of green chemistry. Besides, the substrate scope and product diversity seem limited and deserve an expansion to enrich the azepinoindole library. Therefore, there is an urgent need to explore a versatile method for green and divergent synthesis of azepino[4,3,2-cd]indoles, particularly important spirocyclic frameworks.10
 | ||
| Scheme 1 Background and outline of this work. | ||
Direct C(sp3)–H functionalization enabled by redox-neutral [1,n]-hydride transfer (HT) represents an atom-/step-economic strategy for achieving molecular diversity.11 The past decade has witnessed the endeavours of many reputable groups in exploring [1,n]-HT-involved α-C(sp3)–H functionalization of amines for N-heterocycle synthesis.12–14 However, in contrast to the well-developed synthesis of spirocyclic tetradroquinolines via [1,5]-HT, the access to spirocyclic azepinoindoles via [1,6]-HT is still unknown. The main reason is the lack of a class of suitable substrates that can meanwhile assemble both 7-membered azepinoindole and spirocycle via HT mode. In this context, we designed 4-dialkylamino-indole-3-carbaldehydes as versatile platform molecules and put forward EtOH-mediated aromatization-driven cascade C(sp3)–H functionalization reactions for green and divergent synthesis of spirocyclic azepino[4,3,2-cd]indole derivatives (Scheme 1C). As depicted, the transformation was expected to proceed through the in situ assembly of pre-aromatics, aromatization-driven [1,6]-HT, and cyclization steps. Of note, both [1,6]-HT and the final azepine formation are spatially unfavourable, posing a challenge for the proposal. As a continuous journey in one-step assembly of bioactive molecules via the HT strategy,12 the research findings on this subject are presented herein. This methodology featured green bio-sourced EtOH as the solvent, metal-free and redox-neutral reaction conditions, high step-/atom-economy, water as waste, and a wide substrate scope.
Results and discussion
1-Thioaurones, characterized by their inherent tendency towards aromatization, are reactive pre-aromatic substrates that participate in various aromatization-driven cycloaddition reactions.15 Therefore, we commenced our exploration by performing the model reaction between 4-(pyrrolidin-1-yl)-1-tosyl-1H-indole-3-carbaldehyde 1a and benzo[b]thiophen-3(2H)-one 2a in EtOH (Table 1). It was found that 1-thioaurone 3a was readily afforded in 96% yield at 40 °C (Table 1, entry 1), which confirmed our assumption of in situ assembly of pre-aromatics. When the reactions were performed at higher temperatures, to our delight, the expected benzothiophen-3-one spirocyclic azepino[4,3,2-cd]indole derivative 4a was obtained in up to 73% yield with an excellent diasteromeric ratio (dr) of >20:1 (Table 1, entries 2–4). To improve the reaction efficiency, several protonic acids including PhCO2H (benzoic acid), MsOH (methanesulfonic acid), p-TSA·H2O (p-toluenesulfonic acid monohydrate), and TfOH (triflic acid) were screened (Table 1, entries 6–8). It was found that using 5 mol% TfOH resulted in the production of 4a in 90% yield (Table 1, entry 8). In addition, the elevation of the molar amount of 2a to 0.3 mmol led to a comparative yield (Table 1, entry 9).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Additives | Temp. (°C) | Time (h) | Yield (%) | |
| 3a | 4a | ||||
|
a Reaction conditions: 1a (0.20 mmol), 2a (0.24 mmol), and solvent (2 mL) at the given temperature for the indicated time. Isolated yields after column chromatography. The dr value of 4a was >20:1, which was determined by NMR analysis. PhCO2H = benzoic acid, MsOH = methanesulfonic acid, p-TSA·H2O = p-toluenesulfonic acid monohydrate, and TfOH = triflic acid.
b
2a (0.30 mmol).
|
|||||
| 1 | — | 40 | 1 | 96 | 0 |
| 2 | __ | 60 | 72 | 97 | 0 |
| 3 | — | 90 | 72 | 53 | 42 |
| 4 | — | 120 | 12 | 0 | 73 |
| 5 | PhCO2H | 90 | 72 | 45 | 45 |
| 6 | MsOH | 90 | 21 | 0 | 81 |
| 7 | p-TSA·H2O | 90 | 35 | 0 | 79 |
| 8 | TfOH | 90 | 6 | 0 | 90 |
| 9b | TfOH | 90 | 6 | 0 | 90 |
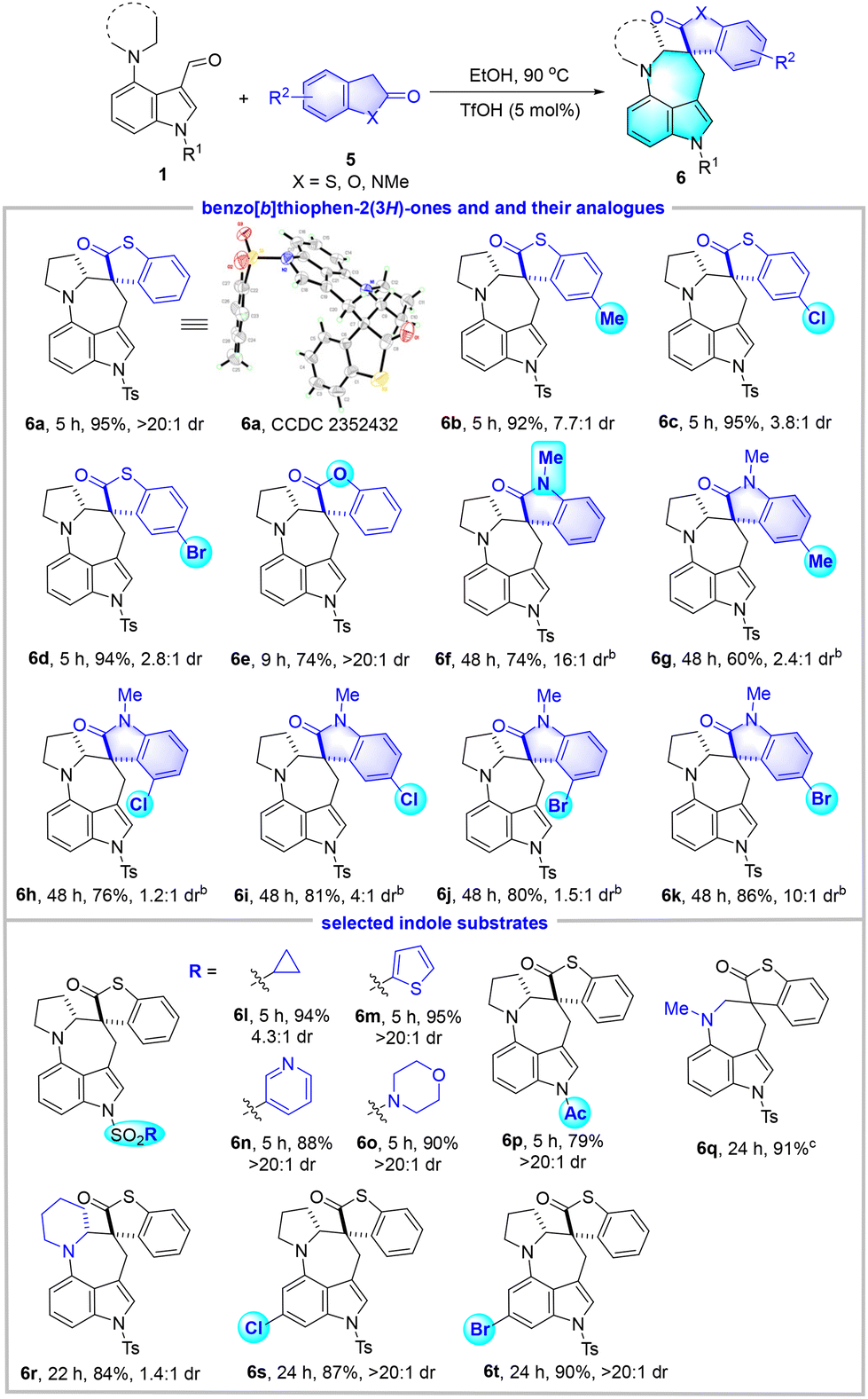
After the establishment of optimized reaction conditions, we turned to investigate the substrate scope by employing a wide range of indole substrates 1, benzo[b]thiophen-3(2H)-ones and their analogues 2 (Table 2). For the variation of benzo[b]thiophen-3(2H)-ones, both electron-donating groups such as Me and electron-withdrawing groups such as Cl and Br on the phenyl ring were well tolerated, and products 4b–4e were delivered in high yields (83%–89%) with excellent diastereoselectivities (>20:1 dr). Benzofuran-3(2H)-one was also a competent candidate for the reaction and gave 4f in 88% yield, albeit with a moderate dr value of 4:1. However, 1-acetylindolin-3-one was found to be incompatible with this system, and the expected 4g was not detected upon the consumption of 1a. Afterwards, various indole substrates 1 were screened. In detail, N-sulfonyl indole-3-carbaldehydes that carry trifluoromethyl, cyclopropyl, n-butyl, pyridyl, thienyl, and morpholinyl substituents were well compatible and provided products 4h–4m in 72%–90% yields and excellent diastereoselectivities (>20:1 dr). The N-Ac and N-Boc substrates were also shown to be feasible to give rise to 4n and 4o in moderate yields. Besides pyrrolidine, both dimethylamine and isoindoline were demonstrated as viable hydride donors, allowing the synthesis of 4p and 4q in good yields. The challenging piperidine was also a competent donor to trigger the [1,6]-HT, although product 4r was afforded in 28% yield with 2.3:1 dr at 110 °C. This could be rationalized by the conformational difference. Moreover, substrates that bear Cl and Br groups at the benzene ring of the indole scaffold reacted to furnish 4s and 4t with good results. Notably, the relative configuration of 4a was unambiguously assigned by X-ray crystallography analysis.

In order to further broaden the substrate scope, we examined the reaction of 1a with benzo[b]thiophen-2(3H)-one 5a under the same conditions as above (Table 3). Satisfyingly, the expected benzothiophen-2-one spirocyclic azepino[4,3,2-cd]indole derivative 6a was successfully accessed in 95% yield with an excellent dr value of >20:1. The structure of 6a was confirmed by X-ray crystallography. Then, various benzo[b]thiophen-2-ones and their analogues 5 as well as selected indole substrates 1 were investigated. Remarkable tolerance to different functional groups was exhibited in this EtOH system. For instance, the introduction of an electron-donating group (Me) or an electron-withdrawing group (Cl and Br) into the phenyl ring of benzo[b]thiophen-2-ones did not influence the reaction efficiency but resulted in much lower diastereoselectivities (6b–6d). Benzofuran-2(3H)-one was also found to be an ideal substrate and gave the desired 6e with excellent diastereoselectivity (>20:1 dr). Moreover, in view of the significance of the spirocyclic oxindole skeleton in both synthetic and medicinal industries,16 1-methylindolin-2-ones that incorporate Me, Cl, and Br groups were tested, which proved competent to furnish spirocyclic oxindoles 6f–6k in 60%–86% yields. Nevertheless, the introduction of substituents, regardless of their electronic effects or positions, led to reduced dr values (1.2:1–16:1). Various N-sulfonyl and N-Ac indoles could survive the reaction conditions and gave 6l–6p in good to high yields (79%–95%). In addition, the acyclic 4-dimethylaminyl indole substrate was well-tolerated, affording the corresponding product 6q in 91% yield, although a higher reaction temperature was required. The substrate with a piperidine motif reacted smoothly and provided 6r in 84% yield, albeit with 1.4:1 dr. The introduction of Cl and Br groups into the indole motif had little effect on the reactions, and products 6s and 6t were synthesized with high yields and excellent diastereoselectivities.
Subsequently, we investigated the reaction between 3-phenylisoxazol-5(4H)-one 7a and 4-(pyrrolidin-1-yl)-1-tosyl-1H-indole-3-carbaldehyde 1a (Scheme 2). It was found that the pre-aromatic 8a could be readily afforded in 99% yield in EtOH at 40 °C after 1.5 h. After adjusting the reaction temperature to 60 °C under additive-free conditions, the isoxazolone spirocyclic azepino[4,3,2-cd]indole derivative 9a, to our gratification, was produced with high efficiency (90% yield, 1.2:1 dr).
 | ||
| Scheme 2 Conditions employed for the reaction of isoxazol-5(4H)-one 7a with indole substrate 1a. | ||
Afterwards, the generality of this reaction was studied (Table 4). A series of isoxazol-5(4H)-ones 7 bearing electron-donating (MeO and Me) and electron-withdrawing (CF3, NO2, F, Cl, and Br) groups on the phenyl ring were subjected to the reactions with 1a. The expected isoxazolone spirocyclic azepino[4,3,2-cd]indole derivatives 9b–9h were furnished in 80%–89% yields with 1.1:1–2.1:1 dr. The structure of product 9e has been confirmed by X-ray diffraction analysis. Furyl- and thienyl-substituted isoxazolones were also found to be suitable substrates to give products 9i and 9j in high yields. In addition, 3-methylisoxazol-5(4H)-one was examined, which afforded the desired 9k in 87% yield with a moderate dr value of 4.8:1. For indole substrates that incorporate diverse N-sulfonyl, N-Ac, and N-Boc groups, good to excellent yields (74%–95%) were obtained for 9l–9s, which indicated the robustness of this method. Moreover, indole substrates that contain the motifs of dimethylamine and isoindoline at the C4 position successfully engaged in cascade C(sp3)–H alkylation to give 9t and 9u in good yields. When the substrate bearing the piperidine moiety was employed, the intermediate 8b was observed and could be isolated; however, we failed to detect and isolate the desired product 9v due to the messy reaction system. Of note, the diastereoselectivity of this transformation was not so satisfactory in spite of its advantages such as excellent functional group tolerance and high yields.

Edaravone (10a), 5-methyl-2-phenyl-2,4-dihydro-3H-pyrazol-3-one, is a kind of free radical scavenger used for protecting mammalian brains.17 The functionalization of such a molecule is of great interest for exploring bioactive drugs. When the reaction between 1a and edaravone 10a was conducted under the same conditions as above, the desired edaravone spirocyclic azepino[4,3,2-cd]indole derivative 11a was successfully delivered in 92% yield with excellent diatereoselectivity (>20:1 dr). With this exciting result in hand, we then screened a broad range of edaravone derivatives 10 (Table 5). The N-aryl pyrazolones 10b–10e that bear MeO, Me, Cl, and Br groups could smoothly participate in the transformations and generated the expected 11b–11e in high yields (90%–94%) with excellent diatereoselectivities (>20:1 dr). In addition, the N-free substrate was applicable to afford 11f in 87% yield with >20:1 dr. Upon changing R2 from the methyl to the cyclopropyl group, the conversion occurred at a higher temperature of 80 °C. Moreover, pyrazolones that contain electron-rich and electron-deficient phenyl and thienyl groups were well tolerated and gave 11h–11m in 90%–94% yields with excellent diatereoselectivities (>20:1 dr). The relative configuration of 11l has been unambiguously determined by X-ray diffraction analysis.

In addition, other substrates, including 2-phenylthiazol-4(5H)-one 12a, 2-phenyloxazol-4(5H)-one 12b, and 2-phenyloxazol-5(4H)-one 14, were investigated to react with 1a (Scheme 3). Pleasingly, the expected thiazolone spirocyclic azepino[4,3,2-cd]indole derivative 13a was furnished in 85% yield with excellent diastereoselectivity (>20:1 dr). However, messy reaction systems were observed for substrates 12b and 14, and we failed to obtain the desired products 13b and 15.
