
Bi-directional charge transfer channels in highly crystalline carbon nitride enabling superior photocatalytic hydrogen evolution†
Runlu
Liu
a,
Siyuan
Liu
a,
Jingyi
Lin
a,
Xiaoxiao
Zhang
a,
Yao
Li
 a,
Hui
Pan
a,
Lingti
Kong
a,
Shenmin
Zhu
*a and
John
Wang
*b
a,
Hui
Pan
a,
Lingti
Kong
a,
Shenmin
Zhu
*a and
John
Wang
*b
aState Key Laboratory of Metal Matrix Composites, School of Materials Science and Engineering, Shanghai Jiao Tong University, Shanghai 200240, China. E-mail: smzhu@sjtu.edu.cn
bDepartment of Materials Science and Engineering, National University of Singapore, Singapore 117574, Singapore. E-mail: msewangj@nus.edu.sg
First published on 7th May 2024
Abstract
Introducing a donor–acceptor (D–A) unit is an effective approach to facilitate charge transfer in polymeric carbon nitride (PCN) and enhance photocatalytic performance. However, the introduction of hetero-molecules can lead to a decrease in crystallinity, limiting interlayer charge transfer and inhibiting further improvement. In this study, we constructed a novel D–A type carbon nitride with significantly higher crystallinity and a bi-directional charge transfer channel, which was achieved through 2,5-thiophenedicarboxylic acid (2,5-TDCA)-assisted self-assembly followed by KCl-templated calcination. The thiophene and cyano groups introduced serve as the electron donor and acceptor, respectively, enhancing in-plane electron delocalization. Additionally, introduced potassium ions are intercalated among the adjacent layers of carbon nitride, creating an interlayer charge transfer channel. Moreover, the highly ordered structure and improved crystallinity further facilitate charge transfer. As a result, the as-prepared photocatalyst exhibits superior photocatalytic hydrogen evolution (PHE) activity of 7.449 mmol h−1 g−1, which is 6.03 times higher than that of pure carbon nitride. The strategy of developing crystalline D–A-structured carbon nitride with controlled in-plane and interlayer charge transfer opens new avenues for the design of carbon nitride with enhanced properties for PHE.
Introduction
With the ever-rapid consumption of limited fossil fuels and consequent environmental problems, there is urgent need to explore alternative clean and renewable energy sources.1–3 Hydrogen, with its high energy density and zero greenhouse gas emissions, is considered the most promising secondary clean energy option.4,5 Semiconductor-based photocatalytic hydrogen evolution (PHE) is believed to be the most viable method for producing high purity hydrogen due to its low cost and scalability.6 Among the various photocatalysts investigated, polymeric carbon nitride (PCN) has garnered significant interest due to its easy synthesis, affordability, relatively high chemical stability and tunable band gap for visible light absorption.7 In 2009, Wang et al. first utilized PCN for PHE, highlighting the advantages of this low-cost, metal-free photocatalyst.8 However, pristine PCN suffers from high recombination rates of photo-generated electron–hole pairs, resulting in a relatively low PHE performance. To enhance the PHE performance of PCN, a spectrum of strategies has been adopted, including element doping,9–11 defect engineering,12–15 co-catalyst compositing,16–18 heterostructure construction,19–21etc.Among these various strategies, incorporating a donor–acceptor (D–A) structure into the PCN skeleton is a facile and effective approach. By introducing electron-donating groups (D) and electron-accepting groups (A), we can promote electron delocalization over the heptazine units of PCN, which can induce intramolecular charge transfer from D-units to A-units under photoexcitation accompanied by efficient electron–hole separation.22–24 Additionally, D-units and A-units can respectively lift up and pull down the HOMO and LUMO energy levels, narrowing the band gap of PCN and expanding the light absorption spectrum.25,26 Nevertheless, the incorporation of the D–A structure does not effectively strengthen the weak interlayer interaction of PCN, limiting charge transfer along the stacking direction. In other words, while the D–A structure improves in-plane charge transfer, it contributes little to interlayer charge transfer, which is critical for enhancing photocatalytic performance.27,28 Furthermore, introducing hetero-molecules can induce the distortion of the pristine crystalline structure and reduce the overall crystallinity of PCN, affecting the mobility and separation rates of photo-generated charge carriers.29,30 Therefore, it is of interest to explore strategies to prepare highly crystalline D–A-structured PCN with enhanced interlayer charge transfer.
Herein, we have designed and synthesized D–A-structured carbon nitride with K ion intercalation (TCCN-K) via the method of 2,5-thiophenedicarboxylic acid (2,5-TDCA)-assisted self-assembly followed by KCl-assisted calcination. On the one hand, self-assembly helps form a highly ordered precursor sustained by hydrogen bonding, thus improving in-plane order in carbon nitride production. Simultaneously, KCl-assisted calcination could accelerate the diffusion rates of reactants and promote the crystallization of heptazine units. On the other hand, the cyano groups induced by KCl-assisted calcination, along with the introduced thiophene groups, form the D–A structure, which promotes electron delocalization. Furthermore, K ions are intercalated into the adjacent layers of the carbon nitride during the calcination process and serve as interlayer electron transfer mediators. These three advantages, induced by a rationally designed synthesis approach, allow a bi-directional electron transfer pathway to be constructed, effectively facilitating charge transfer and separation. As a result, the as-prepared TCCN-K exhibits excellent PHE activity (7.449 mmol h−1 g−1) and service stability. This work details a novel method to synthesize crystalline D–A-structured carbon nitride with both excellent in-plane and interlayer charge transfer, providing a new perspective for designing and developing D–A-structured carbon nitride for PHE applications.
Results and discussion
The formation procedure of TCCN-K is illustrated in Scheme 1. As melamine, cyanuric acid and 2,5-TDCA are all soluble in DMSO and contain abundant H and O/N atoms, these three types of molecules can combine to produce a hydrogen-bonded MCT supramolecular precursor by a self-assembly method, as depicted in Fig. S1.† SEM images at lower magnification (Fig. S2†) reveal that the MCT precursor consists of microspheres with an average diameter of 3–5 μm. At higher magnification (Fig. 1a), it becomes evident that the microspheres are composed of 2D planes stacked via π–π interaction. Fig. S3† shows the XRD patterns and FT-IR spectrum of MCT. Three peaks at 10.6°, 18.4° and 21.6° correspond to the (100), (110) and (200) crystal planes, respectively, indicating a hexagonal lamellar structure. The intense peak at 27.7° arises from the graphite-like stacking of the 2D sheets.31 The FT-IR spectra reveal additional structural details of MCT. The peaks at 1737 and 1781 cm−1 correspond to the stretching vibration peaks of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond in cyanuric acid, while the peak at 769 cm−1 is ascribed to the vibration mode of the triazine ring in melamine. Notably, these three peaks are significantly shifted compared with those in pure cyanuric acid or melamine, indicating the formation of N–H⋯O and N–H⋯N hydrogen bonds between cyanuric acid and melamine.32 The above characterization results confirm the successful self-assembly synthesis of the MCT supramolecular precursor. Comparing it with MC, which is prepared by self-assembly of melamine and cyanuric acid, the FT-IR spectra of MCT exhibit two extra peaks. The signal at ca. 1250 cm−1 is assigned to the characteristic band of the C–O bond,33 while the signal at ca. 1500 cm−1 is attributed to the stretching vibration of the aromatic CC bond.34,35 These two peaks indicate the presence of thiophene molecules in the MCT precursor. Although MC shares a similar morphology and structure with MCT due to the comparable self-assembly process, as evidenced by SEM images, XRD patterns and FT-IR spectra, the catalysts derived from them exhibit distinct properties.
O bond in cyanuric acid, while the peak at 769 cm−1 is ascribed to the vibration mode of the triazine ring in melamine. Notably, these three peaks are significantly shifted compared with those in pure cyanuric acid or melamine, indicating the formation of N–H⋯O and N–H⋯N hydrogen bonds between cyanuric acid and melamine.32 The above characterization results confirm the successful self-assembly synthesis of the MCT supramolecular precursor. Comparing it with MC, which is prepared by self-assembly of melamine and cyanuric acid, the FT-IR spectra of MCT exhibit two extra peaks. The signal at ca. 1250 cm−1 is assigned to the characteristic band of the C–O bond,33 while the signal at ca. 1500 cm−1 is attributed to the stretching vibration of the aromatic CC bond.34,35 These two peaks indicate the presence of thiophene molecules in the MCT precursor. Although MC shares a similar morphology and structure with MCT due to the comparable self-assembly process, as evidenced by SEM images, XRD patterns and FT-IR spectra, the catalysts derived from them exhibit distinct properties.
 | ||
| Scheme 1 The synthesis process of TCCN-K. | ||
 | ||
| Fig. 1 SEM images of MCT (a) and TCCN-K (b). TEM image (c) and the corresponding elemental mapping (d: C, e: N, f: K, g: S) of TCCN-K. | ||
From SEM (Fig. 1b) and TEM (Fig. 1c) images, TCCN-K exhibits a bulk structure constructed by stacking 2D layers, which results from the confinement of KCl crystals, and the corresponding EDS elemental mapping (Fig. 1d–g) shows that the C, N, K and S are rather uniformly distributed within the material. The other three samples display distinct morphology features, as seen in Fig. S2.† TCCN (obtained by removing K ions from TCCN-K) closely resembled the latter in its morphology due to the similar synthesis method. However, DCN (prepared by calcinating an MC precursor without KCl) presents a totally different morphology, namely a hollow microsphere with many large voids, which is formed due to the preferable decomposition of the MC precursor in the center of microsphere.36 KTCCN (prepared by calcinating the mixture of melamine, cyanuric acid and KCl) exhibits a stacking bulk structure, but the size is larger and the surface appears coarser compared to that of TCCN-K. The various morphologies of these samples correlate with their widely different BET specific surface areas. As shown in Fig. S4,† TCCN-K has a specific surface area of 6.472 m2 g−1, which is lower than that of DCN (60.835 m2 g−1), TCCN (9.758 m2 g−1) and KTCCN (26.535 m2 g−1). Interestingly, this result suggests that the specific surface area alone does not account for the enhancement of photocatalytic performance.
The TDCA/KCl-assisted self-assembly synthesis not only affects the morphology but also influences the phase structure of the samples, as observed from the XRD patterns (Fig. 2a). DCN exhibits two characteristic peaks at 13.4° and 27.4°, assigned to the (100) plane of tri-s-triazine units and (002) interplanar packing of the graphitic units.37 TCCN-K, TCCN and KTCCN show three peaks located at ca. 8°, 10° and 28.2°, corresponding to the (110), (010) and (001) peaks of poly(heptazine imide), respectively.38 And the diffraction peaks of these three samples possess smaller full width at half-maximum (FWHM) as compared to DCN, indicating improved crystallinity. The change of XRD patterns indicates that the KCl-assisted calcination could lead to the re-arrangements of the heptazine units. The (001) peak of TCCN slightly shifts to a higher angle when compared with TCCN-K and KTCCN, indicating a smaller interlayer distance. The difference could be due to the varying degrees of intercalation. Since K intercalation could enlarge the interlayer distance and TCCN has lower potassium content than TCCN-K, it is suggested that the potassium ions are intercalated between the layers. Notably, KTCCN exhibits several extra peaks at 15.1°, 23.1°, 32.4° and 35.5°, attributed to the diffraction peaks of the poly(triazine imide) phase.39,40 These peaks are absent in the XRD patterns of TCCN and TCCN-K, possibly because DMSO-assisted self-assembly helped form an ordered structure. To validate this assumption, the crystallinities of the samples are calculated using the formula Xc = Ic/(Ic + Ia), where Ic and Ia refer to the integral intensity of crystalline peaks and amorphous peaks in XRD patterns, respectively. As shown in Fig. S5 and Table S1,† the crystallinities of the samples are 33.6%, 46.2%, 43.8% and 39.0%. These results indicate that the TDCA/KCl-assisted self-assembly synthesis likely promoted the ordered arrangement of heptazine units and enhanced the crystallinity of carbon nitride.41,42
 | ||
| Fig. 2 XRD patterns (a) and FT-IR spectra (b) of DCN, TCCN-K, TCCN and KTCCN; (c) solid-state 13C MAS NMR spectra and the corresponding structure of TCCN-K and DCN. | ||
The molecular structures of the as-prepared samples are characterized using Fourier transform infrared (FT-IR) spectroscopy (Fig. 2b). All samples exhibited three bands at ca. 810, 1200–1700 and 3000–3500 cm−1, corresponding to breathing vibrations of the heptazine units, stretching vibrations of the C–N aromatic heterocycles and stretching vibration modes of the N–H/O–H groups, respectively.43,44 Compared with DCN, the other three samples show several additional peaks. Signals at ca. 1000 and 1150 cm−1 are assigned to the symmetric and asymmetric vibrations of the K–NC2 groups, respectively; a weak signal at ca. 1500 cm−1 is assigned to the stretching vibration of the aromatic CC bond in the thiophene ring;34,35 the peak at ca. 2180 cm−1 corresponds to the terminal cyano groups formed in the presence of KCl.41,45 There are no signals of K–C3, C–KC or N–KN, indicating that K ions will not substitute C or N atoms in heptazine heterocycles. The solid-state 13C NMR spectrum further confirms the molecular structure of TCCN-K. As shown in Fig. 2c, DCN exhibits two peaks at 164.4 and 156.5 ppm, attributed to C1 atoms of the NC–N(NHx) bond and C2 atoms of the NC–(N)2 bond, respectively.46,47 For TCCN-K, the two main peaks slightly shift to 163.5 and 157.1 ppm, and several additional peaks emerge. The two peaks at 118.7 and 170.8 ppm correspond to C4 atoms of the N–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond and the neighboring C3 atoms, respectively.43,48 Besides, the peaks from 130 to 139 ppm are attributed to the carbon atoms in thiophene groups.23 The above results of the solid-state NMR indicate the expected structure tuning effect of the KCl/TDCA-assisted synthesis and confirm the introduction of cyano and thiophene groups.
N bond and the neighboring C3 atoms, respectively.43,48 Besides, the peaks from 130 to 139 ppm are attributed to the carbon atoms in thiophene groups.23 The above results of the solid-state NMR indicate the expected structure tuning effect of the KCl/TDCA-assisted synthesis and confirm the introduction of cyano and thiophene groups.
X-ray photoelectron spectroscopy (XPS) analysis provides valuable insights into the chemical composition and structure of the catalysts. In C 1s spectra (Fig. 3a), DCN exhibits two peaks at binding energies of 284.8 eV and 288.1 eV, assigned to C–C/CC bonds and N–CN bonds, respectively.49 In the case of TCCN-K, the N–CN peak shifts to 288.3 eV, and a new peak at 286.65 eV emerges, corresponding to the binding energy of –CN.13,49 In the C 1s spectrum of TCCN, the binding energy of the N–CN and –CN bonds are 288.45 eV and 286.55 eV, respectively. As for KTCCN, the binding energy of the N–CN and –CN bonds are 288.35 eV and 286.75 eV, respectively. It is noted that the binding energies of TCCN-K, TCCN and KTCCN are higher than those of DCN, indicating lower electron intensity, attributed to the electron-withdrawing effect of the cyano groups. In N 1s spectra (Fig. 3b), the peaks of the samples with D–A structure are also shifted. DCN shows three peaks at 398.9 eV, 400.3 eV and 401.4 eV, attributed to the C–NC, N–(C)3 and C–NHx bonds, respectively.50,51 For TCCN-K, the peaks negatively shift to 398.75 eV, 400.25 eV and 401.4 eV, respectively. For TCCN, the peaks shift to 398.7 eV, 400.15 eV and 401.0 eV, respectively. In the case of KTCCN, the binding energies are 398.8 eV, 400.25 eV and 401.3 eV, respectively. The negatively shifted peaks indicate higher electron intensity of N atoms. In S 2p spectra (Fig. 3c), TCCN-K, TCCN and KTCCN exhibit 2 peaks located at 164.9 and 163.9 eV, distributed to S 2p1/2 and S 2p3/2, respectively. The two peaks show an intensity ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, indicating the introduction of thiophene groups.52 In K 2p spectra (Fig. 3d), TCCN-K, TCCN and KTCCN exhibit 2 peaks at 295.8 and 293.05 eV, close to those of KCl, indicating the existence of K+ ions coordinated with negatively charged N atoms.53,54 The peak intensity of TCCN is obviously weaker than that of TCCN-K, indicating a lower K content. Furthermore, the in-depth XPS was conducted to measure the concentration of K ions at different depth. According to Fig. S6 and Table S2,† the concentration of K ions ranges from 11.28% to 12.42% at depths of 0 to 40 nm, indicating a uniform distribution throughout the bulk. Based on the above characterization studies, we can confirm the formation of D–A units and uniform K ion intercalation.
:2, indicating the introduction of thiophene groups.52 In K 2p spectra (Fig. 3d), TCCN-K, TCCN and KTCCN exhibit 2 peaks at 295.8 and 293.05 eV, close to those of KCl, indicating the existence of K+ ions coordinated with negatively charged N atoms.53,54 The peak intensity of TCCN is obviously weaker than that of TCCN-K, indicating a lower K content. Furthermore, the in-depth XPS was conducted to measure the concentration of K ions at different depth. According to Fig. S6 and Table S2,† the concentration of K ions ranges from 11.28% to 12.42% at depths of 0 to 40 nm, indicating a uniform distribution throughout the bulk. Based on the above characterization studies, we can confirm the formation of D–A units and uniform K ion intercalation.
 | ||
| Fig. 3 High-resolution XPS spectra of C 1s (a) and N 1s (b) of DCN, TCCN-K, TCCN and KTCCN; high-resolution XPS spectra of S 2p (c) and K 2p (d) of TCCN-K, TCCN and KTCCN. | ||
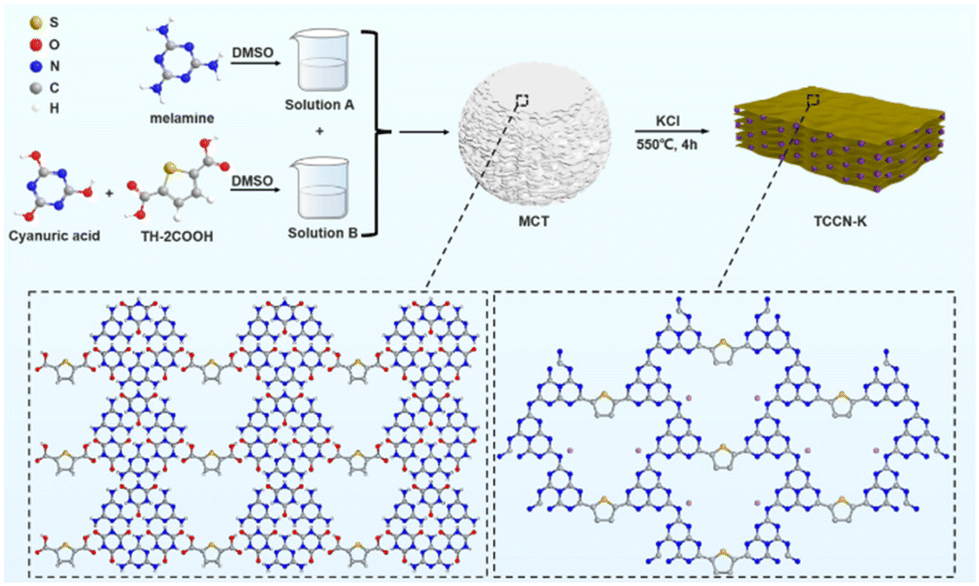
UV-vis diffuse reflectance spectroscopy was conducted to investigate light absorption properties of the samples (Fig. 4a). Compared with DCN, the other three samples exhibit red-shifted absorption edges and enhanced light absorption in the long-wavelength visible light region, which is due to the extended conjugated structure resulting from the introduction of the thiophene and cyano groups, as well as the enhancement of crystallinity.55,56 As shown by the Tauc plots in Fig. 4b, the band gaps of DCN, TCCN-K, TCCN and KTCCN are 2.88, 2.64, 2.72 and 2.76 eV, respectively. The valence band positions measured by XPS-VB spectra are 2.40, 2.47, 2.45 and 2.50 eV, respectively (Fig. S7†). Following the equation EVB–NHE = φ + EVB–XPS − 4.44, (EVB–NHE: VB value in the standard hydrogen electrode potential; φ: the electron work function of the XPS analyzer; EVB–XPS: VB value tested by VB–XPS plots),57,58 the EVB–NHE values of DCN, TCCN-K, TCCN and KTCCN are calculated to be 1.84, 1.91, 1.89 and 1.94 eV, respectively. Therefore, the CB potentials are calculated to be −1.04, −0.73, −0.83 and −0.82 eV, respectively. Consequently, the band structures of the samples can be illustrated as shown in Fig. 4c. Among the 4 samples, the TCCN-K exhibits the narrowest band gap and broadest light absorption range. However, the down-shifted conduction band thermodynamically decreases the reduction ability. Therefore, the adjustment of the energy band structure is not the main reason for the enhancement of PHE activity.
 | ||
| Fig. 4 UV-vis spectra (a), the corresponding Tauc plots derived from the Kubelka–Munk rule (b) and experimental energy band structure diagram (c) of all samples; (d) EPR spectra of DCN, TCCN and TCCN-K under dark (D) and light (L) conditions. | ||
The introduction of the D–A structure would lead to the delocalization of electrons, which is confirmed by an electron paramagnetic resonance (EPR) test. As shown in Fig. 4d, all the samples exhibit one single Lorentzian line with a g value of 2.0034, ascribed to the unpaired electrons in π-conjugated heptazine rings.59,60 Among the four samples, TCCN-K shows the highest EPR signal under dark conditions, indicating the highest concentration of unpaired electrons, which implies that the D–A structure and K ion intercalation extend the delocalization of π-electrons. Under visible-light irradiation, TCCN-K also exhibits the highest signal intensity, indicating the largest number of photo-induced electrons and effective separation of electron–hole pairs.
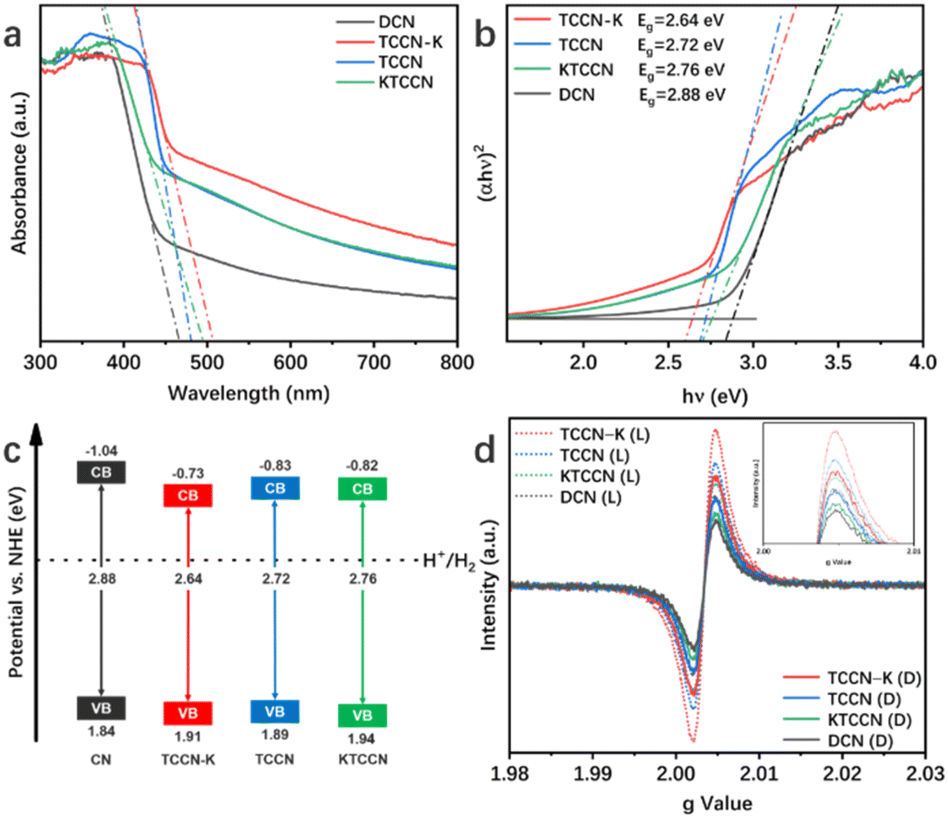
Photocatalytic hydrogen evolution (PHE) performance of the samples was tested under visible light irradiation (λ > 420 nm) with Pt as co-catalyst and TEOA as sacrificial agent. As shown in Fig. 5a, TCCN-K exhibits a PHE rate of 7.449 mmol h−1 g−1 in 6 hours of testing, which is 6.03 times higher than that of DCN (1.236 mmol h−1 g−1) and 2.62 times higher than that of TCCN (2.841 mmol h−1 g−1), respectively. These results suggest that the D–A structure and K ion intercalation can effectively improve the photocatalytic activity of PCN. It is noted that TCCNK has a relatively poor PHE rate performance of 1.251 mmol h−1 g−1, which is close to that of DCN, indicating that the self-assembly synthesis method is critical in enhancing the photocatalytic activity by enhancing the crystallinity to facilitate the charge transfer. Besides, as displayed in Fig. S8,† although DCN displays the lowest H2 generation rate among the four samples, it is still 6.28 times higher than that of bulk carbon nitride (BCN) prepared by traditional thermal calcination, owing to the rich active sites and multiple occurrences of light scattering caused by a large specific surface area and hollow spherical morphology. Nevertheless, the low crystallinity, large band gap and lack of charge transfer channel limit the photocatalytic performance. Fig. 5b shows the AQE values of PHE for TCCN-K under certain wavelengths (420, 435, 450 and 475 nm), which exhibit a similar changing trend with light absorption and reach up to 4.91% at 420 nm. The cycling test was conducted to evaluate the stability of the TCCN-K sample (Fig. 5c). After 4 cycles and reactions for 24 hours in total, the PHE rate was maintained at 7.015 mmol h−1 g−1, which is 94.2% of that in the first cycle. After the cycling test, the sample was collected by centrifugation and washed by deionized water and ethanol several times. TEM, XRD, FT-IR and XPS were then utilized to investigate the structural stability of the sample (Fig. S9 and S10†). It is seen that the morphology, phase components and chemical structure were well maintained after the cycling of photocatalytic reactions. The decrease of photocatalytic activity could result from the consumption of sacrificial agent and the mass loss of the photocatalyst in the degas and reaction progress. From the results of the cycling test and characterization studies, the excellent structural and chemical stabilities of the TCCN-K sample are confirmed.
 | ||
| Fig. 5 The average photocatalytic hydrogen evolution activity of the samples in the 6 hours of testing (a); wavelength-dependent AQE of PHE over TCCN-K (b); and time courses of all samples for photocatalytic hydrogen evolution and the cycling stability test of TCCN-K for 24 h (c). | ||
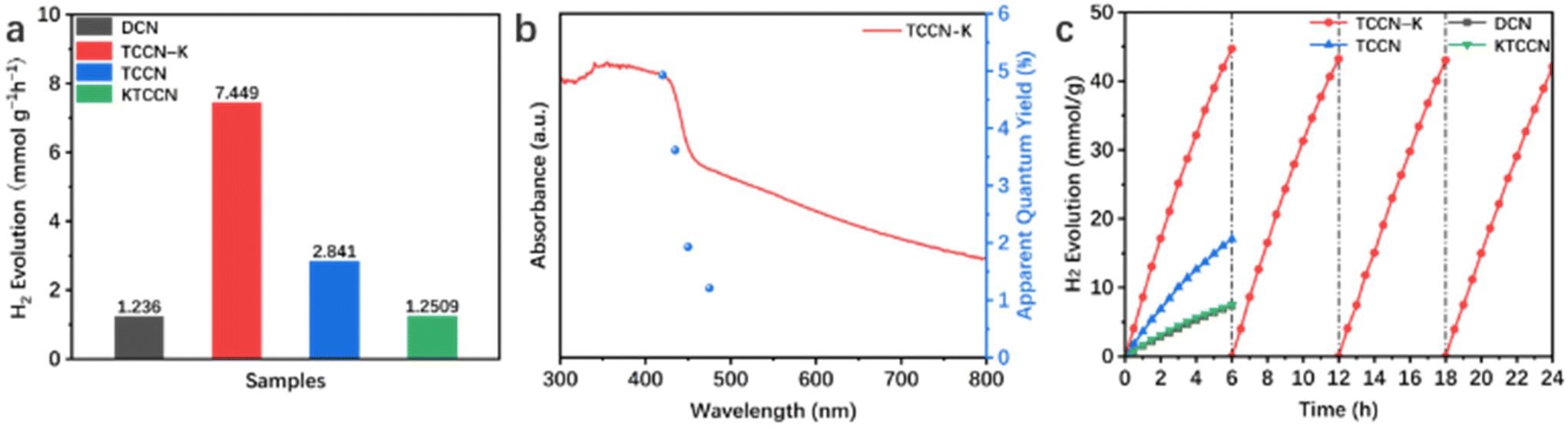
Electrochemical impedance spectra (EIS) and transient photocurrent analysis were employed to describe the charge separation and transfer capability of the samples. Fig. 6a displays the Nyquist plots of the samples and corresponding equivalent circuit. R1, R2 and CPE represent electrolyte resistance, electrode/electrolyte interface resistance and constant phase element, respectively. According to the fitting results, TCCN-K has a smaller interfacial charge transfer resistance (663.4 Ω) than DCN (963.3 Ω), KTCCN (721.1 Ω) and TCCN (692.6 Ω), consistent with the smallest “arc radius”. Fig. 6b shows the photocurrent responses of the samples. The TCCN-K also exhibits the highest photocurrent intensity, suggesting the fastest charge transfer, which is consistent with the results of EIS. Photoluminescence (PL) spectra and time-resolved photoluminescence (TRPL) spectra were employed to analyze the electron–hole recombination behavior. As shown in Fig. 6c, DCN displays one single strong emission peak, indicating the severe recombination of electron–hole pairs. In comparison, TCCN-K, TCCN and KTCCN each exhibits an extra peak at ∼500 nm, which arises from the introduction of thiophene groups.61 The quenched intrinsic emission peak suggests that the recombination of electron–hole pairs is inhibited, indicating the effective separation of electron–hole pairs induced by the presence of a D–A structure. Specifically, TCCN-K exhibits a further reduced emission peak intensity compared with TCCN, implying that K ion intercalation plays a significant role in facilitating charge transfer. Fig. 6d exhibits the TRPL spectra and average lifetime of photo-generated charge carriers obtained by double-exponential fitting. TCCN-K and TCCN display much shorter lifetimes (2.30 and 2.50 ns) than that of DCN (13.89 ns), which reflects the rapid charge carrier transfer to the surface through nonradiative pathways due to the introduction of a donor–acceptor structure and K intercalation as well as the enhanced crystallinity, thus facilitating the photocatalytic redox reaction.41,49,62 Notably, despite the D–A structure and K ion intercalation, KTCCN exhibits a stronger PL emission peak and longer lifetime (7.86 ns) than TCCN-K. This further proves the importance of a self-assembly synthesis method. With the assistance of DMSO, melamine, cyanuric acid and 2,5-TDCA can assemble into highly ordered precursor MCT driven by hydrogen bonding, so that the TCCN-K sample upon calcination can keep its high crystallinity and structure completeness after K ion intercalation, which is advantageous for charge carrier separation and transfer.63 In contrast, KTCCN synthesized by direct calcination suffers from disordered structure and low crystallinity, thus exhibiting poor charge transfer capability.
 | ||
| Fig. 6 EIS (a), transient photocurrent response (b), and PL (c) and TRPL spectra (d) of the samples. | ||
To investigate the contribution of K ions to the enhancement of charge transfer, Hall effect testing of TCCN-K and TCCN was conducted, and the interlayer electrical conductivity of the two samples was measured. As shown in Table 1, TCCN-K exhibits lower resistivity and higher carrier concentration along the in-plane direction, but the Hall mobility is nearly the same as that of TCCN, which indicates that the K ion intercalation does not contribute significantly to the in-plane charge transfer, and the excellent PHE performance of TCCN-K should be explained by some other factor. The interlayer electrical conductivity and resistivity of TCCN-K and TCCN were measured using the four-point probe method. As shown in Fig. S11,† TCCN-K has an obviously lower resistivity (0.189 MΩ cm) and higher conductivity (5.28 μS cm−1) than TCCN (3.33 MΩ cm and 0.30 μS cm−1, respectively). These results suggest that K ion intercalation greatly facilitates the charge transfer between carbon nitride layers, which is the main factor of the enhancement of the PHE performance. Apart from establishing an interlayer charge transfer channel, K ion intercalation also helps with the adsorption of H2O molecules. The static contact angles of a water droplet on the samples were measured. As shown in Fig. S12,† TCCN-K has the smallest contact angle (35.1°) among the three samples, while TCCN also shows a smaller contact angle than DCN (41.4° vs. 51.2°). It confirms that K ion intercalation endows the TCCN-K sample with better hydrophilicity, which is advantageous for the adsorption of reactant, thus promoting the photocatalytic performance.
| Sample | Resistivity (Ω cm) | Hall mobility (cm2 V−1 S−1) | Carrier concentration (cm−3) |
|---|---|---|---|
| TCCN | 835.1 | 5.857 | 1.278 × 1015 |
| TCCN-K | 627.8 | 5.871 | 1.696 × 1015 |
DFT calculations were carried out to confirm the proposed bi-directional charge transfer model. For in-plane charge transfer, the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) of DCN, TCCN and TCCN-K were calculated and are shown in Fig. 7a–f. Apparently, both the HOMO and LUMO of DCN are uniformly delocalized across the heptazine rings, in accordance with the highly symmetric structure. In comparison, TCCN and TCCN-K exhibit an unsymmetric charge density due to the existence of D–A units and K ions. Such electron delocalization could cause the downshift of VB and CB, which is consistent with the observations from the UV-vis and VB-XPS spectra.64 More importantly, the spatial separation of HOMO and LUMO enables in-plane charge separation and inhibits recombination, which is critical for the enhancement of PHE performance. For interlayer charge transfer, the work function and electrostatic potential along the direction perpendicular to the interlayer planes are two important indicators, as they correspond to the energy barrier required for a charge carrier to transfer to the surface and that between the adjacent layers, respectively.65,66 As shown in Fig. 7g and h, TCCN possesses an electrostatic potential of 18.86 eV and a work function of 5.48 eV, while TCCN-K possesses an electrostatic potential of 18.31 eV and a work function of 4.02 eV. The reduced energy barrier and work function of TCCN-K indicate that the interlayer transfer of charge carriers is facilitated. The charge density difference of TCCN-K is further calculated to verify the effect of K ion intercalation. As is seen from Fig. 7i, charge depletion takes place around the intercalated K ions, while charge enrichment is observed nearby the CN layers. This indicated that strong electrostatic interaction exists between the intercalated K ions and the adjacent CN layers, effectively facilitating the interlayer charge transfer and accelerating the separation of photo-generated charge carriers.27,67 Therefore, a bi-directional charge transfer channel is established by a D–A structure and K ion intercalation, as depicted in Scheme 2.
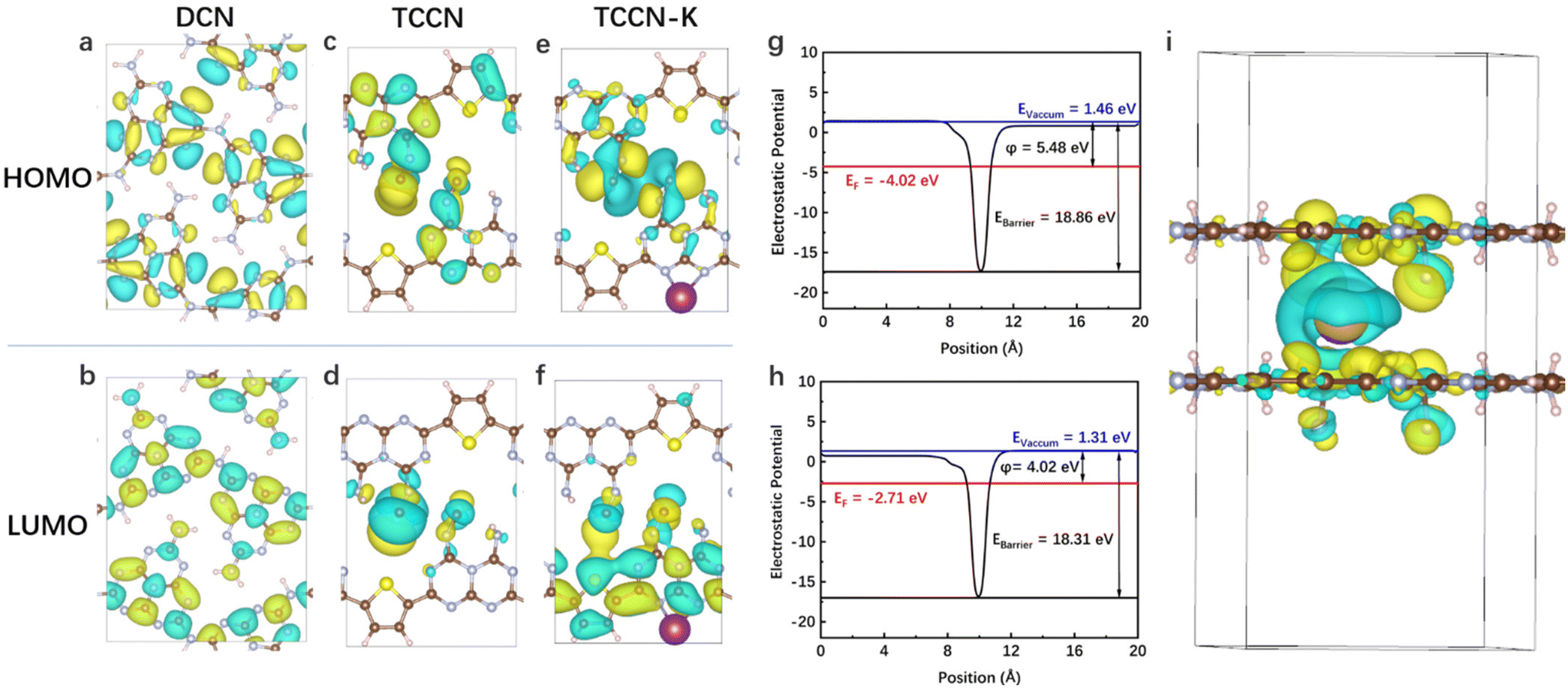 | ||
| Fig. 7 HOMO and LUMO distribution of DCN (a and b), TCCN (c and d) and TCCN-K (e and f); calculated work function and layered electrostatic potential energy of TCCN (g) and TCCN-K (h); and charge density difference of TCCN-K (i). | ||
 | ||
| Scheme 2 Bi-directional charge transfer of TCCN-K. | ||
Conclusions
In summary, a novel D–A type carbon nitride with simultaneous high crystallinity and efficient bi-directional charge transfer channels has been successfully constructed through the method of 2,5-TDCA-assisted self-assembly followed by KCl-templated calcination. The resultant donor–acceptor structure enhances the in-plane electron delocalization, while potassium ions from the KCl template intercalate between the adjacent layers, forming interlayer charge transfer channels. The bi-directional charge transfer pathways, combined with the highly ordered structure and improved crystallinity, significantly facilitate charge transfer. As a result, the as-prepared TCCN-K exhibits over six-fold improvement in PHE activity compared with pure CN. This strategy provides a new perspective for the design and construction of highly crystalline D–A-structured carbon nitride with superior performance in PHE.Author contributions
Runlu Liu: investigation, methodology, formal analysis, visualization, validation, and writing – original draft; Siyuan Liu: investigation; Jingyi Lin: resources; Xiaoxiao Zhang: validation; Yao Li: supervision; Hui Pan: supervision; Lingti Kong: software, visualization and data curation; Shenmin Zhu: supervision, conceptualization, writing – review and editing, project administration and funding acquisition; and John Wang: supervision, conceptualization, and writing – review and editing.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors kindly acknowledge the financial support from the National Natural Science Foundation of China (51672173 and U1733130); the Shanghai Science and Technology Committee (21ZR1435700, 18520744700 and 18JC1410500); and the Shanghai Jiao Tong University Medical Engineering Cross Research Program (YG2023ZD18).References
- M. Yue, H. Lambert, E. Pahon, R. Roche, S. Jemei and D. Hissel, Renewable Sustainable Energy Rev., 2021, 146, 111180 CrossRef
.
- Z. Y. Yu, Y. Duan, X. Y. Feng, X. Yu, M. R. Gao and S. H. Yu, Adv. Mater., 2021, 33, 2007100 CrossRef CAS PubMed
- N. Armaroli and V. Balzani, Chemistry, 2016, 22, 32–57 CrossRef CAS PubMed
- N. Fajrina and M. Tahir, Int. J. Hydrogen Energy, 2019, 44, 540–577 CrossRef CAS
- T. Jafari, E. Moharreri, A. S. Amin, R. Miao, W. Song and S. L. Suib, Molecules, 2016, 21, 900 CrossRef PubMed
- C. Acar, I. Dincer and G. F. Naterer, Int. J. Energy Res., 2016, 40, 1449–1473 CrossRef CAS
- R. Liu, Z. Chen, Y. Yao, Y. Li, W. A. Cheema, D. Wang and S. Zhu, RSC Adv., 2020, 10, 29408–29418 RSC
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed
- W. Jiang, Y. Zhao, X. Zong, H. Nie, L. Niu, L. An, D. Qu, X. Wang, Z. Kang and Z. Sun, Angew. Chem., Int. Ed., 2021, 60, 6124–6129 CrossRef CAS PubMed
- J. Li, D. Wu, J. Iocozzia, H. Du, X. Liu, Y. Yuan, W. Zhou, Z. Li, Z. Xue and Z. Lin, Angew. Chem., Int. Ed., 2019, 58, 1985–1989 CrossRef CAS PubMed
- W. Wang, L. Du, R. Xia, R. Liang, T. Zhou, H. K. Lee, Z. Yan, H. Luo, C. Shang, D. L. Phillips and Z. Guo, Energy Environ. Sci., 2023, 16, 460–472 RSC
- M. Liu, C. Wei, H. Zhuzhang, J. Zhou, Z. Pan, W. Lin, Z. Yu, G. Zhang and X. Wang, Angew. Chem., Int. Ed., 2022, 61, e202113389 CrossRef CAS PubMed
- Z. Tong, L. Huang, H. Liu, W. Lei, H. Zhang, S. Zhang and Q. Jia, Adv. Funct. Mater., 2021, 31, 2010455 CrossRef CAS
- A. Kumar, P. Raizada, A. Hosseini-Bandegharaei, V. K. Thakur, V.-H. Nguyen and P. Singh, J. Mater. Chem. A, 2021, 9, 111–153 RSC
- Y. Liu, M. Tayyab, W. Pei, L. Zhou, J. Lei, L. Wang, Y. Liu and J. Zhang, Small, 2023, 19, 2208117 CrossRef CAS PubMed
- M. Gao, F. Tian, X. Zhang, Z. Chen, W. Yang and Y. Yu, Nanomicro Lett., 2023, 15, 129 CAS
- H. Zhao, Z. Jiang, K. Xiao, H. Sun, H. S. Chan, T. H. Tsang, S. Yang and P. K. Wong, Appl. Catal., B, 2021, 280, 119456 CrossRef CAS
- M. Qian, X. L. Wu, M. Lu, L. Huang, W. Li, H. Lin, J. Chen, S. Wang and X. Duan, Adv. Funct. Mater., 2023, 33, 2208688 CrossRef CAS
- Z. Zhang, L. Ren, H. Li, D. Jiang, Y. Fang, H. Du, G. Xu, C. Zhu, H. Li, Z. Lu and Y. Yuan, Small, 2023, 19, 2207173 CrossRef CAS PubMed
- A. Deng, Y. Sun, Z. Gao, S. Yang, Y. Liu, H. He, J. Zhang, S. Liu, H. Sun and S. Wang, Nano Energy, 2023, 108, 108228 CrossRef CAS
- R. Shen, L. Zhang, N. Li, Z. Lou, T. Ma, P. Zhang, Y. Li and X. Li, ACS Catal., 2022, 12, 9994–10003 CrossRef CAS
- Y. Hu, W. Huang, H. Wang, Q. He, Y. Zhou, P. Yang, Y. Li and Y. Li, Angew. Chem., Int. Ed., 2020, 59, 14378–14382 CrossRef CAS PubMed
- Z. Sun, Y. Jiang, L. Zeng and L. Huang, ChemSusChem, 2019, 12, 1325–1333 CrossRef CAS PubMed
- Z. Sun, Y. Tan, J. Wan and L. Huang, Chin. J. Chem., 2021, 39, 2044–2053 CrossRef CAS
- K. Li and W. D. Zhang, Small, 2018, 14, 1703599 CrossRef PubMed
- R. S. Ashraf, I. Meager, M. Nikolka, M. Kirkus, M. Planells, B. C. Schroeder, S. Holliday, M. Hurhangee, C. B. Nielsen, H. Sirringhaus and I. McCulloch, J. Am. Chem. Soc., 2015, 137, 1314–1321 CrossRef CAS PubMed
- X. Xiao, Y. Gao, L. Zhang, J. Zhang, Q. Zhang, Q. Li, H. Bao, J. Zhou, S. Miao, N. Chen, J. Wang, B. Jiang, C. Tian and H. Fu, Adv. Mater., 2020, 32, 2003082 CrossRef CAS PubMed
- Y. Y. Li, Y. Si, B. X. Zhou, T. Huang, W. Q. Huang, W. Hu, A. Pan, X. Fan and G. F. Huang, Nanoscale, 2020, 12, 3135–3145 RSC
- H. Yu, H. Ma, X. Wu, X. Wang, J. Fan and J. Yu, Sol. RRL, 2020, 5, 2000372 CrossRef
- G. Huang, B. Xiao, L. Bao, D. Wang, Y. Luo, S. Yan and H. Gao, J. Mater. Chem. A, 2024, 12, 3480–3488 RSC
- Y.-S. Jun, E. Z. Lee, X. Wang, W. H. Hong, G. D. Stucky and A. Thomas, Adv. Funct. Mater., 2013, 23, 3661–3667 CrossRef CAS
- G. Arrachart, C. Carcel, P. Trens, J. J. Moreau and M. Wong Chi Man, Chemistry, 2009, 15, 6279–6288 CrossRef CAS PubMed
- G. Wang, Y. Liang, M. Jiang, Q. Zhang, R. Wang, H. Wang and G. Zhou, Polym. Degrad. Stab., 2019, 168, 108942 CrossRef CAS
- J. Shi, M. Tai, J. Hou, Y. Qiao, C. Liu, T. Zhou, L. Wang and B. Hu, Chem. Eng. J., 2023, 456, 141029 CrossRef CAS
- X. Fan, L. Zhang, M. Wang, W. Huang, Y. Zhou, M. Li, R. Cheng and J. Shi, Appl. Catal., B, 2016, 182, 68–73 CrossRef CAS
- W. Yu, T. Zhang and Z. Zhao, Appl. Catal., B, 2020, 278, 118778 CrossRef
- J.-Y. Tang, R.-T. Guo, W.-G. Zhou, C.-Y. Huang and W.-G. Pan, Appl. Catal., B, 2018, 237, 802–810 CrossRef CAS
- J. Kröger, A. Jiménez-Solano, G. Savasci, P. Rovó, I. Moudrakovski, K. Küster, H. Schlomberg, H. A. Vignolo–González, V. Duppel, L. Grunenberg, C. B. Dayan, M. Sitti, F. Podjaski, C. Ochsenfeld and B. V. Lotsch, Adv. Energy Mater., 2020, 11, 2003016 CrossRef
- J. Yang, Y. Liang, K. Li, G. Yang, K. Wang, R. Xu and X. Xie, Appl. Catal., B, 2020, 262, 118252 CrossRef CAS
- J. Zhang, C. Yu, J. Lang, Y. Zhou, B. Zhou, Y. H. Hu and M. Long, Appl. Catal., B, 2020, 277, 119225 CrossRef CAS
- F. Guo, B. Hu, C. Yang, J. Zhang, Y. Hou and X. Wang, Adv. Mater., 2021, 33, 2101466 CrossRef CAS PubMed
- X. Wu, H. Ma, W. Zhong, J. Fan and H. Yu, Appl. Catal., B, 2020, 271, 118899 CrossRef CAS
- W. Wang, H. Zhang, S. Zhang, Y. Liu, G. Wang, C. Sun and H. Zhao, Angew. Chem., Int. Ed., 2019, 58, 16644–16650 CrossRef CAS PubMed
- J. Huang, D. Li, R. Li, Q. Zhang, T. Chen, H. Liu, Y. Liu, W. Lv and G. Liu, Chem. Eng. J., 2019, 374, 242–253 CrossRef CAS
- J. Cheng, Y. Hou, K. Lian, H. Xiao, S. Lin and X. Wang, ACS Catal., 2022, 12, 1797–1808 CrossRef CAS
- L. Lin, H. Ou, Y. Zhang and X. Wang, ACS Catal., 2016, 6, 3921–3931 CrossRef CAS
- G. Zhang, C. Huang and X. Wang, Small, 2015, 11, 1215–1221 CrossRef CAS PubMed
- H. Yu, R. Shi, Y. Zhao, T. Bian, Y. Zhao, C. Zhou, G. I. N. Waterhouse, L. Z. Wu, C. H. Tung and T. Zhang, Adv. Mater., 2017, 29, 1605148 CrossRef PubMed
- L. Chen, C. Chen, Z. Yang, S. Li, C. Chu and B. Chen, Adv. Funct. Mater., 2021, 31, 2105731 CrossRef CAS
- Y. Huang, J. Liu, C. Zhao, X. Jia, M. Ma, Y. Qian, C. Yang, K. Liu, F. Tan, Z. Wang, X. Li, S. Qu and Z. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 52603–52614 CrossRef CAS PubMed
- Z. Pan, M. Zhao, H. Zhuzhang, G. Zhang, M. Anpo and X. Wang, ACS Catal., 2021, 11, 13463–13471 CrossRef CAS
- J. Gong, Z. Xie, B. Wang, Z. Li, Y. Zhu, J. Xue and Z. Le, J. Environ. Chem. Eng., 2021, 9, 104638 CrossRef CAS
- F. Guo, S. Li, Y. Hou, J. Xu, S. Lin and X. Wang, Chem. Commun., 2019, 55, 11259–11262 RSC
- R. Liu, J. Lin, L. Zhu, X. Zhang, Y. Li, H. Pan, L. Kong, S. Zhu and J. Wang, Chem. Eng. J., 2024, 484, 149507 CrossRef CAS
- G. Zhang, Y. Xu, D. Yan, C. He, Y. Li, X. Ren, P. Zhang and H. Mi, ACS Catal., 2021, 11, 6995–7005 CrossRef CAS
- N. Wang, L. Cheng, Y. Liao and Q. Xiang, Small, 2023, 19, 2300109 CrossRef CAS PubMed
- Z. Sun, W. Wang, Q. Chen, Y. Pu, H. He, W. Zhuang, J. He and L. Huang, J. Mater. Chem. A, 2020, 8, 3160–3167 RSC
- J. Yuan, X. Liu, Y. Tang, Y. Zeng, L. Wang, S. Zhang, T. Cai, Y. Liu, S. Luo, Y. Pei and C. Liu, Appl. Catal., B, 2018, 237, 24–31 CrossRef CAS
- Z. Qin, Z. Huang, M. Wang, D. Liu, Y. Chen and L. Guo, Appl. Catal., B, 2020, 261, 118211 CrossRef CAS
- B. Wu, L. Zhang, B. Jiang, Q. Li, C. Tian, Y. Xie, W. Li and H. Fu, Angew. Chem., Int. Ed., 2021, 60, 4815–4822 CrossRef CAS PubMed
- J. Zhang, M. Zhang, S. Lin, X. Fu and X. Wang, J. Catal., 2014, 310, 24–30 CrossRef CAS
- J. Wang, H. Zhao, B. Zhu, S. Larter, S. Cao, J. Yu, M. G. Kibria and J. Hu, ACS Catal., 2021, 11, 12170–12178 CrossRef CAS
- B. Zhao, D. Gao, Y. Liu, J. Fan and H. Yu, J. Colloid Interface Sci., 2022, 608, 1268–1277 CrossRef CAS PubMed
- D. Zhao, C. L. Dong, B. Wang, C. Chen, Y. C. Huang, Z. Diao, S. Li, L. Guo and S. Shen, Adv. Mater., 2019, 31, 1903545 CrossRef CAS PubMed
- Y. Yang, S. Wang, Y. Jiao, Z. Wang, M. Xiao, A. Du, Y. Li, J. Wang and L. Wang, Adv. Funct. Mater., 2018, 28, 1805698 CrossRef
- Y. Kang, Y. Yang, L. C. Yin, X. Kang, L. Wang, G. Liu and H. M. Cheng, Adv. Mater., 2016, 28, 6471–6477 CrossRef CAS PubMed
- J. Li, Z. Zhang, W. Cui, H. Wang, W. Cen, G. Johnson, G. Jiang, S. Zhang and F. Dong, ACS Catal., 2018, 8, 8376–8385 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr00796d |
| This journal is © The Royal Society of Chemistry 2024 |