
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of alcohols: streamlined C1 to Cn hydroxyalkylation through photoredox catalysis†
Francesco
Pasca‡
a,
Yuri
Gelato‡
a,
Michael
Andresini
 a,
Giuseppe
Romanazzi
b,
Leonardo
Degennaro
a,
Marco
Colella
*a and
Renzo
Luisi
*a
a,
Giuseppe
Romanazzi
b,
Leonardo
Degennaro
a,
Marco
Colella
*a and
Renzo
Luisi
*a
aDepartment of Pharmacy-Drug Sciences, Flow Chemistry and Microreactor Technology FLAME-Lab University of Bari “A. Moro”, Via E. Orabona 4, 70125 Bari, Italy. E-mail: marco.colella@uniba.it; renzo.luisi@uniba.it
bDICATECh Politecnico di Bari, Via E. Orabona 4, Bari 70125, Italy
First published on 13th June 2024
Abstract
Naturally occurring and readily available α-hydroxy carboxylic acids (AHAs) are utilized as platforms for visible light-mediated oxidative CO2-extrusion furnishing α-hydroxy radicals proved to be versatile C1 to Cn hydroxyalkylating agents. The direct decarboxylative Giese reaction (DDGR) is operationally simple, not requiring activator or sacrificial oxidants, and enables the synthesis of a diverse range of hydroxylated products, introducing connectivity typically precluded from conventional polar domains. Notably, the methodology has been extended to widely used glycolic acid resulting in a highly efficient and unprecedented C1 hydroxyhomologation tactic. The use of flow technology further facilitates scalability and adds green credentials to this synthetic methodology.
Introduction
The introduction of a single hydroxyl group in a molecular structure can dramatically influence the drug–receptor binding affinity through the creation of an extensive hydrogen bond network.1 Approximately 37% of marketed drugs contain at least one hydroxyl group in their structure (Fig. 1A). Moreover, the –OH group can serve as a handle for further derivatizations. Within the polar domain, direct hydroxyalkylations could be achieved through two different synthetic pathways. In a less explored approach introduced by Tamao, α-silyl carbanions are employed as α-hydroxyalkyl anion equivalents in formal nucleophilic α-hydroxyalkylation reactions (Fig. 1B, path a).2 Moreover, this strategy represents an indirect hydroxyalkylation approach that requires the oxidative cleavage of the carbon–silicon bond at the end of the synthetic sequence. More commonly, hydroxyalkyations are generally achieved through the addition of organolithiums or Grignard reagents to carbonyl compounds (Fig. 1B, path b), and recently the limited functional group tolerance of these highly reactive reagents has been overcome through the use of flow microreactor technology.3 Nevertheless, the direct one-step preparation of γ-hydroxy substituted derivatives is retrosynthetically precluded from the polar mechanisms due to a polarity mismatch (Fig. 1B, path c). In this context, the renaissance of radical chemistry showcased that approaches to C–C bond formation, apart from polar approaches, can offer novel pathways for accessing valuable structures in a more direct manner.4–6 With regard to radical hydroxyalkylations, Leonori reported a hydroxymethylation strategy based on the addition of alkyl radicals generated by halogen-atom transfer (XAT) to formaldehyde from the corresponding iodides (Fig. 1C, path a).7 This approach requires the photochemical generation of an α-aminoalkyl radical that promotes halogen-atom transfer, and an excess of PPh3 as the trapping agent of the transient O-radical. Interestingly, open-shell chemistry also provides a mechanistically inverted strategy for hydroxyalkylations based on the coupling between ketyl radicals and unsaturated acceptors.8,9 In fact, ketyl radicals or ketyl radical anions, characterized by a nucleophilic carbon radical, provide a mechanism for reversing the reactivity of the initial carbonyl compound. This enables their utilization in alternative C–C bond-forming reactions with non-nucleophilic partners. These versatile open-shell species are usually generated from aldehydes or ketones through single electron transfer (SET) reduction, with strong reductants (e.g., Na, K, and Ti) to overcome the large reduction potential of carbonyls. In this context, Kagan's reagent (SmI2) stands out as one of the most frequently employed reagents.10 In recent years, the advent of photoredox catalysis has led to the application of novel photochemical approaches in addressing this thermodynamic challenge as shown by Knowles, Yoon, Rueping, Cozzi, and others.11–16 However, these methodologies require synergistic Lewis acid/photoredox catalysis and/or the use of Hantzsch ester as a sacrificial electron and hydrogen atom donor. Interestingly, an electro-photocatalytic carbonyl reduction has been recently proposed by Wickens and coworkers (Fig. 1C, path b).17 Furthermore, Nagib presented a redox-neutral generation of ketyl radicals through in situ conversion of aldehydes to α-acetoxy iodides followed by Mn-catalyzed atom transfer under visible light irradiation.8 A complementary method for accessing α-hydroxy radicals involves C–H abstraction from alcohols through visible light-promoted hydrogen atom transfer (HAT) (Fig. 1C, path c).18–20 These methodologies require a combination of photoredox and HAT catalysis in the presence of activators capable of overwhelming the bond dissociation energy (BDE) of α-hydroxy C sp3–H bonds.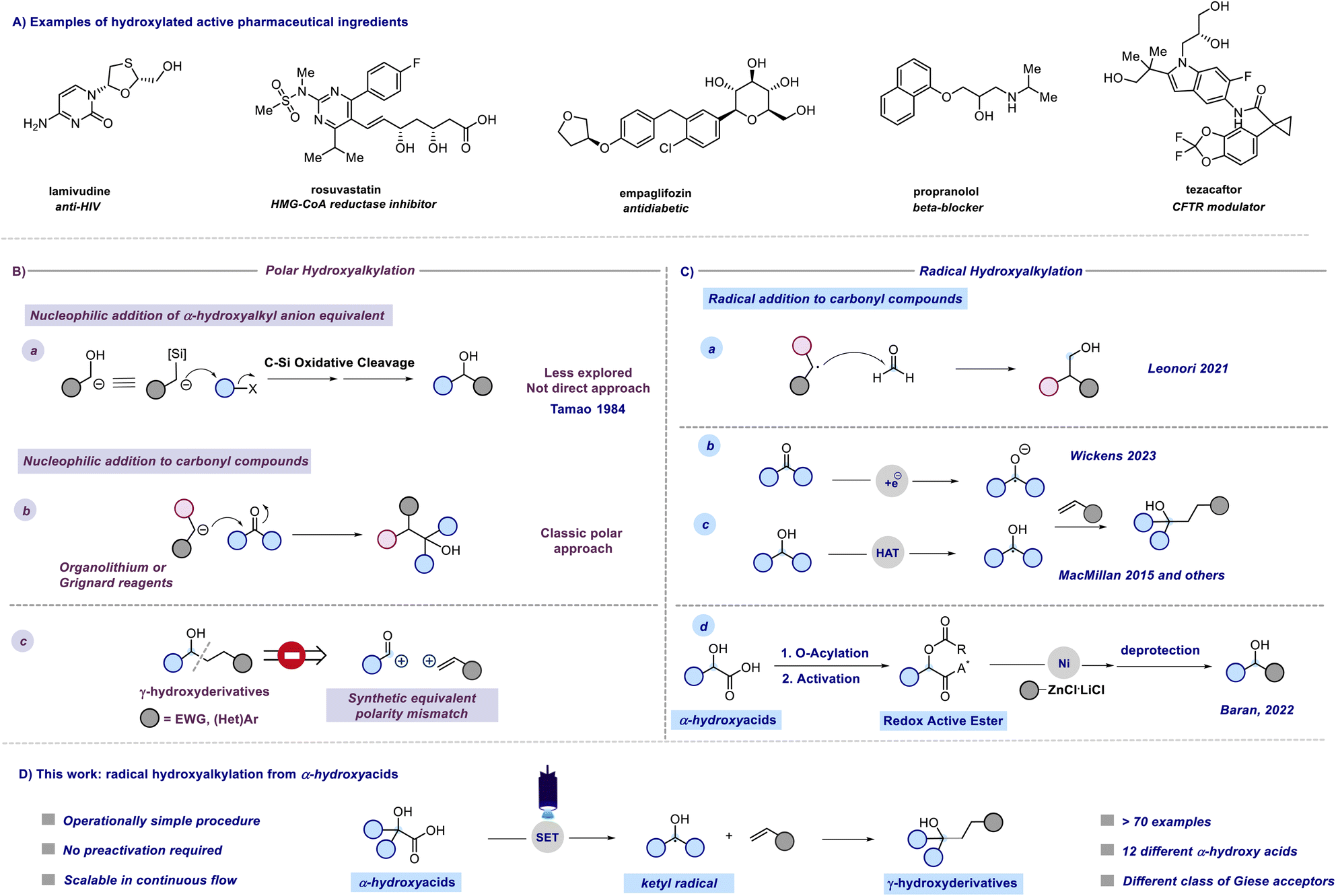 | ||
| Fig. 1 A) Examples of therapeutic agents containing hydroxyalkyl motifs. Overview of the main approaches for incorporating an α hydroxyalkyl motif in both polar (B) and radical (C) domains. (D) This work unlocks the utilisation of α-hydroxy acids as a synthetic platform for radical hydroxyalkylations. | ||
Despite extensive research on the chemistry of α-hydroxyalkyl radicals, we contemplate whether a more direct approach for accessing these versatile intermediates was feasible using readily available starting materials. In other words, we aimed to circumvent the need for complex synergistic catalytic systems or activators, aiming to achieve a more sustainable and easily accessible route to valuable molecules. Recently, Baran reported a radical-based approach for enantioselective decarboxylative Negishi coupling to obtain enantiopure dialkyl carbinols. The methodology relies on the conversion of α-hydroxy acids into redox-active esters (RAEs) using a one-pot O-protection and acid activation protocol (Fig. 1C, path d).21 Inspired by Baran's work and aimed at developing sustainable processes using readily available feedstocks we found α-hydroxy acids to be readily available linchpins for radical hydroxyalkylations.22 Indeed, α-hydroxy acids (AHAs) constitute a group of naturally occurring organic carboxylic acids, finding applications in various fields such as medicine, food processing, cosmetics, polymer synthesis, water treatment, and more.23
Glycolic acid (GA) is the simplest alpha hydroxy acid (AHA) and is naturally occurring.24,25 Lactic acid (LA), found in sour milk and tomato juice, holds the “Generally Recognized As Safe (GRAS)” status, endorsed as harmless by the United States Food and Drug Administration with a growing global demand.26–28 In this context, we questioned whether it would have been possible to leverage AHAs as naturally occurring and readily available reactants to deliver α-hydroxy radicals under visible light irradiation using a green and operationally simple procedure. We report herein a comprehensive study on the generation and use of hydroxyalkyl radicals via the direct decarboxylative Giese reaction (DDGR).29
Results and discussion
A previous attempt to use α-hydroxyacids as ketyl radical precursors was documented by Gonzalez-Gomez and coworkers in 2016. The authors reported a transition-metal-free method for the decarboxylative generation of radicals from carboxylic acids and subsequent coupling with Giese-type acceptors under visible-light irradiation.30 However, in this investigation, the oxidative decarboxylation of lactic acid (2-hydroxypropanoic acid) using 9-mesitylene-10-methylacridinium perchlorate ([Acr-Mes]ClO4) and Na2CO3, respectively as the photocatalyst and base, was reported to be unsuccessful. Despite the disappointing results documented in this study and, to the best of our knowledge, the lack of effective methods for generating hydroxyalkyl radicals from the corresponding hydroxy acids, we maintained confidence in the potential to develop an efficient hydroxyalkylation strategy in order to fill this gap. We started our study using 2-hydroxy-3-phenylpropanoic acid 1a and methyl acrylate as model substrates. Since the generation of ketyl radicals from α-hydroxy acids is unprecedented, we performed cyclic voltammetry investigations and computational studies to evaluate the feasibility of the synthetic plan shown in Fig. 2. The initial cyclic voltammetry (CV) assessment suggested that 2-hydroxy-3-phenylpropanoic acid 1a cannot be readily oxidized (E1/2 = +2.13 V vs. SCE). Conversely, the corresponding carboxylate 1a− exhibited an oxidation potential (Ered1/2 = +1.04 V vs. SCE) that aligns well with the redox window of various organic and metalloorganic photocatalysts.31 From a sustainable standpoint, we looked at an inexpensive and transition-metal-free organic photocatalyst such as 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene (4CzIPN).32 As outlined in Fig. 2, we envisioned an initial excitation of the photocatalyst 4CzIPN (A) to 4CzIPN* (A*) under visible light, followed by a reductive quenching of the excited 4CzIPN* (A*) ( versus SCE) and consequent oxidation of the carboxylate 1− triggering the extrusion of CO2, and formation of the desired α-hydroxy radical 2 and 4CzIPN˙− (A˙−). Addition of the nucleophilic α-hydroxy radical 2 to methyl acrylate would furnish alkyl radical 3. Single-electron reduction of this electron-deficient radical 3 operated by 4CzIPN˙− (A˙−) (
versus SCE) and consequent oxidation of the carboxylate 1− triggering the extrusion of CO2, and formation of the desired α-hydroxy radical 2 and 4CzIPN˙− (A˙−). Addition of the nucleophilic α-hydroxy radical 2 to methyl acrylate would furnish alkyl radical 3. Single-electron reduction of this electron-deficient radical 3 operated by 4CzIPN˙− (A˙−) (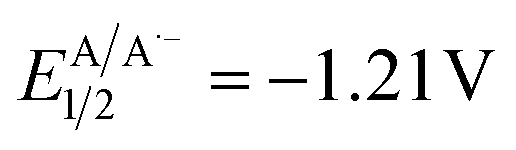 versus SCE) would then furnish the α-hydroxy alkylated product 4 after protonation regenerating the photocatalyst 4CzIPN (A). A chain propagation cycle could operate in addition to the proposed closed catalytic loop. Indeed, the carboxylate 1a−, which should be the most abundant species under basic conditions, could be oxidized by radical 3, thereby generating another ketyl radical 2 and the product 4 after decarboxylation and protonation, respectively. Nevertheless, this SET event should be unlikely due to a mismatch of potentials (vide infra). However, a chain mechanism involving 1a and radical 3 through hydrogen atom transfer (HAT) cannot be ruled out. To gain insight into the reactivity of α-hydroxyalkyl radicals and the feasibility of the proposed catalytic cycle, we conducted a preliminary computational study of the Giese addition reaction utilizing radical 2a as the model substrate computationally coupled to three different Giese acceptors in DMSO as the solvent (Fig. 2B). Initially, calculations were performed at the SMD(DMSO)-ωB97X-D3/def2-TZVP//ωB97X-D3/def2-SVP level of theory (at 298.15 K, 1 M).33–36 The results indicated that the addition reaction of radical 2a to methyl acrylate is moderately exergonic (ΔG° = −18.4 kcal mol−1) with a relatively low energy barrier (ΔG‡ = 10.3 kcal mol−1), making it highly feasible at room temperature (Fig. 2B). The calculation of reduction potentials performed at the SMD(DMSO)-M06-2X/ma-def2-TZVP//ωB97X-D3/def2-SVP level finally suggested that the reaction of 2a with methyl acrylate through the designed catalytic cycle is very likely to occur.37 In fact, the Giese adduct 3a arising from the attack of 2a on methyl acrylate exhibits a calculated reduction potential of −0.76 V versus SCE. Therefore, the single electron transfer process operated by 4CzIPN˙− (
versus SCE) would then furnish the α-hydroxy alkylated product 4 after protonation regenerating the photocatalyst 4CzIPN (A). A chain propagation cycle could operate in addition to the proposed closed catalytic loop. Indeed, the carboxylate 1a−, which should be the most abundant species under basic conditions, could be oxidized by radical 3, thereby generating another ketyl radical 2 and the product 4 after decarboxylation and protonation, respectively. Nevertheless, this SET event should be unlikely due to a mismatch of potentials (vide infra). However, a chain mechanism involving 1a and radical 3 through hydrogen atom transfer (HAT) cannot be ruled out. To gain insight into the reactivity of α-hydroxyalkyl radicals and the feasibility of the proposed catalytic cycle, we conducted a preliminary computational study of the Giese addition reaction utilizing radical 2a as the model substrate computationally coupled to three different Giese acceptors in DMSO as the solvent (Fig. 2B). Initially, calculations were performed at the SMD(DMSO)-ωB97X-D3/def2-TZVP//ωB97X-D3/def2-SVP level of theory (at 298.15 K, 1 M).33–36 The results indicated that the addition reaction of radical 2a to methyl acrylate is moderately exergonic (ΔG° = −18.4 kcal mol−1) with a relatively low energy barrier (ΔG‡ = 10.3 kcal mol−1), making it highly feasible at room temperature (Fig. 2B). The calculation of reduction potentials performed at the SMD(DMSO)-M06-2X/ma-def2-TZVP//ωB97X-D3/def2-SVP level finally suggested that the reaction of 2a with methyl acrylate through the designed catalytic cycle is very likely to occur.37 In fact, the Giese adduct 3a arising from the attack of 2a on methyl acrylate exhibits a calculated reduction potential of −0.76 V versus SCE. Therefore, the single electron transfer process operated by 4CzIPN˙− (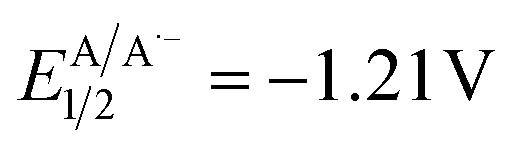 versus SCE) is expected to be thermodynamically favoured corresponding to a cell potential of +0.45 V (ΔG0 = −10.4 kcal mol−1, see the ESI†). In striking contrast, the calculated reduction potential of 2a of −1.93 V versus SCE allowed us to exclude the occurrence of the direct reduction of 2a operated by 4CzIPN˙− which is thermodynamically disfavoured corresponding to a cell potential of −0.72 V (ΔG0 = +16.6 kcal mol−1).
