
Encapsulation of telmisartan inside insulinoma-cell-derived extracellular vesicles outperformed biomimetic nanovesicles in modulating the pancreatic inflammatory microenvironment†
Anjali
Singh
ab,
Subrata Kumar
Pore
 c and
Jayanta
Bhattacharyya
*ab
c and
Jayanta
Bhattacharyya
*ab
aCentre for Biomedical Engineering, Indian Institute of Technology Delhi, New Delhi 110016, India
bDepartment of Biomedical Engineering, All India Institute of Medical Science Delhi, New Delhi 110029, India. E-mail: jayanta@iitd.ac.in
cAmity Institute of Molecular Medicine and Stem Cell Research, Amity University, Noida, 201313, India
First published on 13th September 2024
Abstract
Diabetes mellitus (DM) is a chronic metabolic condition, characterized by hyperglycaemia, oxidative imbalance, pancreatic β-cell death, and insulin insufficiency. Angiotensin II (Ang II) increases oxidative stress, inflammation, and apoptosis, and Ang II type 1 receptor (AT1R) blockers (ARBs) can ameliorate inflammatory response and oxidative stress. However, like other small-molecule drugs, free ARBs show poor in vivo efficacy and dose-limiting toxicities. Hence, in this study, we developed nano-formulations of telmisartan (TEL), an ARB, by encapsulating it inside a murine insulinoma cell-derived extracellular vesicle (nanoTEL) and a bio-mimetic lipid nanovesicle (lipoTEL). Both nano-formulations showed spherical morphology and sustained release of TEL. In vitro, nanoTEL restored oxidative equilibrium, attenuated reactive oxygen species levels, enhanced the uptake of glucose analogue, and increased the expression of glucose transporter protein 4 better than lipoTEL. In a streptozotocin-induced murine model of diabetes, nanoTEL lowered blood glucose levels, improved glucose tolerance, and promoted insulin synthesis and secretion significantly better than lipoTEL. Moreover, nanoTEL was found superior in ameliorating the pancreatic inflammatory microenvironment by regulating NF-κBp65, HIF-1α, and PPAR-γ expression; modulating IL-1β, IL-6, tumor necrosis factor-α, IL-10, and IL-4 levels and inducing the polarization of macrophage from M1 to M2. Further, nanoTEL administration induced angiogenesis and promoted the proliferation of pancreatic cells to restore the structural integrity of the islets of Langerhans more efficiently than lipoTEL. These findings collectively suggest that nanoTEL outperforms lipoTEL in restoring the function of pancreatic β-cells by modulating the pancreatic inflammatory microenvironment and show potential for the treatment of DM.
1. Introduction
Diabetes mellitus (DM) is a metabolic condition with persistent low-grade chronic inflammation and is commonly diagnosed by the presence of high blood glucose levels.1 Insulin resistance and loss of insulin-synthesizing pancreatic β-cells are the major factors responsible for the loss of blood glucose homeostasis.2 Prolonged hyperglycemia increases reactive oxygen species (ROS) levels, induces oxidative imbalance, promotes the accumulation of advanced glycation end products (AGEs), and causes chronic low-grade inflammation. In this pancreatic inflammatory microenvironment, insulin-synthesizing pancreatic β-cells undergo apoptosis, resulting in decreased insulin synthesis and secretion.3–6At the diabetes-defining stage of glucose intolerance and insulin resistance, the expression of the angiotensin II type 1 receptor (AT1R) increases, which enhances oxidative stress in the pancreas.7,8 The increased oxidative stress damages the endoplasmic reticulum and leads to pancreatic β-cell apoptosis.7,8 Previous studies have demonstrated that the AT1R blocker (ARB) can reduce ROS levels and decrease pancreatic hypertrophy.9–11 Telmisartan (TEL), an ARB, has shown anti-diabetes efficacy by reducing insulin resistance, restoring oxidative balance, decreasing ROS levels, and revitalizing the morphology of pancreatic islets.9,12–14 Further, TEL is a partial PPAR-γ agonist and has the potential to maintain glucose homeostasis by modulating AMPK activation, sirtuin expression, translocating glucose transporter 4 (GLUT4)-containing vesicles to the plasma membrane and increasing insulin secretion.12,15 However, like other small-molecule drugs, TEL is associated with few drawbacks, including short plasma half-life, non-specificity, and poor in vivo efficacy. Hence, to achieve the therapeutic effect, administration of frequent and high dosages is required, resulting in dose-dependent toxicities to healthy organs.
Packaging drugs inside nanoparticle-based delivery systems has shown great potential to overcome the limitation of these small-molecule drugs. Cell-derived extracellular vesicles showed promising results as delivery systems, as they can transport a wide range of payloads efficiently.16–20 Extracellular vesicles are non-immunogenic, biodegradable, have long plasma half-life, and low toxicity, making them suitable candidates for drug delivery applications.16–23 MIN6-derived extracellular vesicles (CEVs) have shown the potential to reduce insulin resistance, ameliorate glucose homeostasis, increase insulin level, decrease macrophage infiltration, induce angiogenesis and expression of pancreatic β-cell markers on pluripotent stem cells, and has immune modulatory effects.24–26
By mimicking the composition of MIN6-derived extracellular vesicles, we developed a bio-mimetic lipid nanoparticle (BLN) as an alternative delivery vehicle to check if these synthetically developed vesicles have effects similar to those of the CEV. Hence, we developed two nanoformulations of TEL by encapsulating it inside a CEV (nanoTEL) and a BLN (lipoTEL), to check their effects on restoring the pancreatic microenvironment in the case of type I diabetes. In vitro studies showed that both nanoTEL and lipoTEL ameliorated oxidative balance, reduced ROS levels, and increased GLUT4 expression. Further, in streptozotocin (STZ)-induced diabetic mice, nanoTEL reduced blood glucose levels, increased plasma insulin levels, restored the function of pancreatic islets, and enhanced insulin-positive cells in the islets of Langerhans significantly better than lipoTEL. In addition, nanoTEL was found superior to lipoTEL in increasing the serum levels of anti-inflammatory cytokines, enhancing peroxisome proliferator-activated receptor-γ (PPAR-γ) expression, and inducing cellular proliferation in the pancreas while decreasing the levels of pro-inflammatory cytokines and reducing the expression of nuclear factor-κBp65 (NF-κBp65) and hypoxia-inducible factor-1α (HIF-1α) in the pancreas. Overall, nanoTEL outperformed lipoTEL in restoring the glucoregulatory functions of the pancreas by ameliorating the pancreatic inflammatory microenvironment and promoting cellular proliferation.
2. Methodology
2.1. Materials
(2-(N-(7-Nitrobenz-2-oxa-1,3-diazol-4-yl)amino)-2-deoxyglucose) (2-NBDG), Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), and 1,1-dioctadecyl-3,3,3′,3′-tetramethylindo-carbocyanine perchlorate (DiI) were purchased from Thermo, USA. TEL was purchased from Tokyo Chemical Industry Co. Ltd. Horse serum and 2,7-dichlorofluorescein diacetate (DCF-DA) were purchased from Sigma, Germany. All the other chemicals used were purchased from local manufacturers until or unless mentioned.2.2. Cell line and animal handling
The mouse insulinoma (MIN6) cell line was provided by the National Centre for Cell Science (Pune, India). Additionally, we extend our sincere gratitude to Prof. C. S. Dey (KSBS, IIT Delhi, New Delhi-India) for the gracious gift of the C2C12 murine muscle cell line. Both MIN6 and C2C12 cells were cultured in DMEM with high glucose (25 mM), supplemented with 10% FBS. These cells were maintained in a humidified chamber with 5% carbon dioxide (CO2) to promote cell proliferation and viability. To attain differentiated C2C12 myotubes, C2C12 myoblast cells were cultured until reaching an optimal confluence of 80–90% in DMEM supplemented with 10% FBS. After that cells were cultured in DMEM containing 2% horse serum for 48 hours to induce cellular differentiation.27 During this pivotal stage, the C2C12 cells underwent a differentiation process, from C2C12 myoblast to C2C12 myotubes having tube-like structures of elongated and multinucleated cells. The emergence of these tube-like structures confirmed the successful formation of C2C12 myotubes.For an in vivo study, 4-to-6-week-old male Balb/c mice were procured from the animal breeding facility of the National Institute of Biologicals (NIB), Noida, Uttar Pradesh, India. Strict adherence to ethical principles and guidelines was maintained throughout the experimental procedures. The institutional approval (CPCSEA/IAEC/AIP/2022/12/03) and unwavering compliance with the directives of the institutional animal ethics committee (IAEC) and the committee for control and supervision of experiments on animals (CPCSEA) underscored the ethical integrity of this study.
The mice were housed with a regulated light-darkness cycle of 12 hours each, ensuring the harmonious synchronization of their circadian rhythms. A stable ambient temperature of 23 ± 2 °C and a relative humidity range of 50–60% were maintained within the housing environment. Furthermore, to address their nutritional needs adequately, the mice were provided with a supply of a nutritionally balanced chow diet and access to potable drinking water ad libitum at all times.
2.3. CEV isolation and physicochemical characterization
CEVs were isolated from MIN6 conditioned media by ultra-centrifugation.28 In brief, MIN6 cells were cultured in a 75 mm2 polystyrene flask (Corning, USA) provided with 10% extracellular vesicle-depleted FBS-containing DMEM. For the depletion of extracellular vesicles, FBS was ultra-centrifuged at 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g to pellet down the extracellular vesicles present in it, and its supernatant was used to prepare extracellular vesicle depleted complete media for CEV isolation. MIN6-conditioned media were harvested after 48 hours of cell culture and centrifuged at 300 and 2000g for 10 min each, to eliminate viable and non-viable cells, followed by the removal of debris at 10000g for 30 min at 4 °C. The supernatant obtained in the preceding step was subjected to ultracentrifugation at 100000g for 70 min at 4 °C to pellet down CEVs. The CEV pellet was washed twice by ultracentrifugation, re-suspended in phosphate buffer saline (PBS), and then stored at −80 °C until further experimentation.
000g to pellet down the extracellular vesicles present in it, and its supernatant was used to prepare extracellular vesicle depleted complete media for CEV isolation. MIN6-conditioned media were harvested after 48 hours of cell culture and centrifuged at 300 and 2000g for 10 min each, to eliminate viable and non-viable cells, followed by the removal of debris at 10000g for 30 min at 4 °C. The supernatant obtained in the preceding step was subjected to ultracentrifugation at 100000g for 70 min at 4 °C to pellet down CEVs. The CEV pellet was washed twice by ultracentrifugation, re-suspended in phosphate buffer saline (PBS), and then stored at −80 °C until further experimentation.
To check the purity of isolated CEVs, CEVs and MIN6 cells were lysed in a radioimmunoprecipitation assay (RIPA) buffer and total protein was quantified using a micro-bicinchoninic acid (BCA) kit (Cat #23235, Thermo Scientific, USA) as per the manufacturer's guidelines. The isolated proteins were subjected to western blot analysis to detect the expression of extracellular vesicle-specific (Alix) and cell-specific (Calnexin) markers using a primary antibody for Alix (Cat #634501, from Biolegend) and Calnexin (Cat #MAA280Hu22, from Cloud-Clone Corp.).29–31 Further, the CEV size distribution, average size, and morphology were determined by nanosight tracking analysis (NTA), dynamic light scattering (DLS), and cryo high-resolution transmission electron microscopy (cryoHRTEM). An NTA (NS500 nanoparticle analyzer from Malvern Instruments, UK) was set at camera level 14 and adjusted focus to ensure that the particles appeared as distinct and sharp dots before recording a video of 30 seconds for each sample. All the post-acquisition functions were configured at automatic settings except the detection threshold, which was set to 4. For cryoHRTEM, 2 μL of CEV suspension was drop-cast onto a carbon-coated electron microscopy grid followed by freeze-drying using liquid nitrogen. The cryopreserved nanovesicles were visualized using a Tecnai G2 FEG Twin (Thermo Fisher, USA).
2.4. Lipid isolation from CEVs, synthesis of BLNs, and loading of TEL
Total lipids were isolated from the CEV by the Bligh and Dyer method of lipid isolation.32 In brief, we took the CEV in an amber colour glass vial and added an ice-cold methanol:chloroform solution (1:2) followed by a vigorous vortex. To induce phase separation, we added filtered deionized water into a glass vial, vortexed vigorously, and then centrifuged for 5 min at 4 °C. The top fraction was carefully removed, leaving behind a chloroform fraction containing lipids, which was dried by purging with a stream of nitrogen gas and further analyzed by liquid chromatography–mass spectrometry/mass spectrometry (LC-MS/MS) using 95% methanol and 0.1% formic acid as an extraction solvent. Then, 5 μL of lipid was injected into an LC–MS/MS column (Hibar column with 30 mM length, 2.1 mM internal diameter and 2 μm particle size). The flow rate, temperature, and base bar were maintained at 0.4 mL min−1, 30 °C, and 173 bars, respectively.
Next, lipids present in the CEV were taken in a glass vial, and a thin film was prepared on the inner surface of the glass vial by purging the nitrogen gas and dried thoroughly to remove all the traces of moisture. To synthesize the BLN, we added PBS into the vial containing the lipid layer and vortexed vigorously, followed by size reduction using sonication. Like the CEV, we determined the physicochemical characteristics of the BLN by DLS, NTA, and cryoHRTEM.
The TEL loading into the CEV and BLN was done by probe sonication. Briefly, TEL was added into the vial containing CEVs and sonicated using a Branson Sonifier (Emerson, USA) as follows: amplitude 20%, six cycles of 30 s on/off, with a total on-time of 3 min followed by 1 hour recovery period at 37 °C.31 TEL was loaded inside the BLN following the same steps as mentioned in the synthesis of the BLN where PBS was replaced with a TEL solution. The free TEL was separated from the nanoTEL and lipoTEL by size exclusion chromatography using a Sephadex G-25 column (GE Healthcare, USA), and the TEL loading was determined using the excitation/emission spectra of TEL at 305/365 nm.18,33,34 Moreover, NTA and SEM were used to determine the hydrodynamic diameter and morphology of nanoTEL and lipoTEL. The zeta potential of the nanoTEL and lipoTEL were determined using a Zetasizer Nano-ZS (Malvern Instruments, UK). The release of TEL from nanoTEL and lipoTEL was determined at pH 7.4 for 48 hours at 37 °C.
2.5. In vitro cytotoxicity and uptake assay
The in vitro cytotoxicity of the TEL, nanoTEL, and lipoTEL was determined by a 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT) assay, as described elsewhere.35 Briefly, MIN6 (1 × 104 cells well−1) were seeded, allowed to adhere overnight, and treated for 48 hours with the TEL, nanoTEL, and lipoTEL. Next, the MTT solution was added to the wells containing treated cells and after 3 hours, formazan crystals were dissolved with 100% dimethyl sulfoxide (DMSO). The absorbance was measured at 590 nm using a microplate reader (Biotek, USA), and the cell viability was calculated as follows:| % Viability = (Absorbance at 590 nm of the treated cells/Absorbance at 590 nm of the untreated cells) × 100. |
2.6. In vitro amelioration of oxidative balance, GLUT4 expression, and glucose uptake
The nanoTEL- and lipoTEL-mediated in vitro modulation of oxidative balance and ROS levels was estimated by the NAD+/NADH ratio and DCF-DA assay, as discerning indicators of oxidative stress and redox state dynamics, respectively.9,38 To determine the NAD+/NADH ratio, MIN6 cells (1 × 104) were seeded into a 96-well plate, and the next day, they were treated with free TEL, nanoTEL, and lipoTEL equivalent to 1 and 10 μM free TEL concentration in the presence of 25 mM glucose and 0.5 mM palmitate. After 48 hours of treatment, the concentration of NAD+ and NADH, and the NAD+/NADH ratio were estimated using an NAD+/NADH quantification kit (Cat #K337, BioVision, USA) as per the manufacturer's instructions. Further, to determine the cellular ROS level using a DCF-DA assay,39 MIN6 cells and C2C12 myotubes (1 × 104 and 5 × 103 cells well−1, respectively) were seeded into 96-well plates and treated for 48 h with 1 and 10 μM TEL and its equivalent nanoformulations (nanoTEL, and lipoTEL). These treatments were performed in the presence of 25 mM glucose and 0.5 mM palmitate. Following treatment, cells were treated with 2 μM carboxy DCF-DA in serum-free media at 37 °C for 30 min to estimate the ROS levels. The ROS quantification was done by measuring the fluorescence signal with excitation/emission at 440/520 nm. In addition, representative fluorescent images indicating cellular ROS levels were acquired using a cell imaging multimode reader Cytation 1 (Biotek, USA).To determine the expression levels of GLUT4, 5 × 105 C2C12 myotubes per well were seeded, and the next day, they were treated with TEL, nanoTEL, and lipoTEL for 48 hours. Later, cells were harvested by trypsinization and washed with PBS, followed by lysis with a RIPA buffer containing phosphates and protease inhibitors. The cellular lysate was centrifuged at 14000 rpm for 15 min at 4 °C, the supernatant was obtained, and the protein concentration was quantified using a micro-BCA kit (Thermo, USA) as per the manufacturer's protocol.
For the glucose uptake assay, 5 × 103 C2C12 myotubes were seeded into a 96-well plate and adhered overnight. Subsequently, cells were treated for 48 hours with TEL, nanoTEL, and lipoTEL in the presence of 25 mM glucose and 0.5 mM palmitate, followed by replacement with serum-free low-glucose DMEM media containing 50 μM 2-NBDG, a fluorescent analog of glucose. After 30 min, cells were washed thrice with PBS to remove free 2-NBDG and imaged at excitation and emission wavelengths of 465 nm and 540 nm, respectively. To quantify the glucose uptake, cells were lysed using 0.1 M potassium phosphate buffer (pH 10) containing 1% Triton for 10 min in the darkness, and then 30 μL DMSO was added to each well and the glucose uptake was immediately quantified at an excitation/emission wavelength of 465/540 nm, respectively.40
2.7. In vivo animal study, blood glucose, and serum insulin measurement
Balb/c mice were procured from NIB and were allowed to acclimatize for a week. Diabetes was induced by injecting freshly prepared STZ (40 mg kg−1) intraperitoneally (IP) for 5 consecutive days.41 The fasting blood glucose measurement was carried out to confirm the development of diabetes using a digital glucometer (Glucocare ultima). The mice were randomly divided into five groups, each containing six mice, as follows: (1) Control (PBS treated), (2) TEL, (3) CEV, (4) nanoTEL, and (5) lipoTEL. The animals were IP administrated with (2 mg kg−1) TEL, or its equivalent nanoformulations, on every alternate day. Throughout the study, the blood glucose levels of the mice were recorded and blood samples were collected from the tail vein of mice on days 3, 7, 11, and 12 to determine the serum insulin levels. On day 11, mice were kept on overnight fasting, and the next day, the intraperitoneal glucose tolerance test (IPGTT) was performed by injecting 2 g kg−1 glucose solution IP and the blood glucose levels were determined at 0, 15, 30, 45, 60, 90, and 120 min after glucose administration. After IPGTT, all the mice were sacrificed and organs were harvested for further analyses.2.8. Immunofluorescence (IF) staining of pancreatic tissue sections
The pancreas from mice was preserved in a 4% paraformaldehyde solution and sections of 2 μm thickness were prepared. For IF staining, these sections were blocked with FBS and then incubated with anti-insulin, anti-PPAR-γ, anti-ki-67, anti-Arg1, anti-CD31, anti-F4/80, and anti-CD86 antibodies (Cat #E-AB 70202, Cat #E-AB 60059, Cat #E-AB 22027, and Cat #E-AB-60474 respectively, from Elabscience, and Cat #102402, Cat #123103, and Cat #105002 from Biolegend, USA) followed by washing and detection by fluorescence-labelled secondary antibodies. Finally, the images of IF-stained pancreatic tissue sections were acquired using a cell imaging multimode reader, Cytation 1 (Biotek, USA).2.9. ELISA and western blot analysis
The concentration of insulin and cytokines (IL-1β, IL-6, IL-10, TNF-α, and IL-4) in serum were determined using the commercially available ELISA kits (Cat #E-EL-M1382, Cat #E-EL-M0037, Cat #E-EL-M0044, Cat #E-EL-M0046, Cat #E-EL-M3063, and Cat #E-EL-M0043 from Elabscience, USA) as per the manufacturer's protocol. To check hepatotoxicity, the serum level of alanine transaminase (ALT) and aspartase transaminase (AST) were estimated using a biochemical assay kit (Cat #E-EL-K235M and Cat #E-EL-K236M from Elabscience, USA).For western blot, cells and tissues were lysed into a RIPA buffer containing protease inhibitors, and the protein amount was quantified using a microBCA kit. Then 20 and 80 μg (in vitro and in vivo respectively) of proteins were separated by 12% sodium dodecyl–sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) and then transferred onto a polyvinylidene fluoride (PVDF) membrane. The membrane was blocked with 3% bovine serum albumin and incubated overnight with primary antibodies, followed by washing with Tris-buffered saline with 0.1% Tween 20 (TBST). β-Actin was used as a loading control for proteins. Next, HRP-conjugated secondary antibodies were used for the detection of protein expression. Finally, blots were developed by adding an enhanced chemiluminescence (ECL) reagent, and the images were acquired using a G-box Chemi XRQ imaging system (Syngene, USA). The quantitative densitometry analysis was performed using the Gene Tool, Syngene, USA. The primary antibodies used were purchased from Elabscience, USA: anti-GLUT-4 (Cat #E-AB-30268), anti-β-actin (Cat #E-AB-40338), anti-insulin (Cat #E-AB 70202), anti-PPAR-γ (Cat #E-AB 60059), anti-HIF-α (Cat #E-AB-31662), anti-NF-κBp65 (Cat #E-AB-22016), anti-Arg1 (Cat #E-AB-60474), and anti-CD80 (Cat #E-AB-66663).
2.10 Biodistribution
For the biodistribution study, mice were administered IP with the DiI labelled CEV and BLN where DiI was used as a model drug. To check the biodistribution of nanoformulations, mice were sacrificed at 2 and 24 hours post injection, and different organs (pancreas, heart, lungs, liver, and kidneys) were excised out. The organs were cleaned, cut to small pieces and lysed in a RIPA lysis buffer containing a protease inhibitor using zirconia beads in the mini bead beater (3 cycles of 10 s). The tissue lysates were centrifuged, and the protein amount was quantified in supernatants by a microBCA kit. Lysates prepared using the tissues of mice treated with PBS were used as the control. The tissue concentrations of DiI were determined by the excitation/emission spectra of DiI at 549/565 nm, using a Synergy H1 spectrophotometer, Biotek, USA.2.11. Statistical analysis
All the experiments were performed in triplicate, and results are expressed as mean ± SD. Statistical analysis was performed using GraphPad prism 7. Student's t-test, Turkey's multiple comparison test, one-way analysis of variance (ANOVA) and two-way ANOVA were performed to determine the significance. The difference between the groups was considered significant with a p-value < 0.05.3. Results and discussion
3.1. Development and characterization of TEL nanoformulations
CEVs were obtained from MIN6-conditioned media by ultra-centrifugation (Fig. 1A). To determine whether the isolated particles were CEVs, we checked the expression of Alix, a marker for cell-derived extracellular vesicles and Calnexin, a cell-specific marker by western blot analysis. Fig. 1B shows that Alix was present in CEVs but was absent in the MIN6 cell lysate, whereas Calnexin was not present in CEVs but in the MIN6 cell lysate. This result confirmed that the obtained CEVs were pure and devoid of any cellular contamination. NTA showed the hydrodynamic diameter of various peaks ranging from 75 to 480 nm for CEVs (Fig. 1C). DLS further confirmed the size of CEVs, which revealed an average hydrodynamic diameter of 319 nm (Fig. S1A, ESI†). CryoHRTEM depicted a spherical shape for CEVs (Fig. 1D). To synthesize the BLN, lipids were isolated from CEVs (Fig. 1E) and the compositions were determined by LC–MS/MS analysis (Fig. 1E). LC–MS/MS analysis showed that a total of 33 types of lipids were present in CEVs, where cholesterol (67.1%) was the most abundant lipid (Fig. 1E). Phosphatidic acid (19.2%), phosphatidyl serine (0.3%), phosphatidyl choline (7.6%), sphingomyelin (2.6%), phosphatidyl inositol (0.3%), phosphatidyl ethanolamine (0.1%), and fluorescein (0.003%) were also present in CEVs (Fig. 1E). The lipid mixture isolated from CEVs was further used to synthesize the BLN (Fig. 1E). The average hydrodynamic diameter of the BLN was 97 nm varying from 60 to 650 nm and exhibited a spherical morphology, as indicated by DLS, NTA, and cryoHRTEM (Fig. S1B–D, ESI†).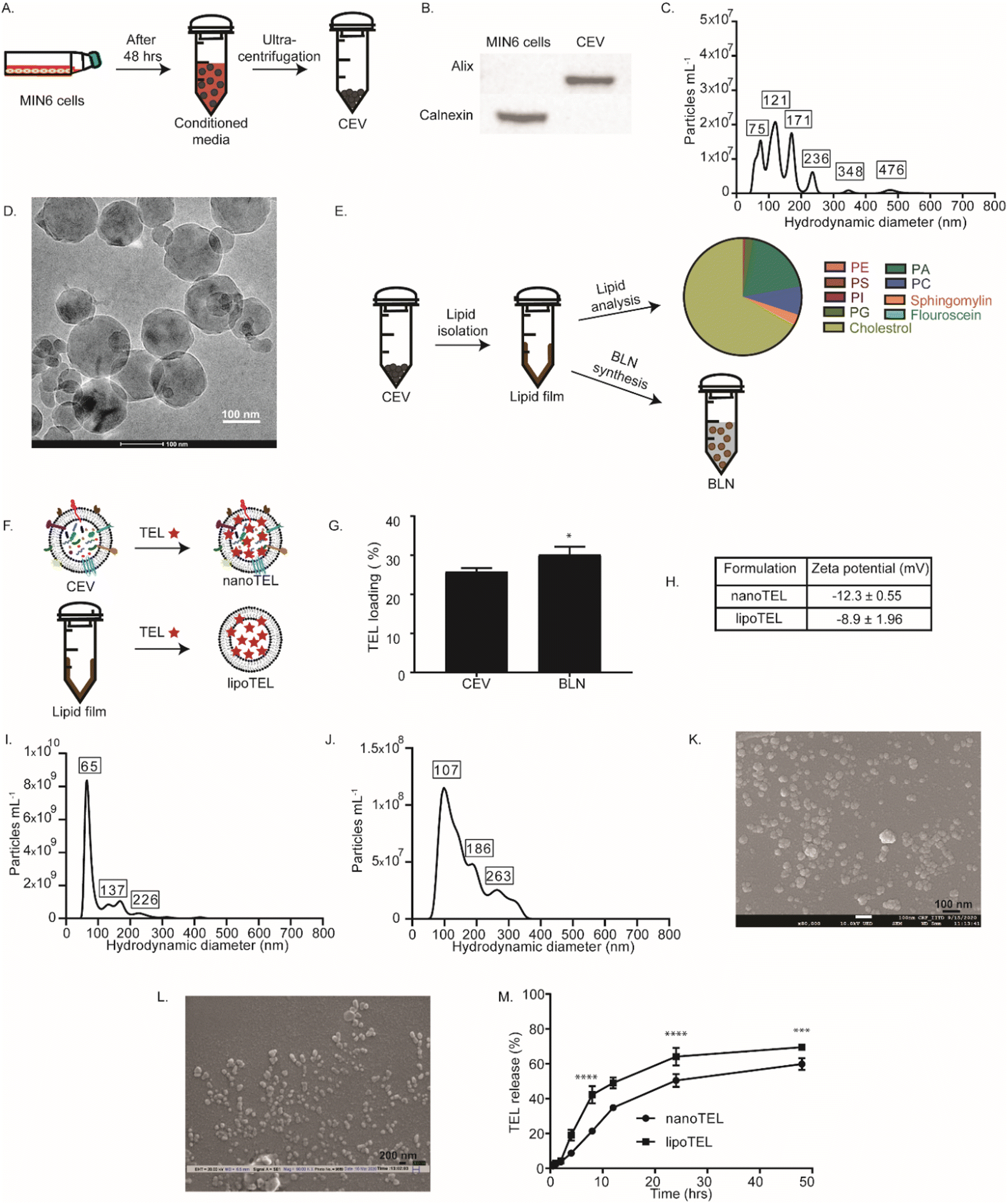 | ||
| Fig. 1 Synthesis and physiochemical characterization of TEL-nanoformulations. (A) Schematic showing the isolation of CEVs from MIN6 conditioned media. (B)–(D) Characterization of CEVs. Western blot analysis (B), NTA (C), and cryoHRTEM images (D) of CEVs. (E) Lipidomic analysis of CEVs and schematic of BLN synthesis. (F) Illustration depicting the encapsulation of TEL inside the CEN and BLN. (G) Percentage loading of TEL into the CEV and BLN. The t-test was performed to check the statistical significance. *P < 0.0257 where * represents w.r.t. CEV, n = 3. (H) Zeta potential of nanoTEL and lipoTEL. (I) and (J) NTA of nanoTEL (I) and lipoTEL (J). (K) and (L) SEM images of nanoTEL (K) and lipoTEL (L). (M) Release profile of TEL from nanoTEL and lipoTEL over a period of 48 hours at pH 7.4. Statistical analysis was determined using two-way ANOVA. *** < 0.0005; **** < 0.0001 where * represents w.r.t. nanoTEL, n = 3. | ||
Next, TEL was encapsulated inside the CEV and BLN with a loading efficiency of 26 ± 1.3 and 29 ± 0.9% respectively (Fig. 1F and G). The zeta potential of the nanoTEL and lipoTEL was found to be −12.3 ± 0.55 and −8.9 ± 1.96 mV, respectively (Fig. 1H and Fig. S1E, F, ESI†). NTA demonstrated that nanoTEL and lipoTEL have hydrodynamic diameters varying from 65 to 236 nm and 107 to 263 nm respectively (Fig. 1I and J). Further, SEM confirmed that after TEL loading, both the CEV and BLN were able to restore the spherical morphology (Fig. 1K and L).
Both nanoTEL and lipoTEL showed an initial burst release of 20 and 40% TEL after 8 hours followed by a slow cumulative release of ∼57 and 60% TEL till 48 hours (Fig. 2M).
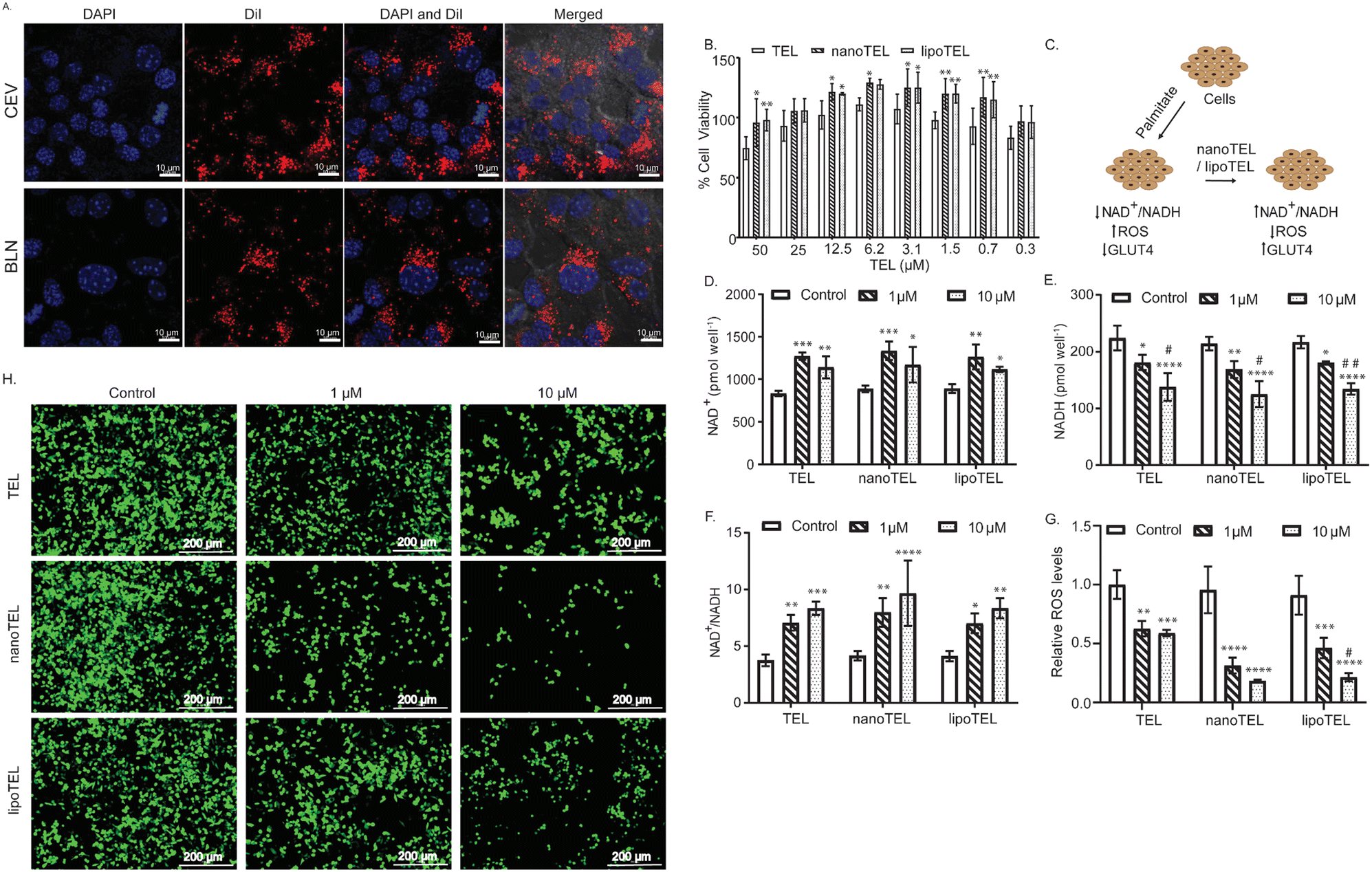 | ||
| Fig. 2 Amelioration of oxidative imbalance in vitro. (A) Confocal microscopy images of cellular uptake of the DiI-labelled CEV and BLN in MIN6 cells at 60× magnification: DiI (red) and nucleus (blue). (B) In vitro viability of MIN6 cells after 48 hours of treatment with TEL, nanoTEL, and lipoTEL determined by the MTT assay. (C) Schematic indicating the in vitro efficacy of nanoTEL and lipoTEL. (D)–(F) Concentration of NAD+ (D), NADH (E), and ratio of NAD+/NADH in MIN6 cells (F). (G) and (H) MIN6 Cells treated with free TEL, lipoTEL, and nanoTEL, followed by carboxy DCF-DA. Relative ROS level in MIN6 cells. The results are expressed as mean ± SD. One-way and two-way ANOVA was performed for statistical significance. *, #P < 0.0463; **, ##P < 0.0087; ***P < 0.0005; ****P < 0.0001, where * represents w.r.t. control and # represents w.r.t. 1 μM TEL concentration, n = 3 (G) and representative fluorescence microscopic images of MIN6 cells. Green fluorescence indicates the intracellular ROS level (H). | ||
3.2. In vitro efficacy
The in vitro uptake of the CEV and BLN was evaluated in both MIN6 cells and C2C12 myotubes. Fig. 2A and Fig. S2, S3A, B (ESI†) demonstrate a significant uptake of the CEV and BLN in both MIN6 cells and C2C12 myotubes. No significant difference in the uptake of the DiI-labelled CEV and BLN was observed in both MIN6 and C2C12 myotubes (Fig. S3A and B, ESI†). Further, the in vitro cyto-compatibility of nanoTEL was compared with lipoTEL and free TEL in MIN6 cells. After 48 hours of treatment, the maximum cell viability for free TEL was found with 6.25 μM, whereas nanoTEL and lipoTEL did not show any toxicity till 25 μM TEL equivalent concentration (Fig. 2B). Since, in diabetes, the increase in NADH levels and the decrease in NAD+/NADH ratio disturb oxidative balance and increase ROS levels and inflammation, we determined the effect of nanoTEL and lipoTEL on the NAD+/NADH ratio, ROS level, GLUT4 expression, and glucose uptake (Fig. 2C).42 Our study showed that free TEL treatment increased the NAD+ level by 1.53 and 1.37 times with 1 and 10 μM respectively (Fig. 2D). Similarly, nanoTEL and lipoTEL treatment also increased the NAD+ level by 1.5 and 1.32 times (nanoTEL) and 1.42 and 1.25 (lipoTEL) times respectively with 1 and 10 μM TEL equivalent dose, indicating that the encapsulation of TEL inside the CEV and BLN did not compromise the efficacy of TEL (Fig. 2D). Further, we observed that free TEL treatment lowered the NADH level by 19.19 (1 μM) and 38.45% (10 μM), whereas 21.25 (1 μM) and 41.63% (10 μM), and 16.93 (1 μM) and 38.08% (10 μM) decrease were observed with nanoTEL and lipoTEL treatment respectively (Fig. 2E). Overall, the NAD+/NADH ratio was increased by 7 and 8.35 times with free TEL, 7 and 8.3 with lipoTEL, and 7.99 and 9.672 times with nanoTEL at 1 and 10 μM TEL equivalent doses, indicating an improved oxidative balance in the insulinoma cells (Fig. 2F).Since, under hyperglycemic condition and exposure to free fatty acids, pancreatic β-cells are prone to generate ROS as they express low levels of anti-oxidative catalase and glutathion,43–45 palmitate and high-glucose-induced ROS levels were analyzed. In MIN6 cells, 1 and 10 μM of free TEL reduced the ROS level by 37.36 and 41.08% whereas 49.12 and 76.46% ROS level were decreased with 1 and 10 μM of TEL equivalent dose of lipoTEL (Fig. 2G). However, nanoTEL further decreased the ROS level by 67.20 and 80.81% with 1 and 10 μM TEL equivalent dose respectively (Fig. 2G). Moreover, the corresponding fluorescence images obtained in the DCF-DA assay further indicated that nanoTEL treatment reduced the ROS level significantly better than TEL and lipoTEL in both MIN6 cells and C2C12 myotubes (Fig. 2H and Fig. S4, ESI†). Next, the effects of free TEL and the nanoformulations on the uptake of glucose and GLUT4 expression in C2C12 myotubes were determined. Free TEL increased the glucose uptake by 1.25 and 1.5 times with 1 and 10 μM TEL concentration respectively (Fig. S5A and B, ESI†). Similarly, treatment with TEL nanoformuations increased the glucose uptake by 1.33 and 1.58 times (nanoTEL) and 1.31 and 1.48 times (lipoTEL), respectively, with 1 and 10 μM TEL equivalent concentrations (Fig. S5A and B, ESI†). Further, we determined the expression of GLUT4, a glucose transporter expressed by murine myotubes. western blot analysis showed that free TEL and nanoformulations significantly enhanced the expression of GLUT4 in C2C12 myotubes (Fig. S5C and D, ESI†). Free TEL increased the GLUT4 expression by 1.2- and 1.4-fold at 1 and 10 μM TEL concentrations (Fig. S5C and D, ESI†). Similarly, nanoTEL and lipoTEL treatment also enhanced the expression of GLUT4 by 1.317- and 1.427-fold (nanoTEL) and 1.184- and 1.287-fold (lipoTEL) in C2C12 myotubes. The above-mentioned data clearly demonstrated that nanoTEL and lipoTEL ameliorated the cellular oxidative imbalance in MIN6 and C2C12 myotubes by increasing the NAD+/NADH ratio, reducing the ROS levels, and enhancing the GLUT4 expression and glucose uptake in the presence of high levels of glucose and palmitate.
3.3. Regulation of blood glucose and serum insulin levels by restoring the structure and function of the islets of Langerhans in the pancreas
To determine and compare the effect of free TEL and the developed nanoformulations, a murine model of STZ-induced T1DM was developed (Fig. 3A). The treatment of nanoTEL significantly reduced the blood glucose levels compared to other groups (Fig. 3B). On day 11, the mean blood glucose level was found to be 528, 462, 446, and 403 mg dL−1 in mice administered with control, free drug, CEV, and lipoTEL respectively. Interestingly, nanoTEL treatment further lowered the mean blood glucose level to 292 mg dL−1 (Fig. 3B). Next, we determined the glucose clearance rate on day 12 by IPGTT and Fig. 3C showed that the blood glucose levels of mice from all groups peaked at 15 min after the glucose administration. However, the blood glucose levels were significantly decreased at all-time points in mice in the nanoTEL treatment group compared to other groups (Fig. 3C).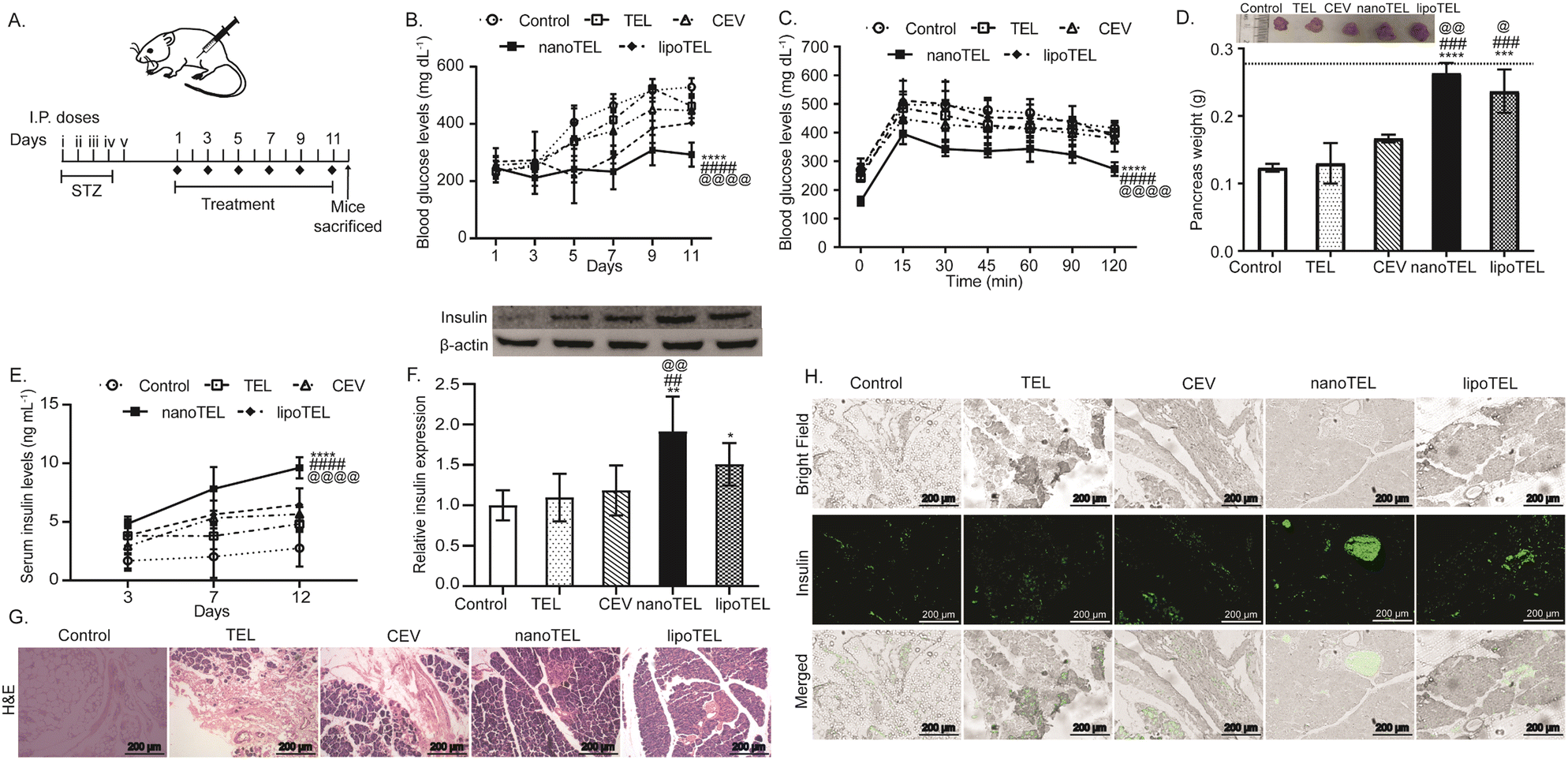 | ||
| Fig. 3 Regulation of blood glucose and insulin levels, and restoration of the structure and function of the islets of Langerhans. (A) Schematic of the in vivo experiment. (B) Blood glucose levels of mice on every alternate day. (C) Fasting blood glucose level during IPGTT on day 12. (D)–(H) On day 12, all the mice were sacrificed. Blood samples were collected and the pancreas were excised out; wet weight and representative images of pancreas. The dashed line indicates the average wet weight of the pancreas from the healthy littermates (D), serum insulin levels on different days (E), relative insulin expression in the pancreas (F), morphological analysis of the isolated pancreas (G), and IF staining indicating insulin-positive cells in the pancreas (H). The data are presented as mean ± SD and the statistical significance was calculated using two-way ANOVA. *, @, P < 0.0350; **, ##, @@ < 0.0046; ***, ###, @@@ < 0.0008; ****, ####, @@@@ < 0.0001 where * represents w.r.t. control and # represents w.r.t. TEL, @ represents w.r.t. CEV, n = 3. | ||
Since, STZ administration induces apoptosis of pancreatic β-cells and damages the pancreatic islets with a drop in pancreatic weight,46–48 and all the mice were sacrificed at the end of the study and the pancreas was collected. On day 12, the mean wet weight of the pancreas in mice treated with nanoTEL was 0.26 g, which is close to the weight of the pancreas of healthy control, as indicated by the dashed line, whereas 0.12, 0.13, 0.16, and 0.23 g were observed respectively with control-, TEL-, CEV-, and lipoTEL-treated mice (Fig. 3D). Fig. 3E shows that the serum insulin levels for nanoTEL and lipoTEL treatment were 9.62 and 6.46 ng mL−1 which are 3.5 and 2.3 times higher than the control group, indicating the increased synthesis and secretion of insulin. Further, insulin expression in the pancreatic tissue was analyzed on day 12 by western blot analysis and we found that nanoTEL- and lipoTEL-treated mice showed 1.9 and 1.5 times higher expression compared to the control (Fig. 3F). At the same time, H&E staining of pancreatic tissue indicated that nanoTEL and lipoTEL treatment restored the islets of Langerhans (Fig. 3G). H&E data showed that control mice had the least or negligible area of the islets of Langerhans in the pancreas, whereas small, distorted, and irregular islets, with few cells were present in mice that received TEL and CEV treatment on day 12 (Fig. 3G). Moreover, lipoTEL-treated mice on day 12 had a significantly large area of islets of Langerhans with more cells, though these islets were not spherical (Fig. 3G). Interestingly, at day 12, nanoTEL treatment restored the structure of pancreatic islets while maintaining close proximity with their surrounding tissues (Fig. 3G). Additionally, the IF staining of pancreas showed that nanoTEL-treated mice had a higher number of insulin-positive cells compared to the other groups on day 12 (Fig. 3H). These results indicated that nanoTEL is superior to lipoTEL and other groups on regulating blood glucose level and improving structure and function of islets of Langerhans in the pancreas of STZ-induced diabetic mice.
3.4. Modulation of NF-κBp65 and PPAR-γ expression and cellular proliferation in the pancreas
In mice, STZ administration enhances the expression of NF-κBp65, which is responsible for the increased secretion of pro-inflammatory cytokines including TNF α and IL-1β and induces apoptosis of the pancreatic β-cells (Fig. 4A).43 Previous studies have demonstrated that the NF-κBp65 inhibitor can restore the structure of islets of Langerhans and enhance the expression of insulin in the pancreas.49–51 In our study, all the mice were sacrificed on day 12 and the expression of NF-κBp65, HIF-1α, PPAR-γ, CD31, and Ki-67 in the pancreatic tissue was measured. On day 12, the CEV and free TEL lowered the expression of NF-κBp65 in the pancreas of STZ-induced diabetic mice by 43 and 52%, respectively compared to control (Fig. 4B). Interestingly, at the same time, 69 and 73% reduction in NF-κBp65 expression in comparison to the control was observed in mice that received lipoTEL and nanoTEL treatment respectively (Fig. 4B). Hyperglycemia-induced hypoxia elevates HIF-1α expression and affects insulin synthesis, and treatment with HIF-1α inhibitor showed the restoration of normoglycemia.52,53 On day 12, free TEL and CEVs reduced the pancreatic HIF-1α expression by 28 and 27% respectively, compared to control, whereas lipoTEL treatment depicted a significant reduction of 48% (Fig. 4C). Interestingly, nanoTEL treatment further reduced the pancreatic HIF-1α expression by 52% compared to the control within 12 days of treatment (Fig. 4C).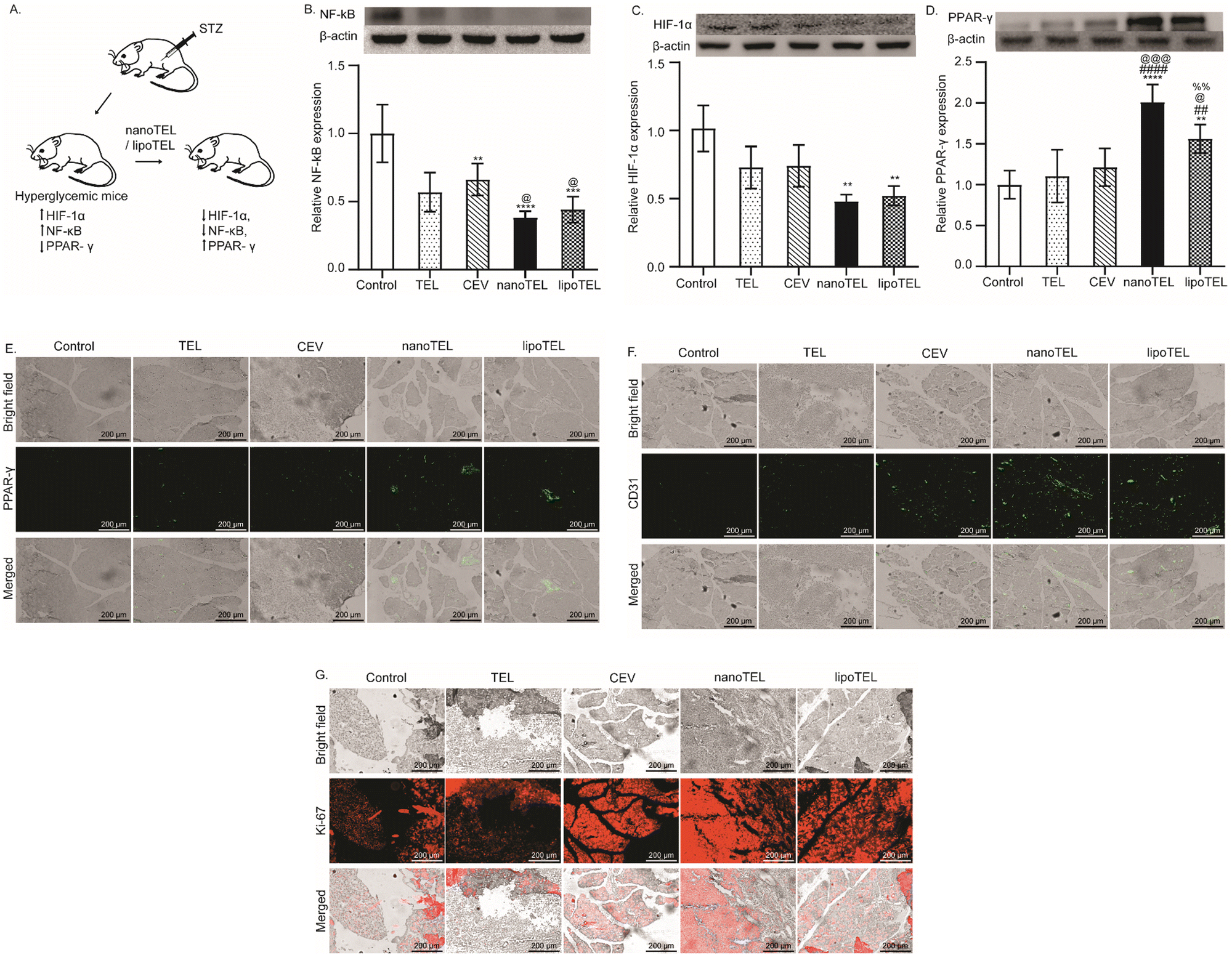 | ||
| Fig. 4 Regulation of inflammation and cellular proliferation in the pancreas of STZ-induced diabetic mice. (A) Schematic depicting the regulation of inflammation and cellular proliferation in the pancreas. (B)–(G) All the mice were sacrificed on day 12 and the pancreas were excised out. Relative expression of NF-κBp65 (B), HIF-1α (C), and PPAR-γ (D) in the pancreatic tissue determined by western blot. Representative IF images indicating the expression of PPAR-γ (E), CD31 (F), and Ki-67 (G) in the pancreatic tissue sections. The results are presented as mean ± SD, and the statistical significance was determined using the two-way ANOVA test. @P < 0.0450; **, ##, %%P < 0.0086; ***, @@@ < P 0.0009; ****, #### < 0.0001 where * represents w.r.t. control, # represents w.r.t. TEL, @ represents w.r.t. CEV, % represents w.r.t. nanoTEL, n = 3. | ||
PPAR-γ protects the pancreatic β-cells from pro-inflammatory cytokine-induced cytotoxicity and also stimulates macrophage polarization from type 1 macrophage (M1) to type 2 macrophage (M2).54–58 However, STZ administration reduces the PPAR-γ expression in the pancreas of STZ-induced diabetic animals.59 On day 12, free TEL and CEV treatment increased the pancreatic expression of PPAR-γ by 10 and 20% respectively compared to control, whereas lipoTEL showed a 50% increase (Fig. 4D). Interestingly, when mice were treated with nanoTEL, a 2-fold increase in PPAR-γ expression over control was observed on day 12 (Fig. 4D). Similarly, IF data also demonstrated that free TEL and CEV treatment moderately increased the PPAR-γ expression in comparison to the control on day 12, whereas lipoTEL treatment significantly enhanced the expression (Fig. 4E). Interestingly, on day 12, nanoTEL treatment outperformed other treatments and depicted the highest PPAR-γ expression in the pancreatic tissue section (Fig. 4E). Hyperglycemia inhibits the pancreatic angiogenesis and its reversal leads to increased proliferation of pancreatic β-cells.60–62Fig. 4F indicates that free TEL and CEVs increased the expression of CD31, an endothelial cell marker used to detect angiogenesis, in the pancreas compared to the control in 12 days, which was further enhanced by lipoTEL. Strikingly, nanoTEL treatment exhibited the highest expression of CD31 in the pancreatic section compared to the control at the same time point (Fig. 4F). Next, we checked the Ki-67 expression to determine the cellular proliferation in the pancreas. Fig. 4G demonstrates that, on day 12, treatment with free TEL and CEVs supported cellular proliferation in the pancreas as the Ki-67 expression was increased in comparison to the control while the expression was further increased with lipoTEL treatment (Fig. 4G). Interestingly among all the groups, nanoTEL depicted the highest pancreatic expression of Ki-67 on day 12 (Fig. 4G). The above-mentioned data clearly indicated that nanoTEL outperformed lipoTEL, free TEL, and CEVs in modulating NF-κBp65 and PPAR-γ expression and promoting cellular proliferation in the pancreas.
To check if the in vivo efficacy of TEL-nanoformulations is due to the better accumulation in different organs, we determined the biodistribution of nanoformulations in mice. The DiI-labelled CEV and BLN showed a 4.7 and 3.8-fold higher accumulation than that of the free DiI in the pancreas 24 hours post administration (Fig. S6A, ESI†). Equally importantly, CEVs lowered the accumulation of DiI in the heart and lung compared to the BLN, whereas the accumulation of CEVs, BLNs and free DiI was found similar in the liver and kidneys (Fig. S6B–E, ESI†). Hyperglycemia-induced inflammation extends beyond the pancreas, affecting various metabolic and biochemical processes in the liver and kidneys.63–65 In addition, the increased ROS level, excessive generation of free radicals, advanced glycation end products and infiltration of proinflammatory macrophages contribute to the liver and kidney dysfunction.63–65 Previous studies have demonstrated that TEL can ameliorate hyperglycemia-induced liver and kidney toxicity.65–70 Hence, the accumulation of CEVs and BLN-based TEL nanoformulations in the liver and kidneys can help in the management of diabetes-associated complications.
3.5. Amelioration of anti-inflammatory-to-pro-inflammatory cytokine ratios in the serum of STZ-induced diabetic mice
Since STZ administration induces pancreatic β-cell apoptosis by increasing pro-inflammatory cytokine IL-6, TNF-α, and IL-1β and decreasing anti-inflammatory cytokine IL-4 and IL-10 levels, we sacrificed all the mice on day 12 and determined the serum concentration of IL-6, TNF-α, IL-1β, IL-4, and IL-10 (Fig. 5A).71–75 In the present study, free TEL and CEV treatment decreased the serum IL-6 and TNF-α levels by 36 and 27% (free TEL) and 41 and 32% (CEV) on day 12 compared to the control, whereas lipoTEL lowered the concentration by 58 and 52% respectively (Fig. 5B and C). Surprisingly, 67 and 69% reduction in serum IL-6 and TNF-α levels over control was observed on day 12, in mice subjected to nanoTEL treatment (Fig. 5B and C). At the same time, the IL-1β level decreased in the serum by 21, 33, and 70% over control, respectively with free TEL, CEV, and lipoTEL treatment; whereas mice treated with nanoTEL showed the least serum IL-1β concentration with 80% reduction compared to the control (Fig. 5D).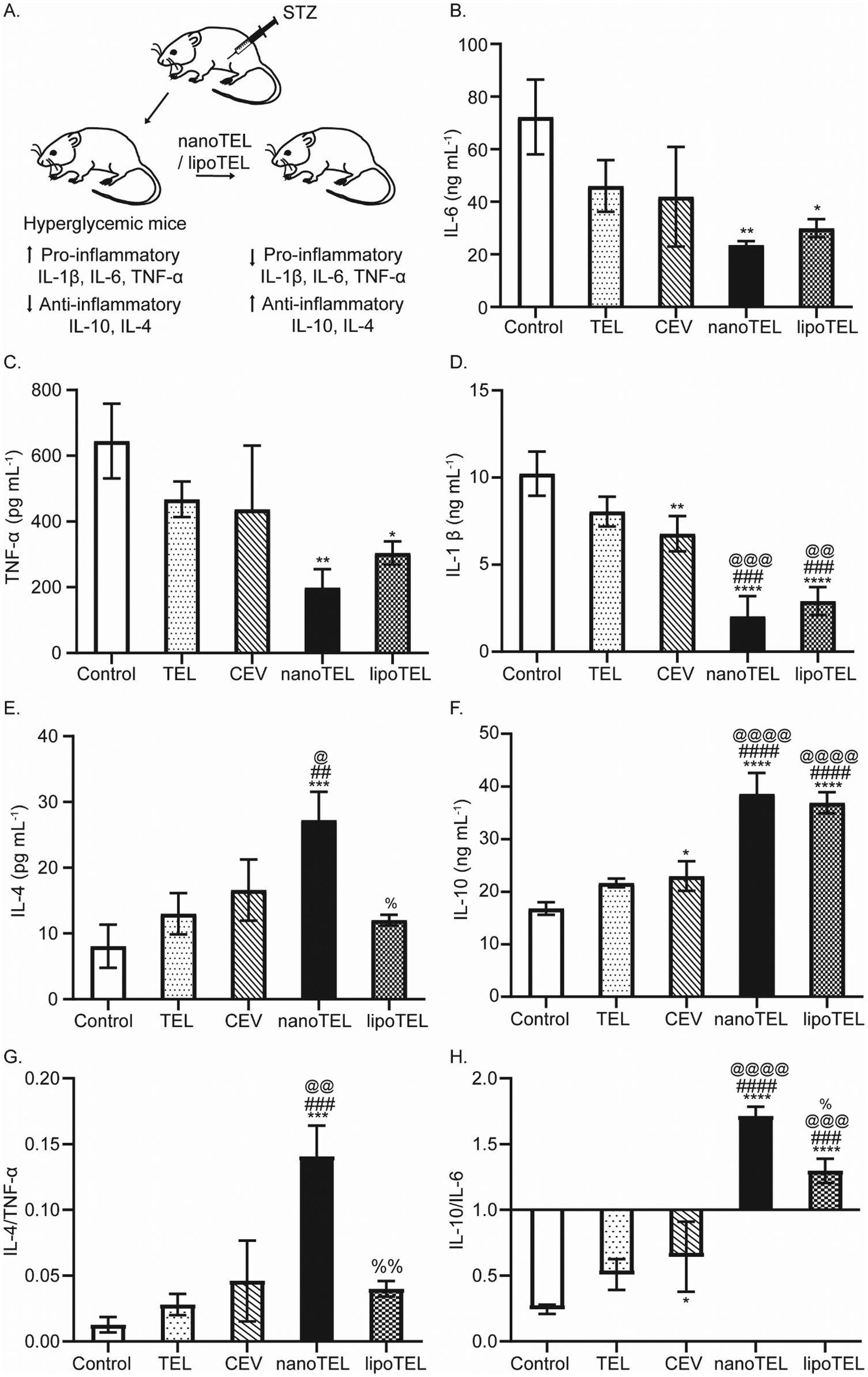 | ||
| Fig. 5 Modulation of anti-inflammatory/pro-inflammatory cytokine ratio in the serum of STZ-induced diabetic mice. (A) Schematic of the TEL-nanoformulation-mediated regulation of cytokine levels. (B)–(H) On day 12, all the mice were sacrificed and blood samples were collected. The serum level of cytokines, IL-6 (B), TNF-α (C), IL-1β (D), IL-4 (E), and IL10 (F). Ratio of serum cytokines, IL-4/TNF-α (G) and IL-10/IL-6 (H). The results are expressed as mean ± SD. Statistical significance among different groups was calculated using the two-way ANOVA test. *, @, %P < 0.0228; **, ##, @@, %%P < 0.0075; ***, ###, @@@ < 0.0009; ****, ####, @@@@ < 0.0001 where * represents w.r.t. control and # represents w.r.t. TEL, @ represents w.r.t. CEV, % represents w.r.t. nanoTEL, n = 3. | ||
In contrast, a 3.37-fold increase in the serum IL-4 level was observed in mice that received nanoTEL treatment compared to control on day 12, whereas only 1.6-, 2-, and 1.4-fold was increased with free TEL, CEV, and lipoTEL respectively (Fig. 5E). Similarly, on day 12, free TEL, CEV, and lipoTEL increased the serum IL-10 levels by 1.3-, 1.4-, and 2.2-fold compared to the control, whereas nanoTEL treatment showed the highest increase of 2.3-fold (Fig. 5F). Since the ratio of anti/pro-inflammatory cytokines indicates the balance of various anti/pro-inflammatory factors associated with pancreatic microenvironment, we determined and compared the IL-4/TNF-α and IL-10/IL-6 ratio. As depicted in Fig. 5G, the IL-4/TNF-α ratios of free TEL-, CEV-, and lipoTEL-treated mice was found as 0.03, 0.043, and 0.04 respectively on day 12. Interestingly, nanoTEL treatment, on day 12, exhibited the highest IL-4/TNF-α ratio of 0.14 (Fig. 5G). Similarly, the IL-10/IL-6 ratio was found significantly better in mice that received nanoTEL compared to other groups post 12 days of treatment (Fig. 5H). At the same time, nanoTEL showed a IL-10/IL-6 ratio of 1.7, whereas the ratio was found to be 1.3, 0.5, and 0.64, respectively for lipoTEL, free TEL, and CEV (Fig. 5H). These results clearly indicated that nanoTEL can ameliorate the pancreatic inflammatory microenvironment by regulating the levels of pro- and anti-inflammatory cytokines significantly better than free TEL, CEV, and lipoTEL.
3.6. Promotion of M2 macrophage in the pancreas of STZ-induced diabetic mice
In diabetic mice, hyperglycemia-induced oxidative stress specifically favors the pro-inflammatory M1 macrophage phenotype (Fig. 6A).76–78 This M1 macrophages induce chronic low-grade inflammation, resulting in insulin resistance and loss of pancreatic β-cells in mice.79 In this study, all the mice were sacrificed on day 12 and the macrophage population and expression of CD80 and Arg1 were determined in the pancreatic tissue. On day 12, free TEL and CEV treatment lowered the M1 macrophage population compared to the control (Fig. 6B). However, treatment with lipoTEL further decreased the M1 population as well as total macrophage population in comparison to the control on day 12 (Fig. 6B). Interestingly, at the same time, both total macrophage (F4/80+) and M1 population (F4/80+CD86+) were significantly reduced in the pancreas of mice that received nanoTEL treatment compared to other groups (Fig. 6B). Similarly, western blot analysis depicted 34 and 25% reduction in CD80 expression in the pancreas of mice that received free TEL and CEV treatment on day 12 compared to the control (Fig. 6C). However, on day 12, lipoTEL treatment demonstrated a reduction of 42% compared to the control, whereas the CD80 expression was lowered by 56% in the pancreas of mice treated with nanoTEL (Fig. 6C).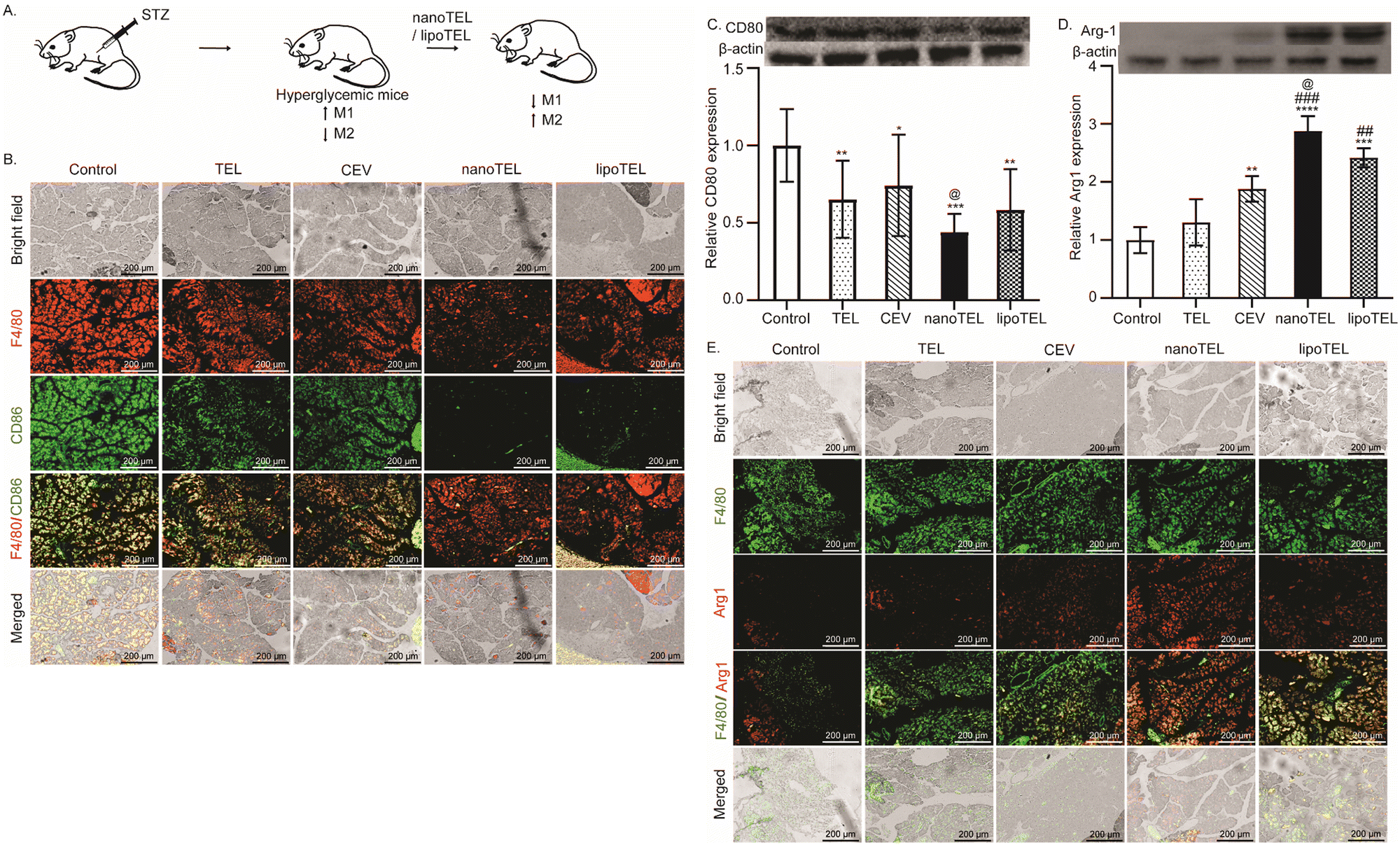 | ||
| Fig. 6 Macrophage population in the pancreas of STZ-induced diabetic mice. (A) Effect of nanoTEL and lipoTEL on macrophages in the pancreas of STZ-induced diabetic mice. (B)–(E) On day 12, all the mice were sacrificed and the pancreas were excised out. Representative IF images of the pancreatic tissue sections indicating F4/80+CD86+ M1 macrophage (B), pancreatic expression of CD80 (C) and Arg1 determined by western blot analysis (D), and representative IF images of the pancreatic tissue sections indicating F4/80+Arg1+ M2 macrophage (E). The results are presented as mean ± SD, and the statistical significance was calculated using two-way ANOVA. **, ##, @@P < 0.0068; ###P < 0.0001; ****P < 0.0001 where * represents w.r.t. control, # represents w.r.t. TEL, @ represents w.r.t. CEV, % w.r.t. nanoTEL, n = 3. | ||
In contrast, free TEL showed no significant change in the expression of Arg1 in the pancreas of mice on day 12, whereas CEV treatment represents a 1.8-fold increase compared to the control (Fig. 6D). At the same time, lipoTEL treatment further increased the expression by 2-fold compared to the control (Fig. 6D), whereas the highest expression of Arg1 was 2.5-fold, observed in mice treated with nanoTEL (Fig. 6D). Moreover, macrophage population on day 12 was further validated by IF analysis, which showed that free TEL and CEV treatment increased the population of F4/80+Arg1+ cells in the pancreas compared to the control (Fig. 6E). Moreover, F4/80+Arg1+ cells were significantly enhanced with lipoTEL treatment and the highest expression was observed in the pancreas of nanoTEL-treated mice on day 12 (Fig. 6E). These findings suggested that nanoTEL has the potential to modulate the macrophage population in the pancreatic microenvironment of STZ-induced diabetic mice significantly better than lipoTEL.
Previous studies have demonstrated that STZ-administration decreases anti-inflammatory macrophage population in mice with an overall increase in total macrophage infiltration in the liver, resulting in hepatotoxicity.80–83 Since our formulation lowered the expression of inflammatory markers (NF-κBp65 and HIF-1α), reduced proinflammatory cytokines, and decreased macrophage population, we checked the effect of our nanoformulations on the serum ALT and AST level. On day 12, serum ALT and AST levels were surged in the control diabetic mice (Fig. S7A and B, ESI†). CEV-treated mice showed a decrease in ALT and AST levels by only 10 and 0.75% respectively, compared to the control (Fig. S7A and B, ESI†). At the same time, treatment with TEL and lipoTEL showed a drop of 64 and 75% respectively, in the serum ALT level compared to the control, whereas AST was decreased by 41 and 45% respectively, which correlate with reduced hepatotoxicity (Fig. S7A and B, ESI†). Interestingly, on day 12, mice that received nanoTEL showed a significant reduction of 75 and 67% respectively in serum ALT and AST levels compared to control (Fig. S7A and B, ESI†). These results clearly suggested that nanoTEL lowered the hepatotoxicity significantly better than lipoTEL, CEV, free TEL and control. These results are consistent with our above-mentioned findings where lipoTEL treatment reduced the macrophage population, proinflammatory cytokines, and expression of inflammatory markers (NF-κBp65 and HIF-1α) while increasing the expression of insulin, PPAR-γ and serum level of anti-inflammatory cytokines.
4. Conclusion
In this study, we have developed two nanoformulations of TEL, nanoTEL and lipoTEL, by encapsulating it inside a CEV and a BLN respectively for the restoration of pancreatic β-cell structure and function. In vitro, nanoTEL improved the oxidative balance, reduced the ROS level, and increased the expression of GLUT4 and uptake of glucose better than lipoTEL and free drug. In a STZ-induced diabetic mice, nanoTEL treatment outperformed lipoTEL by reducing the blood glucose level, improving glucose tolerance, increasing the insulin level, and restoring the pancreatic structure and function through amelioration of the pancreatic inflammatory microenvironment. Hence, our study demonstrated that nanoTEL can be further explored as a potential therapeutic for the amelioration of T1DM and associated hepatotoxicity.Author contributions
A. S. and J. B. designed and conceived the experiments. A. S. performed the experiments, and A. S. and J. B. analyzed the data and wrote the paper. S. P. helped to conduct the animal studies. All authors discussed the results and commented on the manuscript.Ethics approval
All the animal experiments performed in this paper have ethical approval from animal ethical committee, Amity University, Noida.Data availability
The data of this study are available from the corresponding author upon reasonable request.Conflicts of interest
The authors declare that there is no competing interest which could influence the work mentioned in this research article.Acknowledgements
The authors would like to convey their sincere gratitude to the Science and Engineering Research Board (Grant number: ECR/2017/001714), and Indian Council of Medical Research Government of India (Grant no. IIRP-2023-2184) and Indian Institute of Technology Delhi (Grant number: MI02395G) for support of this work. A. S. would like to acknowledge the Council of Scientific and Industrial Research for the doctoral fellowship. We would like to acknowledge the animal facility at Amity University, Noida, U.P. for allowing us to perform the animal studies. We also acknowledge Amit from Institute of Liver and Biliary Sciences for preparing H & E and IF slides. Next, we acknowledgement Witty Tyagi and Prof. Sanjeev Das from National Institute of Immunology for helping us to perform confocal microscopy.References
- A. Chaudhury, C. Duvoor, V. S. Reddy Dendi, S. Kraleti, A. Chada, R. Ravilla, A. Marco, N. S. Shekhawat, M. T. Montales, K. Kuriakose, A. Sasapu, A. Beebe, N. Patil, C. K. Musham, G. P. Lohani and W. Mirza, Front. Endocrinol., 2017, 8, 6 CrossRef PubMed.
- L. Norton, C. Shannon, A. Gastaldelli and R. A. DeFronzo, Metabolism., 2022, 129, 155142 CrossRef CAS PubMed.
- H. Kaneto, N. Katakami, M. Matsuhisa and T. A. Matsuoka, Mediators Inflammation, 2010, 2010, 453892 Search PubMed.
- M. Cnop, N. Welsh, J. C. Jonas, A. Jörns, S. Lenzen and D. L. Eizirik, Diabetes, 2005, 54, S97–S107 CrossRef CAS PubMed.
- S. Tsalamandris, A. S. Antonopoulos, E. Oikonomou, G. A. Papamikroulis, G. Vogiatzi, S. Papaioannou, S. Deftereos and D. Tousoulis, Eur. Cardiol. Rev., 2019, 14, 50–59 Search PubMed.
- M. Kasuga, J. Clin. Invest., 2006, 116, 1756–1760 CrossRef CAS PubMed.
- F. Graus-Nunes and V. Souza-Mello, Biomed. Pharmacother., 2019, 109, 639–645 CrossRef CAS PubMed.
- M. E. Cooper, Am. J. Hypertens., 2004, 17, S16–S20 CrossRef PubMed.
- Y. Saitoh, W. Hongwei, H. Ueno, M. Mizuta and M. Nakazato, Diabetes Metab., 2009, 35, 392–397 CrossRef CAS PubMed.
- A. P. Lakshmanan, K. Watanabe, R. A. Thandavarayan, F. R. Sari, M. Harima, V. V. Giridharan, V. Soetikno, M. Kodama and Y. Aizawa, Free Radical Res., 2011, 45, 575–584 CrossRef CAS PubMed.
- M. A. Ayza, K. A. Zewdie, B. A. Tesfaye, S. T. Gebrekirstos and D. F. Berhe, Diabetes, Metab. Syndr. Obes.: Targets Ther., 2020, 13, 3627–3635 CrossRef CAS PubMed.
- A. Shiota, M. Shimabukuro, D. Fukuda, T. Soeki, H. Sato, E. Uematsu, Y. Hirata, H. Kurobe, N. Maeda, H. Sakaue, H. Masuzaki, I. Shimomura and M. Sata, Cardiovasc. Diabetol., 2012, 11, 139 CrossRef CAS PubMed.
- N. Shigihara, T. Uchida, T. Yorifuji, Y. Toyofuku, M. Tamaki, Y. Fujitani and H. Watada, Diabetol. Int., 2014, 5, 62–68 CrossRef.
- A. J. Battershill and L. J. Scott, Drugs, 2006, 66, 51–83 CrossRef CAS PubMed.
- T. Liu, L. Cui, H. Xue, X. Yang, M. Liu, L. Zhi, H. Yang, Z. Liu, M. Zhang, Q. Guo, P. He, Y. Liu and Y. Zhang, Front. Pharmacol., 2021, 12, 739637 CrossRef CAS PubMed.
- X. Zhao, D. Wu, X. Ma, J. Wang, W. Hou and W. Zhang, Biomed. Pharmacother., 2020, 128, 110237 CrossRef CAS PubMed.
- C. Schindler, A. Collinson, C. Matthews, A. Pointon, L. Jenkinson, R. R. Minter, T. J. Vaughan and N. J. Tigue, PLoS One, 2019, 14, e0214545 CrossRef CAS PubMed.
- M. S. Kim, M. J. Haney, Y. Zhao, V. Mahajan, I. Deygen, N. L. Klyachko, E. Inskoe, A. Piroyan, M. Sokolsky, O. Okolie, S. D. Hingtgen, A. V. Kabanov and E. V. Batrakova, Nanomedicine, 2016, 12, 655–664 CrossRef CAS PubMed.
- D. Sun, X. Zhuang, X. Xiang, Y. Liu, S. Zhang, C. Liu, S. Barnes, W. Grizzle, D. Miller and H. G. Zhang, Mol. Ther., 2010, 18, 1606–1614 CrossRef CAS PubMed.
- X. Zhang, L. Liu, M. Tang, H. Li, X. Guo and X. Yang, Drug Dev. Ind. Pharm., 2020, 46, 1150–1162 CrossRef CAS PubMed.
- X. Zhu, M. Badawi, S. Pomeroy, D. S. Sutaria, Z. Xie, A. Baek, J. Jiang, O. A. Elgamal, X. Mo, K. La Perle, J. Chalmers, T. D. Schmittgen and M. A. Phelps, J. Extracell. Vesicles, 2017, 6, 1324730 CrossRef PubMed.
- Y. Zhao, P. Liu, H. Tan, X. Chen, Q. Wang and T. Chen, Front. Oncol., 2021, 11, 743189 CrossRef CAS PubMed.
- R. Kalluri and V. S. LeBleu, Science, 2020, 367, eaau6977 CrossRef CAS PubMed.
- Y. Sun, Q. Mao, C. Shen, C. Wang and W. Jia, Diabetes, Metab. Syndr. Obes.: Targets Ther., 2019, 12, 2053–2064 CrossRef CAS PubMed.
- Q. Guo, Y. Lu, Y. Huang, Y. Guo, S. Zhu, Q. Zhang, D. Zhu, Z. Wang and J. Luo, Diabetes, Metab. Syndr. Obes.: Targets Ther., 2021, 14, 4767–4782 CrossRef CAS PubMed.
- H. Xu, X. Du, J. Xu, Y. Zhang, Y. Tian, G. Liu, X. Wang, M. Ma, W. Du, Y. Liu, L. Dai, W. Huang, N. Tong, Y. Wei and X. Fu, PLoS Biol., 2020, 18, e3000603 CrossRef CAS PubMed.
- H. Niioka, S. Asatani, A. Yoshimura, H. Ohigashi, S. Tagawa and J. Miyake, Hum. Cell, 2018, 31, 87–93 CrossRef CAS PubMed.
- C. Théry, S. Amigorena, G. Raposo and A. Clayton, Curr. Protoc. Cell Biol., 2006, 30, 3.22.1–3.22.29 Search PubMed.
- S. Bosch, L. de Beaurepaire, M. Allard, M. Mosser, C. Heichette, D. Chrétien, D. Jegou and J.-M. Bach, Sci. Rep., 2016, 6, 36162 CrossRef CAS PubMed.
- C. Guay, V. Menoud, S. Rome and R. Regazzi, Cell Commun. Signaling, 2015, 13, 17 CrossRef PubMed.
- A. Barnwal, S. Ganguly and J. Bhattacharyya, ACS Appl. Mater. Interfaces, 2023, 15, 20012–20026 CrossRef CAS PubMed.
- E. G. Bligh and W. J. Dyer, Can. J. Biochem. Physiol., 1959, 37, 911–917 CrossRef CAS PubMed.
- J. Shen, Z. Jiao, Z. D. Li, X. J. Shi and M. K. Zhong, Pharmazie, 2005, 60, 418–420 CAS.
- N. Torrealday, L. González, R. M. Alonso, R. M. Jiménez and E. Ortiz Lastra, J. Pharm. Biomed. Anal., 2003, 32, 847–857 CrossRef CAS PubMed.
- A. Banerjee, V. Koul and J. Bhattacharyya, Biomacromolecules, 2021, 22, 1885–1900 CrossRef CAS PubMed.
- A. Morales-Kastresana, B. Telford, T. A. Musich, K. McKinnon, C. Clayborne, Z. Braig, A. Rosner, T. Demberg, D. C. Watson, T. S. Karpova, G. J. Freeman, R. H. DeKruyff, G. N. Pavlakis, M. Terabe, M. Robert-Guroff, J. A. Berzofsky and J. C. Jones, Sci. Rep., 2017, 7, 1878 CrossRef PubMed.
- Z. Zhang, J. Yang, W. Yan, Y. Li, Z. Shen and T. Asahara, J. Am. Heart Assoc., 2016, 5, e002856 CrossRef PubMed.
- J. M. Berthiaume, J. G. Kurdys, D. M. Muntean and M. G. Rosca, Antioxid. Redox Signaling, 2019, 30, 375–398 CrossRef CAS PubMed.
- L. Rawat, H. Hegde, S. L. Hoti and V. Nayak, Biomed. Pharmacother., 2020, 128, 110243 CrossRef CAS PubMed.
- P. Manna, A. E. Achari and S. K. Jain, Mol. Cell. Biochem., 2018, 444, 103–108 CrossRef CAS PubMed.
- A. Srivastava, N. Dadheech, M. Vakani and S. Gupta, Biomed. Pharmacother., 2018, 100, 221–225 CrossRef CAS PubMed.
- S. Amjad, S. Nisar, A. A. Bhat, A. R. Shah, M. P. Frenneaux, K. Fakhro, M. Haris, R. Reddy, Z. Patay, J. Baur and P. Bagga, Mol. Metab., 2021, 49, 101195 CrossRef CAS PubMed.
- N. Eguchi, N. D. Vaziri, D. C. Dafoe and H. Ichii, Int. J. Mol. Sci., 2021, 22, 1509 CrossRef CAS PubMed.
- P. Newsholme, K. N. Keane, R. Carlessi and V. Cruzat, Am. J. Physiol.: Cell Physiol., 2019, 317, C420–C433 CrossRef CAS PubMed.
- F. Hao, J. Kang, Y. Cao, S. Fan, H. Yang, Y. An, Y. Pan, L. Tie and X. Li, Apoptosis, 2015, 20, 1420–1432 CrossRef CAS PubMed.
- J. D. Kim, S. M. Kang, B. Il Seo, H. Y. Choi, H. S. Choi and S. K. Ku, Biol. Pharm. Bull., 2006, 29, 477–482 CrossRef CAS PubMed.
- J. A. Koehler, L. L. Baggio, B. J. Lamont, S. Ali and D. J. Drucker, Diabetes, 2009, 58, 2148–2161 CrossRef CAS PubMed.
- D. Zechner, M. Spitzner, A. Bobrowski, N. Knapp, A. Kuhla and B. Vollmar, Diabetologia, 2012, 55, 1526–1534 CrossRef CAS PubMed.
- S. W. Hong, J. Lee, J. H. Cho, H. Kwon, S. E. Park, E. J. Rhee, C. Y. Park, K. W. Oh, S. W. Park and W. Y. Lee, Endocrinol. Metab., 2018, 33, 105–113 CrossRef CAS PubMed.
- A. Farid, P. Moussa, M. Youssef, M. Haytham, A. Shamy and G. Safwat, Saudi J. Biol. Sci., 2022, 29, 103313 CrossRef CAS PubMed.
- E.-K. Kim, K.-B. Kwon, B.-S. Koo, M.-J. Han, M.-Y. Song, E.-K. Song, M.-K. Han, J.-W. Park, D.-G. Ryu and B.-H. Park, Int. J. Biochem. Cell Biol., 2007, 39, 1260–1275 CrossRef CAS PubMed.
- E. Ilegems, G. Bryzgalova, J. Correia, B. Yesildag, E. Berra, J. L. Ruas, T. S. Pereira and P.-O. Berggren, Sci. Transl. Med., 2022, 14, eaba9112 CrossRef CAS PubMed.
- D. Zhang, Y. Liu, Y. Tang, X. Wang, Z. Li, R. Li, Z. Ti, W. Gao, J. Bai and Y. Lv, PLoS One, 2018, 13, 1–13 CAS.
- N. Erfani Majd, M. R. Tabandeh, A. Shahriari and Z. Soleimani, Cell J., 2018, 20, 31–40 Search PubMed.
- D. Gupta, T. Kono and C. Evans-Molina, Diabetes, Obes. Metab., 2010, 12, 1036–1047 CrossRef CAS PubMed.
- H. Kim and Y. Ahn, Diabetes, 2004, 53, S60–S65 CrossRef CAS PubMed.
- Q. Yao, J. Liu, Z. Zhang, F. Li, C. Zhang, B. Lai, L. Xiao and N. Wang, J. Biol. Chem., 2018, 293, 16572–16582 CrossRef CAS PubMed.
- Y. Tian, C. Yang, Q. Yao, L. Qian, J. Liu, X. Xie, W. Ma, X. Nie, B. Lai, L. Xiao and N. Wang, Front. Immunol., 2019, 10, 1895 CrossRef CAS PubMed.
- B. B. Balakrishnan, K. Krishnasamy, V. Mayakrishnan and A. Selvaraj, Biomed. Pharmacother., 2019, 112, 108688 CrossRef CAS PubMed.
- S. Dubois, A. M. Madec, A. Mesnier, M. Armanet, K. Chikh, T. Berney and C. Thivolet, J. Mol. Endocrinol., 2010, 45, 99–105 CAS.
- S. Tan, G. Zang, Y. Wang, Z. Sun, Y. Li, C. Lu and Z. Wang, Diabetes, Metab. Syndr. Obes.: Targets Ther., 2021, 14, 3375–3388 CrossRef PubMed.
- J. Qian, D. Tao, X. Shan, X. Xiao and C. Chen, Lab. Invest., 2022, 102, 290–297 CrossRef CAS PubMed.
- L. Mobasheri, M. Ahadi, A. Beheshti Namdar, M. S. Alavi, A. Bemidinezhad, S. M. Moshirian Farahi, M. Esmaeilizadeh, N. Nikpasand, E. Einafshar and A. Ghorbani, Biomed. Pharmacother., 2023, 167, 115502 CrossRef CAS PubMed.
- R. Jha, S. Lopez-Trevino, H. R. Kankanamalage and J. C. Jha, Biomedicines, 2024, 12, 1098 CrossRef CAS PubMed.
- J. Mohamed, A. H. Nazratun Nafizah, A. H. Zariyantey and S. B. Budin, Sultan Qaboos Univ. Med. J., 2016, 16, e132–e141 CrossRef PubMed.
- P. Balakumar, H. K. Bishnoi and N. Mahadevan, Curr. Diabetes Rev., 2012, 8, 183–190 CrossRef CAS PubMed.
- G. Bakris, E. Burgess, M. Weir, G. Davidai and S. Koval, Kidney Int., 2008, 74, 364–369 CrossRef CAS PubMed.
- Q. Zhang, X. Xiao, M. Li, W. Li, M. Yu, H. Zhang, X. Sun, L. Mao and H. Xiang, J. Mol. Endocrinol., 2012, 49, 35–46 CAS.
- L. M. A. Borém, D. F. Freitas, A. S. Machado, A. F. Paraíso, B. V. Caldas, J. F. R. Neto, J. P. Lima, A. L. S. Guimarães, A. M. B. de Paula and S. H. S. Santos, Egypt. Liver J., 2022, 12, 55 CrossRef.
- T. Hirata, K. Tomita, T. Kawai, H. Yokoyama, A. Shimada, M. Kikuchi, H. Hirose, H. Ebinuma, J. Irie, K. Ojiro, Y. Oikawa, H. Saito, H. Itoh and T. Hibi, Int. J. Endocrinol., 2013, 2013, 587140 Search PubMed.
- F. A. Al-Abbasi, J. King Saud Univ., Sci., 2022, 34, 102249 CrossRef.
- Y. Osada, S. Yamada, A. Nabeshima, Y. Yamagishi, K. Ishiwata, S. Nakae, K. Sudo and T. Kanazawa, Exp. Parasitol., 2013, 135, 388–396 CrossRef CAS PubMed.
- T. M. Preisser, V. P. da Cunha, M. P. Santana, V. B. Pereira, D. C. Cara, B. M. Souza and A. Miyoshi, J. Diabetes Res., 2021, 2021, 6697319 Search PubMed.
- R. Mueller, M.-S. Lee, S. P. Sawyer and N. Sarvetnick, J. Autoimmun., 1996, 9, 151–158 CrossRef CAS PubMed.
- A. Müller, P. Schott-Ohly, C. Dohle and H. Gleichmann, Immunobiology, 2002, 205, 35–50 CrossRef PubMed.
- N. Kattner, Ther. Adv. Endocrinol. Metab., 2023, 14, 20420188231185960 Search PubMed.
- M. Wu, M. Y. Y. Lee, V. Bahl, D. Traum, J. Schug, I. Kusmartseva, M. A. Atkinson, G. Fan and K. H. Kaestner, Cell Rep., 2021, 37, 109919 CrossRef CAS PubMed.
- J. A. Ehses, A. Perren, E. Eppler, P. Ribaux, J. A. Pospisilik, R. Maor-Cahn, X. Gueripel, H. Ellingsgaard, M. K. J. Schneider, G. Biollaz, A. Fontana, M. Reinecke, F. Homo-Delarche and M. Y. Donath, Diabetes, 2007, 56, 2356–2370 CrossRef CAS PubMed.
- E. J. Jeon, D. Y. Kim, N. H. Lee, H.-E. Choi and H. G. Cheon, Sci. Rep., 2019, 9, 1236 CrossRef CAS PubMed.
- T. Abuduyimiti, H. Goto, K. Kimura, Y. Oshima, R. Tanida, K. Kamoshita, N. Leerach, H. Abuduwaili, H. K. Oo, Q. Li, C. M. Galicia-Medina, H. Takayama, K. Ishii, Y. Nakano, Y. Takeshita, T. Iba, H. Naito, M. Honda, K. Harada, Y. Yamamoto and T. Takamura, Am. J. Pathol., 2024, 194, 693–707 CrossRef CAS PubMed.
- S. Niu, Z. Bian, A. Tremblay, Y. Luo, K. Kidder, A. Mansour, K. Zen and Y. Liu, J. Immunol., 2016, 197, 3293–3301 CrossRef CAS PubMed.
- Q. H. Alqahtani, S. Alshehri, A. M. Alhusaini, W. S. Sarawi, S. S. Alqarni, R. Mohamed, M. N. Kumar, J. Al-Saab and I. H. Hasan, Diseases, 2023, 11, 184 CrossRef CAS PubMed.
- A. M. Mahfoz and A. Y. Gawish, Diabetol. Metab. Syndr., 2022, 14, 163 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb00808a |
| This journal is © The Royal Society of Chemistry 2024 |