
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulti-target-directed therapeutic strategies for Alzheimer's disease: controlling amyloid-β aggregation, metal ion homeostasis, and enzyme inhibition
Jeasang
Yoo†
 a,
Jimin
Lee†
a,
Byeongha
Ahn
a,
Jiyeon
Han
*b and
Mi Hee
Lim
*a
a,
Jimin
Lee†
a,
Byeongha
Ahn
a,
Jiyeon
Han
*b and
Mi Hee
Lim
*a
aDepartment of Chemistry, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Republic of Korea. E-mail: miheelim@kaist.ac.kr
bDepartment of Applied Chemistry, University of Seoul, Seoul 02504, Republic of Korea. E-mail: jiyeonhan@uos.ac.kr
First published on 6th January 2025
Abstract
Alzheimer's disease (AD) is the most prevalent neurodegenerative dementia, marked by progressive cognitive decline and memory impairment. Despite advances in therapeutic research, single-target-directed treatments often fall short in addressing the complex, multifactorial nature of AD. This arises from various pathological features, including amyloid-β (Aβ) aggregate deposition, metal ion dysregulation, oxidative stress, impaired neurotransmission, neuroinflammation, mitochondrial dysfunction, and neuronal cell death. This review illustrates their interrelationships, with a particular emphasis on the interplay among Aβ, metal ions, and AD-related enzymes, such as β-site amyloid precursor protein cleaving enzyme 1 (BACE1), matrix metalloproteinase 9 (MMP9), lysyl oxidase-like 2 (LOXL2), acetylcholinesterase (AChE), and monoamine oxidase B (MAOB). We further underscore the potential of therapeutic strategies that simultaneously inhibit Aβ aggregation and address other pathogenic mechanisms. These approaches offer a more comprehensive and effective method for combating AD, overcoming the limitations of conventional therapies.
1. Introduction
Among neurodegenerative diseases, Alzheimer's disease (AD) is the most prevalent form of dementia, characterized by a progressive decline in cognitive function and memory.1 Despite extensive research since its discovery in 1906, effective treatments for this fatal disorder continue to be elusive.2 The pathophysiology of AD remains incompletely understood, highlighting the challenges in identifying its underlying causes and their interconnections.3 Pathological features found in AD-affected brains include amyloid-β (Aβ) aggregate deposition, metal ion dysregulation, oxidative stress, impaired neurotransmission, neuroinflammation, mitochondrial dysfunction, and neuronal cell death, as illustrated in Fig. 1a.4–15 While no single hypothesis can fully explain the complex pathology of AD, it is evident that multiple factors simultaneously contribute to its progression.16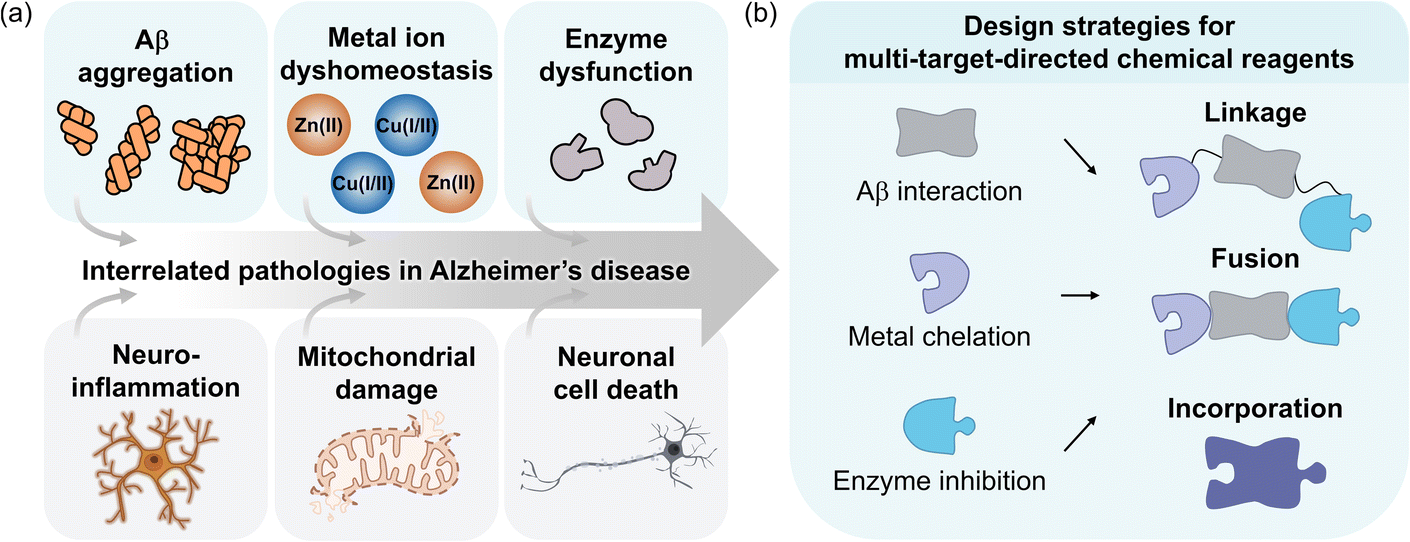 | ||
| Fig. 1 Pathological features in AD and design strategies for multi-target-directed chemical reagents. (a) Multifaceted pathology associated with AD. (b) Schematic illustration of design strategies (linkage, fusion, and incorporation) to develop small molecules against Aβ aggregation, metal ion dyshomeostasis, and enzyme dysfunction simultaneously. | ||
Several U.S. Food and Drug Administration (FDA)-approved or experimental single-target-directed therapies, ranging from small molecules and monoclonal antibodies aimed at disrupting Aβ aggregation17–21 to metal chelators22–27 and neurotransmission enhancers,28–33 have not demonstrated significant disease-modifying effects.3,16,34 Although these treatments effectively modulate specific pathological mechanisms, the multifactorial characteristic of AD likely limits their overall efficacy and can lead to severe side effects. Considering the complexity of AD, it is crucial to prioritize the development of multi-target-directed strategies that engage the multifaceted nature of the disease in drug design and discovery.3,35,36 Given recent advancements in targeting Aβ as a clinical approach and its direct interactions with factors like metal ions and enzymes under pathological conditions, this review primarily illustrates Aβ, metal ions, and AD-related enzymes as pathogenic elements (Fig. 1a). The objective is to provide an overview of integrated therapeutic designs while evaluating the limitations of traditional single-target-directed treatments.17–21 Additionally, this review explores frameworks that simultaneously address these features, focusing on multi-target-directed ligands (MTDLs) and discussing their potential advantages in treating AD, which highlights promising directions for future therapeutic development (Fig. 1b).
2. Single-target-directed drugs and their limitations
2.1 Aβ peptides
In 1984, Aβ peptides were identified as the central component of extracellular amyloid plaques found in the brains of patients.1,37 Since then, they have been recognized as a critical driver of AD pathology, leading to the development of the “amyloid cascade hypothesis,” which remains a prominent theory in understanding AD pathogenesis.1,38 As illustrated in Fig. 2a, Aβ peptides are derived from a transmembrane protein known as amyloid precursor protein (APP).2 APP undergoes processing via non-amyloidogenic and amyloidogenic pathways.44,45 In the amyloidogenic pathway, sequential proteolytic cleavages by β- and γ-secretases generate intrinsically disordered, aggregation-prone Aβ peptides.2,46 Due to the imprecise nature of these cleavages, Aβ peptides are yielded in various forms, such as Aβ1−x, Aβ4−x, Aβ8−x, and Aβ9−x, with Aβ40 and Aβ42 being the most prevalent species found in amyloid plaques (Fig. 2b).46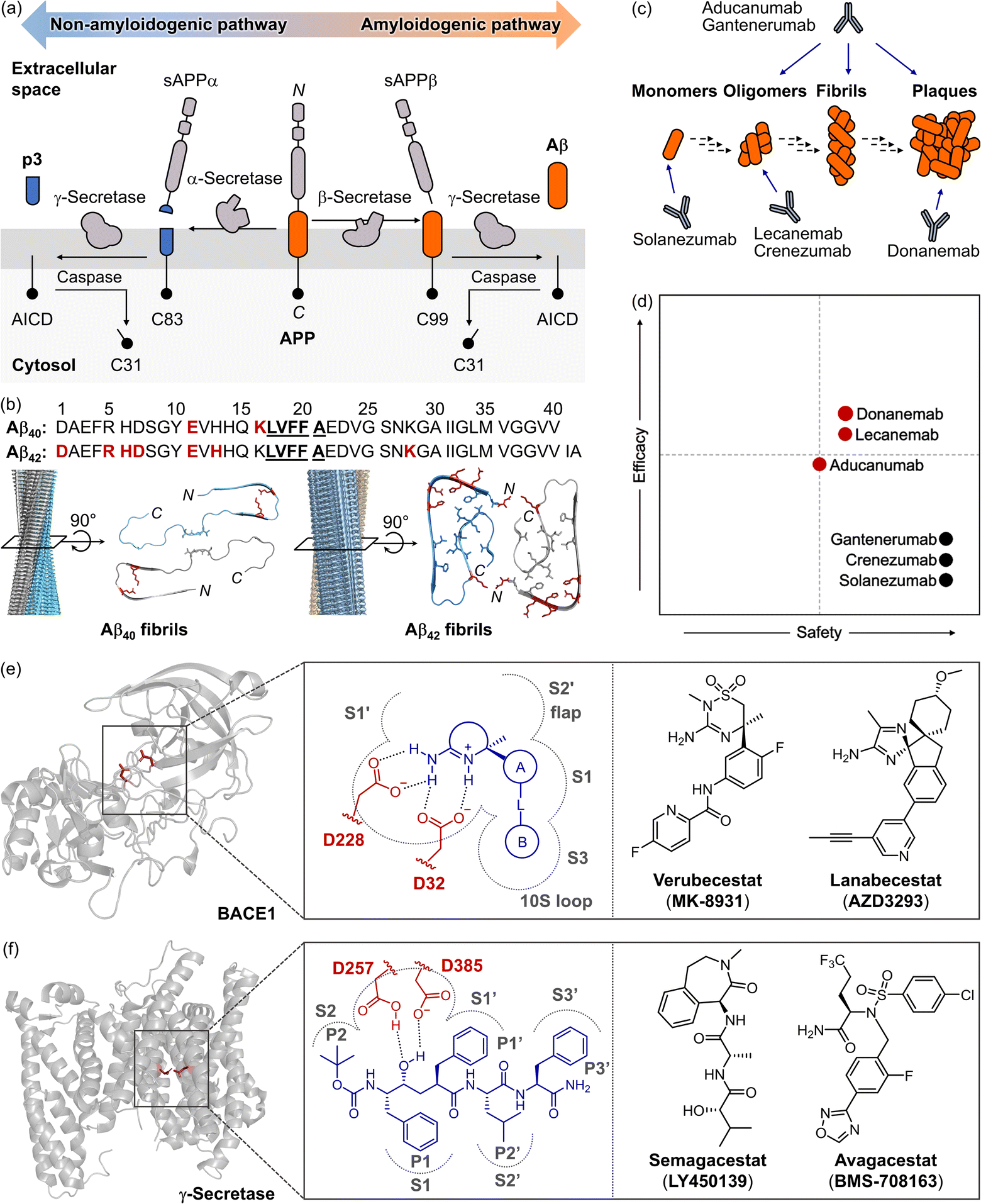 | ||
Fig. 2 Recent drug development against Aβ species or BACE1. (a) Illustration of the proteolytic cleavage of APP. Non-amyloidogenic and amyloidogenic pathways occur depending on the combinational action of α-, β-, and γ-secretases. (b) Amino acid sequences of Aβ40 and Aβ42, with side views of 3D reconstructed Aβ fibrils, and top views of their cross-sections from cryo-EM (for Aβ40, PDB 6SHS39; for Aβ42, PDB 5OQV40). Amino acid residues responsible for the self-recognition sites are highlighted in bold and underlined, while those forming salt bridges in fibrillar structures are indicated in red and depicted as sticks. Core structures of Aβ fibrils are also shown in stick representation. Reproduced with permission from ref. 39 and 40. Copyright© 2019 Springer Nature and 2017 The American Association for the Advancement of Science. (c) Schematic description of Aβ aggregation and Aβ-lowering monoclonal antibodies. (d) Relative safety and efficacy of Aβ-lowering monoclonal antibodies in patients with sporadic early-symptomatic AD. Monoclonal antibodies approved by the U.S. FDA for AD are shown in red dots, while other monoclonal antibodies are presented in black. Reproduced with permission from ref. 41. Copyright© 2023 Springer Nature. (e) Structure of BACE1 (PDB 1W50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 42), zoom-in view of the target site, and BACE1 inhibitors that have reached phase III clinical trials. The catalytic dyad is presented in red and depicted as sticks. (f) Structure of γ-secretase (PDB 5A6343), illustration of the binding pocket, and γ-secretase inhibitors that have progressed to clinical trials. The catalytic dyad is indicated in red with sticks. 42), zoom-in view of the target site, and BACE1 inhibitors that have reached phase III clinical trials. The catalytic dyad is presented in red and depicted as sticks. (f) Structure of γ-secretase (PDB 5A6343), illustration of the binding pocket, and γ-secretase inhibitors that have progressed to clinical trials. The catalytic dyad is indicated in red with sticks. | ||
Under physiological conditions, the levels of Aβ peptides are tightly regulated.47 In the brains of AD patients, however, an imbalance between the production and clearance of Aβ leads to its accumulation.47 Fibrils and plaques are believed to form through intermolecular interactions at the self-recognition and hydrophobic C-terminal regions.39,40 Recent advances in cryogenic electron microscopy (cryo-EM) have elucidated the structures of Aβ fibrils found in the brains of AD patients (Fig. 2b).39,40 Both Aβ40 and Aβ42 fibrils consist of two stacks of peptides, each constructing a right-handed twist along a central axis. In Aβ40 fibrils, the peptide stack typically contains four sets of cross-β sheets spanning residues A2–S8, Y10–H13, Q15–F19, and I32–L34.39 In Aβ42 fibrils, additional interactions, such as salt bridges between D1 and K28, D7 and R5, and E11 with H6 and H13, are observed, with those involving E11 stabilizing a kink in the N-terminal part of the β-sheet around Y10.40 These interactions promote the assembly of monomers into oligomers, protofibrils, and fibrils. Notably, Aβ oligomers are neurotoxic and contribute to cellular dysfunction, inflammation, and synaptic impairment, all of which drive neurodegeneration in AD.48
Given that the accumulation of both soluble and insoluble Aβ aggregates can trigger pathological processes in AD,17 previous research has focused on reducing cerebral Aβ,1 or diminishing its production to alleviate the Aβ burden.49,50 These approaches have been pursued either through the use of anti-amyloid monoclonal antibodies20,41,51 or by inhibiting β- or γ-secretases,49 as illustrated in Fig. 2c–f. Recently, the U.S. FDA has approved three human monoclonal antibodies directed against Aβ (Fig. 2c and d): aducanumab (approved in 2021), lecanemab (approved in 2023), and donanemab (approved in 2024). Each of these therapies has shown potential to slow the progression of AD by targeting specific forms of Aβ: aducanumab addresses soluble oligomers and fibrils;21lecanemab engages with soluble protofibrils;17,18donanemab is designed to target amyloid plaques.19,20 These therapeutics have demonstrated promise in decreasing amyloid plaque burden and potentially delaying cognitive decline.17–21 Despite these encouraging results, the outcomes of clinical trials have often failed to meet expectations.18,20,21,52 Confronting Aβ aggregates with anti-amyloid monoclonal antibodies remains controversial due to mixed treatment outcomes and concerns about adverse effects, such as brain swelling and microhemorrhages, collectively known as amyloid-related imaging abnormalities (ARIA).18,20,21
In recent decades, the inhibition of β-secretases has garnered attention as a therapeutic strategy for AD, with a particular focus on targeting β-site APP cleaving enzyme 1 (BACE1) to reduce the production of Aβ.53 The structure of BACE1, as illustrated in Fig. 2e, includes a protease domain with two catalytic aspartates (D32 and D228) and several sub-pockets within the catalytic clefts (e.g., S1, S1′, S2′, and S3 pockets).42,54 Amidine-based BACE1 inhibitors have been extensively studied, with some advancing to phase II or III clinical trials. These inhibitors interact with the aspartate dyad through salt bridges and specifically bind to the enzyme's active site.54 Notably, verubecestat (MK-8931) and lanabecestat (AZD3293) (shown in Fig. 2e) significantly reduced the levels of Aβ40, Aβ42, and soluble APPβ (sAPPβ) in cerebrospinal fluid (CSF) by up to 80%, accompanied by a modest decrease in plaque load as confirmed by amyloid positron emission tomography (PET) imaging.55,56 Over fourteen β-secretase inhibitors have been developed, showing promising results in lowering Aβ burden in advanced clinical trials. They were discontinued, however, due to adverse effects, including liver toxicity, cognitive worsening, weight loss, sleep disturbances, and skin rashes.52
The inactivation of γ-secretase has also been investigated to control Aβ production and attenuate the progression of AD.43,57,58 As depicted in Fig. 2f, γ-secretase shares a similar structure to that of BACE1, containing two catalytic aspartates (D257 and D385) and five cavities (S1, S1′, S2, S2′, and S3′ pockets) within the active site.43,57,58 Several inhibitors, such as semagacestat (LY450139) and avagacestat (BMS-708163) (Fig. 2f), have been developed to interact with these sites as potential AD treatments.59,60Semagacestat, was the first γ-secretase inhibitor to enter phase III clinical trials, but it was withdrawn due to the lack of cognitive improvement and the emergence of side effects, including further cognitive decline.55,56,59 Additionally, some γ-secretase inhibitors exhibited toxic effects, such as an increased risk of skin cancer, likely resulted from off-target interactions with proteins like the Notch receptor, which is involved in cell proliferation via nuclear signaling.59 Although recent drug approvals targeting Aβ aggregates highlight their relevance as a biomarker, these challenges underscore the limitations of focusing solely on a single pathological factor, such as Aβ species or its associated secretases, to reverse cognitive impairment. The complexity of AD pathogenesis and the unintended effects of these inhibitors emphasize the need for therapeutic strategies that address multiple pathological factors simultaneously.41
2.2 Metal ion dyshomeostasis
Metal ions, such as Fe(II/III), Cu(I/II), and Zn(II), play vital roles in physiological processes, including signal transmission and catalytic reactions, through their coordination to various biological Lewis bases.61–65 These metal ions are distributed between two distinct pools: the static and labile pools. The static pool consists of tightly bound metal ions embedded within proteins, often stabilizing protein structures or occupying catalytic sites of enzymes.61–66 In contrast, the labile pool contains loosely-bound or surface-exposed metal ions that participate in more dynamic processes, such as signal transmission and regulation.61–66The delicate balance between static and labile metal ion pools can be disrupted under pathological conditions, leading to metal ion dyshomeostasis in the brain. In AD-affected regions, such as the hippocampus and amygdala, an increase in unregulated labile copper ions in serum and plasma has been observed.67,68 Additionally, the concentration of Zn(II) in these areas nearly doubles compared to healthy subjects.67 The miscompartmentation of metal ions not only impairs their physiological functions but also induces toxicity, supporting the “metal ion hypothesis” in AD.69 Specifically, the mislocalization of metal ions undermines antioxidant systems against reactive oxygen species (ROS), leading to elevated oxidative stress and severe cytotoxicity.66 Excess ROS can damage biological components, such as lipids and proteins, and induce neuronal death, which further leads to neurodegeneration.69
Extensive research on the regulation of Cu(I/II) and Zn(II) dyshomeostasis has led to the development of several chelators, including trientine, tetrathiomolybdate, dipicolinic acid, ditiocarb, clioquinol, and PBT2, as illustrated in Fig. 3a.22–27 These chelators are designed to remove excess metal ions concentrated in plaques, prevent redox reactions, and potentially offer therapeutic benefits for AD.22–25,72–76 In addition, clioquinol and an 8-hydroxyquinoline derivative, PBT2, were considered ionophores to restore metal ion homeostasis by coordinating metal ions through their nitrogen and oxygen donor atoms, reintroducing them into physiological systems and preventing their pathological interactions with Aβ.26,27 Despite their initial promise, chelation-based therapies have not been successful in clinical trials, with no significant improvement and severe side effects.25,77–79 The failure of these therapies may be attributed to several factors, including (i) non-specific binding to target metal ions, (ii) retinal and ocular toxicity with unclear mechanisms, and (iii) an inaccurate understanding of metal concentrations in AD.25,77–79
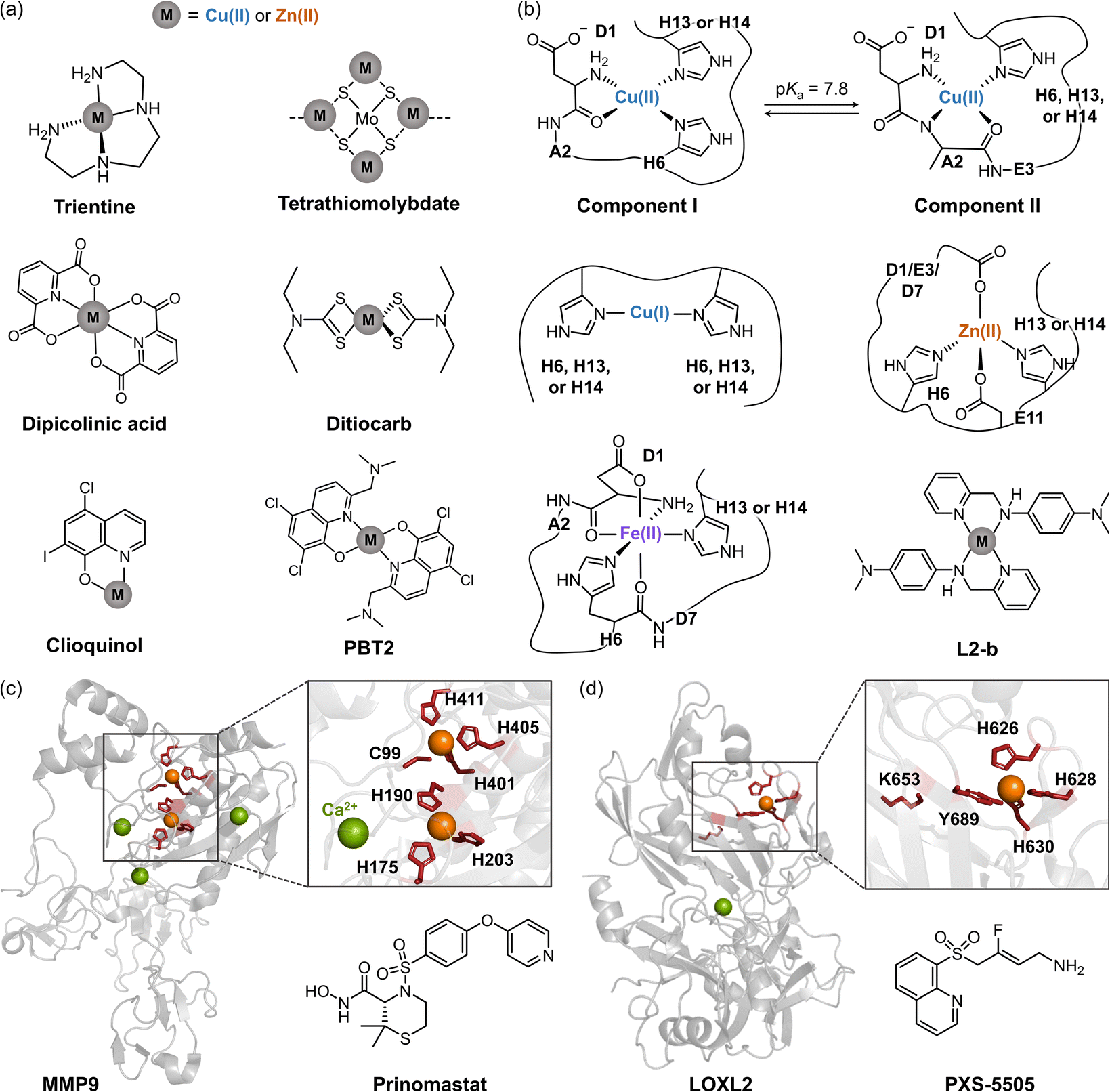 | ||
| Fig. 3 Metal-related pathological factors associated with AD and therapeutic interventions against these components. (a) Examples of metal chelators. (b) Representative coordination modes of Cu(I/II), Zn(II), and Fe(II) to Aβ with a chemical structure of metal-bound L2-b. L2-b, N1,N1-dimethyl-N4-(pyridin-2-ylmethyl)benzene-1,4-diamine. (c) Structures of MMP9 (PDB 1L6J70) and its inhibitor (prinomastat). The Zn(II)-coordination residues are highlighted in red and depicted as sticks. (d) Structures of LOXL2 (PDB 5ZE371) and its inhibitor (PXS-5505). The Cu(II)-coordination residues and catalytic lysine are indicated in red with sticks. Note that Cu(II) occupies the Zn(II)-binding site in human LOXL2, contrary to the crystallographic illustration. | ||
To address these challenges, recent strategies have focused on regulating various pathological features associated with metal ion dyshomeostasis. Excess metal ions can coordinate to amyloidogenic peptides (e.g., Aβ, illustrated in Fig. 3b), accelerating their aggregation and exhibiting cytotoxic effects.69,80,81 The formation of metal–Aβ complexes at the synaptic cleft is well-documented, with metal concentrations in Aβ plaques reaching up to 0.4 mM for copper and 1.0 mM for zinc.82 Specifically, two major Cu(II)-binding modes to Aβ have been identified depending on pH, as shown in Fig. 3b. At pH ≤ 6.5 (Component I), Cu(II) primarily adopts a 3N1O coordination mode involving the N-terminal primary amine, the backbone carbonyl group between D1 and A2, and two imidazole rings from H6 and H13 or H14.83–85 Additional oxygen donor atoms from water molecules or carboxylate groups from D1, E3, D7, or E11 are also proposed.83–85 In contrast, at pH ≥ 8 (Component II), Cu(II) maintains a 3N1O coordination but with different donor atoms: the N-terminal primary amine, a deprotonated amide backbone between D1 and A2, an imidazole ring from H6, H13, or H14, and an oxygen donor atom from the backbone carbonyl group between A2 and E3.69,83,84,86 Cu(I) binds to Aβ in a linear mode, primarily entailing the imidazole rings from H6, H13, or H14, with a preference for engaging H13 and H14.87,88 Moreover, Zn(II) coordinates to Aβ through a 2N2O coordination comprising two oxygen donor atoms from the carboxylate groups of D1, E3, or D7 and E11 and two nitrogen donor atoms from the imidazole rings of H6 and H13 or H14 (Fig. 3b).69,89 Furthermore, although relatively less established, Fe(II) can associate with Aβ through a 3N3O coordination mode. This incorporates three nitrogen donor atoms from the N-terminal primary amine and two imidazoles from H6 and H13 or H14 and three oxygen donor atoms from the backbone carbonyl groups between D1 and A2, between H6 and D7, and the carboxylate group of D1 (Fig. 3b).90,91
These interactions facilitate Aβ aggregation and elevate oxidative stress by either disrupting ROS scavenging systems or overproducing ROS through Fenton chemistry or Fenton-like reactions in the case of redox-active metal ions. For example, the varying coordination modes of Cu(I) and Cu(II), coupled with the conformational flexibility of native peptides, enable redox reactions at the metal center (Eo′ = 0.28 V vs. NHE92), generating ROS.90,93 Note that Fe(II/III) unbound and bound to Aβ is also involved in ROS generation; however, the Eo value could not be accurately determined in aqueous media because of Fe(OH)3 sedimentation.90,91 Considering the impact of metal ions on Aβ aggregation and ROS formation, targeting metal–Aβ complexes, therefore, appears to be a promising therapeutic strategy compared to controlling individual factors.94–96 Lim and coworkers rationally designed a small molecule, L2-b (shown in Fig. 3b), to specifically regulate the reactivities of metal–Aβ complexes.94,97L2-b inhibited metal-induced Aβ aggregation and reduced ROS produced by Cu(I/II)–Aβ complexes.94In vivo studies further demonstrated that L2-b treatment mitigated amyloid pathology and improved cognitive deficits in the 5xFAD mouse model, underscoring the importance of confronting and regulating metal–Aβ interactions.94 Despite the significance of these findings, no clinical trials addressing metal–Aβ species have been reported to date, to the best of our knowledge.
Dysregulated and miscompartmentalized metal ions can significantly impact the structures and functions of metalloenzymes.98–100 Among various metalloenzymes, matrix metalloproteinase 9 (MMP9) and lysyl oxidase-like 2 (LOXL2) have emerged as major pathological factors (depicted in Fig. 3c and d), as they are activated by excess labile zinc or copper ions under pathological conditions.98–105 MMP9, which plays a crucial role in physiological processes, such as embryonic development, angiogenesis, and cell migration, was initially studied for its proteolytic activity against Aβ peptides.103,104
Research in AD mouse models has shown that MMP9 is overexpressed, contributing to neurotoxicity in hippocampal neurons, cognitive deficits, and the degradation of nerve growth factors, thereby implicating MMP9 activation in the progression of AD.104,106,107In vivo studies further suggested that suppressing MMP9 can alleviate neurobehavioral deficits, making it a potential therapeutic target.104,106,107 Current MMP9 inhibitors are designed to bind to its binuclear Zn(II) centers (Fig. 3c). The two Zn(II) ions are coordinated through two main modes: (i) 3N coordination with the imidazole rings of H175, H190, and H205 and (ii) 3N1S coordination with the free thiol of C99 and the imidazole rings from H401, H405, and H411.70 The 3N1S coordination serves as the catalytic center, which becomes activated when the free thiol of C99 dissociates, creating a vacant site. Chelator-based therapeutics, such as prinomastat (shown in Fig. 3c), are being explored for their ability to address this vacant Zn(II)-coordination site and competitively inhibit the active site of MMP9.104,108
In parallel, LOXL2 is another metalloenzyme that contains a catalytic copper center (Fig. 3d).71,109 The LOX family is responsible for the oxidative deamination of ε-amino groups in lysine residues, converting them to hydroxylysine within collagen and elastin chains. This chemical transformation initiates covalent cross-linking in the extracellular matrix, which is crucial for maintaining the tensile strength and elasticity of connective tissues.105,109 The redox-active Cu(I/II) in the LOXL2 active site facilitates this process through the 3N1O coordination with a distorted tetrahedral geometry involving three imidazole rings from H626, H628, and H630 and an additional oxygen donor atom from Y689 (Fig. 3d).71 The catalytic copper then oxidizes the lysine tyrosylquinone (LTQ) cofactor, enabling further deamination of lysines.110
Given the diverse physiological roles of LOXL2, its dysregulation can impact various pathophysiological conditions, such as AD and other vascular diseases.105,114 For example, LOXL2 activation can induce arterial stiffness, leading to brain microvascular damage and Aβ deposition.105 Additionally, suppression of LOXL2 has been shown to enhance the clearance of Aβ aggregates, making it an intriguing therapeutic target.105 LOXL2 inhibitors typically modify the chemical structure of its substrate, the LTQ cofactor, or disrupt the Cu(II)-binding site to halt enzymatic activity.109,110 Fluoroallylamine-based inhibitors, such as PXS-5505 (shown in Fig. 3d), form a covalent bond with the LTQ cofactor, resulting in irreversible termination of its function.110 Other compounds, such as guaiacol derivatives, have also demonstrated modulatory effects against LOXL2 by binding to the catalytic Cu(II) center and showed potential as AD therapeutics.115 Collectively, metal ion dyshomeostasis drives to inappropriate interactions with Aβ and the activation of metalloenzymes, thereby increasing cytotoxicity. These findings suggest that developing multipotent chemical reagents capable of confronting and controlling pathogenic features associated with metal ion dyshomeostasis is a promising therapeutic strategy for AD.116
2.3 Neurotransmitters and related enzymatic systems
Imbalances in neurotransmission caused by the dysregulation of enzymatic systems represent another pathological factor for AD.28,44,103,117–121 Patients with AD often experience diminished signal transmission mediated by neurotransmitters, such as acetylcholine (ACh), dopamine, and serotonin. These neurotransmitters are regulated and degraded by enzymes, including acetylcholinesterase (AChE), monoamine oxidase (MAO), and catechol-O-methyltransferase (COMT).28,117,118 The abnormality in these enzymatic systems has been observed in AD patients, suggesting that targeting neurotransmitter-related enzymes would be a valuable therapeutic strategy.28,117,118AChE is a well-known and extensively studied factor in the pathology of AD (Fig. 4a). According to the “cholinergic hypothesis,” AD is characterized by a significant loss of cholinergic neurons and a decline in ACh levels in the brain, leading to memory loss and cognitive deficits.28,122 As a result, AChE becomes a major target for developing therapeutic inhibitors. The structures of AChE and commonly prescribed AChE inhibitors, such as rivastigmine, galantamine, and donepezil, are presented in Fig. 4a.28–32,123 Recombinant human AChE (rhAChE) contains several essential binding domains. The catalytically active site (CAS) is located at the bottom of a 20 Å deep, narrow binding gorge, and its anionic domain includes amino acid residues W86, Y130, Y337, and F338. The peripheral anionic site (PAS), positioned at the entrance of the binding gorge, comprises amino acid residues Y72, D74, Y124, W286, and Y341.111,112 Current AChE inhibitors work by competitively impeding the enzyme's activity, either by blocking the CAS to prevent substrate interaction or by hindering the access to the active site through the PAS.28–32 For example, donepezil interacts with both the CAS and the PAS of AChE, forming hydrogen bonds with water molecules that connect to the side chains of Y337 and Y341, and engaging in π–π interactions with the side chains of W86 and W286, as illustrated in Fig. 4a.112 These interactions interfere the breakdown of ACh into acetate and choline, thereby increasing the availability of ACh and enhancing cholinergic neurotransmission.28
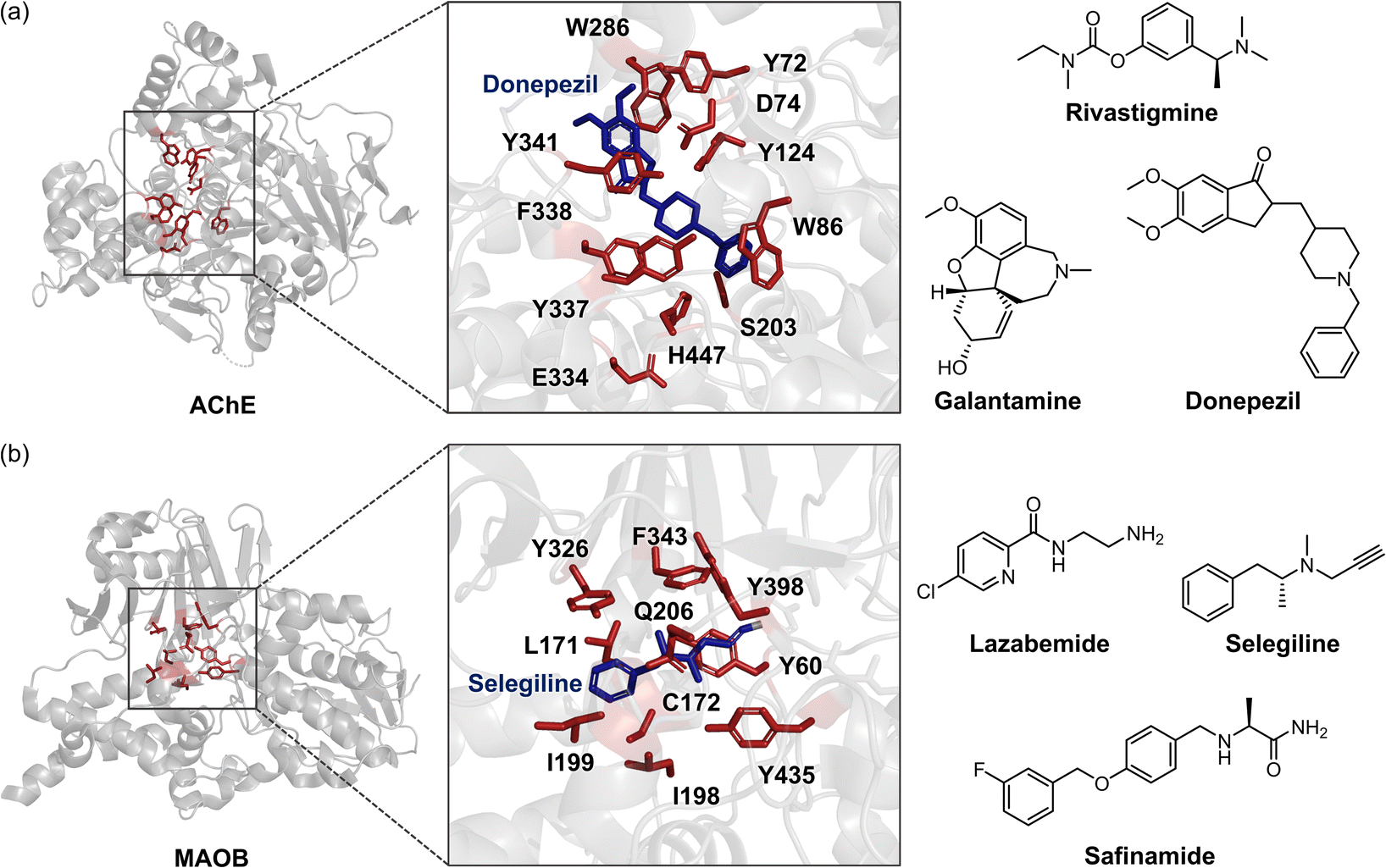 | ||
| Fig. 4 AChE, MAOB, and examples of their inhibitors. (a) Structure of AChE (PDB 4EY4111), binding mode of donepezil at its active site (PDB 7E3H112), and three U.S. FDA-approved AChE inhibitors (donepezil, rivastigmine, and galantamine). The amino acid residues involved in donepezil binding are highlighted in red and depicted as sticks. (b) Structure of MAOB (PDB 1GOS113), interaction with selegiline at its catalytic domain (PDB 2BYB33), and three representative inhibitors (lazabemide, selegiline, and safinamide). The amino acid residues involved in selegiline binding are indicated in red with sticks. | ||
Additionally, the distinctive selectivity of AChE inhibitors against butyrylcholinesterase (BuChE) plays a significant role in their efficacy and suitability for different stages of AD.124–126 Unlike AChE, BuChE shows weaker substrate preference due to the absence of several aromatic residues, retaining only one aspartate and tyrosine residue in its PAS. Interestingly, BuChE activity increases during the late stage of AD, particularly in neurodegenerative regions, which compensates for the decline in AChE activity.124–126 Consequently, non-selective inhibitors may offer advantages in later stages of AD by targeting both enzymes, though they also carry a higher risk of gastrointestinal and cardiovascular side effects.124–126 Currently developed AChE inhibitors exhibit varying selectivity for AChE and BuChE. For example, rivastigmine addresses both AChE and BuChE, while galantamine and donepezil demonstrate moderate and strong selectivity for AChE, respectively.124–126
Along with the cholinergic hypothesis, the loss of dopaminergic and serotonergic neurotransmission represents another pathological factor in AD.127–129 The degeneration of dopaminergic and serotonergic neurons leads to significant local inflammation and exacerbates age-related cell death, as observed in the AD-transgenic Tg2576 mice model.127 This neuronal loss is associated with pathological symptoms, including impairments in hippocampal neuronal excitability, synaptic plasticity, memory, and reward performance.127–129 Furthermore, the activation of enzymes that degrade dopamine and serotonin, such as MAO and COMT, has also been implicated in the pathology of AD.103,117,118,130 Consequently, numerous efforts have focused on targeting these enzymes. Recent studies have highlighted MAO inhibitors, such as lazabemide, selegiline, and safinamide, which are either U.S. FDA-approved or in clinical trials, as displayed in Fig. 4b.131 These inhibitors block the active site of MAO (primarily MAOB), which consists of two cavities: the substrate cavity near the flavin adenine dinucleotide (FAD) and the entrance cavity.113 For instance, selegiline interacts with the substrate/FAD binding site, including amino acid residues L171, C172, I199, Q206, Y326, F343, and Y398, through van der Waals, CH–π, and π–π interactions that are essential for binding.33Selegiline then forms covalent bonds with FAD, irreversibly prohibiting substrate coordination and subsequent reactions, thereby alleviating the loss of dopamine and serotonin under pathological conditions (Fig. 4b).33 Despite their potential to restore neurotransmission, these drugs cannot halt disease progression and provide only modest, short-term cognitive benefits, often accompanied by significant side effects.28,132 While AChE and MAO inhibitors have been the cornerstone of AD treatment, these drawbacks underscore the urgent need for novel therapies. The inadequate efficacy and adverse effects of current treatments emphasize the necessity for disease-modifying therapies that can concurrently address multiple pathophysiological mechanisms of AD.
2.4 Other pathological features
In addition to the abovementioned pathological factors, neuroinflammation, mitochondrial dysfunction, and neuronal cell death are closely associated with the pathogenesis of AD (Fig. 1a).4–7 These features are intricately connected to other pathological elements, such as Aβ and ROS, underscoring the multifaceted nature of the disease.6–15 Under normal conditions, astrocytes and microglia play distinct yet complementary roles in maintaining homeostasis and managing neuroinflammation in the central nervous system (CNS). Astrocytes preserve the integrity of the blood–brain barrier (BBB), regulate cerebral blood flow, and release anti-inflammatory cytokines, contributing to a stable neural environment.133–137 Meanwhile, microglia, the primary immune cells of the CNS, constantly monitor their microenvironment and secrete pro-inflammatory mediators, such as tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β), in response to injury or infection.138,139 Together, these cells balance neuroprotection and tissue repair to support neural homeostasis. In AD, however, this balance is disrupted, leading to chronic neuroinflammation and neuronal death.Aβ aggregates activate microglial receptors, such as Toll-like receptors (TLRs) and nucleotide-binding oligomerization domain-like receptors (NLRs), which stimulate the NLR family pyrin domain-containing 3 (NLRP3) inflammasome.140–144 Once induced, the NLRP3 inflammasome promotes Aβ aggregation by serving as a nucleation platform for fibrillization. It also cleaves pro-caspase-1 into active caspase-1, which processes pro-interleukin-1β (pro-IL-1β) and pro-interleukin-18 (pro-IL-18) into their secretory forms.140–144 The release of cytokines, such as IL-1β and TNF-α, amplifies inflammation and prompts astrocytes to secrete neurotoxic molecules, including ROS and complement components (e.g., complement component 3).145,146 Simultaneously, sustained excitation of nuclear factor-κB (NF-κB) in astrocytes and microglia, triggered by Aβ and cytokine feedback loops, further elevates the production of pro-inflammatory mediators, including interleukin-6 (IL-6) and TNF-α, perpetuating inflammation and oxidative stress.147 Chronic glial activation also impairs microcirculation and disrupts the BBB, exacerbating Aβ accumulation.148,149 Through these interconnected mechanisms, astrocytes and microglia transition from protective roles to active contributors to neuronal damage, driving the progression of AD.
Therapeutic strategies to address astrocyte- or microglia-mediated neuroinflammation in AD primarily focus on modulating the NF-κB pathway, a central regulator of inflammatory responses. These strategies include developing antagonists for membrane receptors that activate NF-κB150–153 and inhibitors of key intracellular signaling molecules within this pathway.154–157 Complementary approaches aim to mitigate Aβ aggregation driven by neuroinflammation.158–164 One promising method involves targeting the active NLRP3 inflammasome, which serves as a nucleation site for Aβ aggregation, thereby attenuating harmful effects of pro-inflammatory responses.158–164 Another strategy focuses on enhancing microglial phagocytosis and autophagy to promote the clearance of Aβ aggregates, offering the dual benefit of reducing inflammation and preventing further neuronal damage.165–169
Mitochondrial dysfunction is another critical pathological feature of AD, closely linked to disease progression through several key mechanisms. One major manifestation is the overproduction of ROS, which induces oxidative stress and damages cellular components, such as lipids, proteins, and DNA.170 Bioenergetic deficits, such as reduced ATP production by disruptions in the electron transport chain (ETC), further compromise neuronal energy supply, ultimately leading to synaptic failure and cell death.171,172 Mitochondrial DNA (mtDNA) degradation exacerbates these effects by impairing metabolic functions and repair mechanisms, intensifying cellular dysfunction.173,174 A key contributor to mitochondrial dysfunction is the diminished clearance of damaged mitochondria, primarily regulated by the phosphatase and tensin homolog-induced kinase 1 (PINK1)-Parkin pathway.175,176 This pathway is altered under pathogenic conditions due to the accumulation of hyperphosphorylated tau and Aβ, resulting in mitochondrial defection and increased cellular stress.177,178 Additionally, the balance between mitochondrial fusion and fission is disturbed. Aβ and ROS overactivate dynamin-related protein 1 (Drp1), causing excessive mitochondrial fragmentation.179–182 This unregulated cleavage compromises energy distribution, weakens synaptic function, and exacerbates neuronal damage.179–182 Mitochondrial biogenesis is also hindered by oxidative stress and neuroinflammation, which suppress peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α), thereby reducing the generation of functional mitochondria and worsening bioenergetic deficits.183–185 Abnormal mitochondrial distribution, coupled with endoplasmic reticulum damage, further amplifies mitochondrial dysfunction, positioning it as a central driver of AD progression.186,187
Strategies to address mitochondrial dysfunction in AD focus on reducing excess ROS, restoring mitochondrial function, and improving bioenergetics. To mitigate ROS overproduction, various therapeutic agents with antioxidant functional groups have been explored.188–190 Efforts are also underway to restore mitochondrial biogenesis, dynamics, and mitophagy by targeting essential proteins involved in these processes, such as nicotinamide adenine dinucleotide-dependent deacetylase sirtuin-1 (SIRT1) and mammalian target of rapamycin (mTOR), which inhibits PGC-1α and the PINK1-Parkin signaling, respectively.191–195 Furthermore, modulating mitochondrial bioenergetics by enhancing components critical to energy production in mitochondria, such as the ETC and Krebs cycle enzymes, may improve energy production in AD.187,196–201
The interconnected pathological systems and signaling cascades drive apoptosis-mediated neuronal cell death through both intrinsic and extrinsic pathways.202–204 Oxidative stress, mitochondrial dysfunction, and Aβ accumulation disrupt intracellular membranes, leading to the release of proteins, such as cytochrome c, into the cytosol.202–206 This triggers the formation of the apoptosome complex and activates caspase-9, further exacerbating cell death and promoting mitochondrial permeabilization.205 Dysregulated survival pathways, including phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt)/mTOR signaling network, further contribute to apoptosis by impairing protective mechanisms and initiating stress-induced responses.15,207,208 Extracellular Aβ aggregates also activate cell surface death receptors (e.g., Fas or TNF), forming a death-inducing signaling complex that induces the extrinsic apoptotic pathway via caspase-8.209,210 Caspase-8 either directly stimulates caspase-3 or amplifies the intrinsic pathway by cleaving the Bcl-2 homology 3 interaction domain death agonist (Bid) into truncated Bid (tBid), which further promotes apoptosis.211,212 Neuroinflammation intensifies these effects via pathways, such as Janus kinase/signal transducer and activator of transcription (JAK-STAT), upregulating the expression of pro-inflammatory and pro-apoptotic genes. Concurrently, alterations in Ras/extracellular signal-regulated kinase (Ras/ERK) signaling compromise neuronal survival, which accelerates apoptosis-directed neurodegeneration.213,214
Non-apoptotic cell death pathways also play a significant role in neurodegeneration associated with AD. Dysregulated autophagy, initiated by Aβ aggregates and impaired lysosomal function, leads to the accumulation of dysfunctional organelles and proteins, worsening neuronal damage.215,216 Pyroptosis, an inflammatory form of cell death, is driven by the activation of the NLRP3 inflammasome, releasing pro-inflammatory cytokines and causing membrane damage via gasdermin-D-mediated pathway.144,217 Cuprotosis, arising from imbalances in copper metabolism, disrupts the mitochondrial function, elevates the oxidative stress, and results in the misprocessing of APP, ultimately contributing to neuronal death and exacerbating Aβ pathology.218 Ferroptosis, an iron-dependent cell death mechanism, is fueled by perturbed iron metabolism, oxidative stress, and lipid peroxidation, thereby promoting neuronal loss.219,220 Collectively, these apoptotic and non-apoptotic pathways form a feedback loop of inflammation, mitochondrial dysfunction, and neuronal damage, which fosters the progression of AD. To regulate apoptosis, inhibitors confronting caspases-3, -8, and -9 are being actively investigated, with caspase-1 inhibitors to mitigate pyroptosis.221–232 Additionally, metal chelators are being explored as a potential therapy to control metal ion dyshomeostasis-mediated cell death, such as cuprotosis and ferroptosis.2,218,232 Taken together, neuroinflammation, mitochondrial damage, and neuronal cell death are increasingly recognized as key pathological targets in AD. In addition to the aggregation of Aβ, the dyshomeostasis of metal ions, and the dysfunction of enzymatic systems, these pathological features highlight the multifaceted nature of AD.4–7 Ongoing research efforts are focused on developing therapeutic interventions to address these interconnected processes, as emphasized in recent literature.6–15
3. Strategies to overcome the complex pathology of AD
Numerous single-target-directed drugs have been developed to address specific pathological factors associated with AD. These treatments have been evaluated at various clinical trial stages, highlighting their potential therapeutic roles within the AD landscape.17,32,55,56 Despite extensive efforts, no single treatment has been able to effectively alter the progression of the disease, achieving limited success.3,18,20,21,25,41,59,77–79,233 This outcome underscores the complexity of AD, where multiple factors contribute to disease onset.16 Therefore, there is a critical need for therapeutics capable of targeting several pathological factors simultaneously.Rational design strategies for multi-target-directed molecules involve combining two or more molecular frameworks, each with established properties, into a single molecular entity, as illustrated in Fig. 1b.35,126,234,235 This integration can be accomplished through three approaches: (i) linkage approach, where the original structures are connected via a linker; (ii) fusion approach, which directly joins two scaffolds without a linker; (iii) incorporation approach, where functional components are merged while preserving structural features essential for their activity.35,126,234,235 Achieving this multifunctionality requires a careful balance of scaffold properties while maintaining their functional integrity.35,126,234,235 These resulting compounds can then undergo structure–activity relationship (SAR) optimization to refine their physicochemical properties, target specificity, and BBB permeability to maximize their therapeutic potential.35,126,234,235
Multifunctional molecules developed to address Aβ aggregation often use frameworks inspired by known imaging agents, e.g., thioflavin-T (ThT), polyphenols, and peptide-based inhibitors.35,126,234,235 These scaffolds are strategically modified to strengthen interactions with the regions of Aβ, including the self-recognition and C-terminal regions, both of which play a crucial role in Aβ aggregation.39 By targeting these regions, the molecules can disrupt aggregation pathways and prevent the formation of toxic oligomers and fibrils.39 These compounds typically incorporate hydrophobic features to enhance binding to Aβ′s hydrophobic domains, as well as π–π stacking or hydrogen bonding capabilities to interact with multiple amino acid residues within the peptide.39
MTDLs designed for confronting Aβ aggregation and metal ion dyshomeostasis simultaneously require the incorporation of nitrogen, oxygen, or sulfur donor atoms for metal chelation into Aβ-binding frameworks to achieve dual function.35,94–96 By precisely tuning dissociation constant (Kd) values and metal-binding geometries, these chelators can prohibit interactions between metal ions and Aβ without disrupting systemic metal ion homeostasis or interfering with the function of metalloenzymes. Additionally, MTDLs can be optimized to block vacant coordination sites within metal-binding domains, thereby preventing redox reactions that produce ROS. Moreover, these molecules can modulate the redox potential of the metal center to mitigate oxidative stress.
To regulate enzyme activity, rational strategies focus on targeting the active sites of enzymes.54,104,108,111,112 A common approach is competitive inhibition, where molecules are designed to mimic natural substrates, enabling them to occupy the active sites of enzymes and block their substrate binding. By integrating computational modeling and crystallographic studies, the development of highly selective and effective MTDLs capable of adjusting enzyme activity is facilitated. Rational design often utilizes backbones from natural products, polyphenols, or established inhibitors, employing interactions, such as hydrogen bonding, π–π stacking, and hydrophobic contacts, to acquire high specificity.236,237
Multifunctionality can be achieved by linking or incorporating key moieties addressing multiple pathways into a single molecular framework. While research on Aβ aggregation remains a central focus, extensive efforts are also being directed toward combining two or more distinct structural motifs within a single molecule to simultaneously target alternative pathological pathways.35,126,234,235 These efforts include inhibiting enzymes, such as BACE1, MMP9, LOXL2, AChE, and MAOB, as well as modulating their interactions with metal ions.35,126,234,235 Such approaches, which have been comprehensively reviewed in several studies, highlight the versatility and therapeutic promise of MTDLs.35,126,234,235 The following sections underscore the critical role of SAR studies, with an emphasis on the development of multi-target-directed compounds that focus on Aβ aggregation with other interconnected pathological factors of AD.
3.1 Chemical reagents capable of controlling Aβ and BACE1
Aβ remains a critical biomarker in AD therapy due to its association with the disease's pathological features and its involvement in neuroinflammation and cognitive decline.1 Consequently, regulating BACE1 activity, the primary enzyme responsible for Aβ production, has become a major focus in the development of AD therapeutics.50,238 Single-target-directed therapies, however, have proven insufficient in slowing disease progression. This has led to a shift in research towards strategies that regulate both the accumulation and production of Aβ to achieve a synergistic clinical effect.233,238 Therapeutic interventions that integrate multiple characteristics can effectively form hydrophobic interactions or hydrogen bonds within the catalytic cleft and aspartate dyad to target BACE1 efficiently. Additionally, these structures can be designed to establish hydrophilic and hydrophobic contacts with Aβ, thereby inhibiting its aggregation. Current approaches with the above factors are widely being explored to address both Aβ and BACE1 using peptides,239 natural products,240,241 and modifications of existing therapeutic frameworks.242,243Peptides naturally contain polar groups that can be modified to improve their ability to cross the BBB and interact efficiently with target enzymes. To optimize these properties, Singh and coworkers designed peptides with hydrophobic groups added at both the N- and C-termini.239 Proline was used as the template to regulate surface area and structural flexibility, with a glycine–proline–alanine (GPA) tripeptide as the model backbone.239 Various derivatives, each featuring different functional groups at the N- and C-termini, were synthesized to achieve an optimal balance between hydrophobicity and hydrophilicity. Among these derivatives, GPA-1 and GPA-2 (shown in Fig. 5a) exhibited significant in vivo efficacy, effectively inhibiting both the activity of BACE1 and the aggregation of Aβ42.239 Both peptides displayed potent ID50 values of 20 nM against BACE1.239 Additionally, GPA-1 and GPA-2 suppressed the formation of β-sheet-rich Aβ42 aggregates by 54% and 34%, respectively.239 Pretreatment with these peptides also alleviated memory deficits in a mouse model with scopolamine-induced memory impairment, as presented in Fig. 5a.239 Behavioral assessments, including the Morris water maze, revealed a significant reduction in latency time to reach the platform, while the passive avoidance task demonstrated decreased latency in reaching the shock-free area.239 Furthermore, peptide-based strategies have been applied to block the β-cleavage site of APP in in vivo systems.244
 | ||
| Fig. 5 Examples of chemical reagents controlling Aβ and BACE1. (a) Small peptide-based compounds (GPA-1 and GPA-2) and their effects on the cognition of mice models after inducing impaired memory with scopolamine on days 12 to 14. GPA-1, NH2-Gly-Pro-Ala-OMe; GPA-2, Cbz-Gly-Pro-Ala-O-cinnamyl. Reproduced with permission from ref. 239. Copyright© 2024 American Chemical Society. (b) A flavonoid-based natural product (YCC31) and its inhibitory impact on the activity of BACE1 and the aggregation of Aβ42. The interactions between YCC31 and BACE1 at the active site, the morphology of the resultant Aβ42 species, and its inhibitory effect on Aβ42 aggregation were analyzed by MD simulations, TEM, and the ThT assay, respectively. YCC31, quercetin-3-O-[β-D-glucopyranosyl-(1→3)-O-α-L-rhamnopyranosyl-(1→6)-O-β-D-glucopyranoside]. Reproduced with permission from ref. 240. Copyright© 2024 Elsevier. (c) A phenothiazine-derived compound (19c) and its effect against the activity of BACE1 and the aggregation of Aβ40. The interactions between 19c and BACE1 at the active site were obtained from docking studies and its inhibitory effect on the formation of β-sheet-rich Aβ40 aggregates was quantitatively measured by the ThT assay. 19c, 4-amino-N-[(4-chlorobenzyl)]-2-[methyl({2-[2-(10H-phenothiazin-10-yl)ethoxy]ethyl})amino]butanamide. Reproduced with permission from ref. 243. Copyright© 2023 Elsevier. | ||
The use of natural products as therapeutic candidates has garnered attention for decades due to their proven efficacy, safety, and increased molecular rigidity compared to synthetic compound libraries, particularly for addressing protein–protein interactions.245 Among these natural compounds, flavonoids are notable for their ability to interact with multiple biological targets through hydrogen bonding and π–π stacking interactions.240,246,247 Building on these properties, Yang and coworkers discovered a natural flavonoid–carbohydrate conjugate, YCC31, which exhibited significant potential for dual inhibition (Fig. 5b). YCC31 reduced Aβ production in 7PA2 cells by modulating the activity of BACE1.240 The compound forms hydrogen bonds with key amino acid residues G95, F169, R189, Y259, K285, and D289 within BACE1 through its hydroxyl groups and ether group oxygen atom. Additionally, its phenyl group participates in CH–π interactions with T292, effectively preventing substrate access and reducing Aβ production, as depicted in Fig. 5b.240 In addition to BACE1 inhibition, YCC31 demonstrated dose-dependent regulation of Aβ42 aggregation, as analyzed by transmission electron microscopy (TEM), ThT assay, and molecular dynamics (MD) simulations.240 The supramolecular equivalent of YCC31 suppressed the fibrillization of Aβ42, generating short fibrils by interacting with amino acid residues D22, S26, N27, and K28, which are crucial for the salt bridging that stabilizes fibrillar structures.240 Moreover, YCC31 decreased the production of ROS and nitric oxide (NO) induced by Aβ-mediated activation of microglia, thereby mitigating oxidative stress and neuroinflammation.240
Modifying existing scaffolds has emerged as a promising approach to accelerate the drug discovery process.248 One notable example is N-benzylbutanamide.243 Bajda and coworkers developed a phenothiazine derivative, 19c, which exhibited the highest hBACE1 inhibitory activity (IC50 = 1.6 μM) and anti-aggregation effects against Aβ40 (99% inhibition at 10 μM), as described in Fig. 5c.243 The inhibition of hBACE1 by 19c is attributed to its ability to block both substrate entry and catalytic proteolysis. The compound interacts with the S2 and S2′ pockets via CH–π interactions with Y71 and a π–oxygen bond with Y198, while forming a cation–π interaction with R128 in the S3′ pocket. Additionally, its amide group establishes a hydrogen bond with the main chain of T72, and its γ-amino group engages in a network of bonds, creating two hydrogen bonds with the side chains of T72 and T329 and an ionic interaction with D328.243 The phenothiazine moiety of 19c fits into the S3 and S4 pockets of BACE1, accompanying in hydrophobic contacts with Q73 and I110.243 Moreover, 19c prevented Aβ40 aggregation by 86%, significantly decreasing both elongation and nucleation rates, as measured by the ThT assay (Fig. 5c). These results indicate that 19c directly associates with fibrillar species to suppress elongation and with soluble nucleic, oligomeric, and prefibrillar forms of Aβ40 during the early stages of polymerization. Beyond its in vitro effects, 19c demonstrated statistically significant anti-amnesic properties and improved non-spatial (contextual and recognition) memory in a scopolamine-induced amnesia mouse model.243 Overall, these findings underscore the potential of multi-targeting strategies in developing effective AD therapeutics, particularly those involving precise molecular interactions with BACE1 and Aβ.
3.2 Chemical reagents capable of regulating Aβ, metal ions, and ROS
Metal ions, such as Cu(I/II), Zn(II), and Fe(II/III) can coordinate to Aβ to form metal–Aβ complexes.82,249 The non-specific removal of these metal ions, however, has been unsuccessful and presents several challenges.25,77–79 As a result, strategies promising alternatives to non-selective metal ion chelation. To achieve this, numerous aspects of each component must be carefully considered. Chemical reagents should incorporate either a redox-active backbone, a metal-chelation moiety, or both to effectively mitigate oxidative stress.25,77 In addition, the structure should include functional groups capable of forming hydrogen bonds and hydrophobic interactions to regulate Aβ aggregation.35,234,235 For these aspects, peptides have been developed.250–256 Sun and coworkers discovered that integrating the Aβ self-recognition site (LVFFA) and a metal-chelating tripeptide (RTH) efficiently modulates metal-mediated Aβ aggregation and ROS generation (Fig. 6a).250–252 Building on this concept, a D-enantiomeric peptide, RTHLVFFARK-NH2 (rk10), was rationally designed and compared to its previously reported L-enantiomer, RK10. The rk10 peptide demonstrated the ability to form stable metal coordination with the amino terminal Cu(II)- and Ni(II)-binding (ATCUN) motif, exhibiting a Kd value of 28 nM, comparable to that of Aβ (logKCu(II) = 10). This Cu(II) chelation prevented redox reactions and excessive ROS production by Cu(II)–Aβ complexes.250 As shown in Fig. 6a, rk10 treatment decreased ROS generation to half the level observed in the Cu(II)–Aβ control without peptide treatment.250 Furthermore, rk10 disrupted the twisted and elongated fibrillar network, suppressing fibril growth and redirecting the aggregation pathway to yield amorphous aggregates, as visualized by atomic force microscopy (AFM).250 A similar trend was observed in the presence of Cu(II), where rk10 addition noticeably decreased the formation of visible structures, compared to RK10.250 These findings suggest that while rk10 possesses metal–chelation properties similar to RK10 (Kd = 31 nM), its enhanced interactions with Aβ further improve the inhibitory effects on Aβ fibrillization and Cu(II)–Aβ-induced ROS generation.250,252
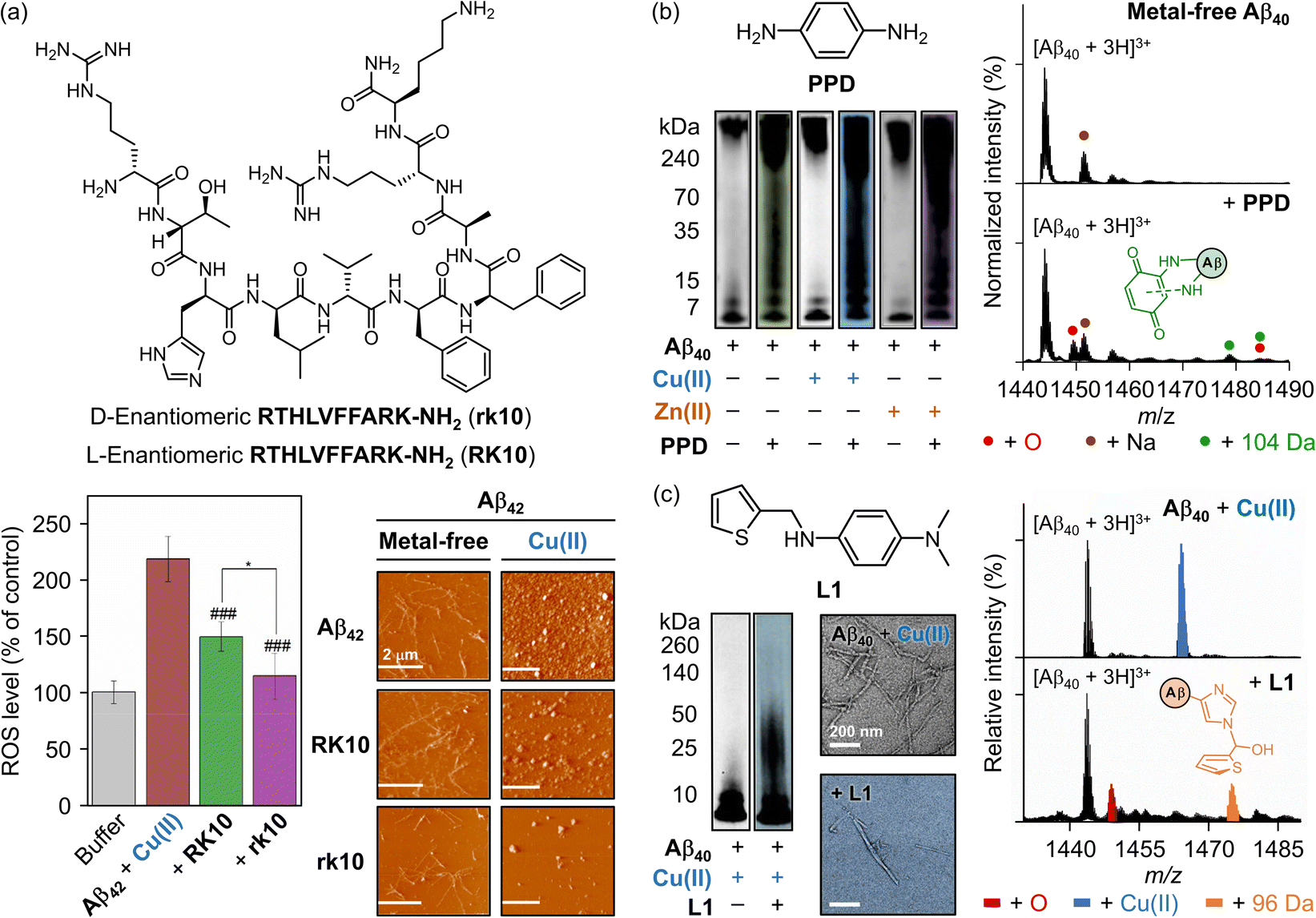 | ||
| Fig. 6 Examples of chemical reagents regulating Aβ peptides and metal ions. (a) Structure of the D-enantiomeric decapeptide RTHLVFFARK-NH2 (rk10) and inhibitory effects of rk10 and its L-enantiomer (RK10) on the generation of ROS and metal-free and Cu(II)–Aβ42 aggregation. The ROS level produced by Cu(II)–Aβ42 in the absence and presence of rk10 or RK10 in SH-SY5Y cell was measured by a fluorescence assay with 2′,7′-dichlorofluorescin diacetate (DCFH-DA). The statistical significance level is expressed by the pound (control as Aβ42 + Cu(II), ###P < 0.001) and asterisk (control as Aβ42 + Cu(II) + RK10, *P < 0.05). The morphology of Aβ42 aggregates generated with and without Cu(II) and rk10 or RK10 was analyzed by AFM. Reproduced with permission from ref. 250. Copyright© 2019 American Chemical Society. (b) Structure of PPD and its impact on the aggregation of both metal-free and metal-added Aβ40. The size distribution of Aβ40 aggregates produced with and without PPD or metal ions was investigated by gel/western blot. The formation of the covalent adduct between Aβ40 and benzoquinone was detected by ESI–MS. PPD, p-phenylenediamine. Reproduced with permission from ref. 257. Copyright© 2020 American Chemical Society. (c) Structure of L1 and its influence on Cu(II)-added Aβ40 aggregation. The size distribution and morphology of the resultant Aβ40 aggregates produced with and without L1 or Cu(II) were investigated by gel/western blot and TEM, respectively. Chemical modifications onto the Cu(II)-coordination sphere of Aβ40 by L1 were monitored by ESI–MS. L1, N1,N1-dimethyl-N4-(thiophen-2-ylmethyl)benzene-1,4-diamine. Reproduced with permission from ref. 258. Copyright© 2020 United States National Academy of Sciences. | ||
An alternative strategy in AD research involves using small molecules that chemically modify Aβ, altering its aggregation pathways. A series of redox-active aromatic compounds has demonstrated reactivities towards metal-free Aβ, metal-bound Aβ, and free organic radicals.257 Among these, p-phenylenediamine (PPD; Fig. 6b) and its derivatives have emerged as promising therapeutic candidates due to their structural simplicity, redox capability, and amphiphilicity.259,260PPD, with two electron-donating groups at the para position, lowered the redox potential and enhanced its ROS scavenging capacity.261,262 Furthermore, PPD was shown to modulate Aβ aggregation, resulting in smaller fibrils and amorphous aggregates, which appeared as smearing bands in the 4–270 kDa range in gel electrophoresis with western blotting (gel/western blot) analyses (Fig. 6b).257 A similar trend was observed in the presence of Cu(II) or Zn(II), indicating that PPD further perturbed metal–Aβ aggregation pathways.257 Mechanistic details revealed that PPD undergoes oxidation to yield benzoquinone-diimine (BQDI) or benzoquinone (BQ)257 during which the M35 residue of Aβ is oxidized.257 In addition, BQ formed covalent adducts with Aβ through primary amino groups, such as K16 (Fig. 6b).257,263,264 This complexation captured Aβ in a relatively compact conformation, likely preventing its involvement in Aβ fibrillization.265 Supported by these in vitro results, PPD significantly reduced cerebral and hippocampal Aβ deposits and improved cognitive function in 5xFAD transgenic mice.257
The structural similarity of N,N-dimethylaniline (DMA) to PPD imparts comparable properties, enabling DMA to interact with both metal-free Aβ and metal–Aβ, and participate in one- or two-electron oxidation processes, thereby contributing to ROS scavenging.266,267 Building on this concept, a derivative named L1 (presented in Fig. 6c), featuring a bidentate ligand attached to the DMA backbone, was developed to directly modify the Cu(II)-coordination sphere in Aβ through copper–O2 chemistry.258 Notably, the H14 residue was modulated when Cu(II)–Aβ was incubated with L1 under aerobic conditions, resulting in mass increments of 16 Da and 96 Da in Aβ, corresponding to oxidation and covalent adduct formation, respectively.258 Since H14 is associated with coordinating both Cu(I) and Cu(II) in Aβ, this chemical transformation effectively disrupted the aggregation of metal–Aβ complexes.258 Moreover, L1 inhibited the aggregation of Cu(II)–Aβ40 by altering the morphology of Aβ aggregates into shorter fibrils, suppressed H2O2 production from the redox cycling of Cu(I/II)–Aβ, and reduced cytotoxicity (Fig. 6c).258
Overall, several small molecules have been developed to address metal-induced Aβ aggregation through three primary mechanisms: (i) directly interacting with both metal ions and Aβ, (ii) chemically modifying Aβ, and (iii) disrupting the coordination sphere of metal–Aβ complexes. These strategies can precisely redirect the binding pattern of redox-active metal ions to Aβ, thereby preventing ROS production. They further suggest that shifting beyond traditional approaches, which solely focus on chelating metal ions, to simultaneously targeting both metal–Aβ interactions and ROS may present a more effective strategy for developing novel AD treatments.
3.3 Chemical reagents capable of modulating Aβ and metalloenzymes
Metal ion dyshomeostasis and the resultant dysregulation of metalloenzymes are emerging as promising therapeutic targets for AD.98,104–107,120,268–272 Among these, MMP9 and LOXL2 have been implicated in the pathogenesis of neurodegenerative diseases due to their involvement in Aβ aggregation.104–107 Consequently, there is growing interest in developing therapies that simultaneously address MMP9 or LOXL2 while inhibiting Aβ aggregation as a combined approach to enhance therapeutic efficacy.As part of metalloenzymes, regulating the activity of MMP9 and LOXL2 requires the inclusion of a metal-coordination motif, with functional groups capable of disrupting Aβ self-assembly. Building on this concept, several polyphenol-based natural products have been investigated for their regulatory properties against MMP9.273,274 Among them, doxycycline (DOX; Fig. 7a), a tetracycline derivative, is a well-known antibiotic that also inhibits MMP9 and reduces neuronal damage in cases of focal brain ischemia.278 This prospective dual-modulatory effect has advanced DOX to phase II clinical trials.279 Kaur and coworkers elucidated the binding mode and antagonistic mechanism of DOX against MMP9 through docking analysis.280DOX primarily interacts with the catalytic domain of MMP9 by forming hydrogen bonds with amino acid residues E402, E416, and L418, and through hydrophobic interactions, such as CH–π and π–π interactions with amino acid residues H401, P421, M422, Y423, and R424 (highlighted green in Fig. 7b).280 Additionally, the coordination of DOX to Zn(II) through the hydroxyl group on its D ring further stabilizes its complexation at the active site, thereby preventing ligand association and catalytic hydrolysis.280
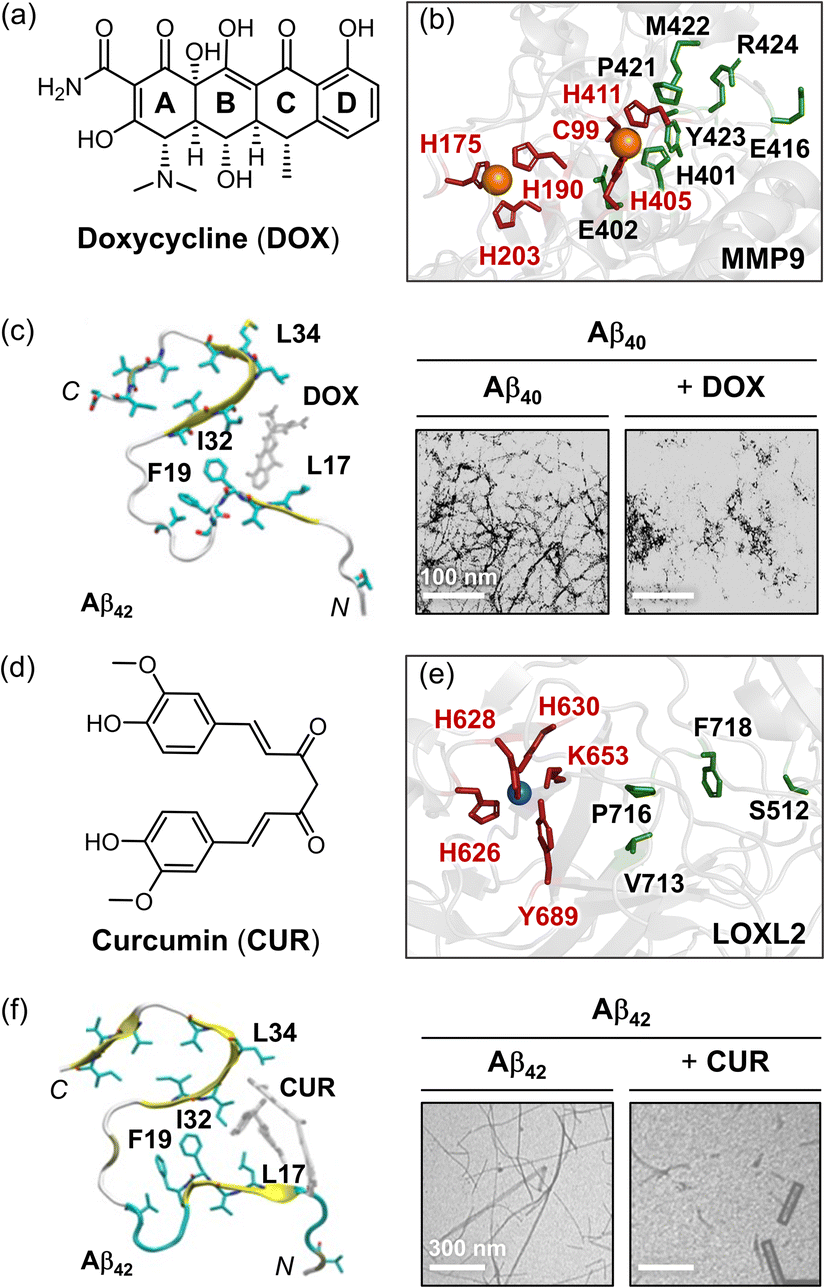 | ||
| Fig. 7 Examples of chemical reagents controlling Aβ and MMP9 or LOXL2. (a) Structure of doxycycline [DOX; (4S,4aR,5S,5aR,6R,12aS)-4-(dimethylamino)-3,5,10,12,12a-pentahydroxy-6-methyl-1,11-dioxo-1,4,4a,5,5a,6,11,12a-octahydrotetracene-2-carboxamide]. (b) Intermolecular interactions between MMP9 (PDB 1L6J70) and DOX. The Zn(II)-coordination and DOX-interacting residues are indicated as sticks, and highlighted in red and green, respectively. (c) Interactions of DOX with Aβ42 fibrils (PDB 2MXU). The morphology of the resultant Aβ42 species was analyzed with TEM. Reproduced with permission from ref. 275 and 276. Copyright© 2019 Multidisciplinary Digital Publishing Institute and 2001 John Wiley and Sons. (d) Structure of curcumin (CUR). CUR, (1E,6E)-1,7-bis(4-hydroxy-3-methoxyphenyl)hepta-1,6-diene-3,5-dione. (e) Intermolecular interactions between LOXL2 (PDB 5ZE371) and CUR. The Cu(II)-coordination and CUR-interacting residues are shown as sticks, and depicted in red and green, respectively. (f) Interactions of CUR with Aβ42 fibrils (PDB 2MXU). The formation of the smaller Aβ42 species was detected by TEM. Reproduced with permission from ref. 275 and 277. Copyright© 2019 Multidisciplinary Digital Publishing Institute and 2017 Elsevier. | ||
Recent studies have also elucidated that DOX directly associates with amyloidogenic fibrils, including those produced by Aβ, and disassembles pre-formed aggregates.275,276,279,281,282 As depicted in Fig. 7c, DOX engages in hydrophobic interactions with key amino acid residues of Aβ42 fibrils, such as L17, F19, I32, and L34, which are critical for self-recognition and fibril stabilization (Fig. 2b).275 Morphological analysis revealed that DOX treatment significantly reduced fibril generation and promoted degradation.276 In addition to its anti-aggregation properties, DOX has been shown to scavenge ROS and other oxidants, such as superoxide anion radicals (O2˙−) and lipid peroxides, diminishing the production of cytotoxic malondialdehyde-acetaldehyde–protein adducts mediated by redox reactions.283 These antioxidant properties of DOX contribute to its anti-inflammatory effects and its ability to alleviate oxidative stress in AD.283
Recent studies have shown that polyphenols can also regulate the activity of LOXL2, highlighting their potential as therapeutic agents.275,277,284 Based on these findings, there is growing interest in identifying noble polyphenols that can simultaneously inhibit the activity of LOXL2 and the aggregation of Aβ. Curcumin (CUR; Fig. 7d), a natural polyphenol found in Curcuma longa plants, has gained recognition as a promising candidate for AD treatment due to its ability to target multiple pathological factors, including LOXL2 and Aβ.275,277,284CUR modulated LOXL2 through mechanisms similar to those of DOX, binding directly to the catalytic domain and preventing ligand complexation and catalytic activity.284 This interaction involves hydrogen bonding between the amide backbone of P716 and a water molecule, which forms a hydrogen bonding network with the side chain of S512, along with hydrophobic interactions with the side chains of V713 and F718, as displayed in Fig. 7e.284 This binding mode resulted in a stable interaction, as confirmed by root mean square deviation (RMSD) analysis.284
Similar to DOX, CUR maintained continuous contact with Aβ fibrils during MD simulations, interacting with the central hydrophobic core of Aβ fibrils, including amino acid residues L17, F19, I32, and L34. CUR also disrupted the salt bridge formed by the side chain of D23. These interactions prohibited the aggregation of Aβ and destabilized fibril structures, leading to shorter, fragmented fibrils, as illustrated in Fig. 7f.275 In addition, CUR has been shown to inhibit the activity and expression of MMP9.285 Furthermore, CUR can chelate several metal ions, such as Cu(II), Fe(II), and Zn(II), through its α,β-unsaturated β-diketo moiety, thereby contributing to the reduction of ROS.286,287 Collectively, addressing metalloenzymes, e.g., MMP9 and LOXL2, using multi-target-directed strategies offers a promising approach to enhancing specificity and therapeutic efficiency in AD treatment. Further research focusing on optimizing polyphenol backbones or modifications could significantly advance the development of AD therapeutics.
3.4 Chemical reagents capable of regulating metal-free Aβ, metal-bound Aβ, ROS, and AChE
The dysregulation of ACh, driven by the upregulation of AChE in senile plaques, is well-known to disrupt cholinergic neurotransmission and contributes to cognitive deficits in AD.35,288 Despite the development of numerous AChE inhibitors, their efficacy has not met clinical expectations.28–32 As a result, recent research has shifted towards multivalent therapeutic approaches that target multiple pathological factors to maximize efficacy while minimizing side effects.101,289–291For AChE inhibition, it is essential to prevent substrate binding to the active site by blocking the CAS, PAS, or both, thereby interrupting enzymatic activity mediated by the catalytic triad. Simultaneously, the molecule should incorporate features that enable interactions with Aβ, such as hydrogen bonding and hydrophobic properties. Flavonoid derivatives have shown promise in modulating several pathological factors, including ROS, metal-free and metal-bound Aβ, and AChE.289,290,292 To enhance multi-targeting capabilities, structural modifications on the isoflavone or flavone core have been explored, as depicted in Fig. 8a.289,290,292 These modifications increased the reactivity of flavonoid by incorporating multiple hydroxyl groups that disrupt substrate complexation at the AChE active site through hydrogen bonding. Additionally, the hydrophobic A and C rings of flavonoids interacted with aromatic side chains, such as W86, F295, and Y341, within the hydrophobic binding pocket (Fig. 8b).292
 | ||
| Fig. 8 Examples of chemical reagents regulating metal-free or metal-bound Aβ and AChE. (a) Structures of flavonoids. Isoflavone, 3-phenyl-4H-chromen-4-one; isoflavone-1, 3-(3,4-dihydroxyphenyl)-5,7-dihydroxy-4H-chromen-4-one; isoflavone-2, 5,6,7-trihydroxy-3-phenyl-4H-chromen-4-one; isoflavone-3, 3-(3,4-dihydroxyphenyl)-5,6,7-trihydroxy-4H-chromen-4-one; flavone, 2-phenyl-4H-chromen-4-one; flavone-1, 2-(3,4-dihydroxyphenyl)-3,5,7-trihydroxy-4H-chromen-4-one; flavone-2, 2-(3,4-dihydroxyphenyl)-5,7-dihydroxy-4H-chromen-4-one; flavone-3, 2-(4-hydroxyphenyl)-3,5,7-trihydroxy-4H-chromen-4-one. (b) Intermolecular interactions between the flavonoids and AChE. Reproduced with permission from ref. 289 and 292. Copyright© 2023 Royal Society of Chemistry and 2020 Royal Society of Chemistry. (c) Impact of isoflavone-3 on the formation of metal-free Aβ42 and metal–Aβ42 aggregates. The morphology of the resultant Aβ42 species was analyzed by TEM. Reproduced with permission from ref. 289. Copyright© 2023 Royal Society of Chemistry. (d) Structures of emodin and its derivatives with different functional groups at the R position. Emodin, 1,3,8-trihydroxy-6-methylanthracene-9,10-dione; emodin-1, 1,8-dihydroxy-3-methyl-6-(2-(piperidin-1-yl)ethoxy)anthracene-9,10-dione; emodin-2, 1,8-dihydroxy-3-methyl-6-(2-(pyrrolidin-1-yl)ethoxy)anthracene-9,10-dione; emodin-3, 1,8-dihydroxy-3-methyl-6-(2-(dimethylamino)ethoxy)anthracene-9,10-dione; emodin-4, 1,8-dihydroxy-3-methyl-6-(2-morpholinoethoxy)anthracene-9,10-dione. (e) Binding of emodin-1 against AChE. Reproduced with permission from ref. 291. Copyright© 2024 Elsevier. (f) Effect of emodin on Aβ42 aggregation. The inhibitory activity of emodin on the production of β-sheet-rich Aβ42 aggregates was quantitatively measured by the ThT assay. Reproduced with permission from ref. 101. Copyright© 2021 John Wiley and Sons. (g) Impact of emodin-1 and emodin-2 on Cu(II)-induced Aβ42 aggregation. The influence of emodin-1 and emodin-2 on the generation of β-sheet-rich Cu(II)–Aβ42 aggregates was analyzed by the ThT assay. Reproduced with permission from ref. 293. Copyright© 2023 Springer Nature. | ||
Among these derivatives, a rationally designed and optimized flavonoid, isoflavone-3 (shown in Fig. 8a), which contains five hydroxyl groups on the isoflavone backbone, exhibited the highest reactivity.289,290 As illustrated in Fig. 8b, the 5-OH group on the A ring formed hydrogen bonds with the backbone carbonyl group of G120–G121 and the hydroxyl group of S203. Additionally, the 3-OH group on the B ring generated a hydrogen bond with the backbone amide group between I294 and F295, while the O1 donor atom on the C ring constructed a hydrogen bond with the hydroxyl group of Y337.289,294 These interactions effectively blocked the cavity located at the AChE active site, inhibiting the enzyme's function with an IC50 value of 0.19 μM.289
Isoflavone-3 significantly modulated the oligomerization and fibrillization of both metal-free Aβ and metal–Aβ. As shown resulted in the formation of shorter and thinner fibrils, whereas in the presence of Cu(II) or Zn(II), it produced fragmented fibrils in Fig. 8c, the addition of isoflavone-3 to metal-free Aβ42 with a mixture of amorphous and fibrillary aggregates.289 These alterations in aggregation were mediated by the interaction of isoflavone-3 with key regions of Aβ42, including the β-turn motif (V12–Q15), self-recognition site (L17–A21), and the C-terminal region (I32–A42), which are critical for Aβ42 fibrillization. Additionally, isoflavone-3 promoted the oxidation of H13, H14, and M35 in the presence of Cu(II), thereby disrupting metal coordination and the hydrophobic interactions between Aβ peptides, which influenced their aggregation behavior.289
The rational design of isoflavone-3 incorporated three hydroxyl groups at the C5–C7 positions on the A ring, a catechol moiety on the B ring, and translocation of the B ring from C2 to C3 on the C ring. This structural modification lowered its redox potential to 1.0 V (vs. SHE), enhancing its scavenging capability against free organic radicals.289 Overall, flavonoids like isoflavone-3 represent a promising platform for developing multifunctional therapeutics for AD by confronting multiple pathological features, including Aβ aggregation, metal ion dyshomeostasis, cholinergic dysfunction, and oxidative stress.
Several studies have suggested that anthraquinone compounds can alleviate AD symptoms by deactivating AChE through simultaneous targeting of the CAS and PAS, while also exhibiting antioxidant and anti-Aβ aggregation properties.101,291Emodin (Fig. 8d), a natural anthraquinone-based polyphenol used in traditional Chinese medicine, is a well-researched chemical reagent that addresses multiple pathological factors in AD.101,291,295–297 Numerous structural modifications have been made to the emodin backbone to improve its dual activity against AChE and Aβ.291 Among these derivatives, emodin-1 (illustrated in Fig. 8d) has demonstrated the most potent AChE inhibitory activity, with an IC50 value of 67 nM.291 The anthraquinone backbone of emodin-1 bound to PAS through CH–π interactions with W279, while additional hydrogen bonds, hydrophobic interactions, and π–π interactions occurred with F331.291 Furthermore, functional groups, such as piperidine and pyrrolidine rings, formed CH–π interactions with W84 and H440, effectively blocking the CAS, as displayed in Fig. 8e.291 These interactions at both the CAS and the PAS contributed to the efficient downregulation of AChE, reducing the degradation of ACh and thereby preserving cholinergic function.
Emodin has also been reported to modulate and inhibit Aβ42 aggregation, both in the absence and presence of Cu(II). As presented in Fig. 8f, emodin reduced the β-sheet-rich aggregation of Aβ42 by up to 80% in a dose-dependent manner,101 which is attributed to interactions between emodin's anthraquinone backbone and the V18 and F19 residues of Aβ, which form the self-recognition site.101Emodin derivatives with various functional groups have demonstrated similar modulatory effects on Aβ42 aggregation, with emodin-1 achieving an inhibition ratio of ca. 76%.293 A comparable trend was observed in the presence of Cu(II), where significant reductions in β-sheet-rich aggregates were noted in samples treated with emodin-1 and emodin-2 (Fig. 8g).293 This effect is likely due to the interaction of emodin-1–4 (Fig. 8d) with the hydrophobic surface of the Aβ42 helix in the N-terminal region, potentially stabilizing the α-helix and reducing the formation of oligomers and fibrils.293 Additionally, emodin possessed chelation properties that can regulate metal ion dyshomeostasis and oxidative stress. Treatment with emodin-1–4 dramatically decreased the formation of hydroxyl radicals (˙OH) and O2˙−, compared to controls with Cu(II) alone.291 These findings suggest that emodin derivatives display multiple reactivities towards pathological factors of AD, including metal chelation, oxidative stress modulation, Aβ aggregation control, and AChE inhibition, indicating their potential as promising MTDLs for AD treatment.291,293
3.5 Chemical reagents capable of controlling Aβ and MAOB
The loss of dopaminergic and serotonergic neurotransmission in AD can be triggered by the increased activity of MAOB.298,299 Inhibitors of MAOB, such as lazabemide, selegiline, and safinamide (Fig. 4b), have been shown to improve cognitive deficits and slow the progression of AD, making them therapeutically relevant candidates for anti-Alzheimer therapies.300,301 Given the limitations in efficacy and potential side effects of MAOB inhibitors, there has been an increasing focus on targeting additional AD pathological features, such as Aβ aggregation and elevated oxidative stress, simultaneously. Several developmental strategies have been employed to achieve this dual functionality, enabling interactions with the active site of MAOB while possessing structural moieties to address Aβ aggregation. For example, modifications of existing therapeutics are emerging as viable candidates for this approach.
Rasagiline (Fig. 9a), a prominent MAOB inhibitor, is currently in phase II clinical trials for AD due to its dual ability to control Aβ aggregation through its 2,3-dihydro-1H-indene moieties.304,305 Building on this framework, Xie and coworkers developed a selective and efficient MAOB inhibitor with the added capacity to target Aβ aggregation by combining the structural advantages of another MAO inhibitor, clorgyline, resulting in rasagiline–clorgyline conjugates.302,306 Among the synthesized conjugates, as described in Fig. 9a, RC-6j, which features a five-carbon linker, exhibited the highest modulatory potency and selectivity, with an IC50 of 4.0 nM and a selectivity index greater than 25000.302RC-6j inhibited MAOB by competitive binding at the active site, interacting with the FAD cofactor and establishing a CH–π interaction with Y435 in the substrate cavity. Compared to rasagiline, the chromanone moiety of RC-6j occupied the entrance cavity, which is composed of amino acid residues W119, F168, L171, C172, I198, I199, I316, and Y326, through van der Waals and hydrophobic interactions, thereby enhancing the binding affinity.302 In addition to MAOB inhibition, RC-6j also prevented Aβ42 aggregation, significantly reducing β-sheet-rich structures by 40% and decreasing overall aggregation (Fig. 9a).302,307 Furthermore, RC-6j mitigated neuronal damage caused by oxidative stress induced by 6-hydroxydopamine, a ROS generator that disrupts mitochondrial function through auto-oxidation.302 These findings suggest that RC-6j is a promising MTDL for simultaneously controlling MAOB activity, Aβ aggregation, and oxidative stress in AD treatment.
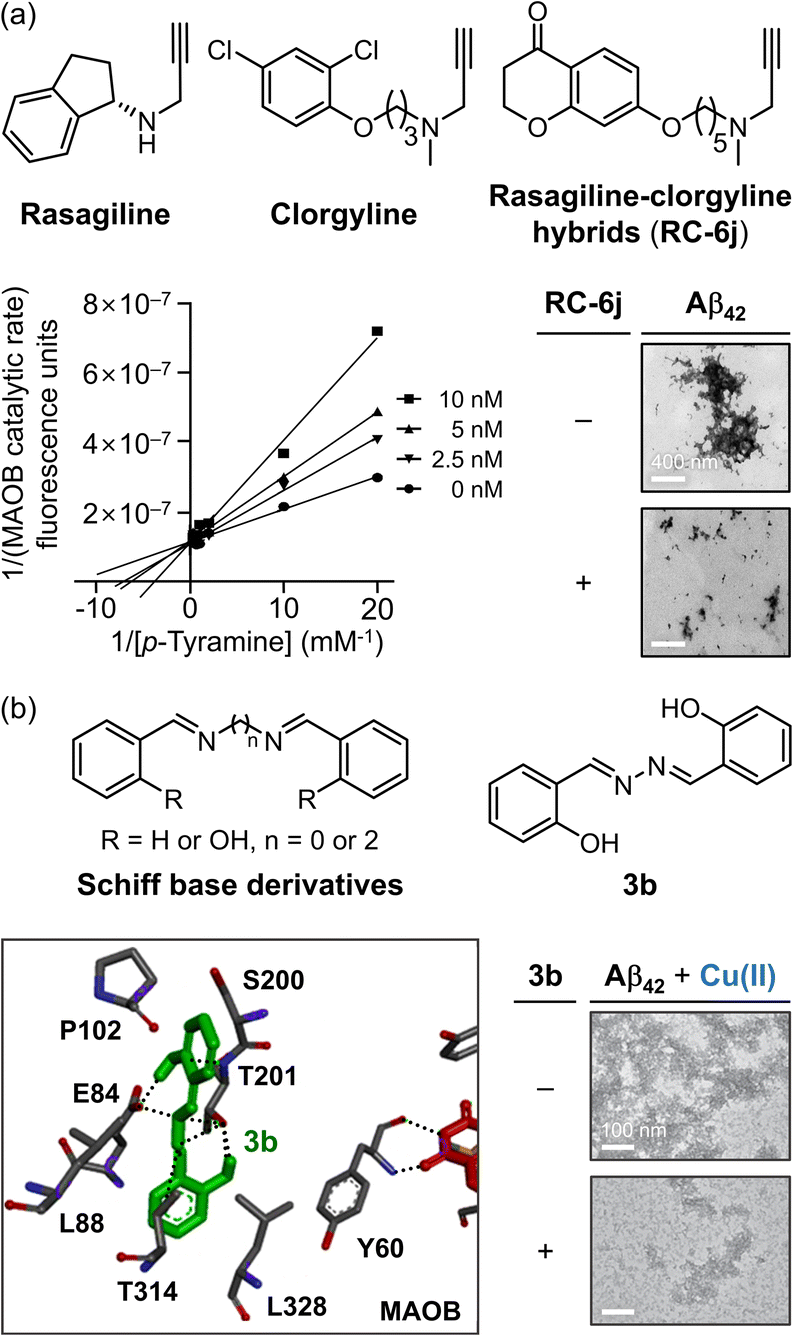 | ||
| Fig. 9 Examples of chemical reagents controlling Aβ and MAOB. (a) Structures of rasagiline, clorgyline, and RC-6j. The inhibitory effect of RC-6j against the activity of MAOB was represented in Lineweaver–Burk reciprocal plots, with its impact on the formation Aβ42 aggregates. The catalytic rate of MAOB was measured through the amount of H2O2 generated by catalytic oxidation of p-tyramine, observed by the Amplex Red H2O2/peroxidase assay. The morphology of the resultant Aβ42 species was monitored by TEM. RC-6j, 7-((5-(methyl(prop-2-yn-1-yl)amino)pentyl)oxy)chroman-4-one. Reproduced with permission from ref. 302. Copyright© 2020 Elsevier. (b) Structures of Schiff base derivatives and 3b, binding mode of 3b towards MAOB, and its effects on Cu(II)-added Aβ42 aggregation. The morphology of the Cu(II)-induced Aβ42 aggregates produced with and without 3b was investigated by TEM. 3b, 2,2′-((1E,1′E)-hydrazine-1,2-diylidenebis(methanylylidene))diphenol. Reproduced with permission from ref. 303. Copyright© 2020 Springer Nature. | ||
Clioquinol (Fig. 3a), which contains nitrogen and oxygen donor atoms for metal chelation, is also a widely utilized therapeutic framework to target metal ions and Aβ aggregation, as discussed in the previous section.26,27 Sang and coworkers designed a combinatorial strategy to address Aβ aggregation, metal ion dyshomeostasis, and MAOB activity by incorporating metal-binding donor atoms like clioquinol and Schiff base moieties into a backbone, as depicted in Fig. 9b.303 Specifically, 3b with 2-hydroxy moiety obtained the lowest IC50 value of 8.4 nM.303 SAR analysis revealed that an increase in the length of the linker domain or elimination of the hydroxyl group from the aromatic ring dramatically decreased their inhibitory activity.303 As illustrated in Fig. 9b, 3b prohibited MAOB activity through binding at its active site; notably, the hydroxyl groups of 3b generated hydrogen bonding with E84 and T201, which are essential for the catalytic activity of MAOB. In the absence of hydroxyl groups or additional methyl linkers, the compound no longer maintains its hydrogen bonding network, dramatically weakening binding affinity.303 Additionally, 3b altered Aβ42 aggregation, reducing fibril growth by 32% and 62% in the presence and absence of Cu(II), respectively. Furthermore, thinner and shorter fibrils were produced in the presence of 3b, supporting the modulatory role of 3b in Aβ42 aggregation.303 These findings suggest that 3b has potential as a multipotent therapeutic agent confronting MAOB, intracellular ROS, and metal-free and Cu(II)-bound Aβ42.
3.6 Chemical reagents capable of controlling Aβ, AChE, and MAOB
While targeting Aβ, AChE, and MAOB individually has shown promise in addressing specific aspects of AD, the complexity of its pathology necessitates a more integrated approach.35,288,298,299 The combinatorial interaction of Aβ, AChE, and MAOB has been shown to induce synaptic dysfunction through synergistic effects, resulting in impaired cholinergic, dopaminergic, and serotonergic neurotransmission.35,288,298,299 Therefore, simultaneously tackling all three mechanisms could improve therapeutic outcomes by alleviating symptoms and slowing disease progression. As discussed earlier, compounds should incorporate structural moieties capable of interacting with the CAS and PAS of AChE, the active site of MAOB, and functional groups that effectively disrupt intermolecular hydrogen bonding and hydrophobic interactions of Aβ.Chromone (Fig. 10a), a heterocyclic compound with a benzopyranone backbone, is a versatile scaffold in drug discovery for various diseases, including AD, due to its antioxidant activity, anti-amyloidogenic properties, and ability to modulate cholinergic and glutaminergic systems.308,310–313Chromone inhibits MAOB activity by forming intermolecular hydrogen bonds between C172 and Y326 through its sp2 oxygen atom, a feature unique to MAOB.314 Several SAR studies have further explored the chromone backbone to enhance its multi-targeting capabilities, as shown in Fig. 10a.308,312,313 Kumar and coworkers found that attaching pyrimidine derivatives to the chromone backbone introduces a tertiary nitrogen moiety as a pharmacophore, enabling additional cation–π interaction with W84 in AChE.308Chromone derivatives (Fig. 10a) demonstrated strong inhibitory activity against MAOB and AChE, while chromone-1, featuring a pyrrolidine group at the C4 position, showed the highest potency (IC50 = 2.1 μM for MAOB and 80 nM for AChE).308 In MAOB, the phenyl ring of chromone-1 engages in π–π stacking with Y236, while pyrrolidine and morpholine rings at C4 form electrostatic interactions with P102, F103, and W119, surrounding the FAD cofactor. These interactions are absent in chromone-3, which has a morpholine ring at the C3 position, emphasizing the importance of functional group positioning in designing multi-target-directed molecules.308 All chromone derivatives exhibited similar interactions with AChE, including π–π stacking with F331 and W279 at the PAS and cation–π interactions with W84 at the CAS, underscoring the critical role of the tertiary nitrogen group in AChE inhibition.308 In addition, treatment with chromone compounds restricted the formation of Aβ fibrils and plaques, as evidenced by the loss of aggregate structures in the images obtained by field emission scanning electron microscopy (FE-SEM) (Fig. 10a).308 These findings emphasize the potential of the chromone scaffold in developing multi-target-directed molecules against MAOB, AChE, and Aβ.
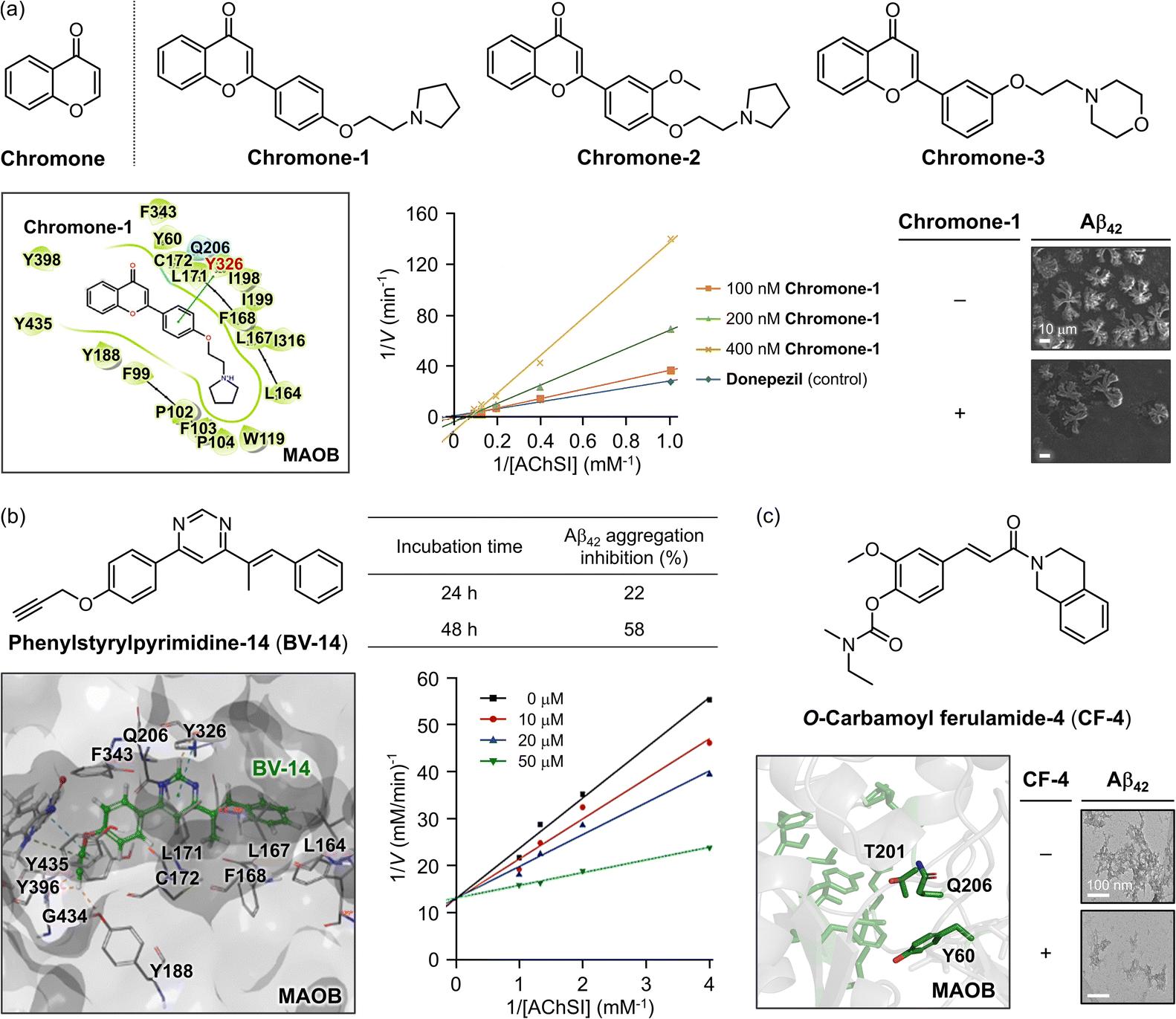 | ||
| Fig. 10 Examples of chemical reagents regulating Aβ, AChE, and MAOB. (a) Structures of chromone and its derivatives, binding of chromone-1 to MAOB, and its impact on AChE activity and Aβ42 aggregation. The inhibitory effect of chromone-1 against the activity of AChE was performed using Ellman's method and represented in a double reciprocal Lineweaver–Burk plot. The morphology of Aβ42 aggregates generated with or without chromone-1 was obtained by FE-SEM. Reproduced with permission from ref. 308. Copyright© 2024 American Chemical Society. Chromone-1, 2-(4-(2-(pyrrolidin-1-yl)ethoxy)phenyl)-4H-chromen-4-one; chromone-2, 2-(3-methoxy-4-(2-(pyrrolidin-1-yl)ethoxy)phenyl)-4H-chromen-4-one; chromone-3, 2-(3-(2-morpholinoethoxy)phenyl)-4H-chromen-4-one. (b) Structure of BV-14, its binding mode towards MAOB, and effects on the Aβ42 aggregation and AChE activity. The extent of the inhibition of Aβ42 aggregation was assessed using the ThT assay. The AChE inhibition by BV-14 was measured using Ellman's method and shown in an overlaid Lineweaver–Burk reciprocal plot. BV-14, (Z)-4-(1-phenylprop-1-en-2-yl)-6-(4-(prop-2-yn-1-yloxy)phenyl)pyrimidine. Reproduced with permission from ref. 309. Copyright© 2024 Royal Society of Chemistry. (c) Structure of CF-4, its binding to MAOB (PDB 1GOS113), and impact on the production of Aβ42 aggregates. The morphology of the resultant Aβ42 species was analyzed by TEM. CF-4, (E)-4-(3-(3,4-dihydroisoquinolin-2(1H)-yl)-3-oxoprop-1-en-1-yl)-2-methoxyphenyl ethyl(methyl)carbamate. Reproduced with permission from ref. 310. Copyright© 2020 Elsevier. | ||
In a separate study, Kumar and coworkers designed phenylstyrylpyrimidine derivatives containing O-propargyl and amidine moieties, a combination of previously known pharmacophores, to evaluate their multi-target potential.309,315,316 The study demonstrated the inhibition of MAOB activity, and a SAR analysis revealed that BV-14 (Fig. 10b), featuring a formamidine group, outperformed other functional derivatives with guanidine, acetamidine, or benzamidine groups, attaining an IC50 of 7.3 μM.309BV-14 suppressed MAOB activity by binding to the active site, with its pyrimidine ring and propargyl group engaging with the Y326 residue and the FAD cofactor of MAOB through hydrophobic interactions, including π–π stacking, and additional hydrogen bonds (not shown in the figure).309 In addition, BV-14 showed the inhibitory effect on AChE, possessing IC50 of 7.3 μM (Fig. 10b), which was achieved by the hydrogen bonding with F288, a residue from the PAS.309 To assess the broader impact of BV-14 on AD-related factors, researchers also evaluated its influence on prohibiting ROS generation and Aβ aggregation. Although detailed insights were not provided, BV-14 lowered ROS production by 48%, as depicted in Fig. 10b. Additionally, BV-14 prevented Aβ42 aggregation, reducing fibril growth by 22%. These findings suggest that BV-14 has potential as a multipotent therapeutic agent targeting MAOB, intracellular ROS, and Aβ42.309
Natural products have also been explored for their potential to modulate Aβ aggregation, along with AChE and MAOB activity. Ferulic acid, a widely distributed plant constituent first isolated from Ferula foetida, has been investigated as a therapeutic agent for AD in this manner.317,318 Chen and coworkers designed O-carbamoyl ferulamide derivatives by combining ferulic acid with a carbamate fragment, a pharmacophore found in rivastigmine (Fig. 4a), to create compounds with multi-targeting potency against MAOB, AChE, and Aβ.310,319 Among these derivatives, CF-4 (Fig. 10c) emerged as a promising and selective MAOB inhibitor with an IC50 of 5.3 μM. CF-4's carbamoyl group interacted with the FAD cofactor, forming intermolecular hydrogen bonds and π–π interactions with the Y60 residue. The carbonyl group of the ferulic acid backbone generated hydrogen bonds with T201 and Q206, emphasizing the importance of the ferulic acid moiety in binding to the active site of MAOB.310 Furthermore, CF-4 not only interacted with the active site of MAOB but also exhibited inhibitory effects on AChE. The benzene ring of the 1,2,3,4-tetrahydroisoquinoline and the ferulic acid created π–π interactions with key amino acid residues in AChE, enabling CF-4 to simultaneously associate with both the CAS and the PAS, leading to effective suppression of AChE.310 Additionally, CF-4 prohibited Aβ42 aggregation by up to 58%,310 with a significant reduction in bulk Aβ42 aggregates (Fig. 10c). Notably, CF-4 also showed self-mediated disaggregation of Aβ42 by 43%, demonstrating its dual capacity to alter and disaggregate Aβ42 aggregates.310CF-4's ability to cross the BBB permeability and its efficacy in improving cognitive defects induced by scopolamine further validated its potential for clinical use.310 These findings on CF-4, with those of chromone derivatives and BV-14, underscore the importance of targeting AChE, MAOB, and Aβ in AD therapy. This comprehensive approach, which combines existing therapeutics to address the complex pathology of AD, offers a more efficient strategy for disease treatment.
4. Conclusions
With the rising prevalence of AD, there is an increasing need for effective therapeutic strategies. The limitations of current single-target-directed treatments highlight the necessity for more comprehensive approaches that address the multifactorial nature of AD. This review illustrates several pathological features in AD, particularly Aβ aggregation, metal ion dysregulation, and enzymes' dysfunction. While Aβ has long been considered a primary therapeutic target due to its role in disrupting cellular functions, inducing inflammation, and impairing synaptic activity, treatments focused solely on Aβ have often demonstrated limited success. Although recent drug approvals aiming at Aβ underscore its continued relevance as a therapeutic biomarker, it is essential to concurrently engage additional neurotoxic factors contributing to AD progression to effectively combat the disease. The complex interplay of metal ion dyshomeostasis, metalloenzyme dysregulation, impaired neurotransmission, and elevated oxidative stress further complicates the pathology of AD and constrains the success of isolated interventions.As such, the development of multi-target-directed molecules capable of modulating multiple pathogenic pathways offers a promising strategy to enhance therapeutic efficacy. Future research should prioritize the refinement and optimization of MTDLs to address the interconnected mechanisms associated with AD. A more systematic therapeutic approach that simultaneously tackles Aβ aggregation, metal ion dyshomeostasis, enzyme dysregulation, and neurotransmitter imbalance holds the potential to overcome the limitations of current single-target-directed treatments. Alongside the multi-target-directed molecules primarily focusing on Aβ, extensive research has also been conducted on therapeutic combinations not covered in this review, which underscores their significance and potential in advancing AD treatment strategies. Furthermore, beyond the pathogenic targets discussed in this review, the development of MTDLs aimed at neuroinflammation, mitochondrial dysfunction, and neuronal cell death could provide new insights into their interconnected roles in AD pathogenesis and therapeutic intervention. A deeper understanding of these processes and their interplay with previously discussed factors could guide the design of MTDLs capable of effectively confronting the complex, multifaceted nature of AD. Expanding the scope of therapeutic targets, as discussed in this review, can serve as a foundation for future research, driving the development of disease-modifying treatments that efficiently address the complexity of AD.
Data availability
This manuscript is a review article; therefore, no new data were generated or analyzed in this study, and a data availability statement is not applicable.Author contributions
All authors contributed to the conceptualization of this review. Jeasang Yoo, Jimin Lee, Byeongha Ahn prepared the draft, which was revised by Professors Mi Hee Lim and Jiyeon Han.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants of the National Research Foundation of Korea funded by the Korean Government {Creative Research Initiative [RS-2022-NR070709 (M. H. L.)]; Global Science Research Center Program [RS-2024-00411134 (M. H. L.)]; RS-2024-00353015 (J. H.)}.References
- Y. Zhang, H. Chen, R. Li, K. Sterling and W. Song, Amyloid β-based therapy for Alzheimer's disease: Challenges, successes and future, Signal Transduct. Targeted Ther., 2023, 8, 1–26 CrossRef PubMed
.
- P. H. Nguyen, A. Ramamoorthy, B. R. Sahoo, J. Zheng, P. Faller, J. E. Straub, L. Dominguez, J.-E. Shea, N. V. Dokholyan, A. De Simone, B. Ma, R. Nussinov, S. Najafi, S. T. Ngo, A. Loquet, M. Chiricotto, P. Ganguly, J. McCarty, M. S. Li, C. Hall, Y. Wang, Y. Miller, S. Melchionna, B. Habenstein, S. Timr, J. Chen, B. Hnath, B. Strodel, R. Kayed, S. Lesné, G. Wei, F. Sterpone, A. J. Doig and P. Derreumaux, Amyloid oligomers: A joint experimental/computational perspective on Alzheimer's disease, Parkinson's disease, Type II diabetes, and amyotrophic lateral sclerosis, Chem. Rev., 2021, 121, 2545–2647 CrossRef PubMed
- O. Benek, J. Korabecny and O. Soukup, A perspective on multitarget drugs for Alzheimer's disease, Trends Pharmacol. Sci., 2020, 41, 434–445 CrossRef PubMed
- F. Leng and P. Edison, Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here?, Nat. Rev. Neurol., 2021, 17, 157–172 CrossRef PubMed
- W. Wang, F. Zhao, X. Ma, G. Perry and X. Zhu, Mitochondria dysfunction in the pathogenesis of Alzheimer's disease: Recent advances, Mol. Neurodegener., 2020, 15, 30 CrossRef PubMed
- P. Goel, S. Chakrabarti, K. Goel, K. Bhutani, T. Chopra and S. Bali, Neuronal cell death mechanisms in Alzheimer's disease: An insight, Front. Mol. Neurosci., 2022, 15, 937133 CrossRef PubMed
- S. Kumari, R. Dhapola and D. H. Reddy, Apoptosis in Alzheimer's disease: Insight into the signaling pathways and therapeutic avenues, Apoptosis, 2023, 28, 943–957 CrossRef PubMed
- I. G. Onyango, G. V. Jauregui, M. Čarná, J. P. Bennett Jr and G. B. Stokin, Neuroinflammation in Alzheimer's disease, Biomedicines, 2021, 9, 524 CrossRef CAS PubMed
- S. Thakur, R. Dhapola, P. Sarma, B. Medhi and D. H. Reddy, Neuroinflammation in Alzheimer's disease: Current progress in molecular signaling and therapeutics, Inflammation, 2023, 46, 1–17 CrossRef CAS
- R. Dhapola, S. S. Hota, P. Sarma, A. Bhattacharyya, B. Medhi and D. H. Reddy, Recent advances in molecular pathways and therapeutic implications targeting neuroinflammation for Alzheimer's disease, Inflammopharmacology, 2021, 29, 1669–1681 CrossRef CAS PubMed
- Z.-Z. Si, C.-J. Zou, X. Mei, X.-F. Li, H. Luo, Y. Shen, J. Hu, X.-X. Li, L. Wu and Y. Liu, Targeting neuroinflammation in Alzheimer's disease: From mechanisms to clinical applications, Neural Regener. Res., 2023, 18, 708–715 CrossRef CAS
- C. Sharma, S. Kim, Y. Nam, U. J. Jung and S. R. Kim, Mitochondrial dysfunction as a driver of cognitive impairment in Alzheimer's disease, Int. J. Mol. Sci., 2021, 22, 4850 CrossRef CAS PubMed
- S. N. Rai, C. Singh, A. Singh, M. Singh and B. K. Singh, Mitochondrial dysfunction: A potential therapeutic target to treat Alzheimer's disease, Mol. Neurobiol., 2020, 57, 3075–3088 CrossRef CAS
- T. Ashleigh, R. H. Swerdlow and M. F. Beal, The role of mitochondrial dysfunction in Alzheimer's disease pathogenesis, Alzheimer's Dementia, 2023, 19, 333–342 CrossRef CAS PubMed
- V. K. Sharma, T. G. Singh, S. Singh, N. Garg and S. Dhiman, Apoptotic pathways and Alzheimer's disease: Probing therapeutic potential, Neurochem. Res., 2021, 46, 3103–3122 CrossRef CAS
- J. Han, Z. Du and M. H. Lim, Mechanistic insight into the design of chemical tools to control multiple pathogenic features in Alzheimer's disease, Acc. Chem. Res., 2021, 54, 3930–3940 CrossRef CAS
- C. H. van Dyck, C. J. Swanson, P. Aisen, R. J. Bateman, C. Chen, M. Gee, M. Kanekiyo, D. Li, L. Reyderman, S. Cohen, L. Froelich, S. Katayama, M. Sabbagh, B. Vellas, D. Watson, S. Dhadda, M. Irizarry, L. D. Kramer and T. Iwatsubo, Lecanemab in early Alzheimer's disease, N. Engl. J. Med., 2023, 388, 9–21 CrossRef CAS PubMed
- J. Cummings, Anti-amyloid monoclonal antibodies are transformative treatments that redefine Alzheimer's disease therapeutics, Drugs, 2023, 83, 569–576 CrossRef CAS
- J. R. Sims, J. A. Zimmer, C. D. Evans, M. Lu, P. Ardayfio, J. Sparks, A. M. Wessels, S. Shcherbinin, H. Wang, E. S. Monkul Nery, E. C. Collins, P. Solomon, S. Salloway, L. G. Apostolova, O. Hansson, C. Ritchie, D. A. Brooks, M. Mintun and D. M. Skovronsky, TRAILBLAZER-ALZ 2 Investigators, Donanemab in early symptomatic Alzheimer disease: The TRAILBLAZER-ALZ 2 Randomized clinical trial, J. Am. Med. Assoc., 2023, 330, 512–527 CrossRef
- L. Söderberg, M. Johannesson, P. Nygren, H. Laudon, F. Eriksson, G. Osswald, C. Möller and L. Lannfelt, Lecanemab, aducanumab, and gantenerumab — Binding profiles to different forms of amyloid-beta might explain efficacy and side effects in clinical trials for Alzheimer's disease, Neurotherapeutics, 2023, 20, 195–206 CrossRef PubMed
- G. C. Alexander, S. Emerson and A. S. Kesselheim, Evaluation of aducanumab for Alzheimer disease: Scientific evidence and regulatory review involving efficacy, safety, and futility, J. Am. Med. Assoc., 2021, 325, 1717–1718 CrossRef PubMed
- I. Mohr and K. H. Weiss, Current anti-copper therapies in management of Wilson disease, Ann. Transl. Med., 2019, 7, S69 CrossRef PubMed
- S. Baldari, G. Di Rocco and G. Toietta, Current biomedical use of copper chelation therapy, Int. J. Mol. Sci., 2020, 21, 1069–1088 CrossRef PubMed
- R. A. Cherny, C. S. Atwood, M. E. Xilinas, D. N. Gray, W. D. Jones, C. A. McLean, K. J. Barnham, I. Volitakis, F. W. Fraser, Y. Kim, X. Huang, L. E. Goldstein, R. D. Moir, J. T. Lim, K. Beyreuther, H. Zheng, R. E. Tanzi, C. L. Masters and A. I. Bush, Treatment with a copper-zinc chelator markedly and rapidly inhibits β-amyloid accumulation in Alzheimer's disease transgenic mice, Neuron, 2001, 30, 665–676 CrossRef
- K. D. Fasae, A. O. Abolaji, T. R. Faloye, A. Y. Odunsi, B. O. Oyetayo, J. I. Enya, J. A. Rotimi, R. O. Akinyemi, A. J. Whitworth and M. Aschner, Metallobiology and therapeutic chelation of biometals (copper, zinc and iron) in Alzheimer's disease: Limitations, and current and future perspectives, J. Trace Elem. Med. Biol., 2021, 67, 126779 CrossRef PubMed
- S. R. Bareggi and U. Cornelli, Clioquinol: Review of its mechanisms of action and clinical uses in neurodegenerative disorders, CNS Neurosci. Ther, 2012, 18, 41–46 CrossRef
- N. Harbison-Price, S. A. Ferguson, A. Heikal, G. Taiaroa, K. Hards, Y. Nakatani, D. Rennison, M. A. Brimble, I. M. El-Deeb, L. Bohlmann, C. A. McDevitt, M. von Itzstein, M. J. Walker and G. M. Cook, Multiple bactericidal mechanisms of the zinc ionophore PBT2, mSphere, 2020, 5, e00157 CrossRef
- G. Marucci, M. Buccioni, D. D. Ben, C. Lambertucci, R. Volpini and F. Amenta, Efficacy of acetylcholinesterase inhibitors in Alzheimer's disease, Neuropharmacology, 2021, 190, 108352 CrossRef
- H. Sugimoto, H. Ogura, Y. Arai, Y. Iimura and Y. Yamanishi, Research and development of donepezil hydrochloride, a new type of acetylcholinesterase inhibitor, Jpn. J. Pharmacol., 2002, 89, 7–20 CrossRef
- R. C. Petersen, R. G. Thomas, M. Grundman, D. Bennett, R. Doody, S. Ferris, D. Galasko, S. Jin, J. Kaye, A. Levey, E. Pfeiffer, M. Sano, C. H. van Dyck and L. J. Thal, Vitamin E and donepezil for the treatment of mild cognitive impairment, N. Engl. J. Med., 2005, 352, 2379–2388 CrossRef
- R. J. Polinsky, Clinical pharmacology of rivastigmine: a new-generation acetylcholinesterase inhibitor for the treatment of Alzheimer’s disease, Clin. Ter., 1998, 20, 634–647 Search PubMed
- C. V. R. Blacker, D. T. Greenwood, K. A. Wesnes, R. Wilson, C. Woodward, I. Howe and T. Ali, Effect of galantamine hydrobromide in chronic fatigue syndrome: A randomized controlled trial, J. Am. Med. Assoc., 2004, 292, 1195–1204 CrossRef PubMed
- L. De Colibus, M. Li, C. Binda, A. Lustig, D. E. Edmondson and A. Mattevi, Three-dimensional structure of human monoamine oxidase A (MAO A): Relation to the structures of rat MAO A and human MAO B, Proc. Natl. Acad. Sci. U.S.A., 2005, 102, 12684–12689 CrossRef PubMed
- 2024 Alzheimer’s disease facts and figures, Alzheimers Dement., 2024, 20, 3708–3821 CrossRef
- M. G. Savelieff, G. Nam, J. Kang, H. J. Lee, M. Lee and M. H. Lim, Development of multifunctional molecules as potential therapeutic candidates for Alzheimer's disease, Parkinson's disease, and amyotrophic lateral sclerosis in the last decade, Chem. Rev., 2019, 119, 1221–1322 CrossRef PubMed
- T. Storr, Multifunctional compounds for the treatment of Alzheimer's disease, Can. J. Chem., 2021, 99, 1–9 CrossRef
- G. G. Glenner and C. W. Wong, Alzheimer's disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein, Biochem. Biophys. Res. Commun., 1984, 120, 885–890 CrossRef PubMed
- J. A. Hardy and G. A. Higgins, Alzheimer's disease: The amyloid cascade hypothesis, Science, 1992, 256, 184–185 CrossRef
- M. Kollmer, W. Close, L. Funk, J. Rasmussen, A. Bsoul, A. Schierhorn, M. Schmidt, C. J. Sigurdson, M. Jucker and M. Fändrich, Cryo-EM structure and polymorphism of Aβ amyloid fibrils purified from Alzheimer's brain tissue, Nat. Commun., 2019, 10, 4760 CrossRef PubMed
- L. Gremer, D. Schölzel, C. Schenk, E. Reinartz, J. Labahn, R. B. G. Ravelli, M. Tusche, C. Lopez-Iglesias, W. Hoyer, H. Heise, D. Willbold and G. F. Schröder, Fibril structure of amyloid-β(1–42) by cryo–electron microscopy, Science, 2017, 358, 116–119 CrossRef
- V. K. Ramanan and G. S. Day, Anti-amyloid therapies for Alzheimer disease: Finally, good news for patients, Mol. Neurodegener., 2023, 18, 42 CrossRef PubMed
- S. Patel, L. Vuillard, A. Cleasby, C. W. Murray and J. Yon, Apo and inhibitor complex structures of BACE (β-secretase), J. Mol. Biol., 2004, 343, 407–416 CrossRef PubMed
- X.-C. Bai, E. Rajendra, G. Yang, Y. Shi and S. HW Scheres, Sampling the conformational space of the catalytic subunit of human γ-secretase, Elife, 2016, 4, e11182 CrossRef
- H. Haukedal and K. K. Freude, Implications of glycosylation in Alzheimer's disease, Front. Neurosci., 2020, 14, 625348 CrossRef PubMed
- E. Nam, Y. Lin, J. Park, H. Do, J. Han, B. Jeong, S. Park, D. Y. Lee, M. Kim, J. Han, M.-H. Baik, Y.-H. Lee and M. H. Lim, APP-C31: An intracellular promoter of both metal-free and metal-bound amyloid-β40 aggregation and toxicity in Alzheimer's disease, Adv. Sci., 2024, 11, 2307182 CrossRef
- M. P. Kummer and M. T. Heneka, Truncated and modified amyloid-beta species, Alzheimer's Res. Ther., 2014, 6, 1–9 CrossRef
- Y. Yi, J. Lee and M. H. Lim, Amyloid-β-interacting proteins in peripheral fluids of Alzheimer's disease, Trends Chem., 2024, 6, 128–143 CrossRef
- S. J. C. Lee, E. Nam, H. J. Lee, M. G. Savelieff and M. H. Lim, Towards an understanding of amyloid-β oligomers: characterization, toxicity mechanisms, and inhibitors, Chem. Soc. Rev., 2017, 46, 310–323 RSC
- N. L. Sikanyika, H. C. Parkington, A. I. Smith and S. Kuruppu, Powering amyloid beta degrading enzymes: A possible therapy for Alzheimer's disease, Neurochem. Res., 2019, 44, 1289–1296 CrossRef PubMed
- J. Zhao, X. Liu, W. Xia, Y. Zhang and C. Wang, Targeting amyloidogenic processing of APP in Alzheimer's disease, Front. Mol. Neurosci., 2020, 13, 137 CrossRef PubMed
- A. Abbott, Could drugs prevent Alzheimer's? These trials aim to find out, Nature, 2022, 603, 216–219 CrossRef
- F. H. Bazzari and A. H. Bazzari, BACE1 Inhibitors for Alzheimer's disease: The past, present and any future?, Molecules, 2022, 27, 8823 CrossRef PubMed
- Y. Luo, B. Bolon, S. Kahn, B. D. Bennett, S. Babu-Khan, P. Denis, W. Fan, H. Kha, J. Zhang, Y. Gong, L. Martin, J.-C. Louis, Q. Yan, W. G. Richards, M. Citron and R. Vassar, Mice deficient in BACE1, the Alzheimer's β-secretase, have normal phenotype and abolished β-amyloid generation, Nat. Neurosci., 2001, 4, 231–232 CrossRef PubMed
- C.-C. Hsiao, F. Rombouts and H. J. M. Gijsen, New evolutions in the BACE1 inhibitor field from 2014 to 2018, Bioorg. Med. Chem. Lett., 2019, 29, 761–777 CrossRef
- M. F. Egan, J. Kost, P. N. Tariot, P. S. Aisen, J. L. Cummings, B. Vellas, C. Sur, Y. Mukai, T. Voss, C. Furtek, E. Mahoney, L. H. Mozley, R. Vandenberghe, Y. Mo and D. Michelson, Randomized trial of verubecestat for mild-to-moderate Alzheimer's disease, N. Engl. J. Med., 2018, 378, 1691–1703 CrossRef PubMed
- A. M. Wessels, P. N. Tariot, J. A. Zimmer, K. J. Selzler, S. M. Bragg, S. W. Andersen, J. Landry, J. H. Krull, A. M. Downing, B. A. Willis, S. Shcherbinin, J. Mullen, P. Barker, J. Schumi, C. Shering, B. R. Matthews, R. A. Stern, B. Vellas, S. Cohen, E. MacSweeney, M. Boada and J. R. Sims, Efficacy and safety of lanabecestat for treatment of early and mild Alzheimer disease: The AMARANTH and DAYBREAK-ALZ randomized clinical trials, J. Am. Med. Assoc. Neurol., 2020, 77, 199–209 Search PubMed
- J.-Y. Hur, G. R. Frost, X. Wu, C. Crump, S. J. Pan, E. Wong, M. Barros, T. Li, P. Nie, Y. Zhai, J. C. Wang, J. TCW, L. Guo, A. McKenzie, C. Ming, X. Zhou, M. Wang, Y. Sagi, A. E. Renton, B. T. Esposito, Y. Kim, K. R. Sadleir, I. Trinh, R. A. Rissman, R. Vassar, B. Zhang, D. S. Johnson, E. Masliah, P. Greengard, A. Goate and Y.-M. Li, The innate immunity protein IFITM3 modulates γ-secretase in Alzheimer's disease, Nature, 2020, 586, 735–740 CrossRef PubMed
- C. C. Shelton, L. Zhu, D. Chau, L. Yang, R. Wang, H. Djaballah, H. Zheng and Y.-M. Li, Modulation of γ-secretase specificity using small molecule allosteric inhibitors, Proc. Natl. Acad. Sci. U.S.A., 2009, 106, 20228–20233 CrossRef PubMed
- R. S. Doody, R. Raman, M. Farlow, T. Iwatsubo, B. Vellas, S. Joffe, K. Kieburtz, F. He, X. Sun, R. G. Thomas, P. S. Aisen, E. Siemers, G. Sethuraman and R. Mohs, A phase 3 trial of semagacestat for treatment of Alzheimer's disease, N. Engl. J. Med., 2013, 369, 341–350 CrossRef CAS
- V. Coric, C. H. van Dyck, S. Salloway, N. Andreasen, M. Brody, R. W. Richter, H. Soininen, S. Thein, T. Shiovitz, G. Pilcher, S. Colby, L. Rollin, R. Dockens, C. Pachai, E. Portelius, U. Andreasson, K. Blennow, H. Soares, C. Albright, H. H. Feldman and R. M. Berman, Safety and tolerability of the γ-secretase inhibitor avagacestat in a phase 2 study of mild to moderate Alzheimer disease, Arch. Neurol., 2012, 69, 1430–1440 CrossRef PubMed
- K. P. Kepp, Bioinorganic chemistry of Alzheimer's disease, Chem. Rev., 2012, 112, 5193–5239 CrossRef CAS
- E. L. Que, D. W. Domaille and C. J. Chang, Metals in neurobiology: Probing their chemistry and biology with molecular imaging, Chem. Rev., 2008, 108, 1517–1549 CrossRef CAS
- J. Kaplan and D. M. Ward, The essential nature of iron usage and regulation, Curr. Biol., 2013, 23, R642–R646 CrossRef CAS PubMed
- C. Livingstone, Zinc: Physiology, deficiency, and parenteral nutrition, Nutr. Clin. Pract., 2015, 30, 371–382 CrossRef CAS
- C. M. Opazo, M. A. Greenough and A. I. Bush, Copper: From neurotransmission to neuroproteostasis, Front. Aging Neurosci., 2014, 6, 143 Search PubMed
- J. Yoo, J. Han and M. H. Lim, Transition metal ions and neurotransmitters: Coordination chemistry and implications for neurodegeneration, RSC Chem. Biol., 2023, 4, 548–563 RSC
- L.-L. Chen, Y.-G. Fan, L.-X. Zhao, Q. Zhang and Z.-Y. Wang, The metal ion hypothesis of Alzheimer's disease and the anti-neuroinflammatory effect of metal chelators, Bioorg. Chem., 2023, 131, 106301 CrossRef CAS PubMed
- S. A. James, I. Volitakis, P. A. Adlard, J. A. Duce, C. L. Masters, R. A. Cherny and A. I. Bush, Elevated labile Cu is associated with oxidative pathology in Alzheimer disease, Free Radicals Biol. Med., 2012, 52, 298–302 CrossRef CAS
- J.-M. Suh, M. Kim, J. Yoo, J. Han, C. Paulina and M. H. Lim, Intercommunication between metal ions and amyloidogenic peptides or proteins in protein misfolding
disorders, Coord. Chem. Rev., 2023, 478, 214978 CrossRef CAS
- P. A. Elkins, Y. S. Ho, W. W. Smith, C. A. Janson, K. J. D'Alessio, M. S. McQueney, M. D. Cummings and A. M. Romanic, Structure of the C-terminally truncated human ProMMP9, a gelatin-binding matrix metalloproteinase, Acta Crystallogr. D, 2002, 58, 1182–1192 CrossRef
- X. Zhang, Q. Wang, J. Wu, J. Wang, Y. Shi and M. Liu, Crystal structure of human lysyl oxidase-like 2 (hLOXL2) in a precursor state, Proc. Natl. Acad. Sci. U.S.A., 2018, 115, 3828–3833 CrossRef
- K. L. Summers, G. Roseman, K. M. Schilling, N. V. Dolgova, M. J. Pushie, D. Sokaras, T. Kroll, H. H. Harris, G. L. Millhauser, I. J. Pickering and G. N. George, Alzheimer's drug PBT2 interacts with the amyloid β 1-42 peptide differently than other 8-hydroxyquinoline chelating drugs, Inorg. Chem., 2022, 61, 14626–14640 CrossRef PubMed
- A. Barbouti, N. Lagopati, D. Veroutis, V. Goulas, K. Evangelou, P. Kanavaros, V. G. Gorgoulis and D. Galaris, Implication of dietary iron-chelating bioactive compounds in molecular mechanisms of oxidative stress-induced cell ageing, Antioxidants, 2021, 10, 491 CrossRef
- C. Liu and X. Luo, Potential molecular and graphene oxide chelators to dissolve amyloid-β plaques in Alzheimer's disease: A density functional theory study, J. Mater. Chem. B, 2021, 9, 2736–2746 RSC
- O. D. Schreiner and T. G. Schreiner, Iron chelators as a therapeutic option for Alzheimer's disease – a mini-review, Front. Aging, 2023, 4, 1234958 CrossRef PubMed
- G. J. Kontoghiorghes and C. N. Kontoghiorghe, Iron and chelation in biochemistry and medicine: New approaches to controlling iron metabolism and treating related diseases, Cells, 2020, 9, 1456 CrossRef PubMed
- M. L. Hegde, P. Bharathi, A. Suram, C. Venugopal, R. Jagannathan, P. Poddar, P. Srinivas, K. Sambamurti, K. J. Rao, J. Scancar, L. Messori, L. Zecca and P. Zatta, Challenges associated with metal chelation therapy in Alzheimer's disease, J. Alzheimer’s Dis., 2009, 17, 457–468 Search PubMed
- S. C. Drew, The case for abandoning therapeutic chelation of copper ions in Alzheimer's disease, Front. Neurosci., 2017, 11, 317 CrossRef PubMed
- V. Chaudhari, S. Bagwe-Parab, H. S. Buttar, S. Gupta, A. Vora and G. Kaur, Challenges and opportunities of metal chelation therapy in trace metals overload-induced Alzheimer's disease, Neurotox. Res., 2023, 41, 270–287 CrossRef
- F. Hane and Z. Leonenko, Effect of metals on kinetic pathways of amyloid-β aggregation, Biomolecules, 2014, 4, 101–116 CrossRef
- A. C. Kim, S. Lim and Y. K. Kim, Metal ion effects on Aβ and tau aggregation, Int. J. Mol. Sci., 2018, 19, 128 Search PubMed
- Y. Yi and M. H. Lim, Current understanding of metal-dependent amyloid-β aggregation and toxicity, RSC Chem. Biol., 2023, 4, 121–131 RSC
- V. Borghesani, B. Alies and C. Hureau, CuII binding to various forms of amyloid-β peptides: Are they friends or foes?, Eur. J. Inorg. Chem., 2018, 1, 7–15 CrossRef PubMed
- C. Hureau, Y. Coppel, P. Dorlet, P. L. Solari, S. Sayen, E. Guillon, L. Sabater and P. Faller, Deprotonation of the Asp1-Ala2 peptide bond induces modification of the dynamic copper(II) environment in the amyloid-β peptide near physiological pH, Angew. Chem., Int. Ed., 2009, 48, 9522–9525 CrossRef CAS
- D. Kim, N. H. Kim and S. H. Kim, 34 GHz pulsed ENDOR characterization of the copper coordination of an amyloid β peptide relevant to Alzheimer's disease, Angew. Chem., Int. Ed., 2013, 52, 1139–1142 CrossRef CAS
- P. Dorlet, S. Gambarelli, P. Faller and C. Hureau, Pulse EPR spectroscopy reveals the coordination sphere of copper(II) ions in the 1-16 amyloid-β peptide: A key role of the first two N-terminus residues, Angew. Chem., Int. Ed., 2009, 48, 9273–9276 CrossRef CAS
- C. Hureau, V. Balland, Y. Coppel, P. L. Solari, E. Fonda and P. Faller, Importance of dynamical processes in the coordination chemistry and redox conversion of copper amyloid-β complexes, J. Biol. Inorg. Chem., 2009, 14, 995–1000 CrossRef CAS PubMed
- N. Yako, T. R. Young, J. M. Cottam Jones, C. A. Hutton, A. G. Wedd and Z. Xiao, Copper binding and redox chemistry of the Aβ16 peptide and its variants: Insights into determinants of copper-dependent reactivity, Metallomics, 2017, 9, 278–291 CrossRef CAS
- B. Alies, A. Conte-Daban, S. Sayen, F. Collin, I. Kieffer, E. Guillon, P. Faller and C. Hureau, Zinc(II) binding site to the amyloid-β peptide: Insights from spectroscopic studies with a wide series of modified peptides, Inorg. Chem., 2016, 55, 10499–10509 CrossRef CAS
- C. Cheignon, M. Tomas, D. Bonnefont-Rousselot, P. Faller, C. Hureau and F. Collin, Oxidative stress and the amyloid beta peptide in Alzheimer's disease, Redox Biol., 2018, 14, 450–464 CrossRef CAS PubMed
- F. Bousejra-ElGarah, C. Bijani, Y. Coppel, P. Faller and C. Hureau, Iron(II) binding to amyloid-β, the Alzheimer's peptide, Inorg. Chem., 2011, 50, 9024–9030 CrossRef CAS
- D. Jiang, L. Men, J. Wang, Y. Zhang, S. Chickenyen, Y. Wang and F. Zhou, Redox reactions of copper complexes formed with different β-amyloid peptides and their neuropathological relevance, Biochemistry, 2007, 46, 9270–9282 CrossRef CAS
- F. Arrigoni, T. Prosdocimi, L. Mollica, L. De Gioia, G. Zampella and L. Bertini, Copper reduction and dioxygen activation in Cu-amyloid beta peptide complexes: Insight from molecular modelling, Metallomics, 2018, 10, 1618–1630 CrossRef CAS PubMed
- M. W. Beck, S. B. Oh, R. A. Kerr, H. J. Lee, S. H. Kim, S. Kim, M. Jang, B. T. Ruotolo, J. Y. Lee and M. H. Lim, A rationally designed small molecule for identifying an in vivo link between metal-amyloid-β complexes and the pathogenesis of Alzheimer's disease, Chem. Sci., 2015, 6, 1879–1886 RSC
- M. R. Jones, E. Mathieu, C. Dyrager, S. Faissner, Z. Vaillancourt, K. J. Korshavn, M. H. Lim, A. Ramamoorthy, V. W. Yong, S. Tsutsui, P. K. Stys and T. Storr, Multi-target-directed phenol-triazole ligands as therapeutic agents for Alzheimer's disease, Chem. Sci., 2017, 8, 5636–5643 RSC
- M. R. Jones, C. Dyrager, M. Hoarau, K. J. Korshavn, M. H. Lim, A. Ramamoorthy and T. Storr, Multifunctional quinoline-triazole derivatives as potential modulators of amyloid-β peptide aggregation, J. Inorg. Biochem., 2016, 158, 131–138 CrossRef CAS PubMed
- S. Hong, Y. K. Go, J. S. Derrick, S. Han, J. Kim, M. H. Lim and S. H. Kim, Advanced electron paramagnetic resonance studies of a ternary complex of copper, amyloid-β, and a chemical regulator, Inorg. Chem., 2018, 57, 12665–12670 CrossRef PubMed
- Z. Xie, H. Wu and J. Zhao, Multifunctional roles of zinc in Alzheimer's disease, Neurotoxicology, 2020, 80, 112–123 CrossRef
- V. Shanbhag, K. Jasmer-McDonald, S. Zhu, A. L. Martin, N. Gudekar, A. Khan, E. Ladomersky, K. Singh, G. A. Weisman and M. J. Petris, ATP7A delivers copper to the lysyl oxidase family of enzymes and promotes tumorigenesis and metastasis, Proc. Natl. Acad. Sci. U.S.A., 2019, 116, 6836–6841 CrossRef
- Z. Zheng, C. White, J. Lee, T. S. Peterson, A. I. Bush, G. Y. Sun, G. A. Weisman and M. J. Petris, Altered microglial copper homeostasis in a mouse model of Alzheimer's disease, J. Neurochem., 2010, 114, 1630–1638 CrossRef PubMed
- L. Wang, S. Liu, J. Xu, N. Watanabe, K. H. Mayo, J. Li and X. Li, Emodin inhibits aggregation of amyloid-β peptide 1–42 and improves cognitive deficits in Alzheimer's disease transgenic mice, J. Neurochem., 2021, 157, 1992–2007 CrossRef
- L. Chen, J. Min and F. Wang, Copper homeostasis and cuproptosis in health and disease, Signal Transduct. Targeted Ther., 2022, 7, 378 CrossRef
- J. Alam and L. Sharma, Potential enzymatic targets in Alzheimer's: A comprehensive review, Curr. Drug Targets, 2019, 20, 316–339 CrossRef PubMed
- P. Zipfel, C. Rochais, K. Baranger, S. Rivera and P. Dallemagne, Matrix metalloproteinases as new targets in Alzheimer's disease: Opportunities and challenges, J. Med. Chem., 2020, 63, 10705–10725 CrossRef
- L. Kelly, M. M. Sharp, I. Thomas, C. Brown, M. Schrag, L. V. Antunes, E. Solopova, J. Martinez-Gonzalez, C. Rodríguez and R. O. Carare, Targeting lysyl-oxidase (LOX) may facilitate intramural periarterial drainage for the treatment of Alzheimer's disease, Cereb. Circ.:Cognit. Behav., 2023, 5, 100171 Search PubMed
- C. Ringland, J. E. Schweig, M. Eisenbaum, D. Paris, G. Ait-Ghezala, M. Mullan, F. Crawford, L. Abdullah and C. Bachmeier, MMP9 modulation improves specific neurobehavioral deficits in a mouse model of Alzheimer's disease, BMC Neurosci., 2021, 22, 39 CrossRef PubMed
- S. Lorenzl, D. S. Albers, N. Relkin, T. Ngyuen, S. L. Hilgenberg, J. Chirichigno, M. E. Cudkowicz and M. F. Beal, Increased plasma levels of matrix metalloproteinase-9 in patients with Alzheimer's disease, Neurochem. Int., 2003, 43, 191–196 CrossRef PubMed
- J. A. Jacobsen, J. L. Major Jourden, M. T. Miller and S. M. Cohen, To bind zinc or not to bind zinc: An examination of innovative approaches to improved metalloproteinase inhibition, Biochim. Biophys. Acta, 2010, 1803, 72–94 CrossRef PubMed
- G. Dong, L.-R. Lin, L.-Y. Xu and E.-M. Li, Reaction mechanism of lysyl oxidase-like 2 (LOXL2) studied by computational methods, J. Inorg. Biochem., 2020, 211, 111204 CrossRef CAS PubMed
- J. L. Chitty, M. Yam, L. Perryman, A. L. Parker, J. N. Skhinas, Y. F. I. Setargew, E. T. Y. Mok, E. Tran, R. D. Grant, S. L. Latham, B. A. Pereira, S. C. Ritchie, K. J. Murphy, M. Trpceski, A. D. Findlay, P. Melenec, E. C. Filipe, A. Nadalini, S. Velayuthar, G. Major, K. Wyllie, M. Papanicolaou, S. Ratnaseelan, P. A. Phillips, G. Sharbeen, J. Youkhana, A. Russo, A. Blackwell, J. F. Hastings, M. C. Lucas, C. R. Chambers, D. A. Reed, J. Stoehr, C. Vennin, R. Pidsley, A. Zaratzian, A. M. Da Silva, M. Tayao, B. Charlton, D. Herrmann, M. Nobis, S. J. Clark, A. V. Biankin, A. L. Johns, D. R. Croucher, A. Nagrial, A. J. Gill, S. M. Grimmond, M. Pajic, P. Timpson, W. Jarolimek and T. R. Cox, A first-in-class pan-lysyl oxidase inhibitor impairs stromal remodeling and enhances gemcitabine response and survival in pancreatic cancer, Nat. Cancer, 2023, 4, 1326–1344 CrossRef CAS PubMed
- J. Cheung, M. J. Rudolph, F. Burshteyn, M. S. Cassidy, E. N. Gary, J. Love, M. C. Franklin and J. J. Height, Structures of human acetylcholinesterase in complex with pharmacologically important ligands, J. Med. Chem., 2012, 55, 10282–10286 CrossRef CAS PubMed
- M. Atanasova, G. Stavrakov, I. Philipova, D. Zheleva, N. Yordanov and I. Doytchinova, Galantamine derivatives with indole moiety: Docking, design, synthesis and acetylcholinesterase inhibitory activity, Bioorg. Med. Chem., 2015, 23, 5382–5389 CrossRef CAS PubMed
- C. Binda, P. Newton-Vinson, F. Hubálek, D. E. Edmondson and A. Mattevi, Structure of human monoamine oxidase B, a drug target for the treatment of neurological disorders, Nat. Struct. Biol., 2002, 9, 22–26 CrossRef CAS
- M. M. M. Wilhelmus, J. G. J. M. Bol, S. G. van Duinen and B. Drukarch, Extracellular matrix modulator lysyl oxidase colocalizes with amyloid-beta pathology in Alzheimer's disease and hereditary cerebral hemorrhage with amyloidosis–Dutch type, Exp. Gerontol., 2013, 48, 109–114 Search PubMed
- M. T. Aqeel, N. Ur-Rahman, A.-U. Khan, Z. Ashraf, S. Khan and M. Arif,
In silico approach for the development of phenolic derivatives as potential anti-angiogenic agents against lysyl oxidase-like 2 enzyme, Front. Aging Neurosci., 2022, 8, 32 Search PubMed
- N. Hewitt and A. Rauk, Mechanism of hydrogen peroxide production by copper-bound amyloid beta peptide: A theoretical study, J. Phys. Chem. B, 2009, 113, 1202–1209 CrossRef CAS
- T. Behl, D. Kaur, A. Sehgal, S. Singh, N. Sharma, G. Zengin, F. L. Andronie-Cioara, M. M. Toma, S. Bungau and A. G. Bumbu, Role of monoamine oxidase activity in Alzheimer's disease: An insight into the therapeutic potential of inhibitors, Molecules, 2021, 26, 3724 CrossRef CAS PubMed
- M. N. Perkovic, D. S. Strac, L. Tudor, M. Konjevod, G. N. Erjavec and N. Pivac, Catechol-O-methyltransferase, cognition and Alzheimer's disease, Curr. Alzheimer Res., 2018, 15, 408–419 CrossRef CAS PubMed
- J. Hugon, F. Mouton-Liger, E. Cognat, J. Dumurgier and C. Paquet, Blood-based kinase assessments in Alzheimer's disease, Front. Aging Neurosci., 2018, 10, 338 CrossRef CAS PubMed
- L. Zhang, S. Sheng and C. Qin, The role of HDAC6 in Alzheimer's disease, J. Alzheimer’s Dis., 2013, 33, 283–295 CAS
- A. Roth, A. Sander, M. S. Oswald, F. Gärtner, U. Knippschild and J. Bischof, Comprehensive characterization of CK1δ-mediated tau phosphorylation in Alzheimer's disease, Front. Mol. Biosci., 2022, 9, 872171 CrossRef CAS PubMed
- R. T. Bartus, R. L. Dean III, B. Beer and A. S. Lippa, The cholinergic hypothesis of geriatric memory dysfunction, Science, 1982, 217, 408–414 CrossRef CAS PubMed
- K. V. Dileep, K. Ihara, C. Mishima-Tsumagari, M. Kukimoto-Niino, M. Yonemochi, K. Hanada, M. Shirouzu and K. Y. J. Zhang, Crystal structure of human acetylcholinesterase in complex with tacrine: Implications for drug discovery, Int. J. Biol. Macromol., 2022, 210, 172–181 CrossRef PubMed
- G. Mushtaq, N. H. Greig, J. A. Khan and M. A. Kamal, Status of acetylcholinesterase and butyrylcholinesterase in Alzheimer's disease and type 2 diabetes mellitus, CNS Neurol. Disord.–Drug Targets, 2014, 13, 1432–1439 CrossRef PubMed
- R. M. Lane, S. G. Potkin and A. Enz, Targeting acetylcholinesterase and butyrylcholinesterase
in dementia, Int. J. Neuropsychopharmacol., 2006, 9, 101–124 CrossRef
- M. Rossi, M. Freschi, L. de Camargo Nascente, A. Salerno, S. de Melo Viana Teixeira, F. Nachon, F. Chantegreil, O. Soukup, L. Prchal, M. Malaguti, C. Bergamini, M. Bartolini, C. Angeloni, S. Hrelia, L. A. Soares Romeiro and M. L. Bolognesi, Sustainable drug discovery of multi-target-directed ligands for Alzheimer's disease, J. Med. Chem., 2021, 64, 4972–4990 CrossRef PubMed
- P. Krashia, A. Nobili and M. D'Amelio, Unifying hypothesis of dopamine neuron loss in neurodegenerative diseases: Focusing on Alzheimer's disease, Front. Mol. Neurosci., 2019, 12, 123 CrossRef PubMed
- A. Nobili, E. C. Latagliata, M. T. Viscomi, V. Cavallucci, D. Cutuli, G. Giacovazzo, P. Krashia, F. R. Rizzo, R. Marino, M. Federici, P. De Bartolo, D. Aversa, M. C. Dell'Acqua, A. Cordella, M. Sancandi, F. Keller, L. Petrosini, S. Puglisi-Allegra, N. B. Mercuri, R. Coccurello, N. Berretta and M. D'Amelio, Dopamine neuronal loss contributes to memory and reward dysfunction in a model of Alzheimer's disease, Nat. Commun., 2017, 8, 14727 CrossRef PubMed
- S. J. Colloby, S. McParland, J. T. O'Brien and J. Attems, Neuropathological correlates of dopaminergic imaging in Alzheimer's disease and Lewy body dementias, Brain, 2012, 135, 2798–2808 CrossRef
- M. B. Leko, M. N. Perković, N. Klepac, D. Š. Štrac, F. Borovećki, N. Pivac, P. R. Hof and G. Šimić, Relationships of cerebrospinal fluid Alzheimer's disease biomarkers and COMT, DBH, and MAOB single nucleotide polymorphisms, J. Alzheimer’s Dis., 2020, 73, 135–145 Search PubMed
- C. Caccia, R. Maj, M. Calabresi, S. Maestroni, L. Faravelli, L. Curatolo, P. Salvati and R. G. Fariello, Safinamide: From molecular targets to a new anti-Parkinson drug, Neurology, 2006, 67, S18–S23 CrossRef PubMed
- K. Sharma, Cholinesterase inhibitors as Alzheimer's therapeutics (Review), Mol. Med. Rep., 2019, 20, 1479–1487 Search PubMed
- D. Attwell, A. M. Buchan, S. Charpak, M. Lauritzen, B. A. MacVicar and E. A. Newman, Glial and neuronal control of brain blood flow, Nature, 2010, 468, 232–243 CrossRef PubMed
- M. Simard and M. Nedergaard, The neurobiology of glia in the context of water and ion homeostasis, Neuroscience, 2004, 129, 877–896 Search PubMed
- M. Pekny, M. Pekna, A. Messing, C. Steinhäuser, J.-M. Lee, V. Parpura, E. M. Hol, M. V. Sofroniew and A. Verkhratsky, Astrocytes: A central element in neurological diseases, Acta Neuropathol., 2016, 131, 323–345 CrossRef CAS
- N. Rouach, A. Koulakoff, V. Abudara, K. Willecke and C. Giaume, Astroglial metabolic networks sustain hippocampal synaptic transmission, Science, 2008, 322, 1551–1555 CrossRef CAS
- C. Eroglu and B. A. Barres, Regulation of synaptic connectivity by glia, Nature, 2010, 468, 223–231 CrossRef CAS
- A. Nimmerjahn, F. Kirchhoff and F. Helmchen, Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo, Science, 2005, 308, 1314–1318 CrossRef PubMed
- D. Davalos, J. Grutzendler, G. Yang, J. V. Kim, Y. Zuo, S. Jung, D. R. Littman, M. L. Dustin and W.-B. Gan, ATP mediates rapid microglial response to local brain injury in vivo, Nat. Neurosci., 2005, 8, 752–758 CrossRef
- D. Tejera and M. T Heneka, Microglia in Alzheimer's disease: The good, the bad and the ugly, Curr. Alzheimer Res., 2016, 13, 370–380 CrossRef PubMed
- T. Wyss-Coray and J. Rogers, Inflammation in Alzheimer disease–a brief review of the basic science and clinical literature, Cold Spring Harb. Perspect. Med., 2012, 30, a006346 Search PubMed
- K. Biber, H. Neumann, K. Inoue and H. W. G. M. Boddeke, Neuronal ‘On’ and ‘Off’ signals control microglia, Trends Neurosci., 2007, 30, 596–602 CrossRef PubMed
- A. Halle, V. Hornung, G. C. Petzold, C. R. Stewart, B. G. Monks, T. Reinheckel, K. A. Fitzgerald, E. Latz, K. J. Moore and D. T. Golenbock, The NALP3 inflammasome is involved in the innate immune response to amyloid-β, Nat. Immunol., 2008, 9, 857–865 CrossRef
- M. T. Heneka, M. P. Kummer, A. Stutz, A. Delekate, S. Schwartz, A. Vieira-Saecker, A. Griep, D. Axt, A. Remus, T.-C. Tzeng, E. Gelpi, A. Halle, M. Korte, E. Latz and D. T. Golenbock, NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice, Nature, 2013, 493, 674–678 CrossRef
- H. Lian, L. Yang, A. Cole, L. Sun, A. C.-A. Chiang, S. W. Fowler, D. J. Shim, J. Rodriguez-Rivera, G. Taglialatela, J. L. Jankowsky, H.-C. Lu and H. Zheng, NFκB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer's disease, Neuron, 2015, 85, 101–115 CrossRef PubMed
- H. Lian, A. Litvinchuk, A. C.-A. Chiang, N. Aithmitti, J. L. Jankowsky and H. Zheng, Astrocyte-microglia cross talk through complement activation modulates amyloid pathology in mouse models of Alzheimer's disease, J. Neurosci., 2016, 36, 577–589 CrossRef PubMed
- A.-L. Hemonnot, J. Hua, L. Ulmann and H. Hirbec, Microglia in Alzheimer disease: Well-known targets and new opportunities, Front. Aging Neurosci., 2019, 11, 233 CrossRef
- E. A. Winkler, Y. Nishida, A. P. Sagare, S. V. Rege, R. D. Bell, D. Perlmutter, J. D. Sengillo, S. Hillman, P. Kong, A. R. Nelson, J. S. Sullvian, Z. Zhao, H. J. Meiselman, R. B. Wenby, J. Soto, E. D. Abel, J. Makshanoff, E. Zuniga, D. C. De Vivo and B. V. Zlokovic, GLUT1 reductions exacerbate Alzheimer's disease vasculo-neuronal dysfunction and degeneration, Nat. Neurosci., 2015, 18, 521–530 CrossRef PubMed
- K. Kisler, A. R. Nelson, A. Montagne and B. V. Zlokovic, Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease, Nat. Rev. Neurosci., 2017, 18, 419–434 CrossRef PubMed
- M. Jian, J. S.-C. Kwan, M. Bunting, R. C.-L. Ng and K. H. Chan, Adiponectin suppresses amyloid-β oligomer (AβO)-induced inflammatory response of microglia via AdipoR1-AMPK-NF-κB signaling pathway, J. Neuroinflammation, 2019, 16, 110 CrossRef
- P. S. Minhas, A. Latif-Hernandez, M. R. McReynolds, A. S. Durairaj, Q. Wang, A. Rubin, A. U. Joshi, J. Q. He, E. Gauba, L. Liu, C. Wang, M. Linde, Y. Sugiura, P. K. Moon, R. Majeti, M. Suematsu, D. Mochly-Rosen, I. L. Weissman, F. M. Longo, J. D. Rabinowitz and K. I. Andreasson, Restoring metabolism of myeloid cells reverses cognitive decline in ageing, Nature, 2021, 590, 122–128 CrossRef PubMed
- R. Ullah, M. Ikram, T. J. Park, R. Ahmad, K. Saeed, S. I. Alam, I. U. Rehman, A. Khan, I. Khan, M. G. Jo and M. O. Kim, Vanillic acid, a bioactive phenolic compound, counteracts LPS-induced neurotoxicity by regulating c-Jun N-terminal kinase in mouse brain, Int. J. Mol. Sci., 2020, 22, 361 CrossRef PubMed
- Y. N. Paudel, E. Angelopoulou, C. Piperi, I. Othman, K. Aamir and M. F. Shaikh, Impact of HMGB1, RAGE, and TLR4 in Alzheimer's disease (AD): From risk factors to therapeutic targeting, Cells, 2020, 9, 383 CrossRef PubMed
- Y. Xiao, Y. Dai, L. Li, F. Geng, Y. Xu, J. Wang, S. Wang and J. Zhao, Tetrahydrocurcumin ameliorates Alzheimer's pathological phenotypes by inhibition of microglial cell cycle arrest and apoptosis via Ras/ERK signaling, Biomed. Pharmacother., 2021, 139, 111651 CrossRef
- H. Mollazadeh, A. F. G. Cicero, C. N. Blesso, M. Pirro, M. Majeed and A. Sahebkar, Immune modulation by curcumin: The role of interleukin-10, Crit. Rev. Food Sci. Nutr., 2019, 59, 89–101 CrossRef CAS PubMed
- K.-A. Chuang, M.-H. Li, N.-H. Lin, C.-H. Chang, I.-H. Lu, I.-H. Pan, T. Takahashi, M.-D. Perng and S.-F. Wen, Rhinacanthin C alleviates amyloid-β fibrils' toxicity on neurons and attenuates neuroinflammation triggered by LPS, amyloid-β, and interferon-γ in glial cells, Oxid. Med. Cell. Longev., 2017, 2017, 5414297 CrossRef
- M. Xu, X. Zhang, F. Ren, T. Yan, B. Wu, K. Bi, W. Bi and Y. Jia, Essential oil of Schisandra chinensis ameliorates cognitive decline in mice by alleviating inflammation, Food Funct., 2019, 10, 5827–5842 RSC
- M. Aminzadeh, M. Roghani, A. Sarfallah and G. H. Riazi, TRPM2 dependence of ROS-induced NLRP3 activation in Alzheimer's disease, Int. Immunopharmacol., 2018, 54, 78–85 CrossRef
- F. La Rosa, M. Saresella, I. Marventano, F. Piancone, E. Ripamonti, N. Al-Daghri, C. Bazzini, C. P. Zoia, E. Conti, C. Ferrarese and M. Clerici, Stavudine reduces NLRP3 inflammasome activation and modulates amyloid-β autophagy, J. Alzheimer's Dis., 2019, 72, 401–412 Search PubMed
- J. Yin, F. Zhao, J. E. Chojnacki, J. Fulp, W. L. Klein, S. Zhang and X. Zhu, NLRP3 inflammasome inhibitor ameliorates amyloid pathology in a mouse model of Alzheimer's disease, Mol. Neurobiol., 2018, 55, 1977–1987 CrossRef
- N. Lonnemann, S. Hosseini, C. Marchetti, D. B. Skouras, D. Stefanoni, A. D'Alessandro, C. A. Dinarello and M. Korte, The NLRP3 inflammasome inhibitor OLT1177 rescues cognitive impairment in a mouse model of Alzheimer's disease, Proc. Natl. Acad. Sci. U.S.A., 2020, 117, 32145–32154 CrossRef
- Z. Lv and Y. Guo, Metformin and its benefits for various diseases, Front. Endocrinol., 2020, 11, 191 CrossRef
- X.-Y. Lu, S. Huang, Q.-B. Chen, D. Zhang, W. Li, R. Ao, F. C.-Y. Leung, Z. Zhang, J. H. Y. Tang and S.-J. Zhang, Metformin ameliorates Aβ pathology by insulin-degrading enzyme in a transgenic mouse model of Alzheimer's disease, Oxid. Med. Cell. Longev., 2020, 2020, 2315106 Search PubMed
- I. Y. Tamboli, E. Barth, L. Christian, M. Siepmann, S. Kumar, S. Singh, K. Tolksdorf, M. T. Heneka, D. L. P. Wunderlich and J. Walter, Statins promote the degradation of extracellular amyloid β-peptide by microglia via stimulation of exosome-associated insulin-degrading enzyme (IDE) secretion, J. Biol. Chem., 2010, 285, 37405–37414 CrossRef PubMed
- S. Wang, M. Mustafa, C. M. Yuede, S. V. Salazar, P. Kong, H. Long, M. Ward, O. Siddiqui, R. Paul, S. Gilfillan, A. Ibrahim, H. Rhinn, I. Tassi, A. Rosenthal, T. Schwabem and M. Colonna, Anti-human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer's disease model, J. Exp. Med., 2020, 217, e20200785 CrossRef PubMed
- R. Luo, L.-Y. Su, G. Li, J. Yang, Q. Liu, L.-X. Yang, D.-F. Zhang, H. Zhou, M. Xu and Y. Fan, Activation of PPARA-mediated autophagy reduces Alzheimer disease-like pathology and cognitive decline in a murine model, Autophagy, 2020, 16, 52–69 CrossRef PubMed
- J. Ji, T.-F. Xue, X.-D. Guo, J. Yang, R.-B. Guo, J. Wang, J.-Y. Huang, X.-J. Zhao and X.-L. Sun, Antagonizing peroxisome proliferator-activated receptor γ facilitates M1-to-M2 shift of microglia by enhancing autophagy via the LKB1–AMPK signaling pathway, Aging Cell, 2018, 17, e12774 CrossRef PubMed
- N. Wang, H. Wang, L. Li, Y. Li and R. Zhang, β-Asarone inhibits amyloid-β by promoting autophagy in a cell model of Alzheimer's disease, Front. Pharmacol, 2020, 10, 1529 CrossRef PubMed
- Y.-S. Lee, D. P. Gupta, S. H. Park, H.-J. Yang and G. J. Song, Anti-inflammatory effects of dimethyl fumarate in microglia via an autophagy dependent pathway, Front. Pharmacol, 2021, 12, 612981 CrossRef PubMed
- D. A. Butterfield and B. Halliwell, Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease, Nat. Rev. Neurosci., 2019, 20, 148–160 CrossRef CAS
- W. S. Liang, E. M. Reiman, J. Valla, T. Dunckley, T. G. Beach, A. Grover, T. L. Niedzielko, L. E. Schneider, D. Mastroeni, R. Caselli, W. Kukull, J. C. Morris, C. M. Hulette, D. Schmechel, J. Rogers and D. A. Stephan, Alzheimer's disease is associated with reduced expression of energy metabolism genes in posterior cingulate neurons, Proc. Natl. Acad. Sci. U.S.A., 2008, 105, 4441–4446 CrossRef CAS
- W. M. Brooks, P. J. Lynch, C. C. Ingle, A. Hatton, P. C. Emson, R. L. M. Faull and M. P. Starkey, Gene expression profiles of metabolic enzyme transcripts in Alzheimer's disease, Brain Res., 2007, 1127, 127–135 CrossRef CAS PubMed
- M. H. Yan, X. Wang and X. Zhu, Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease, Free Radic. Biol. Med., 2013, 62, 90–101 CrossRef CAS PubMed
- V. Boczonadi, G. Ricci and R. Horvath, Mitochondrial DNA transcription and translation: Clinical syndromes, Essays Biochem., 2018, 62, 321–340 CrossRef
- T. N. Nguyen, B. S. Padman and M. Lazarou, Deciphering the molecular signals of PINK1/Parkin mitophagy, Trends Cell Biol., 2016, 26, 733–744 CrossRef CAS PubMed
- F. Fischer, A. Hamann and H. D. Osiewacz, Mitochondrial quality control: An integrated network of pathways, Trends Biochem. Sci., 2012, 37, 284–292 CrossRef CAS
- P. I. Moreira, S. L. Siedlak, X. Wang, M. S. Santos, C. R. Oliveira, M. Tabaton, A. Nunomura, L. I. Szweda, G. Aliev, M. A. Smith, X. Zhu and G. Perry, Autophagocytosis of mitochondria is prominent in Alzheimer disease, J. Neuropathol, Exp. Neurol., 2007, 66, 525–532 CAS
- R. A. Nixon and D.-S. Yang, Autophagy failure in Alzheimer's disease—locating the primary defect, Neurobiol. Dis., 2011, 43, 38–45 CrossRef CAS
- X. Wang, B. Su, H.-G. Lee, X. Li, G. Perry, M. A. Smith and X. Zhu, Impaired balance of mitochondrial fission and fusion in Alzheimer's disease, J. Neurosci., 2009, 29, 9090–9103 CrossRef CAS PubMed
- M. Manczak, M. J. Calkins and P. H. Reddy, Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer's disease: Implications for neuronal damage, Hum. Mol. Genet., 2011, 20, 2495–2509 CrossRef CAS PubMed
- D.-H. Cho, T. Nakamura, J. Fang, P. Cieplak, A. Godzik, Z. Gu and S. A. Lipton, S-nitrosylation of Drp1 mediates β-amyloid—related mitochondrial fission and neuronal injury, Science, 2009, 324, 102–105 CrossRef CAS
- M. Manczak and P. H. Reddy, Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer's disease neurons: Implications for mitochondrial dysfunction and neuronal damage, Hum. Mol. Genet., 2012, 21, 2538–2547 CrossRef CAS
- W. Qin, V. Haroutunian, P. Katsel, C. P. Cardozo, L. Ho, J. D. Buxbaum and G. M. Pasinetti, PGC-1α expression decreases in the Alzheimer disease brain as a function of dementia, Arch. Neurol., 2009, 66, 352–361 CrossRef PubMed
- A. C. Rice, P. M. Keeney, N. K. Algarzae, A. C. Ladd, R. R. Thomas and J. P. Bennett Jr, Mitochondrial DNA copy numbers in pyramidal neurons are decreased and mitochondrial biogenesis transcriptome signaling is disrupted in Alzheimer's disease hippocampi, J. Alzheimer's Dis., 2014, 40, 319–330 CAS
- M. Sajan, B. Hansen, R. Ivey III, J. Sajan, C. Ari, S. Song, U. Braun, M. Leitges, M. Farese-Higgs and R. V. Farese, Brain insulin signaling is increased in insulin-resistant states and decreases in FOXOs and PGC-1α and increases in Aβ1–40/42 and phospho-tau may abet Alzheimer development, Diabetes, 2016, 65, 1892–1903 CrossRef CAS
- U. Cagin, O. F. Duncan, A. P. Gatt, M. S. Dionne, S. T. Sweeney and J. M. Bateman, Mitochondrial retrograde signaling regulates neuronal function, Proc. Natl. Acad. Sci. U.S.A., 2015, 112, E6000–E6009 CrossRef CAS
- M. Ying, X. Sui, Y. Zhang, Q. Sun, Z. Qu, X. Luo, R. C.-C. Chang, J. Ni, J. Liu and X. Yang, Identification of novel key molecules involved in spatial memory impairment in triple transgenic mice of Alzheimer's disease, Mol. Neurobiol., 2017, 54, 3843–3858 CrossRef PubMed
- E. Giraldo, A. Lloret, T. Fuchsberger and J. Viña, Aβ and tau toxicities in Alzheimer's are linked via oxidative stress-induced p38 activation: Protective role of vitamin E, Redox Biol., 2014, 2, 873–877 CrossRef PubMed
- S.-Y. Kook, K.-M. Lee, Y. Kim, M.-Y. Cha, S. Kang, S.-H. Baik, H. Lee, R. Park and I. Mook-Jung, High-dose of vitamin C supplementation reduces amyloid plaque burden and ameliorates pathological changes in the brain of 5XFAD mice, Cell Death Dis., 2014, 5, e1083 CrossRef PubMed
- V. Frazzini, S. Guarnieri, M. Bomba, R. Navarra, C. Morabito, M. Mariggiò and S.-L. Sensi, Altered Kv2.1 functioning promotes increased excitability in hippocampal neurons of an Alzheimer's disease mouse model, Cell Death Dis., 2016, 7, e2100 CrossRef
- W. Zhang, G.-J. Gu, X. Shen, Q. Zhang, G.-M. Wang and P.-J. Wang, Neural stem cell transplantation enhances mitochondrial biogenesis in a transgenic mouse
model of Alzheimer's disease–like pathology, Neurobiol. Aging, 2015, 36, 1282–1292 CrossRef PubMed
- X. Gan, S. Huang, L. Wu, Y. Wang, G. Hu, G. Li, H. Zhang, H. Yu, R. H. Swerdlow, J. X. Chen and S. S. Yan, Inhibition of ERK-DLP1 signaling and mitochondrial division alleviates mitochondrial dysfunction in Alzheimer's disease cybrid cell, Biochim. Biophys. Acta, Mol. Basis Dis., 2014, 1842, 220–231 CrossRef PubMed
- X. Qi, N. Qvit, Y.-C. Su and D. Mochly-Rosen, A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity, J. Cell Sci., 2013, 126, 789–802 Search PubMed
- A. U. Joshi, A. E. Ebert, B. Haileselassie and D. Mochly-Rosen, Drp1/Fis1-mediated mitochondrial fragmentation leads to lysosomal dysfunction in cardiac models of Huntington's disease, J. Mol. Cell. Cardiol., 2019, 127, 125–133 CrossRef PubMed
- E. F. Fang, Y. Hou, K. Palikaras, B. A. Adriaanse, J. S. Kerr, B. Yang, S. Lautrup, M. M. Hasan-Olive, D. Caponio, X. Dan, P. Rocktäschel, D. L. Croteau, M. Akbari, N. H. Greig, T. Fladby, H. Nilsen, M. Z. Cader, M. P. Mattson, N. Tavernarakis and V. A. Bohr, Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer's disease, Nat. Neurosci., 2019, 22, 401–412 CrossRef PubMed
- T. Jiang, F. Yin, J. Yao, R. D. Brinton and E. Cadenas, Lipoic acid restores age-associated impairment of brain energy metabolism through the modulation of Akt/JNK signaling and PGC1α transcriptional pathway, Aging Cell, 2013, 12, 1021–1031 CrossRef
- Z. Liu, I. Patil, H. Sancheti, F. Yin and E. Cadenas, Effects of lipoic acid on high-fat diet-induced alteration of synaptic plasticity and brain glucose metabolism: A PET/CT and 13C-NMR study, Sci. Rep., 2017, 7, 5391 CrossRef PubMed
- E. Martin, R. E. Rosenthal and G. Fiskum, Pyruvate dehydrogenase complex: Metabolic link to ischemic brain injury and target of oxidative stress, J. Neurosci. Res., 2005, 79, 240–247 CrossRef PubMed
- M. J. McManus, M. P. Murphy and J. L. Franklin, The mitochondria-targeted antioxidant MitoQ prevents loss of spatial memory retention and early neuropathology in a transgenic mouse model of Alzheimer's disease, J. Neurosci., 2011, 31, 15703–15715 CrossRef PubMed
- N. A. Stefanova, N. A. Muraleva, V. P. Skulachev and N. G. Kolosova, Alzheimer's disease-like pathology in senescence-accelerated OXYS rats can be partially retarded with mitochondria-targeted antioxidant SkQ1, J. Alzheimer's Dis., 2014, 38, 681–694 Search PubMed
- J. Yao, L. Zhao, Z. Mao, S. Chen, K. C. Wong, J. To and R. D. Brinton, Potentiation of brain mitochondrial function by S-equol and R/S-equol estrogen receptor β-selective phytoSERM treatments, Brain Res., 2013, 1514, 128–141 CrossRef
- D. Averill-Bates, M. Grondin and F. Ouellet, Activation of apoptosis signaling pathways by reactive oxygen species, Cryobiology, 2018, 80, 170 CrossRef
- D. P. Del Re, D. Amgalan, A. Linkermann, Q. Liu and R. N. Kitsis, Fundamental mechanisms of regulated cell death and implications for heart disease, Physiol. Rev., 2019, 99, 1765–1817 CrossRef PubMed
- D. Tang, R. Kang, T. V. Berghe, P. Vandenabeele and G. Kroemer, The molecular machinery of regulated cell death, Cell Res., 2019, 29, 347–364 CrossRef
- R. Jan and G.-S. Chaudhry, Understanding apoptosis and apoptotic pathways targeted cancer therapeutics, Adv. Pharm. Bull., 2019, 9, 205–218 CrossRef PubMed
- Z. Du, E. Nam, Y. Lin, M. Hong, T. Molnár, I. Kondo, K. Ishimori, M.-H. Baik, Y.-H. Lee and M. H. Lim, Unveiling the impact of oxidation-driven endogenous protein interactions on the dynamics of amyloid-β aggregation and toxicity, Chem. Sci., 2023, 14, 5340–5349 RSC
- R. Sugiura, R. Satoh and T. Takasaki, ERK: A double-edged sword in cancer. ERK-dependent apoptosis as a potential therapeutic strategy for cancer, Cells, 2021, 10, 2509 CrossRef
- K. Maiese, Z. Z. Chong, S. Wang and Y. C. Shang, Oxidant stress and signal transduction in the nervous system with the PI 3-K, Akt, and mTOR cascade, Int. J. Mol. Sci., 2012, 13, 13830–13866 CrossRef PubMed
- N. S. Erekat, Apoptosis and its therapeutic implications in neurodegenerative diseases, Clin. Anat., 2022, 35, 65–78 CrossRef
- C.-K. Wu, L. Thal, D. Pizzo, L. Hansen, E. Masliah and C. Geula, Apoptotic signals within the basal forebrain cholinergic neurons in Alzheimer's disease, Exp. Neurol., 2005, 195, 484–496 CrossRef CAS
- Y. Kiraz, A. Adan, M. K. Yandim and Y. Baran, Major apoptotic mechanisms and genes involved in apoptosis, Tumor Biol., 2016, 37, 8471–8486 CrossRef
- D. R. McIlwain, T. Berger and T. W. Mak, Caspase functions in cell death and disease, Cold Spring Harb. Perspect. Biol., 2015, 5, a008656 Search PubMed
- T.-J. Fan, L.-H. Han, R.-S. Cong and J. Liang, Caspase family proteases and apoptosis, Acta Biochim. Biophys. Sin., 2005, 37, 719–727 CrossRef PubMed
- Z. Cai, B. Zhao and A. Ratka, Oxidative stress and β-amyloid protein in Alzheimer's disease, NeuroMolecular Med., 2011, 13, 223–250 CrossRef PubMed
- R. A. Nixon, The role of autophagy in neurodegenerative disease, Nat. Med., 2013, 19, 983–997 CrossRef
- R. A. Nixon, J. Wegiel, A. Kumar, W. H. Yu, C. Peterhoff, A. Cataldo and A. M. Cuervo, Extensive involvement of autophagy in Alzheimer disease: An immuno-electron microscopy study, J. Neuropathol. Exp. Neurol., 2005, 64, 113–122 CrossRef
- A. Abbott, Is “friendly fire” in the brain provoking Alzheimer's disease?, Nature, 2018, 556, 426–428 CrossRef
- Z. Yang, R. Feng and H. Zhao, Cuproptosis and Cu: A new paradigm in cellular death and their role in non-cancerous diseases, Apoptosis, 2024, 29, 1330–1360 CrossRef PubMed
- A. M. Daugherty, E. M. Haacke and N. Raz, Striatal iron content predicts its shrinkage and changes in verbal working memory after two years in healthy adults, J. Neurosci., 2015, 35, 6731–6743 CrossRef
- C. Ghadery, L. Pirpamer, E. Hofer, C. Langkammer, K. Petrovic, M. Loitfelder, P. Schwingenschuh, S. Seiler, M. Duering, E. Jouvent, H. Schmidt, F. Fazekas, J.-F. Mangin, J. Chabriant, M. Dichgans, S. Ropele and R. Schmidt, R2* mapping for brain iron: Associations with cognition in normal aging, Neurobiol. Aging, 2015, 36, 925–932 CrossRef PubMed
- S. Dhani, Y. Zhao and B. Zhivotovsky, A long way to go: caspase inhibitors in clinical use, Cell Death Dis., 2021, 12, 949 CrossRef
- A. Justin-Thenmozhi, M. S. Bharathi, R. Kiruthika, T. Manivasagam, A. Borah and M. M. Essa, Attenuation of aluminum chloride-induced neuroinflammation and caspase activation through the AKT/GSK-3β pathway by hesperidin in wistar rats, Neurotox. Res., 2018, 34, 463–476 CrossRef
- H. Wen, Z. Fu, Y. Wei, X. Zhang, L. Ma, L. Gu and J. Li, Antioxidant activity and neuroprotective activity of stilbenoids in rat primary cortex neurons via the PI3K/Akt signalling pathway, Molecules, 2018, 23, 2328 CrossRef PubMed
- S. Lei, S. Wu, G. Wang, B. Li, B. Liu and X. Lei, Pinoresinol diglucoside attenuates neuroinflammation, apoptosis and oxidative stress in a mice model with Alzheimer's disease, Neuroreport, 2021, 32, 259–267 CrossRef PubMed
- S.-H. Huang, S.-T. Fang and Y.-C. Chen, Molecular mechanism of vitamin K2 protection against amyloid-β-induced cytotoxicity, Biomolecules, 2021, 11, 423 CrossRef PubMed
- R. I. Moreno, V. O. Zambelli, G. Picolo, Y. Cury, A. C. Morandini, A. C. Marques and J. M. Sciani, Caspase-1 and cathepsin B inhibitors from marine invertebrates, aiming at a reduction in neuroinflammation, Mar. Drugs, 2022, 20, 614 CrossRef PubMed
- J. Flores, A. Noël, B. Foveau, J. Lynham, C. Lecrux and A. C. LeBlanc, Caspase-1 inhibition alleviates cognitive impairment and neuropathology in an Alzheimer's disease mouse model, Nat. Commun., 2018, 9, 3916 CrossRef PubMed
- L. Zhou, J. Flores, A. Noël, O. Beauchet, P. J. Sjöström and A. C. LeBlanc, Methylene blue inhibits Caspase-6 activity, and reverses Caspase-6-induced cognitive impairment and neuroinflammation in aged mice, Acta Neuropathol. Commun., 2019, 7, 1–18 CrossRef PubMed
- A. Tubeleviciute-Aydin, A. Beautrait, J. Lynham, G. Sharma, A. Gorelik, L. J. Deny, N. Soya, G. L. Lukacs, B. Nagar, A. Marinier and A. C. LeBlanc, Identification of allosteric inhibitors against active caspase-6, Sci. Rep., 2019, 9, 5504 CrossRef PubMed
- M. Bresinsky, J. M. Strasser, B. Vallaster, P. Liu, W. M. McCue, J. Fuller, A. Hubmann, G. Singh, K. M. Nelson, M. E. Cuellar, C. M. Wilmot, B. C. Finzel, K. H. Ashe, M. A. Walters and S. Pockes, Structure-based design and biological evaluation of novel caspase-2 inhibitors based on the peptide AcVDVAD-CHO and the caspase-2-mediated tau cleavage sequence YKPVD314, ACS Pharmacol. Transl. Sci., 2022, 5, 20–40 CrossRef PubMed
- J. Cummings, G. Lee, P. Nahed, M. E. Z. N. Kambar, K. Zhong, J. Fonseca and K. Taghva, Alzheimer's disease drug development pipeline: 2022, Alzheimer's Dement. Transl. Res. Clin. Interv., 2022, 8, e12295 CrossRef PubMed
- G. Plascencia-Villa and G. Perry, Preventive and therapeutic strategies in Alzheimer's disease: Focus on oxidative stress, redox metals, and ferroptosis, Antioxid. Redox Signaling, 2021, 34, 591–610 CrossRef PubMed
- D. Jeremic, L. Jiménez-Díaz and J. D. Navarro-López, Past, present and future of therapeutic strategies against amyloid-β peptides in Alzheimer's disease: A systematic review, Ageing Res. Rev., 2021, 72, 101496 CrossRef PubMed
- B. Kumar, A. Thakur, A. R. Dwivedi, R. Kimar and V. Kumar, Multi-target-directed ligands as an effective strategy for the treatment of Alzheimer's disease, Curr. Med. Chem., 2022, 29, 1757–1803 CrossRef CAS PubMed
- N. Kumar, V. Kumar, P. Anand, V. Kumar, A. Ranjan Dwivedi and V. Kumar, Advancements in the development of multi-target directed ligands for the treatment of Alzheimer's disease, Bioorg. Med. Chem., 2022, 61, 116742 CrossRef CAS PubMed
- F. I. Baptista, A. G. Henriques, A. M. S. Silva, J. Wiltfang and O. A. B. da E. e Silva, Flavonoids as therapeutic compounds targeting key proteins involved in Alzheimer's disease, ACS Chem. Neurosci., 2014, 5, 83–92 CrossRef CAS PubMed
- P. Williams, A. Sorribas and M.-J. R. Howes, Natural products as a source of Alzheimer's drug leads, Nat. Prod. Rep., 2011, 28, 48–77 RSC
- H. Hampel, J. Hardy, K. Blennow, C. Chen, G. Perry, S. H. Kim, V. L. Villemagne, P. Aisen, M. Vendruscolo, T. Iwatsubo, C. L. Masters, M. Cho, L. Lannfelt, J. L. Cummings and A. Vergallo, The amyloid-β pathway in Alzheimer's disease, Mol. Psychiatr., 2021, 26, 5481–5503 CrossRef CAS PubMed
- B. Kaur, R. Kaur, Vivesh, S. Rani, R. Bhatti and P. Singh, Small peptides targeting BACE-1, AChE, and A-β reversing scopolamine-induced memory impairment: A multitarget approach against Alzheimer's disease, ACS Omega, 2024, 9, 12896–12913 CAS
- S. Tan, L. Wu, J. Liu, Z. Wu, Q. Cheng, Q. Qu, L. Zhu, Y. Yan, H. Wu, T.-J. Ling, R.-T. Liu and S. Yang, Quercetin-3-O-glc-1-3-rham-1-6-glucoside decreases Aβ production, inhibits Aβ aggregation and neurotoxicity, and prohibits the production of inflammatory cytokines, Eur. J. Pharmacol., 2024, 970, 176491 CrossRef CAS PubMed
- S. Chakraborty, J. Rakshit, J. Bandyopadhyay and S. Basu, Multi-target inhibition ability of neohesperidin dictates its neuroprotective activity: Implication in Alzheimer's disease therapeutics, Int. J. Biol. Macromol., 2021, 176, 315–324 CrossRef CAS PubMed
- I. D. Garía Marín, R. H. Camarillo López, O. A. Martínez, I. I. Padilla-Martínez, J. Correa-Basurto and M. C. Rosales-Hernández, New compounds from heterocyclic amines scaffold with multitarget inhibitory activity on Aβ aggregation, AChE, and BACE1 in the Alzheimer disease, PLoS One, 2022, 17, e0269129 CrossRef PubMed
- P. Zaręba, K. Łątka, G. Mazur, B. Gryzło, A. Pasieka, J. Godyń, D. Panek, A. Skrzypczak-Wiercioch, G. C. Höfner, G. Latacz, M. Maj, A. Espargaró, R. Sabaté, K. Jóźwiak, K. T. Wanner, K. Sałat, B. Malawska, K. Kulig and M. Bajda, Discovery of novel multifunctional ligands targeting GABA transporters, butyrylcholinesterase, β-secretase, and amyloid β aggregation as potential treatment of Alzheimer's disease, Eur. J. Med. Chem., 2023, 261, 115832 CrossRef PubMed
- W. Sato, M. Watanabe-Takahashi, T. Murata, N. Utsunomiya-Tate, J. Motoyama, M. Anzai, S. Ishihara, N. Nishioka, H. Uchiyama, J. Togashi, S. Nishihara, K. Kawasaki, T. Saito, T. C. Saido, S. Funamoto and K. Nishikawa, A tailored tetravalent peptide displays dual functions to inhibit amyloid β production and aggregation, Commun. Biol., 2023, 6, 383 CrossRef CAS PubMed
- A. G. Atanasov, S. B. Zotchev, V. M. Dirsch and C. T. Supuran, International natural products sciences taskforce, Nat. Rev. Drug Discovery, 2021, 20, 200–216 CrossRef CAS PubMed
- S. R. Alizadeh and M. A. Ebrahimzadeh,
O-Glycoside quercetin derivatives: Biological activities, mechanisms of action, and structure-activity relationship for drug design, a review, Phytother Res., 2022, 36, 778–807 CrossRef CAS PubMed
- H. Hampel, R. Vassar, B. De Strooper, J. Hardy, M. Willem, N. Singh, J. Zhou, R. Yan, E. Vanmechelen, A. De Vos, R. Nisticò, M. Corbo, B. P. Imbimbo, J. Streffer, I. Voytyuk, M. Timmers, A. A. Tahami Monfared, M. Irizarry, B. Albala, A. Koyama, N. Watanabe, T. Kimura, L. Yarenis, S. Lista, L. Kramer and A. Vergallo, The β-secretase BACE1 in Alzheimer's disease, Biol. Psychiatr., 2021, 89, 745–756 CrossRef CAS PubMed
- V. S. Kulkarni, V. Alagarsamy, V. R. Solomon, P. A. Jose and S. Murugesan, Drug repurposing: An effective tool in modern drug discovery, Russ. J. Bioorg. Chem., 2023, 49, 157–166 CrossRef CAS PubMed
- M. Lovell, J. D. Robertson, W. J. Teesdale, J. L. Campbell and W. R. Markesbery, Copper, iron and zinc in Alzheimer's disease senile plaques, J. Neurol. Sci., 1998, 158, 47–52 CrossRef CAS
- W. Liu, X. Dong and Y. Sun, D-Enantiomeric RTHLVFFARK-NH2: A potent multifunctional decapeptide inhibiting Cu2+-mediated amyloid β-protein aggregation and remodeling Cu2+-mediated amyloid β aggregates, ACS Chem. Neurosci., 2019, 10, 1390–1401 CrossRef CAS PubMed
- X. Li, W. Wang, X. Dong and Y. Sun, Conjugation of RTHLVFFARK to human lysozyme creates a potent multifunctional modulator for Cu2+-mediated amyloid β-protein aggregation and cytotoxicity, J. Mater. Chem. B, 2020, 8, 2256–2268 RSC
- K. Singh, A. Kaur, D. Goyal and B. Goyal, Mechanistic insights into the mitigation of Aβ aggregation and protofibril destabilization by D-enantiomeric decapeptide rk10, Phys. Chem. Chem. Phys., 2022, 24, 21975–21994 RSC
- S. Mann, A. Kaur, A. Kaur, N. Priyadarshi, B. Goyal, N. K. Singhal and D. Goyal, Triazole-peptide conjugate as a modulator of Aβ-aggregation, metal-mediated Aβ-aggregation, and cytotoxicity, ACS Chem. Neurosci., 2023, 14, 1631–1645 CrossRef CAS
- R. Roy, K. Pradhan, J. Khan, G. Das, N. Mukherjee, D. Das and S. Ghosh, Human serum albumin-inspired glycopeptide-based multifunctional inhibitor of Amyloid-β toxicity, ACS Omega, 2020, 5, 18628–18641 CrossRef CAS PubMed
- L. Lei, Z. Zou, J. Liu, Z. Xu, Y. Fu, Y. Tian and W. Zhang, Multifunctional peptide-assembled micelles for simultaneously reducing amyloid-beta and reactive oxygen species, Chem. Sci., 2021, 12, 6449–6457 RSC
- S. Kalita, S. Kalita, A. H. Kawa, S. Shill, A. Gupta, S. Kumar and B. Mandal, Copper chelating cyclic peptidomimetic inhibits Aβ fibrillogenesis, RSC Med. Chem., 2022, 13, 761–774 RSC
- M. Kim, J. Kang, M. Lee, J. Han, G. Nam, E. Tak, M. S. Kim, H. J. Lee, E. Nam, J. Park, S. J. Oh, J.-Y. Lee, J.-Y. Lee, M.-H. Baik and M. H. Lim, Minimalistic principles for designing small molecules with multiple reactivities against pathological factors in dementia, J. Am. Chem. Soc., 2020, 142, 8183–8193 CrossRef PubMed
- J. Han, H. J. Lee, K. Y. Kim, G. Nam, J. Chae and M. H. Lim, Mechanistic approaches for chemically modifying the coordination sphere of copper–amyloid-β complexes, Proc. Natl. Acad. Sci. U.S.A., 2020, 117, 5160–5167 CrossRef
- S. J. L. Lauw, X. Xu and R. D. Webster, Primary-colored electrochromism of 1, 4-phenylenediamines, ChemPlusChem, 2015, 80, 1288–1297 CrossRef PubMed
- Y. Fan, J.-H. Liu, C.-P. Yang, M. Yu and P. Liu, Graphene–polyaniline composite film modified electrode for voltammetric determination of 4-aminophenol, Sensor. Actuator. B Chem., 2011, 157, 669–674 CrossRef
- R. van den Berg, G. R. M. M. Haenen, H. van den Berg and A. Bast, Applicability of an improved Trolox equivalent antioxidant capacity (TEAC) assay for evaluation of antioxidant capacity measurements of mixtures, Food Chem., 1999, 66, 511–517 CrossRef
- G. Kim, E. Lelong, J. Kang, J.-M. Suh, N. Le Bris, H. Bernard, D. Kim, R. Tripier and M. H. Lim, Reactivities of cyclam derivatives with metal–amyloid-β, Inorg. Chem. Front., 2020, 7, 4222–4238 RSC
- A. Meyer and K. Fischer, Oxidative transformation processes and products of para-phenylenediamine (PPD) and para-toluenediamine (PTD)—a review, Environ. Sci. Eur., 2015, 27, 1–16 CrossRef
- J. S. Derrick, R. A. Kerr, Y. Nam, S. B. Oh, H. J. Lee, K. G. Earnest, N. Suh, K. L. Peck, M. Ozbil and K. J. Korshavn, A redox-active, compact molecule for cross-linking amyloidogenic peptides into nontoxic, off-pathway aggregates: In vitro and in vivo efficacy and molecular mechanisms, J. Am. Chem. Soc., 2015, 137, 14785–14797 CrossRef
- G. Bitan, B. Tarus, S. S. Vollers, H. A. Lashuel, M. M. Condron, J. E. Straub and D. B. Teplow, A molecular switch in amyloid assembly: Met35 and amyloid β-protein oligomerization, J. Am. Chem. Soc., 2003, 125, 15359–15365 CrossRef PubMed
- M. W. Beck, J. S. Derrick, R. A. Kerr, S. B. Oh, W. J. Cho, S. J. C. Lee, Y. Ji, J. Han, Z. A. Tehrani, N. Suh, S. Kim, S. D. Larsen, K. S. Kim, J.-Y. Lee, B. T. Ruotolo and M. H. Lim, Structure-mechanism-based engineering of chemical regulators targeting distinct pathological factors in Alzheimer's disease, Nat. Commun., 2016, 7, 13115 CrossRef PubMed
- D. J. Hayne, S. Lim and P. S. Donnelly, Metal complexes designed to bind to amyloid-β for the diagnosis and treatment of Alzheimer's disease, Chem. Soc. Rev., 2014, 43, 6701–6715 RSC
- N. Kim and H. J. Lee, Redox-active metal ins and amyloid-degrading enzymes in Alzheimer's disease, Int. J. Mol. Sci., 2021, 22, 7697 CrossRef PubMed
- B. R. Sahoo, P. K. Panda, W. Liang, W.-J. Tang, R. Ahuja and A. Ramamoorthy, Degradation of Alzheimer's amyloid-β by a catalytically inactive insulin-degrading enzyme, J. Mol. Biol., 2021, 433, 166993 CrossRef
- T. Saido and M. A. Leissring, Proteolytic degradation of amyloid β-protein, Cold Spring Harb Perspect. Med., 2012, 2, a006379 Search PubMed
- T. Silva, J. Reis, J. Teixeira and F. Borges, Alzheimer's disease, enzyme targets and drug discovery struggles: From natural products to drug prototypes, Ageing Res. Rev., 2014, 15, 116–145 CrossRef PubMed
- Y. Cheng and H. Chen, Aberrance of zinc metalloenzymes-induced human diseases and its potential mechanisms, Nutrients, 2021, 13, 4456 CrossRef PubMed
- T. Latronico, T. Petraglia, C. Sileo, D. Bilancia, R. Rossano and G. M. Liuzzi, Inhibition of MMP-2 and MMP-9 by dietary antioxidants in THP-1 macrophages and sera from patients with breast cancer, Molecules, 2024, 29, 1718 CrossRef PubMed
- T. Latronico, R. Rossano, D. V. Miniero, E. Casalino and G. M. Liuzzi, Neuroprotective effect of resveratrol against manganese-induced oxidative stress and matrix metalloproteinase-9 in an "In Vivo" model of neurotoxicity, Int. J. Mol. Sci., 2024, 25, 2142 Search PubMed
- A. Gautieri, M. Beeg, M. Gobbi, F. Rigoldi, L. Colombo and M. Salmona, The anti-amyloidogenic action of doxycycline: A molecular dynamics study on the interaction with Aβ42, Int. J. Mol. Sci., 2019, 20, 4641 CrossRef PubMed
- G. Forloni, L. Colombo, L. Girola, F. Tagliavini and M. Salmona, Anti-amyloidogenic activity of tetracyclines: Studies in vitro, FEBS Lett., 2001, 487, 404–407 CrossRef
- B. Chandra, V. S. Mithu, D. Bhowmik, A. K. Das, B. Sahoo, S. Maiti and P. K. Madhu, Curcumin dictates divergent fates for the central salt bridges in amyloid-β40 and amyloid-β42, Biophys. J., 2017, 112, 1597–1608 CrossRef PubMed
- H. Lee, J.-W. Park, S.-P. Kim, E. H. Lo and S.-R. Lee, Doxycycline inhibits matrix metalloproteinase-9 and laminin degradation after transient global cerebral ischemia, Neurobiol. Dis., 2009, 34, 189–198 CrossRef PubMed
- A. D'Souza, A. Szabo, K. E. Flynn, B. Dhakal, S. Chhabra, M. C. Pasquini, D. Weihrauch and P. N. Hari, Adjuvant doxycycline to enhance anti-amyloid effects: Results from the dual phase 2 trial, EClinicalMedicine, 2020, 23, 100361 CrossRef PubMed
- S. Patnaik, M. Rai, S. Jalali, K. Agarwal, A. Badakere, L. Puppala, S. Vishwakarma, D. Balakrishnan, P. K. Rani, R. Kekunnaya, P. P. Chhablani, S. Chakrabarti and I. Kaur, An interplay of microglia and matrix metalloproteinase MMP9 under hypoxic stress regulates the opticin expression in retina, Sci. Rep., 2021, 11, 7444 CrossRef CAS PubMed
- L. Medina, F. Gonzalez-Lizárraga, A. Dominguez-Meijide, D. Ploper, V. Parrales, S. Sequeira, M.-S. Cima-Omori, M. Zweckstetter, E. Del Bel, P. P. Michel, T. F. Outeiro, R. Raisman-Vozari, R. Chehin and S. B. Socias, Doxycycline interferes with tau aggregation and reduces its neuronal toxicity, Front. Aging Neurosci., 2021, 13, 635760 CrossRef CAS PubMed
- F. V. Santa-Cecilia, C. A. Leite, E. Del-Bel and R. Raisman-Vozari, The neuroprotective effect of doxycycline on neurodegenerative diseases, Neurotox. Res., 2019, 35, 981–986 CrossRef CAS PubMed
- D. L. Clemens, M. J. Duryee, C. Sarmiento, A. Chiou, J. D. McGowan, C. D. Hunter, S. L. Schlichte, J. Tian, L. W. Klassen, J. R. O'Dell, G. M. Thiele, T. R. Mikuls, M. C. Zimmerman and D. R. Anderson, Novel antioxidant properties of doxycycline, Int. J. Mol. Sci., 2018, 19, 4078 CrossRef
- C. H. Mehta, S. Paliwal, M. S. Muttigi, R. N. Seetharam, A. S. B. Prasad, Y. Nayak, S. Acharya and U. Y. Nayak, Polyphenol-based targeted therapy for oral submucous fibrosis, Inflammopharmacology, 2023, 31, 2349–2368 CrossRef CAS
- S. Swarnakar, K. Ganguly, P. Kundu, A. Banerjee, P. Maity and A. V. Sharma, Curcumin regulates expression and activity of matrix metalloproteinases 9 and 2 during prevention and healing of indomethacin-induced gastric ulcer, J. Biol. Chem., 2005, 280, 9409–9415 CrossRef CAS PubMed
- T. Ak and İ. Gülçin, Antioxidant and radical scavenging properties of curcumin, Chem. Biol. Interact., 2008, 174, 27–37 CrossRef
- S. Prasad, D. DuBourdieu, A. Srivastava, P. Kumar and R. Lall, Metal–Curcumin complexes in therapeutics: An approach to enhance pharmacological efects of curcumin, Int. J. Mol. Sci., 2021, 22, 7094 CrossRef
- M. R. Picciotto, M. J. Higley and Y. S. Mineur, Acetylcholine as a neuromodulator: Cholinergic signaling shapes nervous system function and behavior, Neuron, 2012, 76, 116–129 CrossRef
- S. Park, M. Kim, Y. Lin, M. Hong, G. Nam, A. Mieczkowski, J. Kardos, Y.-H. Lee and M. H. Lim, Designing multi-target-directed flavonoids: A strategic approach to Alzheimer's disease, Chem. Sci., 2023, 14, 9293–9305 RSC
- G. Nam, Y. Ji, H. J. Lee, J. Kang, Y. Yi, M. Kim, Y. Lin, Y.-H. Lee and M. H. Lim, Orobol: An isoflavone exhibiting regulatory multifunctionality against four pathological features of Alzheimer's disease, ACS Chem. Neurosci., 2019, 10, 3386–3390 CrossRef
- X. Kou, J. Liu, Y. Chen, A. Yang and R. Shen, Emodin derivatives with multi-factor anti-AD activities: AChE inhibitor, anti-oxidant and metal chelator, J. Mol. Struct., 2021, 1239, 130459 CrossRef
- G. Nam, M. Hong, J. Lee, H. J. Lee, Y. Ji, J. Kang, M.-H. Baik and M. H. Lim, Multiple reactivities of flavonoids towards pathological elements in Alzheimer's disease: Structure-activity relationship, Chem. Sci., 2020, 11, 10243–10254 RSC
- R. Shen, W. Zhao, X. Li, J. Liu, A. Yang and X. Kou, Emodin derivatives as promising multi-aspect intervention agents for amyloid aggregation: Molecular docking/dynamics simulation, bioactivities evaluation, and cytoprotection, Mol. Divers., 2023, 2023(28), 3085–3099 Search PubMed
- E. Bendary, R. R. Francis, H. M. G. Ali, M. I. Sarwat and S. El Hady, Antioxidant and structure–activity relationships (SARs) of some phenolic and anilines compounds, Ann. Agric. Sci., 2013, 58, 173–181 CrossRef
- A. Baghiani, N. Charef, M. Djarmouni, H. A. Saadeh, L. Arrar and M. S. Mubarak, Free radical scavenging and antioxidant effects of some anthraquinone derivatives, Med. Chem., 2011, 7, 639–644 CrossRef PubMed
- D. Shrimali, M. K. Shanmugam, A. P. Kumar, J. Zhang, B. K. Tan, K. S. Ahn and G. Sethi, Targeted abrogation of diverse signal transduction cascades by emodin for the treatment of inflammatory disorders and cancer, Cancer Lett., 2013, 341, 139–149 CrossRef PubMed
- X. Di, X. Wang, X. Di and Y. Liu, Effect of piperine on the bioavailability and pharmacokinetics of emodin in rats, J. Pharm. Biomed. Anal., 2015, 115, 144–149 CrossRef PubMed
- J. Saura, J. M. Luque, A. M. Cesura, M. Da Prada, V. Chan-Palay, G. Huber, J. Löffler and J. Richards, Increased monoamine oxidase B activity in plaque-associated astrocytes of Alzheimer brains revealed by quantitative enzyme radioautography, Neuroscience, 1994, 62, 15–30 CrossRef CAS PubMed
- M. Nebbioso, A. Pascarella, C. Cavallotti and N. Pescosolido, Monoamine oxidase enzymes and oxidative stress in the rat optic nerve: Age-related changes, Int. J. Exp. Pathol., 2012, 93, 401–405 CrossRef CAS
- M. Sano, C. Ernesto, R. G. Thomas, M. S. Klauber, K. Schafer, M. Grundman, P. Woodbury, J. Growdon, C. W. Cotman, E. Pfeiffer, L. S. Schneider and L. J. Thal, A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer's disease, N. Engl. J. Med., 1997, 336, 1216–1222 CrossRef CAS
- M. Naoi and W. Maruyama, Monoamine oxidase inhibitors as neuroprotective agents in age-dependent neurodegenerative disorders, Curr. Pharm. Des., 2010, 16, 2799–2817 CrossRef CAS
- S.-S. Xie, J. Liu, C. Tang, C. Pang, Q. Li, Y. Qin, X. Nong, Z. Zhang, J. Guo, M. Cheng, W. Tang, N. Liang and N. Jiang, Design, synthesis and biological evaluation of rasagiline-clorgyline hybrids as novel dual inhibitors of monoamine oxidase-B and amyloid-β aggregation against Alzheimer's disease, Eur. J. Med. Chem., 2020, 202, 112475 CrossRef CAS PubMed
- J. Shi, Y. Zhou, K. Wang, Q. Ma, R. Wei, Q. Li, Y. Zhao, Z. Qiao, S. Liu, Y. Leng, W. Liu and Z. Sang, Design, synthesis and biological evaluation of Schiff's base derivatives as multifunctional agents for the treatment of Alzheimer's disease, Med. Chem. Res., 2021, 30, 624–634 CrossRef CAS
- C. Binda, F. Hubálek, M. Li, Y. Herzig, J. Sterling, D. E. Edmondson and A. Mattevi, Binding of rasagiline-related inhibitors to
human monoamine oxidases: A kinetic and crystallographic analysis, J. Med. Chem., 2005, 48, 8148–8154 CrossRef CAS PubMed
- D. C. Matthews, A. Ritter, R. G. Thomas, R. D. Andrews, A. S. Lukic, C. Revta, J. W. Kinney, B. Tousi, J. B. Leverenz, H. Fillit, K. Zhong, H. H. Feldman and J. Cummings, Rasagiline effects on glucose metabolism, cognition, and tau in Alzheimer's dementia, Alzheimer's Dementia, 2021, 7, e12106 CrossRef PubMed
- M. D. Mertens, S. Hinz, C. E. Müller and M. Gütschow, Alkynyl–coumarinyl ethers as MAO-B inhibitors, Bioorg. Med. Chem., 2014, 22, 1916–1928 CrossRef CAS PubMed
- M. Biancalana and S. Koide, Molecular mechanism of Thioflavin-T binding to amyloid fibrils, Biochim. Biophys. Acta, Proteins Proteomics, 2010, 1804, 1405–1412 CrossRef CAS
- N. Kumar, K. Jangid, V. Kumar, R. P. Yadav, J. Mishra, S. Upadhayay, V. Kumar, B. Devi, V. Kumar, A. R. Dwivedi, P. Kumar, S. Baranwal, J. S. Bhatti and V. Kumar, In vitro and in vivo investigations of chromone derivatives as potential multitarget-directed ligands: Cognitive amelioration utilizing a scopolamine-induced zebrafish model, ACS Chem. Neurosci., 2024, 15, 2565–2585 CrossRef CAS PubMed
- B. Devi, K. Jangid, V. Kumar, T. Arora, N. Kumar, A. R. Dwivedi, J. Parkash and V. Kumar, Phenylstyrylpyrimidine derivatives as potential multipotent therapeutics for Alzheimer's disease, RSC Med. Chem., 2024, 15, 2922 RSC
- Z. Sang, K. Wang, P. Bai, A. Wu, J. Shi, W. Liu, G. Zhu, Y. Wang, Y. Lan, Z. Chen, Y. Zhao, Z. Qiao, C. Wang and Z. Tan, Design, synthesis and biological evaluation of novel O-carbamoyl ferulamide derivatives as multi-target-directed ligands for the treatment of Alzheimer's disease, Eur. J. Med. Chem., 2020, 194, 112265 CrossRef PubMed
- A. Nakajima, Y. Ohizumi and K. Yamada, Anti-dementia activity of nobiletin, a citrus flavonoid: A review of animal studies, Clin. Psychopharmacol. Neurosci., 2014, 12, 75–82 CrossRef PubMed
- N. Kumar, K. Jangid, V. Kumar, B. Devi, T. Arora, J. Mishra, V. Kumar, A. R. Dwivedi, J. Parkash, J. S. Bhatti and V. Kumar, Mannich reaction mediated derivatization of chromones and their biological evaluations as putative multipotent ligands for the treatment of Alzheimer's disease, RSC Med. Chem., 2024, 15, 4206–4221 Search PubMed
- L. Ismaili, J. Monnin, A. Etievant, R. L. Arribas, L. Viejo, B. Refouvelet, O. Soukup, J. Janockova, V. Hepnarova, J. Korabecny, T. Kucera, D. Jun, R. Andrys, K. Musilek, A. Baguet, E. M. García-Frutos, A. De Simone, V. Andrisano, M. Bartolini, C. de los Ríos, J. Marco-Contelles and E. Haffen, (±)-BIGI-3h: Pentatarget-directed ligand combining cholinesterase, monoamine oxidase, and glycogen synthase kinase 3β inhibition with calcium channel antagonism and antiaggregating properties for Alzheimer's Disease, ACS Chem. Neurosci., 2021, 12, 1328–1342 CrossRef PubMed
- B. Mathew, G. E. Mathew, J. P. Petzer and A. Petzer, Structural exploration of synthetic chromones as selective MAO-B inhibitors: A mini review, Comb. Chem. High Throughput Screen., 2017, 20, 522–532 Search PubMed
- B. Kumar, M. Kumar, A. R. Dwivedi and V. Kumar, Synthesis, biological evaluation and molecular modeling studies of propargyl-containing 2,4,6-trisubstituted pyrimidine derivatives as potential anti-Parkinson agents, ChemMedChem, 2018, 13, 705–712 CrossRef PubMed
- B. Kumar, V. Kumar, V. Prashar, S. Saini, A. R. Dwivedi, B. Bajaj, D. Mehta, J. Parkash and V. Kumar, Dipropargyl substituted diphenylpyrimidines as dual inhibitors of monoamine oxidase and acetylcholinesterase, Eur. J. Med. Chem., 2019, 177, 221–234 CrossRef PubMed
- A. Sgarbossa, D. Giacomazza and M. Di Carlo, Ferulic acid: A hope for Alzheimer's disease therapy from plants, Nutrients, 2015, 7, 5764–5782 CrossRef PubMed
- S. F. Nabavi, K. P. Devi, D. S. Malar, A. Sureda, M. Daglia and S. M. Nabavi, Ferulic acid and Alzheimer's disease: Promises and pitfalls, Mini Rev. Med. Chem., 2015, 15, 776–788 CrossRef PubMed
-
S. M. Ezzat, M. A. Salem, N. M. El Mahdy and M. F. Ragab, Rivastigmine, in Naturally Occurring Chemicals Against Alzheimer's Disease, Elsevier, 2021, pp. 93–108 Search PubMed
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |