
Mitochondria-targeting nanozyme alleviating temporomandibular joint pain by inhibiting the TNFα/NF-κB/NEAT1 pathway†
Qian
Bai‡
a,
Yaoyao
Zhou‡
a,
Xiaona
Cui‡
ab,
Haichao
Si
e,
Tingting
Wu
a,
Abdul
Nasir
ac,
Heng
Ma
ac,
Junyue
Xing
d,
Yingying
Wang
d,
Xiaolei
Cheng
d,
Xiaojun
Liu
*b,
Shaoyan
Qi
*b,
Zhisong
Li
c and
Hao
Tang
 *d
*d
aMedical Research Center, The Second Affiliated Hospital of Zhengzhou University, Zhengzhou, Henan 450052, China
bDepartment of Critical Care Medicine, The Second Affiliated Hospital of Zhengzhou University, Zhengzhou, Henan, China. E-mail: liuxiaojunzzu@163.com; qishaoyan1970@163.com
cDepartment of Anesthesiology and Perioperative Medicine, The Second Affiliated Hospital of Zhengzhou University, Zhengzhou, Henan, China
dNational Health Commission Key Laboratory of Cardiovascular Regenerative Medicine, Heart Center of Henan Provincial People's Hospital, Central China Fuwai Hospital of Zhengzhou University, Fuwai Central China Cardiovascular Hospital & Central China Branch of National Center for Cardiovascular Diseases, Zhengzhou, Henan 451464, China. E-mail: tangpku_zzuhao@zzu.edu.cn
eDepartment of Anesthesiology, Nanyang Central Hospital, Nanyang, Henan, China
First published on 4th August 2023
Abstract
Inflammatory cytokines that are secreted into the spinal trigeminal nucleus caudalis (Sp5C) may augment inflammation and cause pain associated with temporomandibular joint disorders (TMD). In a two-step process, we attached triphenylphosphonium (TPP) to the surface of a cubic liposome metal–organic framework (MOF) loaded with ruthenium (Ru) nanozyme. The design targeted mitochondria and was designated Mito-Ru MOF. This structure scavenges free radicals and reactive oxygen species (ROS) and alleviates oxidative stress. The present study aimed to investigate the effects and mechanisms by which Mito-Ru MOF ameliorates TMD pain. Intra-temporomandibular joint (TMJ) injections of complete Freund's adjuvant (CFA) induced inflammatory pain for ≥10 d in the skin areas innervated by the trigeminal nerve. Tumor necrosis factor-alpha (TNF-α), nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), long non-coding RNA nuclear paraspeckle assembly transcript 1 (lncRNA NEAT1), and ROS also have been proved to be significantly upregulated in the Sp5C of TMD mice. Moreover, a single Mito-Ru MOF treatment alleviated TMD pain for 3 d and downregulated TNF-α, NF-κB, lncRNA NEAT1, and ROS. NF-κB knockdown downregulated NEAT1 in the TMD mice. Hence, Mito-Ru MOF inhibited the production of ROS and alleviated CFA-induced TMD pain via the TNF-α/NF-κB/NEAT1 pathway. Therefore, Mito-Ru MOF could effectively treat the pain related to TMD and other conditions associated with severe acute inflammatory activation.
1. Introduction
In patients with TMD, abnormal pain can result in depression, insomnia, and low work efficiency. Pain and other symptoms associated with TMD are the consequences of more than merely the histopathology of the temporomandibular joint (TMJ) and its surrounding structures.1 TMD pain may arise from altered nociception processing (involving inflammatory cytokines and ROS) in the trigeminal ganglia (TG) and the Sp5C.2,3LncRNA NEAT1 is a recently discovered type of long non-coding RNA. It is widely expressed in various tissues and is implicated in numerous pain responses.4 As NEAT1 deficiency significantly attenuates inflammatory responses, NEAT1 might stimulate inflammasomes.5 Our sequencing data showed that NEAT1 was strongly upregulated in the Sp5C of mice exhibiting TMD pain. We aimed to explore whether NEAT1 plays an important role in TMD pain. Silencing NEAT1 suppressed ROS and malondialdehyde (MDA) and enhanced superoxide dismutase (SOD) activity in vitro.6 Mechanical stress and inflammatory reactions increased ROS levels and destroyed the cartilage in TMJ.7 Therefore, inhibiting ROS generation during inflammation could mitigate cartilage and bone degeneration in TMJ. Antioxidant biomolecules and nanoparticles have been proposed as therapeutic approaches for TMD.8 However, free antioxidant enzymes often have inadequate therapeutic efficacy as they are readily inactivated and have short half-lives and low transmembrane transport capacity.9,10 Carbogenic nanozyme effectively treated acute traumatic brain injury (TBI) in mice by effectively reducing ROS levels.11 Antioxidant nanoparticles can modulate the inflammatory response. Nevertheless, no prior study has investigated the analgesic effect or underlying mechanisms of antioxidant nanoparticles in TMD pain management. Thus, further research is required to clarify the roles of antioxidants in the Sp5C and in TMD pain.
In this study, bovine serum albumin (BSA)-loaded cube zeolitic imidazolate framework (ZIF)-8 (Cube MOF) was synthesized by biomimetic mineralization. Ru nanozymes were immobilized in the MOF by physical adsorption and reduced in situ to Ru MOF. Liposomes were then encapsulated in the Ru MOF to improve their biocompatibility, functionally modify the TPP ligands, and synthesize mitochondria-targeting nanozyme (Mito-Ru MOF). The catalytic capacity of Mito-Ru MOF to degrade ROS was verified. TNF-α, NF-κB, NEAT1, and ROS were substantially upregulated in the Sp5C of the mice induced with TMD by CFA injection. In addition, a single intravenous (i.v.) Mito-Ru MOF injection alleviated TMD pain through downregulated TNF-α, NF-κB, NEAT1, and ROS. Therefore, we hypothesized that Mito-Ru MOF alleviates TMD pain by inhibiting the TNF-α/NF-κB/NEAT1 pathways in the Sp5C (Scheme 1).
 | ||
| Scheme 1 TPP-liposome-encapsulated MOF and its therapeutic mechanisms in temporomandibular joint pain based on oxidative stress regulation. | ||
2. Results
2.1. Intra-TMJ CFA injection caused abnormal pain and upregulated NF-κB, NEAT1, and ROS in Sp5C
Compared with the mice injected with saline (control), the mice administered intra-TMJ CFA injection presented with significantly lower head withdrawal thresholds in their trigeminal nerve-innervated skin areas (Fig. 1A and Fig. S2, ESI†). Representative hematoxylin–eosin (H&E) staining of the TMJ cartilage from the saline and CFA groups 3 d after TMJ injection is shown in Fig. 1B. Immune cell infiltration and bone destruction were observed in the CFA mice compared with the control (Fig. 1B; scale bar: 50 μm). Immunofluorescence staining disclosed increased 8-hydroxy-2′-deoxyguanosine (8-OHGD) protein (ROS marker) immunoreactivity in the Sp5C of the TMD pain mice on day 3 after TMJ injection (Fig. 1C and D; ***P < 0.001 vs. saline group; t-test). High sequencing analysis of lncRNA expression in the entire Sp5C transcriptome was conducted on the CFA-TMJ injection mouse model (Fig. 1E). We focused on lncRNA NEAT1 as it was reported to play an important role in neuropathic pain.4,12 We performed quantitative real-time polymerase chain reactions (qRT-PCR) to measure NEAT1 expression in the Sp5C on day 3 after CFA injection. NEAT1 mRNA was upregulated in the Sp5C following CFA injection (Fig. 1F). It was reported that NF-κB binds the NEAT1 promoter region after lipopolysaccharide (LPS)-stimulated p65 nuclear translocation.13 Here, we investigated whether NF-κB is implicated in TMD pain. We harvested Sp5C tissues on day 3 after intra-TMJ injection and used western blotting to compare the total p65 and phosphorylated p-p65 [modified and phosphorylated NF-κB] protein levels in the CFA- and saline-injected sites. Relative to the control, the CFA did not significantly alter the total protein p65 (Fig. 1H; nsP > 0.05 vs. saline; t-test). However, CFA induction strongly upregulated p-p65 compared to the control (Fig. 1G;). Thus, CFA promotes p65 nuclear translocation and increases phosphorylation. These responses are positively correlated with NEAT1 expression (Fig. 1F). CFA administration increased the pp-65 fluorescence intensity. Immunofluorescence staining showed increased p-p65 protein immunoreactivity in the Sp5C on day 3 after TMJ injection in the CFA mice (Fig. 1I and J; ***P < 0.001 vs. saline; one-way ANOVA). | ||
| Fig. 1 Effects of intra-TMJ CFA injection on pain behavior and molecular changes. (A) Threshold of response to mechanical stimuli in von Frey test. ***P < 0.001 vs. saline. ****P < 0.0001 vs. saline group, N = 5; each time point after TMJ injection; two-way analysis of variance (ANOVA). (B) H&E staining of TMJ cartilage from saline and CFA groups (scale bar = 50 μm) revealed that CFA injection affected articular cartilage cellularity and cell morphology and inflammatory cell infiltration in TMJ. (C) Representative immunofluorescence staining images showing an increase in 8-OHGD (ROS marker) expression in Sp5C on day 3 after intra-TMJ injection (scale bar = 50 μm). (D) Quantitative 8-OHGD expression in SP5C in (C). ***P < 0.001 vs. saline, N = 3. (E) Heat map displaying hierarchical clustering of differentially expressed (DE) lncRNAs in saline and CFA groups 3 d post-TMJ injection (C_TG, TG from the CFA group; S_TG, TG from the saline control group; C_Sp5C, Sp5C from CFA group; S_Sp5C, Sp5C from the saline control group. Red indicates upregulation and blue indicates downregulation). (F) Validation of DE lncRNA Neat1 in Sp5C on day 3 post-TMJ injection. ****P < 0.0001 vs. saline, N = 3. (G) CFA increased P-p65 in Sp5C on day 3 post-TMJ injection. ****P < 0.0001 vs. saline, N = 56; t-test (H) CFA did not alter p65 expression in Sp5C on day 3 post-TMJ injection. nsP > 0.05 vs. saline, N = 33. (I) Representative immunofluorescence staining images showing an increase in p-p65 (ROS marker) expression in SP5C on day 3 after intra-TMJ injection (scale bar = 400 μm). (J) Quantitative p-p65 expression in SP5C in (I), N = 20. | ||
2.2. Mito-Ru MOF synthesis and characterization
Mito-Ru MOF synthesis is shown in Fig. 2A. Cube MOF with wrapped BSA was synthesized by biomimetic mineralization. Ru3+ was adsorbed to the MOF pores and the encapsulated BSA could enhance adsorption. Ru MOF was then obtained by in situ NaBH4 reduction and encapsulated into liposomes by mixing it with lecithin, the amine phospholipid DSPE-PEG-NH2, and cholesterol. TPP was then conjugated onto the surface of the amine-activated liposome-Ru MOF to enable mitochondrial targeting. Transmission electron microscopy (TEM) disclosed that Ru MOF (Fig. 2B) and Mito-Ru MOF nanozyme (Fig. 2C) both had monodispersed cube morphology and diameter ∼100 nm. Dynamic light scattering (DLS) revealed that the hydrated Ru MOF and Mito-Ru MOF nanozyme particles were in the ranges of 94.36–100.55 nm and 110.58–116.12 nm, respectively (Fig. 2D). After the Ru MOF nanozymes were coated with the liposomes, their zeta potentials decreased from the range of 16.2–18.2 to the range of −4.68 to −5.03. The zeta potentials of Lip@Ru MOF and Mito-Ru MOF increased from the range of −4.68 to −5.03 to −1.12 to −1.57. Hence, TPP was successfully conjugated on the MOF nanozymes (Fig. 2E). Powder X-ray diffraction (PXRD) of the MOF, Ru MOF, and Mito-Ru MOF nanozymes confirmed that the Ru nanozymes formed in the crystalline zeolitic imidazolate framework (ZIF)-8 shell (Fig. 2F). The PXRD patterns of Ru MOF and Mito-Ru MOF matched those of simulated ZIF-8 and me-ZIF-8. Thus, the predicted Ru MOF and Mito-Ru MOF formed successfully and remained stable in the presence of the Ru nanozymes and liposomes. | ||
| Fig. 2 Mito-Ru MOF preparation and characterization. (A) Schematic of Mito-Ru MOF preparation. (B) Photographs of cube ZIF and Ru MOF. (C) and (D) TEM images of Ru MOF and Mito-Ru MOF. (E) Zeta potentials of me-MOF, Ru MOF, and Mito-Ru MOF. (F) Diameters of me-MOF, Ru MOF, and Mito-Ru MOF. (G) PXRD patterns of me-MOF, Ru MOF, and Mito-Ru MOF. | ||
Ru MOF and Mito-Ru MOF had multienzyme-like activity under neutral conditions. Ru MOF and Mito-Ru MOF exhibited superoxide dismutase (SOD)/catalase (CAT)-like activity in phosphate buffer (pH 7.4) (Fig. 3A–C). Therefore, they can generate O2 (Fig. 3A), decompose H2O2 (Fig. 3B), and eliminate O2˙− (Fig. 3C), and mimic the cascade activity of natural endogenous SOD/CAT. We also examined the nitrogen radical-scavenging ability of Ru MOF and Mito-Ru MOF via 2,2-azinobis-(3-ethylbenzothiazoline-6-sulfonate) (ABTS) and 1,1-diphenyl-2-picrylhydrazyl (DPPH) free radical-scavenging assays (Fig. 3D–F). Both Ru MOF and Mito-Ru MOF inhibited ABTS and DPPH free radical formation, demonstrated strong antioxidant activity, and could be administered as antioxidant agents in PD therapy. Ru MOF had stronger antioxidant activity than Mito-Ru MOF. In addition to enzyme-like catalytic radical-scavenging activity, Ru MOF might also be able to scavenge free radicals by physical adsorption. Liposome encapsulation would shield the active site on Ru MOF. However, liposome encapsulation could also reduce protein adsorption by Ru MOF and enhance its circulation and stability in vivo. Both ZIF-8 and Ru MOF retain their acid sensitivity. Furthermore, acidic environments improve the CAT-like activity of Ru MOF (Fig. 3G).
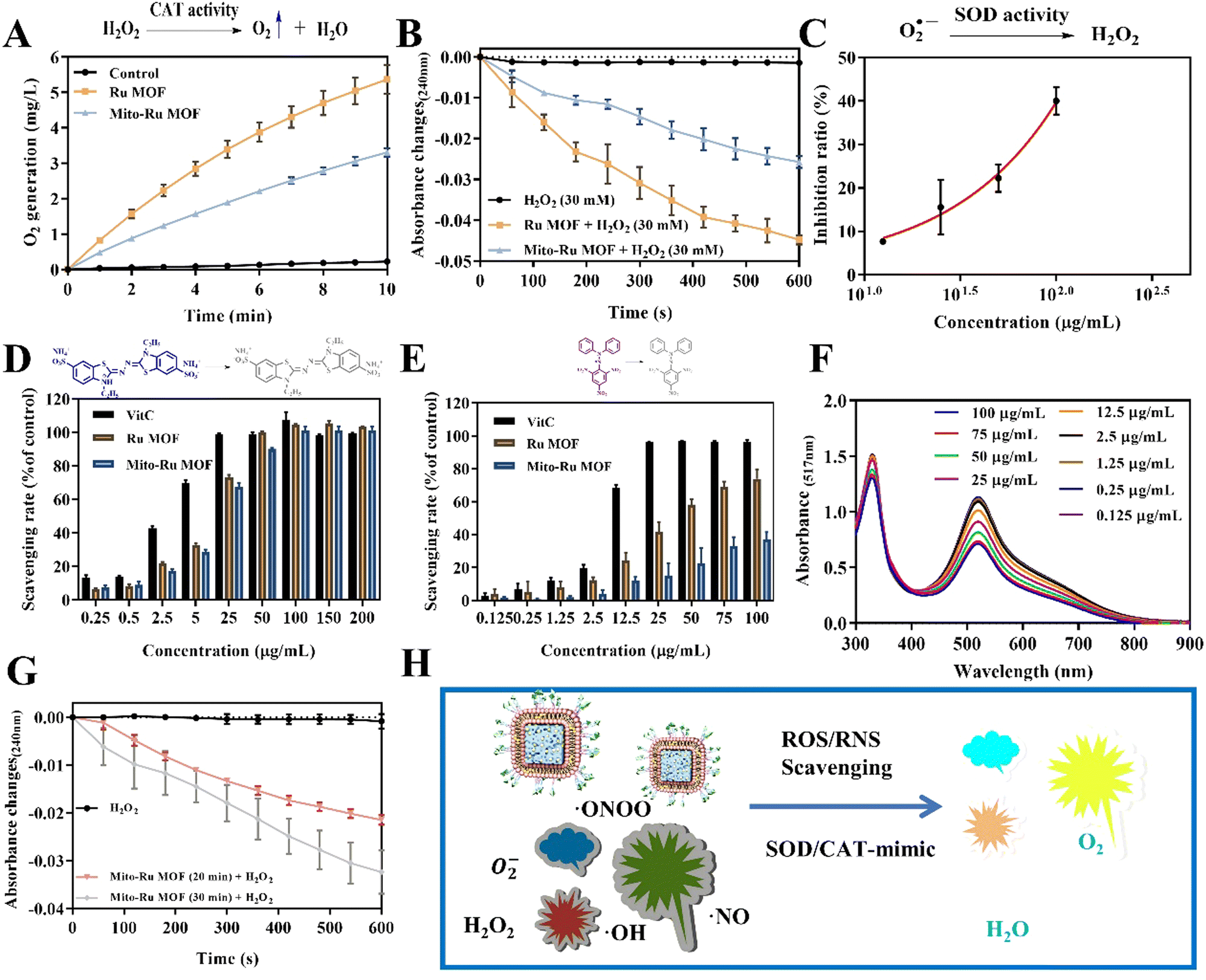 | ||
| Fig. 3 ROS scavenging by Mito-Ru MOF. (A) and (B) CAT-like activity of Ru MOF and Mito-Ru MOF. (C) SOD-like activity of Mito-Ru MOF and Mito-Ru MOF. (D) ABTS˙+ rates of vitamin C (ascorbic acid), Ru MOF, and Mito-Ru MOF. (E) DPPH˙+ rates of vitamin C, Ru MOF, and Mito-Ru MOF. (F) DPPH˙+ spectra of 0.125–75 μg mL−1 Mito-MOF at 516 nm. (G) CAT-like activity of Mito-Ru MOF and Mito-Ru MOF after acid disintegration. (H) Schematic of antioxidant activity of nanozyme-integrated chiral E Mito-Ru MOF. | ||
2.3. In vitro neuroprotective efficacy assessment
In the cell counting kit 8 (CCK8) assay, negligible cytotoxicity was observed in response to ≤60 μg mL−1 Mito-Ru MOF or Ru MOF (Fig. 4A). Hence, 60 μg mL−1 was designated the appropriate dosage for the subsequent studies. Nevertheless, Mito-Ru MOF pre-treatment increased relative cell viability. The fluorescent probe 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) indicated that Mito-Ru MOF pre-treatment markedly lowered relative intracellular ROS levels (Fig. 4C). Mito-Ru MOF displayed significantly better ROS scavenging and cytoprotective effects than Ru MOF. Live/dead staining was performed to evaluate the relative neuroprotective efficacy of Mito-Ru MOF and Ru MOF. Live cells stain green while dead cells stain red. There were more viable Mito-Ru MOF-treated cells than H2O2-treated or Ru MOF-treated cells (Fig. 4B). Meanwhile, flow cytometry was used to verify once again that Mito-Ru MOF could alleviate nerve damage caused by hydrogen peroxide, with a remission effect of 14.4% (Fig. 4E). The Membrane permeable JC-1 dyes are widely used to monitor mitochondrial health. The JC-1 dye exhibited potentio-dependent accumulation in mitochondria, and the fluorescence emission shifted from green (∼529 nm) to red (∼590 nm), Compared with H2O2-treated or Ru MOF-treated cells, Mito-Ru MOF can effectively relieve mitochondrial damage caused by hydrogen peroxide (Fig. 4D). Mito-Ru MOF displayed significantly better ROS scavenging and cytoprotective effects than Ru MOF. Thus, Mito-Ru MOF had superior neuroprotective efficacy in vitro as they could eliminate ROS, alleviate oxidative damage, and improve neuron viability.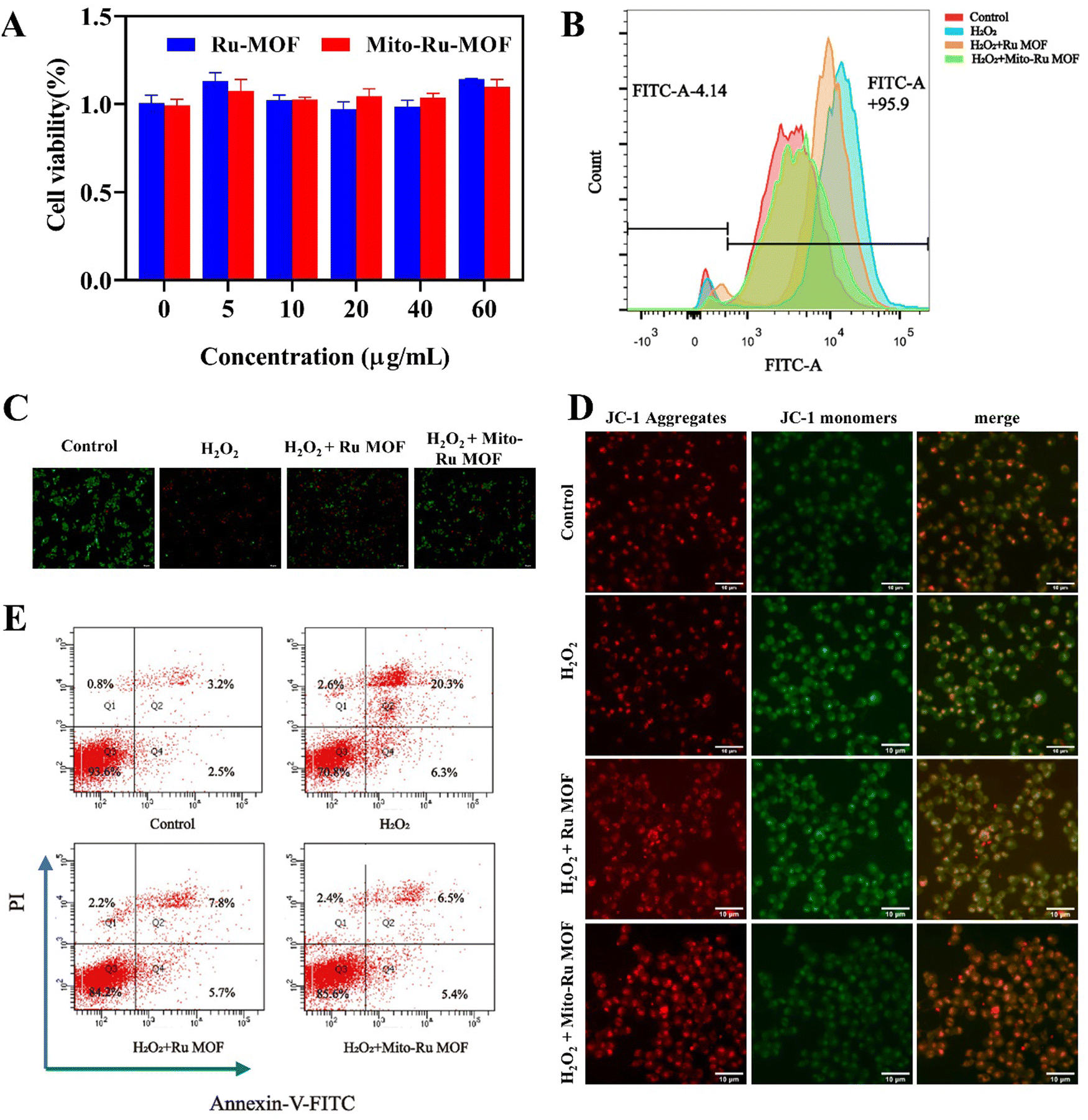 | ||
| Fig. 4 Mito-Ru MOF removes intracellular ROS and alleviates oxidative damage in neurons. (A) SH-SY5Y cell viability under various treatment strategies and concentrations. (B) Intracellular ROS stained by DCFH-DA and detected by flow cytometry (FACS). (C) Live–dead cell-staining of SH-SY5Y cell after treating with H2O2, Ru MOF + H2O2, and Mito-Ru MOF + H2O2. (D) JC-1 cell-staining of SH-SY5Y cell after treating with H2O2, Ru MOF + H2O2, and Mito-Ru MOF + H2O2. (E) The cell apoptosis analysis using the flow cytometry based on the H2O2, Ru MOF + H2O2, and Mito-Ru MOF + H2O2. Annexin V-FITC/PI staining. | ||
2.4. In vivo Mito-Ru MOF administration attenuates CFA-induced TMD pain
We performed pain behavior, oxidative stress, tissue staining, and molecular detection analyses on mouse TMD pain models subjected to Mito-Ru MOF. To determine whether Mito-Ru MOF effectively treats TMD pain, we administered a single i.v. injection of 8 mg kg−1 Mito-Ru MOF at 30 min after intra-TMJ CFA injection. The degree of inflammatory TMJ pain induced by the CFA was assessed based on mechanical hypersensitivity with von Frey filaments. Unilateral CFA injection dramatically lowered the head withdrawal threshold at the injection side of the trigeminal nerve-innervated facial skin area (Fig. 2A; ****P < 0.0001 vs. saline; two-way ANOVA). In the CFA-induced TMD model, i.v. Mito-Ru MOF injection reversed the CFA-induced reduction in head withdrawal threshold compared to the saline control after 3 d (Fig. 5A; ####P < 0.0001 vs. CFA + vehicle; two-way ANOVA). In the Mito-Ru MOF group, the pain threshold reverted to that of the CFA group in the mouse TMD pain model after 4 d. Mito-Ru MOF treatment alone did not affect the head withdrawal threshold in the mice that were not pre-injected with CFA (Fig. 5A; nsP > 0.05 vs. saline; two-way ANOVA). Histopathological examinations were then conducted to assess the effects of intra-TMJ CFA injection and Mito-Ru MOF treatment on the synovial tissues. Fig. 2B shows the CFA injection caused hypertrophy and increased leukocyte infiltration in the synovial tissues of the TMJ. However, a single i.v. Mito-Ru MOF injection 30 min after the intra-TMJ CFA injection reversed inflammatory cell infiltration in the mouse TMD pain model after 3 d (Fig. 5B). Moreover, the post-CFA Mito-Ru MOF injection reversed 8-OHGD upregulation in the mouse TMD pain model after 3 d (Fig. 5C and D and Fig. S3, ESI†).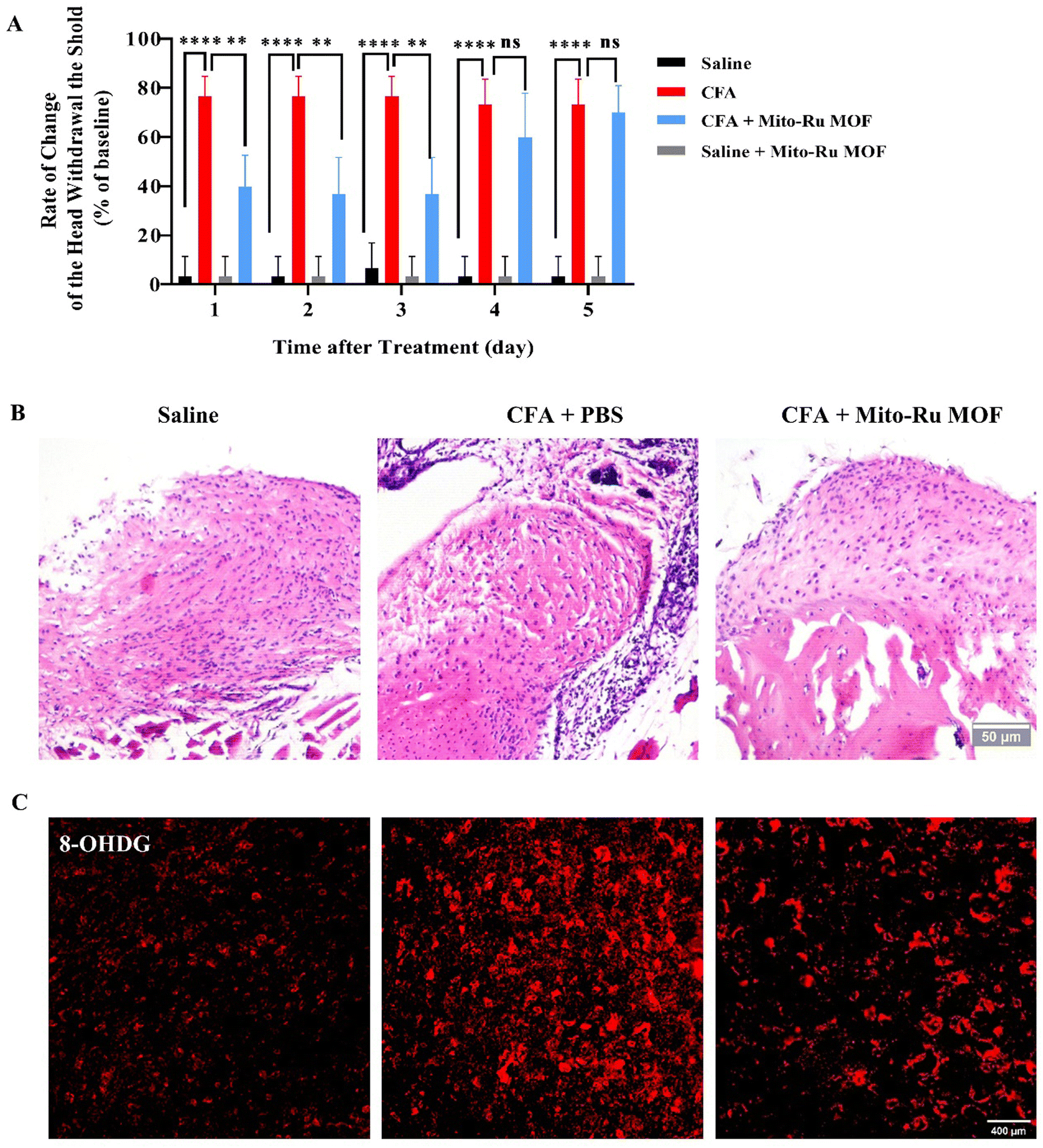 | ||
| Fig. 5 Roles of i.v. Mito-Ru MOF injection 30 min after intra-TMJ CFA injection and TMD pain induction. (A) Single i.v. Mito-Ru MOF injection 30 min after intra-TMJ CFA injection reversed the decrease in the head withdrawal threshold in the mouse TMD pain model after 3 d. ****P < 0.0001 vs. saline; ####P < 0.0001 vs. CFA + PBS, N = 5. (B) Single i.v. Mito-Ru MOF injection 30 min after intra-TMJ CFA injection reversed the increase in inflammatory cell infiltration in the mouse TMD pain model after 3 d (scale bar = 50 μm). (C) Single i.v. Mito-Ru MOF injection 30 min after intra-TMJ CFA injection reversed 8-OHGD upregulation in Sp5C of mouse TMD pain model Sp5C after 3 d (scale bar = 400 μm). (D) Quantitative 8-OHGD expression in Sp5C in (C); ****P < 0.0001, N = 3. | ||
2.5. Intravenous Mito-Ru MOF injection 30 min after intra-TMJ CFA injection downregulated TNFα/NF-κB/Neat1 pathways
We then explored the molecular mechanism by which Mito-Ru MOF alleviates inflammatory pain in the TMJ. At 30 min after the intra-TMJ CFA injection, we intravenously injected 8 mg kg−1 Mito-Ru MOF, harvested the Sp5C tissues after 3 d, and used RT-q-PCR and western blotting (WB) to measure the NEAT1, total protein p65, and phosphorylated protein p-p65 levels in the CFA and saline sites. The NEAT1 levels were higher in the Sp5C of the mice administered intra-TMJ CFA injections than they were in the Sp5C of the mice injected with saline. Moreover, the Mito-Ru MOF treatment reversed the CFA-induced increase in NEAT1 (Fig. 6A and Fig. S4, ESI†). Single systemic Mito-Ru MOF administration reversed p-p65 upregulation (Fig. 6B). DAPI (4′,6-diamidino-2-phenylindole) nuclear staining showed that the Mito-Ru MOF treatment counteracted the increase in p-p65 colocalization (Fig. 6D and E) in the Sp5C of the mouse TMD pain model after 3 d. This observation indicates that the p-p65 entered the nuclei where it functioned as a transcription factor (TF). We then injected NF-κB-siRNA into the Sp5C to determine whether NF-κB knockdown would downregulate NEAT1 in vivo. Intra-Sp5C NF-κB injection downregulated Neat1 in naive mice (Fig. 6F). TNF-α may activate NF-κB phosphorylation and promote translocation of phosphorylated NF-κB to the nuclei where it functions as a TF.14 TNF-α plays a key role in CFA-induced inflammatory pain in the TMD.15 The results of the present work suggest that CFA upregulates TNF-α while Mito-Ru MOF counteracts this effect (Fig. 6G). In summary, the injection of CFA into the TMJ upregulated TNF-α in the Sp5C, the TNF-α activated p-p65, and the p-p65 entered the nuclei where it functioned as a TF and induced NEAT1. In contrast, the Mito-Ru MOF counteracted all the foregoing responses, thereby mitigating CFA-induced TMJ pain.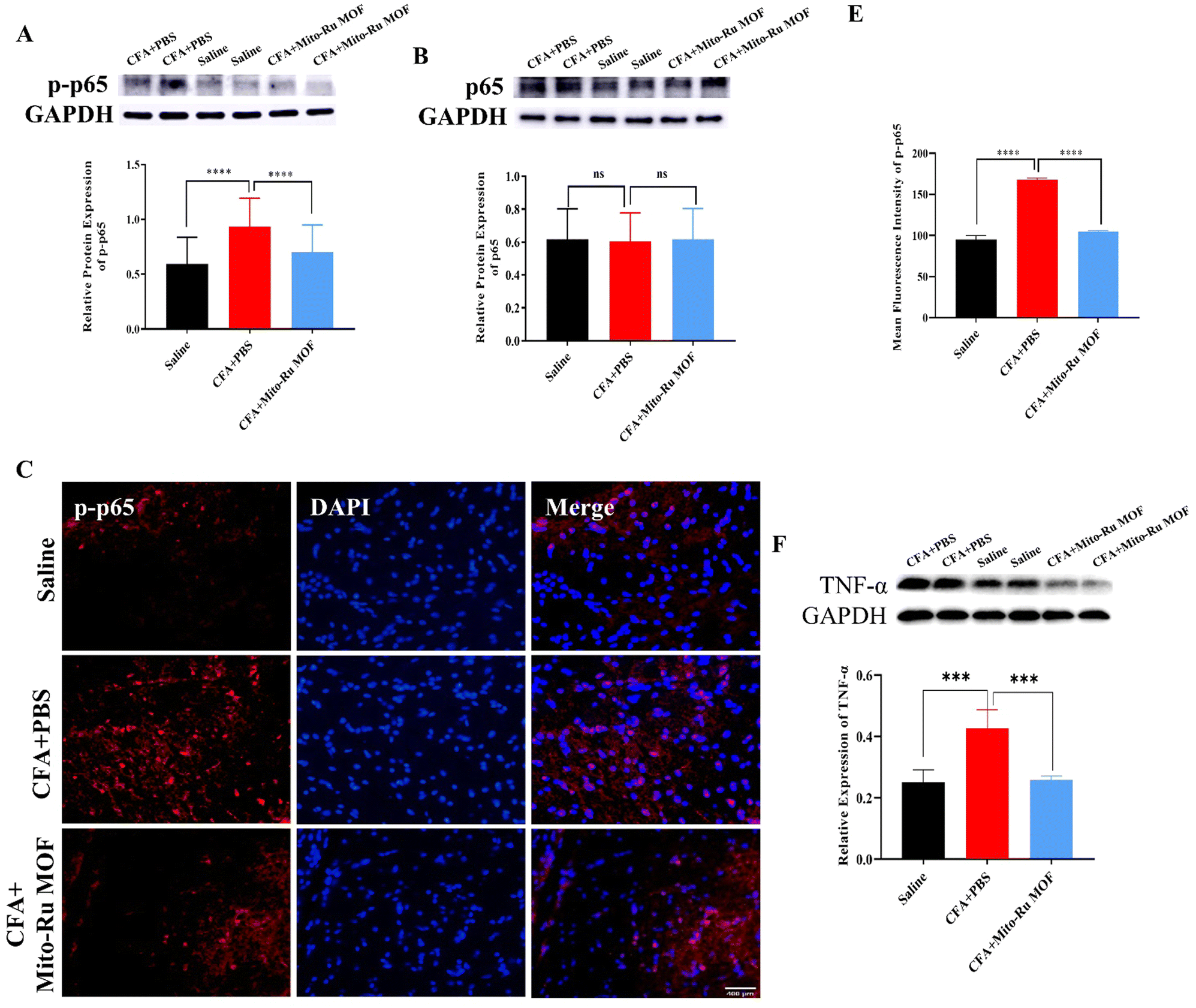 | ||
| Fig. 6 Intravenous Mito-Ru MOF injection 30 min after intra-TMJ CFA injection downregulated the TNFα/NF-κB/Neat1 pathways in a mouse TMD pain model. (A) Single i.v. Mito-Ru MOF injection 30 min after intra-TMJ CFA injection downregulated Neat1 in Sp5C in mouse TMD pain model after 3 d; **P < 0.01; ****P < 0.0001, two-way ANOVA, N = 18. (B) Single i.v. Mito-Ru MOF injection 30 min after TMJ CFA injection downregulated p-p65 in Sp5C in mouse TMD pain model after 3 d; ****P < 0.0001 vs. CFA + vehicle; two-way ANOVA, N = 39. (C) Single i.v. Mito-Ru MOF injection 30 min after TMJ CFA/saline injection did not alter p65 expression in Sp5C after 3 d; nsP > 0.05, N = 27. (D) Single i.v. Mito-Ru MOF injection 30 min after TMJ CFA injection counteracted the increase in p-p65 immunofluorescence intensity in Sp5C in mouse TMD pain model after 3 d (scale bar = 400 μm). (E) Statistical analysis of data in (D) ****P < 0.0001 vs. CFA + vehicle group, N = 3, two-way ANOVA. (F) Intra-Sp5C NF-κB injection in downregulated Neat1 in naive mice; **P < 0.01 vs. Scramble control, N = 5; t-test. (G) Single i.v. Mito-Ru MOF injection 30 min after intra-TMJ CFA injection downregulated TNF-α in Sp5C in mouse TMD pain model after 3 d; ***P < 0.001 vs. CFA + vehicle, N = 4; two-way ANOVA. | ||
3. Discussion
The number of patients presenting with TMD-related pain is increasing while the average age of this patient subpopulation is decreasing.16 Proinflammatory activation in the nervous system might be a key etiological factor in the pathogenesis of TMD pain.1 In the present study, we established a TMD pain model by injecting the TMJ of mice with 10 μL of 5 mg mL−1 CFA. This treatment elicits minimal indications of tissue damage while addressing the key features of TMD.13 CFA-TMJ injection-induced mechanical hypersensitivity starts on day 1 and continues for ≥10 d.15 CFA injection affected articular cartilage cellularity and cell morphology and inflammatory cell infiltration in the TMJ (Fig. 1B). Hence, this treatment successfully established the mouse TMD pain model. Proinflammatory activation and aberrant ROS generation in the CNS play critical roles in provoking inflammatory pain. ROS inhibition with antioxidants blocks acute inflammatory pain.17 H2O2-induced oxidative stress elevated intracellular ROS and caused apoptosis and functional impairment in cultured TMJ-derived chondrocytes.18 Here, we observed ROS upregulation in the Sp5C of the TMD mice (Fig. 1C and D). We designed a mitochondria-targeting, ZIF-8-tailored nanozyme and explored the antinociception efficacy of Mito-Ru MOF against TMD pain.After the TMJ CFA injection, we performed RNA-Seq and genome-wide read mapping on the Sp5C. A clustered heatmap of the DE lncRNAs (Fig. 1E) revealed distinct gene expression patterns in the Sp5C 3 d after the TMJ injection. We focused on lncRNA NEAT1 as it played an important role in prior rat chronic constriction injury (CCI)12 and spinal cord injury (SCI)4 models. RT-PCR showed that NEAT1 was upregulated by 50% in the Sp5C at 3 d after the CFA injection (Fig. 1F). NF-κB may bind the NEAT1 promoter and induce NEAT1 after LPS-stimulated p65 nuclear translocation.11,19 The results suggested that CFA increases p65 phosphorylation to p-p65 (Fig. 1G and J) and this reaction is positively correlated with NEAT1 expression (Fig. 1F).
We used the von Frey test to investigate whether Mito-Ru MOF treatment reduces mechanical allodynia in the mouse TMD pain model. Recent studies have found that neurotrophic factors which play an important role in inflammatory and neuropathic pain and their singaling such as MAPK, PI3K-AKT, and Src are involved in neuropathic pain, Mito Ru MOF may affect temporomandibular joint pain through neurotrophic factors.20 A single i.v. Mito-Ru MOF injection 30 min after the intra-TMJ CFA injection reversed the decline in the head withdrawal threshold 3 d after induction of the mouse TMD pain model (Fig. 2A). The single i.v. Mito-Ru MOF injection 30 min after the intra-TMJ CFA injection also counteracted the increases in 8-OHGD and NEAT1 as well as p-p65 colocalization (disclosed by DAPI staining) in the Sp5C in the mouse TMD pain model after 3 d. Increasing p-p65 colocalization indicates increasing nuclear p-p65 translocation. In the nuclei, the p-p65 functions as a TF. We also injected NF-κB-siRNA into the Sp5C and found that it downregulated Neat1 in naive mice (Fig. 3).
4. Conclusion
Our previous study confirmed that TNF-α upregulation plays a crucial role in TMD pain.13 TNF-α signaling also promotes ROS/RNS generation which, in turn, controls TNF-α signaling downstream of its receptors.21 TNF-α-mediated NF-kB activation and ROS are essential for NF-kB signaling downstream of TNF-α.17 Whereas cytokines can induce ROS generation, ROS can stimulate proinflammatory cytokine production by modulating the DNA-binding properties of NF-κB proteins. NF-κB binds the NEAT1 promoter and promotes NEAT1 expression after LPS-stimulated p65 nuclear translocation.11 NEAT1 silencing decreases ROS and MDA activity while increasing SOD activity in vitro.6 In this positive feedback loop, TNF-α-induced ROS production triggers NEAT1 or TNF expression via NF-kB. Hence, proper ROS regulation is vital to a successful TNF-α-mediated innate response.17 Previous studies have found that NF-kB is activated by two separate pathways, both classical and alternative pathways of NF-kB, the activation of NF-kB is known to be central for the regulation of the synthesis and activity of inflammatory cytokines, Mito Ru MOF may affect NF-kB signaling through classical and alternative pathways in bone and joint microenvironments.22 The interactions of ROS with the downstream TNF-α and NF-κB (NEAT1) pathways are complex as ROS can participate in various reactions at different sites simultaneously.23 Furthermore, numerous ROS effects and interactions are cell-specific. Future research should endeavor to elucidate the mechanisms by which ROS influences downstream signaling.The present work demonstrated that a single administration of the oxidative stress reducer Mito-Ru MOF could alleviate TMD pain for 3 d possibly by inhibiting the TNF-α/NF-κB/NEAT1 pathways in the Sp5C. Mito-Ru MOF is promising as an analgesic agent against inflammatory pain. However, further experimental studies and clinical trials are required to validate the dosages, intervals, and administration routes of Mito-Ru MOF that are safe and efficacious for the management of inflammatory pain in humans.
Experimental section
The detailed experimental processes are available in the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This study was supported by Young and middle-aged academic leaders of health in Henan Province (No. HNSWJW-2021001); Program for Science & Technology Innovation Talents in Universities of Henan Province (No. 22HASTIT047); National Natural Science Foundation of China (No. 82071577); Natural Science Foundation of Henan Province (No. 222300420073, 222301420051).References
- D. E. Harper, A. Schrepf and D. J. Clauw, Pain Mechanisms and Centralized Pain in Temporomandibular Disorders, J. Dent. Res., 2016, 95(10), 1102–1108 CrossRef CAS.
- J. G. Chichorro, F. Porreca and B. Sessle, Mechanisms of craniofacial pain, Cephalalgia, 2017, 37(7), 613–626 CrossRef.
- K. Dashnyam, J. H. Lee, N. Mandakhbayar, G. Z. Jin, H. H. Lee and H. W. Kim, Intra-articular biomaterials-assisted delivery to treat temporomandibular joint disorders, J. Tissue Eng., 2018, 9, 2041731418776514 Search PubMed.
- S. Xian, R. Ding, M. Li and F. Chen, LncRNA NEAT1/miR-128-3p/AQP4 axis regulating spinal cord injury-induced neuropathic pain progression, J. Neuroimmunol., 2021, 351, 577457 CrossRef CAS.
- P. Zhang, L. Cao, R. Zhou, X. Yang and M. Wu, The lncRNA Neat1 promotes activation of inflammasomes in macrophages, Nat. Commun., 2019, 10(1), 1495 CrossRef PubMed.
- D. D. Chen, L. L. Hui, X. C. Zhang and Q. Chang, NEAT1 contributes to ox-LDL-induced inflammation and oxidative stress in macrophages through inhibiting miR-128, J. Cell. Biochem., 2018, 2493–2501 Search PubMed.
- M. C. Lee, Y. Kawai, H. Shoji, F. Yoshino, H. Miyazaki, H. Kato, M. Suga and E. Kubota, Evidence of reactive oxygen species generation in synovial fluid from patients with temporomandibular disease by electron spin resonance spectroscopy, Redox Rep., 2004, 9(6), 331–336 CrossRef CAS.
- Q. Li, Y. Liu, X. Dai, W. Jiang and H. Zhao, Nanozymes Regulate Redox Homeostasis in ROS-Related Inflammation, Front. Chem., 2021, 9, 740607 CrossRef.
- H. Tang, Q. Li, W. Yan and X. Jiang, Angew. Chem., Int. Ed., 2021, 60, 13829 CrossRef CAS.
- Y.-Q. Liu, Y. Mao, E. Xu, H. Jia, S. Zhang, V. L. Dawson, T. M. Dawson, Y.-M. Li, Z. Zheng, W. He and X. Mao, Nano Today, 2021, 36 CAS.
- X. Mu, H. He, J. Wang, W. Long, Q. Li, H. Liu, Y. Gao, L. Ouyang, Q. Ren, S. Sun, J. Wang, J. Yang, Q. Liu, Y. Sun, C. Liu, X. D. Zhang and W. Hu, Carbogenic Nanozyme with Ultrahigh Reactive Nitrogen Species Selectivity for Traumatic Brain Injury, Nano Lett., 2019, 19(7), 4527–4534 CrossRef CAS PubMed.
- L. X. Xia, C. Ke and J. M. Lu, NEAT1 contributes to neuropathic pain development through targeting miR-381/HMGB1 axis in CCI rat models, J. Cell. Physiol., 2018, 233(9), 7103–7111 CrossRef CAS.
- W. Zhou, X. Chen, Q. Hu, X. Chen, Y. Chen and L. Huang, Galectin-3 activates TLR4/NF-kappaB signaling to promote lung adenocarcinoma cell proliferation through activating lncRNA-NEAT1 expression, BMC Cancer, 2018, 18(1), 580 CrossRef.
- Z.-G. Liu, Molecular mechanism of TNF signaling and beyond, Cell Res., 2005, 15(1), 24–27 CrossRef CAS PubMed.
- Q. Bai, S. Liu, H. Shu, Y. Tang, S. George, T. Dong, B. L. Schmidt and F. Tao, TNFalpha in the Trigeminal Nociceptive System Is Critical for Temporomandibular Joint Pain, Mol. Neurobiol., 2019, 56(1), 278–291 CrossRef CAS.
- M. Pihut, A. Gala and M. Kulesa-Mrowiecka, Temporomandibular disorders and their impact on the development of the overloading changes within temporomandibular joints, Folia Med. Cracov., 2022, 62(4), 45–56 Search PubMed.
- M. Mittal, M. R. Siddiqui, K. Tran, S. P. Reddy and A. B. Malik, Reactive oxygen species in inflammation and tissue injury, Antioxid. Redox Signaling, 2014, 20(7), 1126–1167 CrossRef CAS PubMed.
- T. Ueno, M. Yamada, Y. Sugita and T. Ogawa, N-acetyl cysteine protects TMJ chondrocytes from oxidative stress, J. Dent. Res., 2011, 90(3), 353–359 CrossRef CAS PubMed.
- S. Liu, Y. Tang and H. Shu, et al., Dopamine receptor D2, but not D1, mediates descending dopaminergic pathway-produced analgesic effect in a trigeminal neuropathic pain mouse model, Pain, 2019, 160(2), 334–344 CrossRef CAS PubMed.
- S. Zhu, Y. Li, S. Bennett, J. Chen, I. Z. Weng, L. Huang, H. Xu and J. Xu, The role of glial cell line-derived neurotrophic factor family member artemin in neurological disorders and cancers, Cell Proliferation, 2020, 53(7), e12860 CrossRef CAS.
- H. Blaser, C. Dostert, T. W. Mak and D. Brenner, TNF and ROS Crosstalk in Inflammation, Trends Cell Biol., 2016, 26(4), 249–261 CrossRef CAS.
- J. Xu, H. F. Wu, E. S. Ang, K. Yip, M. Woloszyn, M. H. Zheng and R. X. Tan, NF-kappaB modulators in osteolytic bone diseases, Cytokine Growth Factor Rev., 2009, 20(1), 7–17 CrossRef CAS.
- M. J. Morgan and Z. G. Liu, Crosstalk of reactive oxygen species and NF-kappaB signaling, Cell Res., 2011, 21(1), 103–115 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tb00929g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |