
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning melanin: theoretical analysis of functional group impact on electrochemical and optical properties†
Florian
Heppner
 a,
Noah
Al-Shamery
b,
Pooi See
Lee
b and
Thomas
Bredow
*a
a,
Noah
Al-Shamery
b,
Pooi See
Lee
b and
Thomas
Bredow
*a
aMulliken Center for Theoretical Chemistry, Clausius-Institute for Physical and Theoretical Chemistry, University of Bonn, Beringstr. 4, D-53115 Bonn, Germany. E-mail: bredow@thch.uni-bonn.de
bSchool of Materials Science and Engineering, Nanyang Technological University, 50 Nanyang Ave, #01-30 General Office Block N4.1, 639798 Singapore, Singapore
First published on 16th May 2024
Abstract
The poly indolequinone eumelanin has been found to be a useful material for organic energy storage systems as a compelling, eco-friendly, and renewable alternative to sparse, inorganic materials. Current research has spotlighted the tuning of electrochemical properties of the material through chemical derivatization. The porphyrin-like tetrameric protomolecule of eumelanin presents an ideal candidate for analyzing the influence of adding functional groups on the electronic and optical properties of the system. In this study, after verifying the stability of the tetrameric protomolecule as a model structure, derivatives of F, Cl, Br, I and NH2-substituted melanin were analyzed to investigate the impact of different functional groups. Additionally, for the first time, the new protomolecule adsorbed on a graphene surface could be modeled. One-electron properties were obtained with the Green function method combined with screened Coulomb interaction (GW). Excited-state properties were calculated using the GW–BSE (Bethe–Salpeter equation) method. The most significant shift in the gap was observed for di-aminated melanin with a difference of 0.45 eV (16%). The optical absorption spectra for the halogenides exhibited an increasing red-shift with increasing amount of substitution. The aminated melanin shows a much larger shift in the main absorption band of around 100 nm while reducing the absorption band intensity by 50%. These results can serve as a guide for future applications of the derivatives and offer insights on what other eumelanin derivatives may be of interest.
1 Introduction
For over a decade, the poly indolequinone eumelanin (named just “melanin” in the following) has been the subject of studies for materials scientists and chemists due to its manifold occurrences in nature and the functions of melanin inside the human body.1,2 Metal ion chelation, radical scavenging, and photoprotection are beneficial properties, not only for biological systems, but for applications outside the human body like energy storage and sensing.3,4One goal of recent research endeavors has been the optimization of the material properties in electrochemical applications. Previous studies have concentrated mainly on increasing the dispersability of the material in polar solvents in order to increase the film processability of melanin.5–7 This has resulted in functionalization approaches like galactosyl-thio or poly(vinyl alcohol) substitution by Corani et al. for water-soluble melanin analogues, or the sulfonated melanin soluble in dimethyl sulfoxide (DMSO) by Bronze-Uhle et al.8–10 The new approach investigated in this work focuses on the possibility of functionalization for directly influencing and improving the electronic and optical properties of the synthesized structures. Properties of interest are e.g., the energy gap between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) for indicating the electronic conductivity and the optical absorption range for potential usage in solar cell applications. The most recent work investigating this type of melanin functionalization is a work covering melanin protected by tert-butyloxycarbonyl (Boc) and nitrated melanin, giving the so-called materials Mel–Boc and Mel–NO2.11
This work has suggested that by theoretical approaches using geometry and energy optimization, vital information can be obtained about the potential usefulness of specifically substituted melanin derivatives aimed for electrochemical applications. The work presented here will build on these new foundations by further introducing and validating the previously discussed tetrameric eumelanin protomolecule and deliberating the usefulness of halogenated melanin tetramers and melanin substituted with NH2 groups for electrochemical applications. These specific functionalizations are explicitly of interest, as from a synthesis standpoint, they can be obtained easily using Mel–NO2 as an educt. For discussing the new functionalizations, first, the material, existing and novel derivatizations, and the basis of the theoretical modeling of the tetramers need to be discussed.
1.1 Eumelanin
The material class of melanin consists of different species like neuromelanin, pheomelanin, and allomelanin,1 but material scientists have mostly focused on the so-called eumelanin for electrochemical applications. It is a diffusive, macromolecular polymer, consisting of the monomers 5,6-dihyroxyindole (DHI) and 5,6-dihydroxyindole-2-carboxylic acid (DHICA).12 At the top of Fig. 1, the molecular structures of eumelanin are shown. Depicted are the quinone iminium (M), indole quinone (I), semiquinone (Q), and hydroquinone (H) forms that can be concurrently present. | ||
| Fig. 1 Comproportionation reaction of eumelanin (top), literature tetramer by Kaxiras et al.13,14 (left), optimized 6H structure (right).11 The arrows indicate positions for potential functionalization. | ||
Engagement in comproportionation reactions is one of the principal contributors to electron storage in melanin.6,15 Synthetic melanin that is used as a precursor molecule for certain derivative synthesis can be obtained by polymerization of DHI and DHICA under biomimetic conditions.16 One main attribute of synthetic melanin is its hydration dependence, particularly in terms of conductivity arising from ionic and proton transport.17
Also shown at the bottom of Fig. 1 are the porphyrin-like tetrameric protomolecules that have been suspected to be a possible energetic minimum structure of melanin.18 While the structure of the polymer can manifest as a disordered model featuring diverse-sized oligomers,14 the π–π stacking of the depicted tetramers with a plane spacing of 3.7 Å has been shown to be a structural leitmotif that can be used when attempting to conceptualize theoretical models of the material while also matching experimental findings.19
1.2 Melanin derivatives
Some of the melanin derivatives and melanin-mimics found in literature build upon this tetrameric base structure. Other works try to either isolate specific monomeric structure motifs of melanin or create an analogue material trying to benefit from the hydroquinone/quinone equilibrium of the base melanin structure. Derivatives found in literature intend to increase the polarity of the material by introducing highly polar galactosyl-thio and sulfonyl groups that drastically affect the structural integrity of the polymer and increase solubility in water or DMSO.8,20 In the case of the sulfonated melanin, depending on the reaction conditions, the degree of sulfonation can be varied which reduces the amount of present DHICA species. Additionally, some of the quinone/hydroquinone moieties are replaced by sulfonyl groups.7,9,10,20 Tetrameric protomolecule analysis for this material focusing on electronic and optical properties can be found in the literature, further justifying the selected approach in the work presented here.18On the other hand, melanin-mimic structures like the indole-5,6-quinones observed by Kohler and Lumb et al. are much easier to simulate,21 as the structure explicitly isolates the indolequinone state of the melanin redox equilibrium. While the structural integrity of the melanin monomer is preserved using this synthesis approach, allowing for in-depth analysis of the quinone structure, the complex diffusive nature of polymerized melanin is not taken into account. The comparability to other melanin systems found in literature for electrochemical applications is reduced, but the optical and paramagnetic properties are comparable.21
In between these two approaches stands the post-polymerization derivatization approach by Al-Shamery et al. that intends to maintain the main structure motifs of the melanin polymers by selecting synthesis pathways that aim to target only free spots on the oligomers after complete polymerization.11 Thus, the synthesized nitrated derivative Mel–NO2 was successfully studied experimentally and theoretically, with the results showing a good match between theory and experiments when using the tetrameric protomolecule of melanin in vacuum as a basis for calculations. Learning from these different synthesis approaches for derivatives and how comparable the obtained products are to theoretical calculations, it becomes obvious that the work presented here focuses on further observing post-polymerization affected tetramers. The introduction of the halogenides F, Cl, Br, I, and the electron donating group NH2, giving the derivatives Mel–F, Mel–Cl, Mel–Br, Mel–I, and Mel–NH2, will be analyzed theoretically on the basis of quantum-chemical calculations. As noted earlier, the selection of these derivatives is a logical conclusion when considering the potential simple synthetical accessibility. The aminated melanin could be accessed by reducing the Mel–NO2 by catalytic hydrogenation.22 The halogenides can then be introduced by using a Sandmeyer-type reaction.23 Furthermore, introducing halogenides can give insights on trends regarding substituent size and polarity affecting the overall electronic structure of the system.24
1.3 Theoretical models of eumelanin
The theoretical modeling of eumelanin has been an object of discussion since the early 2000s, as the enigmatic diffuse polymer structure imposes a great challenge for calculations.13 What has been agreed on, is that a selection of protomolecules can be made for simplifying the structure and making it accessible for simulations. Kaxiras et al. have proposed in their works from 2006 and 2008 a porphyrin-like tetramer structure to use as one potential basis for melanin modeling.13,14 The so-called “HMIM” structure shown in Fig. 1 on the left derives its name from the used monomeric units, being two semiquinone, one indole quinone, and one hydroquinone unit. It has been shown to be usable as a basis for comparison to electrochemical data when, e.g., investigating the metal ion chelation between two of these tetrameric structures.19 Furthermore, Karttunen et al. used this tetrameric structure for modeling the π–π stacking of melanin protomolecules.25 Additionally, Cardia et al. recently used both short chains and the HMIM structure for the calculation of absorption spectra of unmodified eumelanin, further validating the importance of the porphyrin-like tetramer structure for melanin simulations.26However, the accuracy of the stability of the base structure could be vastly improved by using modern DFT tools that have been developing rapidly in the years since the initial proposal of the structure. Crescenci et al. conducted a manual in-depth analysis of all different tetrameric units that can be constructed using the four monomeric units, providing insights into the stability, electronic, and optical properties of tetramers of different oxidation state and possible transitions between them. This computationally intensive “by hand” manual conformational and tautomer search found the most stable to be conformers of the “six electron oxidation state”.27 In our previous work, we have independently obtained the “6H” structure shown in Fig. 1 on the left, deriving its name from the six protons located in the four semiquinone units and the center of the tetramer.11 This structure is in accordance with the previous findings of Crescenci et al., proving the viability of utilizing the “tautomerize” function of the Conformer-Rotamer Ensemble Sampling Tool (CREST),28 to study the different structural permutations of M/I/Q/H monomers in a computationally efficient manner.
The 6H structure was previously used as a basis for calculating the formation energies and HOMO–LUMO gaps of Mel–NO2. It will be further built upon in the present work in order to obtain information about the potential usefulness of the derivatives Mel–F, Mel–Cl, Mel–Br, Mel–I, and Mel–NH2 in electrochemical applications while also learning about how the substituents with trends in different sizes and polarity will affect the overall electronic and optical properties of melanin.
2 Results and discussion
2.1 Validation of the 6H structure using coupled-cluster methods
The “6H” tetrameric protomolecule,11 was reviewed once more by comparing the total energy of the literature-known HMIM tetramer with the 6H tetramer using the coupled-cluster method with single, double and perturbative triple excitations (CCSD(T), approximated by the domain-based local pair natural orbital scheme implemented in ORCA).29 CCSD(T) represents the so-called “gold standard” in modern quantum chemistry and is expected to achieve chemical accuracy. At this theoretical level, the 6H tetramer is 53 kJ mol−1 more stable than the literature-known HMIM tetramer (additional data in Table S1, ESI†), verifying its usage for the further analysis of functionalized melanin.During our work concerning this tetrameric model with CREST, an intriguing behaviour was observed. One or more of the monomeric subunits underwent a ring reduction of the six-membered ring of the indol-like monomeric structure, extruding CO. Further DFT calculations at the r2SCAN-3c30 level validated this “CO extrusion” to be energetically favourable. Further information is presented in the ESI† (see Fig. S1).
2.2 Adsorption of 6H on graphite
Additionally, the adsorption of the 6H tetramer on a graphite surface was calculated. This surface was chosen as representative of many carbon-based and graphite paper substrates found in the literature for melanin thin-film applications.7,31,32 Furthermore, these model calculations can give insights on the potential differences in absorption spectra of melanin in vacuum and of melanin adsorbed to surfaces. The 6H tetramer was adsorbed on the graphite (001) surface in order to calculate the effect of surface interaction on the electronic properties. The adsorption structure of an isolated 6H tetramer was optimized on a single-layer (−66)(−9−9) supercell using the r2SCAN functional33 augmented by the D4 dispersion correction34 as implemented in the plane-wave code VASP version 6.4.1.35 Further computational details can be found in Section 4.4. The optimized structure is shown in Fig. 2. | ||
| Fig. 2 Optimized adsorption structure of 6H on graphite (001) (r2SCAN-D4). | ||
The 6H tetramer adsorbs parallel to the surface. It is slightly rotated with respect to the graphite lattice, apparently in order to maximize the interaction between the benzene rings with the surface. The intramolecular structure is only slightly changed with respect to the optimized gas-phase geometry. This indicates that the molecule is physisorbed, as expected on graphite. The calculated adsorption energy is relatively large, −287 kJ mol−1. However, the interaction is dominated by London dispersion (approximated by the D4 correction) which contributes 187 kJ mol−1, corresponding to 65% of the total adsorption energy.
The 6H molecular and adsorbate structures and the graphite lattice parameters are taken from the VASP35 r2SCAN-D4 optimizations.
First, it has to be mentioned that the calculated quasiparticle HOMO–LUMO gaps of gas-phase 6H obtained with WEST/G0W0 and MOLGW/evGW0 are very similar, 2.79 eV and 2.78 eV (see Table 1), respectively. This indicates that the two approaches have a similar accuracy. It is observed that adsorption has a strong effect on the HOMO and LUMO energies. The 6H HOMO energy increases from −5.73 eV in the gas phase to −2.67 eV after adsorption. A smaller but still pronounced effect is visible for the LUMO energy which is shifted from −2.93 eV in the gas phase to −1.55 eV on graphite. This strong upshift of the HOMO energy is not due to any orbital interaction between 6H and the surface. Fig. 3 shows that the 6H HOMO is essentially unaltered by adsorption, and no contributions from surface C atoms can be seen. Shifts in orbital energies for similar molecules on surfaces have been reported in the literature.36 These shifts may be explained by Pauli repulsion between the π-electron systems of the molecule and the surface,37 or by polarization originating from both the surface or the molecule.38 However, these shifts are typically less pronounced than those observed for the 6H tetramer.
| Substituents | HOMO–LUMO gap [eV] | Difference to 6H [%] | |
|---|---|---|---|
| F | 1 | 2.87 | 3.2 |
| 2-A | 2.93 | 5.7 | |
| 2-B | 2.88 | 3.9 | |
| 3 | 2.95 | 6.3 | |
| 4 | 2.96 | 6.6 | |
| Cl | 1 | 2.87 | 3.3 |
| 2-A | 2.91 | 4.9 | |
| 2-B | 2.82 | 1.4 | |
| 3 | 2.88 | 3.6 | |
| 4 | 2.81 | 1.1 | |
| Br | 1 | 2.86 | 3.2 |
| 2-A | 2.90 | 4.5 | |
| 2-B | 2.80 | 0.9 | |
| 3 | 2.86 | 2.9 | |
| 4 | 2.77 | 0.0 | |
| I | 1 | 2.85 | 2.7 |
| 2-A | 2.88 | 3.7 | |
| 2-B | 2.78 | 0.0 | |
| 3 | 2.82 | 1.5 | |
| 4 | 2.73 | −1.7 | |
| NH2 | 1 | 2.58 | −7.1 |
| 2-A | 2.33 | −16.1 | |
| 2-B | 2.7 | −0.1 | |
| 3 | 2.39 | −13.9 | |
| 4 | 2.40 | −13.4 | |
| OMe | 2-A | 2.83 | 2.0 |
| 6H | 2.78 |
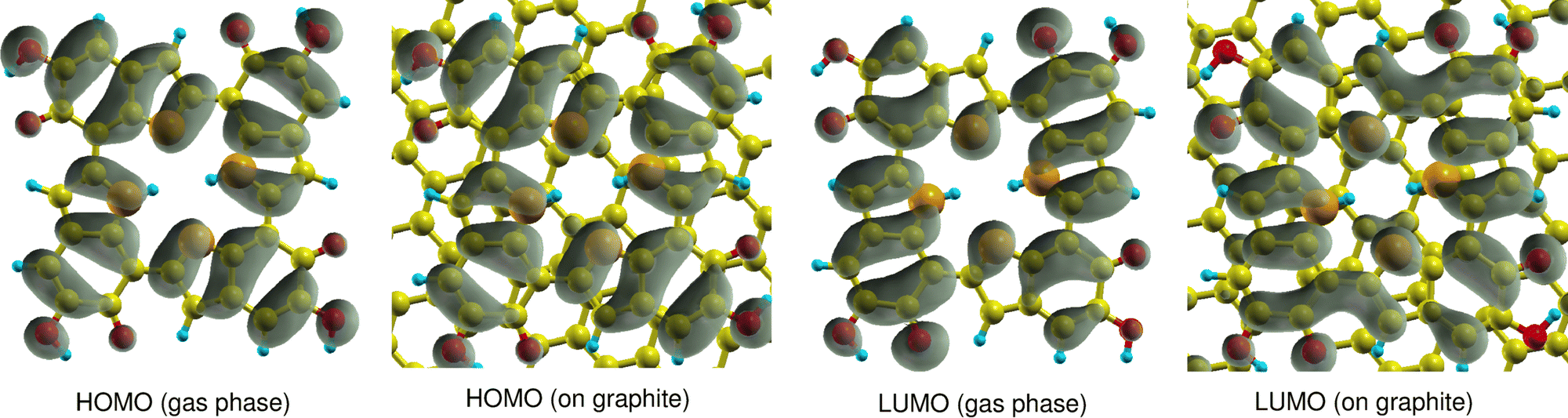 | ||
| Fig. 3 Comparison of HOMO and LUMO orbital densities of 6H calculated in the gas phase and on graphite (001). | ||
The 6H HOMO is 0.6 eV below the Fermi level of the graphite surface, the LUMO 0.5 eV above. Thus, the two orbitals are almost symmetrically aligned to the graphite Fermi level, so that no electron transfer between the molecule and the surface takes place. The electron density of the LUMO is different in gas phase and on the surface. This can explain the smaller energy shift due to stabilization by polarization effects. The overall effect of adsorption of graphite (001) is a significant reduction of the 6H HOMO–LUMO gap from 2.79 eV to 1.12 eV. It is expected that this would have a significant effect on the electrochemical properties of melanin, if monolayers of tetramers were to be produced and observed. In the systems discussed in literature using melanin-films as working electrodes, the main observed electrochemical contributions would stem from the top bulk layer-interaction of the film with the electrolyte due to fabrication methods like drop-casting being used.5 As the melanin species at the electrolyte interface are relatively far away from the substrate surface, the observed influences of adsorption on the graphite model surface from the work presented here come not into play as strongly, but will be of high relevance for future work if even thinner melanin-films or tetramer monolayers can be produced.
2.3 Semiempirical conformer search and DFT energy evaluation
Having re-evaluated and confirmed the 6H structure, we proceed with using it as the basis for the derivatives analysis. The first step for analyzing the influence of the functional groups is identifying the energetical minimum structure for all selected substituents and different degrees of functionalization. This was achieved with the help of the tautomerize function of CREST.39 By using this function, all tautomers and conformers of a given structure are analyzed and energetically ordered at the semiempirical GFN2-xTB level.40 In the present case, only five different input structures were needed in order to cover all possibilities for the introduction of one (1X) to four (4X) substituents. For one, three, and four substituted 6H tetramers, only a single CREST run is required. For two substituents, two different cases need to be considered, substitution on opposite monomer units (2X-A) or substitution on adjacent monomer units (2X-B). The starting monomer selected for the substitution is not relevant, the “tautomerize” function ensures that the functional group will end up at the most stable monomer.It is important to note that there is a high likelihood that the more reactive C4 position41 will be (partly) occupied by other monomers during the polymerization process in the actual system and the functionalization was done post-polymerization.11 Thus, to stay close to the experimental conditions while still utilizing the tetrameric protomolecule, only substitution at the C3 position was analyzed in this work.
The respective model input structures and used nomenclature are presented in Fig. 4(a). All structures obtained by CREST calculations and used for further calculations are also presented in Fig. S2 (ESI†). The resulting minimum structures were then verified using DFT on the r2SCAN-3c30 level of theory. It is notable that the geometry optimization on this higher level of theory did not lead to significantly different geometries compared to the GFN2-xTB results. The geometries obtained with CREST applying the “tautomerize” option are therefore validated for this system.
 | ||
| Fig. 4 (a) The analyzed starting structures for the CREST conformer and tautomer search (X = F, Cl, Br, I, NH2). By using the “tautomerize” function of CREST, only the presented five starting structures needed to be analyzed to account for all possible configurations for the 4 degrees of functionalization. (b) The resulting formation energies for the post-polymerization functionalized 6H tetramers with the analyzed substituents F, Cl, Br, I and NH2, calculated on the r2SCAN-3c30 level of theory. | ||
In the experiments conducted by Al-Shamery et al.,11 functional groups were introduced by post-polymerization synthesis approaches. Therefore, as a model reaction for calculating formation energies, the following reactions were chosen (reaction eqn (1) for the halogenides, reaction eqn (2) for amines).
 | (1) |
 | (2) |
The resulting formation energies are depicted in Fig. 4(b). For the introduction of fluorine and chlorine, the formation energy decreases with increasing amount of substitution. For bromine, iodine, and amine, the formation energies increase with increasing amount of substitution. Notably, only the formation energies for fluorine and chlorine are negative. These reactions are expected to be exothermic and can therefore be assumed to be experimentally accessible. However, for bromine and amine, the formation energies are only slightly positive.
2.4 Comparing HOMO–LUMO gaps
The electronic structure of all found minima structures was subsequently calculated applying the GW method as implemented in MOLGW.42 The eigenvalue-self consistent GW variant was applied without update of initial wavefunction (in this case obtained with the hybrid functional BHLYP43 and cc-pVDZ basis sets44,45), denoted as evGW0. The calculated quasiparticle gaps of corrected HOMO and LUMO are presented in Fig. 5. For all halogenated tetramers, a slight increase in the HOMO–LUMO gap is observed, with a maximum increase of 6.1%. In contrast, for Mel–NH2, with exception of the double substitution of adjacent monomer units, a substantial decrease in the HOMO–LUMO gap is observed, with a maximal decrease of over 16%. All values and their difference to the 6H tetramer are presented in Table 1.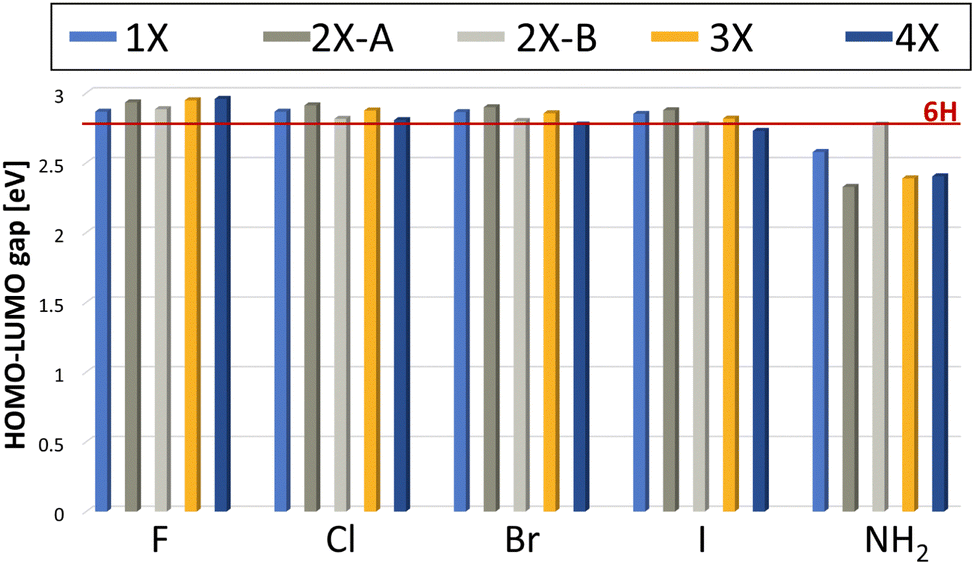 | ||
| Fig. 5 Calculated HOMO–LUMO gap using the evGW0 approximation. The red line indicates the HOMO–LUMO gap calculated for the 6H tetramer, the unsubstituted reference. | ||
Previous experimental studies have shown that a reduced gap is desirable, as it is associated with less constrained electron movement, increased conductivity and redox center availability.46 The correlation between a reduced HOMO–LUMO gap and improved energy storage performance was also demonstrated for this system by Al-Shamery et al. with Mel–NO2 showing a reduced band gap and increased electrochemical performance.11 Therefore, Mel–NH2 is expected to potentially show improved energy storage performance.
The results presented here further help rationalizing the systematic influence that functional groups can have on the melanin electronic structure. Based on our selected substituent scope, it can be deduced that electronic withdrawing groups with mainly a negative inductive (−I) effect lead to a slight increase in the HOMO–LUMO gap. Electron donating groups, like the amine group as a positive mesomeric (+M) substituent on the other hand, lead to a decrease in the HOMO–LUMO gap. This is in contrast to previous work, in which a reduced HOMO–LUMO gap for the strong −M substituent, the nitro group, was observed.11
As chemical intuition predicts, electron donating groups (EDGs) are expected to raise the HOMO while electron withdrawing groups (EWGs) can lower the LUMO for conjugated systems,47 indicating that the HOMO raising effect observed for the amine group could be stronger than the LUMO lowering effect observed for the nitro group.
Comparing the influence of the different substitution pattern for di-substitution, 2X-A and 2X-B on the gap, substitution on opposite monomers always shows a stronger influence (either increasing the gap for all halogenides, or decreasing the gap for NH2). This could be a hint that steric aspects must be considered as well for evaluating the influence of a functional group, as the functional groups are closer together for adjacent monomer di-substituted species, leading to potential sterically repulsive interaction. Future work could also consider the change of chelating properties of the material with the substituent effects of the functional groups as an additional factor to consider for predicting electrochemical performance. The small overall band gap influence of the halogenide substituents when compared to substituents with a mesomeric effect is to be expected, since inductive effects are considered to be less strong than mesomeric effects.
A different explanation for the positive influence of the NH2 groups on the HOMO–LUMO gap could be the formation of hydrogen bonds between the amines and the carbonyl functions of the melanin monomers. Hydrogen bonding has shown to strongly affect the band gaps in e.g., hybrid organic–inorganic perovskites.48 This kind of interaction was observed for all aminated melanin derivatives, an example is depicted in Fig. 6.
 | ||
| Fig. 6 Energetic minimum structure for the 4NH2 tetramer. The hydrogen bonds between the NH2 substituents and the carbonyl functions are marked (black dotted lines). | ||
The possibility to form hydrogen bonds is a characteristic of the amine group. Since 2NH2-A substituted melanin shows the most pronounced decrease in the HOMO–LUMO gap, this substitution pattern was chosen for analyzing another +M substituent, which is unable to form such hydrogen bonds. Consequently, 2OMe-A melanin with methoxy groups was additionally analyzed (Mel–OMe, see Fig. S3, ESI†). The HOMO–LUMO gap was calculated to 2.83 eV, showing an increased HOMO–LUMO gap compared to the 6H tetramer (by 2.0%), thus demonstrating the opposite effect of 2NH2-A melanin. This strongly suggests that the observed reduction of the HOMO–LUMO gap for the Mel–NH2 is not primarily caused by the electronic effects of the substituents, but rather due to the interaction via the hydrogen bonds.
Another factor could be the visible loss of planarity in the above structure. This loss of planarity is also visible in the case of di-aminated Mel–NH2. However, it is worth noting that this is also observed in Mel–OMe (Fig. S9, ESI†), yet no reduction in the HOMO–LUMO gap occurred. Therefore, hydrogen bonding is expected to be the primary contributing factor. This opens up possibilities for future melanin derivatives to be engineered specifically with the exploitation of the hydrogen bonding effects in mind.
2.5 UV-Vis spectra of new derivatives
Finally, the UV-Vis spectra of all substituents and all degrees of functionalization were calculated with evGW0–BSE implemented in MOLGW using the same basis sets as mentioned above. The computed spectra are presented in Fig. 7 (for more details see Fig. S4–S8, ESI†). In all UV-Vis spectra, two main bands are visible, one at around 280 nm and a second one at around 430 nm, as well as the characteristic exponential decay-like curve when moving to high wavelengths. This characteristic shape has been discussed in the literature to be the result of the structural disorder of melanin resulting in both Mie-scattering and direct absorption of the emitted light.49 For a direct comparison of absolute shifts to experimental results, one must also consider shifts caused by solvation effects.50 The relative trends observed in the shifts and intensity changes in the data should be considered for the selection of derivatives to further analyze. Within each substituent, with increasing substitution degree, the bands shift slightly to higher wave lengths, matching literature findings.51 Comparing the band positions for each different substituent, there is also an overall bathochromic shift going from fluorine to iodine, while the absorption peak calculated for the amine function is most strongly shifted. This reduction of the excited state energy is in line with the trend of the decreasing band gaps. This trend in increasing bathochromic shift going from F to I was also observed in DFT investigations of organic–inorganic hybrid systems.52,53 For further information on the absolute band positions, see Table S2 (ESI†). These results can be of use for future endeavours incorporating melanin and melanin derivatives into organic solar cell devices, aiming to maximize the absorption in the vis and near-vis regions.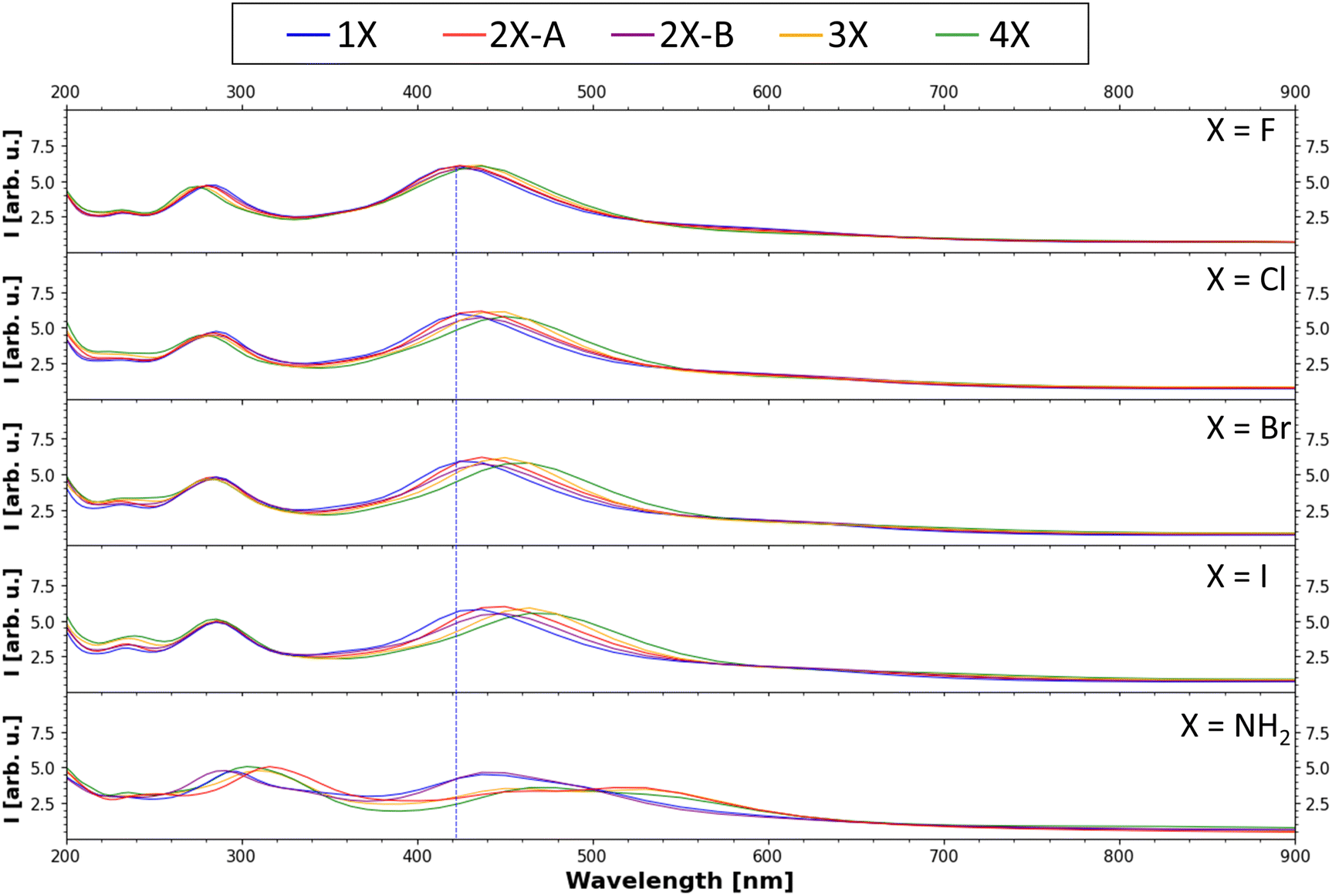 | ||
| Fig. 7 Calculated UV-Vis absorption spectrum (evGW0–BSE results). The peak with the highest intensity of the 1F tetramer (one fluorinated monomer in the tetramer, for nomenclature see Fig. 4(a)) is marked with a blue line to emphasize the shift of the band position due to the introduction of the other functional groups. | ||
3 Conclusion
In this work, we successfully demonstrated an application of the CREST “tautomerize” function and validated the structures obtained by this approach. Using the nitrated melanin as a potential starting point, the halogenides and the amine were analyzed as new easily accessible derivatives of synthetic melanin. It was shown that the aminated melanin has a significantly smaller HOMO–LUMO gap with respect to the previously found minima structures, with a maximal reduction of over 16%. This indicates that these derivatives could potentially have higher conductivity values when being practically synthesized and used as thin-film electrodes which is useful for electrochemical applications.46On the contrary, the halogenated melanin protomolecules showed a slight increase in the HOMO–LUMO gap, with a maximum increase of 6.6% for the tetra-substituted melanin with fluorine. On basis of the data presented, Mel–NH2 is an interesting protomolecule for melanin polymers with improved properties for energy storage applications. However, this also shows the potential of fine-tuning the band gap in both directions by using different substituents with a different degree of substitution, which is interesting for e.g., dye-sensitized and multi-dye dye-sensitized solar cell applications.54,55
This is further supported by the calculated absorption spectra that show that the already high absorption range of melanin in the range of the solar light spectrum can be increased even further through chemical derivatization. This could make future work using melanin as a dye in dye-sensitized solar cell (DSSC) applications more interesting, as in previous works, devices using unmodified nature-based pigments including melanin have often shown lower efficiency but higher overall stability compared to other DSSC systems.56 The aminated melanin showed a decrease in absorption band intensity, thus covering less of the solar spectrum, underlining that different derivatives specialized for different application need to be selected depending on if lower band gaps or UV-Vis absorption at higher wavelengths with a specific intensity are required.
Future work that aims to synthesize the observed derivatives will be of high interest in order to fully prove their potential usefulness in electrochemical applications and to validate the observed trend of the influence of substituent effects on the HOMO–LUMO gap spectroscopically. Finally, the calculations indicate that adsorption has a strong effect on the HOMO and LUMO energies and can also strongly modify the band gap. This effect has to be taken into account in future theoretical studies of melanin and other protomolecules as electrode materials.
4 Methods
4.1 Semiempirical conformer search with CREST
The conformal space of all possible tautomers of one, two, three, and four times substituted melanin derivatives for using fluorine, chlorine, bromine, iodine, and amine as substituents was screened. Additionally, 2OMe-A di-substituted melanin was analyzed. CREST version 2.1128,39 was used, applying the tautomerize function and constraining the atom position of all oxygens. The latter was needed to prevent cyclizations, ranked energetically favourable by CREST, but proved to be energetically unfavourable by DFT calculations.4.2 Validation and formation energy calculations with DFT
The tautomers yielded by the CREST calculations were validated by DFT calculation using ORCA version 5.0.2.29 The composite method r2SCAN-3C30 was used for all calculations, if not stated otherwise. For the further validation of the new 6H model for synthetic melanin, the in ORCA implemented DLPNO-CCSD(T) scheme was applied using the default parameters.4.3 Electronic structure calculations with MOLGW
GW–BSE calculations were performed using MOLGW version 3.2.42 First, an eigenvalue-selfconsistent G6W0 calculation was performed, directly followed by a BSE calculation. Both were performed on the BHLYP/cc-pVDZ(-RI)43–45 level of theory. For calculations with iodine, an effective core potential was applied (specifying the auxiliary basis set cc-pVDZ(-RI)-PP, as implemented in MOLGW). For all calculations, the frozen core approximation was applied.4.4 Adsorption structure calculations with VASP
The adsorption structure of an isolated 6H tetramer was optimized on a single-layer (− 66)(−9−9) supercell using the r2SCAN functional33 augmented by the D4 dispersion correction34 as implemented in the plane-wave code VASP version 6.4.1.35 The kinetic energy cutoff was set to 400 eV, and the Γ point approximation was applied for the integration in reciprocal space. This approximation is justified by the size of the supercell. The vacuum distance between the slabs was set to 20 Å which is generally considered as converged.4.5 Electronic structure calculations with WEST
The electronic structure of 6H in gas phase and adsorbed on graphite (001) was calculated at G0W0 level using the WEST code version 5.5.57 The level of theory is slightly lower compared to the gas-phase calculations presented above due to the large site of the adsorbate system. The quasiparticle energy calculation was based on a PBE wavefunction obtained with Quantum Espresso version 7.2.58 Here, the kinetic energy cutoff was 30 Ry (408 eV), and again the Γ point approximation was applied for the integration in reciprocal space. In the G0W0 calculation, 1024 bands were considered.Author contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication. F. H., T. B., N. A.-S., and P. S. L. conceived the original work under the main supervision of T. B., F. H. and N. A.-S. discussed what functional groups to analyze. F. H. and T. B. selected computational methods and performed all calculations and data analysis. F. H. and N. A.-S. wrote the original draft; all authors reviewed and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the funding for N. A.-S. that is provided by the Singapore International Graduate Award from the Nanyang Technological University Singapore. T. B. thanks the High Performance Computing and Analytics Lab of the university of Bonn for computational resources on the bonna cluster.References
- M. d'Ischia, K. Wakamatsu, F. Cicoira, E. Di Mauro, J. C. Garcia-Borron, S. Commo, I. Galván, G. Ghanem, K. Kenzo, P. Meredith, A. Pezzella, C. Santato, T. Sarna, J. D. Simon, L. Zecca, F. A. Zucca, A. Napolitano and S. Ito, Pigm. Cell Melanoma Res., 2015, 28, 520–544 CrossRef PubMed
.
- F. Solano, New J. Sci., 2014, 2014, 1–28 CrossRef
- M. d'Ischia, K. Wakamatsu, A. Napolitano, S. Briganti, J.-C. Garcia-Borron, D. Kovacs, P. Meredith, A. Pezzella, M. Picardo, T. Sarna, J. D. Simon and S. Ito, Pigm. Cell Melanoma Res., 2013, 26, 616–633 CrossRef PubMed
- N. Al-Shamery, T. Benselfelt and P. S. Lee, ACS Appl. Mater. Interfaces, 2023, 15, 25966–25979 CrossRef CAS PubMed
- P. Kumar, E. D. Mauro, S. Zhang, A. Pezzella, F. Soavi, C. Santato and F. Cicoira, J. Mater. Chem. C, 2016, 4, 9516–9525 RSC
- A. B. Mostert, S. B. Rienecker, M. Sheliakina, P. Zierep, G. R. Hanson, J. R. Harmer, G. Schenk and P. Meredith, J. Mater. Chem. B, 2020, 8, 8050–8060 RSC
- J. V. Paulin, A. P. Coleone, A. Batagin-Neto, G. Burwell, P. Meredith, C. F. O. Graeff and A. B. Mostert, J. Mater. Chem. C, 2021, 9, 8345–8358 RSC
- A. Corani, A. Huijser, A. Iadonisi, A. Pezzella, V. Sundström and M. d'Ischia, J. Phys. Chem. B, 2012, 116, 13151–13158 CrossRef CAS PubMed
- E. S. Bronze-Uhle, A. Batagin-Neto, P. H. P. Xavier, N. I. Fernandes, E. R. de Azevedo and C. F. O. Graeff, J. Mol. Struct., 2013, 1047, 102–108 CrossRef CAS
- M. Piacenti da Silva, E. S. Bronze-Uhle, J. V. Paulin and C. F. O. Graeff, J. Mol. Struct., 2014, 1056–1057, 135–140 CrossRef
- N. Al-Shamery, F. Heppner, C. Dosche, S. Morgenschweis, T. Bredow, G. Wittstock and P. S. Lee, manuscript in preparation, 2024.
- M. d'Ischia, A. Napolitano, A. Pezzella, P. Meredith and M. Buehler, Angew. Chem., Int. Ed., 2020, 59, 11196–11205 CrossRef PubMed
- E. Kaxiras, A. Tsolakidis, G. Zonios and S. Meng, Phys. Rev. Lett., 2006, 97, 218102 CrossRef PubMed
- S. Meng and E. Kaxiras, Biophys. J., 2008, 94, 2095–2105 CrossRef CAS PubMed
- K. A. Motovilov, V. Grinenko, M. Savinov, Z. V. Gagkaeva, L. S. Kadyrov, A. A. Pronin, Z. V. Bedran, E. S. Zhukova, A. B. Mostert and B. P. Gorshunov, RSC Adv., 2019, 9, 3857–3867 RSC
- S. Reale, M. Crucianelli, A. Pezzella, M. d'Ischia and F. De Angelis, J. Mass Spectrom., 2012, 47, 49–53 CrossRef CAS PubMed
- M. Sheliakina, A. B. Mostert and P. Meredith, Adv. Funct. Mater., 2018, 28, 1805514 CrossRef
- J. V. Paulin, M. P. Pereira, B. A. Bregadiolli, J. P. Cachaneski-Lopes, C. F. O. Graeff, A. Batagin-Neto and C. C. B. Bufon, J. Mater. Chem. C, 2023, 11, 6107–6118 RSC
- Y. J. Kim, A. Khetan, W. Wu, S.-E. Chun, V. Viswanathan, J. F. Whitacre and C. J. Bettinger, Adv. Mater., 2016, 28, 3173–3180 CrossRef CAS PubMed
- E. S. Bronze-Uhle, J. V. Paulin, M. Piacenti-Silva, C. Battocchio, M. L. M. Rocco and C. F. D. O. Graeff, Polym. Int., 2016, 65, 1339–1346 CrossRef CAS
- X. Wang, L. Kinziabulatova, M. Bortoli, A. Manickoth, M. A. Barilla, H. Huang, L. Blancafort, B. Kohler and J.-P. Lumb, Nat. Chem., 2023, 15, 787–793 CrossRef CAS PubMed
- H.-U. Blaser, H. Steiner and M. Studer, ChemCatChem, 2009, 1, 210–221 CrossRef CAS
- R. Akhtar, A. F. Zahoor, N. Rasool, M. Ahmad and K. G. Ali, Mol. Diversity, 2022, 26, 1837–1873 CrossRef CAS PubMed
- Q. Liu, B. Sun, Z. Liu, Y. Kao, B.-W. Dong, S.-D. Jiang, F. Li, G. Liu, Y. Yang and F. Mo, Chem. Sci., 2018, 9, 8731–8737 RSC
- S. Soltani, S. Sowlati-Hashjin, C. G. Tetsassi Feugmo and M. Karttunen, J. Phys. Chem. B, 2022, 126, 1805–1818 CrossRef CAS PubMed
- R. Cardia, N. Dardenne, G. Mula, E. Pinna, G.-M. Rignanese, J.-C. Charlier and G. Cappellini, J. Phys. Chem. A, 2023, 127, 10797–10806 CrossRef CAS PubMed
- O. Crescenzi, M. DIschia and A. Napolitano, Biomimetics, 2017, 2, 21 CrossRef PubMed
- P. Pracht, F. Bohle and S. Grimme, Phys. Chem. Chem. Phys., 2020, 22, 7169–7192 RSC
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed
- S. Grimme, A. Hansen, S. Ehlert and J.-M. Mewes, J. Chem. Phys., 2021, 154, 064103 CrossRef CAS PubMed
- M. I. N. da Silva, S. N. Dezidério, J. C. Gonzalez, C. F. O. Graeff and M. A. Cotta, J. Appl. Phys., 2004, 96, 5803–5807 CrossRef CAS
- S. N. Dezidério, C. A. Brunello, M. I. N. da Silva, M. A. Cotta and C. F. O. Graeff, J. Non-Cryst. Solids, 2004, 338–340, 634–638 CrossRef
- J. W. Furness, A. D. Kaplan, J. Ning, J. P. Perdew and J. Sun, J. Phys. Chem. Lett., 2020, 11, 8208–8215 CrossRef CAS PubMed
- E. Caldeweyher, S. Ehlert, A. Hansen, H. Neugebauer, S. Spicher, C. Bannwarth and S. Grimme, J. Chem. Phys., 2019, 150, 154122 CrossRef PubMed
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed
- A. J. Kny, M. Reimer, N. Al-Shamery, R. Tomar, T. Bredow, S. Olthof, D. Hertel, K. Meerholz and M. Sokolowski, Nanoscale, 2023, 15, 10319–10329 RSC
- F. Ortmann, W. G. Schmidt and F. Bechstedt, Phys. Rev. Lett., 2005, 95, 186101 CrossRef CAS PubMed
- J. B. Neaton, M. S. Hybertsen and S. G. Louie, Phys. Rev. Lett., 2006, 97, 216405 CrossRef CAS PubMed
- S. Grimme, J. Chem. Theory Comput., 2019, 15, 2847–2862 CrossRef CAS PubMed
- C. Bannwarth, S. Ehlert and S. Grimme, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef CAS PubMed
- C.-T. Chen, F. J. Martin-Martinez, G. S. Jung and M. J. Buehler, Chem. Sci., 2017, 8, 1631–1641 RSC
- F. Bruneval, T. Rangel, S. M. Hamed, M. Shao, C. Yang and J. B. Neaton, Comput. Phys. Commun., 2016, 208, 149–161 CrossRef CAS
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS
- D. E. Woon and T. H. Dunning, Jr., J. Chem. Phys., 1993, 98, 1358–1371 CrossRef CAS
- T. H. Dunning, Jr., J. Chem. Phys., 1989, 90, 1007–1023 CrossRef
- F. Wang, J. Cai, C. Yang, H. Luo, X. Li, H. Hou, G. Zou and D. Zhang, Small, 2023, 19, 2300510 CrossRef CAS PubMed
- Y. Mao, M. Head-Gordon and Y. Shao, Chem. Sci., 2018, 9, 8598–8607 RSC
- C. Tian, Y. Liang, W. Chen, Y. Huang, X. Huang, F. Tian and X. Yang, Phys. Chem. Chem. Phys., 2020, 22, 1841–1846 RSC
- A. Pezzella, A. Iadonisi, S. Valerio, L. Panzella, A. Napolitano, M. Adinolfi and M. dIschia, J. Am. Chem. Soc., 2009, 131, 15270–15275 CrossRef CAS PubMed
- R. Tomar, L. Bernasconi, D. Fazzi and T. Bredow, J. Phys. Chem. A, 2023, 127, 9661–9671 CrossRef CAS PubMed
- Z. Fang, F. Wu, Q. Tao, Q. Qin, C. Au, Y. Li, H. Zhang, N. Wang and B. Yi, Spectrochim. Acta, Part A, 2019, 215, 9–14 CrossRef CAS PubMed
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed
- Z.-H. Gao, J. Dong, Q.-F. Zhang and L.-S. Wang, Nanoscale Adv., 2020, 2, 4902–4907 RSC
- M. Dürr, S. Rosselli, A. Yasuda and G. Nelles, J. Phys. Chem. B, 2006, 110, 21899–21902 CrossRef PubMed
- M. Akhtaruzzaman, M. Shahiduzzaman, V. Selvanathan, K. Sopian, M. I. Hossain, N. Amin and A. K. M. Hasan, Appl. Mater. Today, 2021, 25, 101204 CrossRef
- G. Nandan Arka, S. Bhushan Prasad and S. Singh, J. Sol. Energy, 2021, 226, 192–213 CrossRef CAS
- M. Govoni and G. Galli, J. Chem. Theory Comput., 2015, 11, 2680–2696 CrossRef CAS PubMed
- P. Giannozzi, O. Andreussi, T. Brumme, O. Bunau, M. B. Nardelli, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, M. Cococcioni, N. Colonna, I. Carnimeo, A. D. Corso, S. de Gironcoli, P. Delugas, R. A. DiStasio, A. Ferretti, A. Floris, G. Fratesi, G. Fugallo, R. Gebauer, U. Gerstmann, F. Giustino, T. Gorni, J. Jia, M. Kawamura, H.-Y. Ko, A. Kokalj, E. Küçükbenli, M. Lazzeri, M. Marsili, N. Marzari, F. Mauri, N. L. Nguyen, H.-V. Nguyen, A. O. de-la Roza, L. Paulatto, S. Poncé, D. Rocca, R. Sabatini, B. Santra, M. Schlipf, A. P. Seitsonen, A. Smogunov, I. Timrov, T. Thonhauser, P. Umari, N. Vast, X. Wu and S. Baroni, J. Phys.: Condens. Matter, 2017, 29, 465901 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00192c |
| This journal is © The Royal Society of Chemistry 2024 |