 | ||
| Scheme 3 Investigation of other substrates. | ||
In order to demonstrate the synthetic utility of the developed EtOH-mediated cascade C(sp3)–H alkylation, gram-scale syntheses and transformations of products were performed (Scheme 4). As shown in Scheme 4A, the preparation of 4e, 6a, 9a, and 11e on a gram scale could be achieved in high yields with the maintenance of diastereoselectivities. Various derivatization reactions are depicted in Scheme 4B. The Wittig reaction of 4a gave rise to alkene 16 in 89% yield (Scheme 4B, a), while the reduction of 4a by LiAlH4 furnished alcohol 17 in 90% yield (Scheme 4B, b). The Grignard reactions of 4a gave tertiary alcohols 18 and 19 in high yields (Scheme 4B, c and d). The treatment of 4o with TFA (trifluoroacetic acid) in DCM afforded the N–H free product 20 (Scheme 4B, e), which could be further functionalized into 21 and 22via N-propargylation and N-allylation (Scheme 4B, f and g). Surprisingly, the reduction of 6a by LiAlH4 afforded the stable thiohemiacetal 23 in 93% yield (Scheme 4B, h), which could undergo an interesting cascade acid-catalyzed dehydration/aromatization-driven carbocation rearrangement/reduction reaction to provide 3-(benzo[b]thiophen-3-ylmethyl)-indole 24 in 75% yield (Scheme 4B, i). Moreover, the nucleophilic addition of MeMgBr resulted in the successful formation of thiohemiketal 25 in 86% yield (Scheme 4B, j). The N-H products 26 and 27 could be accessed via N-Ts and N-Boc deprotection (Scheme 4B, k and l).
 | ||
| Scheme 4 Gram-scale synthesis and derivatization of products. | ||
To gain insights into the reaction mechanism, mechanistic studies were conducted (Scheme 5). As clearly shown in Scheme 5A, the proposed pre-aromatic intermediates 3a, 28a, 8a, and 29a were efficiently assembled in EtOH at 40 °C and isolated in nearly quantitative yields. These pre-aromatics could successfully convert into the expected products 4a, 6a, 9a and 11a under the corresponding standard conditions. These results confirmed the in situ generation of pre-aromatic intermediates and their capability to undergo cascade C(sp3)–H alkylation for synthesizing spirocyclic azepino[4,3,2-cd]indoles. Then, control experiments were performed to monitor the formation of intermediate 3a and product 4a in the model reaction (Scheme 5B), which provided further confirmation for the assembly of pre-aromatics and their subsequent conversions. Furthermore, the comparison of the model reaction with the control reaction between 1a and 2,3-dihydro-1H-inden-1-one 30 was investigated under the standard conditions (Scheme 5C). It was found that no desired spirocyclic product 31 was detected in the control reaction, thus strongly supporting our proposal that aromatization acted as the driving force to facilitate and enable the cascade C(sp3)–H alkylation reaction. In addition, deuterated labelling experiments were carried out using the reactions of the deuterated substrate [D]-1a with 2a and 7a, respectively (Scheme 5D). Remarkably, the complete transfer (100%) of the [D]-label to the benzylic position of the indole was observed, which provided compelling evidence for the occurrence of the intramolecular [1,6]-HT step.
 | ||
| Scheme 5 Mechanistic studies. | ||
Based on the above experimental results, a plausible reaction mechanism was proposed for the synthesis of 4a (Scheme 6). As illustrated, the EtOH-promoted condensation between 4-(pyrrolidin-1-yl)-1-tosyl-1H-indole-3-carbaldehyde 1a and benzo[b]thiophen-3(2H)-one 2a achieved the in situ assembly of pre-aromatic 1-thioaurone 3a, with the release of H2O as waste. Aided by the catalytic amount of TfOH in EtOH, the aromatization-driven intramolecular [1,6]-HT of 3a occurred and yielded aromatic Int. A, which finally underwent intramolecular cyclization to afford product 4a.
 | ||
| Scheme 6 Proposed mechanism. | ||
Conclusions
In conclusion, the EtOH-mediated cascade C(sp3)–H alkylation reactions of well-designed 4-dialkylamino-indole-3-carbaldehydes have been realized in this work. The green and divergent synthesis of spirocyclic azepino[4,3,2-cd]indole derivatives, including benzothiophenone, benzofuranone, oxindole, isoxazolone, pyrazolone, thiazolone, and oxazolone spirocyclic azepino[4,3,2-cd]indole derivatives, was realized with good to high yields and excellent diastereoselectivities (up to >20:1 dr). The gram-scale synthesis and derivatization of products demonstrated the synthetic application potential of this methodology. The mechanistic studies proved that the transformation proceeded through a cascade in situ assembly of pre-aromatics/aromatization-driven [1,6]-HT/cyclization process. Many advantages of this protocol were exhibited, such as green bio-sourced EtOH as the reaction medium, metal-free and redox-neutral conditions, H2O as waste, high yields, excellent diastereoselectivities, a wide substrate scope, high step-/atom-economy, gram-scale synthesis, and diverse transformations. We were optimistic that this method would provide some inspiration for both green synthetic chemistry and drug discovery.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from the Taishan Scholars Construction Projects of Shandong (No. tsqn201909131), the National Natural Science Foundation of China (No. 22301157), and the Natural Science Foundation of Shandong Province (No. ZR2023QB218).References
- For selected reviews on green chemistry, see:
(a) S. Kar, H. Sanderson, K. Roy, E. Benfenati and J. Leszczynski, Chem. Rev., 2022, 122, 3637–3710 CrossRef PubMed
; (b) P. T. Anastas and J. B. Zimmerman, Green Chem., 2019, 21, 6545–6566 RSC
- For selected reviews on bio-sourced EtOH as a green solvent, see:
(a) C. J. Clarke, W.-C. Tu, O. Levers, A. Bröhl and J. P. Hallett, Chem. Rev., 2018, 118, 747–800 CrossRef CAS
- For selected reviews on azepinoindole, see:
(a) C.-B. Ji, X. Hu, S.-Z. Jiang and J.-H. Yang, Eur. J. Org. Chem., 2022, e202201000 CrossRef
- For selected recent reports on azepinoindole synthesis, see:
(a) H. Tanaka, T. Yasui, M. Uyanik and K. Ishihara, Org. Lett., 2023, 25, 2377–2381 CrossRef PubMed
-
(a) J. M. Matthews, M. N. Greco, L. R. Hecker, W. J. Hoekstra, P. Andrade-Gordon, L. de Garavilla, K. T. Demarest, E. Ericson, J. W. Gunnet, W. Hageman, R. Look, J. B. Moore and B. E. Maryanoff, Bioorg. Med. Chem. Lett., 2003, 13, 753–756 CrossRef
- W. Wang, M. Yang, D. Han, Q. He and R. Fan, Adv. Synth. Catal., 2020, 362, 1281–1285 CrossRef
-
(a) Z. Shafiq, L. Liu, Z. Liu, D. Wang and Y.-J. Chen, Org. Lett., 2007, 9, 2525–2528 CrossRef PubMed
- B. Zou, C. Chen, S. Y. Leong, M. Ding and P. W. Smith, Tetrahedron, 2014, 70, 578–582 CrossRef
- B.-Y. Xue, C.-Y. Hou, X.-B. Wang, M.-S. Xie and H.-M. Guo, Org. Chem. Front., 2023, 10, 1910–1914 RSC
- For selected reviews on applications of spirocycles, see:
(a) K. Hiesinger, D. Dar'in, E. Proschak and M. Krasavin, J. Med. Chem., 2021, 64, 150–183 CrossRef PubMed
- For selected reviews on direct C(sp3)–H functionalization via [1,n]-HT, see:
(a) K. Mori, Bull. Chem. Soc. Jpn., 2022, 95, 296–305 CrossRef
- For selected [1,n]-HT works of our group, see:
(a) X.-D. An, D.-Y. Shao, B. Qiu and J. Xiao, Org. Lett., 2023, 25, 2432–2437 Search PubMed
- For selected [1,n]-HT works of other groups, see:
(a) K. Amano, T. Kawasaki-Takasuka and K. Mori, Org. Lett., 2024, 26, 1824–1827 CrossRef
-
(a) F. I. M. Idiris, C. E. Majesté, G. B. Craven and C. R. Jones, Chem. Sci., 2018, 9, 2873–2878 RSC
- For a selected review that includes cycloaddition of pre-aromatic 1-thioaurones, see:
(a) J. Huang, W. Wang, L. Zhang and X. Meng, Chin. Chem. Lett., 2023, 34, 108003 CrossRef
- For selected reviews on biologically active spirocyclic oxindoles and their synthesis, see:
(a) Y. Wang, A. A. Cobo and A. K. Franz, Org. Chem. Front., 2021, 8, 4315–4348 RSC
-
(a) K. Okamura, T. Tsubokawa, H. Johshita, H. Miyazaki and Y. Shiokawa, Neurol. Res., 2014, 36, 65–69 CrossRef PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2305982, 2352432, 2300404 and 2352430. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4gc04534c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |