
Constructing fecal-derived electrocatalysts for CO2 upcycling: simultaneously tackling waste and carbon emissions†
Yanxi
Hu‡
a,
Xintian
Wang‡
a,
Juan
Liu‡
 a,
Xuanzhao
Lu‡
a,
Li
Huang
a,
Yujing
Jiang
a,
Lingyu
Zhao
a,
Jinfeng
Li
b,
Zhizhen
Yin
c,
Jian
Cui
*b,
Wenlei
Zhu
*a and
Yuanyuan
Wang
*a
a,
Xuanzhao
Lu‡
a,
Li
Huang
a,
Yujing
Jiang
a,
Lingyu
Zhao
a,
Jinfeng
Li
b,
Zhizhen
Yin
c,
Jian
Cui
*b,
Wenlei
Zhu
*a and
Yuanyuan
Wang
*a
aSchool of Chemistry and Chemical Engineering, School of the Environment, State Key Laboratory of Pollution Control & Resource Reuse, State Key Laboratory of Coordination Chemistry, Nanjing University, Nanjing 210023, China. E-mail: wenleizhu@nju.edu.cn; wangyy@nju.edu.cn
bInstitute of Botany, Jiangsu Province and Chinese Academy of Sciences, Nanjing Botanical Garden, Mem. Sun Yat-Sen, Jiangsu Engineering Research Center of Aquatic Plant Resources and Water Environment Remediation, Nanjing 210014, China. E-mail: jcui@cnbg.net
cKey Laboratory of Pollutant Chemistry and Environmental Treatment, School of Resources and Environment, Yili Normal University, Xinjiang 835000, Yining, China
First published on 15th January 2025
Abstract
The escalating global fecal waste and rising CO2 levels present dual significant environmental challenges, further intensified by urbanization. Traditional fecal waste management methods are insufficient, particularly in addressing the related health risks and environmental threats. This study explores the synthesis of biochar from pig manure as a carbon substrate to disperse and stabilize Cu nanoparticles, resulting in the formation of an efficient Cu-NB-2000 electrocatalyst for electrocatalytic CO2 reduction (ECR). Through characterization analyses and electrolysis tests, the structure–activity relationships were evaluated, revealing enhanced catalytic activity and selectivity towards value-added multicarbon products. At an optimal potential, the catalyst achieved a remarkable faradaic efficiency for ECR (87.14%) and multicarbon products (44.80%), outperforming previously reported biochar-supported Cu catalysts. The high selectivity was attributed to the substantial presence of pyridine N and the distinctive Cu–N coordination structure. This work introduces, for the first time, a method for preparing an ECR catalyst by loading Cu nanoparticles onto biochar derived from pig manure. This approach not only promotes the valorization of organic waste but also efficiently converts CO2, offering an effective strategy for an integrated “pollution reduction–carbon mitigation” management system.
Introduction
As the worldwide population continues to expand and urbanization progresses, fecal waste has become an increasingly pressing environmental problem. By 2030, the global generation of fecal waste is projected to reach considerable volumes.1 Statistical data show that China produces about 3.8 billion tons of livestock manure each year.2 Fecal waste primarily contains pathogenic and organic pollutants; improper management and subsequent exposure to human and animal feces are closely associated with impaired growth, gastrointestinal illnesses, reduced cognitive abilities, and zoonotic infections.1 Traditional methods for treating fecal sludge include composting, drying beds, digestion, and constructed wetlands, yet these methods do not sufficiently mitigate ecological concerns.3 For instance, serious health risks, such as helminthic diseases, diarrhea, and environmental threats, make its use in irrigation unsuitable.4 Additionally, manure significantly contributes to greenhouse gas emissions, releasing CO2, methane, and nitrous oxide when not properly handled.5,6 Addressing the challenges of global warming and climate change necessitates innovative approaches for converting CO2 into value-added chemicals and feedstocks to lower net CO2 emissions.7,8 In this context, integrated strategies that combine manure pollution treatment with CO2 conversion technologies offer a promising dual benefit of reducing both fecal pollution and carbon emissions.Electrocatalytic CO2 reduction (ECR) technology offers an effective approach for mitigating CO2 emissions by converting CO2 into valuable chemicals and fuels under relatively mild conditions. This method contributes to closing the carbon loop and promoting a circular economy.9–18 Various products, including carbon monoxide (CO), formate (HCOO−), methane (CH4), ethylene (C2H4), ethanol (C2H5OH), and acetate (CH3COO−),19 have been produced. Among these carbonaceous products, multicarbon (C2+) compounds have gained significant attention due to their high energy density and unique chemical functionalities.20 Currently, Cu-based materials are regarded as the most promising catalysts for generating C2+ products, owing to their moderate interaction with reaction intermediates on the Cu surface. However, enhancing C2+ selectivity and improving the kinetics of proton-coupled electron transfer remain essential challenges. Therefore, developing highly efficient Cu-based catalysts to improve their activity towards C2+ product formation is of great importance.21,22
In recent years, significant efforts have been made to enhance the catalytic activity for C2+ products in ECR using Cu-based catalysts, resulting in the development of various approaches for catalyst design, such as surface oxide states, chemical compositions, particle sizes, crystal facets, coordination structures, grain boundaries, and reaction microenvironments.23–26 Notably, the activity and selectivity of carbon-supported Cu catalysts for CO2 electroreduction differ markedly at the triple-phase boundary compared to those at the polycrystalline Cu/solution interface.27 Firstly, one reason for this is the increase in the surface area of the carbon support, which aids in dispersing Cu nanoparticles and improves the diffusion and adsorption of CO2 on the catalyst surface,28 resulting in more effective contact between the active site and the reactant molecule.29 Furthermore, because of sensitivity to oxidation and experimental difficulties associated with the synthesis of small-size Cu nanoparticles,27 heteroatoms like N or O in the carbon support can interact with Cu nanoparticles, stabilizing them on the carbon substrate.30 Additionally, studies have shown that N heteroatoms can lower the reaction energy barrier of CO*, speed up the rate-limiting step, and boost overall catalyst performance.31–34 For instance, N-doped graphene, due to its strong electron-donating properties, not only facilitates C–C coupling but also inhibits the deoxygenation of HOCCH* to C2H4 on the Cu surface, thereby increasing selectivity for C2H5OH formation.35 Lastly, the type of N has a great influence on the selectivity of the product. Experimental and theoretical analyses have demonstrated that pyrrolic N in N-doped carbon can accelerate the hydrogenation of *CO to the *CHO intermediate, leading to high current density and excellent selectivity for CH4, while pyridinic or graphitic N has the opposite effect.36 Therefore, it is crucial to explore specific types of N-doped carbon-supported Cu catalysts for high selectivity to C2+ products.
Activated carbon and carbon black (CB) are commonly used types of carbon supports that exhibit high conductivity and hydrophobicity, making them promising adjustable H2O diffusion media.37 Kong et al.38 regulated H2O diffusion by controlling the thickness of the carbon black layer between the gas diffusion electrode (GDE) and the electrolyte. Since the molar ratio of H2O to CO2 (H2O/CO2) around Cu can effectively adjust the hydrogenation degree of CO2 and regulating H2O diffusion is an effective strategy for adjusting the H2O/CO2 ratio, a volcano-type relationship was observed between the CB layer thickness and the faradaic efficiency (FE) of C2+ products on the Cu catalyst. Biochar, which is a carbon-rich material synthesized through the thermochemical conversion of biomass, exhibits high porosity, cation exchange capacity, specific surface area, and active functional sorption sites. These characteristics make biochar highly effective as a versatile adsorbent and support the development of technologies aimed at achieving negative carbon emissions.39 The straightforward carbonization of fecal sludges to produce biochar adsorbents or catalysts promotes a circular economy by reducing the costs associated with sludge processing. Notably, biochar derived from manure has a larger surface area that effectively adsorbs CO2, thereby enhancing the reactivity of catalyst active sites with CO2.29,40,41 Furthermore, manure inherently contains various nutrients, such as N, that can serve as an intrinsic nitrogen source.42,43 In this study, we embraced the “Pollution Control and Carbon Reduction” design concept by utilizing a hydrothermal method to process pig manure, an organic solid waste, to synthesize porous, high surface area biochar as a carbon substrate. By applying a thermal decomposition method, Cu nanoparticles were loaded on the carbon substrate to create the Cu-NB-2000 electrocatalyst, which is efficient for ECR. We used scanning electron microscopy (SEM), transmission electron microscopy (TEM), X-ray diffraction spectroscopy (XRD), and X-ray photoelectron spectroscopy (XPS) to characterize the catalyst's morphology, composition, crystal structure, and surface valence states. We then assessed the ECR performance of the Cu-NB-2000 catalyst with various Cu loadings and N doping contents by measuring performance parameters such as FE and partial current density for ECR and evaluating their stability through prolonged electrolysis testing.
Results and discussion
Synthesis and characterization
As shown in Fig. 1a, we synthesized fecal-derived biochar using a modified hydrothermal method.28 Cu-NB-2000 was then obtained by the pyrolysis of derived biochar, copper(II) acetylacetonate, and KOH at 800 °C under an Ar atmosphere, with urea as the extra N source (Fig. 1a). Cu-B-2000 was prepared using the same procedure as Cu-NB-2000 but without urea. The SEM images of the Cu-B-2000 catalyst showed many pores on the biochar surface with diameters of approximately 80–120 nm (Fig. S1a and S1b†), belonging to the macroporous range. In comparison, the SEM images of Cu-NB-2000 showed more and smaller pores (Fig. 1b). Most of the pores had diameters of 15–50 nm, falling within the mesoporous range (Fig. 1c), suggesting that N doping increased the proportion of pores on the substrate and reduced the pore size. The presence of abundant mesopores on the surface of Cu-NB-2000 can effectively promote CO2 diffusion to the active sites and enhance the catalyst's performance.44 From the TEM images of Cu-NB-2000, it was observed that Cu nanoparticles were well dispersed on the biochar substrate (Fig. 1d), with particle sizes mainly concentrated between 3 and 5 nm (Fig. 1d inset). Energy-dispersive X-ray spectroscopy (EDS) elemental mapping images of Cu-NB-2000 (Fig. 1e and f) further confirmed a relatively uniform distribution of Cu, N, O, and C elements on the biochar surface, although a small amount of Cu element was aggregated. We synthesized Cu-NC-2000 following the same procedure as Cu-NB-2000 but using activated carbon instead of biochar as the carbon substrate. The SEM image of Cu-NC-2000 showed the aggregated morphology of activated carbon with some mesopores between the particles (Fig. S1c†). There were few nanoparticles on the activated carbon, as shown in the TEM images (Fig. S1d†). EDS elemental mapping images of Cu-NC-2000 indicated a relatively uniform distribution of Cu, N, O, and C elements on the activated carbon substrate (Fig. S2†); however, the content of the N element was extremely low. | ||
| Fig. 1 (a) Scheme of the synthetic process for preparing Cu-NB-2000. SEM images of Cu-NB-2000 at the scale of (b) 10 μm and (c) 10 nm. (d) TEM image of Cu-NB-2000. (e) SEM maps showed the distribution of (f) Cu, N, O, and C elements. | ||
According to the BET characterization results, the surface area of manure-derived biochar was approximately 2.4 m2 g−1 (Table S1† and Fig. 2a). After KOH activation, the surface area of Cu-B-2000 increased nearly 100-fold, reaching 257.2 m2 g−1. Further N doping modification of the biochar substrate resulted in an additional increase, with Cu-NB-2000 reaching a surface area of 421.9 m2 g−1, about twice that of Cu-B-2000. The increase was consistent with the observation in SEM images, where Cu-NB-2000 showed more mesopores than Cu-B-2000. Additionally, Cu-NC-2000, which was synthesized based on activated carbon, had a surface area of 325.5 m2 g−1, which can be attributed to the pores between connected activated carbon. Further pore size and volume analysis (Fig. 2b) revealed that the internal pore diameters of Cu-B-2000, Cu-NB-2000, and Cu-NC-2000 are mainly within the mesoporous range. The larger surface area and pore volume of Cu-NB-2000 favor subsequent Cu nanoparticle loading and adsorption of reactant molecules.
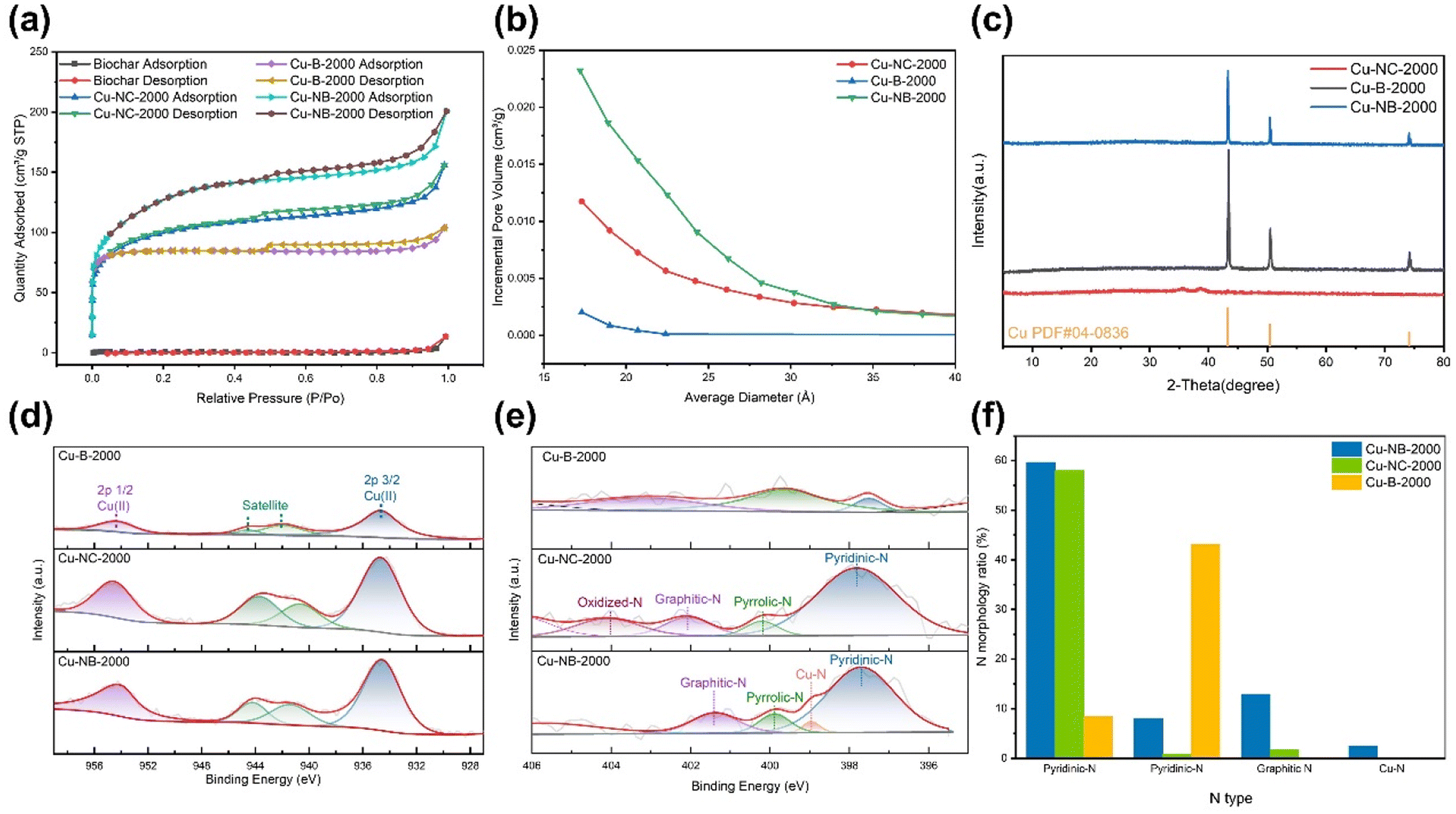 | ||
| Fig. 2 (a) N2 adsorption and desorption isotherms of fecal-derived biochar, Cu-NB-2000, Cu-NC-2000 and Cu-B-2000. (b) The incremental pore volume of Cu-NC-2000, Cu-B-2000, and Cu-NB-2000. (c) XRD patterns of Cu-NC-2000, Cu-B-2000 and Cu-NB-2000. The high-resolution (d) Cu 2p and (e) N 1s XPS spectra of Cu-B-2000, Cu-NC-2000 and Cu-NB-2000. (f) The proportion of different N types for Cu-NB-2000, Cu-NC-2000, and Cu-B-2000 catalysts. | ||
The XRD patterns of Cu-B-2000 and Cu-NB-2000 showed characteristic peaks at 43.3°, 50.4°, and 74.1°, corresponding to the (111), (100), and (110) planes of Cu (Fig. 2c), respectively. Moreover, the facet distributions of Cu-B-2000 and Cu-NB-2000 were similar, with the main plane being (111). The relatively strong intensity of the plane peaks indicated a high loading of Cu nanoparticles for Cu-B-2000 and Cu-NB-2000, consistent with the extensive distribution of Cu elements observed in the EDS map (Fig. 1f). In contrast, the XRD pattern of Cu-NC-2000 showed weaker peaks corresponding to the (111), (100), and (110) planes of Cu, suggesting a lower Cu loading. Therefore, pig manure-derived biochar can be more effective than activated carbon for loading Cu nanoparticles at the same Cu addition level due to its larger surface area and pore volume. Surface elemental valence states and coordination environments of Cu were further revealed by the XPS analysis (Fig. 2d, e and Fig. S3†). The Cu 2p spectra of Cu-B-2000, Cu-NB-2000, and Cu-NC-2000 revealed that the copper nanoparticles on the surface were primarily in the Cu(II) state (Fig. 2d). Since the XRD results for all three catalysts did not show the presence of CuO, the Cu(II) state was attributed to oxidation occurring at the catalyst surface. The N 1s XPS spectra showed five peaks at 405.7 eV, 401.4 eV, 400.0 eV, 399.0 eV, and 397.8 eV, which can respectively be assigned to oxidized N, graphitic N, pyrrolic N, Cu–N, and pyridinic N, considering reasonable experimental errors and noise interference (Fig. 2e).45,46 The N 1s XPS spectra of Cu-B-2000 showed that N mainly existed in the form of graphitic N and pyrrolic N, with a low proportion of pyridinic N (Fig. 2f). For Cu-NB-2000, the N 1s XPS spectra presented pyridinic N, pyrrolic N, and graphitic N, with pyridinic N constituting a significant proportion of the total nitrogen content, alongside a unique Cu–N coordination structure. Compared to the other forms of N, the N atom in pyridinic N can act as a Lewis base, and it is easier to provide the lone pair of electrons and form coordination bonds with metal ions. The N 1s XPS spectra of Cu-NC-2000 showed pyridinic N, pyrrolic N, graphitic N, and N–O, with the proportion of pyridinic N similar to that in Cu-NB-2000. However, no Cu–N coordination structure was observed in Cu-NC-2000. The Cu loading of Cu-NC-2000 was much less than that of Cu-NB-2000, which may result in the disappearance of Cu–N coordination in Cu-NC-2000.
ECR performances
The ECR activity and selectivity of Cu-B-2000, Cu-NB-2000, and Cu-NC-2000 were evaluated in a flow cell configuration with 1 M KOH as the electrolyte. Steady-state chronoamperometry curves for CO2 electrolysis were recorded at different potentials between −0.71 V and −1.51 V versus the reversible hydrogen electrode (vs. RHE). After 10 minutes of electrolysis, the XPS spectra of Cu-NB-2000 showed that the oxidation state of Cu immediately shifted from Cu(II) to a mixture of Cu(II), Cu(I) and Cu(0) (Fig. S4 and S5†). As shown in Fig. 3a, the FE of ECR (FECO2) for Cu-B-2000, Cu-NB-2000, and Cu-NC-2000 all exhibited an increasing-then-decreasing trend in the voltage range of −0.71 V to −1.51 V (vs. RHE). At −0.91 V (vs. RHE), the FECO2 of Cu-NB-2000 reached 87.14%, higher than that of Cu-NC-2000 (66.40%) and Cu-B-2000 (54.68%). Meanwhile, the FE of the competing HER reaction (FEH2) for Cu-NB-2000 was only 12.48%, indicating that Cu-NB-2000 effectively suppressed the side reaction. At a relatively low voltage range, the monocarbon products of Cu-NB-2000 and Cu-NC-2000 were mainly CO, with small amounts of HCOOH and methane (Fig. 3b). However, at a higher voltage range, the proportion of methane increased, which was consistent with the reported results that large overpotentials always favor methane production.47 However, Cu-B-2000 exhibited a higher proportion of CH4 among its single-carbon products at small overpotentials. The significant production of CH4 may be related to the higher proportion of pyrrolic-N in Cu-B-2000. Pyrrolic-N can accelerate the hydrogenation of *CO to *CHO intermediates, which is likely to enhance the formation of methane.36,48 At an electrode potential of −0.91 V (vs. RHE), the multicarbon products of Cu-B-2000, Cu-NB-2000, and Cu-NC-2000 began to emerge in large quantities (Fig. 3c). Among these, Cu-B-2000 only produced ethylene with a relatively low FE (16.88%), whereas Cu-NB-2000 yielded a variety of multicarbon products including ethylene, ethanol, and acetic acid, demonstrating excellent selectivity for C2+ products with a total FE of 41.37%. The multicarbon products for Cu-NC-2000 included ethylene, ethanol, acetic acid, and propanol, with a total FE of 34.35%. Simultaneously, the current densities of C2+ products (jC2+) for Cu-B-2000, Cu-NB-2000, and Cu-NC-2000 gradually increased. At −1.1 V vs. RHE for the Cu-NB-2000 catalyst, the FEC2+ reached 44.80% and jC2+ rose to 109.8 mA cm−2, which were 1.3 times and 3.1 times higher than those of Cu-B-2000 and Cu-NC-2000, respectively. Furthermore, at a more negative potential of −1.51 V, Cu-NB-2000 achieves a higher FEC2+ of 47.35%, with a jC2+ of 241.2 mA cm−2, demonstrating excellent selectivity for C2+ products. Therefore, it can be concluded that N-doped biochar is more advantageous than N-doped activated carbon for the preparation of ECR catalysts to produce multicarbon products. Compared with the previous research where the products on the C-supported Cu catalyst were mainly C1 products,49–51 Cu-NB-2000 efficiently produces C2+ products, surpassing the performance of most carbon-supported Cu catalysts and all the previously reported biochar-supported Cu catalysts52–57 (Fig. 3d and Table S2†). Due to the mentioned fact that the proportion of pyridinic N in Cu-NB-2000 is similar to that in Cu-NC-2000, and the facet distribution remains consistent, it is speculated that the enhanced Cu loading and the resulting unique Cu–N coordination structure in Cu-NB-2000 contribute to the higher selectivity for multicarbon products.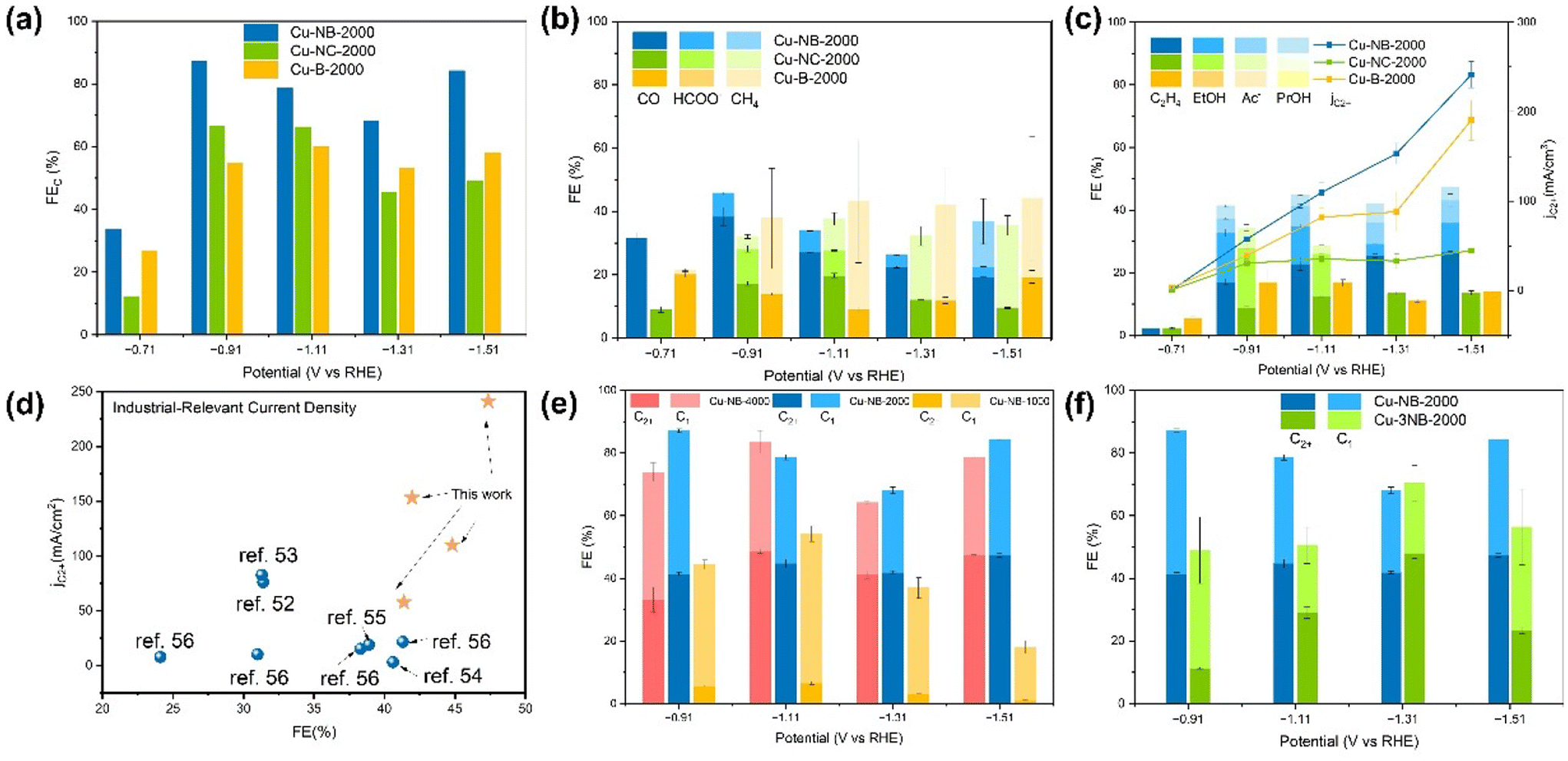 | ||
| Fig. 3 FEs of (a) ECR, (b) respective C1 products, and (c) multicarbon products as a function of applied potential over the Cu-NB-2000, Cu-NC-2000, and Cu-B-2000 samples. (d) jC2+ and FEC2+ were compiled from the literature on carbon or biochar-supported Cu catalysts for ECR. (e) FEs of C2+ and C1 products as a function of applied potential over the Cu-NB-4000, Cu-NB-2000, and Cu-NB-1000 samples. (f) FEs of C2+ and C1 products as a function of applied potential over the Cu-3NB-2000 and Cu-NB-2000 samples. | ||
Since the metal loading of the catalyst is generally approximately positively correlated with the amount of metal precursor, we adjusted the amount of Cu precursor used in the synthesis process, producing Cu-NB-4000 and Cu-NB-1000 catalysts to further investigate the impact of Cu loading on the catalytic performance of Cu-NB-2000. According to the results of inductively coupled plasma optical emission spectrometry (ICP-OES), the copper content in Cu-NB-1000, Cu-NB-2000, and Cu-NB-4000 increased sequentially (Table S3†). Compared to Cu-NB-2000, the FE of C1 products was higher for the Cu-NB-1000 catalyst, which has the lowest Cu loading, but both the FECO2 and FEC2+ were much lower (Fig. 3e). The Cu loading in Cu-NB-4000 was higher than that in Cu-NB-2000, but the FECO2 and FEC2+ were similar. Since reducing Cu loading from Cu-NB-2000 negatively impacted ECR, while increasing Cu loading led to metal waste and decreased electrochemical mass activity, the Cu content in Cu-NB-2000 was demonstrated to be optimal. Moreover, while the ECR selectivity for C2+ products does not fully depend on the Cu loading content, it becomes a significant factor when the loading is insufficient. At the same time, the content of pyridine N might affect the formation of distinct Cu–N bonds, which has a significant impact on its catalytic performance. The Cu-3NB-2000 catalyst was obtained by only increasing the amount of N source by a factor of three. According to the N 1s XPS spectra of Cu-3NB-2000, the proportion of pyridine N was reduced, and Cu–N bonds were not formed (Fig. S5†). Accordingly, the ECR performance significantly decreased, with FEC2+ being only about one-quarter of that of Cu-NB-2000, indicating that the selectivity to C2+ products was highly related to the formation of Cu–N bonds (Fig. 3f).
We conducted a long-duration constant potential test to assess the stability of the Cu-NB-2000 catalyst. During the 8-hour ECR test at −0.91 V (vs. RHE), the current density and FEC2+ remained relatively stable within a certain range (Fig. 4a). The high stability of the Cu-NB-2000 catalyst could be attributed to the formation of Cu–N bonds, where N-doped carbon played a key role in stabilizing the active Cu nanoparticles with high surface energy. The gas diffusion electrode (GDE) with the Cu-NB-2000 catalyst was washed and characterized directly. SEM images revealed that after the long-term test, the surface morphology of Cu-NB-2000 remained constant without aggregated Cu nanoparticles (Fig. 4b). Due to the disturbance of GDE, the intensity of characteristic XRD peaks was not prominent. However, the XRD results of the catalyst after testing also showed the same facets corresponding to the (111), (100), and (110) planes of Cu (Fig. 4c), and no significant changes appeared in facet distribution. The XPS spectra (Fig. 4d) showed that the surface copper mainly existed in the 0 and +1 oxidation states, which were the same as the observed oxidation states before the tests.
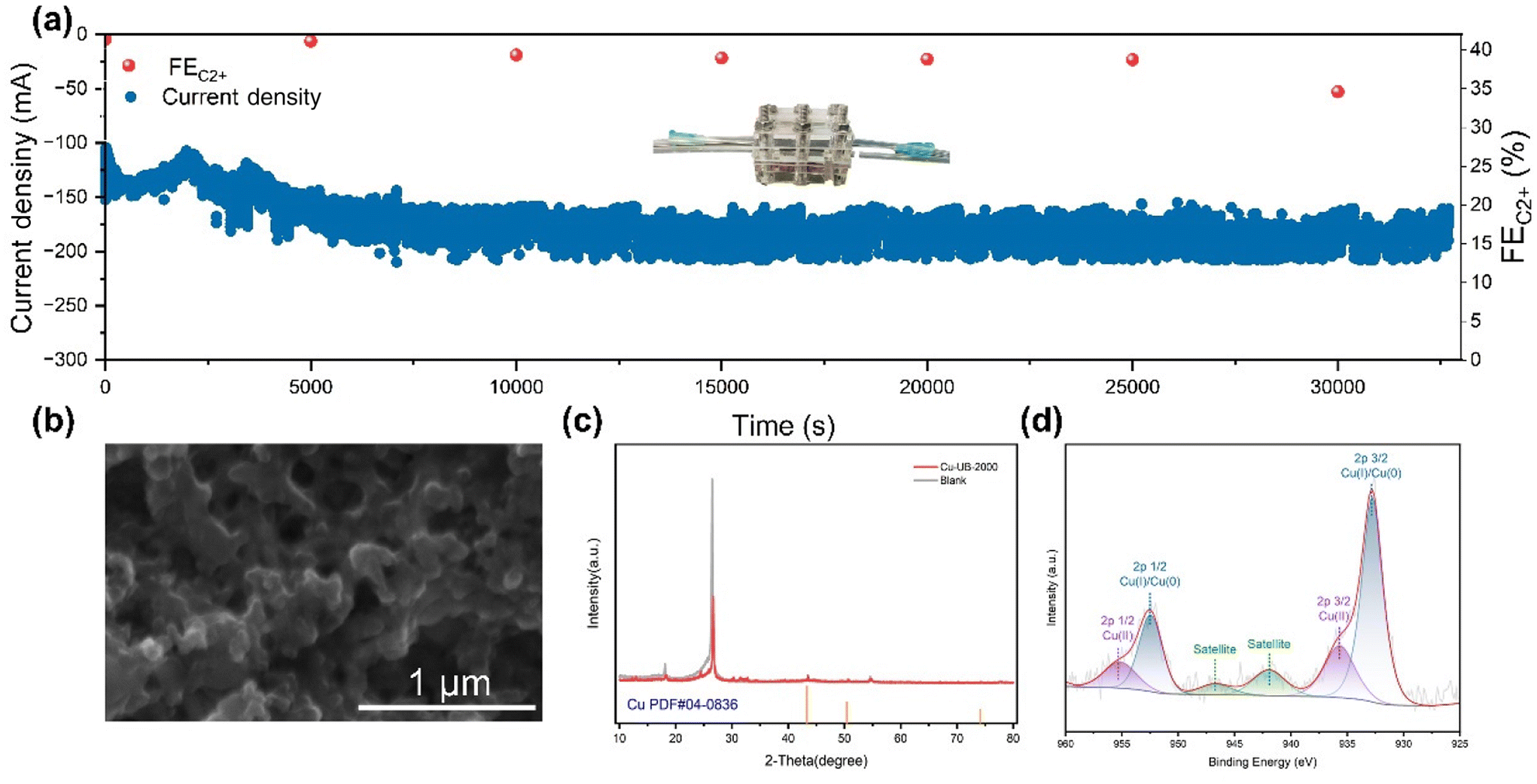 | ||
| Fig. 4 (a) Long-time stability of Cu-NB-2000 in 1 M KOH at an applied j of 150 mA cm−2 in a flow cell reactor, showing stable activity over 542 min. (b) SEM image, (c) XRD and (d) XPS patterns of the GDE (drop-coated with Cu-NB-2000) after the stability test. | ||
Conclusion
In summary, we designed and synthesized a novel manure-derived Cu-NB-2000 electrocatalyst for ECR, which demonstrates a large specific surface area, pore volume, and a higher content of pyridinic N, which innovatively constructs Cu–N bonds. These features improved the catalytic activity and stability of the Cu-NB-2000 electrocatalyst, enhancing the selectivity for ECR and multicarbon products. At −0.91 V, Cu-NB-2000 achieved an FECO2 of 87.14%, and at −1.11 V, it achieved an FEC2+ of 44.80%, surpassing all biochar-based Cu catalysts. Hence, in this work, we have developed an effective electrocatalyst from large quantities of available ultra-low-cost environmental pollutants, capable of efficiently reducing the greenhouse gas CO2 under mild reaction conditions and producing economically valuable chemical products. This is a viable “kill three birds with one stone” strategy with potential economic and environmental benefits. However, the aspects of this material can still be improved: the amount of copper raw material added to Cu-NB-2000 remains relatively high. In the future, we plan to optimize the synthesis method and raw material ratios to reduce copper usage and lower the material's raw material cost. Furthermore, the material's selectivity for specific carbon products is still low. We aim to further enhance the interaction between Cu and biochar to achieve highly selective catalytic conversion of CO2 into a single product.Data availability
Data will be made available on request.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by the Program for Innovative Talents and Entrepreneurs in Jiangsu (020513006012 and 020513006014) and the Zijin Scholars Foundation (0205181022). Y. H. would like to acknowledge the support from College Students’ Innovative Entrepreneurial Training Plan Program. W. Z. would like to acknowledge the support from the National Natural Science Foundation of China (22176086), the Research Funds for the Jiangsu Distinguished Professor Program, the Carbon Peaking and Carbon Neutrality Technological Innovation Foundation of Jiangsu Province (BE2022861), the Research Funds from the Frontiers Science Center for Critical Earth Material Cycling of Nanjing University, the State Key Laboratory of Pollution Control and Resource Reuse, and the Fundamental Research Funds for the Central Universities (021114380214 and 021114380222). Y. J. would like to acknowledge the support from the Fundamental Research Funds for the Central Universities-Cemac “GeoX” Interdisciplinary Program and the Frontiers Science Center for Critical Earth Material Cycling of Nanjing University (2024QNXZ07). J. L. would like to acknowledge the support from the China Postdoctoral Science Foundation (2024M751389), the Natural Science Foundation of Jiangsu Province (BK20241024), and the Jiangsu Provincial Department of the Natural Resources Marine Science and Technology Innovation Project (JSZRHYKJ202311).References
- D. M. Berendes, P. J. Yang, A. Lai, D. Hu and J. Brown, Nat. Sustain., 2018, 1, 679–685 CrossRef PubMed.
- Q. Meng, S. Wang, Q. Niu, H. Yan and Q. Li, Waste Manage., 2021, 127, 1–9 CrossRef CAS PubMed.
- K. Fakkaew, T. Koottatep and C. Polprasert, J. Environ. Manage., 2018, 216, 421–426 CrossRef CAS PubMed.
- B. C. Krueger, G. D. Fowler, M. R. Templeton and B. Moya, Water Res., 2020, 169, 115253 CrossRef CAS PubMed.
- H. Chen, S. K. Awasthi, T. Liu, Y. Duan, X. Ren, Z. Zhang, A. Pandey and M. K. Awasthi, J. Hazard. Mater., 2020, 389, 121908 CrossRef CAS.
- S. Cheng, J. Long, B. Evans, Z. Zhan, T. Li, C. Chen, H. P. Mang and Z. Li, Environ. Res., 2022, 212, 113468 CrossRef CAS.
- F. Wang, J. D. Harindintwali, K. Wei, Y. Shan, Z. Mi, M. J. Costello, S. Grunwald, Z. Feng, F. Wang and Y. Guo, Innov. Geosci., 2023, 1, 100015 CrossRef.
- R. A. Simmer, E. J. Jansen, K. J. Patterson and J. L. Schnoor, ACS Environ. Au, 2023, 3, 195–208 CrossRef CAS PubMed.
- H. Song, J. T. Song, B. Kim, Y. C. Tan and J. Oh, Appl. Catal., B, 2020, 272, 119049 CrossRef.
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef PubMed.
- M. B. Ross, P. De Luna, Y. Li, C.-T. Dinh, D. Kim, P. Yang and E. H. Sargent, Nat. Catal., 2019, 2, 648–658 CrossRef.
- Q. Gong, P. Ding, M. Xu, X. Zhu, M. Wang, J. Deng, Q. Ma, N. Han, Y. Zhu, J. Lu, Z. Feng, Y. Li, W. Zhou and Y. Li, Nat. Commun., 2019, 10, 2807 CrossRef PubMed.
- X.-F. Qiu, J.-R. Huang, C. Yu, Z.-H. Zhao, H.-L. Zhu, Z. Ke, P.-Q. Liao and X.-M. Chen, Angew. Chem., Int. Ed., 2022, 61, e202206470 CrossRef PubMed.
- Y. Hu, Y. Ding, L. Xie, H. Li, Y. Jiang, K. Gong, A. Zhang, W. Zhu and Y. Wang, Carbon, 2024, 230, 119574 CrossRef.
- R. Yang, Y. Cai, Y. Qi, Z. Tang, J. J. Zhu, J. Li, W. Zhu and Z. Chen, Nat. Commun., 2024, 15, 7140 CrossRef PubMed.
- B.-Q. Miao, W.-S. Fang, B. Sun, F.-M. Li, X.-C. Wang, B.-Y. Xia and Y. Chen, Chin. J. Struct. Chem., 2023, 42, 100095 CrossRef.
- T.-J. Wang, W.-S. Fang, Y.-M. Liu, F.-M. Li, P. Chen and Y. Chen, J. Energy Chem., 2022, 70, 407–413 CrossRef.
- J. Ding, T. Wei, T. Hou, W. Liu, Q. Liu, H. Zhang, J. Luo and X. Liu, Nanoscale, 2024, 16, 10628–10636 RSC.
- Y. Cai, R. Yang, J. Fu, Z. Li, L. Xie, K. Li, Y.-C. Chang, S. Ding, Z. Lyu and J.-R. Zhang, Nat. Synth., 2024, 3, 891–902 CrossRef.
- H. Du, L.-X. Liu, P. Li, Q. Min, S. Guo and W. Zhu, ACS Nano, 2023, 17, 8663–8670 CrossRef PubMed.
- U. Nwosu and S. Siahrostami, Catal. Sci. Technol., 2023, 13, 3740–3761 RSC.
- T. Wang, S. Zhang, Q. Liu, Y. Qiu, J. Luo and X. Liu, Acta Phys. -Chim. Sin., 2023, 39, 2207026 Search PubMed.
- W. Ma, X. He, W. Wang, S. Xie, Q. Zhang and Y. Wang, Chem. Soc. Rev., 2021, 50, 12897–12914 RSC.
- M. Kuang and G. Zheng, Chem. Catal., 2023, 3, 100565 CrossRef CAS.
- S. Wei, W. Liu, P. Bai, C. Yang, X. Kong and L. Xu, Eco Environ. Health, 2024, 3, 247–255 CrossRef PubMed.
- X. Li, Y. Chen, X. Zhan, Y. Xu, L. Hao, L. Xu, X. Li, M. Umer, X. Tan, B. Han, A. W. Robertson and Z. Sun, The Innov. Mater., 2023, 1, 100014 CrossRef.
- O. A. Baturina, Q. Lu, M. A. Padilla, L. Xin, W. Li, A. Serov, K. Artyushkova, P. Atanassov, F. Xu, A. Epshteyn, T. Brintlinger, M. Schuette and G. E. Collins, ACS Catal., 2014, 4, 3682–3695 CrossRef CAS.
- T. Zheng, K. Jiang, N. Ta, Y. Hu, J. Zeng, J. Liu and H. Wang, Joule, 2019, 3, 265–278 CrossRef CAS.
- M. Sevilla, J. B. Parra and A. B. Fuertes, ACS Appl. Mater. Interfaces, 2013, 5, 6360–6368 CrossRef CAS.
- P. Hou, W. Song, X. Wang, Z. Hu and P. Kang, Small, 2020, 16, 2001896 CrossRef CAS PubMed.
- L. Dong, D. Feng, Y. Zhang, Z. Wang, Y. Zhao, Q. Du, J. Gao and S. Sun, Carbon Capture Sci. Technol., 2024, 13, 100250 CrossRef.
- A. Vijayakumar, Y. Zhao, K. Wang, Y. Chao, H. Chen, C. Wang and G. G. Wallace, ChemElectroChem, 2023, 10, e202200817 CrossRef.
- H. Tao, F. Wang, Z. Zhang and S. Min, Sustainable Energy Fuels, 2023, 7, 2991–2996 RSC.
- S. Gao, T. Wang, M. Jin, S. Zhang, Q. Liu, G. Hu, H. Yang, J. Luo and X. Liu, Sci. China Mater., 2023, 66, 1013–1023 CrossRef.
- X. Wang, Z. Wang, F. P. García de Arquer, C.-T. Dinh, A. Ozden, Y. C. Li, D.-H. Nam, J. Li, Y.-S. Liu, J. Wicks, Z. Chen, M. Chi, B. Chen, Y. Wang, J. Tam, J. Y. Howe, A. Proppe, P. Todorović, F. Li, T.-T. Zhuang, C. M. Gabardo, A. R. Kirmani, C. McCallum, S.-F. Hung, Y. Lum, M. Luo, Y. Min, A. Xu, C. P. O'Brien, B. Stephen, B. Sun, A. H. Ip, L. J. Richter, S. O. Kelley, D. Sinton and E. H. Sargent, Nat. Energy, 2020, 5, 478–486 CrossRef.
- Y. Wu, C. Chen, X. Yan, R. Wu, S. Liu, J. Ma, J. Zhang, Z. Liu, X. Xing, Z. Wu and B. Han, Chem. Sci., 2022, 13, 8388–8394 RSC.
- X. Kong, C. Wang, Z. Xu, Y. Zhong, Y. Liu, L. Qin, J. Zeng and Z. Geng, Nano Lett., 2022, 22, 8000–8007 CrossRef PubMed.
- X. Kong, C. Wang, Z. Xu, Y. Zhong, Y. Liu, L. Qin, J. Zeng and Z. Geng, Nano Lett., 2022, 22, 8000–8007 CrossRef PubMed.
- N. Mayilswamy, A. Nighojkar, M. Edirisinghe, S. Sundaram and B. Kandasubramanian, Appl. Phys. Rev., 2023, 10, 031308 Search PubMed.
- B. Zhang, D. Song, Y. Liu, Q. Deng, Y. Yuan, Q. Liu and J. Huo, AIChE J., 2023, 69, e18175 CrossRef.
- J. Wang, H. Wang, Z. Han and J. Han, Front. Chem. Sci. Eng., 2015, 9, 57–63 CrossRef.
- E. A. Odey, Z. Li, X. Zhou and L. Kalakodio, Environ. Sci. Pollut. Res. Int., 2017, 24, 23441–23452 CrossRef PubMed.
- R. Harder, R. Wielemaker, T. A. Larsen, G. Zeeman and G. Öberg, Crit. Rev. Environ. Sci. Technol., 2019, 49, 695–743 CrossRef.
- Y. Zhao, Z. Shi, F. Li, C. Jia, Q. Sun, Z. Su and C. Zhao, ACS Catal., 2024, 14, 3926–3932 CrossRef CAS.
- Q. Lai, J. Zhu, Y. Zhao, Y. Liang, J. He and J. Chen, Small, 2017, 13, 1700740 CrossRef.
- H. Miao, S. Li, Z. Wang, S. Sun, M. Kuang, Z. Liu and J. Yuan, Int. J. Hydrogen Energy, 2017, 42, 28298–28308 CrossRef CAS.
- S. Matsuda, M. Osawa and M. Umeda, Electrocatalysis, 2024, 15, 318–328 CrossRef CAS.
- Y. Wu, C. Chen, X. Yan, R. Wu, S. Liu, J. Ma, J. Zhang, Z. Liu, X. Xing, Z. Wu and B. Han, Chem. Sci., 2022, 13, 8388–8394 RSC.
- H. Shang, Z. Zuo, L. Li, F. Wang, H. Liu, Y. Li and Y. Li, Angew. Chem., Int. Ed., 2018, 57, 774–778 CrossRef CAS.
- X. Meng, G. Pan, H. Liu, Y. Qian, X. Wang, C. Wang, L. Hu, H. Wang and Q. Chen, ACS Appl. Mater. Interfaces, 2022, 14, 17240–17248 CrossRef CAS.
- W. Ni, Z. Yixiang, Y. Yao, X. Wang, R. Zhao, Z. Yang, X. Li and Y.-M. Yan, ACS Appl. Mater. Interfaces, 2022, 14, 13261–13270 CrossRef CAS PubMed.
- Y. Zhang, J. Gui, D. Wang, J. Mao, C. Zhang and F. Li, Nanoscale, 2023, 15, 1092–1098 RSC.
- J. Gui, L. Li, B. Yu, D. Wang, B. Yang, Q. Gu, Y. Zhao, Y. Zhu and Y. Zhang, ACS Appl. Mater. Interfaces, 2023, 15, 25516–25523 CrossRef CAS.
- M. Sun, Z. Tao, X. Xu, S. Min and L. Kang, Appl. Catal., A, 2023, 666, 119406 CrossRef CAS.
- Z. Yu, M. Gu, H. Wang, L. Jiang, P. Sun, X. Liu, Y. Pei, G. Wang and X. Zhang, J. Phys. Chem. C, 2024, 128, 4171–4179 CrossRef CAS.
- C. Chen, J. Zhang, Y. Qi, J. Zhang, T. Guo and H. Wang, Fuel, 2024, 372, 132245 CrossRef.
- W. Liu, S. Chen, Z. Mei, L. Li, H. Li, W. Zhao and H. Tao, Surf. Interfaces, 2024, 44, 103608 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr04943h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |