
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDeriving kinetic insights from mechanochemically synthesized compounds using multivariate analysis (MCR-ALS) of powder X-ray diffraction data†
Laura
Macchietti
 a,
Lucia
Casali
ab,
Franziska
Emmerling
*b,
Dario
Braga
a and
Fabrizia
Grepioni
*a
a,
Lucia
Casali
ab,
Franziska
Emmerling
*b,
Dario
Braga
a and
Fabrizia
Grepioni
*a
aDipartimento di Chimica “Giacomo Ciamician”, Università di Bologna, Via F. Selmi 2, 40126 Bologna, Italy. E-mail: fabrizia.grepioni@unibo.it
bBAM Federal Institute for Materials Research and Testing, Richard-Willstätter-Strasse 11, 12489 Berlin, Germany. E-mail: franziska.emmerling@bam.de
First published on 14th February 2024
Abstract
Kinetics information on the progress of the mechanochemical reactions is key to their understanding and subsequent scale-up. For crystalline materials, the most robust and tested method for obtaining kinetic data is the Quantitative Phase Analysis (QPA) via Rietveld refinement. In this work, we tested the feasibility of the Multivariate Curve Resolution-Alternating Least Squares (MCR-ALS) method on powder X-ray diffraction (PXRD) data of mechanochemical processes by studying the system theophylline (TP) and malonic acid (MA) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric ratio at different milling conditions. We have highlighted the strengths and weaknesses of the MCR-ALS method, and we demonstrated why it may be an alternative route to obtain quantitative information on mechanochemical kinetics.
:1 stoichiometric ratio at different milling conditions. We have highlighted the strengths and weaknesses of the MCR-ALS method, and we demonstrated why it may be an alternative route to obtain quantitative information on mechanochemical kinetics.
1. Introduction
Mechanically-activated reactions – classified as mechanochemistry – are attracting increasing attention because of their inherently sustainable nature.1 The minimized use of solvents and energy required, together with performances generally fast and quantitative, make these reactions a valid alternative route for the preparation of various classes of compounds.2–4 Apart from all the merits of this synthetic methodology, the limit of mechanochemistry is the poor mechanistic understanding of the solid-state transformations involved. Since the reactions may follow different paths with respect to conventional solution chemistry, the outcome of these reactions cannot be taken for granted, and scale-up of laboratory reactions is not an obvious procedure.5 The urge to gain a deeper understanding of mechanistic behaviour paved the way for the development of methodologies aimed at monitoring the mechanochemical reactions, without (ideally) interfering with the milling processes.6 Of all the methods, time-resolved in situ (TRIS) methods for real-time monitoring via X-ray diffraction are outstanding.7 By recording powder diffractograms with the progress of the mechanochemical reactions it is possible to obtain otherwise not accessible information on crystalline intermediates or new products, or on reaction time, thus optimizing – and ideally controlling – the outcome of a synthetic procedure.8–11 The collection of such data may be also used for obtaining a kinetic profile: along with the information on the evolution time of the reaction, it would also represent a further step in the understanding of the mechanochemical reactions and their following scale-up at the industrial level.12 By finding the best fitting of kinetic equations with the experimental data, it is possible to gain insight into the microscopic features of mechanical activation.13 When dealing with crystalline materials, the most powerful method for obtaining kinetic data is the Quantitative Phase Analysis (QPA) via Rietveld refinement. Such a method provides a quantitative identification of the crystalline phases (reactants, intermediates, products) appearing and/or disappearing in the course of a reaction, with concomitant information on microstructural parameters such as crystal size and microstrain. For in situ data collected with a synchrotron source (and with the ball milling setup) the quality of the powder diffractograms is significantly worsened, thus affecting the reliability of the Rietveld method. However, the implementation of work strategies to deal with synchrotron data represents a turning point in performing Rietveld analysis on synchrotron data,14 as pointed out in recent work by Lampronti et al.15 When these work strategies are not easily applicable, or if more in general we are dealing with unknown crystal structures or with poorly crystalline materials, the Rietveld method may be no longer feasible, and the strategy of the single peak analysis can instead be utilized.16 The calculation of the normalized areas under the Bragg peaks may represent a good alternative strategy for the evaluation of the reaction rates, but a necessary condition for this method to work is that the peaks under investigation do not interfere with other peaks, i.e. the peaks of reagents and products need to be in different angular regions of the diffractogram. However, this ideal condition is seldom achieved, especially with low symmetry crystals, therefore the use of a method that instead relies on all the peaks in the diffractogram or on chemometric techniques could provide a valid help, particularly in assisting the in situ monitoring. Multivariate analysis, with its ability of elaborating big data set, is already a strategic tool for process monitoring,17 and can help analysing the entire XRD profile, with the possibility of uncovering hidden information otherwise difficult to detect. Multivariate analysis, at the same time, offers fast calculations able to process the data without prior knowledge of the crystalline phases and features,18 or in the case of complex profiles, as in the presence of amorphous phases. To this aim, dimensionality reduction algorithms, like principal component analysis (PCA), can be fundamental to explore and perform cluster analysis of big datasets, as in high-throughput screening,19 to correlate macroscopic properties20 and to extract kinetic information also in the presence of shifts in the peak position.21 In this work we present an application of the MCR-ALS analysis to powder X-ray diffraction (PXRD) data. The MCR-ALS (multivariate curve resolution – alternating least squares)22,23 is a method that enables to deconstruct a dataset of composite signals into two separate matrices, the first representing the signals of k “pure” components, the second their respective concentrations profiles. The number of components, k, is a variable of the method defined during the computational process; its ideal value is the number of pure chemical entities present in the sample that generates the signal of interest, which evolves with time, e.g., following a chemical reaction. In such conditions the mathematical deconvolution acquires a chemical meaning and can provide direct information on the evolution of the system. As MCR-ALS is a self-modelling method, the calculation can be performed directly on the experimental data, without the need for a calibration step or a prior knowledge of the signal composition; this makes it a suitable method for monitoring chemical transformations where multiple or unknown phases may be present at different times, as is the case during a mechanochemical synthesis.While there have been several reports on the application of such a method to the monitoring of mechanochemical processes, it has generally been used for the treatment of spectroscopic data.24–26 In this work we aim to prove its validity in the context of the powder diffraction data, for which to our knowledge there has been a limited application,27,28 by studying a mechanochemical reaction characterized by a kinetics suitable for ex situ monitoring, namely the mechanochemical reaction between theophylline (TP) and malonic acid (MA) in a 1:1 stoichiometric ratio (see Scheme 1; symbols for milling frequency, temperature and added solvent are taken from Michalchuk et al.29).
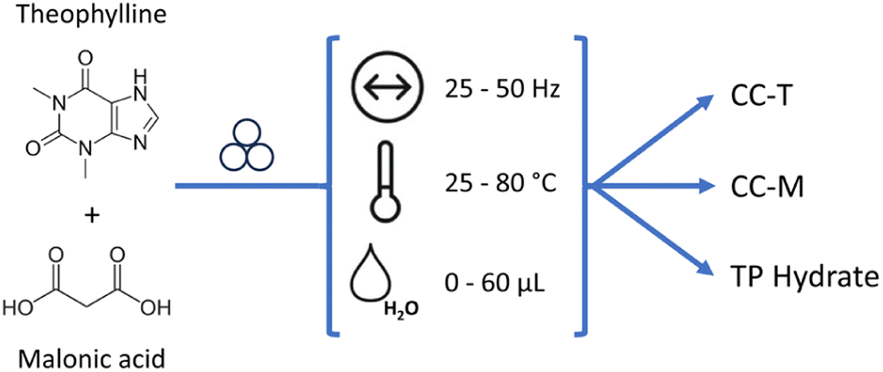 | ||
| Scheme 1 Co-crystallization of theophylline (TP) and malonic acid (MA): ball milling conditions tested in this work (variable frequency, temperature and water amount), and the possible products, i.e., the triclinic co-crystal TP·MA (CC-T), the monoclinic co-crystal TP·MA (CC-M) and hydrated theophylline (TP hydrate). | ||
2. Materials and methods
Theophylline and malonic acid were purchased from Sigma Aldrich and used without further purification.2.1 Co-crystal synthesis via ball-milling
2.2 X-ray powder diffraction (XRD)
2.3 Single crystal X-ray diffraction
Single crystals for the triclinic polymorph of the theophylline·malonic acid co-crystal (CC-T) were obtained dissolving 0.4 mmol of theophylline and 0.4 mmol of malonic acid in 8 mL of chloroform/ethanol 10:1, heated at 40 °C to facilitate the dissolution. The solution was then cooled and left to evaporate.31 The same procedure applied changing the solvent with a 10:1 chloroform/methanol mixture produced single crystals of both triclinic e monoclinic polymorphs.
Single crystal data for the triclinic and monoclinic TP·MA co-crystals was collected at room temperature on an Oxford Xcalibur using Mo-Kα radiation, equipped with a graphite monochromator and a CCD Sapphire detector. The SHELXT32 and SHELXL33 programs were used for the solution and refinement of the structures based on F2, implemented in the Olex2 software.34 All non-H atoms were refined anisotropically. Hydrogen atoms were added in calculated positions, and refined riding on their respective C, N and O atoms.
The program mercury35 was used for the calculation of the powder X-ray patterns based on single-crystal data, either retrieved from the Cambridge structural database (CSD)36 or obtained in this work.
2.4 Multivariate analysis
The MCR method allows the decomposition of the data matrix D(s×n) (n variables, s observations) into the k-components matrix S(k×n) and the respective concentration profiles C(s×k), according to the equation:| D(s×n) = C(s×k) × S(k×n) + E(s×n) |
The alternating least squares calculation (ALS) allows to optimize the output through an iterative process. Starting from the result of the first step, at each iteration the above equation is solved alternately for the C or S matrix, until a convergence criterion is met. The first iteration requires an initial estimate of either the concentrations or the pure profiles, that can be evaluated with different methods.23,37 The SIMPLISMA algorithm38 was chosen here for the initial calculation of the pure components profiles, but the number of these components needs first to be defined; in this work models were computed starting from two and up to four components, evaluating for each dataset the plausibility of the results by matching the pure profiles with the reference X-ray diffraction patterns calculated on the basis of single crystal data.
To obtain a chemically acceptable solution it is possible to impose a number of constraints;23 in our case non-negativity on both the concentration and the pure profiles, and the closure of the concentrations were employed. At the end of the computational process the concentration values obtained were normalized in the 0–1 range.
All calculations were performed with Python (v3.10.11)39 scripts to integrate a SIMPLISMA algorithm developed in-house and the MCR-ALS from the Spectrochempy library (a complete documentation of the module, with tutorials, is available at https://www.spectrochempy.fr/stable).40
The MCR-ALS method was applied to powder X-ray diffraction data collected in both ex situ and in situ experiments. The ex situ data was corrected for baseline to shift the diffractograms vertically, as close as possible to the zero axis, and allow a better deconvolution of the profiles during the calculation. The correction was achieved with a linear interpolation of the two extremes of the XRD profile followed by subtraction of the minimum value of the linearly corrected profile. Lastly the data was area normalized during the MCR calculation through the ‘normSpec’ parameter of the function.
The in situ data was pre-treated at first with the software DPDAK for the integration of the data, then with a Python script for the subtraction of the background.
2.5 Rietveld analysis
Phase quantification analysis was performed using the software TOPAS 6;41 a Chebyshev function and a pseudo-Voigt (TCHZ type) were used to fit the background and the peak shape, respectively. The zero error, displacement of the sample, and the lattice parameters of the phases were allowed to fit.3. Results and discussion
3.1 Preliminary test and intermediate identification
To test the feasibility of applying the MCR-ALS method to a mechanochemical process, we selected from the literature a mechanochemical reaction characterized by a kinetics suitable for ex situ monitoring. The system of choice is a 1:1 stoichiometric mixture of theophylline (TP) and malonic acid (MA). A co-crystal of theophylline and malonic acid is known in the literature in two polymorphic forms: a monoclinic form (CC-M), reported by Trask et al.42 [refcode XEJXAM, T = 180 K], obtained by manual grinding, and a triclinic form (CC-T), recently reported by Stanton et al.31 [refcode XEJXAM01, T = 150 K], obtained by solvent slow evaporation. Single crystals of both monoclinic and triclinic co-crystals, depicted in Fig. 1, were reprepared in this work and their structures redetermined at room temperature (see Section 2.3); the corresponding calculated patterns could thus be compared directly with those measured at room temperature for the products of the grinding experiments.
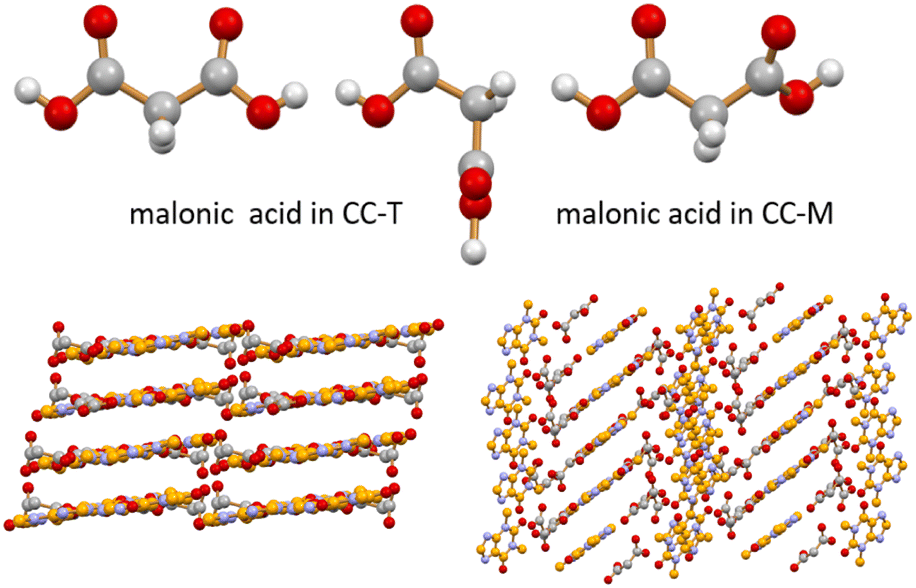 | ||
| Fig. 1 Different conformations observed for malonic acid in CC-T (top left, two independent molecules) and in CC-M (top right) at room temperature (see ESI†). Different packing arrangements are associated with these different molecular conformations in the CC-T (bottom left) and CC-M (bottom right) theophylline·malonic acid co-crystals at room temperature. H atoms omitted for clarity in the two packings. | ||
A preliminary test of one hour milling under neat grinding (NG) conditions at 20 Hz with two 5 mm balls resulted in a powder that was analysed by XRD. As can be seen from the blue pattern in Fig. 2, the reagents are still present, together with traces of CC-M. Additional peaks, however, marked with the green arrows, were found to partially match the pattern simulated for the triclinic polymorph of theophylline·malonic acid, CC-T,31 although the complex mixture profile did not allow for an easy identification.
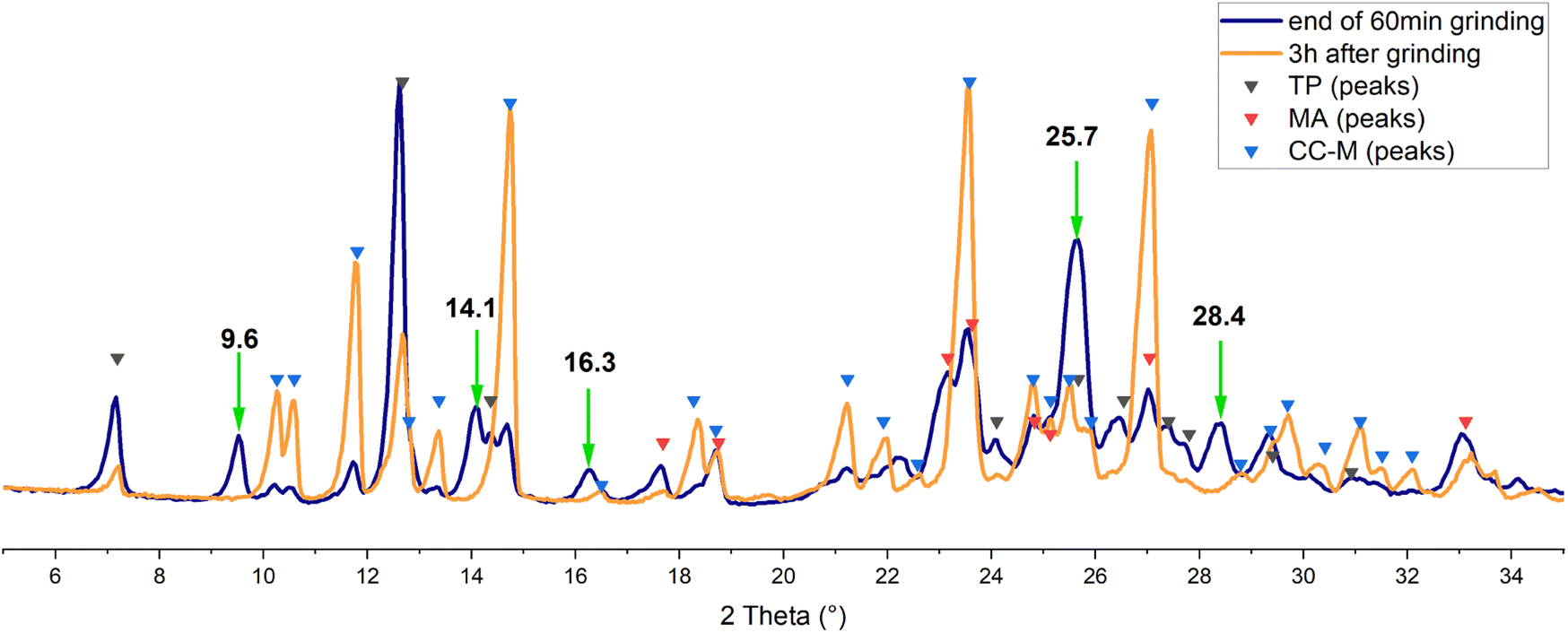 | ||
| Fig. 2 Experimental XRD patterns acquired at the end of the grinding process (blue) and after 3 h (orange). Black, red and blue triangles marks the angular positions of the reflections calculated, on the basis of single crystal data, for theophylline, malonic acid and CC-M, respectively. Green arrows, with 2θ values in bold, evidence the unassigned reflections. | ||
The same mixture was analysed again three hours after the end of the grinding process (orange line in Fig. 2): there is an evident increase in the intensity of the signals for CC-M, along with the disappearance of the additional peaks mentioned above, suggesting the presence, in the pristine grinding product, of a transient phase. Theophylline is reported to spontaneously form co-crystals with nicotinamide in tablets,43 and water and humidity were suggested to be the key factors in the transformation.44 The most recent study on this matter45 also analysed the theophylline-malonic acid system, but in the absence of grinding of the reagents the formation of the triclinic co-crystal was not observed.
This preliminary finding of a possible transient phase made the reaction between theophylline and malonic acid the perfect multi-component system to test with the MCR method, and the spontaneous transformations accompanying the grinding process allowed an easy setup for the monitoring.
The data acquired, as described in Section 2.4, was analysed with the MCR-ALS method, and the best model was obtained with three components. From the concentration profiles (Fig. 3a) it is possible to follow the evolution of the calculated components. In this experiment the component 1 (orange line) progressively increases with time, being thus consistent with the product, i.e., the co-crystal. The component 2 (blue line) follows a reverse trend, therefore we can assume it represents the reagents. Finally, the component 3 (red line) shows a rapid increase in the first hours of the monitoring, then slowly decreases to zero, in agreement with the appearance of a transient phase and its subsequent transformation into the final product.
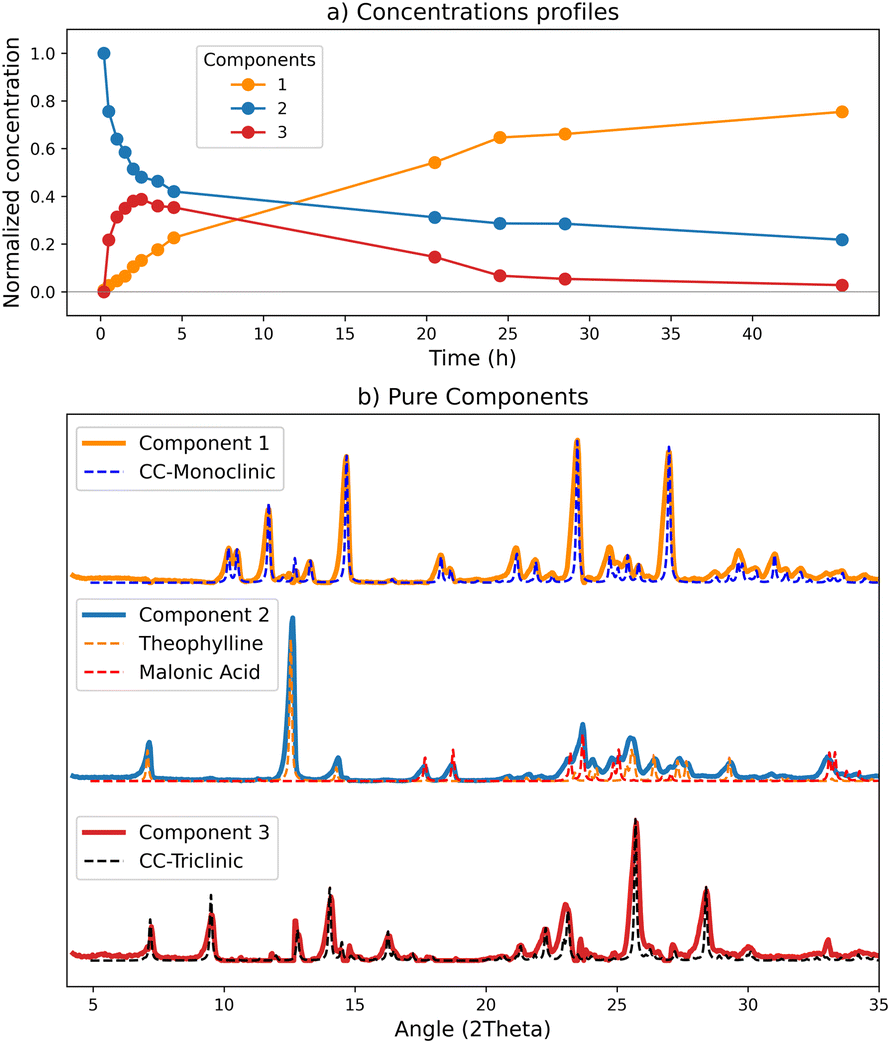 | ||
| Fig. 3 Results of the MCR-ALS calculation with 3 components for the ex situ data set: (a) normalized concentration corresponding to the C matrix; (b) comparison of the pure profiles resulting from the deconvolution (S matrix) in solid lines, and, in dotted lines, the reference patterns calculated from single crystal data. | ||
To evaluate the goodness of the deconvolution and the reliability of the kinetics, the patterns calculated with the MCR method for the pure components were compared with the reference patterns (Fig. 3(b)) based on our new room temperature (see ESI†) single crystal data. As it is evident from Fig. 3(b), there is almost a perfect match between the two sets of patterns. The ‘reagents’ component (n°2) fits the combination of theophylline form II (refcode BAPLOT01)46 and of malonic acid form β (refcode MALNAC),47 corresponding to the starting materials; since the deconvolution originates from the variations of the data set, and the reagents simultaneously disappear upon co-crystal formation, the signals due to theophylline and malonic acid are detected by the method as a single component. The ‘product’ component (n°1), as expected, fits the pattern of the monoclinic co-crystal.
A relevant result is the profile behaviour of component 3, accompanying the transient kinetics, that perfectly matches the XRD pattern of CC-T, the triclinic polymorph of the theophylline·malonic acid co-crystal. We have evidence that the patterns obtained through the model data allow a reliable and easy identification of all the phases constituting the solid mixture.
Concentrations and pure patterns returned by the calculation are in arbitrary units:23 when normalized, concentration values represent the weight of the contribution of each pure component to the corresponding mixture signal.48 Therefore it is important, for a correct interpretation of the results, to be aware of the characteristics of the pure component matching each concentration profile. The presence of mixed components for example, will affect the quantitative calculation: the concentration profiles can still be diagnostic of the evolution of the process, but the returned values will not be associable to a single solid form.
3.2 Quantitative analysis on ex situ data
If the deconvolution results in chemically pure profiles, the concentration values obtained can be used for quantitative analysis. For spectroscopic data an additional calibration step is usually required to convert the model output into meaningful concentration units, and several approaches are possible, either internal (correlation constraint49) or external50 to the MCR-ALS algorithm. With powder diffraction data, phase quantification is evaluated as relative percentage of each solid form in the mixture; in terms of measurement units, the normalized concentrations of the calculation can potentially represent the percentage of the corresponding solid phase in the sample.In the calculation previously shown the components were consistent with chemically pure phases, therefore we tested the quantitative application of the method by comparing the normalized percentages obtained with phase quantification computed via Rietveld analysis.
The Rietveld quantification was applied to the experimental patterns, registered starting from the end of the 30 min grinding (t0), as described in Section 2.5.
The results, divided by component, are reported in Fig. 4(a), compared with the normalized concentrations of the MCR method. The comparison shows a good match between the two methods. The box plots in Fig. 4(b) visualize the absolute differences between the results of the two calculations; a maximum difference of seven percentage points can be observed, registered on the reagents data, while the mean values (red line) are of 4% and 3% for the reagents and CC-T respectively, and lower than 2% for the monoclinic co-crystal. The comparison highlights a negative offset on all the Rietveld values for the reagents and an over-quantification on all the triclinic co-crystal values.
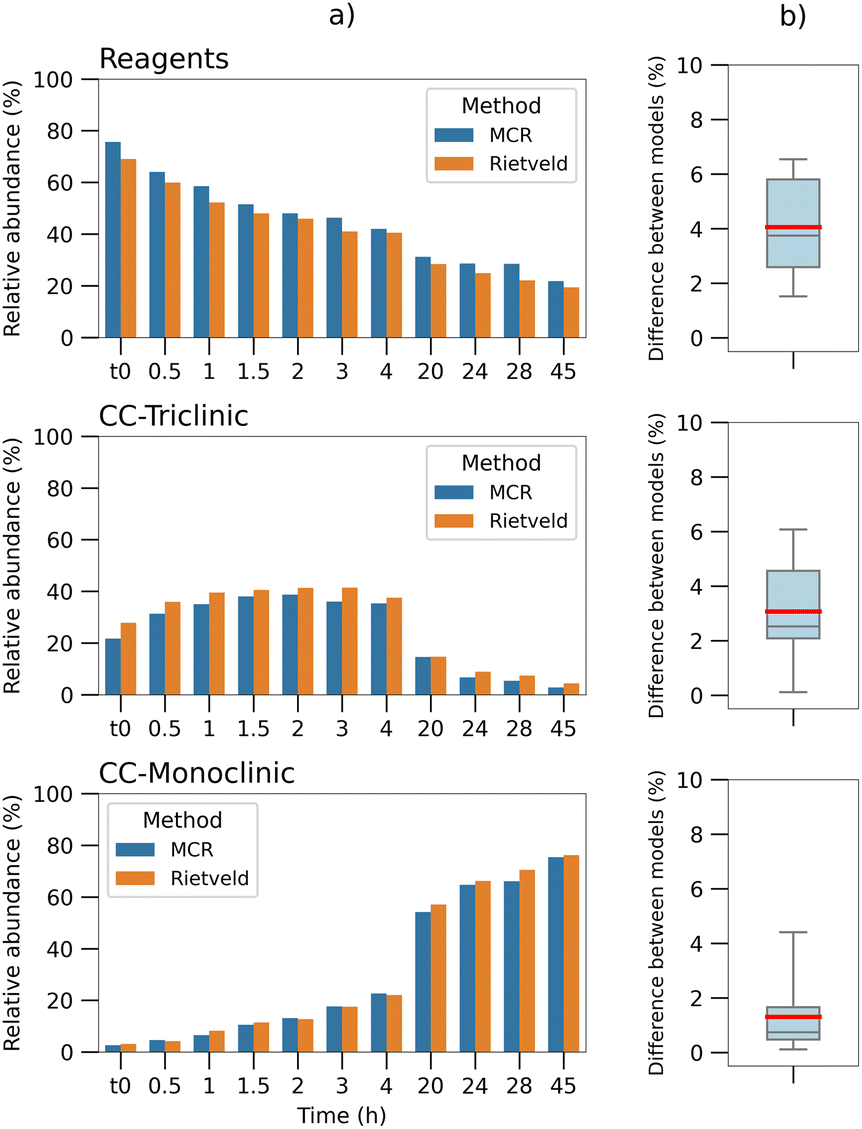 | ||
| Fig. 4 (a) Barplots comparison between the normalized concentrations calculated for each time point by the MCR-ALS method (blue), with phase quantification performed via Rietveld analysis on the diffractogram of the respective time point (orange), divided by component. Rietveld values for the “reagents” correspond to the sum of the theophylline and malonic acid results. (b) Boxplots of the absolute differences between the quantification of the two methods (data in column “a”), divided by component. The upper and lower bars correspond to the minimum and maximum values of the distribution; the red line shows the mean value. | ||
Quantitative analysis by means of power diffraction data has limits in its accuracy, due to numerous factors affecting the diffraction pattern, such as preferred orientation, peak broadening and amorphization of the solid sample (as an example, an analysis of preferred orientation effects has been added to ESI, Section 4†).
Taking into account these effects and a mean error of around 10% that has been reported for the Rietveld method,51 the normalized MCR values obtained for the dataset shown here appear reasonable in estimating (i) the concentrations of each component in the system and (ii) their evolution.
3.3 Study of the process kinetics via in situ monitoring
These preliminary findings allowed a first characterization of the system and of the phases involved. On the basis of these results, we decided to investigate further the application of the MCR-ALS method to in situ synchrotron data. To evaluate the effect of process variables on the kinetics, both neat grinding (NG) and liquid assisted grinding (LAG) conditions were explored: grinding frequency and temperature were varied for the NG tests, while for the LAG experiments frequency and water volume were changed. Each condition was tested in duplicate, but, for sake of clarity, only one experiment will be discussed when no significant difference between the two was observed. A summary of the conditions analysed can be found in Table 1.| Name | Condition | Frequency (Hz) | Temperature (°C) | Water (μL) |
|---|---|---|---|---|
| a L: low frequency (25 Hz), H: high frequency (50 Hz); W60, W30, W10: LAG condition with relative water volume (60, 30 and 10 μL, respectively); T50, T80: experiments at 50 °C and 80 °C, respectively. | ||||
| L1 | NG | 25 | RT | — |
| H1 | NG | 50 | RT | — |
| H2 | NG | 50 | RT | — |
| T50 | NG | 25 | 50 | — |
| T80 | NG | 25 | 80 | — |
| HW60 | LAG | 50 | RT | 60 |
| HW30 | LAG | 50 | RT | 30 |
| LW30 | LAG | 25 | RT | 30 |
| LW10 | LAG | 25 | RT | 10 |
It is important to notice, though, that the characteristic diffraction peaks of the triclinic form are detectable only on the intermediate component (component 2); the corresponding kinetic profile, therefore, is representative of its evolution, even if the quantification cannot be extremely accurate, since it includes traces of other phases.
Once the correct solid phase has been assigned to each calculated component, it is possible to qualitatively compare the kinetics of the different NG experiments, as shown in Fig. 5.
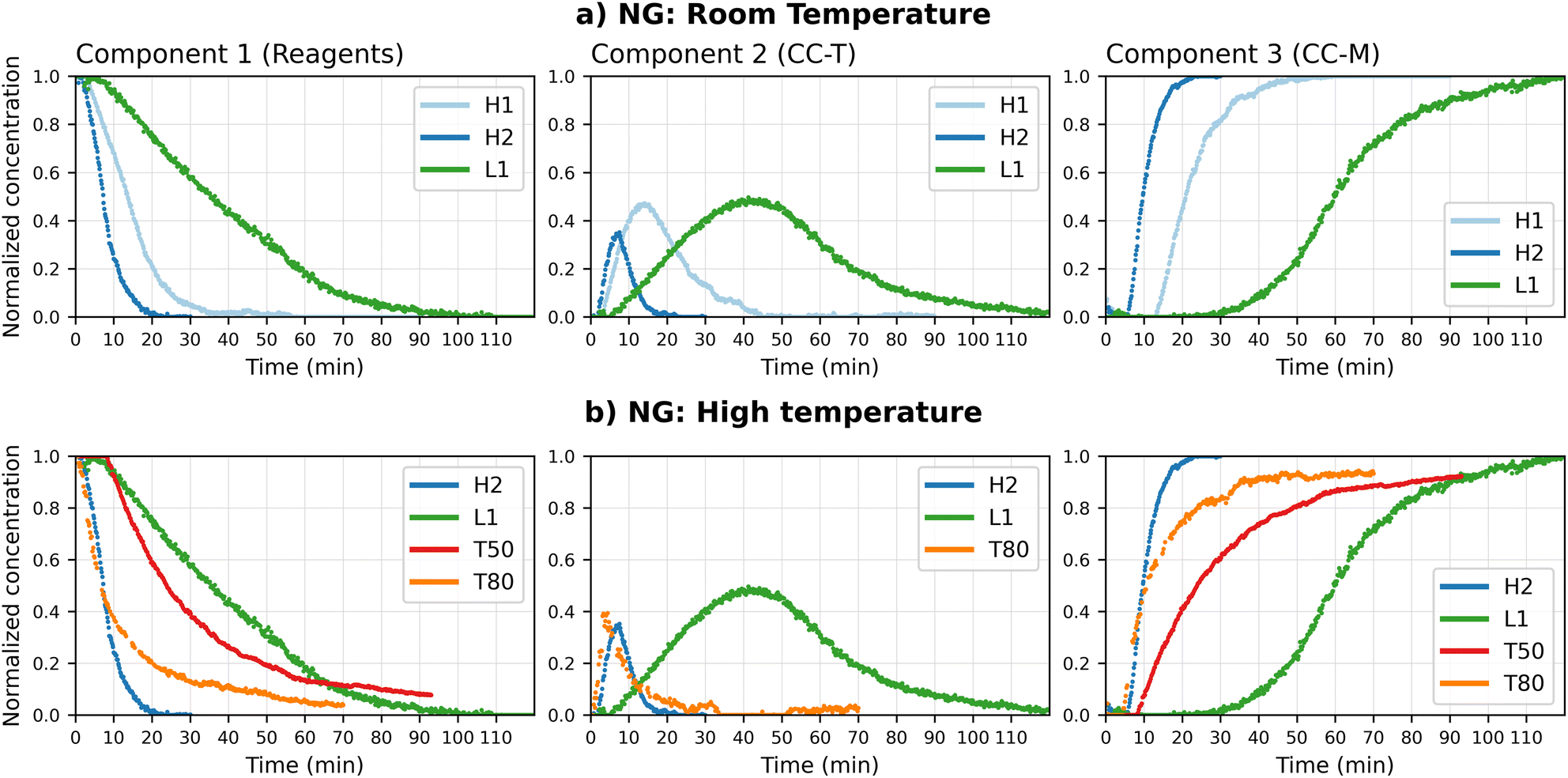 | ||
| Fig. 5 Normalized concentration profiles obtained by the MCR-ALS calculation for the in situ neat grinding (NG) data, divided by component (a: RT experiments, b: effect of temperature). The profiles are grouped based on the comparison of the relative pure profile with the reference XRD patterns. All tests at higher temperature were performed at the grinding frequency of 25 Hz, while the H2 and L1 tests correspond to RT tests at 50 and 25 Hz, respectively. | ||
An analysis of the two 50 Hz curves (H1 and H2) evidences a faster kinetics with respect to the lower ball milling frequency experiments (L1). However, H1 and H2 show a markedly different behaviour, starting at ca. 10 min, for all components, suggesting a greater variability of the process in this milling conditions. The variability affects in particular the intermediate phase, that in H2 not only decreases at an earlier stage but reaches a lower concentration at its peak. To exclude that the observed differences were due to computing inaccuracies, the profiles were compared with the experimental XRD pattern. Fig. ESI 13† reports the progression, in the first 20 min, of the characteristic peaks of CC-T and CC-M, next to the calculated profiles; the times of appearance and relative heights of the peaks match the curves returned by the model, supporting the hypothesis of a variable kinetics, and validating the model results. This case, while supporting the known and obvious importance of repetitions, presents a clear example of how in situ monitoring, combined with a method that allows an easy visualization of the data, can be a valid tool to rapidly identify variations in the process.
For these data sets the best models were obtained with two components for the experiment at 50 °C, and with three components for the experiment at 80 °C.
The analysis of the pure profiles for the 50 °C test (Fig. ESI 7†) shows a good match of the decreasing component with the profiles of the reagents, while the “product” component matches the monoclinic polymorph (CC-M). No traces of CC-T seem to be detected by the model, but a closer inspection of the experimental data (Fig. ESI 14†) shows, between 5 and 14 minutes, the characteristic peak of the triclinic form at 6.8°, although its intensity is too low to be quantified by the model.
The components profiles for the 80 °C experiment (Fig. ESI 8†) shows a separation into three components, analogous to the room temperature tests. Similarly to the RT 50 Hz case, the intermediate component unambiguously includes the triclinic form, but shows also evident contributions from both the monoclinic form and theophylline. This suggests a fast transition, coherent with the increase in temperature, as observed with the 50 °C test, but, contrary to the previous result, the intermediate phase gives rise to a higher signal that was indeed detected by the method.
The faster transformation kinetics, due to the higher temperature, can be clearly observed in Fig. 5(b), where the corresponding concentration profiles are compared with the RT results previously discussed. On the basis of these observations, a hypothesis can be formulated for the conversion, namely a two-steps reaction mechanism: the reagents combine first to give CC-T (step I), then CC-T transforms into the more stable CC-M (step II). The variation in the CC-T concentration with the increase in temperature would suggest an influence of this variable on the rate of the first reaction: at RT (green line) the formation of CC-T is the rate determining step, and the appearance of the more stable co-crystal CC-M depends on the formation of the triclinic polymorph; upon raising the temperature to 50 °C (red line) formation of CC-T occurs more rapidly and immediately converts to CC-M, i.e. only traces of CC-T can be detected and the appearance of CC-M occurs 20 minutes earlier; at 80 °C (orange line) a further acceleration of the first reaction results in the accumulation of CC-T, which is again observed, and its transformation in CC-M is now the rate determining step.
The profiles obtained show that, while the co-crystal kinetics is accelerated by the increase in temperature, a full conversion is still not achieved after 60 min of grinding. In the present system, therefore, an increase in the grinding frequency appears to be a better option, compared to an increase in temperature, to improve the conversion time.
The addition of water introduces a further variable, since theophylline is known to easily transform, in the presence of water,52 to its hydrated form. The hydrated form of theophylline would thus need to be considered as a potential, additional constituent of the reaction mixture.
The best results were obtained with the MCR-ALS deconvolution method when two components were used for all collected datasets. An analysis of the pure profiles shows that the products component matches the profile for CC-M, but theophylline hydrate is also present in almost all experiments. The only exception is the 10 μL and 25 Hz test, which shows a barely noticeable signal of hydrated theophylline. Fig. 6(c) shows an example of the deconvolution process (see ESI Section 4† for the complete set of results). The reagent component in the tests with higher water content, for both 25 Hz and 50 Hz (30 μL and 60 μL, respectively), matches the pure theophylline pattern (Fig. 6(a)), while the tests with lower water content, 10 μL for the 25 Hz test and 30 μL for the 50 Hz, also show the signal of CC-T (Fig. 6(b)).
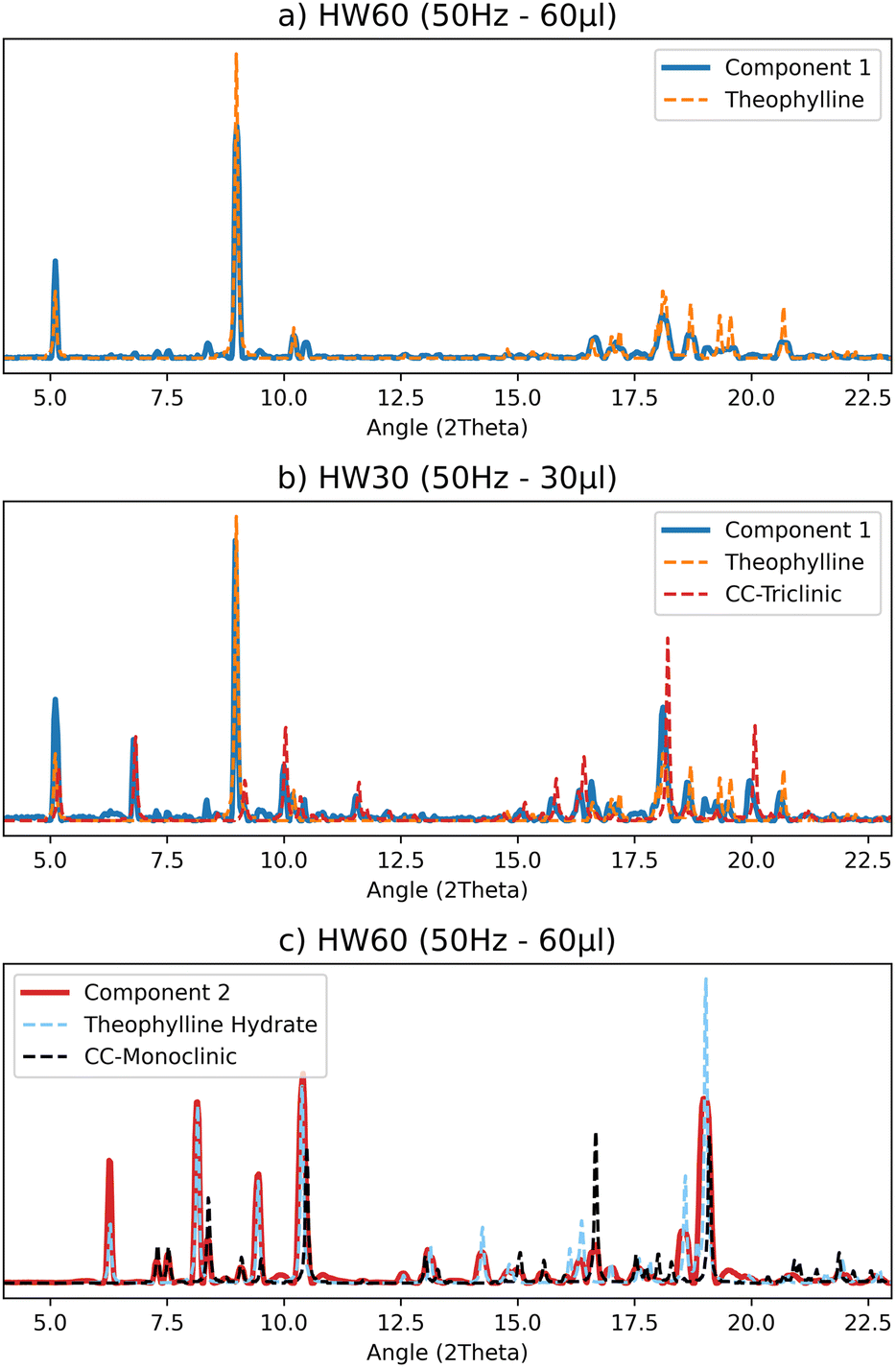 | ||
| Fig. 6 Selected comparisons of the pure profiles (S matrix) obtained by the MCR-ALS calculation for the LAG data set with the reference pattern from single crystal structures: (a) “reagents” component containing exclusively the theophylline profile, (b) “reagents” component matching theophylline and the triclinic co-crystal, (c) “products” component matching the monoclinic polymorph and the theophylline hydrate. | ||
Based on the pure profiles analysis, we can assume that the formation of CC-T is an extremely rapid event (compared to the acquisition rate employed), occurring at the very beginning of the process, therefore the CC-T diffraction peaks are interpreted by the computational method as part of the reagents profile. Moreover, this phase is detected by the method only when lower volumes of water are added to the reagents mixture, suggesting that water has an active role in the kinetics of the solid phase transformation, leading to the formation of CC-T only at low water contents.
The metastable form CC-T rapidly transforms into the stable form CC-M; however, the appearance of CC-T affects the kinetics of the process. In Fig. 7 the reagents concentration profiles are compared, and a noticeable difference emerges between the experiments with lower (LW10, HW30) and higher (LW30, HW60) water content. The lower water content tests show a slower decreasing profile, coherent with the formation of the triclinic polymorph that, with its appearance, delays the formation of the products.
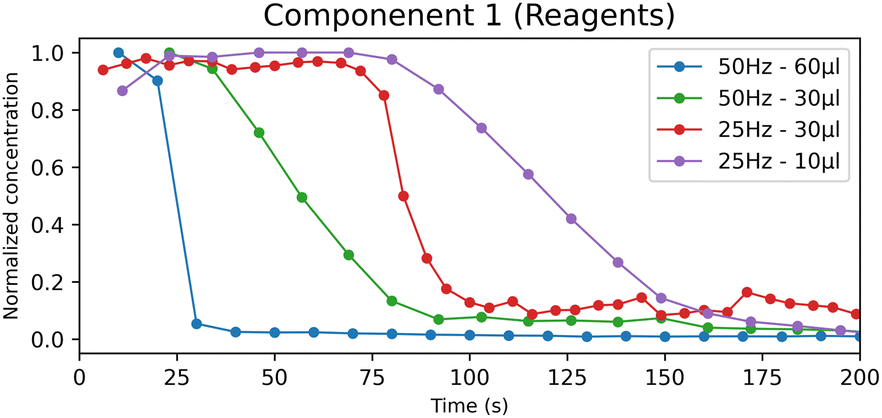 | ||
| Fig. 7 Normalized concentration profiles of the “reagents” component obtained by the MCR-ALS calculation for the LAG experiments. | ||
From a model evaluation perspective, this result shows that, even if the formation of CC-T does not result into a separate component, the concentration profiles still indicate the presence of an additional chemical process. A more detailed analysis of the effects of the CC-T kinetics on the concentration profiles can be found in the ESI† (ESI Section 7).
To further explore the influence of water on the process, we need to consider, in addition to the solvent role in the formation of the co-crystal, the potential formation of theophylline hydrate.
For all experiments, except for the LW10 test, the characteristic peaks of the hydrate could be recognized in the PXRD patterns, and the hydrate is also correctly observed in the product component. After 20 to 60 minutes of grinding the pattern of the hydrated theophylline is still identifiable, suggesting that the hydrate is a stable product competing with the CC-M form. Evidence of this can be seen in Fig. 8(a), where the profiles for the second component (the products) are compared with the characteristic peaks, highlighted in light blue and orange, of theophylline hydrate and CC-M, respectively.
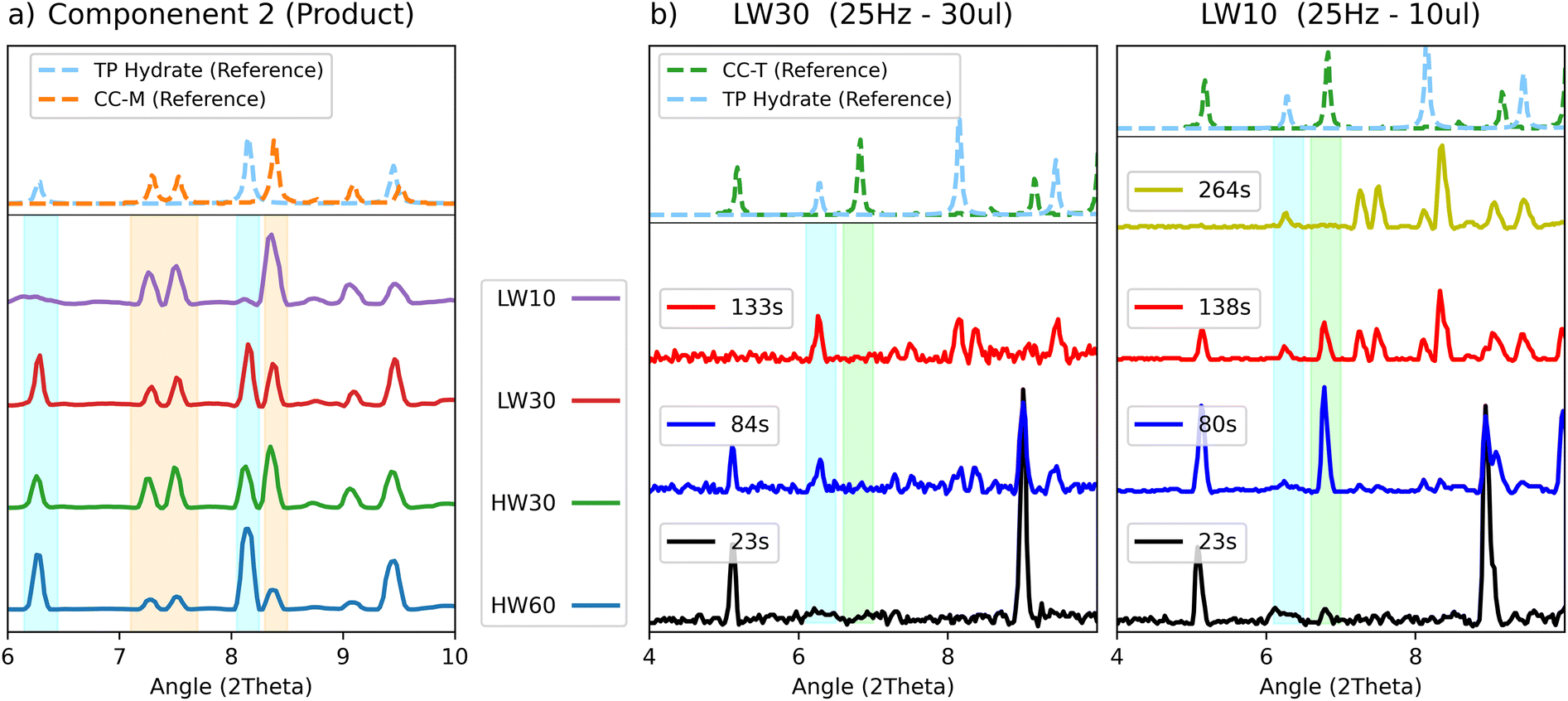 | ||
| Fig. 8 (a) Comparison of the “product” component obtained by the MCR-ALS calculation for the LAG data. The characteristic peaks of theophylline hydrate and monoclinic co-crystal are highlighted in light blue and orange respectively, with in dotted line the calculated pattern as reference. (b) Experimental patterns of the LW30 and LW10 test at selected time points of 20 s (black), 80 s (blue), 130 s (red) and 260 s (yellow), with the characteristic peaks of theophylline hydrate and triclinic co-crystal highlighted in light blue and green, respectively. | ||
The product component for the ball milling experiments with lower water content, (LW10 - purple and HW30 – green) also show a lower relative intensity of the hydrate peaks with respect to those of CC-M, supporting the hypothesis of the competition between the two transformations, as an increase in water favours the formation of the hydrate. Moreover, under equal water volume conditions, as in the 30 μL of the 25 Hz (LW30 – red) compared to 30 μL of the 50 Hz (HW30 – green) test, a higher milling frequency results in a higher CC-M content.
Competition also seems to take place between theophylline hydrate and CC-T, as can be seen in Fig. 8(b), where the patterns for the two 25 Hz experiments (LW10–LW30) are compared. The two tests differ only for the added water volume and show different behaviours with respect to the appearance of CC-T. The plot collects the diffractograms sampled at equal time points for both tests. It is noticeable that with 10 μL of water the triclinic form appears and is fully formed after 80 s, while the hydrate starts appearing between the 80 s and 130 s. Upon increasing the volume to 30 μL, no traces of CC-T are visible between 20 s and 80 s, despite the slower kinetics formation of the hydrate, that only appears after 80 s of grinding. This suggests an inhibition of CC-T formation in the presence of an excess of water.
4. Conclusions
In this work we tested the feasibility of the MCR-ALS method on PXRD data of mechanochemical processes by studying the system theophylline (TP) and malonic acid (MA) in a 1:1 stoichiometric ratio.
We first applied the MCR-ALS method on data collected ex situ at different milling times, and then we tested the quantitative information of the method by comparing the normalized percentages obtained with the phase quantification computed via Rietveld analysis on the same data set. Considering that a mean error of around 10% has been reported for the Rietveld method, the normalized MCR values obtained appear reasonable in estimating the concentrations of each component in the system and their evolution over time.
By applying the MCR-ALS method on data collected in situ, we then managed to better evaluate the strengths and the limits of the method. In fact, through the investigation of mechanochemical reactions performed at different milling conditions (frequency, water amount, temperature), we observed that the method is no longer reliable for obtaining quantitative information when the solid-state transformations involved are too fast compared to the acquisition rate. However, the method is still capable to detect fast changes over time, therefore we can still have a qualitative information on the evolution of the reaction and obtain a kinetic profile.
Finally, in situ monitoring helped us to gain insight into the solid-state reactivity of the system under study. For example, we observed for the first time the triclinic polymorph as transient phase in the synthesis via grinding; we also explored the effect of temperature on the transformation, showing how the increase in temperature, while inducing an initial speeding of the conversion, does not allow the reaction to be quantitative. By comparing the NG and LAG conditions, the in situ experiments reveal that the presence of water in the solid-state transformation is the key to speed up the process, even with a potential competitive hydrate formation, that was in this case possible to investigate. Depending on the milling frequency a specific amount of water was needed to obtain the desired product; also a higher ball milling frequency, with the same amount of water, results in a more quantitative reaction. It is evident, therefore, that this method could be employed as a tool to optimize a mechanochemical process.
In conclusion, this work has demonstrated the validity of the MCR-ALS method as an alternative way of obtaining quantitative information on mechanochemical kinetics. In fact, the monitoring of the mechanochemical reactions may highlight the appearance of unknown or poorly crystalline phases for which the Rietveld method would be no longer suitable. Moreover, the application of the MCR-ALS method on in situ data represents a powerful tool to gain insight into the mechanochemical behaviour and thus optimize the mechanochemical processes.
Author contributions
LM, FG and LC conceived the project; LM and LC planned the experiments and analysed data. FG and EF supervised the project. All authors discussed the results, wrote and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The support of the University of Bologna (RFO scheme, FG, DB), a PNRR DM 352/2022 project co-financed by CHEMESSENTIA S.r.l (LM) are acknowledged. The EU Horizon project IMPACTIVE is also acknowledged (LC, EF).Notes and references
- O. Galant, G. Cerfeda, A. S. McCalmont, S. L. James, A. Porcheddu, F. Delogu, D. E. Crawford, E. Colacino and S. Spatari, ACS Sustain. Chem. Eng., 2022, 10, 1430–1439 CrossRef CAS.
- C. Blazquez-Barbadillo, J. F. González, A. Porcheddu, D. Virieux, J. C. Menéndez and E. Colacino, Green Chem. Lett. Rev., 2022, 15, 881–892 CrossRef CAS.
- Y. Zhang, Y. Wang, X. Yang, L. Zhao, R. Su, J. Wu, D. Luo, S. Li, P. Chen, M. Yu, Q. Gong and R. Zhu, Adv. Mater., 2022, 34, 2107420 CrossRef CAS PubMed.
- J. Alić, T. Stolar, Z. Štefanić, K. Užarević and M. Šekutor, ACS Sustain. Chem. Eng., 2023, 11, 617–624 CrossRef.
- T. Stolar, S. Lukin, M. Tireli, I. Sović, B. Karadeniz, I. Kereković, G. Matijašić, M. Gretić, Z. Katančić, I. Dejanović, M. D. Michiel, I. Halasz and K. Užarević, ACS Sustain. Chem. Eng., 2019, 7, 7102–7110 CrossRef CAS.
- P. A. Julien and T. Friščić, Cryst. Growth Des., 2022, 22, 5726–5754 CrossRef CAS.
- A. A. L. Michalchuk and F. Emmerling, Angew. Chem., Int. Ed., 2022, 61, e202117270 CrossRef CAS PubMed.
- K. Linberg, B. Röder, D. Al-Sabbagh, F. Emmerling and A. A. L. Michalchuk, Faraday Discuss., 2023, 241, 178–193 RSC.
- M. Rautenberg, B. Bhattacharya, J. Witt, M. Jain and F. Emmerling, CrystEngComm, 2022, 24, 6747–6750 RSC.
- A. M. Belenguer, G. I. Lampronti, A. A. L. Michalchuk, F. Emmerling and J. K. M. Sanders, CrystEngComm, 2022, 24, 4256–4261 RSC.
- L. Casali, T. Feiler, M. Heilmann, D. Braga, F. Emmerling and F. Grepioni, CrystEngComm, 2022, 24, 1292–1298 RSC.
- M. Carta, L. Vugrin, G. Miletić, M. J. Kulcsár, P. C. Ricci, I. Halasz and F. Delogu, Angew. Chem., Int. Ed., 2023, 62, e202308046 CrossRef CAS PubMed.
- M. Carta, E. Colacino, F. Delogu and A. Porcheddu, Phys. Chem. Chem. Phys., 2020, 22, 14489–14502 RSC.
- I. Halasz, T. Friščić, S. A. J. Kimber, K. Užarević, A. Puškarić, C. Mottillo, P. Julien, V. Štrukil, V. Honkimäki and R. E. Dinnebier, Faraday Discuss., 2014, 170, 203–221 RSC.
- G. I. Lampronti, A. A. L. Michalchuk, P. P. Mazzeo, A. M. Belenguer, J. K. M. Sanders, A. Bacchi and F. Emmerling, Nat. Commun., 2021, 12, 6134 CrossRef CAS PubMed.
- I. C. B. Martins, M. Carta, S. Haferkamp, T. Feiler, F. Delogu, E. Colacino and F. Emmerling, ACS Sustain. Chem. Eng., 2021, 9, 12591–12601 CrossRef CAS.
- M. Nawaz, A. S. Maulud, H. Zabiri and H. Suleman, IEEE Access, 2022, 10, 49708–49724 Search PubMed.
- P. Guccione, M. Lopresti, M. Milanesio and R. Caliandro, Crystals, 2020, 11, 12 CrossRef.
- G. Barr, W. Dong and C. J. Gilmore, J. Appl. Crystallogr., 2004, 37, 243–252 CrossRef CAS.
- J. Oddershede, K. Nielsen and K. Stahl, Z. fur Krist. – Cryst. Mater., 2007, 222, 186–192 CrossRef CAS.
- P. Guccione, L. Palin, M. Milanesio, B. D. Belviso and R. Caliandro, Phys. Chem. Chem. Phys., 2018, 20, 2175–2187 RSC.
- R. Tauler, B. Kowalski and S. Fleming, Anal. Chem., 1993, 65, 2040–2047 CrossRef CAS.
- A. de Juan, J. Jaumot and R. Tauler, Anal. Methods, 2014, 6, 4964–4976 RSC.
- S. J. Mazivila, R. A. E. Castro, J. M. M. Leitão and J. C. G. Esteves Da Silva, J. Pharm. Biomed. Anal., 2019, 169, 235–244 CrossRef CAS PubMed.
- S. J. Mazivila, R. A. E. Castro, J. M. M. Leitão and J. C. G. Esteves Da Silva, Vib. Spectrosc., 2020, 106, 102992 CrossRef CAS.
- A. Bjelopetrović, S. Lukin, I. Halasz, K. Užarević, I. Đilović, D. Barišić, A. Budimir, M. Juribašić Kulcsár and M. Ćurić, Chem. - Eur. J., 2018, 24, 10672–10682 CrossRef PubMed.
- M. A. Rodriguez, M. R. Keenan and G. Nagasubramanian, J. Appl. Crystallogr., 2007, 40, 1097–1104 CrossRef CAS.
- S. Ishihara, Y. Hattori and M. Otsuka, Spectrochim. Acta, Part A, 2019, 221, 117142 CrossRef CAS PubMed.
- A. A. L. Michalchuk, E. V. Boldyreva, A. M. Belenguer, F. Emmerling and V. V. Boldyrev, Front. Chem., 2021, 9, 685789 CrossRef CAS PubMed.
- G. Benecke, W. Wagermaier, C. Li, M. Schwartzkopf, G. Flucke, R. Hoerth, I. Zizak, M. Burghammer, E. Metwalli, P. Müller-Buschbaum, M. Trebbin, S. Förster, O. Paris, S. V. Roth and P. Fratzl, J. Appl. Crystallogr., 2014, 47, 1797–1803 CrossRef CAS PubMed.
- S. A. Stanton, J. J. Du, F. Lai, G. Stanton, B. A. Hawkins, J. A. Ong, P. W. Groundwater, J. A. Platts and D. E. Hibbs, J. Phys. Chem. A, 2021, 125, 9736–9756 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. a. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- J. L. Atwood, Encyclopedia of Supramolecular Chemistry - Two-Volume Set (Print), CRC Press, 2013 Search PubMed.
- R. Tauler, S. Lacorte and D. Barceló, J. Chromatogr. A, 1996, 730, 177–183 CrossRef CAS.
- W. Windig and J. Guilment, Anal. Chem., 1991, 63, 1425–1432 CrossRef CAS.
- Anaconda Software Distribution. Computer software. Vers. 2-2.4.0. Anaconda, Nov. 2016. Web, https://anaconda.com Search PubMed.
- A. Travert and C. Fernandez, SpectroChemPy, a framework for processing, analyzing and modeling spectroscopic data for chemistry with Python (version 0.4.10) Zenodo, 2023, DOI:10.5281/zenodo.3823841, URL: https://www.spectrochempy.fr.
- A. A. Coelho, J. Appl. Crystallogr., 2018, 51, 210–218 CrossRef CAS.
- A. Trask, W. Motherwell and W. Jones, Int. J. Pharm., 2006, 320, 114–123 CrossRef CAS PubMed.
- T. Ervasti, J. Aaltonen and J. Ketolainen, Int. J. Pharm., 2015, 486, 121–130 CrossRef CAS PubMed.
- A. Jayasankar, D. J. Good and N. Rodríguez-Hornedo, Mol. Pharm., 2007, 4, 360–372 CrossRef CAS PubMed.
- R. D. Davies, N. J. Vigilante, A. D. Frederick, V. S. Mandala and M. A. Mehta, J. Chem. Crystallogr., 2022, 52, 479–484 CrossRef CAS.
- Y. Ebisuzaki, P. D. Boyle and J. A. Smith, Acta Crystallogr. C, 1997, 53, 777–779 CrossRef.
- J. A. Goedkoop and C. H. MacGillavry, Acta Crystallogr., 1957, 10, 125–127 CrossRef CAS.
- M. A. Rodriguez, M. R. Keenan and G. Nagasubramanian, J. Appl. Crystallogr., 2007, 40, 1097–1104 CrossRef CAS.
- M. C. Antunes, J. E. J. Simão, A. C. Duarte and R. Tauler, Analyst, 2002, 127, 809–817 RSC.
- M. B. Mamián-López and R. J. Poppi, Anal. Chim. Acta, 2013, 760, 53–59 CrossRef PubMed.
- K. Ufer and M. D. Raven, Clays Clay Miner., 2017, 65, 286–297 CrossRef CAS.
- H. Zhu, C. Yuen and D. J. W. Grant, Int. J. Pharm., 1996, 135, 151–160 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Single crystal data, pure components resulting from the MCR-ALS calculations, additional data on Rietveld phase quantification, details of the XRD patterns from the in situ experiments. CCDC 2305331 and 2305332. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3mr00013c |
| This journal is © The Royal Society of Chemistry 2024 |