versus SCE) is expected to be thermodynamically favoured corresponding to a cell potential of +0.45 V (ΔG0 = −10.4 kcal mol−1, see the ESI†). In striking contrast, the calculated reduction potential of 2a of −1.93 V versus SCE allowed us to exclude the occurrence of the direct reduction of 2a operated by 4CzIPN˙− which is thermodynamically disfavoured corresponding to a cell potential of −0.72 V (ΔG0 = +16.6 kcal mol−1).
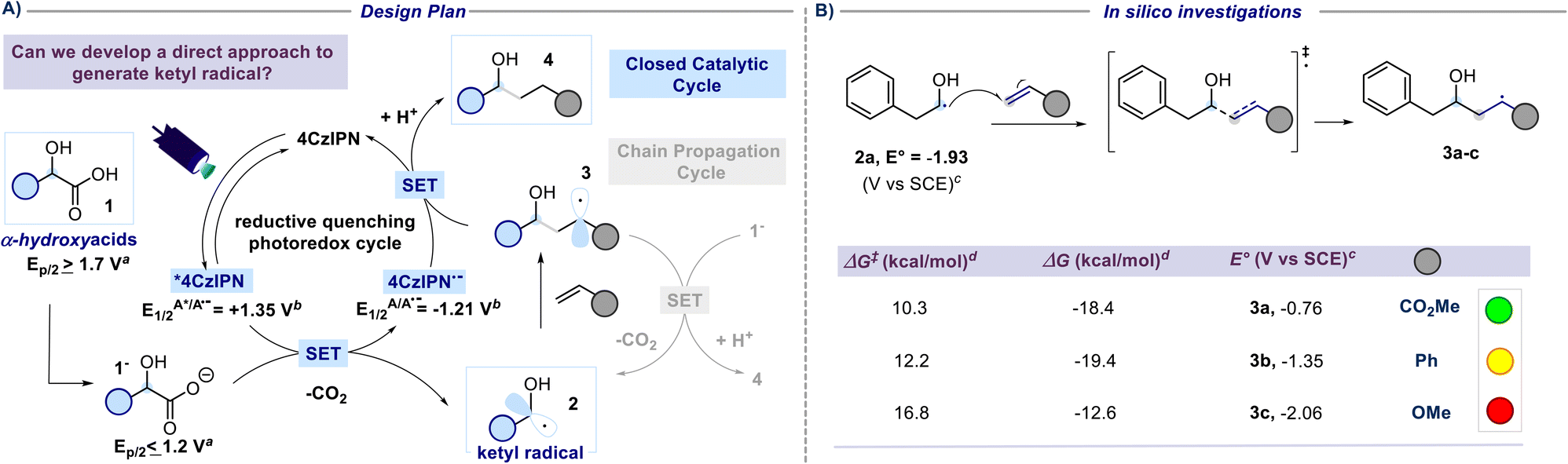 | ||
| Fig. 2 Reaction development. (A) Design plan for the direct generation of a ketyl radical from α-hydroxy acids. SET, single-electron transfer. aOxidation potentials measured by cyclic voltammetry in a 0.1 M solution of NBu4PF6 in MeCN at 25 °C with 100 mV s−1 scan rate and reported vs. SCE. Carboxylates generated in situ through the addition of a solution of NBu4OH. Here, the potentials at half the peak (Ep/2) are reported. See the ESI† for further experimental details. bLiterature values vs. SCE.38 (B) Computational studies on the addition of the ketyl radicals to unsaturated acceptors. cValues calculated at the SMD(DMSO)-M06-2X/ma-def2-TZVP//ωB97X-D3/def2-SVP level of theory (298.15 K, 1 M). dValues calculated at the SMD(DMSO)-ωB97X-D3/def2-TZVP//ωB97X-D3/def2-SVP level of theory (298.15 K, 1 M). | ||
Additionally, the reaction of 2a with other SOMOphiles such as styrene and methyl vinyl ether was also investigated in silico, and such reactions were similarly found to be exergonic (ΔG° = −19.4 and −12.6 kcal mol−1 respectively) but with slightly higher activation barriers (ΔG‡ = 12.2 and 16.8 kcal mol−1 respectively). However, the calculated reduction potential of the Giese adducts 3c (−2.06 V versus SCE) was found to mismatch the potential required for the closure of the catalytic cycle, suggesting that such electron-rich substrates might prove ineffective in performing the desired coupling reaction. Differently, the calculated reduction potential for adduct 3b (−1.35 V versus SCE) is only slightly different from the one of 4CzIPN˙− implying that the effectiveness of such a SOMOphile in participating in the catalytic reaction could not be completely ruled out. The nucleophilic character of the ketyl radicals reported in this study was proved by calculating the global electrophilicity index ω whose value was <1 eV (see the ESI† for details).39,40 Delighted by these computational results, and bearing in mind that hydroxylakylations were predicted to be successful on electron-poor acceptors, we began the optimization campaign by subjecting 2-hydroxy-3-phenylpropanoic acid 1a and methyl acrylate to blue light irradiation in the presence of 4CzIPN (2.5 mol%) and 2,6-lutidine, and using dimethyl sulfoxide as the reaction solvent. Disappointingly, the expected α-hydroxy alkylated product 4a was observed only in traces (Fig. 3). Similarly, the use of DABCO furnished very low yield of 4a, while switching to the inorganic base Na2HPO4 to promote the formation of the corresponding carboxylate 1a− returned 4a in 23% yield. An excellent result was observed using K3PO4 which provided 4a in 90% yield (Fig. 3, chart 1). Similar results were observed with the use of potassium tert-butoxide (88% yield). A substoichiometric amount of the base was detrimental for the yield. Particular attention was given to the reaction time. Indeed, extending the duration from 2 to 3 hours led to a significant increase in yield, achieving complete conversion of the starting reagents (Fig. 3, chart 5). Interestingly, when the duration was extended to 16 hours, intramolecular lactonization of 4a occurred, leading to the complete formation of lactone 5. Consequently, the developed transformation could result in two distinct products by merely adjusting the reaction time. The preliminary optimization study used a concentration of 0.31 M in DMSO that could be increased up to 0.77 M without compromising the efficiency of the reaction (Fig. 3, chart 4). Furthermore, increasing the light source power from 40 to 128 W did not adversely affect the reaction performance (refer to Fig. 3, chart 6). A solvent screening revealed that polar aprotic solvents, such as dimethylformamide (DMF) or dimethyl sulfoxide, yielded superior results, whereas the use of acetonitrile, 2-methyltetrahydrofuran and dichloromethane hindered the reaction (see Fig. 3, chart 3). However, DMSO was chosen as a preferred solvent for this process because, comparatively, it offers a less hazardous alternative to DMF.41 Evaluation of alternative photocatalysts commonly employed for decarboxylation reactions (see Fig. 3, chart 2) established 4CzIPN as the most effective catalyst.
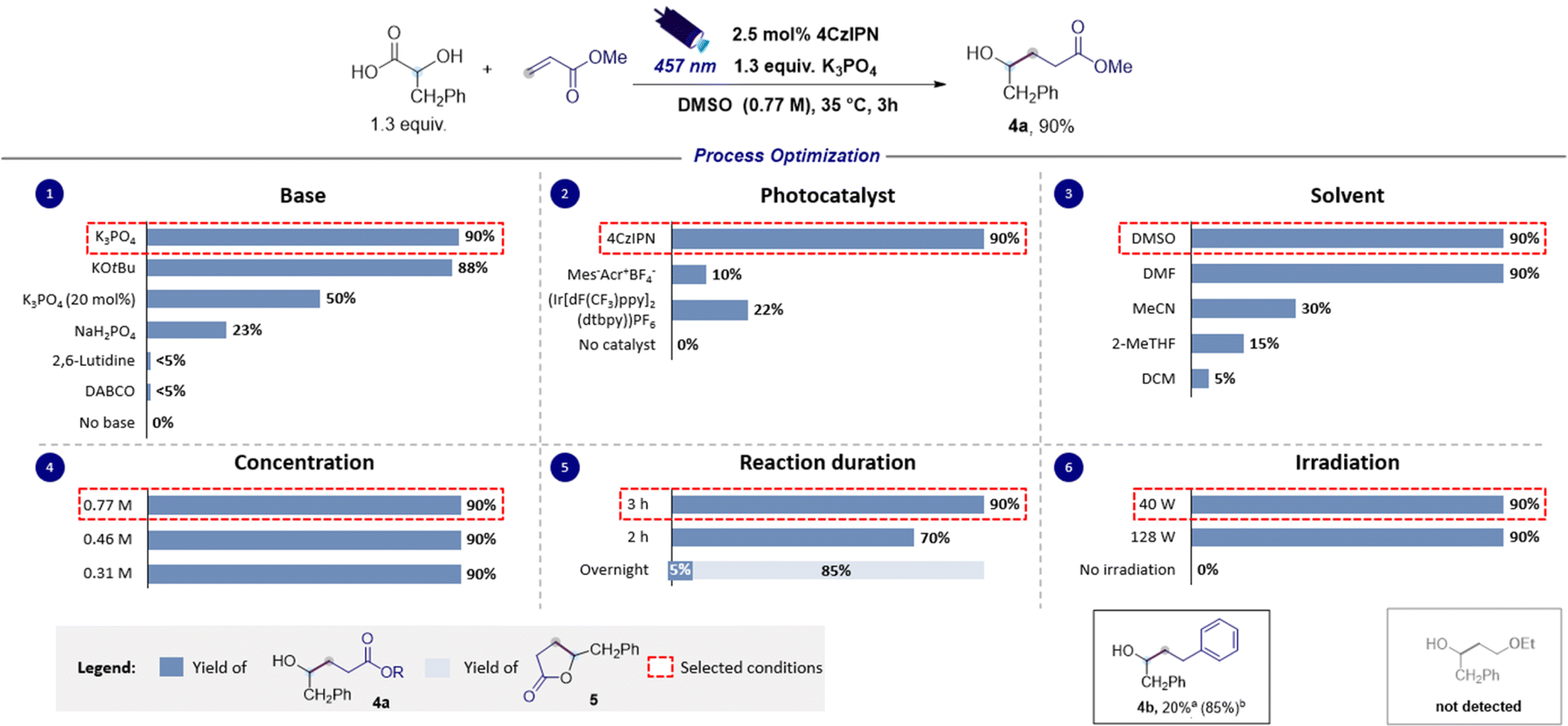 | ||
| Fig. 3 Optimization of radical hydroxyalkylation involving ketyl radicals. Yields were determined by proton NMR spectroscopy using dibromomethane as an internal standard. aReaction performed using 40 W and a reaction time of 16 hours. bReaction performed using 128 W and a reaction time of 16 hours. Abbreviations: Me, methyl; Ph, phenyl; equiv., equivalents; DABCO, 1,4-diazabicyclo[2.2.2]octane; 4CzIPN, 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene; ppy, 2-phenylpyridyl; dtbbpy, 4,4'-di-tert-butyl-2,2'-bipyridyl; DMSO, dimethyl sulfoxide; DMF, N,N′-dimethylformamide; 2-MeTHF, 2-methyltetrahydrofuran; DCM, dichloromethane; M, molarity; h, hours; W, watt. | ||
Under the optimized conditions, electron rich SOMOphiles such as vinylethyl ether and styrene were tested as reaction partners in the radical hydroxyalkylation. To our delight, and according to the computational results (Fig. 2), hydroxyalkylation occurred only with styrene leading to 4b while no reaction was observed with vinylethyl ether. This result further supports the nucleophilic nature of the ketyl radical 2a. Subsequently, armed with the optimal conditions, we explored the range of α-hydroxy carboxylic acids suitable for this radical addition protocol. Aiming to establish a robust and broadly applicable synthetic method, we tested 12 different α-hydroxy carboxylic acids (1a–l). Additionally, cyclic voltammetry measurements were conducted on each of the 12 distinct α-hydroxy acids (refer to the ESI†) to confirm their compatibility with the synthetic approach outlined in Fig. 2. As reported in Scheme 1, a range of structurally diverse α-hydroxy acids (1a–j) underwent efficient coupling with readily available acrylates furnishing products 4a and 6–22 in good to excellent yields (70–95%). Moreover, this decarboxylative protocol can be also applied to the addition of tertiary α-hydroxy radicals to acrylates leading to products 18–22 in excellent yields (75–95%). Interestingly, the presence of a substituent at the β position of the acrylate did not affect the reaction outcome (product 18b, 75%). Not surprisingly, a competing lactonization was observed with α-substituted acrylates after 3 hours of reaction time. However, prolonging the reaction duration to 16 hours provided lactone derivatives 9, 10, 17 and 22 as exclusive products in very good yields (70–93%). Next, varied Giese-type acceptors were evaluated in the radical hydroxyalkylation reaction. As reported in Scheme 1 optimized conditions could be successfully applied to acrylamide acceptors furnishing interesting and hard to make γ-hydroxylated amides 23–27 in good to excellent yields (70–95%). Remarkably, the unprotected indole core was fully compatible with the optimized protocol (Scheme 1, compound 26). Notably, electron-poor alkenes, such as 4-chlorostyrene and 2-vinylpyridine could also be adopted as coupling partners of ketyl radicals producing the corresponding hydroxyalkylated derivatives 28–32 in moderate to excellent yields (51–90%). Notably, the synthesis of hydroxylated adduct 29 can be scaled up to 1 mmol without compromising reaction efficiency. α,β-Unsaturated ketones, including carvone and a naproxen derivative, were found as competent SOMOphiles for this transformation leading to products 33–36 (85–91%). Radical hydroxyalkylation could be executed on heterosubstituted Giese-type acceptors. We were pleased to find that γ-hydroxy phosphonates 37–39 could be prepared in excellent yields (91–93%) through the addition of α-hydroxy radicals to diethyl vinylphosphonate (Scheme 1).42 Moreover, the optimized mild reaction conditions are compatible with the use of several sulfur-containing Giese-type acceptors, including vinyl sulfoxides (products 40 and 41, 79–91%), sulfoximines (products 43 and 45–47, 89–91%), and sulfones (products 42 and 44, 85–88%). Remarkably, the study of the scope of the reaction was extended to naturally occurring AHAs such as lactic acid (1d), mandelic acid (1c) and quinic acid (1k). To our delight, lactic acid smoothly reacted with acrylates giving the corresponding adducts 12-13 in excellent yields (87–91%). Similarly, mandelic acid reacted with methyl acrylate furnishing product 11 in 77% yield (Scheme 1). Remarkably, the cyclic polyol quinic acid, readily obtainable from coffee beans and other natural sources,43 was found to be effective in the radical hydroxylation of pentafluoro styrene producing the corresponding adduct 32 in 89% yield (Scheme 1). Products 15 and 29 were selected as representative examples for further manipulation of the synthesized hydroxylated derivatives (see the ESI† for further details). However, the protocol proved ineffective for certain Giese acceptors (see the ESI† for further details).
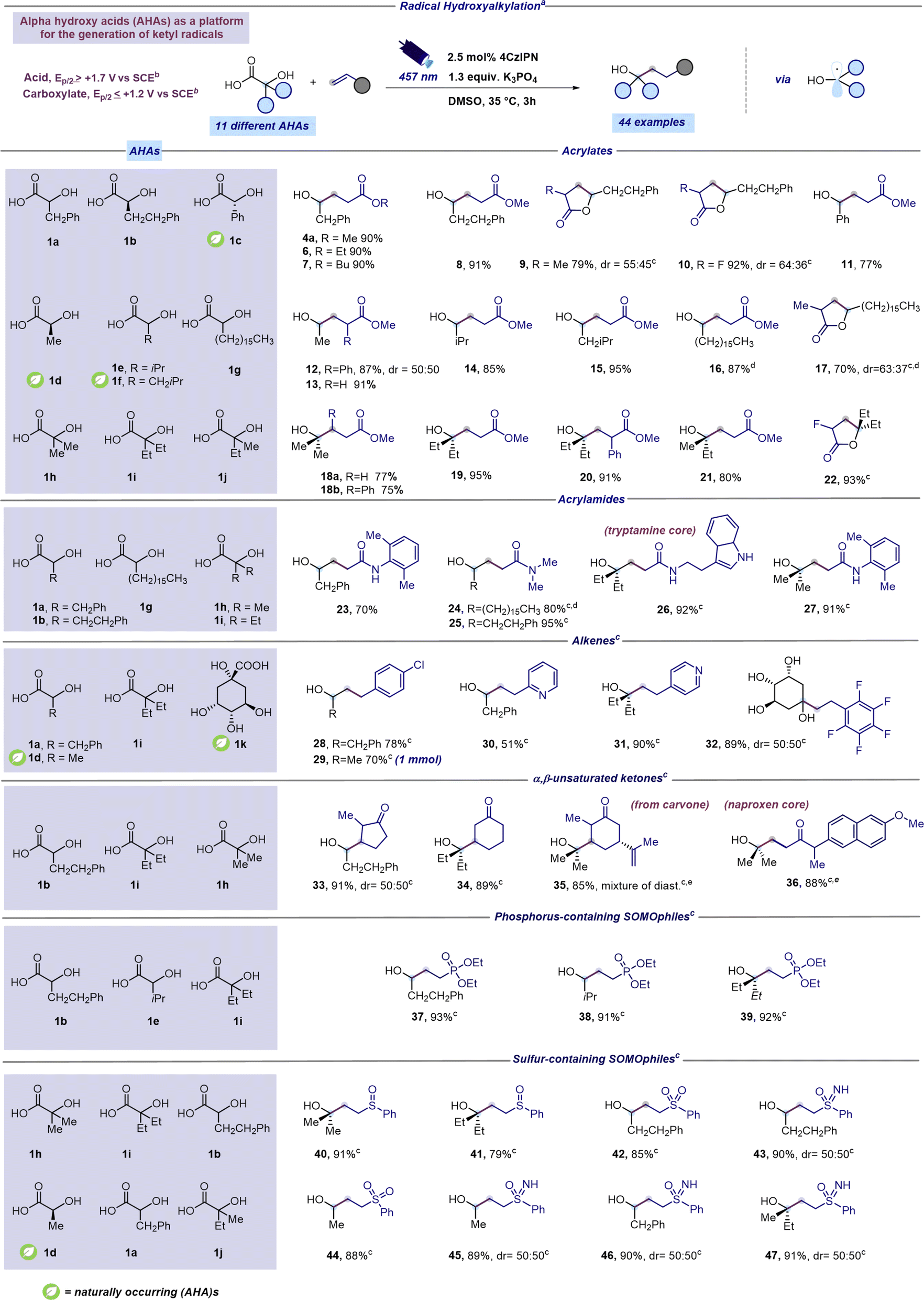 | ||
| Scheme 1 Scope for radical hydroxyalkylation from AHAs. aReaction performed under optimized reaction conditions: unsaturated acceptor (0.23 mmol, 1 equiv.), α-hydroxy acid (1.3 equiv.), K3PO4 (1.3 equiv.), and 4CzIPN (2.5 mol%) in DMSO (0.46 M) as the solvent. The mixture was irradiated with 457 nm light (40 W) for 3 h unless otherwise specified. Isolated yields are reported. bOxidation potentials measured by cyclic voltammetry. Refer to the ESI† for further details. cA reaction time of 16 hours was used instead of 3 hours. dReaction performed using a concentration of 0.15 M instead of 0.46 M. eReaction performed using 128 W instead of 40 W. Abbreviations: Ph, phenyl; Me, methyl; iPr, isopropyl; Et, ethyl. | ||
Encouraged by these findings and the broad applicability of the methodology, we pondered whether it would be feasible to achieve a more challenging and unprecedented C1 hydroxyalkylation using the readily available glycolic acid (GA). Indeed, GA stands out as the simplest alpha hydroxy acid and occurs naturally in plants such as sugarcane, pineapple, and sugar beets. While chemical synthesis continues to be the primary method for GA production, considerable efforts are being made to establish sustainable GA production processes. This positions GA as an economical and easily accessible chemical with significantly lower toxicity compared to an alternative radical C1 synthon, such as methanol.44 In order to satisfy our curiosity and introduce a new and elegant method for hydroxymethylation of organic scaffolds, we subjected GA (1j) and methyl acrylate to blue light irradiation under optimized reaction conditions. This preliminary test was executed utilizing d6-DMSO as the solvent for a direct NMR analysis of the reaction crude and to mitigate issues associated with the volatility and water solubility of the expected product 48. Astonishingly, 1H NMR of the reaction crude revealed the almost quantitative formation of the desired hydroxymethylated product 48 (Scheme 2). Encouraged by this result, we next sought to assess the generality of this new radical C1 hydroxyhomologation strategy testing various unsaturated acceptors. According to the previously reported α-hydroxy acids, high yields were obtained in C1-hydroxyhomologation using acrylates (products 49–56, 55–95% in Scheme 2). Remarkable chemoselectivity was observed in the presence of additional unsaturations, exemplified by the use of allyl acrylate furnishing 54 in 85% yield. Moreover, the C1-hydroxyhomologation of a menthol derivative led to 55 in 95% yield, while an azetidine-containing acrylate provided 56 in 93% yield. This hydroxy methyl radical could be also successfully coupled with N-aryl acrylamides resulting in compounds 57 and 58 formed in 55 and 70%, respectively. Notably, this approach facilitates the introduction of the hydroxymethyl fragment onto a derivative of the bioactive amantadine (product 59, 93% yield) and enables the efficient preparation of derivative 60, which contains the structural core of the antihistaminic bepotastine, in 91% yield. Lastly, C1-hydroxyhomologation with sulfur-containing SOMOphiles was tested. Specifically, we succeeded in the hydroxymethylation of two vinylic sulfones giving products 61 and 62 in 50 and 93% respectively, and a vinylic sulfoximine forming 63 in 38% yield.
 | ||
| Scheme 2 Scope of the radical hydroxymethylation protocol. aReaction performed under optimized reaction conditions: unsaturated acceptor (0.23 mmol, 1 equiv.), glycolic acid (1.3 equiv.), K3PO4 (1.3 equiv.), and 4CzIPN (2.5 mol%) in DMSO (0.15 M) as the solvent. The mixture was irradiated with 457 nm light (40 W) for 3 h unless otherwise specified. Isolated yields are reported. bOxidation potentials measured by cyclic voltammetry. Refer to the ESI† for further details. cValues calculated at the SMD(DMSO)-M06-2X/ma-def2-TZVP//ωB97X-D3/def2-SVP level of theory (298.15 K, 1 M). dA reaction time of 16 hours was used instead of 3 hours. eReaction performed using 128 W instead of 40 W. Abbreviations: Me, methyl; Et, ethyl; Bu, butyl; Hex, hexyl; Bn, benzyl; Ph, phenyl; Tf, triflyl. | ||
Next, the C1-hydroxyhomologation of electron-deficient alkenes was evaluated (Scheme 2). Vinyl pyridines smoothly reactswith the putative hydroxymethyl radical giving the corresponding adducts 64 and 65 in high yields (80 and 85% respectively). Similarly, the reaction with styrenes bearing electron withdrawing substituents produced 66 and 67 in 87% and 80% yield, respectively. Interestingly, the use of α-(trifluoromethyl) styrene returned gem-difluoroalkene 68 in 85% yield. Formation of 68 could be explained considering the catalytic cycle described in Fig. 2, where reduction of radical 3 is followed by a fluoride anion elimination which likely occurs faster than protonation (vide infra).45 Furthermore, a hydroxymethyl radical could be coupled with α,β-unsaturated ketones, including carvone and a naproxen derivative, producing compounds 69–72 in good to excellent yields (60–95%). However, during the investigation of the scope of the reaction we encountered in ineffective SOMOphiles (Scheme 2).
Mindful of the inherent challenges posed by batch photocatalytic methods at scale, our subsequent objective was to transition this hydroxyalkylation reaction to a continuous-flow platform.46–48 Based on our expertise in microfluidic technology,49–51 we identified some issues in the batch protocol that would hinder transitioning to a flow process. Firstly, the heterogeneous nature of the reaction mixture would result in non-uniform irradiation and limited light penetration when operating at larger scales. Consequently, we replaced K3PO4 with potassium tert-butoxide (KOtBu) to handle a homogeneous reaction mixture. Secondly, the light power needed to be increased to 128 W to achieve a more efficient irradiation. Adopting these adjustments, we set out to explore the feasibility of the radical hydroxyalkylation in continuous flow focusing on naturally occurring AHAs such as glycolic, lactic, and mandelic acids (Scheme 3). We were pleased to find that the continuous flow protocol not only yielded similar or higher yields compared to batch processing but also allowed for a significant reduction in reaction time (see the ESI† for further examples under continuous flow conditions). It is worth pointing out that the benzylic radical, generated from mandelic acid (1c), which was thought to be unreactive under photoredox conditions,52 was effectively employed using our SET protocol. Pleasingly, the continuous flow platform facilitates the efficient production of 29 at the 5 mmol scale in high yields. Furthermore, the assessment of productivity and STY (space time yield) for the preparation of 29 in flow (204 mg h−1 and 25.4 g L−1 h−1, respectively) and batch (1.9 mg h−1 and 1.7 g L−1 h−1, respectively) clearly demonstrated the advantage of the flow protocol compared to batch.
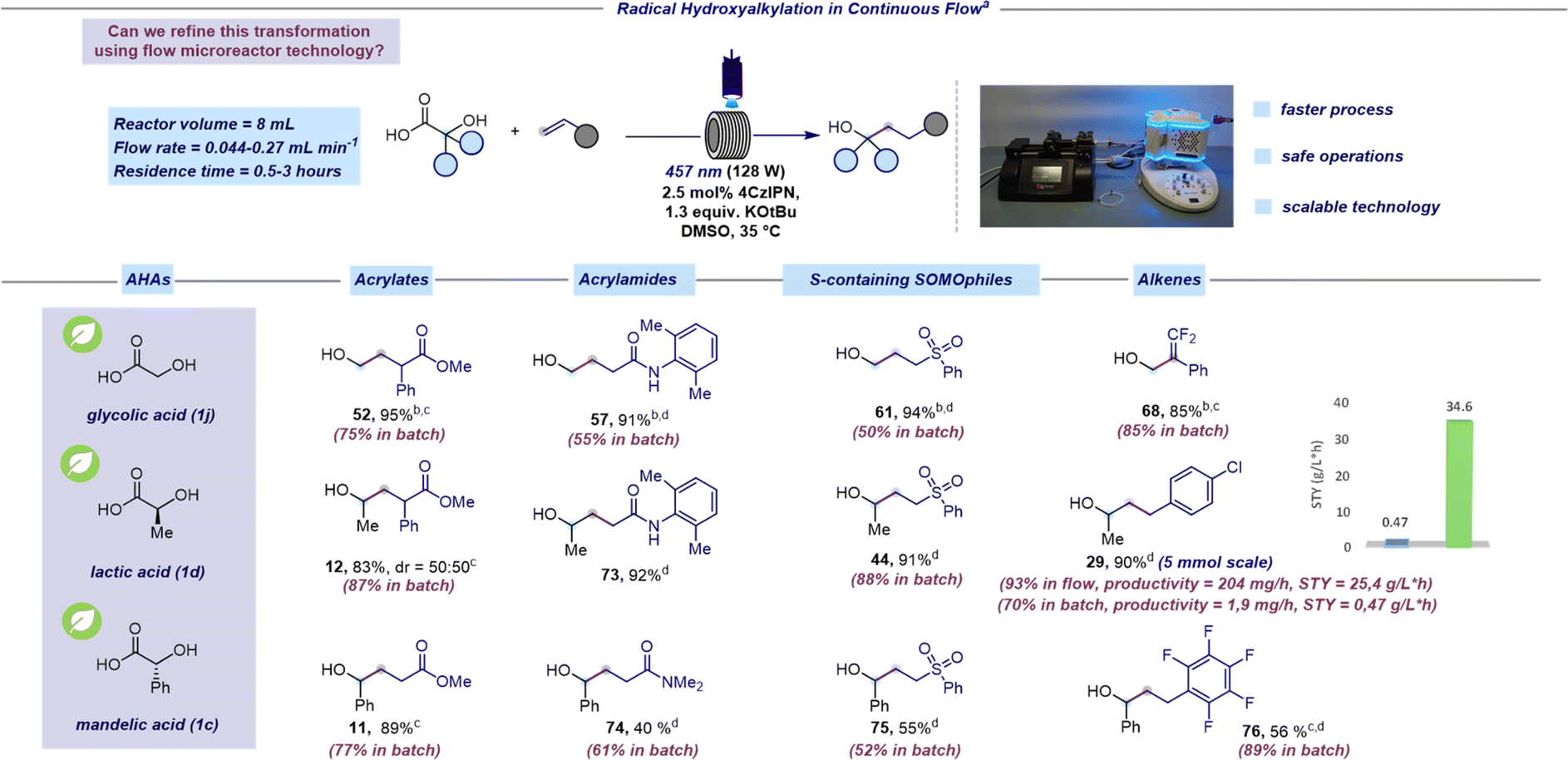 | ||
| Scheme 3 Radical hydroxyalkylation in continuous flow. aReaction performed under optimized reaction conditions: unsaturated acceptor (0.23 mmol, 1 equiv.), α-hydroxy acid (1.3 equiv.), KOtBu (1.3 equiv.), and 4CzIPN (2.5 mol%) in DMSO (0.46 M) as the solvent. The mixture was irradiated with 457 nm light (128 W) using a residence time of 0.5 hours unless otherwise specified. Isolated yields are reported. bThe reaction was performed by premixing the hydroxy acid and the base before adding the photocatalyst (see the ESI† for further details). A concentration of 0.08 M was used. cReaction performed using 40 W instead of 128 W. dA residence time of 3 hours was used. | ||
After assessing the synthetic versatility of this C1 to Cn radical hydroxyalkylation, a mechanistic investigation was undertaken with the aim to further support the synthetic design proposed in Fig. 2. The addition of radical inhibitors like TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxy) or BHT (butylated hydroxytoluene) resulted in complete inhibition of the reaction (Fig. 4A). Moreover, on–off experiments revealed that no conversion occurred in the dark, and the reaction resumed in the presence of light, showcasing the requirement for continuous light irradiation to achieve complete conversion of the reactants (Fig. 4B). Nevertheless, as shown in Fig. 2, this experiment does not exclude the possibility that a chain process could operate in addition to the closed catalytic cycle. In fact, as suggested by Yoon, the observation that the conversion does not increase during periods of darkness might be related to the fact that the chain processes terminate faster than the timescale of the analytical measurements adopted.53 Additionally, Stern–Volmer quenching studies reveal that the carboxylate of the alpha-hydroxy acid is capable of quenching the excited state of the photocatalyst (4CzIPN*). On the other hand, we did not observe any luminescence decrease in 4CzIPN* in the presence of the unsaturated SOMOphile (Fig. 4C). These observations collectively support the mechanistic proposal that the excited state 4CzIPN* behaves as the oxidant of α-hydroxy acids in the photoredox catalytic cycle (see Fig. 2).
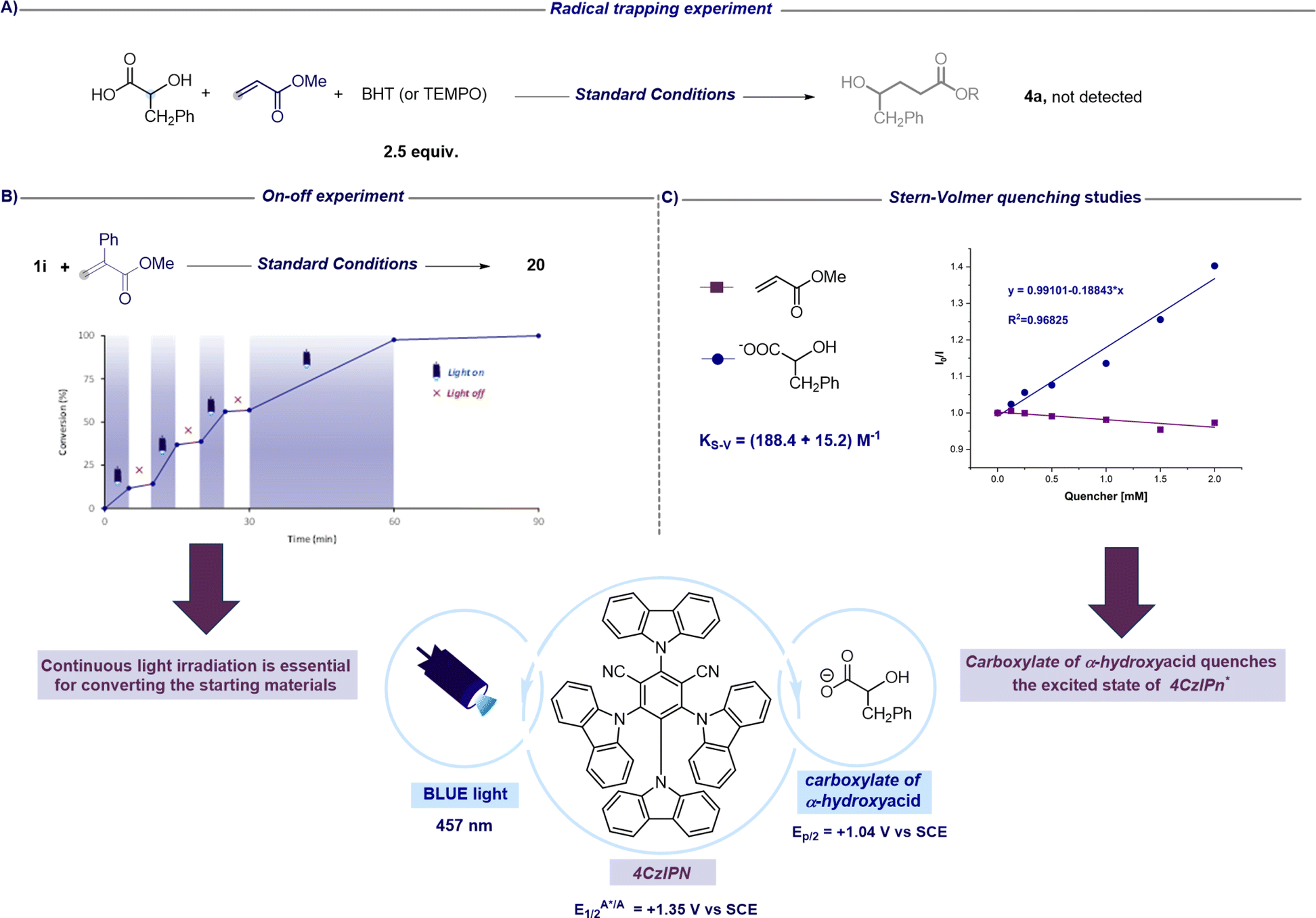 | ||
| Fig. 4 A) Radical trapping experiment using 2,6-bis(1,1-dimethylethyl)-4-methylphenol (BHT) or (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO). (B) Light on–off cycles. (C) Stern–Volmer quenching studies of the *4CzIPN excited state by using the carboxylate of acid 1a and methyl acrylate. | ||
Conclusions
In summary, we have pioneered a straightforward photoredox γ-hydroxyalkylation method by seamlessly coupling α-hydroxyalkyl radicals with unsaturated Giese-type acceptors. This operationally user-friendly technique reveals a wide generality, exhibiting a broad applicability to both α-hydroxy carboxylic acids (AHAs) and Giese-type acceptors. Furthermore, it facilitates late-stage functionalization of biorelevant structures. Of particular significance is the utilization of naturally occurring AHAs, including widely available glycolic and lactic acids, as precursors for generating the corresponding C1 and C2 α-hydroxy radicals. Notably, the flow protocol has outperformed conventional batch procedures, underscoring the remarkable capabilities of flow microreactor technology in intensifying and scaling up photochemical processes. This pioneering disclosure represents a rare demonstration of the use of abundant α-hydroxy carboxylic acids (AHAs) as a platform for direct generation of versatile ketyl radicals. We anticipate that this direct methodology will not only offer operational simplicity but also contribute to improved sustainability and broaden the applicability in the synthetic utilization of α-hydroxy radicals.Data availability
All data supporting this article are available as part of the article and its ESI file.†Author contributions
M. C. and R. L. conceived and designed the project. F. P. and Y. G. performed most of the experiments with input from L. D., M. C. and R. L. The computational investigations were carried out by M. A. The CV and Stern–Volmer experiments were carried out by G. R. All authors contributed to the writing and editing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Marilisa Pia Riganti for experimental support. We thank the European Commission – Horizon Europe Framework, project SusPharma grant agreement no. 101057430 for financial support. M. C. acknowledges the Italian MUR for funding under the framework of the Action IV.6 PON R&I 2014–2020 – DM 1062. We acknowledge the CINECA award under the ISCRA initiative, for the availability of high performance computing resources and support (M. A., project code HP10CKZH2A).Notes and references
- J. Cramer, C. P. Sager and B. Ernst, J. Med. Chem., 2019, 62, 8915–8930 CrossRef CAS PubMed.
- K. Tamao, T. Iwahara, R. Kanatani and M. Kumada, Tetrahedron Lett., 1984, 25, 1909–1912 CrossRef CAS.
- M. Spennacchio, P. Natho, M. Andresini and M. Colella, J. Flow Chem., 2024, 14, 43–83 CrossRef CAS.
- A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Chem. Rev., 2021, 122, 1485–1542 CrossRef PubMed.
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed.
- G. E. M. Crisenza, D. Mazzarella and P. Melchiorre, J. Am. Chem. Soc., 2020, 142, 5461–5476 CrossRef CAS PubMed.
- L. Caiger, C. Sinton, T. Constantin, J. J. Douglas, N. S. Sheikh, F. Juliá and D. Leonori, Chem. Sci., 2021, 12, 10448–10454 RSC.
- L. Wang, J. M. Lear, S. M. Rafferty, S. C. Fosu and D. A. Nagib, Science, 2018, 362, 225–229 CrossRef CAS PubMed.
- Á. Péter, S. Agasti, O. Knowles, E. Pye and E. D. J. Procter, Chem. Soc. Rev., 2021, 50, 5349–5365 RSC.
- N. Kern, M. P. Plesniak, J. J. W. McDouall and D. J. Procter, Nat. Chem., 2017, 9, 1198–1204 CrossRef CAS PubMed.
- M. Nakajima, E. Fava, S. Loescher, Z. Jiang and M. Rueping, Angew. Chem., Int. Ed., 2015, 54, 8828–8832 CrossRef CAS PubMed.
- L. J. Rono, H. G. Yayla, D. Y. Wang, M. F. Armstrong and R. R. Knowles, J. Am. Chem. Soc., 2013, 135, 17735–17738 CrossRef CAS PubMed.
- J. Du, K. L. Skubi, D. M. Schultz and T. P. Yoon, Science, 2014, 344, 392–396 CrossRef CAS PubMed.
- F. Calogero, G. Magagnano, S. Potenti, F. Pasca, A. Fermi, A. Gualandi, P. Ceroni, G. Bergamini and P. G. Cozzi, Chem. Sci., 2022, 13, 5973–5981 RSC.
- E. Pinosa, Y. Gelato, F. Calogero, M. M. Moscogiuri, A. Gualandi, A. Fermi, P. Ceroni and P. G. Cozzi, Adv. Synth. Catal., 2023, 366, 798–805 CrossRef.
- L. Qi and Y. Chen, Angew. Chem., Int. Ed., 2016, 55, 13312–13315 CrossRef CAS PubMed.
- J. M. Edgecomb, S. N. Alektiar, N. G. W. Cowper, J. A. Sowin and Z. K. Wickens, J. Am. Chem. Soc., 2023, 145, 20169–20175 CrossRef CAS PubMed.
- For examples of accessing α-hydroxy radicals using quinuclidinium radicals as HAT catalysts, see: (a) J. L. Jeffrey, J. A. Terrett and D. W. C. MacMillan, Science, 2015, 349, 1532–1536 CrossRef CAS PubMed; (b) J. Twilton, M. Christensen, D. A. DiRocco, R. T. Ruck, I. W. Davies and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2018, 57, 5369–5373 CrossRef CAS PubMed; (c) K. Sakai, K. Oisaki and M. Kanai, Adv. Synth. Catal., 2019, 362, 337–343 CrossRef; (d) K. Merkens, N. Sanosa, I. Funes-Ardoiz and A. Gómez-Suárez, ACS Catal., 2022, 12, 13186–13192 CrossRef CAS; (e) S. Paul, D. Filippini, F. Ficarra, H. Melnychenko, C. Janot and M. Silvi, J. Am. Chem. Soc., 2023, 145, 15688–15694 CrossRef CAS PubMed.
- For examples of the generation of α-hydroxy radicals using other HAT catalysts, see: (a) L. Niu, J. Liu, X.-A. Liang, S. Wang and A. Lei, Nat. Commun., 2019, 10, 467 CrossRef PubMed; (b) C. J. Oswood and D. W. C. MacMillan, J. Am. Chem. Soc., 2021, 144, 93–98 CrossRef PubMed.
- For examples of accessing α-hydroxy radicals from carbohydrates through visible light-promoted hydrogen atom transfer (HAT), see: (a) Y. Wang, H. M. Carder and A. E. Wendlandt, Nature, 2020, 578, 403–408 CrossRef CAS PubMed; (b) D. J. Gorelik, V. Dimakos, T. Adrianov and M. S. Taylor, Chem. Commun., 2021, 57, 12135–12138 RSC; (c) H. M. Carder, C. E. Suh and A. E. Wendlandt, J. Am. Chem. Soc., 2021, 143, 13798–13805 CrossRef CAS PubMed; (d) Y.-A. Zhang, X. Gu and A. E. Wendlandt, J. Am. Chem. Soc., 2022, 144, 599–605 CrossRef CAS PubMed.
- Y. Gao, B. Zhang, L. Levy, H.-J. Zhang, C. He and P. S. Baran, J. Am. Chem. Soc., 2022, 144, 10992–11002 CrossRef CAS PubMed.
- L. Chu, C. Ohta, Z. Zuo and Z. D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 10886–10889 CrossRef CAS PubMed.
- T. C. Bhalla, V. Kumar, and S. K. Bhatia, Hydroxy acids: production and applications, Advances in Industrial Biotechnology, IK International Publishing House Pvt Ltd, 2014, ch. 4, pp 56–76 Search PubMed.
- X. Zhou, M. Zha, J. Cao, H. Yan, X. Feng, D. Chen and C. Yang, ACS Sustainable Chem. Eng., 2021, 9, 10948–10962 CrossRef CAS.
- C. Lachaux, C. J. R. Frazao, F. Krauβer, N. Morin, T. Walther and J. M. François, Frontiers in Bioengineering and Biotechnology., 2019, 7, 359 CrossRef PubMed.
- F. A. Castillo Martinez, E. M. Balciunas, J. M. Salgado, J. M. Domínguez González, A. Converti and R. P. d. S. Oliveira, Trends Food Sci. Technol., 2013, 30, 70–83 CrossRef.
- A. O. Ojo and O. de Smidt, Processes, 2023, 11, 688 CrossRef CAS.
- Y. Huang, Y. Wang, N. Shang and N. P. Li, Foods, 2023, 12, 2311 CrossRef CAS PubMed.
- D. M. Kitcatt, S. Nicolle and A.-L. Lee, Chem. Soc. Rev., 2022, 51, 1415–1453 RSC.
- N. P. Ramirez and J. C. Gonzalez-Gomez, Eur. J. Org Chem., 2017, 2017, 2154–2163 CrossRef CAS.
- J. D. Nicewicz, H. Roth and N. Romero, Synlett, 2015, 27, 714–723 CrossRef.
- T.-Y. Shang, L.-H. Lu, Z. Cao, Y. Liu, W.-M. He and B. Yu, Chem. Commun., 2019, 55, 5408–5419 RSC.
- M. A. J. Dubois, J. J. Rojas, A. J. Sterling, H. C. Broderick, M. A. Smith, A. J. P. White, P. W. Miller, C. Choi, J. J. Mousseau, F. Duarte and J. A. Bull, J. Org. Chem., 2023, 88, 6476–6488 CrossRef CAS PubMed.
- Y.-S. Lin, G.-D. Li, S.-P. Mao and J.-D. Chai, J. Chem. Theory Comput., 2012, 9, 263–272 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- F. De Vleeschouwer, V. Van Speybroeck, M. Waroquier, P. Geerlings and F. De Proft, Org. Lett., 2007, 9, 2721–2724 CrossRef CAS PubMed.
- A. J. Fernandes, R. Giri, K. N. Houk and D. Katayev, Angew. Chem., Int. Ed., 2024, 63, e202318377 CrossRef CAS PubMed.
- S. Kar, H. Sanderson, K. Roy, E. Benfenati and J. Leszczynski, Chem. Rev., 2021, 122, 3637–3710 CrossRef PubMed.
- A recent example of the use of vinylphosphonate as the radical acceptor can be found here: J. D. Grayson and A. J. Cresswell, Tetrahedron, 2021, 81, 131896 CrossRef CAS.
- A. Barco, S. Benetti, C. D. Risi, P. Marchetti, G. P. Pollini and V. Zanirato, Tetrahedron: Asymmetry, 1997, 8, 3515–3545 CrossRef CAS.
- L. Capaldo, D. Ravelli and M. Fagnoni, Chem. Rev., 2021, 122, 1875–1924 CrossRef PubMed.
- A similar generation of gem-difluoroalkenes from trifluoromethyl alkenes has been previously documented, as detailed here: X. Lu, X.-X. Wang, T.-J. Gong, J.-J. Pi, S.-J. He and Y. Fu, Chem. Sci., 2019, 10, 809–814 RSC.
- S. D. A. Zondag, D. Mazzarella and T. Noël, Annu. Rev. Chem. Biomol. Eng., 2023, 14, 283–300 CrossRef CAS PubMed.
- A. Slattery, Z. Wen, P. Tenblad, J. Sanjosé-Orduna, D. Pintossi, T. den Hartog and T. Noël, Science, 2024, 383, eadj1817 CrossRef CAS PubMed.
- M. González-Esguevillas, D. F. Fernández, J. A. Rincón, M. Barberis, O. de Frutos, C. Mateos, S. García-Cerrada, J. Agejas and D. W. C. MacMillan, ACS Cent. Sci., 2021, 7, 1126–1134 CrossRef PubMed.
- M. Colella, Y. Gelato, M. Andresini, E. Graziano, G. Vilé, L. Degennaro and R. Luisi, Eur. J. Org Chem., 2023, 26, e202300413 CrossRef CAS.
- M. Andresini, M. Colella, R. S. Dibenedetto, E. Graziano, G. Romanazzi, A. Aramini, L. Degennaro and R. Luisi, React. Chem. Eng., 2023, 8, 3203–3209 RSC.
- M. Spennacchio, M. Colella, M. Andresini, R. S. Dibenedetto, E. Graziano, A. Aramini, L. Degennaro and R. Luisi, Chem. Commun., 2023, 59, 1373–1376 RSC.
- L. Capaldo, L. Buzzetti, D. Merli, M. Fagnoni and D. Ravelli, J. Org. Chem., 2016, 81, 7102–7109 CrossRef CAS PubMed.
- M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426–5434 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc02696a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |