
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances and perspectives in synthetic applications of silylboronates as silyl radical precursors†
Zhihua
Cai‡
 ab,
Qing-Qing
Bu‡
ab,
Xi-Yu
Wang
c,
Shengchao
Yang
*ab,
Jian
Zhou
c and
Jin-Sheng
Yu
*c
ab,
Qing-Qing
Bu‡
ab,
Xi-Yu
Wang
c,
Shengchao
Yang
*ab,
Jian
Zhou
c and
Jin-Sheng
Yu
*c
aSchool of Chemistry and Chemical Engineering, State Key Laboratory Incubation Base for Green Processing of Chemical Engineering, Shihezi University, Shihezi, Xinjiang 832003, P. R. China. E-mail: shengchao.yang@shzu.edu.cn
bXinjiang Key Laboratory of Organosilicon Functional Molecules and Materials, Turpan, Xinjiang 838200, P. R. China
cState Key Laboratory of Petroleum Molecular & Process Engineering, Shanghai Engineering Research Center of Molecular Therapeutics and New Drug Development, School of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200062, P. R. China. E-mail: jsyu@chem.ecnu.edu.cn
First published on 26th December 2024
Abstract
Silylboronates, as powerful and versatile reagents, have been widely used in synthetic chemistry over the past few decades, due to their ability to incorporate silicon and boron atoms into organic molecules. With the rapid development of radical chemistry, the use of silylboronates as silyl radical precursors has recently become a research focus in organic synthesis. Significant achievements have been made in the synthetic applications of silylboronates as silyl radical sources for various C–Si and C–X bond forming transformations. This review summarizes these recent advances, discusses their advantages and limitations, and illustrates the synthetic chances still open for further research and applications in this emerging area.
 Zhihua Cai | Zhihua Cai received his BS and MS degrees in 2009 and 2012 from Shihezi University under the guidance of Prof. Lin He. He received his PhD degree from the University of Chinese Academy of Sciences in 2019 under the guidance of Prof. Gang Li. Then he joined Shihezi University, where he is now an associate professor. His research interests include organosilicon and benzyne chemistry. |
 Qing-Qing Bu | Qing-Qing Bu received his PhD degree from Goettingen University in 2018 under the guidance of Prof. Lutz Ackermann. Then she joined Shihezi University as a Young Scholar. Her research interests include the hydrosilylation reaction, the synthesis of silane coupling agents and functional silicone oil, and the application of calcium carbide. |
 Xi-Yu Wang | Xi-Yu Wang was born in Shaanxi, China. She received her BSc degree from Xi'an Medical University in 2020 and obtained her Master's degree from the Guizhou University of Traditional Chinese Medicine in 2023. Currently, she is pursuing her Doctoral degree under the guidance of Prof. Jin-Sheng Yu. Her thesis work focuses on the catalytic asymmetric construction of Si-stereogenic chiral organosilicon compounds. |
 Shengchao Yang | Shengchao Yang received his BS degree from Central South University and Monash University (2 + 2 joint-supervision) in 2013. He received his PhD degree from Monash University in 2018. Then he joined Shihezi University as an associate professor in 2019 and was promoted to full professor in 2023. His research interests include organosilicon chemistry and silicon-based functional materials. |
 Jian Zhou | Jian Zhou obtained his PhD degree in 2004 from the Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, under the guidance of Prof. Yong Tang. After spending one year working as a postdoctoral fellow with Professor Shū Kobayashi at the University of Tokyo and three years with Professor Benjamin List at the Max-Planck-Institut für Kohlenforschung, he joined the Shanghai Key Laboratory of Green Chemistry and Chemical Processes at ECNU as a Professor at the end of 2008. His research interests include the development of new chiral catalysts and asymmetric reactions for the construction of tetrasubstituted carbon stereocenters. |
 Jin-Sheng Yu | Jin-Sheng Yu received his PhD degree from East China Normal University (ECNU) in 2016 under the guidance of Prof. Jian Zhou. After two years as a JSPS postdoctoral fellow with Prof. Masakatsu Shibasaki at the Institute of Microbial Chemistry, he joined ECNU as a Zijiang Young Scholar and was later promoted to full professor. His research interests include organosilicon and organofluorine chemistry, asymmetric catalysis, and biochemical pesticides. |
1 Introduction
Organosilicon compounds, owing to their unique chemical and physical properties, have found widespread applications in the fields of synthetic chemistry, pharmaceuticals, agrochemicals, and materials science.1–3 Considerable efforts have hence been focused on synthesizing value-added organosilanes with structural diversity, and plenty of efficient silylation protocols have been accordingly developed by using various organosilicon reagents.4 Among them, the use of silylboronates,5 powerful and versatile reagents to incorporate silicon and/or boron atoms into organic molecules, has increased exponentially over the past few decades,6 since the pioneering discovery that the Si–B bond could oxidatively add to low-valent transition metals (e.g. platinum, palladium, nickel, etc.) by the groups of Ito7a,b and Tanaka.7c Several major strategies for Si–B bond activation, including oxidative addition, transmetalation, Lewis base activation, and carbenoid insertion, have been subsequently established, which enabled a large number of elegant transformations involving silylboronates,6a,d such as 1,n-additions of C–C multiple bonds,8 1,2-additions of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) X bonds,9 1,4-additions of α,β-unsaturated carbonyl and related compounds,10 allylic and propargylic substitution,11 functionalization of strained-ring compounds or carbenoids and related compounds,12 cycloadditions of multiple-bond systems,13 C–H bond silylations,14a,b cross-coupling of C(sp2)–X or C(sp3)–X bonds,14c,d and others.15
X bonds,9 1,4-additions of α,β-unsaturated carbonyl and related compounds,10 allylic and propargylic substitution,11 functionalization of strained-ring compounds or carbenoids and related compounds,12 cycloadditions of multiple-bond systems,13 C–H bond silylations,14a,b cross-coupling of C(sp2)–X or C(sp3)–X bonds,14c,d and others.15
In parallel with these impressive advances,6 the synthetic applications of silylboronates as silyl radical precursors are also gaining increasing attention and become a hot research topic in recent years, along with the renaissance of radical chemistry,16 because silyl radical species17 are valuable intermediates for accessing diverse organosilanes via regio- and chemoselective silylation in organic synthesis.
As a consequence, a variety of silyl radical generation methods from silylboronates through photochemical, electrochemical or base activation strategies and their synthetic applications in various silyl radical involved transformations have been elegantly established for constructing C–Si or C–X bonds in the past few years. As illustrated in Fig. 1, these protocols can be classified into the following four categories, according to the kind of reaction developed: (1) hydrosilylation of alkenes or alkynes; (2) silyl radical involved silylfunctionalization; (3) selective radical silylation via C–X bond cleavage; (4) silyl radical mediated cross-coupling reactions.
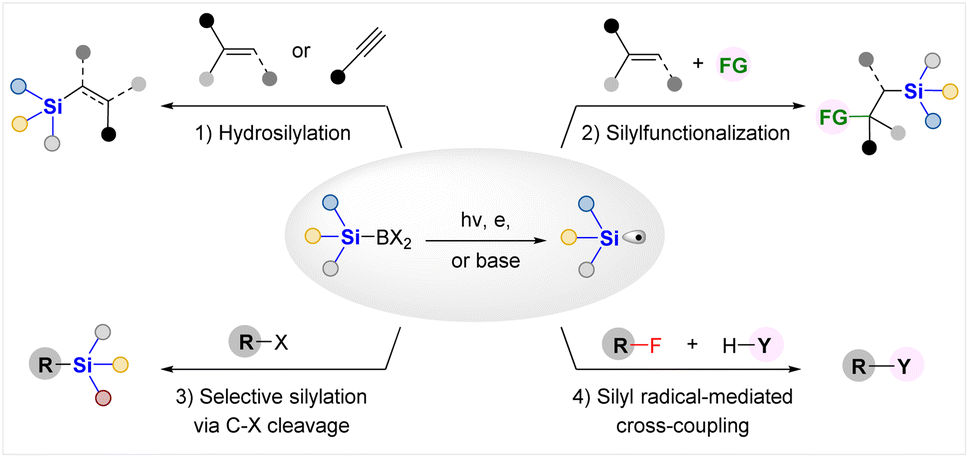 | ||
| Fig. 1 . Synthetic applications of silylboronates as silyl radical precursors. | ||
Despite great advances made in this emerging area, there lacks a timely review article to summarize these latest advances in exploring the synthetic potential of silylboronates as silyl radical precursors in organic synthesis; in sharp contrast, several elegant reviews have been published by Oestreich and coworkers to introduce the applications of silylboronates as silyl anion equivalents.6a,d In light of this, together with the flourishing of Si–B chemistry, we feel that it is necessary to present a review article that focuses on elucidating the latest technological innovations and the reactions for synthetic applications of silylboronates as silyl radical precursors, outlining the remaining synthetic opportunities, thereby facilitating the researchers with some references and inspiration to develop more efficient approaches for silyl radical generation from Si–B agents and diverse synthetic applications of the thus obtained silyl radicals. In this review, we will introduce the advances according to the classification shown in Fig. 1.
2 Hydrosilylation of alkenes or alkynes with silylboronates
Hydrosilylation of alkenes or alkynes is an important and powerful way of preparing organosilicon compounds in industry and the laboratory.18 With the rapid development of radical chemistry,16 silyl radical-mediated hydrosilylation has been extensively explored in the past decade and has been one of the hot research areas in organic synthesis.17a,b In this context, the use of silylboronates as silyl radical precursors for developing hydrosilylation has recently received increasing attention, since the seminal work by Ito, who demonstrated the generation of silyl radicals from a silylaminoboronate via homolysis under UV irradiation.19 Accordingly, a series of hydrosilylation reactions of alkenes or alkynes with silylboronates have been developed by using photochemical or electrochemical methods, allowing the access of various silylated alkanes or alkenes.Early in 2000, the Ito group pioneered the generation of silyl radicals from organosilylboronate by using a photochemical method. Under UV irradiation of a high pressure Hg lamp, a dimethylphenylsilyl radical was effectively generated from PhMe2Si–B(NiPr2)2 (1a) via homolytic photolysis of the Si–B bond, which was confirmed by trapping experiments with TEMPO (Scheme 1).19 The thus generated organosilyl radical enabled the regioselective hydrosilylation of monosubstituted alkenes 2 and delivered the desired organosilanes 3 with moderate yields. Interestingly, under UV irradiation, PhMe2Si–B(NiPr2)2 (1a) could promote the radical polymerization of vinyl acetate, methyl methacrylate and acrylate, affording the corresponding polymers containing a dimethylphenylsilyl at the polymer termini. Moreover, the radical pathway was confirmed by the experimental fact that no polymerization occurred in the presence of TEMPO or without irradiation.
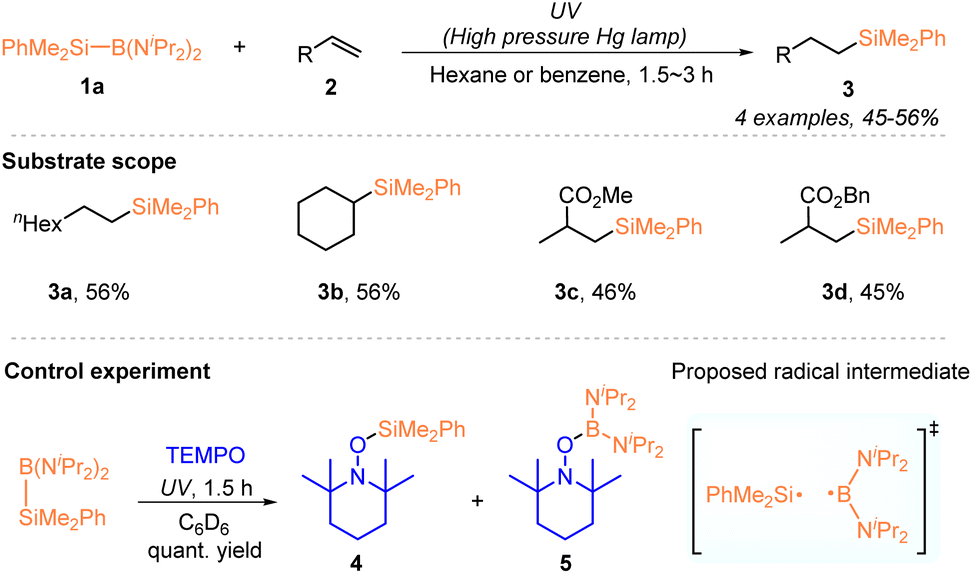 | ||
| Scheme 1 Photochemically induced silyl radical hydrosilylation of alkenes with PhMe2Si–B(NiPr2)2 (1a). | ||
In 2021, by using organosilylboronate as the radical precursor, Poisson and coworkers developed a copper-photocatalyzed hydrosilylation of alkenes under continuous flow (eqn (1), Scheme 2).20 Under blue LED irradiation, the use of 0.2 mol% of their developed [Cu(dmp)(XantphosTEPD)]PF6 effectively enabled the hydrosilylation of various monosubstituted alkenes 2 with the Suginome reagent PhMe2Si–Bpin (1b), in the presence of 2 equiv. K2CO3, which afforded the desired silylated alkanes 3 with 99% yields in all cases. Furthermore, the authors established the hydrosilylation of alkynes 6 with PhMe2Si–Bpin (1b) catalyzed by a Cu photocatalyst under blue LED irradiation and continuous flow conditions, allowing access to a variety of silylated alkenes 7 with excellent yields and good to excellent linear selectivities (eqn (2), Scheme 2). Based on the mechanistic studies, a possible reaction mechanism is proposed in Scheme 2. A borate species I was first generated from PhMe2Si–Bpin under the action of the Lewis base hydroxide, which underwent oxidation with an excited Cu photocatalyst [CuI]* to give the radical cation II. Subsequently, the Si–B bond cleavage of intermediate II delivered the dimethylphenylsilyl radical III, which then reacted with alkenes or alkynes. The thus formed β-silylated carbon-centered radical species IV delivered the desired products after undergoing H-abstraction from HO–Bpin species or chain propagation.
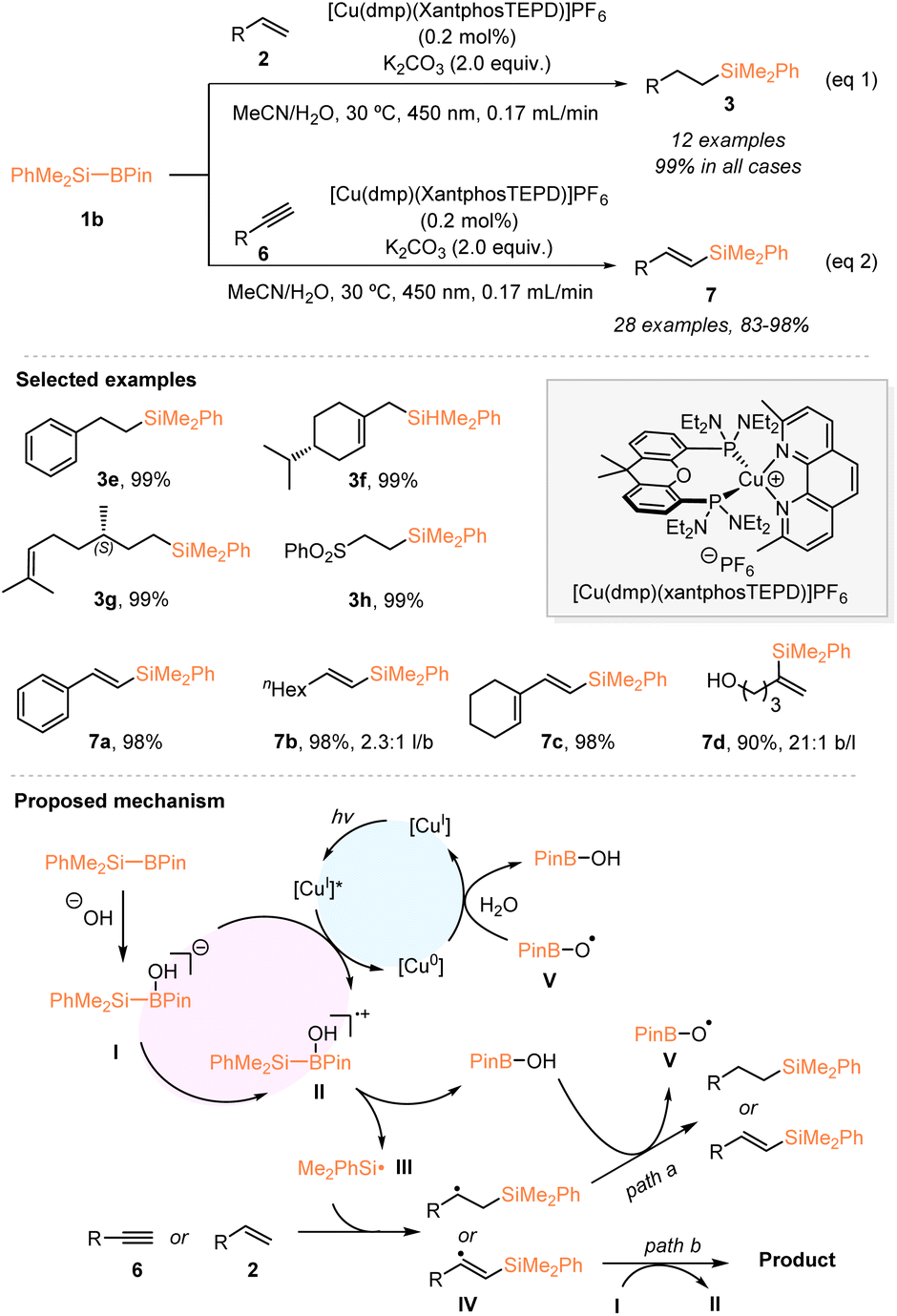 | ||
| Scheme 2 Copper-photocatalyzed hydrosilylation of alkenes or alkynes with PhMe2Si–Bpin. | ||
One year later, Ohmiya, Sumida, and coworkers described a visible-light-driven organosilyl radical generation method from silylboronates (Scheme 3).21 By using 1 mol% 4CzlPN, they realized the hydrosilylation of alkenes 2 with organosilylboronates 1 under blue LED irradiation, in the presence of 10 mol% Lewis base DMAP, affording the desired silylated products 3 in 30–93% yields. Notably, the corresponding deuterated organosilane 3n was achieved with 48% yield and 95% deuterium incorporation, when methanol-d4 was used instead of MeOH.
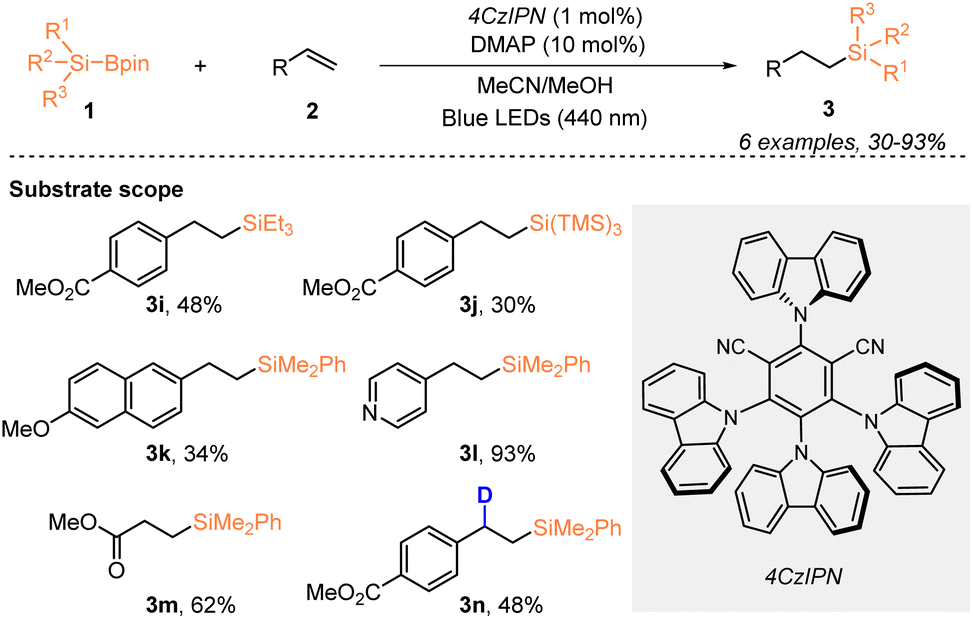 | ||
| Scheme 3 Alkene hydrosilylation with silylboronate catalyzed by 4CzlPN. | ||
In 2023, Tanaka and Nagashima reported a photocatalytic protocol for in situ generation of both secondary and tertiary organosilyl radicals from silylboronates via one-electron oxidation of ate complexes of silylboronates and alkoxide co-catalysts (Scheme 4).22 By employing such an “anionic SET strategy”, highly efficient hydrosilylation and deuterosilylation of both electron-deficient alkenes 8 and electron-rich alkenes 10 with tertiary or secondary silylboronates 1 were achieved. It was found that a series of electron-deficient alkenes 8 reacted well with silylboronates by using a combination of 1 mol% 4CzlPN and 10 mol% NaOEt, under blue LED irradiation and afforded the corresponding organosilanes 9 with 42–99% yields, whilst the use of [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 (2 mol%) and NaOEt (20 mol%) proved to be more efficient for the hydrosilylation of electron-rich alkenes 10, furnishing the desired products 11 with 25–99% yields. Notably, such a catalytic system was suitable for the generation of secondary silyl radicals, although the use of secondary silyl radicals has been problematic in silylation due to their instability. As shown in Scheme 4, the plausible reaction mechanism was proposed according to the experimental results and DFT calculations. In addition, the elaboration of the product via the functionalization of the Si–H bond further highlighted the synthetic usefulness of this method.
 | ||
| Scheme 4 Organophotocatalytic hydrosilylation of electron-rich and -deficient alkenes with silylboronates. | ||
Almost simultaneously, Zhang, Wang, and coworkers reported a visible light-induced organophotocatalytic hydrosilylation of electron-deficient alkenes 8 with organosilylboronates 1 (Scheme 5).23 In the presence of 3 mol% 4CzlPN, a variety of hydrosilylation products 9 were obtained in 30–85% yields, under the irradiation of 24 W blue LEDs (eqn (1), Scheme 5). The necessity of both a photocatalyst and visible light was demonstrated by control experiments, since no target was detected in the absence of a photocatalyst or in darkness. The mild reaction conditions, broad substrate scope, and late-stage functionalization of three plant terpenoids further highlighted its practicability. A possible reaction mechanism was proposed, in which the in situ generated redox-active complex I from silylboronate and methanol via O → B coordination underwent a SET process with the photocatalyst in a photoexcited state (PC*) to deliver the silyl radical II and reduced PC˙− species, accompanied by the formation of MeO–Bpin (III). Subsequently, the formed silyl radical II reacted with alkenes to afford the desired products after photoreduction and protonation, along with the regeneration of the photocatalyst 4CzlPN. Moreover, this strategy was successfully extended to the hydrosilylation of electron-rich alkenes 10, by slightly tuning the reaction conditions (eqn (2), Scheme 5). Notably, the authors explored the hydrosilylation of alkynes with PhMe2Si–Bpin, under the catalysis of 3 mol% 4CzlPN and blue LED irradiation, allowing the access of monosilylated alkenes via hydrosilylation or bis-silylated alkanes via bis-silylation depending on the amount of PhMe2Si–Bpin employed.
 | ||
| Scheme 5 Photochemically induced homolytic cleavage of Si–B bonds. | ||
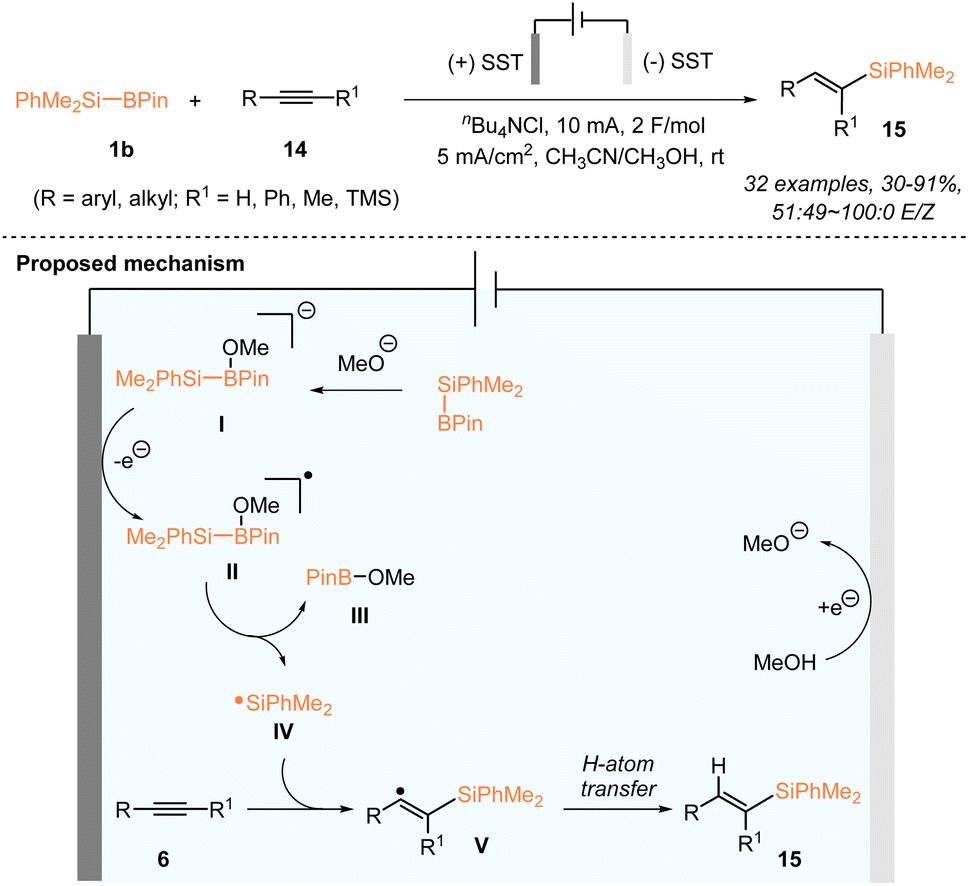
In addition to photocatalysis, the generation of silyl radicals from silylboronates proves to be feasible under electrochemical conditions. The electrochemical hydrosilylation of both alkenes and alkynes with PhMe2Si–Bpin (1b) has been accordingly established. In 2022, the Poisson group reported the electrochemical generation of silyl radicals from silylboronates for the first time and realized the electrochemical hydrosilylation of alkynes 14 with PhMe2Si–Bpin (Scheme 6).24
 | ||
| Scheme 6 Electrochemical hydrosilylation of alkynes. | ||
Various terminal or internal alkynes 14 worked smoothly, in an undivided cell using stainless steel electrodes at both the anode and the cathode, at a constant current of 5 mA and a total charge of 2 F mol−1 in a 0.1 M solution of nBu4NCl in a mixed solvent of MeCN/MeOH, delivering a series of monosilylated alkenes 15 in 30–91% yields with a 51![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :49 ∼ 100:0 E/Z ratio. The mechanistic study suggested the involvement of a dimethylphenylsilyl radical in this alkyne hydrosilylation. A plausible reaction mechanism is proposed in Scheme 6. The methoxide (MeO) anion, generated in situ from the reduction of MeOH at the cathode, first reacted with PhMe2Si–Bpin to give a borate species I, which underwent anodic oxidation to give the radical species II. The silyl radical IV was subsequently generated from II, accompanied by the production of MeO–Bpin. The addition of the silyl radical to alkyne substrates 14 produced the reactive vinyl radical V, which delivered the targets 15 after undergoing H abstraction from MeOH.
:49 ∼ 100:0 E/Z ratio. The mechanistic study suggested the involvement of a dimethylphenylsilyl radical in this alkyne hydrosilylation. A plausible reaction mechanism is proposed in Scheme 6. The methoxide (MeO) anion, generated in situ from the reduction of MeOH at the cathode, first reacted with PhMe2Si–Bpin to give a borate species I, which underwent anodic oxidation to give the radical species II. The silyl radical IV was subsequently generated from II, accompanied by the production of MeO–Bpin. The addition of the silyl radical to alkyne substrates 14 produced the reactive vinyl radical V, which delivered the targets 15 after undergoing H abstraction from MeOH.
In the same year, Poisson and coworkers developed a facile electrochemical protocol for the synthesis of gem-difluoro and γ-fluoroallyl silanes 17 and 19 through a silyl radical mediated transformation between α-trifluoromethylstyrenes 16 or α-difluoromethylstyrenes 18 and PhMe2Si–Bpin (Scheme 7).25
 | ||
| Scheme 7 Electrochemical synthesis of gem-difluoro- and γ-fluoroallyl silanes. | ||
It was found that the in situ electro-generated silyl radical reacted smoothly with a series of α-trifluoromethylstyrenes 16 to afford the desired gem-difluoroallyl silanes 17 with 36–90% yields, in an undivided cell using a stainless steel electrode at the anode and a Pt electrode at the cathode, in a constant current of 25 mA and a total charge of 3 F mol−1. By slightly varying the reaction conditions, α-difluoromethylstyrenes 18 could be effectively converted into the corresponding γ-fluoroallyl silanes 19 with 50–86% yields and a 63:37 ∼ 90:10 E/Z ratio. A similar mechanism is proposed in Scheme 7. Additionally, the excellent functional group tolerance, diversity and versatility of the thus obtained fluorinated building blocks further highlighted the synthetic value of this method, as exemplified by the preparation of α-difluoroalkyl substituted styrenes 18a and 20, difluoromethylated homoallylic alcohol 22, and β-difluoromethyl silane 23.
Later in 2023, also by using an electrochemical method, Tang, Pan, and coworkers realized a highly efficient radical hydrosilylation of electron-withdrawing alkenes 8 with PhMe2Si–Bpin 1b (Scheme 8).26 In an undivided cell using stainless steel electrodes at both the anode and the cathode, a large panel of electron-withdrawing alkenes 8 successfully reacted with reactive dimethylphenylsilyl radical IV, which were electro-generated from PhMe2Si–Bpin, enabling the formation of various hydrosilylated adducts 24 with 60–87% yields. This direct electrochemical hydrosilylation did not require exogenous oxidants and catalysts, making it a green, efficient and sustainable approach for the synthesis of organosilanes. In addition, this method was applied for the late-stage hydrosilylation of natural product acrylate derivatives such as piperitol, Boc-L-prolinol, and cholesterol, allowing the access of the corresponding silylated natural product derivatives. The mechanistic study revealed that a silyl radical was involved in this electrochemical alkene hydrosilylation. As depicted in Scheme 8, a reaction mechanism was proposed, which was similar to that of Poisson's work shown in Schemes 6 and 7.
 | ||
| Scheme 8 Electrochemical hydrosilylation of alkenes with PhMe2Si–Bpin. | ||
3 Silyl radical involved silylfunctionalization with silylboronates
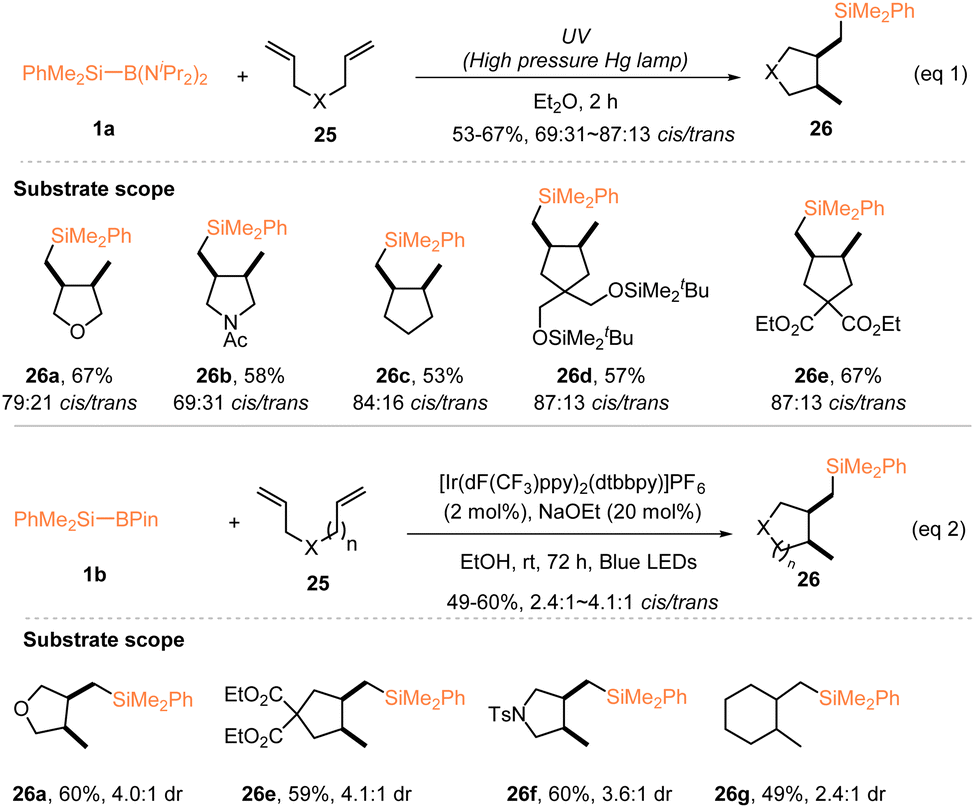
Along with the development of silyl radical hydrosilylations with silylboronates, a large number of radical silylfunctionalizations have been established using silylboronates as the radical sources, for the diverse synthesis of functionalized organosilanes. Early in 2000, Matsumoto and Ito investigated a photochemical intramolecular cyclization of 1,6-dienes 25 with PhMe2Si–B(NiPr2)2 (1a), during their study of the silyl radical-induced alkene hydrosilylation. In the presence of 1.2 equiv. PhMe2Si–B(NiPr2)2 (1a), irradiation of various 1,6-dienes 25 with a high pressure Hg lamp afforded 5-exo-cyclized products 26 exclusively in 53–67% yields with 69:31 ∼ 87:13 cis/trans selectivities (eqn (1), Scheme 9).19 It should be mentioned that different functional groups, such as ester, silyl ether, ether, and amide, were tolerated in this radical-induced silylative cyclization. 23 years later, by using a combination of 2 mol% [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 and 20 mol% NaOEt, Tanaka and Nagashima reported a similar radical silylative cyclization of oxygen-, tosylamide-, and malonate-linked 1,6-dienes or methylene-linked 1,7-diene 25 with PhMe2Si–Bpin 1b, allowing the preparation of five- or six-membered cycles featuring a PhMe2Si group with 49–60% yields and 2.4:1 ∼ 4.1:1 cis/trans selectivities, under the irradiation of blue LEDs (eqn (2), Scheme 9).22
 | ||
| Scheme 9 Photochemical silylative cyclization of dienes with silylboronates. | ||
In 2019, Suginome, Ohmura, and coworkers developed a regioselective syn-1,2-silaboration of terminal alkynes 6 with hexylene glycol-derived silylboronic ester PhMe2Si–B(hex) (1c) by using a pyridine-based organocatalyst (Scheme 10).27 Under the catalysis of 1–5 mol% 4-cyanopyridine 27a, a series of aromatic alkynes and alkyl propiolates 6 underwent regio- and stereoselective addition with PhMe2Si–B(hex), to afford (Z)-3-boryl-2-silylacrylates 28 with 18–83% yields. Based on the initial experimental studies, a plausible reaction mechanism is proposed in Scheme 10. PhMe2Si–B(hex) was first activated using 4-cyanopyridine 27avia coordination with the B atom, leading to a homolytic cleavage of the Si–B bond to deliver a radical pair II consisting of a silyl radical and boron radical stabilized by 4-cyanopyridine. Subsequently, the radical pair was added to the C–C triple bond of alkynes 6 in a syn fashion, and the formed species III would deliver the targets 28, accompanied by the regeneration of 27a. The observed regioselectivity might stem from the fact that the more nucleophilic pyridine-boryl radical preferentially attacked the more electron-deficient alkyne terminus. Interestingly, terminal alkyl allenes were also suitable substrates in this 4-cyanopyridine catalysed silaboration with PhMe2Si–B(hex) 1c, furnishing β-borylallylsilanes 30 as the major products. Additionally, the thus obtained silaboration adducts were useful synthetic intermediates in the synthesis of stereo-defined functionalized organosilanes via the elaboration of the boryl group, as exemplified by the preparation of (Z)-3-aryl-2-silylacrylate 33 through Suzuki–Miyaura coupling.
 | ||
| Scheme 10 Regioselective syn-1,2-silaboration of alkynes catalyzed by 4-cyanopyridine. | ||
In 2020, Uchiyama, Kanazawa, and coworkers reported a photochemical silaboration of [1.1.1]propellane 35 with PhMe2Si–Bpin 1b, which enabled the direct incorporation of B and Si functionalities onto the bicyclo[1.1.1]pentane (BCP) scaffold (Scheme 11).28 Under UV irradiation, the generated dimethylphenylsilyl radical I from PhMe2Si–Bpin 1b and O2 underwent the addition of [1.1.1]propellane 35 to give the BCP radical species II, which then reacted with another molecular PhMe2Si–Bpin to deliver the product 36, accompanied by the formation of silyl radical III. Control experiments, together with radical trapping experiments, demonstrated the involvement of a radical chain mechanism in this silaboration, probably initiated by oxygen. Remarkably, the synthetic utility of this methodology was highlighted by its diverse elaborations toward various BCP scaffolds featuring a silyl group. For example, a BCP analogue 38 of the bioactive compound, as a potential bioisostere of the biaryloxy scaffold,29 was effectively prepared from 36via six step reactions.
 | ||
| Scheme 11 Silaboration of [1.1.1]propellane with PhMe2Si–Bpin. | ||
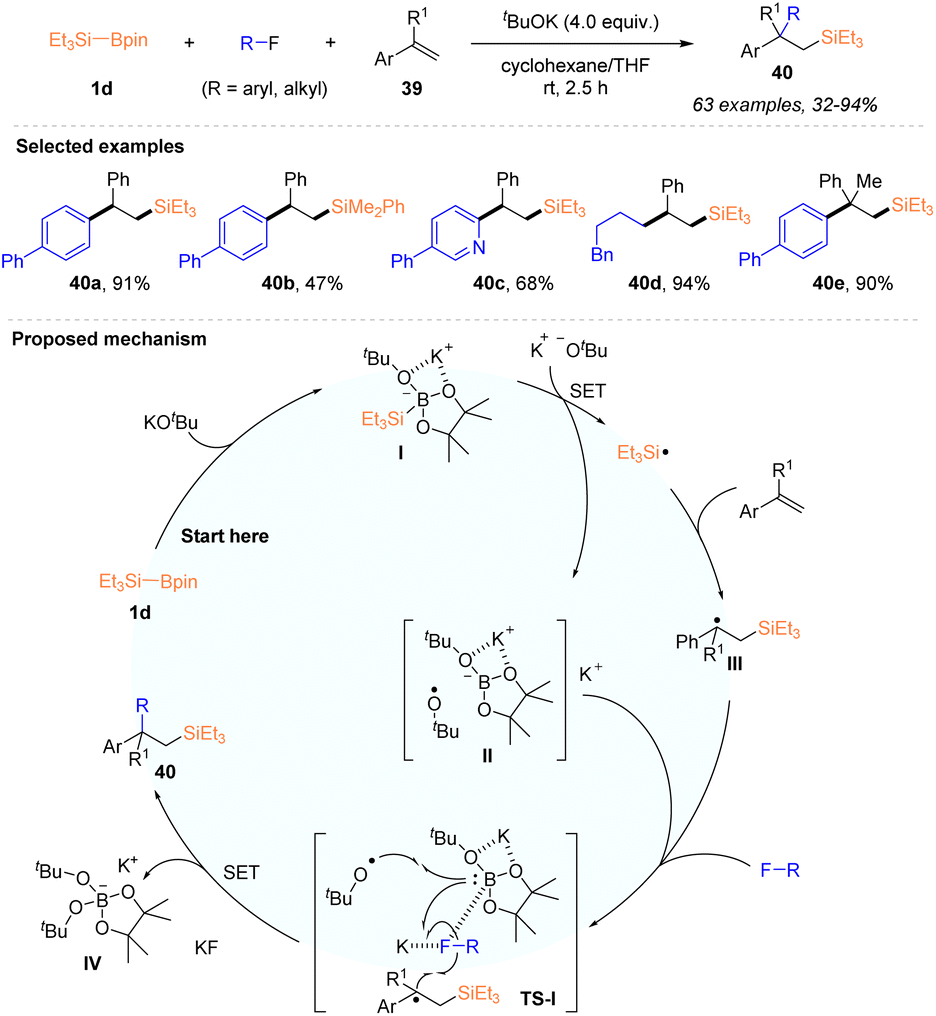
In 2021, the Shibata group disclosed a catalyst-free and tBuOK enabled regioselective carbosilylation of alkenes using silylboronate and organic fluorides, allowing the introduction of silyl and carbon groups across the C–C double bond of alkenes (Scheme 12).30 Under the action of 4 equiv. tBuOK, a variety of silylated alkanes 40 with β-tertiary or quaternary carbon centers were synthesized in 32–94% yields from styrenes or α-substituted styrenes 39, in the presence of Et3Si–Bpin 1d and aryl or alkyl fluorides. Moreover, an intramolecular carbosilylation of fluoroarenes featuring an alkenyl side chain with Et3Si–Bpin 1d was also established. The mild conditions at room temperature, broad substrate scope and excellent chemo- and site-selectivity demonstrated the practicability of this protocol. It should be mentioned that electron-deficient acrylates and acrylamides, as well as internal styrenes, were not suitable under this reaction system. As depicted in Scheme 12, the authors proposed a possible mechanism involving the silyl radical pathway, according to the initial mechanistic study. Et3Si–Bpin 1d was first activated by tBuOK, and the formed intermediate I underwent a SET process with a t-butoxy anion (tBuO−) to deliver a triethylsilyl radical (˙SiEt3) via the cleavage of the Si–B bond, accompanied by the generation of boron species II. The addition of ˙SiEt3 to styrene gave rise to a carbon-centered radical adduct III, which underwent radical reactions with fluorides, ˙OBut, and borate anions, as shown in the transition state TS-I, where the C–F bond of fluorides was activated by both K+ and boron atoms. Finally, the desired carbosilylated adducts were obtained after forming a C–C bond, with the concomitant generation of a stable borate complex [Bpin(OtBu)2]K IV and KF.
 | ||
| Scheme 12 t BuOK enabled regioselective carbosilylation of alkenes. | ||
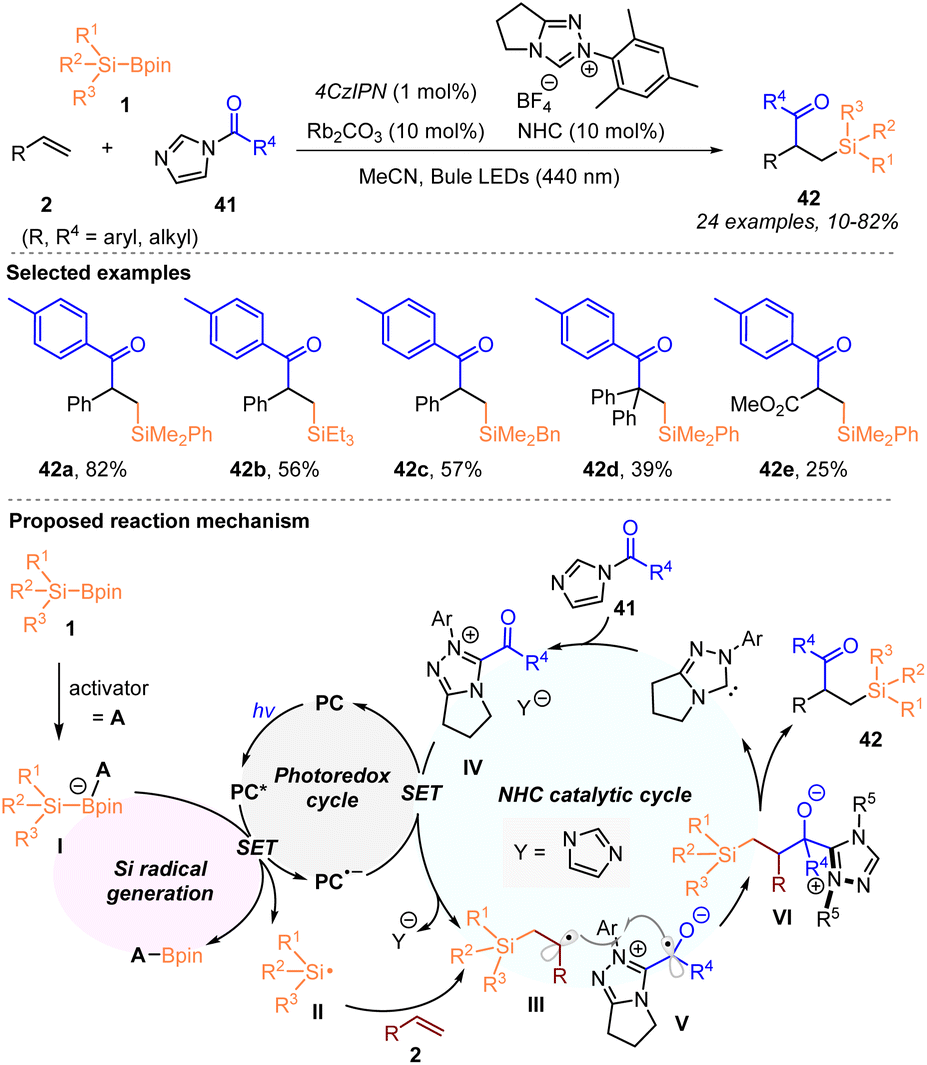
In 2022, by combining visible-light-induced silyl radical generation from silylboronates with radical NHC catalysis, Ohmiya, Sumida, and coworkers accomplished the acylsilylation of alkenes 2 with silylboronates 1 and acylimidazoles 41 (Scheme 13).21
 | ||
| Scheme 13 Radical relay three-component coupling of alkenes with silylboronates and acylimidazoles. | ||
In the presence of 4CzIPN (1 mol%), the NHC catalyst and Rb2CO3 (10 mol%, each), the radical relay three-component coupling process involving a silyl radical allows the introduction of acyl and organosilyl moieties into alkenes, affording a variety of β-acyl substituted silanes 42 with 10–82% yields, under blue LED irradiation. A possible reaction mechanism is illustrated in Scheme 13, in which the silyl radical II was first formed via SET oxidation of the activated silylboronates I with the excited photocatalyst (PC*). The radical addition of silyl radical II to alkenes 2 led to the generation of carbon-centered radical species III. The generated acyl azolium complex IV underwent a SET reduction with the reduced photocatalyst (PC˙−) in the NHC catalytic cycle, to deliver the persistent ketyl radical V. Subsequently, radical–radical coupling between III and V occurred to give the intermediate VI, which produced the targets 42 with the regeneration of the NHC catalyst. Notably, as mentioned by the authors, such acylsilylation was difficult to achieve with HAT-promoted silyl radical generation methods.
Also in 2022, Li, Wang and co-workers described a metal-free 1,2-silylpyridylation of alkenes 43 with PhMe2Si–Bpin (1b) and 4-cyanopyridines 27via pyridine-mediated B–B and B–Si bond activation (Scheme 14).31a In the presence of DABCO and B2pin2 (50 mol%, each), a wide range of alkenes 43 and substituted 4-cyanopyridines 27 were tolerated and provided various C4-silylalkylated pyridines 44 in 30–76% yields with excellent functional group compatibility.
 | ||
| Scheme 14 1,2-Silylpyridylation of aryl alkenes with PhMe2Si–Bpin. | ||
More importantly, this method could be easily applied to the late-stage modification of complex bioactive molecules. A proposed mechanism with a silyl radical addition and sequential radical–radical coupling process is demonstrated in Scheme 14, in which the addition of the silyl radical to the alkene was the rate-limiting step. The author proposed that the 4-cyanopyridine might play dual roles in this 1,2-silylpyridylation, including the homolytic cleavage of the Si–B bond to give the silyl radical and pyridine-boryl radical, and the homolytic cleavage of the B–B bond of B2pin2 to produce a persistent pyridine-boryl radical for the subsequent radical–radical coupling process. Additionally, the presence of DABCO might inhibit the competitive Si–C bonding events of the silyl radical and 4-cyanopyridinyl-boryl radical.
Later on, the same group reported a visible-light catalyzed arylsilylation of alkenes 39 with silylboronates 1 and (hetero)aryl nitriles 27, which enabled the diverse construction of valuable silicon-containing 1,1-diaryl derivatives (Scheme 15).31b Under blue LED irradiation, the arylsilylation of mono- or di-substituted alkenes 39 proceeded smoothly to deliver various silylated 1,1-diaryl compounds 45 with 30–84% yields, in the presence of 2 mol% Ir(ppy)3 and 1 equiv. Rb2CO3. Based on control experiments, the authors proposed a plausible mechanism, in which aryl nitriles underwent single-electron reduction with excited  to generate aryl nitrile radical anion I and oxidizing Ir(ppy)3+. The activated silylborane II was oxidized by Ir(ppy)3+ to afford the silyl radical III after undergoing Si–B bond cleavage, along with the regeneration of Ir(ppy)3. Subsequently, the addition of silyl radical III to alkenes led to the formation of carbon-centered radical IV, which coupled with aryl nitrile radical anion I to give the desired products 45.
to generate aryl nitrile radical anion I and oxidizing Ir(ppy)3+. The activated silylborane II was oxidized by Ir(ppy)3+ to afford the silyl radical III after undergoing Si–B bond cleavage, along with the regeneration of Ir(ppy)3. Subsequently, the addition of silyl radical III to alkenes led to the formation of carbon-centered radical IV, which coupled with aryl nitrile radical anion I to give the desired products 45.
 | ||
| Scheme 15 Visible light-catalyzed arylsilylation of alkenes. | ||
In 2024, Wang, Jia and coworkers disclosed a novel strategy to generate silyl radicals from silylboronates 1via nucleohomolytic substitution of boron with aminyl radicals and applied such a strategy to realize an efficient silyl-oximation of alkenes 2 utilizing silylboronates 1 and N-nitrosamine 46 (Scheme 16).32 Under the irradiation of 425 nm LEDs, a variety of α-oximinoesters 47 bearing a silyl group were obtained in 31–87% yields. The radical-trapping experiment with TEMPO and the radical clock experiment with vinyl cyclopropane supported the involvement of a silyl radical intermediate in this visible-light-induced catalyst-free silyl-oximation of alkenes. Accordingly, a plausible mechanism is proposed in Scheme 16, whereby photo-induced homolytic cleavage of the N–NO bond in N-nitrosamine produces an amino radical I and a persistent NO˙ radical. Subsequently, a nucleohomolytic substitution of silylboronates with amino radical I released a silyl radical III, which then reacted with an alkene to afford carbon-centered radical IV that was captured by the persistent NO˙ radical to deliver the target after tautomerization.
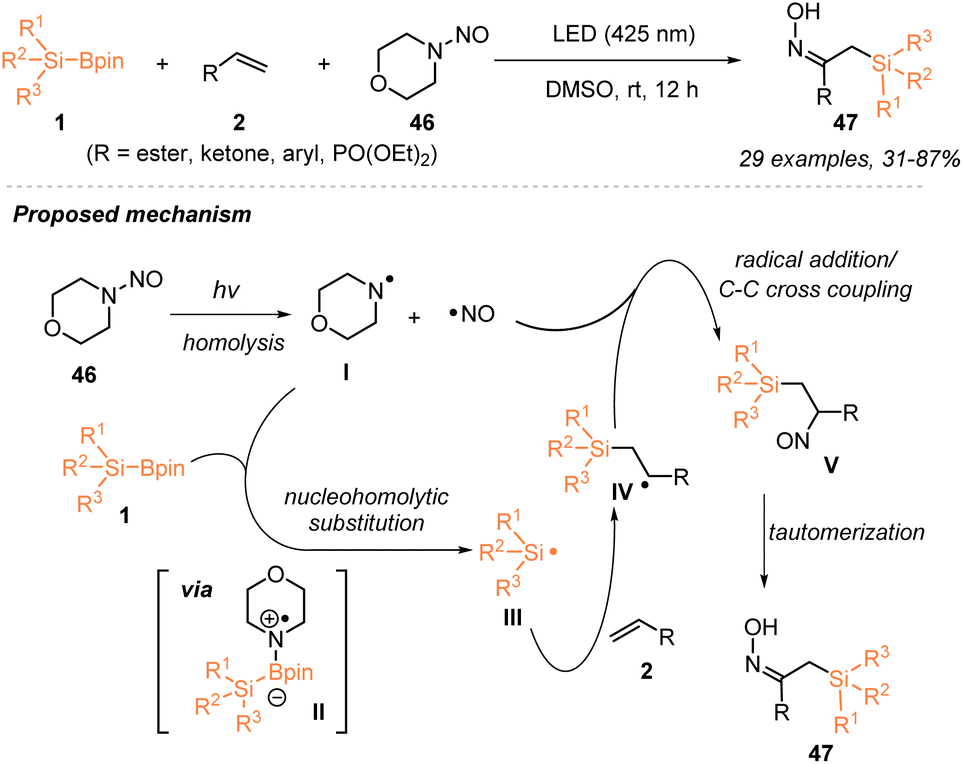 | ||
| Scheme 16 Silyl-oximation of alkenes with silylboronate and N-nitrosamine. | ||
Very recently, Lian and coworkers developed a triphasic 1,2-hydroxysilylation of alkenes 39 with PhMe2Si–Bpin 1b under an oxygen atmosphere by mechanically piezoelectric catalysis through a single-electron-transfer (SET) pathway (Scheme 17).33 A series of aromatic and aliphatic alkenes 39 worked well with PhMe2Si–Bpin 1b under an oxygen atmosphere to deliver the corresponding 1,2-hydroxysilylation products 48 with 32–94% yields. Moreover, this method could be used for the late-stage modification of drug-derived alkenes. Notably, this strategy pioneers the research in mechanic force-induced solid–liquid–gas triphase reactions under ambient conditions and indicates that silylboronate can be transformed into a silyl radical by highly polarized Li2TiO3 particles and oxygen under ball-milling conditions for the first time. The excellent substrate scope with good functionality tolerance, simple operation, multiple recycles of the catalyst, and solvent-free conditions further highlighted the practicability of the current protocol. A silyl radical process involving reaction pathway was proposed according to the mechanistic studies, as illustrated in Scheme 17. The highly polarized Li2TiO3 particles were first generated by agitating Li2TiO3 particles via ball-milling, which reduced oxygen to give superoxide radicals (˙O2−). Subsequently, the intermediate I, which was produced from superoxide radicals (˙O2−) via coordinating with the boron center of silylboronate 1b, underwent SET oxidation to afford the silyl radical species II, accompanied by the reduction of the oxidized Li2TiO3 particles. The trapping of the silyl radical II with alkenes delivered alkyl radical intermediate III, which then interacted with triplet oxygen to give the peroxyl radical species IVvia the formation of the C–O bond. Finally, the target 48 was generated from intermediate IV after undergoing a HAT process and reduction with silylboronate.
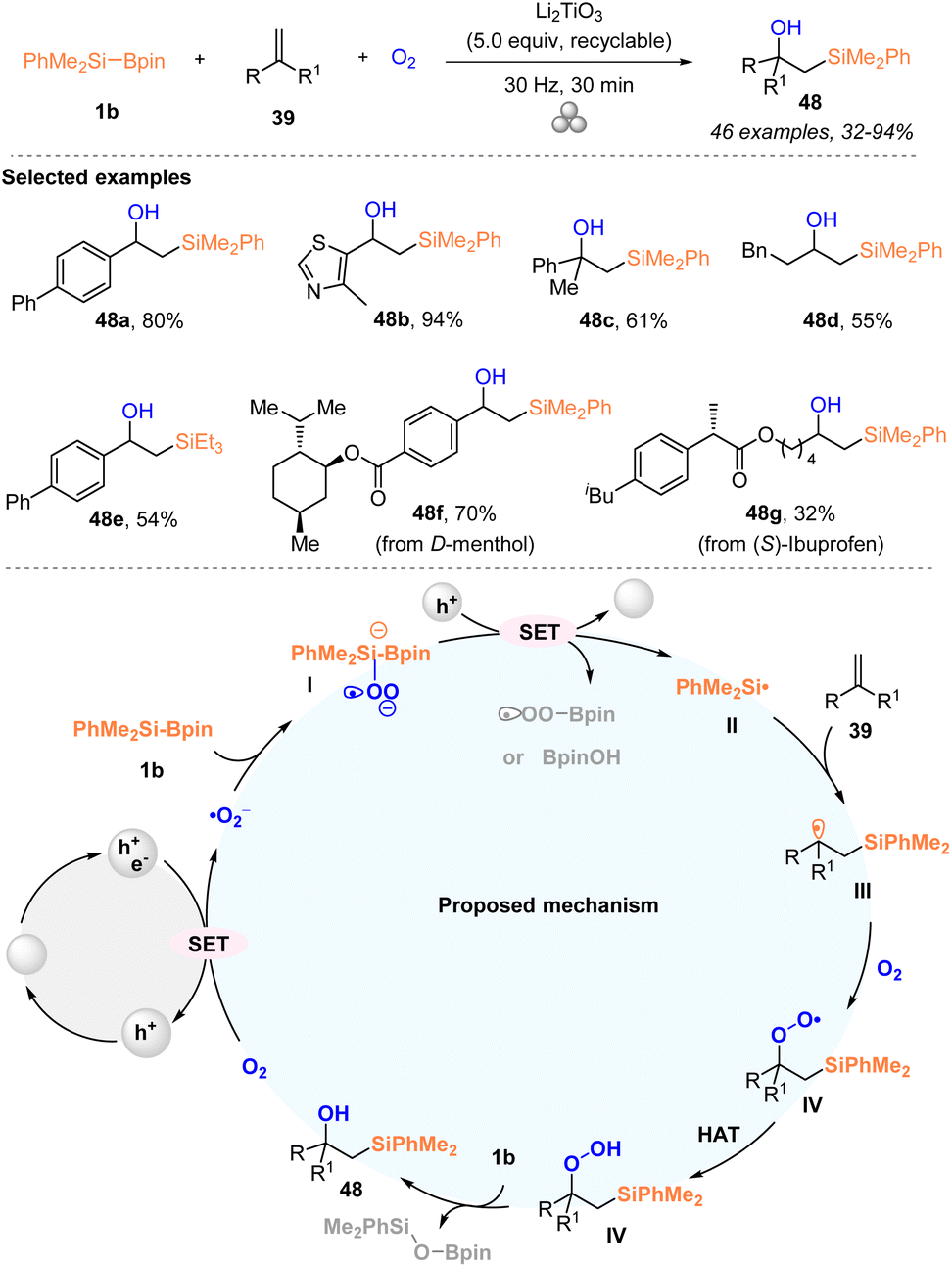 | ||
| Scheme 17 Triphasic hydroxysilylation of alkenes by mechanically piezoelectric catalysis. | ||
In 2023, Tanaka and coworkers developed a chemo-, regio- and stereo-selective dearomatic triple elementalization (carbo-sila-boration) of quinolines through the addition of organolithium and sequential visible light-induced silaboration, allowing the efficient synthesis of various carbo-sila-borated tetrahydroquinolines 50 that are expected to be versatile synthetic platforms (Scheme 18).34 The tandem sequence started with the addition of organolithium to quinolines 49. The generated lithium anilides then reacted with silylboronates to afford the anilide-silylborane ate complexes II, which underwent Si–B bond cleavage to form silyl radicals under blue LED irradiation, thus enabling the occurrence of carbo-sila-boration to deliver the desired products 50. Control experiments and DFT calculation studies revealed the involvement of a Si–B bond activation process in this sequence, in which visible light excitation of anilide–silylborane ate complexes II produced a silyl radical rather than a silyl anion. Notably, the synthetic utility of this method was highlighted by the potential of carbo-sila-borated tetrahydroquinolines as synthetic platforms, as exemplified by the chemo- and stereospecific conversions of C–B/C–Si bonds to C–C, C–O, and C–H/D bonds. In addition, the asymmetric carbo-sila-boration of quinoline was explored by combining an asymmetric alkylation of quinoline with n-BuLi enabled by chiral ligand L1, delivering chiral carbo-sila-borated tetrahydroquinoline (2S,3R,4S)-50a.
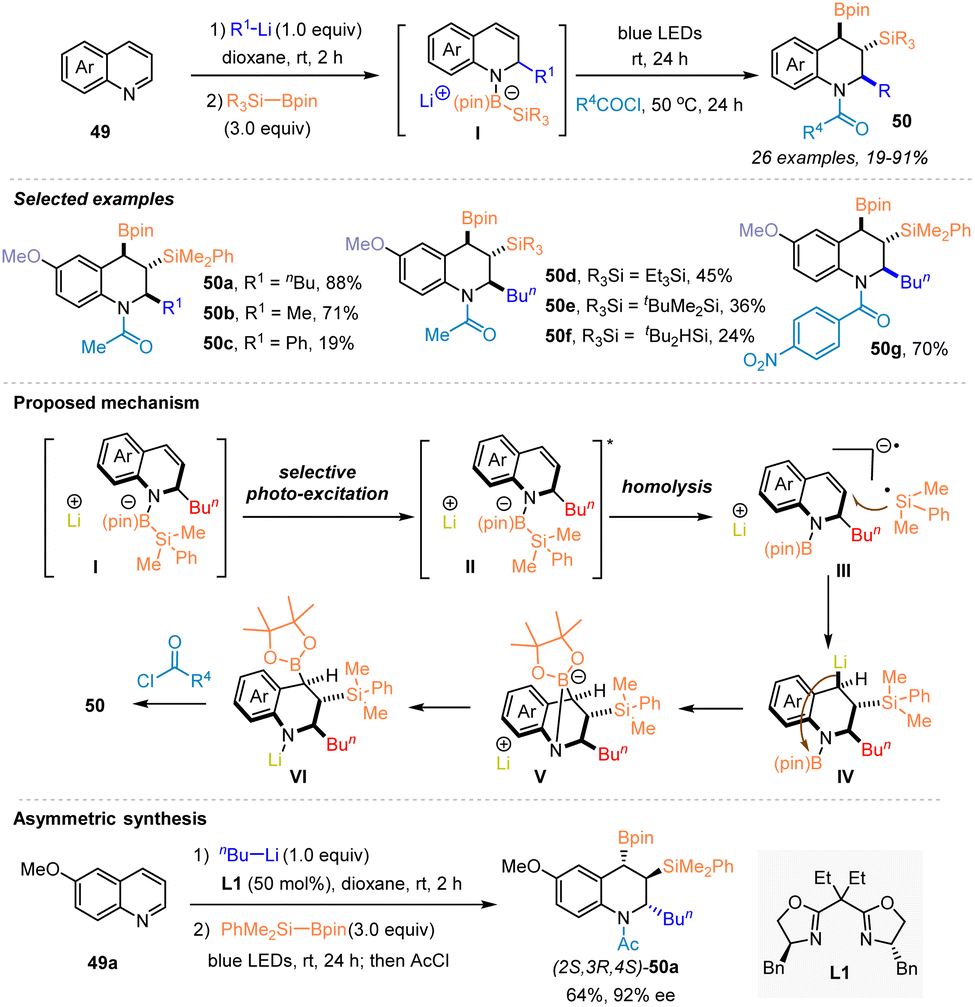 | ||
| Scheme 18 Dearomative triple elementalization (carbo-sila-boration) of quinolines. | ||
4 Selective silylation via C–X bond cleavage
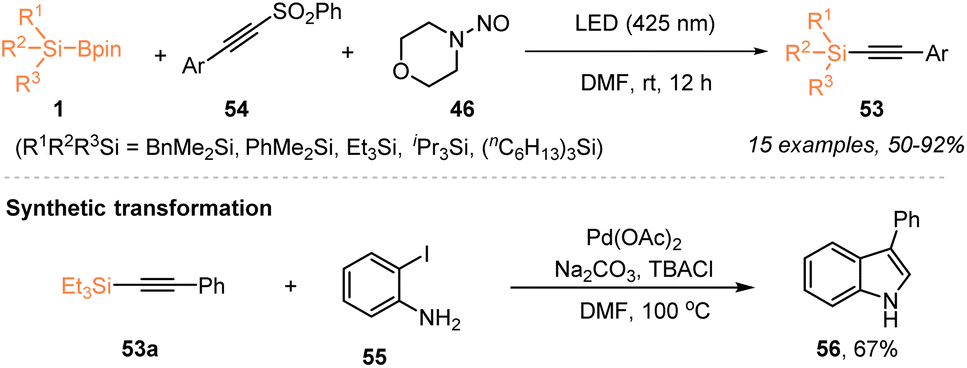
Aside from the radical silylfunctionalization of unsaturated hydrocarbons, e.g. alkenes or alkynes, the use of silylboronate as a silyl radical precursor has recently been applied for selective silylation via C–X bond cleavage. In 2023, Zhang, Wang, and coworkers employed a visible-light-induced organophotocatalytic strategy to realize a radical Minisci-type C–H silylation of N-heteroarenes with silylboronates 1 (Scheme 19).23 The use of 3 mol% 4CzlPN enabled the facile synthesis of silylated N-heteroarenes 51 with 48–72% yields, in the presence of TFA (1.0 equiv.), under 24 W blue LED irradiation. Moreover, the authors found that the silyl radicals generated via such an organophotocatalytic strategy could react with ethynylbenziodoxolones (EBXs) 52 through C–I bond cleavage, thus realizing the alkynylation of silylboronates 1 with EBXs 52 for the preparation of alkynylsilanes 53 with 56–82% yields.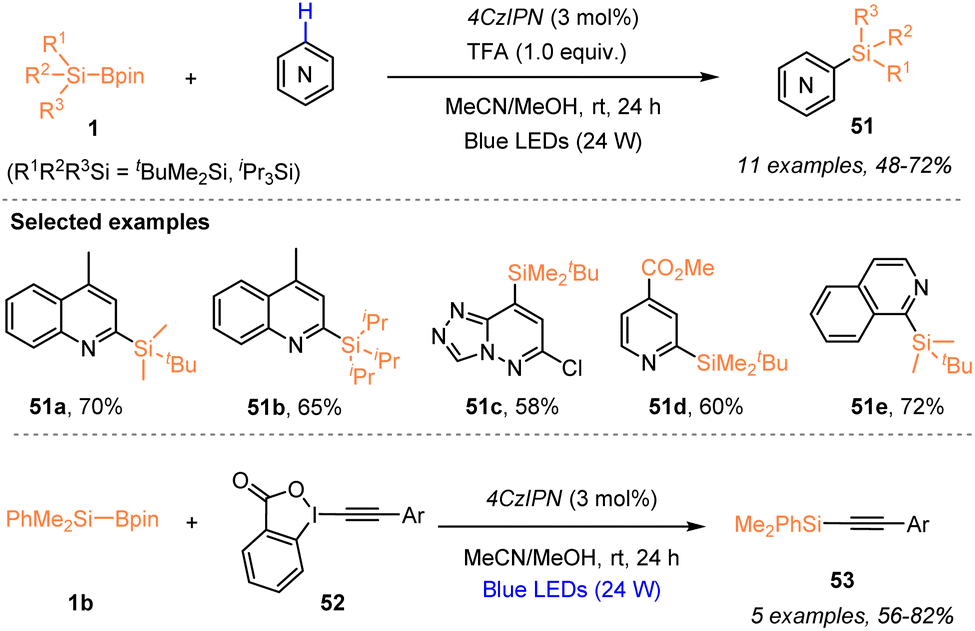 | ||
| Scheme 19 Organophotocatalytic silylation via C–H or C–I bond cleavage. | ||
A year later, Wang and Jia et al. also reported the preparation of synthetically useful alkynylsilanes by using silyl radicals generated from silylboronates via nucleohomolytic substitution of boron with aminyl radicals (Scheme 20).32 Under 425 nm LED irradiation, the alkynylation of silylboronates 1 with ethynyl phenyl sulfones (EPSs) 54 proceeded smoothly to afford a series of silylated alkynes 53 with 50–92% yields, in the presence of N-nitrosamine 46. The process for silyl radical generation was the same as the pathway of Scheme 16. The transformation of alkynylsilane 53a into 3-phenylindole further highlighted the utility of this protocol.
 | ||
| Scheme 20 Visible light-induced alkynylation of silylboronates. | ||
In 2023, Feng, Xue, and Liu accomplished a tBuONa enabled site-selective silylation of alkyl/aryl thioethers 57 with Et3Si–Bpin (1d) via C(alkyl)–S bond cleavage (Scheme 21).35
 | ||
| Scheme 21 Site-selective silylation of thioethers with Et3Si–Bpin via C–S bond cleavage. | ||
It was found that a series of primary or secondary alkyl aryl sulfides 57 were tolerated and afforded various alkylsilanes 3 with 40–98% yields, in the presence of 4.0 equiv. tBuONa. Et3Si–Bpin was activated by tBuONa to form complex I, which further interacted with another tBuONa molecule to produce the active silyl radical via a single-electron transfer process. Subsequently, the resulting silyl radical prefers to work with the C(alkyl)–S bond, facilitating the C(alkyl)–S bond cleavage to yield alkylsilanes. Furthermore, the gram scale synthesis and late-stage silylation of bioactive molecules highlighted the usefulness of this protocol. Radical inhibition and radical clock experiments confirmed the involvement of silyl radical species in this reaction system.
Also in 2023, Cui, Cao, Shi, and coworkers reported a dearomatization silylation of benzofurans or furopyridines via silyl radical addition and subsequent C–O bond scission (Scheme 22).36 Under the action of 2.0 equiv. tBuOK, a series of benzofurans or furopyridines without substituents at the C2 or C3 position worked well with Et3Si–Bpin 1d to deliver the ring-opening vinylsilanes 61 with 43–92% yields. Benzofurans, featuring an aryl at the C2 position or a methyl at the C3 position, were tolerated as well and afforded the corresponding vinylsilanes with moderated to good yields.
 | ||
| Scheme 22 Dearomatization radical silylation of benzofurans or furopyridines. | ||
It should be mentioned that benzofurans with a bulky tert-butyl group at the C2-position or an aryl group at the C3-position underwent1,5 Brook rearrangement following silyl radical addition and subsequent C–O bond scission, producing vinylphenoxyl silanes 63 with 80–93% yields. Based on initial mechanistic studies, the authors proposed a reaction mechanism, as shown in Scheme 22. The triethylsilyl radical EtSi˙ was first generated via the reaction of Et3Si–Bpin with a tBuO radical that was formed from tBuOK and O2. The addition of EtSi˙ to benzofurans or furopyridines proceeded to give the carbon-centered radical species I, whilst the direct interaction of Et3Si–Bpin with tBuOK led to the formation of complex II. Subsequently, radical species I underwent a SET process with complex II to afford intermediate III-A or III-B. Then III-A underwent β-elimination to give the product 61 after protonation, while III-B underwent sequential β-elimination and [1,5]-Brook rearrangement to furnish vinylphenoxyl silanes 63. Moreover, the diverse product elaboration demonstrated the practicability of this protocol, as evidenced by the preparation of trimethoxyl oxyresveratrol 64 and doxepin analogue 65.
5 Silyl radical mediated cross-coupling with silylboronates
With the application of silylboronates as silyl radical precursors for constructing organosilanes via C–Si bond forming reactions, silylboronates have also been identified as an effective medium for enabling C–F bond cleavage of organic fluorides to enable cross coupling reactions. In 2023, the Shibata group pioneered this study and established a transition metal-free silylboronate-mediated cross-coupling of organic fluorides 66 with amines 67via inert C–F bond activation under mild conditions (Scheme 23).37 A series of (hetero)aryl or alkyl fluorides reacted smoothly with cyclic or acyclic N-alkylanilines or secondary dialkylamines 67, in the presence of 2 equiv. Et3Si–Bpin and 4 equiv. tBuOK, to afford various tertiary amines 68 with 18–98% yields. The coordination of Et3Si–Bpin and tBuOK enabled the cross-coupling to proceed at room temperature, thus avoiding the high potential barriers associated with thermally induced SN2 or SN1 amination. A single-electron-transfer/radical-mediated defluorinative amination involving a frustrated radical pair chemistry mechanism is illustrated in Scheme 23, according to the mechanistic studies. The intermediate I was first generated from Et3Si–Bpin with KOtBu, which then produced a frustrated radical pair II that comprises Et3Si˙ and a boron-radical species via the homolytic cleavage of the Si–B bond. The formed Et3Si˙ underwent hydrogen abstraction from amines 67 to provide a frustrated radical pair III consisting of an amino radical and the boron radical species, along with the generation of HSiEt3. The frustrated radical pair III subsequently interacted with organic fluorides 66via transition state IV, where the C–F bond was activated by the interaction between the F atom and B center, and the amino radical selectively attacked the carbon center of the C–F bond, to deliver the desired cross-coupling product, accompanied by the formation of stable [Bpin(OtBu)2]K (VI). The significant feature of this transformation included the selective activation of the C–F bond of organofluorides by silylboronate without affecting potentially cleavable C–O, C–Cl, C–H or C–N bonds and the CF3 group, mild reaction conditions, broad substrate scope, and late-stage modification of fluorine-containing drugs. | ||
| Scheme 23 Silylboronate-mediated radical cross-coupling of fluorides and amines. | ||
Shortly after, by employing the strategy of silyl radical-mediated cross-coupling of fluorides, the same group realized an efficient cross-coupling of aryl fluorides 66 with arylalkanes 69 or 71 enabled by a combination of Et3Si–Bpin 1d with tBuOK, allowing the diverse access of various triaryl- or diarylalkanes 70 or 72, which are promising scaffolds for pharmaceuticals and functional materials, with moderate to excellent yields under very mild reaction conditions (Scheme 24).38 A salient feature of this protocol was that the activation of the C–F and C–H bonds occurred at room temperature. The practicability was further highlighted by the fact that no transition metal and specialized ligands with high temperature, which are usually required in common cross-coupling reactions, were used in this method. A similar silyl radical-involved reaction mechanism is proposed in Scheme 24, based on the ESR analysis and experimental studies.
 | ||
| Scheme 24 Silylboronate-mediated cross-coupling of benzylic C–H with arylfluorides. | ||
Subsequently, they further utilized a silylboronate-mediated cross-coupling strategy to enable the cross coupling of alkyl fluorides and aryl alkanes for the formation of C(sp3)–C(sp3) bonds (Scheme 25).39 Under the action of Et3Si–Bpin with tBuOK, a variety of alkyl fluorides 66 could react efficiently with mono- or diaryl alkanes 71 at 60 °C, producing diaryl or aryl alkanes 73 bearing a tertiary or quaternary carbon center in 22–98% yields. Moreover, alkyl chlorides, bromides, and iodides were also suitable substrates for this transformation. Although a radical-involving pathway was supported by radical trapping and ESR experiments, radical ring-opening experiments and DFT calculations suggested that an ionic process might occur in this cross-coupling. Notably, mechanistic studies using DFT calculations were conducted on silylboronate-mediated cross-coupling for the first time and supported the crucial role of diglyme in enhancing the reaction efficiency via encapsulation of potassium cations (Scheme 25).
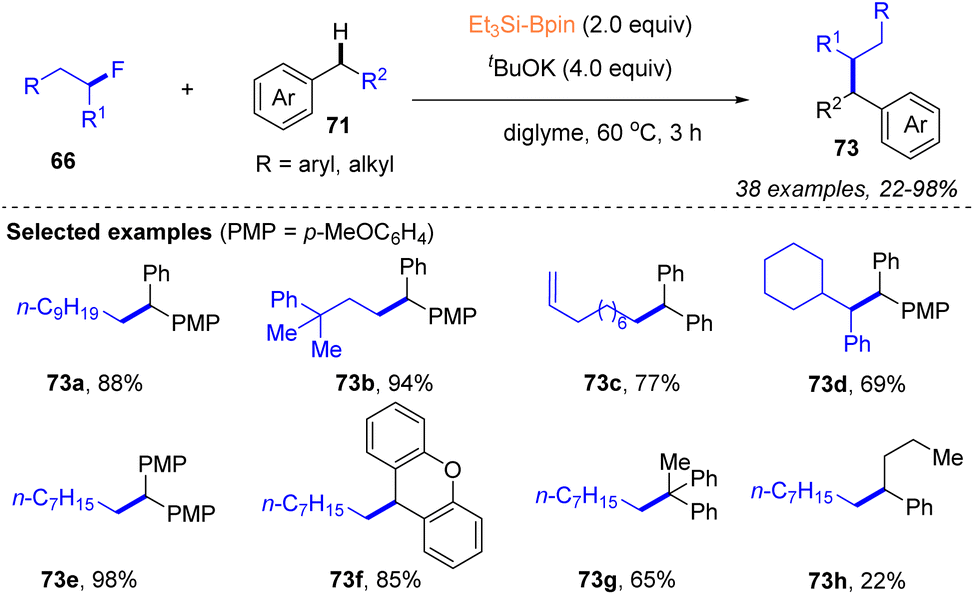 | ||
| Scheme 25 Silylboronate-mediated cross-coupling of alkylfluorides and aryl alkanes. | ||
More recently, Shibata and coworkers developed an efficient silylboronate-mediated cross-coupling between organic fluorides 66 and allenes 74via a catalyst-free silyl radical relay strategy, which provided a facile protocol for the construction of α-alkynyl substituted all-carbon quaternary centers (Scheme 26).40
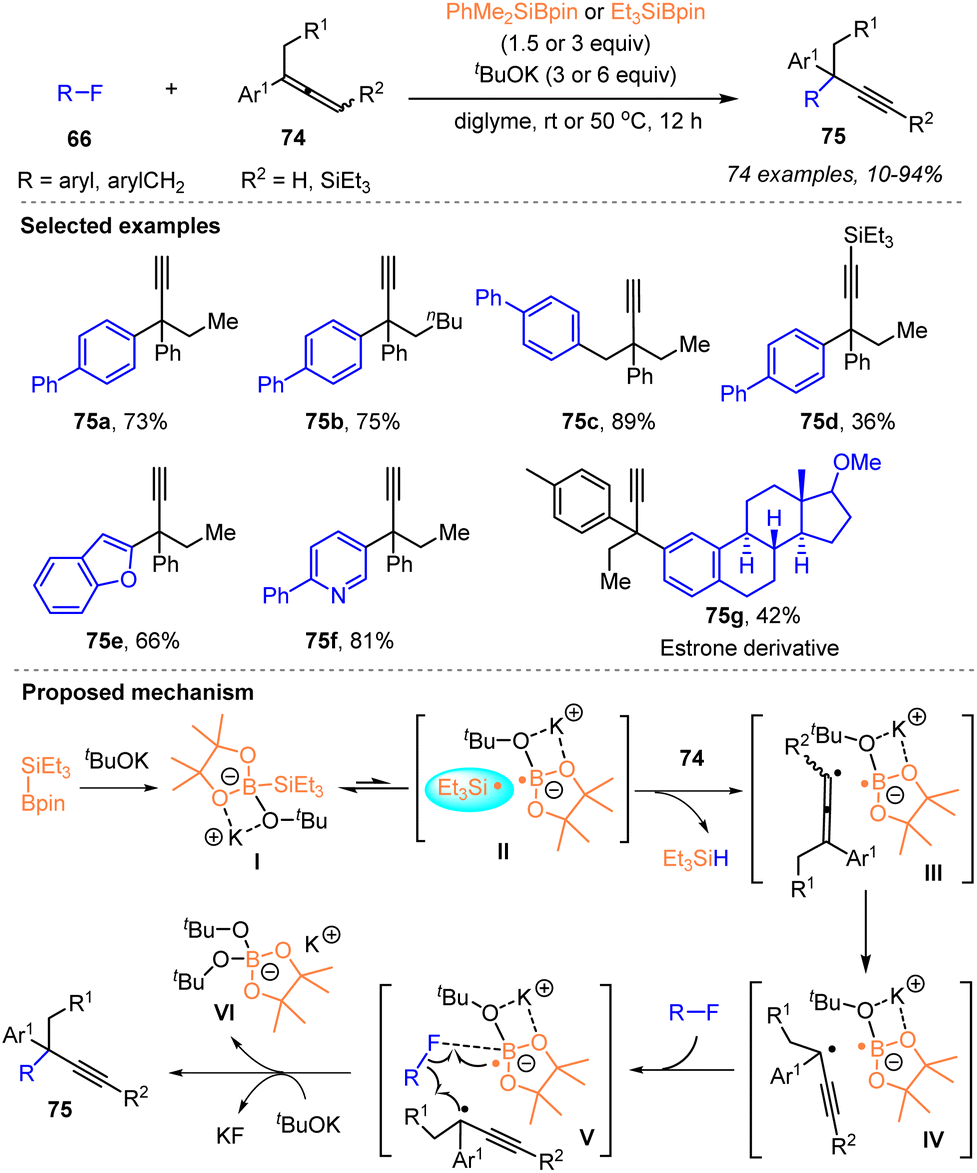 | ||
| Scheme 26 Radical cross-coupling of organic fluorides and allenes mediated by silylboronates. | ||
Under the action of tBuOK and Et3Si–Bpin 1d or PhMe2Si–Bpin 1b, a variety of aryl or benzyl fluorides 66 worked well with disubstituted allenes 74 to afford a library of coupling products 75 featuring an α-ethynyl-containing all-carbon quaternary center with up to 94% yields. The key to success was in situ radical rearrangement of the formed allenyl radicals to give bulky tertiary propargyl radicals. A salient feature of this cross-coupling was the ability of the in situ generated silyl radical to abstract a proton directly from the C(sp2)–H bond of allenes to form an allenyl radical, which was subsequently isomerized to a propargylic radical, as depicted in the proposed mechanism of Scheme 26. Furthermore, mild reaction conditions without a transition metal, wide substrate scope, the late-stage functionalization of bioactive molecules and the modification of a liquid crystalline material demonstrated the practical utility of the current protocol.
6 Conclusion and outlook
The past few years have witnessed significant advancements in synthetic applications of silylboronates as silyl radical precursors, with the rapid development of radical chemistry. As summarized in this review article, a variety of C–Si or C–X bond forming transformations have been established for the diverse construction of organosilanes or other valuable molecules (e.g. tertiary amines, triaryl- or diarylalkanes and α-alkynyl substituted quaternary centers), including hydrosilylation of alkenes or alkynes; silyl radical involved silylfunctionalization; selective radical silylation via C–X bond cleavage; silyl radical mediated cross-coupling.Despite these impressive achievements, there is still ample room for further exploration in synthetic applications of silylboronates as silyl radical precursors toward valuable chemicals. First, more successful and diverse transformations are needed to demonstrate the synthetic practicability and potential of silylboronates as silyl radical precursors, since most of the currently reported examples focus on the hydrosilylation or silylfunctionalization. Second, the generation of silyl radical species from silylboronates mainly relies on the use of photochemical, electrochemical or stoichiometric base activation. Therefore, the development of new and efficient methods for the generation of silyl radicals from silylboronates will be highly desirable. Third, no asymmetric reactions using silylboronates as silyl radical sources have been reported, which provides another important direction to explore radical asymmetric transformations involving silylboronates for accessing chiral organosilanes, in particular, silicon-stereogenic chiral silanes.41 With the continuous emergence of new silylboronate reagents, catalytic systems, and synthetic strategies, it can be anticipated that more and more attractive and facile silyl radical transformations involving silylboronates will be explored, which will greatly broaden the synthetic applications of silylboronates in the area of radical chemistry.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
All authors co-wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 22471073 and 22171087), the International Cooperation Project (No. DL2023047001L), the Major R&D projects (No. 2023B01019 and 2023AA004), the Key Laboratory Project of Shihezi City (No. 2023PT01), the Innovation Team Project of Shihezi City (No. 2023TD03), the Technology Innovation Project of Shanghai Municipal Agricultural Committee [No. HNK(T2023302)], the Innovation Program of Shanghai Municipal Education Commission (No. 2023ZKZD37), and the Ministry of Education (PCSIRT) and the Fundamental Research Funds for the Central Universities. We also acknowledge Prof. Kai Hong at East China Normal University for valuable discussion.References
- (a) S. E. Denmark and R. F. Sweis, Acc. Chem. Res., 2002, 35, 835–846 CrossRef PubMed; (b) T. Hiyama and M. Oestreich, Organosilicon Chemistry: Novel Approaches and Reactions, Wiley-VCH, Weinheim, Germany, 1st edn, 2019 CrossRef; (c) T. Komiyama, Y. Minami and T. Hiyama, ACS Catal., 2017, 7, 631–651 CrossRef.
- (a) A. K. Franz and S. O. Wilson, J. Med. Chem., 2013, 56, 388–405 CrossRef PubMed; (b) R. Ramesh and D. S. Reddy, J. Med. Chem., 2018, 61, 3779–3798 CrossRef PubMed; (c) E. Remond, C. Martin, J. Martinez and F. Cavelier, Chem. Rev., 2016, 116, 11654–11684 CrossRef PubMed.
- (a) L. Gai, J. Mack, H. Lu, T. Nyokong, Z. Li, N. Kobayashi and Z. Shen, Coord. Chem. Rev., 2015, 285, 24–51 CrossRef CAS; (b) E. A. Marro and R. S. Klausen, Chem. Mater., 2019, 31, 2202–2211 CrossRef CAS.
- For a book: (a) Reagents for silicon-mediated organic synthesis (Handbook of Reagents for Organic Synthesis), ed. P. L. Fuchs, Wiley, 2013, For reviews: Search PubMed; (b) C. Chatgilialoglu, C. Ferreri, Y. Landais and V. Timokhin, Chem. Rev., 2018, 118, 6516–6572 CrossRef CAS PubMed; (c) H. Ottosson and P. G. Steel, Chem.–Eur. J., 2006, 12, 1576–1585 CrossRef CAS; (d) W. P. Weber, Silicon Reagents for Organic Synthesis, Springer, Berlin 1983 CrossRef; (e) L. A. Paquette, Science, 1982, 217, 793–800 CrossRef CAS PubMed; (f) Z.-T. Ye, Z.-W. Wu, X.-X. Zhang, J. Zhou and J.-S. Yu, Chem. Soc. Rev., 2024, 53, 8546–8562 RSC; (g) X.-S. Hu, J.-S. Yu and J. Zhou, Chem. Commun., 2019, 55, 13638 RSC , For selected examples: ; (h) M. Liu, K. Dong, B. Xu, Z.-M. Zhang, Z. Wei and J. Zhang, Org. Chem. Front., 2024, 11, 3821–3826 RSC; (i) X.-X. Zhang, Y. Gao, Y.-X. Zhang, J. Zhou and J.-S. Yu, Angew. Chem., Int. Ed., 2023, 62, e202217724 CrossRef CAS; (j) W.-B. Wu, X. Yu, J.-S. Yu, X. Wang, W.-G. Wang and J. Zhou, CCS Chem., 2022, 4, 2140 CrossRef CAS; (k) X.-S. Hu, J.-X. He, S.-Z. Dong, Q.-H. Zhao, J.-S. Yu and J. Zhou, Nat. Commun., 2020, 11, 5500 CrossRef PubMed; (l) B.-S. Mu, Y. Gao, F.-M. Yang, W.-B. Wu, Y. Zhang, X. Wang, J.-S. Yu and J. Zhou, Angew. Chem., Int. Ed., 2022, 61, e202208861 CrossRef; (m) C.-W. Lei, X.-Y. Wang, B.-S. Mu, J.-S. Yu, Y. Zhou and J. Zhou, Org. Lett., 2022, 24, 8364–8369 CrossRef; (n) W.-B. Wu, B.-S. Mu, J.-S. Yu and J. Zhou, Chem. Sci., 2022, 13, 3519–3525 RSC.
- For typical synthetic methods, see: (a) T. Takeuchi, R. Shishido, K. Kubota and H. Ito, Chem. Sci., 2021, 12, 11799–11804 RSC; (b) R. Shishido, M. Uesugi, R. Takahashi, T. Mita, T. Ishiyama, K. Kubota and H. Ito, J. Am. Chem. Soc., 2020, 142, 14125–14133 CrossRef; (c) T. A. Boebel and J. F. Hartwig, Organometallics, 2008, 27, 6013–6019 CrossRef; (d) M. Suginome, T. Matsuda and Y. Ito, Organometallics, 2000, 19, 4647–4649 CrossRef , Also see: ; (e) T. Ohmura and M. Suginome, Bull. Chem. Soc. Jpn., 2009, 82, 29–49 CrossRef.
- (a) M. Oestreich, E. Hartmann and M. Mewald, Chem. Rev., 2013, 113, 402–441 CrossRef PubMed; (b) J. R. Wilkinson, C. E. Nuyen, T. S. Carpenter, S. R. Harruff and R. Van Hoveln, ACS Catal., 2019, 9, 8961–8979 CrossRef; (c) W. Xue and M. Oestreich, ACS Cent. Sci., 2020, 6, 1070–1081 CrossRef; (d) J.-J. Feng, W. Mao, L. Zhang and M. Oestreich, Chem. Soc. Rev., 2021, 50, 2010–2073 RSC.
- (a) M. Suginome, H. Nakamura and Y. Ito, Chem. Commun., 1996, 2777–2778 RSC; (b) M. Suginome, T. Matsuda, H. Nakamura and Y. Ito, Tetrahedron, 1999, 55, 8787–8800 CrossRef; (c) S. Onozawa, Y. Hatanaka and M. Tanaka, Chem. Commun., 1997, 1229–1230 RSC.
- For selected recent examples, see: (a) M. Suginome, H. Nakamura and Y. Ito, Angew. Chem., Int. Ed. Engl., 1997, 36, 2516–2518 CrossRef; (b) T. Ohmura, K. Oshima, H. Taniguchi and M. Suginome, J. Am. Chem. Soc., 2010, 132, 12194–12196 CrossRef PubMed; (c) T. Fujihara, Y. Tani, K. Semba, J. Terao and Y. Tsuji, Angew. Chem., Int. Ed., 2012, 51, 11487–11490 CrossRef PubMed; (d) Y. Tani, T. Fujihara, J. Terao and Y. Tsuji, J. Am. Chem. Soc., 2014, 136, 17706–17709 CrossRef; (e) Z.-T. He, X.-Q. Tang, L.-B. Xie, M. Cheng, P. Tian and G.-Q. Lin, Angew. Chem., Int. Ed., 2015, 54, 14815–14818 CrossRef; (f) T. Iwamoto, T. Nishikori, N. Nakagawa, H. Takaya and M. Nakamura, Angew. Chem., Int. Ed., 2017, 56, 13298–13301 CrossRef; (g) H. Sakaguchi, M. Ohashi and S. Ogoshi, Angew. Chem., Int. Ed., 2018, 57, 328–332 CrossRef; (h) Z. Liu, J. Chen, H.-X. Lu, X. Li, Y. Gao, J. R. Coombs, M. J. Goldfogel and K. M. Engle, Angew. Chem., Int. Ed., 2019, 58, 17068–17073 CrossRef; (i) Y. Gu, Y. Duan, Y. Shen and R. Martin, Angew. Chem., Int. Ed., 2020, 59, 2061–2065 CrossRef; (j) T. R. Pradhan, M. Paudel, T. Feoktistova, P. H.-Y. Cheong and J. K. Park, Angew. Chem., Int. Ed., 2022, e202116154 Search PubMed; (k) G. Wang, M. Wei, T. Liu, W. Jin, Y. Zhang, B. Wang, Y. Xia and C. Liu, Adv. Synth. Catal., 2022, 364, 909–913 CrossRef; (l) Q. Chen, Z. Li and Y. Nishihara, Org. Lett., 2022, 24, 385–389 CrossRef PubMed; (m) Z.-Y. Chen, M.-W. Yang, Z.-L. Wang and Y.-H. Xu, Org. Lett., 2023, 25, 5242–5247 CrossRef PubMed; (n) H. Moniwa, M. Yamanaka and R. Shintani, J. Am. Chem. Soc., 2023, 145, 23470–23477 CrossRef PubMed; (o) Y. Ozawa, H. Koriyama, Y. Shiratori and H. Ito, ACS Org. Inorg. Au, 2023, 3, 104–108 CrossRef PubMed; (p) Q. Li, Z.-L. Wang and Y.-H. Xu, Chin. Chem. Lett., 2023, 34, 108150 CrossRef; (q) C. Ding, Y. Ren, Y. Yu and G. Yin, Nat. Commun., 2023, 14, 7670 CrossRef PubMed.
- For selected recent examples, see: (a) V. Cirriez, C. Rasson, T. Hermant, J. Petrignet, J. Díaz Alvarez, K. Robeyns and O. Riant, Angew. Chem., Int. Ed., 2013, 52, 1785–1788 CrossRef PubMed; (b) A. Hensel, K. Nagura, L. B. Delvos and M. Oestreich, Angew. Chem., Int. Ed., 2014, 53, 4964–4967 CrossRef PubMed; (c) Y. Huo, P. Shen, W. Duan, Z. Chen, C. Song and Y. Ma, Chin. Chem. Lett., 2018, 29, 1359–1362 CrossRef; (d) M. Takeda, A. Mitsui, K. Nagao and H. Ohmiya, J. Am. Chem. Soc., 2019, 141, 3664–3669 CrossRef PubMed; (e) K. Yabushita, A. Yuasa, K. Nagao and H. Ohmiya, J. Am. Chem. Soc., 2019, 141, 113–117 CrossRef PubMed; (f) Z.-Y. Zhao, M. Cui, E. Irran and M. Oestreich, Angew. Chem., Int. Ed., 2023, e202215032 Search PubMed.
- For selected recent examples, see: (a) M. O'Brien and A. H. Hoveyda, J. Am. Chem. Soc., 2011, 133, 7712–7715 CrossRef PubMed; (b) R. T. H. Linstadt, C. A. Peterson, D. J. Lippincott, C. I. Jette and B. H. Lipshutz, Angew. Chem., Int. Ed., 2014, 53, 4159–4163 CrossRef CAS PubMed; (c) J. A. Calderone and W. L. Santos, Angew. Chem., Int. Ed., 2014, 53, 4154–4158 CrossRef CAS; (d) H. Wu, J. M. Garcia, F. Haeffner, S. Radomkit, A. R. Zhugralin and A. H. Hoveyda, J. Am. Chem. Soc., 2015, 137, 10585–10602 CrossRef CAS PubMed; (e) T. Kitanosono, L. Zhu, C. Liu, P. Xu and S. Kobayashi, J. Am. Chem. Soc., 2015, 137, 15422–15425 CrossRef CAS; (f) M. Wang, Z.-L. Liu, X. Zhang, P.-P. Tian, Y.-H. Xu and T.-P. Loh, J. Am. Chem. Soc., 2015, 137, 14830–14833 CrossRef CAS PubMed; (g) C. Rasson, A. Stouse, A. Boreux, V. Cirriez and O. Riant, Chem.–Eur. J., 2018, 24, 9234–9237 CrossRef CAS; (h) W. Mao, W. Xue, E. Irran and M. Oestreich, Angew. Chem., Int. Ed., 2019, 58, 10723–10726 CrossRef CAS; (i) Q. Li, Z.-L. Wang, H.-X. Lu and Y.-H. Xu, Org. Lett., 2022, 24, 2832–2836 CrossRef CAS; (j) Y. Xiao, Z.-Y. Zhao, S. Kemper, E. Irran and M. Oestreich, Angew. Chem., Int. Ed., 2024, e202407056 CAS , For reviews, see: ; (k) X. Tang, L. Xie, Y. Chen, P. Tian and G. Lin, Chin. J. Org. Chem., 2016, 36, 2011–2023 CrossRef CAS; (l) B. Han, W. Li, S. Chen, Z. Zhang, X. Zhao, Y. Zhang and L. Zhu, Chin. J. Org. Chem., 2023, 43, 555–572 CrossRef CAS , and ref. 6..
- For selected examples, see: (a) D. J. Vyas and M. Oestreich, Angew. Chem., Int. Ed., 2010, 49, 8513–8515 CrossRef CAS; (b) L. B. Delvos, D. J. Vyas and M. Oestreich, Angew. Chem., Int. Ed., 2013, 52, 4650–4653 CrossRef CAS PubMed; (c) R. Shintani, R. Fujie, M. Takeda and K. Nozaki, Angew. Chem., Int. Ed., 2014, 53, 6546–6549 CrossRef CAS; (d) Z.-L. Liu, C. Yang, Q.-Y. Xue, M. Zhao, C.-C. Shan, Y.-H. Xu and T.-P. Loh, Angew. Chem., Int. Ed., 2019, 58, 16538–16542 CrossRef CAS.
- For selected examples, see: (a) M. Suginome, T. Matsuda and Y. Ito, J. Am. Chem. Soc., 2000, 122, 11015–11016 CrossRef; (b) H. Lee, J. T. Han and J. Yun, ACS Catal., 2016, 6, 6487–6490 CrossRef; (c) T. Yamamoto, R. Murakami, S. Komatsu and M. Suginome, J. Am. Chem. Soc., 2018, 140, 3867–3870 CrossRef PubMed; (d) L. Zhang and M. Oestreich, Chem.–Eur. J., 2019, 25, 14304–14307 CrossRef PubMed; (e) M. Cui and M. Oestreich, Org. Lett., 2020, 22, 3684–3687 CrossRef PubMed; (f) M. Kondo, J. Kanazawa, T. Ichikawa, T. Shimokawa, Y. Nagashima, K. Miyamoto and M. Uchiyama, Angew. Chem., Int. Ed., 2020, 59, 1970–1974 CrossRef PubMed; (g) K. Sekine, D. Akaishi, K. Konagaya and S. Ito, Chem.–Eur. J., 2022, e202200657 CrossRef PubMed; (h) B. Yang, K. Cao, G. Zhao, J. Yang and J. Zhang, J. Am. Chem. Soc., 2022, 144, 15468–15474 CrossRef PubMed.
- (a) T. Ohmura, I. Sasaki and M. Suginome, Org. Lett., 2019, 21, 1649–1653 CrossRef PubMed; (b) I. Sasaki, T. Ohmura and M. Suginome, Org. Lett., 2020, 22, 2961–2966 CrossRef PubMed; (c) Y. Zhang, W. Xu, T. Gao, M. Guo, C.-H. Yang, H. Xie, X. Kong, Z. Yang and J. Chang, Org. Lett., 2022, 24, 7021–7025 CrossRef PubMed.
- (a) Y. Gu, Y. Shen, C. Zarate and R. Martin, J. Am. Chem. Soc., 2019, 141, 127–132 CrossRef PubMed; (b) J. Zheng, H. Zhang, S. Kong, Y. Ma, Q. Du, B. Yi, G. Zhang and R. Guo, ACS Catal., 2024, 14, 1725–1732 CrossRef CAS; (c) S. Kamio, M. Nakamoto, T. Yamagishi, M. Oestreich and H. Yoshida, Chem. Commun., 2024, 60, 6379–6382 RSC; (d) K. Asai, K. Hirano and M. Miura, Eur. J. Org Chem., 2022, e202101535 CrossRef CAS.
- (a) T. Takeuchi, A. Roy and H. Ito, J. Am. Chem. Soc., 2023, 145, 16249–16260 CrossRef CAS; (b) I. Sasaki, A. Maebashi, J. Li, T. Ohmura and M. Suginome, Eur. J. Org Chem., 2022, e202101573 CrossRef CAS; (c) L. Zhou, J. Qiu, C. Wang, F. Zhang, K. Yang and Q. Song, Org. Lett., 2022, 24, 3249–3253 CrossRef CAS PubMed.
- (a) T. Kubo and M. Abe, Chem. Rev., 2024, 124, 4541–4542 Search PubMed; (b) S. Crespi and M. Fagnoni, Chem. Rev., 2020, 120, 979–9833 CrossRef PubMed; (c) J. P. Barham and B. König, Angew. Chem., Int. Ed., 2020, 59, 11732–11747 CrossRef CAS PubMed; (d) M. Yan, J. C. Lo, J. T. Edwards and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 12692–12714 CrossRef CAS; (e) A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2015, 55, 58–102 CrossRef; (f) C. Chatgilialoglu and A. Studer, Encyclopedia of Radicals in Chemistry, Biology and Materials, Wiley-VCH, Weinheim, 2012 CrossRef.
- (a) X. Zhang, J. Fang, C. Cai and G. Lu, Chin. Chem. Lett., 2021, 32, 1280–1292 CrossRef CAS; (b) L.-Q. Ren, N. Li, J. Ke and C. He, Org. Chem. Front., 2022, 9, 6400–6415 RSC; (c) J.-S. Li and J. Wu, ChemPhotoChem, 2018, 2, 839–846 CrossRef; (d) X. Shang and Z.-Q. Liu, Org. Biomol. Chem., 2016, 14, 7829–7831 RSC; (e) C. Chatgilialoglu, Chem. Rev., 1995, 95, 1229–1251 CrossRef.
- (a) J. Sun and L. Deng, ACS Catal., 2016, 6, 290–300 CrossRef; (b) B. Marciniec, H. Maciejewski, C. Pietraszuk and P. Pawluć, In Hydrosilylation: A Comprehensive Review on Recent Advances, ed. B. Marciniec, Springer, Berlin, 2009, ch. 1 CrossRef; (c) S. Díez-González and S. P. Nolan, Acc. Chem. Res., 2008, 41, 349–358 CrossRef PubMed; (d) A. K. Roy, Adv. Organomet. Chem., 2007, 55, 1–59 CrossRef; (e) F. Buch, H. Brettar and S. Harder, Angew. Chem., Int. Ed., 2006, 45, 2741–2745 CrossRef PubMed; (f) L. D. Almeida, H.-L. Wang, K. Junge, X.-J. Cui and M. Beller, Angew. Chem., Int. Ed., 2021, 60, 550–565 CrossRef PubMed.
- A. Matsumoto and Y. Ito, J. Org. Chem., 2000, 65, 5707–5711 CrossRef.
- M. Zhong, X. Pannecoucke, P. Jubault and T. Poisson, Chem.–Eur. J., 2021, 27, 11818–11822 CrossRef PubMed.
- N. Takemura, Y. Sumida and H. Ohmiya, ACS Catal., 2022, 12, 7804–7810 CrossRef.
- R. Arai, Y. Nagashima, T. Koshikawa and K. Tanaka, J. Org. Chem., 2023, 88, 10371–10380 Search PubMed.
- Y. Wan, Y.-M. Zhao, J.-J. Zhu, Q.-Y. Yuan, W. Wang and Y.-Q. Zhang, Green Chem., 2023, 25, 256–263 RSC.
- T. Biremond, P. Jubault and T. Poisson, ACS Org. Inorg. Au, 2022, 2, 148–152 CrossRef CAS PubMed.
- M. Aelterman, T. Biremond, P. Jubault and T. Poisson, Chem.–Eur. J., 2022, 28, e202202194 CrossRef CAS PubMed.
- H.-Y. Zhou, L.-Q. Fei, J.-L. Zhang, Y.-M. Pan and H.-T. Tang, Adv. Synth. Catal., 2023, 365, 1591–1595 CrossRef CAS.
- Y. Morimasa, K. Kabasawa, T. Ohmura and M. Suginome, Asian J. Org. Chem., 2019, 8, 1092–1096 CrossRef CAS.
- M. Kondo, J. Kanazawa, T. Ichikawa, T. Shimokawa, Y. Nagashima, K. Miyamoto and M. Uchiyama, Angew. Chem., Int. Ed., 2020, 59, 1970–1974 CrossRef CAS.
- F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS.
- (a) J. Zhou, B. Jiang, Y. Fujihira, Z. Zhao, T. Imai and N. Shibata, Nat. Commun., 2021, 12, 3749 CrossRef CAS , For a related defluorosilylation of fluoroarenes or fluoroalkanes with silylboronates, see: ; (b) B. Cui, S. Jia, E. Tokunaga and N. Shibata, Nat. Commun., 2018, 9, 4393 CrossRef PubMed.
- (a) L. Gao, X. Liu, G. Li, S. Chen, J. Cao, G. Wang and S. Li, Org. Lett., 2022, 24, 5698–5703 CrossRef; (b) J. Cao, L. Gao, G. Wang and S. Li, Green Chem., 2024, 26, 4785–4791 RSC.
- H. Lan, X. Huo, Y. Jia and D. Wang, Org. Lett., 2024, 26, 1011–1016 CrossRef.
- X. Wang, X. Zhang, X. He, G. Guo, Q. Huang, F. You, Q. Wang, R. Qu, F. Zhou and Z. Lian, Angew. Chem., Int. Ed., 2024, 63, e202410334 CrossRef.
- S. Ishigaki, Y. Nagashima, D. Yukimori, J. Tanaka, T. Matsumoto, K. Miyamoto, M. Uchiyama and K. Tanaka, Nat. Commun., 2023, 14, 652 CrossRef PubMed.
- (a) S. Chen, X. Guo, H. Hou, S. Geng, Z. Liu, Y. He, X. Xue and Z. Feng, Angew. Chem., Int. Ed., 2023, 62, e202303470 CrossRef , For their related work using silylboronates, see: ; (b) H. Zhang, E. Wang, S. Geng, Z. Liu, Y. He, Q. Peng and Z. Feng, Angew. Chem., Int. Ed., 2021, 60, 10211–10218 CrossRef; (c) S. Wang, M. Sun, H. Zhang, J. Zhang, Y. He and Z. Feng, CCS Chem., 2020, 2, 2164–2173 Search PubMed.
- B. Cui, Y. Tian, Y. Gao, S. Hua, Y. Shi and C. Cao, J. Org. Chem., 2023, 88, 11173–11185 CrossRef PubMed.
- J. Zhou, Z. Zhao and N. Shibata, Nat. Commun., 2023, 14, 1847 CrossRef.
- J. Zhou, Z. Zhao, B. Jiang, K. Yamamoto, Y. Sumiib and N. Shibata, Chem. Sci., 2023, 14, 4248–4256 Search PubMed.
- J. Zhou, Z. Zhao, T. Kiyono, A. Matsuno, J. Escorihuela and N. Shibata, Chem. Sci., 2024, 15, 17418–17424 Search PubMed.
- J. Zhou, Z. Zhao, S. Mori, K. Yamamoto and N. Shibata, Chem. Sci., 2024, 15, 5113–5122 RSC.
- X. Wang, C. Feng, J. Jiang, S. Maeda, K. Kubota and H. Ito, Nat. Commun., 2023, 14, 5561 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Professor Yong Tang on the occasion of his 60th birthday. |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |