
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnveiling the photophysical mechanistic mysteries of tetrazine-functionalized fluorogenic labels†
Tianruo
Shen
 and
Xiaogang
Liu
*
and
Xiaogang
Liu
*
Science, Mathematics and Technology Cluster, Singapore University of Technology and Design, 8 Somapah Road, Singapore 487372, Singapore. E-mail: xiaogang_liu@sutd.edu.sg
First published on 23rd January 2025
Abstract
Tetrazine-based fluorogenic labels are widely utilized in medical and biological studies, exhibiting substantial fluorescence enhancement (FE) following tetrazine degradation through bio-orthogonal reactions. However, the underlying mechanisms driving this fluorogenic response remain only partially resolved, particularly regarding the diminished FE efficiency in the deep-red and near-infrared (NIR) regions. This knowledge gap has impeded efforts to optimize these labels for extended emission wavelengths and improved FE ratios. This review offers a photophysical perspective, discussing the fluorescence quenching pathways (i.e., energy flows and charge separation) that regulate the fluorogenic properties exhibited in various types of tetrazine labels. Moreover, this work examines the emerging role of intramolecular rotations in certain tetrazine-based structures and the integration of additional quencher units. The proposed alternative quenching channel offers the potential to surpass traditional wavelength constraints while achieving improved FE. By examining these photophysical mechanisms, this review aims to advance the understanding of tetrazine-functionalized fluorogenic labels and provide guiding principles for their future design and practical applications.
 Tianruo Shen | Tianruo Shen is a PhD candidate at the Singapore University of Technology and Design (SUTD), under the supervision of Associate Professor Xiaogang Liu. He earned his MEng in Innovation by Design at SUTD in 2021 and continued to pursue his doctoral studies at the same institution, where he applies quantum chemical calculations to facilitate interdisciplinary research. In 2024, he received the “CAS Registry® Innovator” accolade from the Chemical Abstracts Service (CAS) for his contributions to the design of novel chemical molecules. Additionally, for recognizing his outstanding performance in academic excellence and social commitment, he was honored with the “RSC Excellent Student Award” by the Royal Society of Chemistry (RSC) in 2024. |
 Xiaogang Liu | Xiaogang Liu is an Associate Professor at the Singapore University of Technology and Design (SUTD) and a council member of the Singapore National Institute of Chemistry (SNIC). He obtained his PhD in physics from the University of Cambridge in 2014 under the mentorship of Professor Jacqueline M. Cole. Since joining SUTD in 2017, he has led the Fluorescence Research Group, which focuses on developing functionalized dyes by elucidating structure-property relationships. The research of his team seeks to shift dye chemistry from traditional trial-and-error methods to precise molecular engineering. In recognition of his contributions, he received the “Shimadzu-SNIC Industry Award” in Applied and Translational Chemistry from SNIC in 2022 and was named a “Materials Chemistry Frontiers Emerging Investigator” by RSC in 2023. |
1. Introduction
In recent decades, tetrazine-functionalized fluorogenic labels have found widespread utilization in biological and medical contexts.1–5 This popularity arises from their remarkable chemo-selectivity, rapid reaction kinetics, exceptional biocompatibility, and tunable optical properties.6–11 These attributes make them ideal for diverse employments in living systems, including in bioimaging,12–16 drug delivery,17–21 disease diagnosis,22–24 pathophysiological therapy,25–30 and hazardous substance detection.31–33 Notably, bio-orthogonal imaging, leveraging the unique fluorogenicity of these tetrazine labels, stands out among numerous in vivo applications. In their native form, these labels are typically non-fluorescent due to the presence of a tetrazine fragment. However, upon tetrazine degradation via different bio-orthogonal reactions (Fig. S1, ESI†), such as inverse electron-demand Diels–Alder (iEDDA) reactions with dienophiles,34–36 [4 + 1] cycloadditions with isocyanides,37–39 superoxide-responsive triggering,40 and light-activated tetrazine photo-fragmentation,41–43 a significant fluorescence enhancement (FE) can be observed. This unique fluorogenic response effectively avoids the requirement for the tedious washing steps and mitigates the interference from the background emissions, thereby increasing both convenience and the signal-to-noise ratio in bio-orthogonal imaging.44–47 Notably, bio-orthogonal activations with different counterparts result in diverse changes in physical and chemical properties, including reaction kinetics, molar absorption coefficients, ultraviolet-visible (UV-vis) absorption peaks, emission maxima, quantum yields, and FE ratios.48–51 These variations arise from the distinct products generated during the reactions. Consequently, selecting suitable triggers to degrade tetrazine is critical for optimizing FE ratios and improving other key parameters.Despite their advantages, persistent challenges restrict the optimization of high-performance tetrazine-derived labels with enhanced fluorogenicity. A primary issue is the undetermined mechanistic basis for their activable photoluminescence effect, particularly the fluorescence quenching that occurs in the precursors before bio-orthogonal reactions. Additionally, extending fluorescence into the deep-red and near-infrared (NIR) regions while maintaining high FE ratios remains challenging. It is, therefore, necessary to understand the photophysical mechanisms that drive the fluorogenicity of these labels for advancing their versatile bio-orthogonal utilizations. This knowledge will be foundational in developing next-generation labels with superior fluorogenic performance.
This review systematically analyzes over a decade of research on tetrazine-based fluorogenic labels from a distinctive standpoint of photophysics (Scheme 1). Section 2 categorizes these labels according to their linking modes and emission wavelengths, offering a foundation for the detailed analysis. Sections 3 and 4 highlight the photophysical mechanisms governing their fluorogenic properties across the visible and deep-red-to-NIR spectra. In the visible range, fluorescence quenching in precursors is mainly induced by energy flows (Scheme 1a), i.e., energy transfer (EnT) and internal conversion (IC).52,53 As emission wavelengths extend into deep-red and NIR regions, charge separation (Scheme 1b), including photoinduced electron transfer (PET) and photoinduced charge centralization (PCC),54,55 predominates fluorescence quenching. The effectiveness and limitations of these mechanisms are subsequently analyzed by considering various factors affecting these photophysical processes and their fluorogenic behaviors. Section 5 brings focus to two emerging phenomena associated with (dis)aggregation: the “Matthew effect” and torsion-induced disaggregation (TIDA),56,57 providing unique opportunities for label development through the modulation of aggregation–disaggregation processes. Section 6 explores how the intramolecular motions, e.g., twisted intramolecular charge transfer (TICT), introduce additional quenching channels that deactivate the NIR fluorescence of flexible tetrazine labels. Incorporating additional quenchers for NIR dyes to modulate their fluorescence intensities is also proposed as a future direction. These strategies hold the potential to transcend the traditional wavelength dependence of these labels, henceforward broadening their bio-orthogonal imaging applicability in the NIR range.
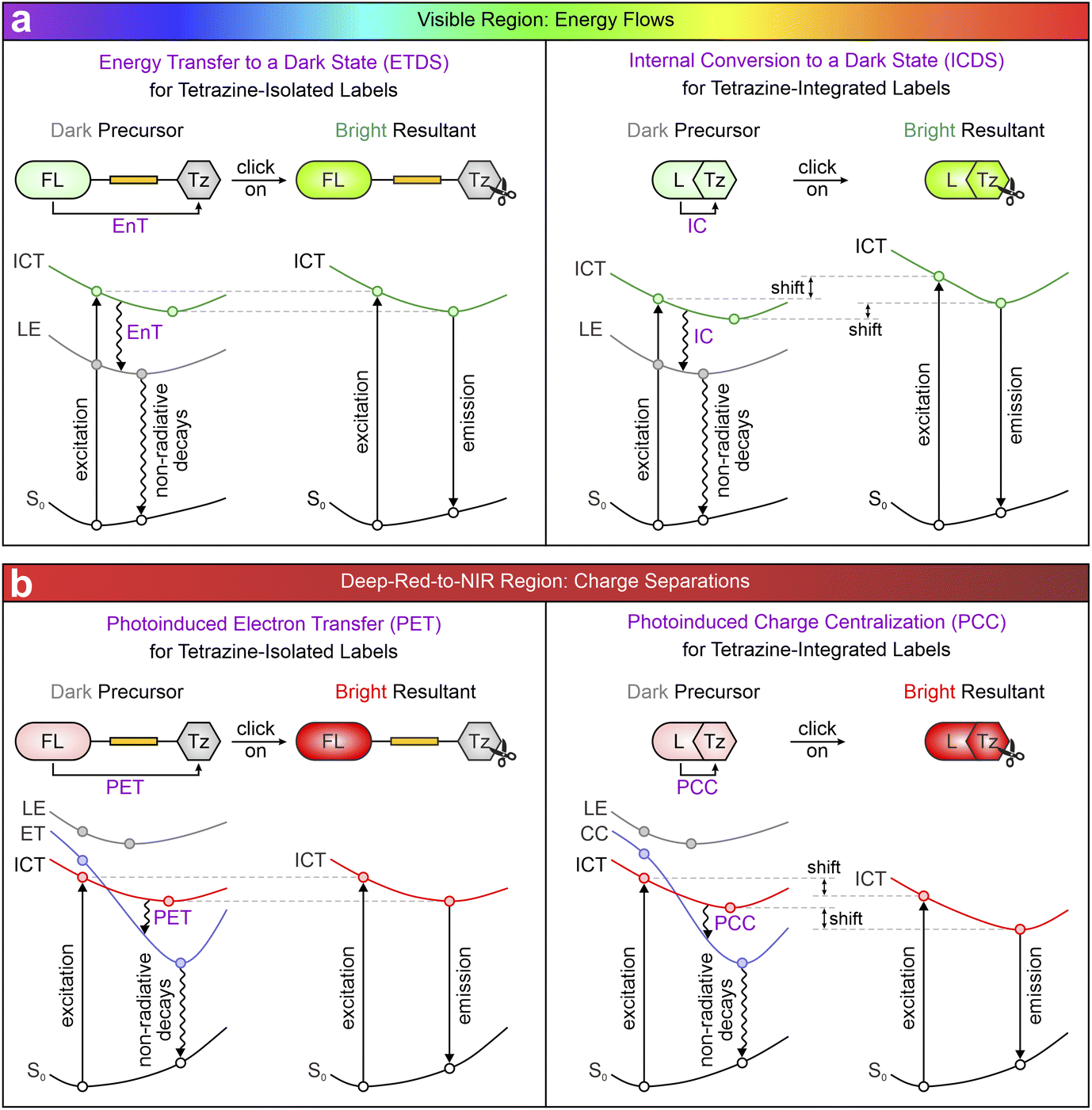 | ||
| Scheme 1 Illustration of four fluorescence working mechanisms in tetrazine labels across (a) visible and (b) deep-red-to-NIR spectral regions. FL – fluorophore; Tz – tetrazine; L – entire label (FL and Tz share a unified π-conjugation network); S0 – ground state; ICT – intramolecular charge transfer; LE – locally excited; EnT – energy transfer; IC – internal conversion; ET – electron transfer; CC – charge centralization; PET – photoinduced electron transfer; PCC – photoinduced charge centralization. | ||
2. Classification of tetrazine labels
Tetrazine labels are generally categorized based on the connection mode between the tetrazine fragment and the main fluorophore, specifically whether the two maintain continuous π-conjugation (Scheme 2). The most prevalent arrangement involves linking the former to the latter through non-conjugated spacers with varying lengths, effectively isolating their conjugation systems (Scheme 2a). The non-conjugated linking bridge spatially separates tetrazine and the main fluorophore, allowing each to retain its electronic properties. Upon tetrazine degradation, the fluorescence of the main fluorophore is greatly restored. For instance, Weissleder and co-workers developed a series of tetrazine-isolated labels by attaching tetrazine fragments to various fluorescent derivatives, including boron-dipyrromethene (BODIPY), coumarin, and fluorescein, via long non-conjugated linkers (Scheme 2a left).58 The bio-orthogonal activation of these labels via trans-cyclooctenol (TCO-OH) effectively restored their fluorescence, yielding emission peaks between 480 and 573 nm and FE ratios from 3.3 to 20.6-fold. In another study, Weissleder et al. reduced the linker length by attaching a tetrazine moiety to a BODIPY derivative via an orthogonal phenyl ring (Scheme 2a middle),59 achieving a 1600-fold FE ratio upon bio-orthogonal reactions with TCO-OH. Besides, Wombacher and co-workers engineered a series of rhodamine–tetrazine labels displaying a “face-to-face” configuration (Scheme 2a right), effectively minimizing the distance between the rhodamine and tetrazine units. This design maximizes the quenching effect and enables efficient fluorescence recovery upon bio-orthogonal reactions.60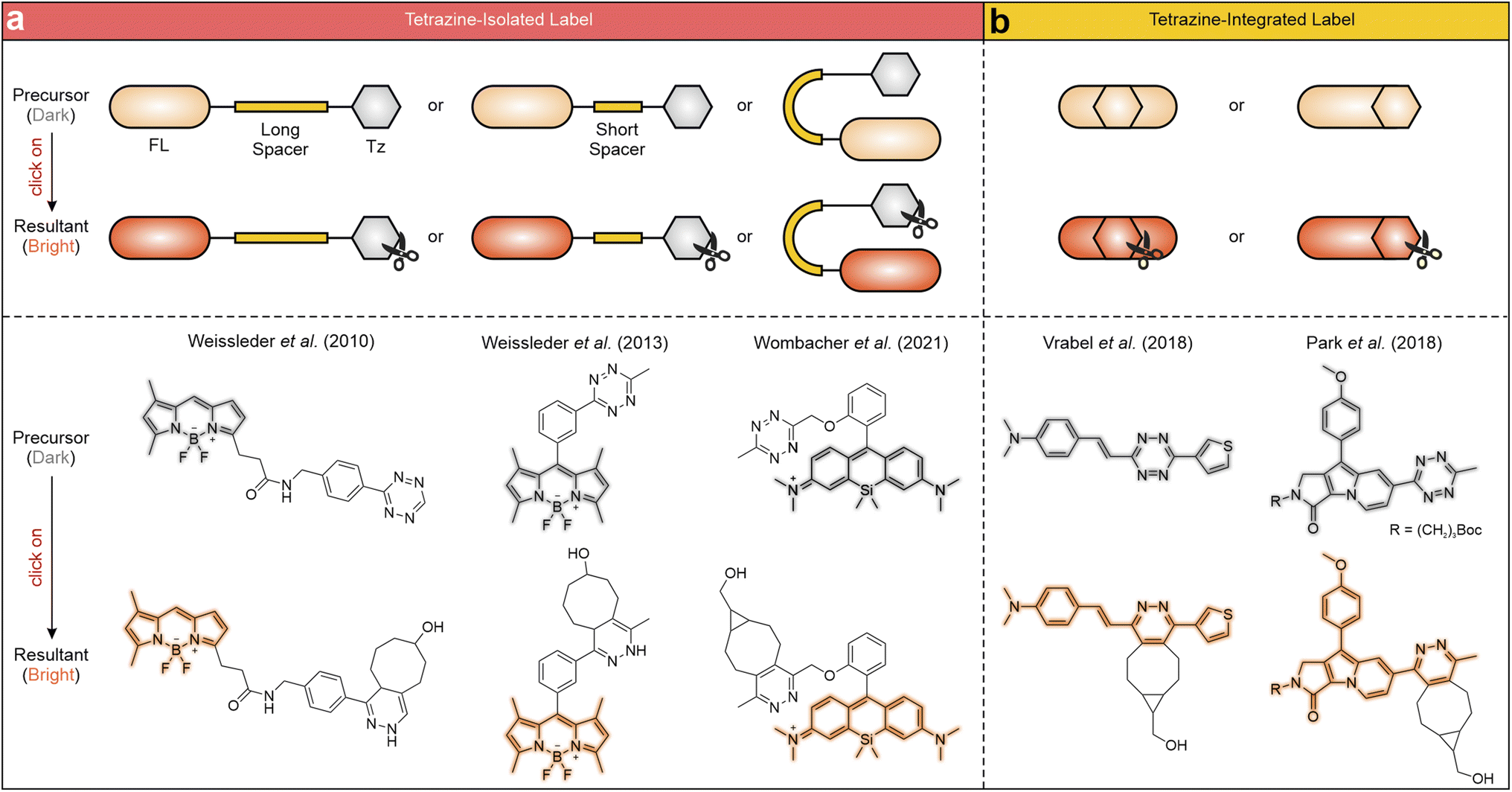 | ||
| Scheme 2 General features (top) and representative structures (bottom) of (a) tetrazine-isolated and (b) tetrazine-integrated labels before and after iEDDA reactions with TCO-OH or bicyclononyne (BCN). FL – fluorophore; Tz – tetrazine. | ||
Alternatively, recent studies have introduced another way to incorporate tetrazine into the main fluorophore, using either a single bond or a conjugated linker (i.e., alternating single and double bonds; Scheme 2b). Besides, tetrazine can also be directly incorporated into the backbone of the fluorophore, allowing it to become a fully integrated component with continuous π-conjugation. Such a molecule exhibits high planarity, characterized by a small dihedral angle between tetrazine and the main fluorophore. This structural arrangement endows seamless fusion of the tetrazine's conjugation with that of the main fluorophore, facilitating their electronic interactions. After the bio-orthogonal reactions, fluorescence is emitted from the entire label, involving the residual tetrazine component. For example, the group of Vrabel grafted different substituents onto the 3-position and 6-position of tetrazine, creating various conjugation-fused tetrazine-integrated labels (Scheme 2b left).61–65 The entire labels emitted multiple color fluorescence upon the iEDDA reactions with different dienophiles, as tetrazine moieties (in precursors) and their residues (in resultants) have non-negligible contributions to the π-conjugations of the labels. Park and co-workers further explored monochromophoric tetrazine-fused labels by directly attaching tetrazine fragments to various SF fluorophores (Scheme 2b right).66
The development of tetrazine-derived fluorogenic labels has progressed into the deep-red and NIR regions, enhancing their suitability, e.g., reduced phototoxicity, minimized autofluorescence interference, increased tissue penetration, and improved biocompatibility, for bio-orthogonal imaging applications. Consequently, the peak emission wavelength (λem) has emerged as an additional criterion for classifying these labels on another scale.
Although these labels generally exhibit strong FE ratios post-bio-orthogonal reactions (with reduced FE in the NIR region), the fluorescence quenching mechanisms in the pre-reacted states vary distinctly. These mechanisms under different categories are detailed and discussed in Sections 3 and 4.
3. Energy flows drive the fluorogenicity in tetrazine labels within the visible spectrum
3.1 Energy transfer to a dark state (ETDS) in tetrazine-isolated labels
It is well established that connecting tetrazine to various fluorescent dyes via different spacers effectively quenches fluorescence. As depicted in Fig. 1, this quenching phenomenon has been described across a diverse range of conventional dye structures, including BODIPY (1-1a, 1-3c, 1-2a–c, 1-8a,b, and 1-5c,d),58,59,67–70 fluorescein (1-1b, 1-3a, 1-6a–d),47,58,67,71 rhodamine (1-3b, 1-5a and 1-11a–c),60,67,72 coumarin (1-4-d and 1-10a–d),73,74 naphthalene (1-9a–d),75 naphthalimide (1-12a–d),76 cyanine (1-13a–b),77 and nitrobenzoxadiazole (NBD; 1-15a,b).69 It also extends to specially synthesized chromophores like Seoul-Fluor (SF; 1-7a–c) and carbon-dipyrromethene (C-DIPY; 1-14a).66,78 Together, these dyes cover a wide range of the visible spectrum, making tetrazine-based quenching applicable to diverse fluorophore families (Fig. 1). | ||
| Fig. 1 Representative molecular structures of tetrazine-isolated fluorogenic labels with visible fluorescence. The numbers in each bracket show the λem (nm) and FE ratios (-fold), respectively. The reaction conditions are listed in Table S1, ESI.† | ||
The low background emissions in precursors are mostly considered quenching by energy transfer (EnT). In a visible tetrazine-isolated label, the main fluorophore serves as an energy donor, whereas the tetrazine functionalizes as an energy acceptor. Energy absorbed by the main fluorophore is transferred to the tetrazine through EnT, dissipating its fluorescence via non-radiative decays. Choi et al. summarized that the EnT processes observed in tetrazine-isolated labels can be classified into three types:79 Förster resonance EnT (FRET),80 Dexter EnT (DET),81 and through-bond EnT (TBET).82 FRET occurs when the excited-state donor (main fluorophore) transfers energy to the ground-state acceptor (tetrazine) via dipole–dipole coupling interactions. This process requires significant overlap between the fluorophore's emission spectrum and tetrazine's absorption spectrum, along with matched transition dipole moments. The FRET efficiency is highly sensitive to the donor–acceptor distance, following an inverse sixth power dependency. It often takes place when the donor–acceptor separation is from 10–100 Å (1–10 nm). In comparison, DET requires direct electronic interactions, which necessitate close spatial proximity and orbital overlap between the fluorophore and tetrazine, typically requiring distances below 10 Å (1 nm). It is worth noting that in FRET/DET-type labels, tetrazine is often linked through a non-conjugated and flexible bridge. The inherent flexibility of these non-conjugated bridges allows the resulting labels to adopt multiple configurations, featuring various distances between the fluorophore and tetrazine. Consequently, the same label could exhibit both FRET and DET quenching processes, depending on the configurations. Conversely, a TBET-type label utilizes a rigid conjugated spacer to connect the main fluorophore to tetrazine with an orthogonal configuration, enhancing the EnT efficiency dramatically.83 Currently, distinguishing TBET from FRET or DET experimentally remains challenging, as TBET likely represents a specialized form of EnT (including both FRET and DET) facilitated by the rigid conjugated bridges.
To streamline the photophysical models for visible tetrazine-isolated labels, Liu and co-workers proposed a unified fluorescence quenching mechanism, i.e., energy transfer to a dark state (ETDS), emphasizing the inherent non-radiative nature of tetrazine. Taking 1-7b′ (a simplified version of 1-7b with a methyl substitution; Fig. 2a) as a representative, they demonstrated how a long linker isolates tetrazine from the main fluorophore, preserving the independence of their conjugated systems.53 This molecule delineates two contrasting states induced by the two independent conjugations. The computational results revealed that during vertical excitation, the main fluorophore, i.e., the SF part, introduces a bright state with a strong oscillator strength (f = 0.427; Fig. 2b left), dominating the light absorption upon photoexcitation. This bright π–π* transition state demonstrates a substantial intramolecular charge transfer (ICT) feature with a significant charge transfer distance (dCT = 1.604 Å; Fig. 2c left). Below this bright state, a locally excited (LE) state (dCT = 0.012 Å) emerges as a dark one (f = 0.005), arising from the tetrazine-centred n–π* transition. Following excited-state geometric relaxation, the energy of this dark state stabilizes at 2.097 eV, lying 1.046 eV below that of the bright one. The negligible overlap in hole–electron distributions between the SF and tetrazine fragments further characterizes 1-7b′ as a dual-conjugation system, with two electronically independent π-systems. Consequently, EnT from the SF fragment (energy donor) to tetrazine (energy acceptor) effectively quenches the fluorescence of 1-7b′ by non-radiative decays, resulting in feeble background emissions. In contrast, the subsequent bio-orthogonal reaction with BCN realizes the transformation from 1-7b′ into 1-7b′-BCN, removing tetrazine's dark state and activating the fluorescence of the SF fluorophore (Fig. 2 right). Due to the negligible involvement of tetrazine in SF's conjugation, 1-7b′ and 1-7b′-BCN share similarities in terms of bright state (de)excitation energies and hole–electron distributions. Therefore, both the peak absorption wavelength (λabs) and λem of 1-7b remain nearly constant regardless of the bio-orthogonal reactions.66 This ETDS mechanism is applicable across various tetrazine-isolated labels, independent of fluorophore types.
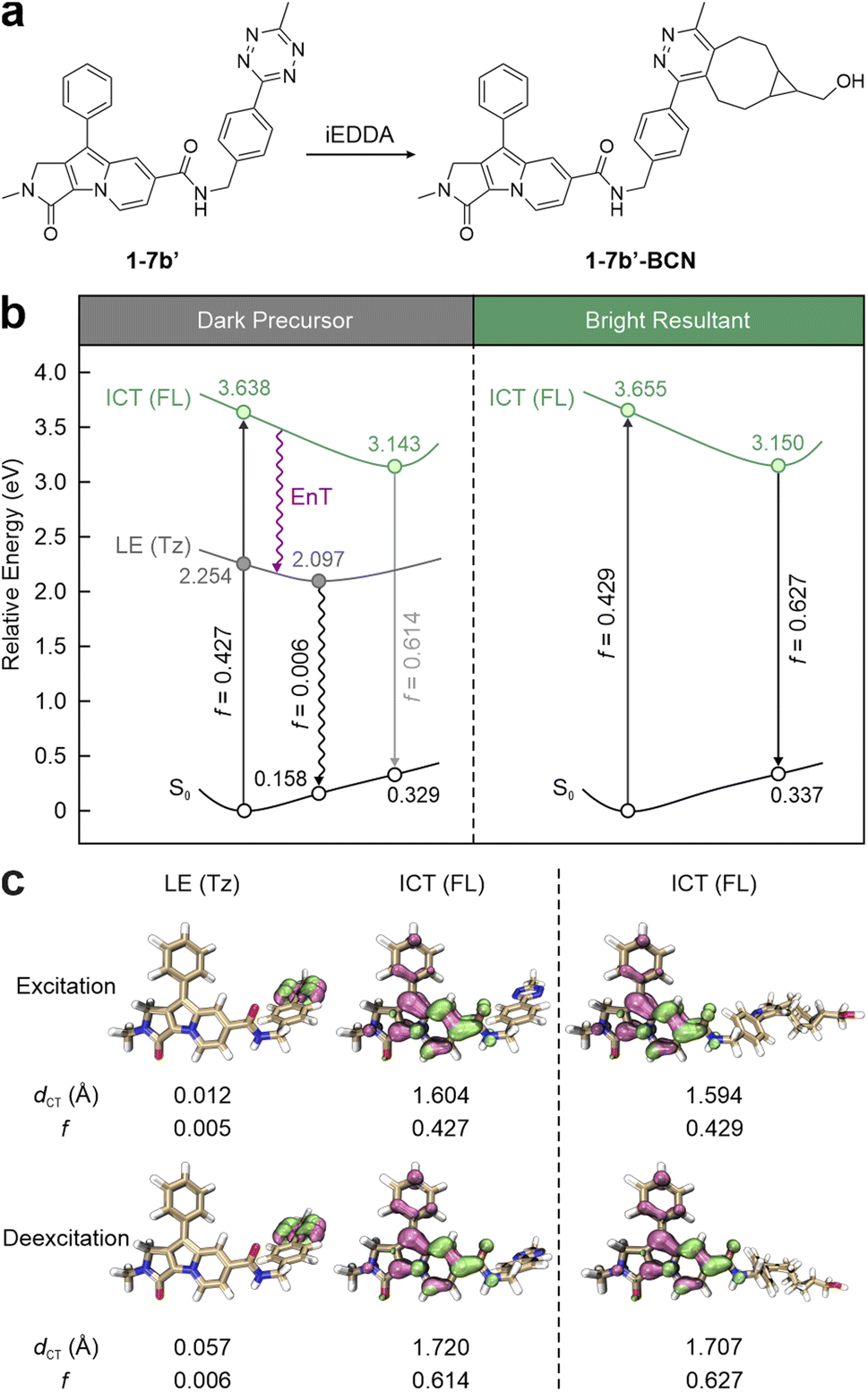 | ||
| Fig. 2 (a) Molecular structures and (b) photophysical processes with (c) the corresponding hole–electron distributions, charge transfer distances (dCT), and oscillator strength (f) values for different states of 1-7b′ (left) and 1-7b′-BCN (right) in ethanol (EtOH). Hole – highlighted in mauve; electron – highlighted in green; FL – fluorophore; Tz – tetrazine; S0 – ground state; LE – locally excited; ICT – intramolecular charge transfer. Reproduced with permission from ref. 53. Copyright 2023, The Royal Society of Chemistry. | ||
To improve the EnT quenching efficiency and achieve a higher FE ratio after bio-orthogonal reactions, regulating the distance between the main fluorophore and tetrazine is essential. Sauer and co-workers proposed using a flexional spacer to induce a stacking conformation, minimizing the fluorophore-tetrazine distance.84 Similarly, Wombacher and co-workers developed rhodamine-based tetrazine labels with a “face-to-face” stacking arrangement between the main fluorophore and tetrazine.60 Employing a short and flexible oxymethyl spacer significantly reduced the donor–acceptor distance from 9.2 Å in 1-11a to 4.2 Å in 1-11b (Fig. 3a), thereby enabling higher EnT efficiency in 1-11b. Following bio-orthogonal activations, this design boosted the FE ratio by approximately 13.8-fold.
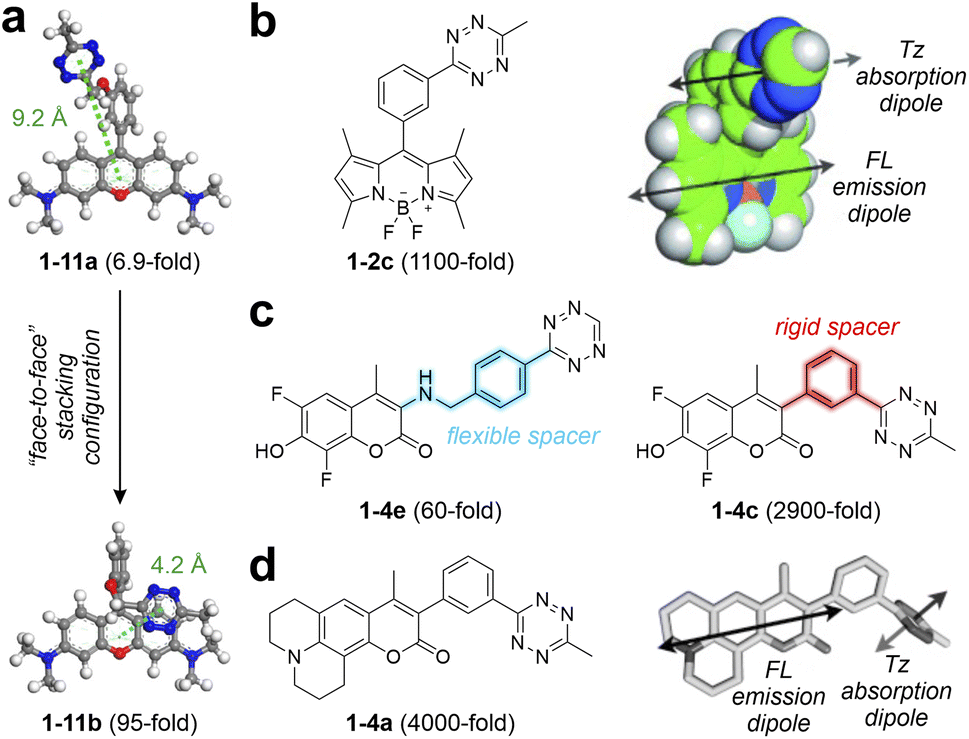 | ||
| Fig. 3 (a) Optimized ground-state geometries of 1-11a (left) and 1-11b (right) with corresponding centroid distances between the main fluorophore and tetrazine in water. (b) Molecular structure (left) and orientation of the FL-Tz transition dipoles (right) of 1-2c. (c) Molecular structures of 1-4e (left) and 1-4c (right). (d) Molecular structure (left) and orientation of the FL-Tz transition dipoles (right) of 1-4a. FL – fluorophore; Tz – tetrazine. Reproduced with permission from ref. 59, 60 and 73. Copyright 2021, American Chemical Society. Copyright 2013, Wiley-VCH. Copyright 2014, Wiley-VCH. | ||
Moreover, Weissleder and co-workers reported a series of BODIPY-derived tetrazine-isolated labels with robust fluorogenic effects by carefully engineering the spacer group.59 By connecting the tetrazine moiety to the BODIPY core through a rigid benzene ring, this design minimizes vibrations and long EnT distances typically associated with flexible linkers. This structural choice not only brings the donor and acceptor closer but also optimizes the orientation of their transition dipoles.
The EnT efficiency is enhanced by the parallel alignment of the BODIPY emission and tetrazine absorption dipoles (Fig. 3b). Unlike the modest 15-fold FE ratio displayed by the previous 1-1a,58 featuring a long flexible linker, these new labels demonstrated exceptional fluorogenic properties, evidenced by the FE ratio reaching up to 1600-fold post-iEDDA reactions.59 The excellent fluorogenic signal underscores the potential of spacer engineering in designing tetrazine-isolated BODIPY labels. Inspired by these findings, Weissleder and colleagues extended this approach to different coumarin-based tetrazine labels.73 Their iEDDA products exhibited a broad emission range from 455 to 502 nm, with ultrahigh FE ratios ranging from 2500 to 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold. For instance, 1-4c and 1-4e share the same core fluorescent skeleton but differ in the spacer (Fig. 3c). The highly rigid spacer in 1-4c enables a remarkable FE ratio (2500-fold) following bio-orthogonal reactions, in stark contrast to only 60-fold in the post-reacted 1-4e (using a flexible spacer). This study also emphasized the importance of aligning fluorophore-tetrazine transition dipoles to enhance fluorescence quenching, as illustrated in Fig. 3d. Further expanding this design strategy, New and co-workers76 applied it to naphthalimide dyes, yielding red-emitting labels with FE ratios up to 200-fold in post-triggered 1-12d. These studies further underscore the broad applicability of short and rigid spacers, essential for the TBET mechanism, in achieving high fluorogenicity across various tetrazine-isolated systems.
000-fold. For instance, 1-4c and 1-4e share the same core fluorescent skeleton but differ in the spacer (Fig. 3c). The highly rigid spacer in 1-4c enables a remarkable FE ratio (2500-fold) following bio-orthogonal reactions, in stark contrast to only 60-fold in the post-reacted 1-4e (using a flexible spacer). This study also emphasized the importance of aligning fluorophore-tetrazine transition dipoles to enhance fluorescence quenching, as illustrated in Fig. 3d. Further expanding this design strategy, New and co-workers76 applied it to naphthalimide dyes, yielding red-emitting labels with FE ratios up to 200-fold in post-triggered 1-12d. These studies further underscore the broad applicability of short and rigid spacers, essential for the TBET mechanism, in achieving high fluorogenicity across various tetrazine-isolated systems.
Besides, the FE ratio is also associated with the different bio-orthogonal activation modes for disabling the tetrazine moiety. For example, Weissleder and co-workers demonstrated that after cycloaddition with TCO-OH through the iEDDA reaction, 1-2b exhibited a 340-fold increment in the FE ratio.59 In contrast, Xiao and colleagues reported a distinct 178-fold FE ratio resulting from the photo-fragmentation of the tetrazine moiety in 1-2b.41 Similarly, 1-4b displayed variable FE ratios when subjected to different bio-orthogonal activations.41,73 These findings highlight the importance of selecting appropriate reaction modes for the bio-orthogonal disintegration of tetrazine to achieve optimal FE ratios.
In designing ETDS-dominated tetrazine labels, reducing the fluorophore-tetrazine distance is essential for efficient EnT, thereby effectively minimizing background noise and enabling substantial FE ratios upon bio-orthogonal reactions. This regulation of donor–acceptor distances stimulated the emergence of next-generation tetrazine labels, specifically tetrazine-integrated designs with “zero” donor–acceptor distance. These innovative labels introduce a distinct quenching pathway that diverges from traditional ETDS mechanisms, representing a significant advancement in fluorogenic labelling technology.
3.2 Internal conversion to a dark state (ICDS) in tetrazine-integrated labels
The labels discussed in Section 3.1 require spacers of varying sizes to attach the tetrazine to different fluorophores, effectively isolating the π-conjugations of the two fragments. While a short EnT distance facilitates fluorescence quenching before and enhancement after bio-orthogonal reactions, a novel approach has emerged to further minimize the distance in traditional tetrazine labels. This method directly incorporates tetrazine into the skeleton of the main fluorophore, creating a fused π-conjugated system with “zero” donor–acceptor distance (Fig. 4). | ||
| Fig. 4 Representative molecular structures of tetrazine-integrated fluorogenic labels with visible fluorescence. The numbers in each bracket show the λem (nm) and FE ratios (-fold), respectively. The reaction conditions are listed in Table S2, ESI.† | ||
Vrabel et al. proposed a design approach to develop tetrazine-merged fluorogenic labels by regulating the substituent groups on both sides of the tetrazine (2-1a–i).61 This method effectively integrated the tetrazine fragments into the conjugated systems of the generated labels.62–65 These multi-coloured labels exhibited strong potential for bio-orthogonal imaging applications. Other groups have reported similar tetrazine-fused fluorophores. For instance, Park et al. introduced the monochromophoric design rule by directly attaching tetrazine fragments to various SF fluorophores (2-2a–d),66 yielding labels with emission ranges from 484 to 581 nm and FE ratios exceeding 600-fold. This fusion strategy also finds its generalizability in various dye families (Fig. 4 and S2, ESI†), covering coumarin (2-3a–d),85 kaleidolizine (KIz; 2-4a–c),86 Huaxi-Fluor (HX; 2-5a,b),87 acridine (2-6a),88 BODIPY (2-7a–e),89 cyanine (2-8a,b),77 cyanine-styryl (2-9a,b),90,91 and 2-(4-(dimethylamino)styryl)-4H-chromen-4-one (DMAC; 2-10a,b).42
Liu and co-workers identified a generalizable mechanism behind the fluorogenicity of tetrazine-integrated labels, termed internal conversion to a dark state (ICDS).53 The simplified 2-2a (2-2a′) was selected as a representative example (Fig. 5a). This example demonstrates how the direct attachment of tetrazine to the SF fluorophore enables significant electronic interactions between the two moieties. Unlike 1-7b′, where the tetrazine is geometrically and electronically isolated from the SF fluorophore, 2-2a′ exhibits a planar conformation, allowing efficient electronic communication across the entire label. The photophysical analysis revealed that the most stable state is still the tetrazine-induced dark LE state (f = 0.005; dCT = 0.051 Å) during the photoexcitation, similar to that of 1-7b′ (Fig. 5 left). Situated 1.020 eV above this dark state is a bright state (f = 0.377; Fig. 5b left), which dominates the light absorbance. Yet, in contrast to the tetrazine-isolated configuration, where the ICT state is merely induced by the SF fragment in 1-7b′, the π–π* transition of 2-2a′ extends across the entire label. This indicates a significant contribution of tetrazine to the bright state transition. The integrated nature of tetrazine and SF in 2-2′ is also reflected by a 0.349 eV reduction in the S2 vertical excitation energy compared to that of 1-7b′, where only the SF moiety participates in the bright state transition. This alteration is also corroborated by the experimental data, i.e., the λabs of 2-2a (415 nm) is longer than that of 1-7b (375 nm) by 40 nm.66 Hence, the integrated π-conjugation renders 2-2a′ as a single-entity system. Following geometric relaxation, the bright state energy is positioned 0.754 eV above the dark one. The energy absorbed by the entire label (involving the SF fragment and tetrazine) transitions to its tetrazine fragment via internal conversion (IC), resulting in the fluorescence quenching of 2-2a′.
 | ||
| Fig. 5 (a) Molecular structures and (b) photophysical processes with (c) corresponding hole–electron distributions, dCT, and f values for different states of 2-2a′ (left) and 2-2a′-BCN (right) in EtOH. Hole – highlighted in mauve; electron – highlighted in green; L – entire label; Tz – tetrazine; S0 – ground state; LE – locally excited; ICT – intramolecular charge transfer; ET – electron transfer; PET – photoinduced electron transfer; CI – conical intersection. Reproduced with permission from ref. 53. Copyright 2023, The Royal Society of Chemistry. | ||
In contrast, the BCN-triggered bio-orthogonal reaction disrupts the dark state associated with tetrazine, unleashing the fluorescence through a new bright π–π* transition state (f = 0.735; Fig. 5b right). It is worth noting that the tetrazine residue in 2-2a′-BCN also has clear contributions to this bright state transition (Fig. 5c right). The bio-orthogonal reactions modify the structure of tetrazine, thus increasing the bright state energy from 3.289 eV in the precursor to 3.780 eV in the post-reacted label. This elevation is corroborated by the hypsochromically shifted λabs from 415 nm in 2-2a to 386 nm in its iEDDA product.66 This spectral variation distinguishes these ICDS labels from ETDS-type tetrazine labels. In ICDS labels, tetrazine residues contribute to bright state transitions, considerably impacting the absorption spectrum post-reaction.
Inspired by the ICDS mechanism and the integrated conjugation characteristic, Liu's group proposed several fluorophore design strategies. In one approach, they introduced an additional coumarin molecule at the right end of 2-3a, positioning the tetrazine centrally within the entire label to extend the π-conjugation network (2-11a; Fig. 6a).53 Following the BCN adduction, this approach leads to bathochromic shifts in both λabs and λem compared to those of 2-3a-BCN, as seen in 2-11a-BCN (Fig. 6b and S3, ESI†). Meanwhile, they modified the π-bridge structure of 2-5b by swapping the positions of the –CH2– unit with the thiophene ring, yielding its constitutional isomer (2-11b; Fig. 6c).53 This alteration promotes quinoidization within the π-bridge, greatly enhancing ICT and reducing the bright state energy.92 As a result, both λabs and λem of 2-11b-BCN are significantly redshifted compared to those of 2-5b-BCN (Fig. 6d and S3, ESI†).
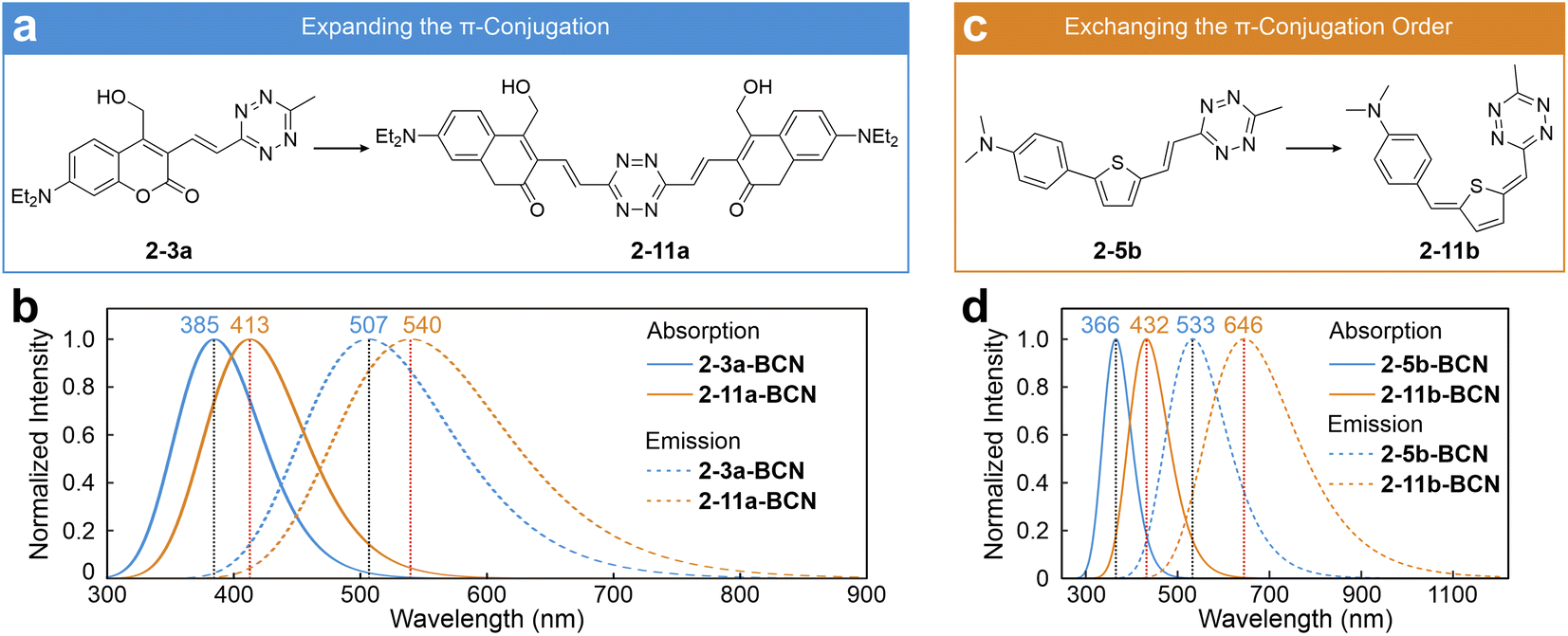 | ||
| Fig. 6 Design strategies of (a) expanding the π-conjugation and (c) exchanging the π-conjugation order to induce redshifted tetrazine dyes. Calculated absorption and emission spectra of (b) 2-3a-BCN/2-11a-BCN and (d) 2-5b-BCN/2-11b-BCN in EtOH. Reproduced with permission from ref. 53. Copyright 2023, The Royal Society of Chemistry. | ||
Although the tetrazine-based fluorogenic labels generally exhibit favorable FE ratios in the visible spectrum, these ratios significantly decrease when their emission wavelengths shift to deep-red and NIR regions.93,94 This reduction is due to the inhibition of the energy flows, either via EnT or IC.54 As discussed in Section 3, both ETDS and ICDS mechanisms rely on a stable dark state, generated by the tetrazine's n–π* transition, as an energy “sink”, with the bright π–π* state of the fluorophore acting as the energy “source”. This energy positioning allows effective fluorescence quenching through sufficient EnT/IC from the main fluorophore (or the entire label) to the tetrazine, resulting in minimal background emissions (Scheme 3 left). Nevertheless, the excitation/deexcitation energy of the bright state drops on bathochromically shifting the λabs/λem of the main fluorophore (or the entire label). Yet, the tetrazine-induced dark LE state remains almost constant. When the bright state approaches the dark one, state crossing during geometric relaxation may invert their positions (Scheme 3 middle). This swap reduces energy flow efficacy and suppresses the fluorescence quenching effect. Furthermore, in a typical NIR dye, the bright state is usually more stable than tetrazine's dark state (Scheme 3 right), making EnT and IC energetically unfavorable and decreasing the fluorescence quenching efficiency. This analysis highlights the intrinsic challenges in developing tetrazine-functionalized labels with high fluorogenicity in the deep-red and NIR regions.
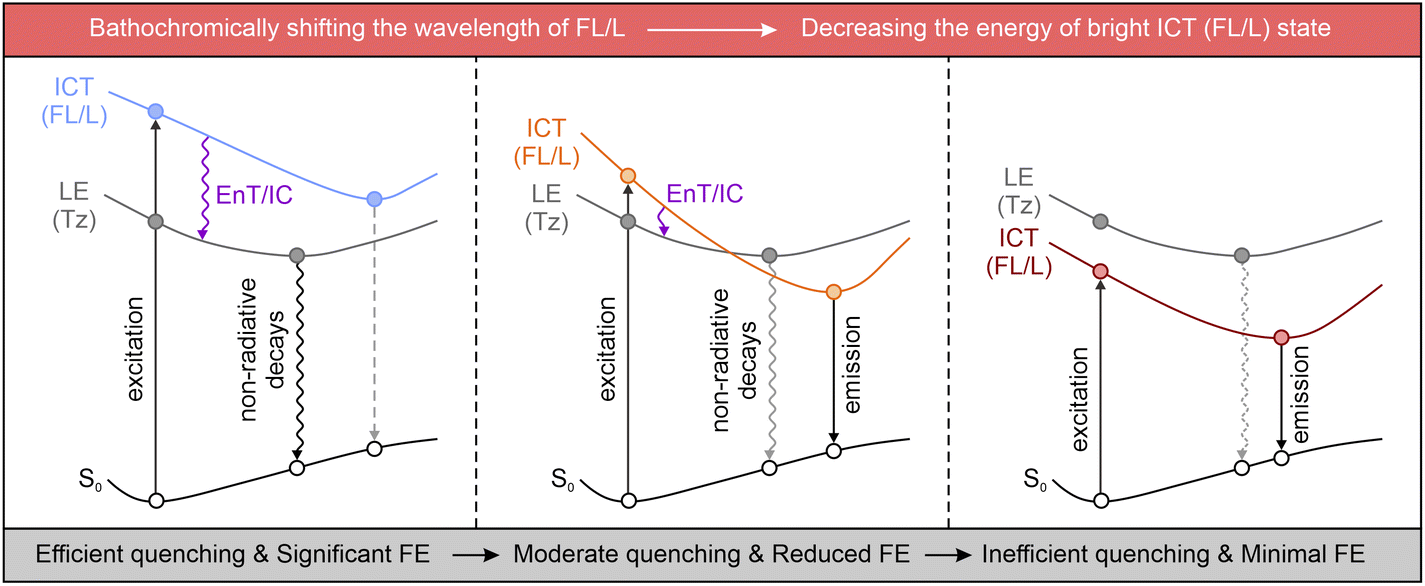 | ||
| Scheme 3 Wavelength/energy level-dependent photophysical processes in tetrazine-based labels. FL – fluorophore; Tz – tetrazine; L – entire label; S0 – ground state; ICT – intramolecular charge transfer; LE – locally excited; EnT – energy transfer; IC – internal conversion. | ||
Nonetheless, several tetrazine-isolated and tetrazine-integrated fluorogenic labels with deep-red to NIR emissions have been developed in recent years, effectively circumventing the energy requirements described above. These intriguing advancements have motivated researchers to expand upon existing mechanisms, further enhancing our understanding of fluorescence quenching processes in long-wavelength tetrazine labels and improving their fluorogenic properties.
4. Charge separation dominates fluorogenicity in deep-red and near-infrared (NIR) tetrazine labels
4.1 Photoinduced electron transfer (PET) in tetrazine-isolated labels
Several studies have reported tetrazine-isolated labels featuring deep-red to NIR emissions following iEDDA reactions (Fig. 7). In 2017, Wu and O'Shea pioneeringly proposed the generation of NIR-fluorescent tetrazine-isolated labels by linking tetrazine to different aza-BODIPY skeletons (3-1a–c).95 These labels displayed moderate FE ratios from 7 to 48-fold with average λem values of around 708 nm. They suggested that photoinduced electron transfer (PET) might contribute to the fluorescence quenching in these precursors. Sauer et al. subsequently reported rhodamine, phenoxazine, and cyanine-based tetrazine-isolated dyes (3-2a–j), showcasing a broad emission range from 637 to 712 nm and discernible FE ratios (2–13-fold).84 Chen and co-workers successfully developed BODIPY-derived tetrazine labels emitting across deep-red to NIR regions and achieving impressive FE ratios between 158 and 647-fold (3-3a–e).96 Xu and colleagues later designed a BODIPY-cyanine tetrazine label (3-4a), displaying a redshifted λem at 733 nm and a remarkable FE ratio (1073-fold).97 Mao et al. carried forward this approach to rhodamine and cyanine families, producing compounds with far-red to NIR emissions up to 806 nm and FE ratios as high as 1459-fold (3-5a–e).94 Yet, the quenching mechanism of these tetrazine-isolated labels in deep-red and NIR spectra remains controversial. The unclear correlation between their fluorogenicity and molecular structures hampers further amelioration.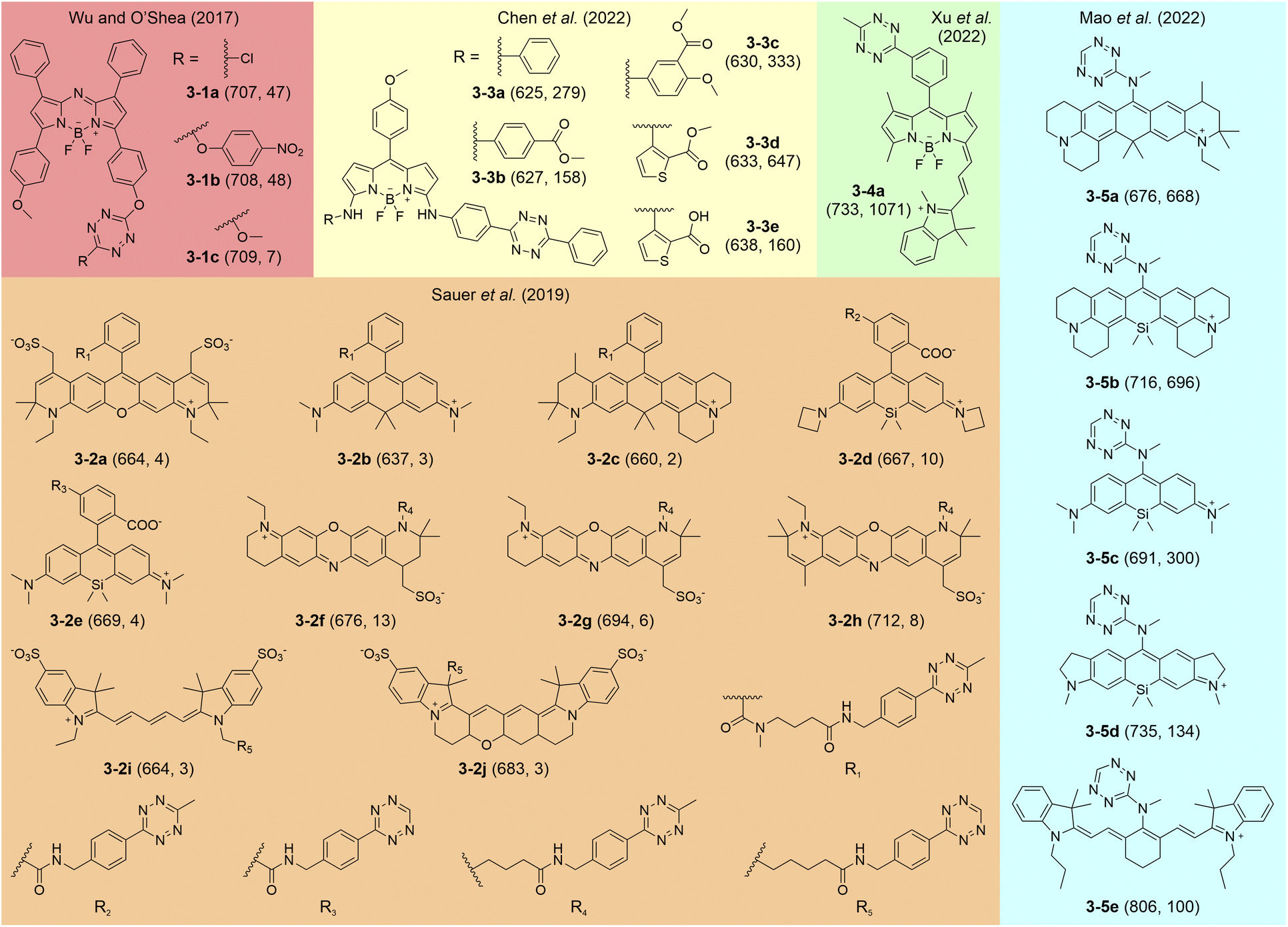 | ||
| Fig. 7 Molecular structures of representative tetrazine-isolated labels with deep-red-NIR emissions. The numbers in each bracket show the λem (nm) and FE ratios (-fold), respectively. The reaction conditions are listed in Table S3, ESI.† | ||
In 2024, Shen and co-workers conducted comprehensive computational investigations on these labels, rationalizing that PET is the dominant mechanism responsible for fluorescence quenching in tetrazine-isolated labels emitting deep-red-to-NIR fluorescence.54 Their findings suggest that even when the ETDS is suppressed, the fluorescence of these precursors can still be quenched by PET. Upon removal of the tetrazine fragments, this quenching channel is blocked, enabling fluorescence recovery in the post-reacted labels.
For instance, in label 3-4a reported by Xu and co-workers (Fig. 8a),97 the tetrazine is installed on the main fluorophore, i.e., a NIR BODIPY-cyanine fragment, via a benzene spacer, forming a dual-entity system (with two separate π-conjugation networks: one for the fluorophore and the other for the tetrazine). During photoexcitation and deexcitation, the tetrazine-induced dark state lies at higher energies than the fluorophore-caused bright state (Fig. 8b left). To be specific, the tetrazine leads to an elevated dark LE state, described by a negligible f value (0.007) and a small dCT (0.038 Å) upon vertical excitation (Fig. 8c left). Conversely, the π–π* transition of the BODIPY-cyanine skeleton generates a bright ICT state (f = 1.148), contributing to light absorption. This bright state is beneath the dark one by 0.258 eV and stabilizes at 2.279 eV following geometric relaxation, prohibiting EnT to the tetrazine. However, another dark state (f = 0) is discovered 0.245 eV below the bright ICT state. In its transition, the hole is delocalized throughout the main fluorophore. In contrast, the electron concentrates on the tetrazine fragment, illustrating a complete charge transfer feature with a large dCT (9.930 Å), indicative of the electron transfer (ET) phenomenon. Hence, BODIPY-cyanine and tetrazine can be viewed as the electron-donor and electron-acceptor, respectively. The PET pathway is activated to quench the fluorescence of the main fluorophore, resulting in 3-4a emitting almost no fluorescence. The subsequent iEDDA reaction with tetrazine eliminates the dark state (Fig. 8 right). Moreover, both excitation and deexcitation/emission energies remain almost the same after the iEDDA reaction, as the tetrazine moiety and residue do not participate in the conjugated system of the BODIPY-cyanine dye.
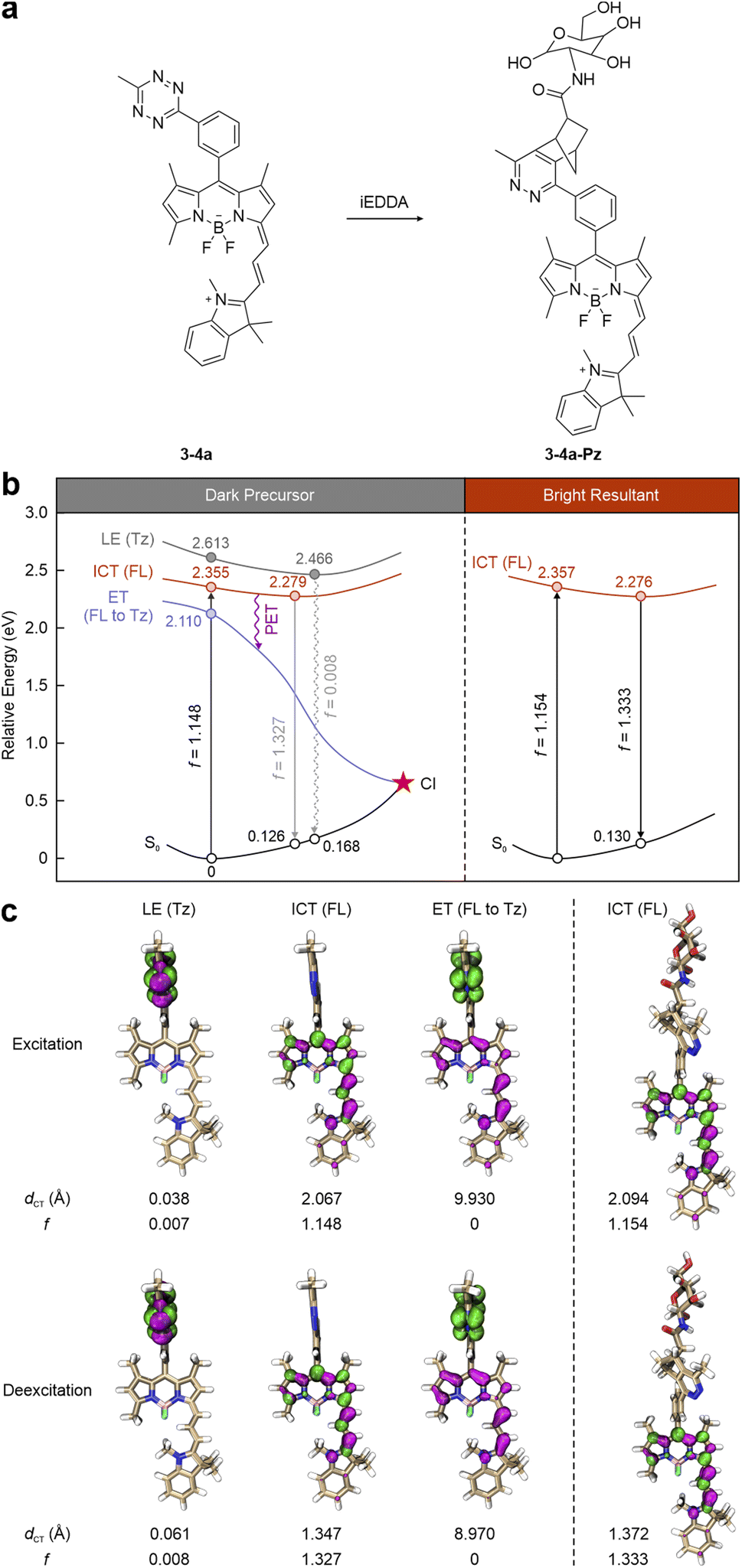 | ||
| Fig. 8 (a) Molecular structures and (b) photophysical processes with (c) corresponding hole–electron distributions, dCT, and f values for different states of 3-4a (left) and 3-4a-Pz (right) in dimethyl sulfoxide (DMSO). Hole – highlighted in pink; electron – highlighted in green; FL – fluorophore; Tz – tetrazine; S0 – ground state; LE – locally excited; ICT – intramolecular charge transfer; ET – electron transfer; PET – photoinduced electron transfer; CI – conical intersection. Reproduced with permission from ref. 54. Copyright 2024, The Royal Society of Chemistry. | ||
It is worth noting that PET has been identified as the leading quenching mechanism in the deep-red-to-NIR tetrazine-isolated labels. However, its efficiency remains restricted in some scenarios. This efficiency, often reflected by the FE ratios upon bio-orthogonal activations, is strongly influenced by the electronic energy difference (ΔEL) between the lowest unoccupied molecular orbitals (LUMOs) of the main fluorophore (acting as an electron-donating group) and tetrazine (serving as an electron-withdrawing group; Fig. 9a top).54 A large ΔEL suggests that the LUMO energy of tetrazine is much higher than that of the main fluorophore, thereby inhibiting the ET and reducing the quenching effect (Fig. 9b top). For example, due to variations in ΔEL, 3-4a and 3-1c bio-orthogonal resultants displayed starkly different FE ratios, i.e., 1079-fold and 7-fold, respectively. With a small ΔEL of 0.171 eV, 3-4a shows more effective PET quenching (Fig. 9a bottom). In comparison, the ΔEL value of 3-1c reaching 0.692 eV (Fig. 9b bottom) results in a less efficient PET, leading to higher background emissions and a lower FE ratio. Besides, shortening the ET distance is also crucial for promoting sufficient PET quenching.
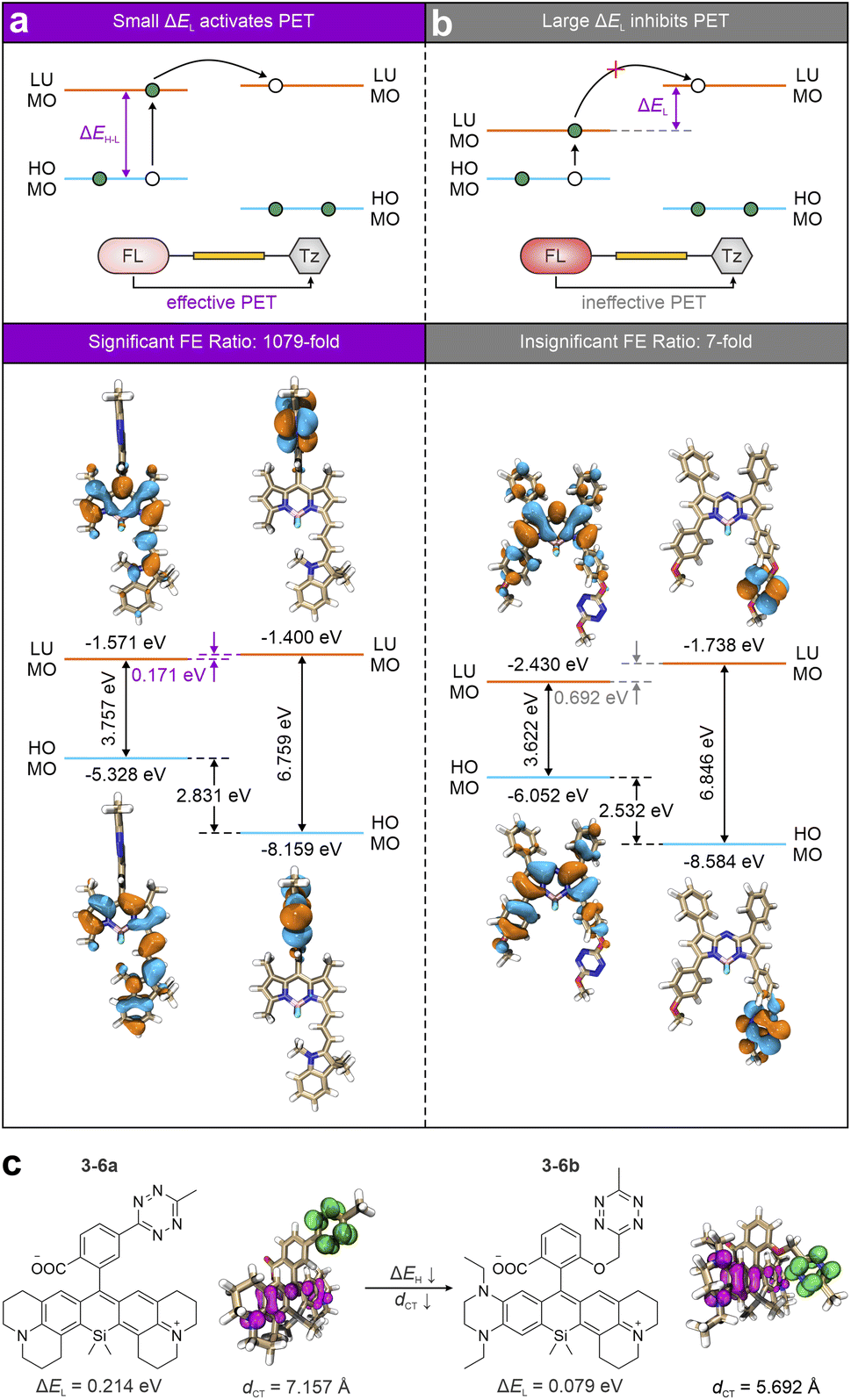 | ||
| Fig. 9 Schematic illustrations of (a) effective and (b) ineffective PET activations based on the frontier molecular orbitals (top) with corresponding representatives (bottom) of deep-red-to-NIR tetrazine-isolated labels. (c) Molecular structures and hole–electron distributions (top) with corresponding ΔEL and dCT (bottom) of the ET states for 3-6a and 3-6b during the photoexcitation processes in DMSO. Hole – highlighted in pink; electron – highlighted in green. Reproduced with permission from ref. 54. Copyright 2024, The Royal Society of Chemistry. | ||
These two factors, i.e., minimal ΔEL and reduced ET distance, are essential for systematically optimizing the PET efficiency in NIR tetrazine-isolated labels. To achieve this, Shen et al. demonstrated a design strategy by structurally modifying silicon–rhodamine (Si–rhodamine; Fig. 9c).54 By introducing dual electron-donating groups to the main skeleton of 3-6a, the gap between the highest occupied molecular orbital (HOMO) and LUMO (ΔEH-L) is reduced to the redshifted λabs and λem in the generated 3-6b. Meanwhile, this modification also lowers ΔEL to 0.079 eV. Additionally, the close stacking configuration between the modified Si–rhodamine and tetrazine reduces the ET distance from 7.157 Å to 5.692 Å. Therefore, this approach, addressing both the energy level alignment and electron transfer distance, shows promise for enhancing the wavelength and FE ratios of tetrazine-isolated labels. Similarly, Mao and co-workers engineered a mono-substituted amino-tetrazine at the meso-position of various rhodamine and cyanine scaffolds to further minimize the ET distance (Fig. 7).94 The resulting molecules (3-5a–e) exhibit substantial turn-on ratios, demonstrating the efficacy of this design strategy.
4.2 Photoinduced charge centralization (PCC) in tetrazine-integrated labels
Recently, Shen et al. unveiled a distinctive charge separation pathway called photoinduced charge centralization (PCC; Fig. 10a).54 This pathway was serendipitously discovered while investigating the photophysical properties of tetrazine-integrated fluorogenic labels in the visible region. Further studies confirmed that the PCC mechanism predominantly quenches deep-red-to-NIR fluorescence in tetrazine-integrated labels with fused π-conjugations (Fig. 10b).55 | ||
| Fig. 10 (a) Comparison between PET and PCC. (b) Molecular structures of 4-1a and 4-1b. The numbers in each bracket show the λem (nm) and FE ratios (-fold), respectively. The reaction conditions are listed in Table S3, ESI.† (c) Photophysical processes with (d) corresponding hole–electron distributions, dCT, and f values for different states of 4-1a before (left) and after (right) the iEDDA reaction in water. Hole – highlighted in pink; electron – highlighted in green; L – entire label; Tz – tetrazine; S0 – ground state; LE – locally excited; ICT – intramolecular charge transfer; ET – electron transfer; PET – photoinduced electron transfer; CI – conical intersection. (e) Molecular structure of 4-2a; (f) demonstration of bio-orthogonal reactions between 4-2a with various counterparts; (g) calculated normalized emission spectra of seven post-reacted 4-2a in water. Reprinted with permission from ref. 55. Copyright 2024, American Chemical Society. | ||
For example, in the NIR tetrazine-merged label 4-1b, reported by the group of Park,89 tetrazine is directly bonded to the main fluorophore, forming a fused π-conjugated system. During vertical excitation, the most stable state is the bright ICT state generated by the π–π* transition spanning the entire label (Fig. 10c and d left). The tetrazine-induced dark LE state sits above the bright state by 0.252 eV during excitation and 0.212 eV during deexcitation, thereby suppressing IC from the entire label to the tetrazine. Yet, after geometric relaxation, a low-energy dark charge centralization (CC) state turns out to be the most stable one at 1.713 eV. In this CC state, the hole is delocalized throughout the entire label, while the electron concentrates on the tetrazine, demonstrating tetrazine's significant role in the charge distribution of both the hole and the electron. Consequently, photoexcitation drives charge transfer from the entire label to concentrate on its internal tetrazine. The dark CC state effectively quenches the fluorescence of the entire label. Like other tetrazine labels, upon activation by BCN, the resulting compound exhibits strong fluorescence due to the inhibition of PCC (Fig. 10c and d right and Fig. S4, ESI†).
Although the PCC process is similar to the well-known PET, it possesses several distinguishing features. PET necessitates the isolation of tetrazine from the main fluorophore, ensuring that their π-conjugations remain independent (Fig. 10a left). Upon photoexcitation, almost complete charge separation occurs from the main fluorophore to the tetrazine, with almost no spatial overlap in the hole–electron distribution of the ET state. In contrast, PCC involves a unified π-conjugation system, where tetrazine is fused with the fluorophore both geometrically and electronically (Fig. 10a right). Upon photoexcitation, charge transfer occurs from the entire label to tetrazine, forming a CC state with considerable overlap between hole and electron distributions. This overlap region typically resides in the tetrazine fragment.
This unique PCC pathway not only enhances the mechanistic understanding of fluorogenicity in long-wavelength tetrazine-fused labels but also enriches photophysics and physical chemistry by introducing a novel photoinduced charge separation process. However, reports on PCC-driven tetrazine-integrated dyes are scarce. The applicability and generalizability of this mechanism in regulating the fluorogenicity of long-emission tetrazine labels remains an area that warrants further experimental investigations.
To this end, building on the PCC mechanism and the conjugation-fused structure, Shen and Liu theoretically designed a tetrazine-integrated label featuring a quinoidal π-conjugation bridge to achieve redshifted wavelengths (Fig. 10e).55 Their simulations modelled interactions between the label and various counterparts (Fig. 10f), and produced seven distinct compounds with varied absorption and emission wavelengths (Fig. 10g and S5, ESI†). These differences stem from the diverse π–π* bright state transitions, affected by the electron-withdrawing strengths of the tetrazine residues. Interestingly, post-reaction labels emit multicolor fluorescence based on their specific triggers, potentially enabling multiplex NIR bio-orthogonal imaging and the simultaneous visualization of various intracellular processes. This approach highlights the versatility of the PCC mechanism and its potential to advance intracellular NIR-imaging technologies.
5. Influence of aggregation/disaggregation on the fluorescence modulation of tetrazine labels
5.1 “Matthew effect”: aggregation-enhanced fluorogenicity in tetrazine labels
In recent years, researchers have explored the attachment of tetrazine to various aggregation-induced emission luminogens (AIEgens), including 2-4a–c as shown in Fig. 4, as well as 5-1a–i, 5-2a–f, and 5-3a–c in Fig. 11. Kim and co-workers proposed that tetrazine-modified AIE labels could serve as effective fluorogenic agents for film-state and in vivo imaging applications.86 These labels were synthesized by conjugating tetrazine at different positions on KIz skeletons (Fig. 4), leading to distinctive behaviors that deviate from conventional click activation responses. For instance, 2-4a does not display a strong FE ratio in tetrahydrofuran (THF) after the iEDDA reaction (Fig. 12a). However, adding water promotes aggregation, producing a blueshifted λem and a 22.5-fold FE increment (Fig. 12a and b). In solid-state applications, tetrazine-AIE labels exhibited substantial FE responses (up to 67.8-fold) following reactions with trans-cyclooctene (TCO) in spin-coated films, further validating their aggregation-induced emission properties. Coincidentally, Tian et al. advanced this area by developing a set of tetrazine-linked tetraphenylethylene (TPE) fluorophores as AIE bio-orthogonal fluorogenic labels (5-1a–j; Fig. 11).98 Upon activation, these labels exhibited tunable emissions ranging from 472 to 597 nm and remarkable FE ratios of up to 2400-fold in aggregate phases. For instance, after undergoing BCN adduction in DMSO, 5-1d displayed minimal fluorescence. However, increasing the water fraction caused a dramatic FE increase, reaching a 1486-fold enhancement (Fig. 12c and d). Meanwhile, they further innovated by designing a “double-quenched” system in which two tetrazine groups were attached to distinct phenyl rings on a TPE fragment (5-1j–i; Fig. 11). In this setup, the destruction of one tetrazine group did not yield significant FE. A strong FE response can be observed only by disrupting both tetrazine groups, demonstrating a sequential activation strategy. Moreover, these BCN adducts showed marked AIE effects, with FE ratios increasing alongside water content. Building on this foundation, Tian and collaborators developed naphthalimide-tetrazine AIEgens,99 expanding the utility of tetrazine-based AIEgens and showcasing the adaptability of this approach across different fluorophores (5-2a–f; Fig. 11, 12e and f). This cumulative work underscores the versatility of tetrazine-modified AIE systems and their potential for bioimaging and other fluorogenic applications. | ||
| Fig. 11 Molecular structures of representative tetrazine labels with aggregation features. The numbers in each bracket show the λem (nm) and FE ratios (-fold), respectively (at the aggregate phases). The reaction conditions are listed in Table S4, ESI.† | ||
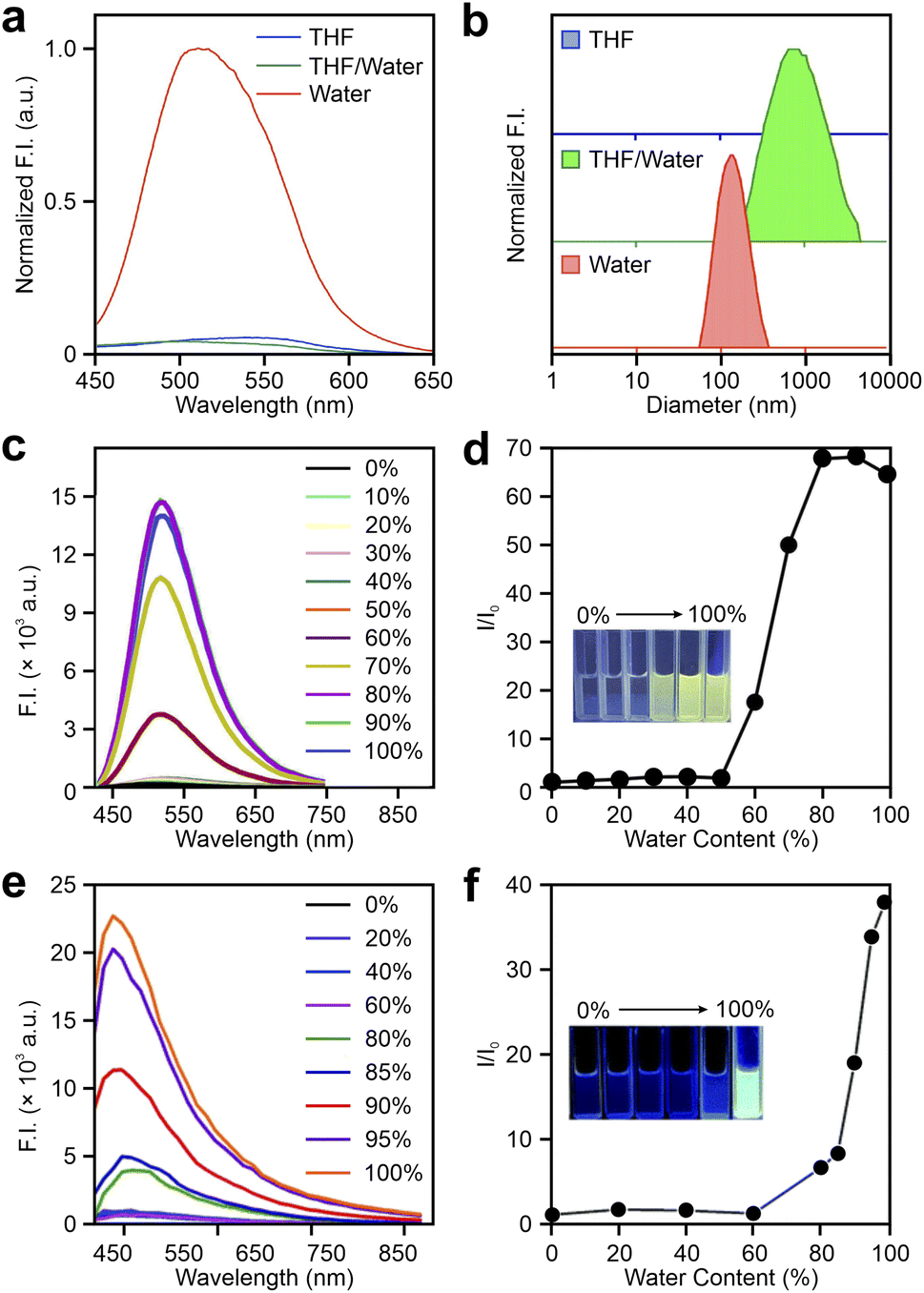 | ||
| Fig. 12 (a) Emission spectra and (b) dynamic light scattering results of 2-4a after reacting with TCO in THF, a THF/water mixture (v/v = 1/1), and water. Emission spectra of (c) 5-1d and (e) 5-2a after reacting with BCN in DMSO–water mixtures with various water contents; the water content-dependence of relative fluorescence intensities of BCN-triggered (d) 5-1d and (f) 5-2a; the inset image shows the physical appearances of BCN-triggered labels in different DMSO–water mixtures under UV light. Reproduced with permission from ref. 86, 98 and 99. Copyright 2021, Elsevier. Copyright 2022, The Royal Society of Chemistry. Copyright 2022, The Royal Society of Chemistry. | ||
To decipher the photophysical mechanism that drives the fluorogenic properties of tetrazine-based labels with aggregation-induced characteristics, Tang and co-workers conducted a detailed mechanistic analysis of these labels (5-3a–c; Fig. 11). Inspired by the “Matthew effect” from the psychosociological theory,100 they adapted this concept to describe the photophysical processes unique to these labels (Scheme 4).56
 | ||
| Scheme 4 Illustration of the “Matthew effect” in tetrazine labels represented by 5-3c and its bio-orthogonal product triggered by BCN in the monomer and aggregate phases. Hole – highlighted in pink; electron – highlighted in cyan; L – entire label; Tz – tetrazine; S0 – ground state; LE – locally excited; ICT – intramolecular charge transfer; TICT – twisted intramolecular charge transfer; IC – internal conversion; ET – electron transfer; PET – photoinduced electron transfer. Reproduced with permission from ref. 56. Copyright 2024, Wiley-VCH. | ||
Before the bio-orthogonal reactions, in the monomer form of 5-3c, as with other tetrazine-integrated labels (Scheme 4a), a tetrazine-generated low-lying dark LE state is observed underneath the label-caused bright ICT state. Additional fluorescence quenching occurs due to the label's structural flexibility, which allows intramolecular rotations that bring the molecule into a twisted configuration, creating a twisted intramolecular charge transfer (TICT) state. Thus, the fluorescence of 5-3c is synergistically quenched by multiple non-radiative decay channels, i.e., IC and TICT, enhancing quenching efficiency prior to the iEDDA reactions. Even when the dye unintentionally binds to biomolecules and restricts intramolecular rotations, energy dissipation through IC can still minimize background emission.
Next, due to low solubility in water, most monomers of 5-3c tend to aggregate prior to iEDDA reactions (Scheme 4b). In these aggregates, pairs of molecules form dimers with complex electronic states, including four dark states below the bright ICT state. The two lowest states, S1 and S2, are well-known LE states associated with n–π* transitions in the tetrazine fragments. Above these states, two additional dark ET states (S3 and S4) emerge due to interactions between neighboring molecules, displaying significantly larger dCT values than S1 and S2. After photoexcitation, fluorescence in aggregated 5-3c is suppressed through both IC within each monomer and PET between neighboring molecules. Thus, multiple dark states in aggregates synergistically enhance quenching, yielding extremely low background emissions. Notably, the dark ET states of S3 and S4 are specific to the aggregated form.
After undergoing iEDDA reactions, most tetrazine labels regain fluorescence as the reaction disrupts the tetrazine-induced dark LE state. Nonetheless, fluorescence recovery is limited in the BCN adduct of 5-3. Despite the elimination of the tetrazine-caused dark LE state, the TICT state and other non-radiative decay channels, persisting due to intramolecular rotations, continue to quench fluorescence in the post-reacted label (Scheme 4c and Fig. S6, ESI†). In contrast, when aggregation occurs in the products, these intramolecular rotations are hindered by restricted free volume and reduced local polarity, which prevents the formation of the non-emissive TICT state (Scheme 4d). This aggregation thus triggers robust fluorescence activation, accompanied by dramatic FE responses.
Therefore, aggregation in tetrazine precursors provides additional quenching pathways via PET on top of tetrazine's intrinsic dark states, effectively reducing background emissions. Following bio-orthogonal reactions, aggregation enhances emission in the resulting fluorophores by inhibiting intramolecular rotations, markedly boosting FE values. The developed labels displayed unique aggregation-enhanced fluorogenic behaviors, i.e., aggregation-caused quenching (ACQ) before bio-orthogonal reactions and AIE afterwards. This strategy advances the design, functionality, and versatility of tetrazine-based labels, paving the way for innovative bio-orthogonal imaging applications.
5.2 Torsion-induced disaggregation (TIDA) in tetrazine-isolated NIR labels
Pu and collaborators discovered a unique torsion-induced disaggregation (TIDA) phenomenon in NIR cyanine-tetrazine-based dyes (Fig. 13).57 In these dyes, tetrazine does not act as a typical energy or electron acceptor. Instead, tetrazine ligation and subsequent bio-orthogonal reactions enhance steric hindrance, promoting disaggregation and utilizing disaggregation-induced-emission to achieve the FE effect. In this configuration, tetrazine is linked to a cyanine fluorophore (CyP7), which exhibits NIR fluorescence and a small Stokes shift through a non-conjugated spacer. Owing to its flat configuration, CyP7 exhibits a pronounced ACQ effect, as evidenced by the diminished peak fluorescence intensity with concentrations exceeding 2 μM (Fig. 13b). However, unlike conventional tetrazine-based dyes, attaching tetrazine to CyP7 does not induce significant fluorescence quenching. This is because ETDS does not apply to NIR dyes, and the ΔEL between tetrazine and CyP7 is too large to activate effective PET quenching. In contrast, tetrazine attachment counteracts the inherent ACQ effect of CyP7. The fluorescence maximum of tetrazine-linked CyP7 (6-1a) remains relatively constant even at higher concentrations (Fig. 13b), though some partial aggregation likely persists. Upon bio-orthogonal reaction with TCO derivatives, the increased steric hindrance further promotes disaggregation, leading to a 2.5-fold FE ratio at a dye concentration of 8 μM.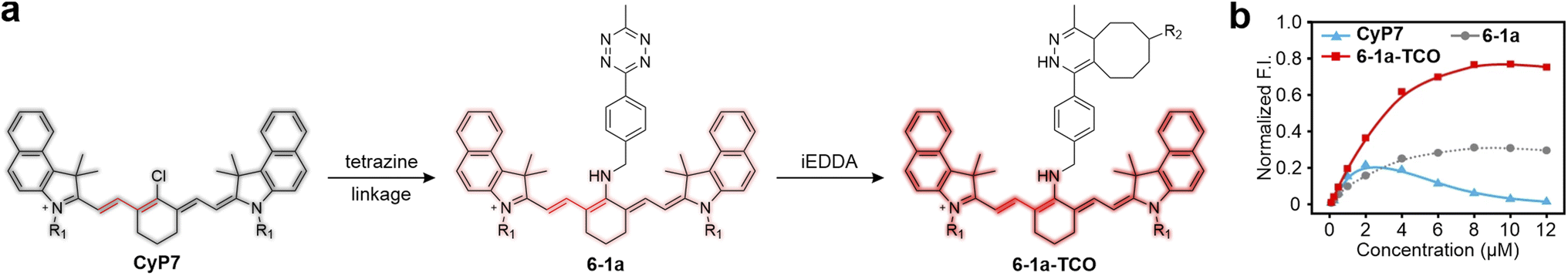 | ||
| Fig. 13 (a) Structural variations associated with the TIDA process. Molecular structures of R1 and R2 are shown in Fig. S7, ESI.† (b) Concentration-dependence of peak F. I. of CyP7, 6-1a, and 6-1a-TCO in methanol. Reproduced with permission from ref. 57. Copyright 2022, Springer Nature. | ||
This remarkable TIDA effect broadens our understanding of tetrazine-based systems, challenging the perception of tetrazine solely as a fluorescence quencher. Although direct experimental data, such as crystal structures, are lacking to confirm cis–trans isomerization in the CyP7 scaffold, it is convincing that bio-orthogonal reactions with tetrazine increase steric hindrance and promote disaggregation. This strategy may offer a pathway to convert many ACQ-prone fluorophores into fluorogenic systems.
6. Future directions
This section further proposes two design strategies to overcome the conventional wavelength limitations of tetrazine labels in the NIR spectrum by introducing additional novel quenching mechanisms to enable effective fluorogenicity. One approach involves linking tetrazine to a flexible NIR fluorophore (Scheme 5a). In the deep-red-to-NIR region, fluorescence quenching through the aforementioned energy flows or charge separation from the main fluorophore to tetrazine is generally restricted. However, rotatable bonds in the main fluorophore can induce a TICT state (or other dark states or conical intersection points), effectively quenching fluorescence in the monomeric phase. Following bio-orthogonal activations, the post-reacted labels would exhibit weak luminescence due to intramolecular rotations (Scheme 5b). As the reactions progress, the post-reacted labels accumulate, forming aggregated complexes (Scheme 5c). This aggregation restricts intramolecular rotations, thus deactivating the TICT quenching pathway and enabling strong fluorescence signals, i.e., a favorable FE effect. Alternatively, bio-orthogonal reactions may anchor the labels to specific cellular locations, such as protein cavities or organelle membranes. Such binding could similarly restrict intramolecular rotations, restoring fluorescence and producing a marked FE effect.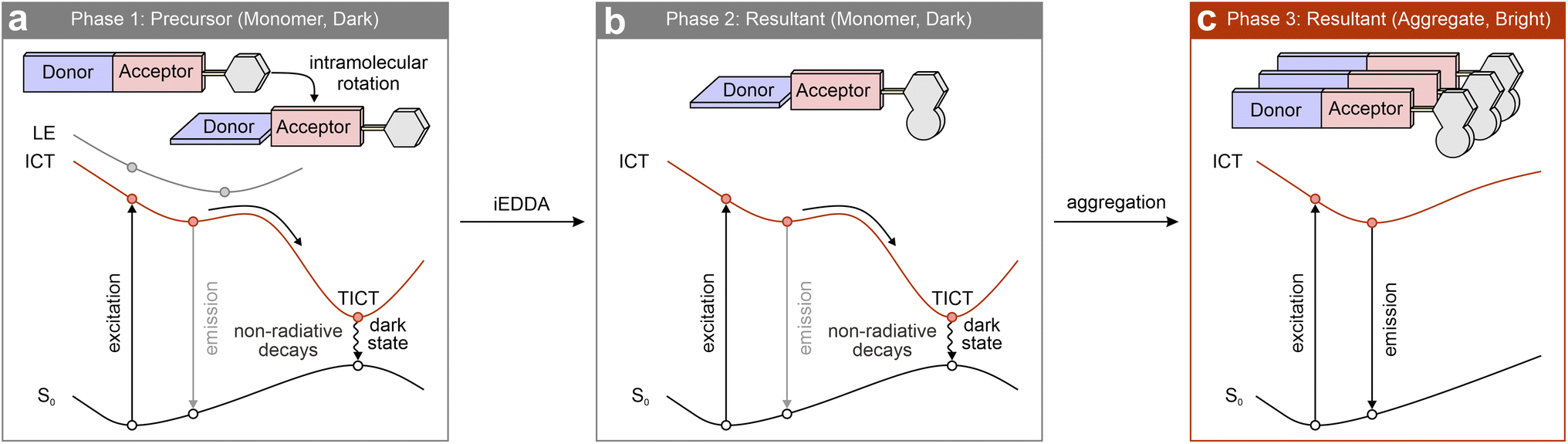 | ||
| Scheme 5 Proposed design strategy and working mechanism. S0 – ground state; ICT – intramolecular charge transfer; LE – locally excited; TICT – twisted intramolecular charge transfer. | ||
An alternative strategy lies in attaching a quencher to a tetrazine-linked NIR fluorophore and designing a chemical reaction that specifically cleaves the quencher from the label.101
Initially, this quencher can substantially reduce the fluorescence of the tetrazine-conjugated fluorophore. Upon cleavage of the quencher, the quenching pathway is interrupted, leading to a marked increment in fluorescence intensity. Simultaneously, the tetrazine group can interact bio-orthogonally with its counterparts to achieve targeted localization. This approach facilitates controlled and reaction-dependent fluorescence activation, making it ideal for applications that demand high specificity in fluorescence signaling.
In both strategies discussed, the tetrazine moiety functions primarily as a targeting agent rather than an energy or electron acceptor governing fluorogenicity. Specifically, the bio-orthogonal reactivity of tetrazine is employed to guide the label to the targeted location, while fluorescence is regulated through modulating intramolecular motions or cleaving the attached quencher. By decoupling the roles of bio-orthogonal targeting and fluorescence regulation, these methods also provide opportunities to integrate alternative quenching mechanisms into tetrazine labels, overcoming the traditional wavelength-dependent limitations in fluorogenicity.
7. Conclusions
Overall, this review comprehensively summarized the photophysical mechanisms of fluorogenicity in tetrazine-based fluorogenic labels, detailing label categorization by using connection modes and wavelengths. Within the visible spectrum, energy transfer (EnT) and internal conversion (IC) dominate quenching in tetrazine-isolated and tetrazine-integrated labels, respectively, while photoinduced charge separation processes, including photoinduced electron transfer (PET) and photoinduced charge centralization (PCC), become central in the deep-red and NIR regions. The review also highlights how molecular aggregation/disaggregation phenomena, such as the “Matthew effect” and torsion-induced disaggregation (TIDA), can modulate fluorogenic properties. Looking ahead, the review suggests advancing the fluorogenic behaviors of tetrazine labels by optimizing intramolecular rotations and integrating alternative quenching mechanisms to achieve NIR fluorogenicity without compromising bio-orthogonal functionality. These photophysical insights aim to inspire research into next-generation tetrazine-functionalized labels with enhanced fluorogenic properties, paving the way for expanded applications in bio-orthogonal contexts.Data availability
No primary research results, software, or code have been included, and no new data were generated or analyzed as part of this review.Author contributions
T. S.: conceptualization, methodology, investigation, formal analysis, visualization, writing – original draft, and writing – review & editing; X. L.: software, project administration, supervision, funding acquisition, resources, data curation, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Singapore University of Technology and Design (SUTD; SKI 2021_04_09). T. S. acknowledges the scholarship support from the Doctor of Philosophy program at SUTD.Notes and references
- N. K. Devaraj and R. Weissleder, Acc. Chem. Res., 2011, 44, 816–827 CrossRef CAS PubMed.
- H. Wu and N. K. Devaraj, Acc. Chem. Res., 2018, 51, 1249–1259 CrossRef CAS PubMed.
- J. Mondal and A. Sivaramakrishna, Top. Curr. Chem., 2022, 380, 34 CrossRef CAS PubMed.
- N. K. Devaraj, R. Weissleder and S. A. Hilderbrand, Bioconjugate Chem., 2008, 19, 2297–2299 CrossRef CAS PubMed.
- R. E. Bird, S. A. Lemmel, X. Yu and Q. A. Zhou, Bioconjugate Chem., 2021, 32, 2457–2479 CrossRef CAS PubMed.
- G. N. Lipunova, E. V. Nosova, G. V. Zyryanov, V. N. Charushin and O. N. Chupakhin, Org. Chem. Front., 2021, 8, 5182–5205 RSC.
- A. Darko, S. Wallace, O. Dmitrenko, M. M. Machovina, R. A. Mehl, J. W. Chin and J. M. Fox, Chem. Sci., 2014, 5, 3770–3776 RSC.
- M. L. Blackman, M. Royzen and J. M. Fox, J. Am. Chem. Soc., 2008, 130, 13518–13519 CrossRef CAS PubMed.
- E. M. Sletten and C. R. Bertozzi, Acc. Chem. Res., 2011, 44, 666–676 CrossRef CAS PubMed.
- J. Šečkutė and N. K. Devaraj, Curr. Opin. Chem. Biol., 2013, 17, 761–767 CrossRef PubMed.
- Y. Chen, H. Jiang, T. Hao, N. Zhang, M. Li, X. Wang, X. Wang, W. Wei and J. Zhao, Chem. Biomed. Imaging, 2023, 1, 590–619 CrossRef CAS PubMed.
- J. Yang, B. Zhu and C. Ran, Chem. Biomed. Imaging, 2023, 1, 434–447 CrossRef CAS PubMed.
- Y. Liao, Y. Liang, Y. Huang, X. Zeng, T. He and J. Yin, Chin. Chem. Lett., 2024, 35, 109092 CrossRef CAS.
- E. Kozma and P. Kele, Top. Curr. Chem., 2024, 382, 7 CrossRef CAS PubMed.
- Kenry and B. Liu, Trends Chem., 2019, 1, 763–778 CrossRef CAS.
- M. C. Giel and Y. Hong, Aggregate, 2023, 4, e336 CrossRef CAS.
- Q. Min and X. Ji, J. Med. Chem., 2023, 66, 16546–16567 CrossRef CAS PubMed.
- Y. Wang, C. Zhang, H. Wu and P. Feng, Molecules, 2020, 25, 5640 CrossRef CAS PubMed.
- R. Pareja Tello, S. Wang, F. Fontana, A. Correia, G. Molinaro, S. Lopez Cerda, S. Hietala, J. Hirvonen, G. Barreto and H. A. Santos, Biomater. Sci., 2023, 11, 4972–4984 RSC.
- X. Ji, Z. Pan, B. Yu, L. K. De La Cruz, Y. Zheng, B. Ke and B. Wang, Chem. Soc. Rev., 2019, 48, 1077–1094 RSC.
- R. M. Versteegen, R. Rossin, W. ten Hoeve, H. M. Janssen and M. S. Robillard, Angew. Chem., Int. Ed., 2013, 52, 14112–14116 CrossRef CAS PubMed.
- L. Zhou, Z. Wang, L. Wang, X. Zhang and Y. Xiao, J. Am. Chem. Soc., 2023, 145, 28296–28306 CrossRef CAS PubMed.
- E. G. Tomarchio, R. Turnaturi, E. Saccullo, V. Patamia, G. Floresta, C. Zagni and A. Rescifina, Bioorg. Chem., 2024, 150, 107573 CrossRef CAS PubMed.
- Z. Li, Q. Chen, J. Wang, X. Pan and W. Lu, Top. Curr. Chem., 2021, 379, 39 CrossRef CAS PubMed.
- X. Xie, B. Li, J. Wang, C. Zhan, Y. Huang, F. Zeng and S. Wu, ACS Appl. Mater. Interfaces, 2019, 11, 41875–41888 CrossRef PubMed.
- T. He, C. Ren, Y. Luo, Q. Wang, J. Li, X. Lin, C. Ye, W. Hu and J. Zhang, Chem. Sci., 2019, 10, 4163–4168 RSC.
- Z. Tan, M. Lin, J. Liu, H. Wu and H. Chao, Dalton Trans., 2024, 53, 12917–12926 RSC.
- J. Huang, Z. Yao, B. Li and Y. Ping, J. Controlled Release, 2023, 361, 270–279 CrossRef CAS PubMed.
- J. Wu, T. Sun, C. Yang, T. Lv, Y. Bi, Y. Xu, Y. Ling, J. Zhao, R. Cong, Y. Zhang, J. Wang, H. Wen, H. Jiang, F. Li and Z. Huang, Biomater. Sci., 2021, 9, 1816–1825 RSC.
- L. K. B. Tam and D. K. P. Ng, Mater. Chem. Front., 2023, 7, 3184–3193 RSC.
- I. Ortiz-Gomez, M. Ortega-Munoz, A. Salinas-Castillo, J. A. Alvarez-Bermejo, M. Ariza-Avidad, I. de Orbe-Paya, F. Santoyo-Gonzalez and L. F. Capitan-Vallvey, Talanta, 2016, 160, 721–728 CrossRef CAS PubMed.
- T. W. Myers, C. J. Snyder, D. E. Chavez, R. J. Scharff and J. M. Veauthier, Chem. - Eur. J., 2016, 22, 10590–10596 CrossRef CAS PubMed.
- Z. Zhao, L. Cao, T. Zhang, R. Hu, S. Wang, S. Li, Y. Li and G. Yang, ChemistrySelect, 2016, 1, 2581–2585 CrossRef CAS.
- A.-C. Knall and C. Slugovc, Chem. Soc. Rev., 2013, 42, 5131–5142 RSC.
- S. Mayer and K. Lang, Synthesis, 2017, 49, 830–848 CAS.
- K. Kang, J. Park and E. Kim, Proteome Sci., 2016, 15, 1–13 CrossRef PubMed.
- J. Tu, M. Xu, S. Parvez, R. T. Peterson and R. M. Franzini, J. Am. Chem. Soc., 2018, 140, 8410–8414 CrossRef CAS PubMed.
- H. Stockmann, A. A. Neves, S. Stairs, K. M. Brindle and F. J. Leeper, Org. Biomol. Chem., 2011, 9, 7303–7305 RSC.
- J. Tu, D. Svatunek, S. Parvez, H. J. Eckvahl, M. Xu, R. T. Peterson, K. N. Houk and R. M. Franzini, Chem. Sci., 2020, 11, 169–179 RSC.
- X. Jiang, M. Li, Y. Wang, C. Wang, Y. Wang, T. Shen, L. Shen, X. Liu, Y. Wang and X. Li, Nat. Commun., 2023, 14, 1401 CrossRef CAS PubMed.
- A. Loredo, J. Tang, L. Wang, K. L. Wu, Z. Peng and H. Xiao, Chem. Sci., 2020, 11, 4410–4415 RSC.
- R. Zhang, Y. Teng, G. Shao, Y. Wang, H. Yang and Y. Tian, Mater. Chem. Front., 2024, 8, 2341–2349 RSC.
- C. Wang, C. Liu, Q. Wei, L. Yang, P. Yang, Y. Li and Y. Cheng, Research, 2020, 6563091 CAS.
- H. Sun, Q. Xue, C. Zhang, H. Wu and P. Feng, Org. Chem. Front., 2022, 9, 481–498 RSC.
- A. Hamsath, O. L. Lederberg, Q. Cui, M. Shieh, Y. Lam, B. J. Brummett, S. Xu, J. R. Robinson and M. Xian, Chem. Sci., 2022, 13, 10336–10341 RSC.
- R. Garcia-Vazquez, U. M. Battisti, J. T. Jorgensen, V. Shalgunov, L. Hvass, D. L. Stares, I. N. Petersen, F. Crestey, A. Loffler, D. Svatunek, J. L. Kristensen, H. Mikula, A. Kjaer and M. M. Herth, Chem. Sci., 2021, 12, 11668–11675 RSC.
- A. Wieczorek, P. Werther, J. Euchner and R. Wombacher, Chem. Sci., 2017, 8, 1506–1510 RSC.
- S. Segawa, X. He and B. Z. Tang, Luminescence, 2024, 39, e4619 CrossRef PubMed.
- X. Chen and Y.-W. Wu, Org. Biomol. Chem., 2016, 14, 5417–5439 Search PubMed.
- H. Son, D. Kim, S. Kim, W. G. Byun and S. B. Park, Angew. Chem., Int. Ed., 2024, e202421982, DOI:10.1002/anie.202421982.
- B. Pinto-Pacheco, W. P. Carbery, S. Khan, D. B. Turner and D. Buccella, Angew. Chem., Int. Ed., 2020, 59, 22140–22149 CrossRef CAS PubMed.
- W. Chi, L. Huang, C. Wang, D. Tan, Z. Xu and X. Liu, Mater. Chem. Front., 2021, 5, 7012–7021 RSC.
- T. Shen, W. Zhang, P. Yadav, X. W. Sun and X. Liu, Mater. Chem. Front., 2023, 7, 1082–1092 RSC.
- T. Shen, X. Li and X. Liu, Mater. Chem. Front., 2024, 8, 2135–2141 RSC.
- T. Shen and X. Liu, Bioconjugate Chem., 2024, 35, 1024–1032 CrossRef CAS PubMed.
- S. Segawa, X. Ou, T. Shen, T. Ryu, Y. Ishii, H. H. Y. Sung, I. D. Williams, R. T. K. Kwok, K. Onda, K. Miyata, X. He, X. Liu and B. Z. Tang, Aggregate, 2024, 5, e499 CrossRef CAS.
- X. Zhang, J. Gao, Y. Tang, J. Yu, S. S. Liew, C. Qiao, Y. Cao, G. Liu, H. Fan, Y. Xia, J. Tian, K. Pu and Z. Wang, Nat. Commun., 2022, 13, 3513 Search PubMed.
- N. K. Devaraj, S. Hilderbrand, R. Upadhyay, R. Mazitschek and R. Weissleder, Angew. Chem., Int. Ed., 2010, 49, 2869–2872 CrossRef CAS PubMed.
- J. C. Carlson, L. G. Meimetis, S. A. Hilderbrand and R. Weissleder, Angew. Chem., Int. Ed., 2013, 52, 6917–6920 CrossRef CAS PubMed.
- P. Werther, K. Yserentant, F. Braun, K. Grussmayer, V. Navikas, M. Yu, Z. Zhang, M. J. Ziegler, C. Mayer, A. J. Gralak, M. Busch, W. Chi, F. Rominger, A. Radenovic, X. Liu, E. A. Lemke, T. Buckup, D. P. Herten and R. Wombacher, ACS Cent. Sci., 2021, 7, 1561–1571 CrossRef CAS PubMed.
- M. Vrabel, P. Kolle, K. M. Brunner, M. J. Gattner, V. Lopez- Carrillo, R. de Vivie-Riedle and T. Carell, Chem. - Eur. J., 2013, 19, 13309–13312 CrossRef CAS PubMed.
- A. Vazquez, R. Dzijak, M. Dracinsky, R. Rampmaier, S. J. Siegl and M. Vrabel, Angew. Chem., Int. Ed., 2017, 56, 1334–1337 CrossRef CAS PubMed.
- S. J. Siegl, A. Vazquez, R. Dzijak, M. Dracinsky, J. Galeta, R. Rampmaier, B. Klepetarova and M. Vrabel, Chem. - Eur. J., 2018, 24, 2426–2432 Search PubMed.
- S. J. Siegl, J. Galeta, R. Dzijak, A. Vázquez, M. Del Río- Villanueva, M. Dračínský and M. Vrabel, ChemBioChem, 2019, 20, 886–890 CrossRef CAS PubMed.
- S. J. Siegl, J. Galeta, R. Dzijak, M. Dracinsky and M. Vrabel, ChemPlusChem, 2019, 84, 493–497 Search PubMed.
- Y. Lee, W. Cho, J. Sung, E. Kim and S. B. Park, J. Am. Chem. Soc., 2018, 140, 974–983 CrossRef CAS PubMed.
- H. Wu, J. Yang, J. Seckute and N. K. Devaraj, Angew. Chem., Int. Ed., 2014, 53, 5805–5809 CrossRef CAS PubMed.
- G. Linden, L. Zhang, F. Pieck, U. Linne, D. Kosenkov, R. Tonner and O. Vazquez, Angew. Chem., Int. Ed., 2019, 58, 12868–12873 CrossRef CAS PubMed.
- M. Işık and M. A. Kisaçam, J. Org. Chem., 2024, 89, 6513–6519 CrossRef PubMed.
- H. Wu, B. T. Cisneros, C. M. Cole and N. K. Devaraj, J. Am. Chem. Soc., 2014, 136, 17942–17945 CrossRef CAS PubMed.
- J. Šečkutė, J. Yang and N. K. Devaraj, Nucleic Acids Res., 2013, 41, e148 CrossRef PubMed.
- P. Werther, J. S. Mohler and R. Wombacher, Chem. - Eur. J., 2017, 23, 18216–18224 CrossRef CAS PubMed.
- L. G. Meimetis, J. C. Carlson, R. J. Giedt, R. H. Kohler and R. Weissleder, Angew. Chem., Int. Ed., 2014, 53, 7531–7534 CrossRef CAS PubMed.
- J. Galeta, R. Dzijak, J. Oboril, M. Dracinsky and M. Vrabel, Chem. - Eur. J., 2020, 26, 9945–9953 CrossRef CAS PubMed.
- D. Kim, J. H. Lee, J. Y. Koo, H. M. Kim and S. B. Park, Bioconjugate Chem., 2020, 31, 1545–1550 CrossRef CAS PubMed.
- M. E. Graziotto, L. D. Adair, A. Kaur, P. Verite, S. R. Ball, M. Sunde, D. Jacquemin and E. J. New, RSC Chem. Biol., 2021, 2, 1491–1498 Search PubMed.
- E. Albitz, K. Nemeth, G. Knorr and P. Kele, Org. Biomol. Chem., 2023, 21, 7358–7366 RSC.
- L. K. B. Tam, P. C. Lo, P. C. K. Cheung and D. K. P. Ng, Chem. - Asian J., 2023, 18, e202300562 CrossRef CAS PubMed.
- S. K. Choi, J. Kim and E. Kim, Molecules, 2021, 26, 1868 CrossRef CAS PubMed.
- T. Forster, Naturwissenschaften, 1946, 33, 166–175 Search PubMed.
- D. L. Dexter, J. Chem. Phys., 1953, 21, 836–850 Search PubMed.
- S. Speiser, Chem. Rev., 1996, 96, 1953–1976 CrossRef CAS PubMed.
- J. Fan, M. Hu, P. Zhan and X. Peng, Chem. Soc. Rev., 2013, 42, 29–43 RSC.
- G. Beliu, A. J. Kurz, A. C. Kuhlemann, L. Behringer-Pliess, M. Meub, N. Wolf, J. Seibel, Z. D. Shi, M. Schnermann, J. B. Grimm, L. D. Lavis, S. Doose and M. Sauer, Commun. Biol., 2019, 2, 261 CrossRef PubMed.
- M. Bojtar, K. Nemeth, F. Domahidy, G. Knorr, A. Verkman, M. Kallay and P. Kele, J. Am. Chem. Soc., 2020, 142, 15164–15171 CrossRef CAS PubMed.
- S.-K. Choi, Y. Lee, S. E. Yoon, H. Choi, J. Kim, J. H. Kim, S. Lee, W. Kim and E. Kim, Sens. Actuators, B, 2021, 340, 129966 Search PubMed.
- W. Mao, J. Tang, L. Dai, X. He, J. Li, L. Cai, P. Liao, R. Jiang, J. Zhou and H. Wu, Angew. Chem., Int. Ed., 2021, 60, 2393–2397 CrossRef CAS PubMed.
- M. O. Loehr and N. W. Luedtke, Angew. Chem., Int. Ed., 2022, 61, e202112931 CrossRef CAS PubMed.
- D. Kim, H. Son and S. B. Park, Angew. Chem., Int. Ed., 2023, 62, e202310665 CrossRef CAS PubMed.
- P. Geng, E. List, F. Ronicke and H. A. Wagenknecht, Chem. - Eur. J., 2023, 29, e202203156 CrossRef CAS PubMed.
- B. Pfeuffer, P. Geng and H. A. Wagenknecht, ChemBioChem, 2024, 25, e202300739 CrossRef CAS PubMed.
- T. Shen, Y. Gao, C. Wang, Z. Xu and X. Liu, J. Phys. Chem. B, 2021, 125, 1447–1452 CrossRef CAS PubMed.
- L. Chen, F. Li, M. Nandi, L. Huang, Z. Chen, J. Wei, W. Chi, X. Liu and J. Yang, Dyes Pigm., 2020, 177, 108313 CrossRef CAS.
- W. Mao, W. Chi, X. He, C. Wang, X. Wang, H. Yang, X. Liu and H. Wu, Angew. Chem., Int. Ed., 2022, 61, e202117386 CrossRef CAS PubMed.
- D. Wu and D. F. O'Shea, Chem. Commun., 2017, 53, 10804–10807 RSC.
- L. Chen, F. Li, Y. Li, J. Yang, Y. Li and B. He, Chem. Commun., 2022, 58, 298–301 RSC.
- Z. He, T. Ishizuka, Y. Hishikawa and Y. Xu, Chem. Commun., 2022, 58, 12479–12482 RSC.
- Y. Teng, R. Zhang, B. Yang, H. Yang, X. Li, D. Yin, X. Feng and Y. Tian, J. Mater. Chem. B, 2022, 10, 8642–8649 RSC.
- Y. Wang, Y. Teng, H. Yang, X. Li, D. Yin and Y. Tian, Chem. Commun., 2022, 58, 949–952 RSC.
- R. K. Merton, Science, 1968, 159, 56–63 CrossRef CAS.
- Y. Wang, D. Torres-García, T. P. Mostert, L. Reinalda and S. I. Van Kasteren, Angew. Chem., Int. Ed., 2024, 63, e202401733 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc07018f |
| This journal is © The Royal Society of Chemistry 2025 |