
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThorium metal–organic framework crystallization for efficient recovery from rare earth element mixtures†
Madeleine A.
Gaidimas
 a,
Courtney S.
Smoljan
b,
Zi-Ming
Ye
a,
Charlotte L.
Stern
a,
Christos D.
Malliakas
a,
Kent O.
Kirlikovali
a and
Omar K.
Farha
*ab
a,
Courtney S.
Smoljan
b,
Zi-Ming
Ye
a,
Charlotte L.
Stern
a,
Christos D.
Malliakas
a,
Kent O.
Kirlikovali
a and
Omar K.
Farha
*ab
aDepartment of Chemistry and International Institute for Nanotechnology, Northwestern University, Evanston, IL 60208, USA. E-mail: o-farha@northwestern.edu
bDepartment of Chemical and Biological Engineering, Northwestern University, Evanston, IL 60208, USA
First published on 31st January 2025
Abstract
Rare earth (RE) elements are critical materials that underpin many modern technologies, particularly in the clean energy industry. Despite their importance, these vital resources are difficult to obtain due to the presence of numerous metals and radioactive contaminants, such as thorium, that are present in RE ores. Current processing methods, which are dominated by homogeneous solvent extraction, are inefficient and produce substantial hazardous waste. In this work, we describe an alternative strategy to separate thorium from REs through metal–organic framework (MOF) crystallization. Starting from a mixture of thorium and rare earth ions in solution, we utilize the simple carboxylate ligand trimesic acid to selectively crystallize a novel thorium MOF, NU-2500, leaving the remaining rare earth ions in solution. By leveraging the increased oxophilicity of Th(IV) compared to RE(III) ions, we observe the exclusive formation of the thermodynamically preferred Th-MOF product. This valence-selective crystallization strategy occurs rapidly (within 30 minutes) at mild temperatures (80 °C) with an environmentally-friendly ethanol/water solvent system to produce phase-pure NU-2500 containing >98% molar fraction of thorium. Sequestering the radioactive Th(IV) ions within a solid framework enables facile separation of REs through simple filtration. We demonstrate that our selective crystallization platform retains its high selectivity for Th crystallization even at low initial Th concentrations and in complex mixtures with multiple different REs. We anticipate that further insights into the kinetics and thermodynamics of MOF crystallization can be applied to additional challenging industrial separations.
Introduction
Rare earth (RE) elements, which include the 15 lanthanides as well as scandium and yttrium, are crucial components in many modern technologies.1–3 Their 4f orbitals endow them with unique electronic, magnetic, and optical properties that make them desirable for a range of applications. For instance, neodymium alloys are among the highest performing permanent magnets available, cerium-based catalysts enable CO oxidation within catalytic converters, and RE phosphors are responsible for producing the colors displayed on phone screens.4–6 Notably, REs are critical for many clean energy technologies including wind turbines,7,8 LED lighting,9 and electric vehicles.10 In most applications, REs cannot be substituted with more readily available transition metal alternatives. Consequently, the demand for REs has been rising in recent years and is expected to continue increasing to fulfill the growing needs of the clean energy industry.11–16Despite their name, REs are abundant within the earth's crust, though they are typically present in low concentrations.2,3 The difficulty in obtaining pure REs originates from the complexity of their processing. RE ores typically contain a mixture of multiple different REs, in addition to numerous transition metal and actinide impurities which must be separated and removed.17,18 Radioactive contaminants such as thorium are of particular concern. For example, monazite, one of the most commonly mined RE minerals, can contain up to 20 wt% ThO2.19 Improper treatment of these ores during RE mining and processing can therefore result in contamination of surrounding land and water sources with hazardous radioactive waste.20–22
RE processing is currently dominated by solvent extraction methods, which rely on the preferential distribution of metals between aqueous and organic phases.23,24 Extractants are commonly organophosphorus compounds such as tributyl phosphate (TBP)25 or di(2-ethylhexyl)phosphoric acid (D2EHPA),26 which chelate one metal and concentrate it in the organic phase, allowing it to be separated from other metals remaining in the aqueous phase (Fig. 1a). However, depending on the selectivity of the extractant, numerous cycles may be required to produce pure REs.22,24 These repeated extraction cycles, which frequently employ hazardous solvents including benzene and kerosene, result in the production of extensive chemical waste.23 As such, there are serious environmental concerns associated with current RE processing techniques, especially when considering the presence of radioactive thorium in RE processing waste streams.27–29 The development of more efficient and environmentally friendly RE purification strategies is therefore critical to ensuring a stable supply of these vital resources.30
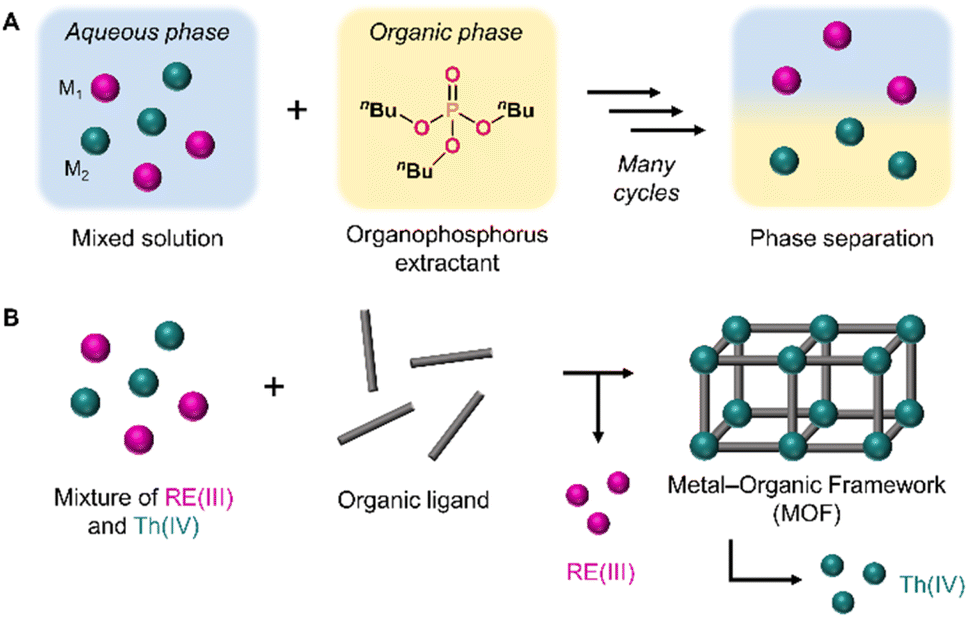 | ||
| Fig. 1 (A) Scheme depicting traditional solvent extraction methods using an organophosphorus extractant to separate metals through two liquid phases. (B) Selective crystallization strategy employed in this work to separate Th(IV) ions from a mixture with RE(III) ions through metal–organic framework synthesis. | ||
With this goal in mind, we aspired to move away from liquid–liquid separation approaches and instead investigated a heterogeneous strategy to increase the efficiency and selectivity of RE purification, in addition to being an environmentally-friendly alternative. We opted to utilize metal–organic frameworks (MOFs), which are multidimensional nanostructures assembled from metal ion or cluster nodes and organic linkers.31 MOFs have a high degree of synthetic tunability, meaning a vast range of coordination modes and framework topologies can be accessed through the judicious selection of appropriate linkers and nodes.32,33 Furthermore, MOFs are crystalline, allowing their structures to be characterized at the atomic level. MOFs have been previously investigated as sorbent materials for the selective capture of ions from solution, including transition metals,34,35 REs,36–38 and actinides.39–41 We instead sought to purify REs through the underexplored strategy of leveraging MOF crystallization itself as a separation method. With this approach, we aimed to selectively crystallize a single-metal MOF product by adding an organic linker to a mixture of metals, leaving the remaining metal ions in solution (Fig. 1b).
Previously reported crystallization-based strategies to purify REs have utilized materials such as polyoxometalates,42 selenites,43 and borates44 to amplify differences in coordination between lanthanides and actinides. Separation strategies involving MOFs specifically have capitalized on variations in metal ion size45–47 or valency48 to favor the formation of a MOF containing a single metal from an initial mixture of metals. The design of our separation platform was inspired by the nuances of the MOF crystallization process, which is often highly sensitive to small deviations in synthetic parameters such as reagent concentration, solvent, temperature, and reaction time.49 Through manipulation of these synthetic conditions, we sought to gain control over crystallization kinetics and thermodynamics for frameworks constructed from different metal ions to exploit their varying formation rates for a crystallization-based separation scheme. We were particularly interested in employing coordination modulators, which are typically monotopic ligands that compete with the linker for binding at the inorganic node, slowing down crystallization.50 Utilizing acidic or basic modulators also results in changing the pH of the reaction mixture, thus influencing the acid–base equilibrium of the linker. Through these mechanisms, modulators can control the framework crystallization and dictate the formation of MOFs with different topologies, particle morphologies, and crystal sizes.51–54 The use of coordination modulators is well-documented in the literature, particularly with zirconium-based MOFs.55 There are several examples of MOF systems where multiple distinct frameworks, or polymorphs, can be formed from identical node and linker starting materials.56–59 By changing the identity of the modulator or tuning other synthetic conditions, MOF crystallization can be finely controlled to isolate various kinetic and thermodynamic products, facilitating the selective synthesis of individual frameworks.60
In the context RE purification, a selective MOF crystallization approach has several advantages over traditional homogenous solvent extraction methods.61 The solid MOF products can be easily separated from the remaining ions in solution through simple filtration, eliminating the need for mixing and settling steps required for liquid–liquid extraction techniques. The potentially higher selectivities of a crystallization-based approach would also reduce the total number of separation cycles necessary to achieve a desired RE purity threshold. This would significantly decrease both the solvent demands and hazardous waste production currently associated with RE purification processes.
To capitalize on these advantages, we developed a selective crystallization strategy to separate thorium from REs by leveraging control of MOF formation. We anticipated that the hard bases of carboxylate-based organic linkers would be ideal to coordinate with thorium(IV) and RE(III) ions, which are hard Lewis acids, allowing for the rapid formation of crystalline MOFs from solution.62 We selected a range of monotopic carboxylic acid modulators commonly used in MOF synthesis to investigate their potential influence on crystallization kinetics that could be leveraged in a separation. Considering the different valencies of Th(IV) and RE(III) ions, we predicted the formation of unique framework structures for each metal under similar synthetic conditions. This would allow us to exploit the potential differences in their framework crystallization kinetics and thermodynamics to favor the formation of a MOF product containing a single metal.
In this work, we investigate a range of framework synthesis conditions to develop a selective crystallization platform to efficiently separate Th from REs. Utilizing the simple carboxylate-based trimesic acid (BTC) linker, we identified distinct framework products from identical synthesis conditions depending on the identity of the metal: [La(BTC)(H2O)6] or a novel Th-MOF, NU-2500. By leveraging insights into crystallization thermodynamics, we demonstrate the exclusive formation of the Th-MOF product from Th/RE mixtures, achieving >98% molar fraction Th in the MOF product from a single pass. Notably, our platform utilizes a eco-friendly ethanol and water solvent system, and selective crystallization occurs rapidly with mild heating. This crystallization strategy enables Th(IV) ions to be removed from solution and safely immobilized within a solid framework, preventing their further release into the environment. By utilizing crystalline MOFs, we can precisely characterize the structure of our separation products through X-ray diffraction methods. More broadly, these structural insights will shed new light on the thermodynamics and kinetics of f-block element crystallization.
Results and discussion
Synthesis of single-metal structures
Before studying MOF crystallization from a mixture of metals, we first investigated framework crystallization with a single metal at a time to identify the different phases that each metal will form under a particular set of synthetic conditions. To promote variation in crystallization kinetics and thermodynamics that could be leveraged in a separation, we aimed to identify a set of synthetic conditions in which unique MOFs could be crystallized depending on the identity of the metal. For an organic linker, we selected the tritopic carboxylate ligand 1,3,5-benzenetricarboxylic acid (BTC), which is inexpensive and frequently used in MOF syntheses. We also selected an environmentally-friendly solvent system consisting of ethanol and water to avoid the use of more hazardous solvents such as N,N-dimethylformamide (DMF). We first selected lanthanum as a representative element to identify MOF products resulting from the crystallization of a trivalent RE.Initially, we intended to take advantage of coordination modulation for a separation strategy. We selected a range of monotopic carboxylic acid modulators: benzoic acid, acetic acid, formic acid, 2-fluorobenzoic acid, and 4-fluorobenzoic acid. We anticipated that some modulators would result in MOF formation, while others would not yield crystalline products. By testing the same modulators with both La(III) and Th(IV), we planned to identify a modulator that could selectively form a MOF with just one of the two metals, which would be promising for a crystallization-based separation of a La and Th mixture. However, following the solvothermal reaction of La(NO3)3·6H2O with BTC linker and each of the aforementioned modulators (see ESI† for complete synthetic details), we observed the formation of only one crystalline product, regardless of which modulator was used (Fig. S1†). Through powder X-ray diffraction (PXRD) measurements, we identified this product as [La(BTC)(H2O)6], a previously reported La framework (Fig. 2a).63 In this framework, each La(III) ion is 9-coordinated by three carboxylate oxygen atoms from three separate BTC linkers and six water molecules, forming a tricapped trigonal prismatic geometry. The structure forms a parallel ribbon motif extending along the a axis, and assembles into a three-dimensional framework through noncovalent interactions (Fig. 2b). These include π–π stacking between the phenyl groups of the BTC linkers, as well as hydrogen bonding between coordinated water molecules and free carboxylate groups. We further corroborated the formation of [La(BTC)(H2O)6] through scanning electron microscopy (SEM) measurements (Fig. S2†), which revealed a sheaf-like particle morphology consistent with previous reports of [La(BTC)(H2O)6].63,64
 | ||
| Fig. 2 (A) Scheme demonstrating crystallization of either [La(BTC)(H2O)6] or NU-2500 from 1,3,5-benzenetricarboxylic acid linker with La(III) or Th(IV), respectively. Schematic representations of 3-D structures and individual metal ion nodes of (B) [La(BTC)(H2O)6] and (C) NU-2500. To account for the occupational disorder of BTC ligands within the NU-2500 crystal, this schematic is simplified to demonstrate average linker occupancy. Color code: La (magenta), Th (teal), C (gray), and O (red). Hydrogen atoms are omitted from the structures for clarity. Experimental PXRD patterns and simulated patterns from single crystal data for (D) [La(BTC)(H2O)6] and (E) NU-2500. | ||
Using the same synthetic conditions, we next investigated the crystallization preferences of thorium with our chosen modulators. Similarly to La crystallization, the effects of modulator identity on the formation of Th-MOFs were limited; all of the modulators resulted in the formation of crystalline products. Through PXRD analysis, we identified the primary product resulting from 2-fluorobenzoic acid modulation as the thorium analogue of MOF-808 (Fig. S3†).65,66 MOF-808 is constructed from hexanuclear metal cluster nodes; the use of fluorinated modulators has been previously observed to enable the crystallization of cluster-based f-block MOFs through the formation of μ3-F bridging groups within the cluster.67,68 For the remaining four modulators, we observed the formation of a novel Th-MOF, NU-2500 (Fig. 2a). SEM imaging revealed that crystalline NU-2500 powder consisted of spherical particles approximately 5 μm in diameter (Fig. S4a†). Despite this unusual morphology, we confirmed the crystallinity of NU-2500 through PXRD measurements (Fig. 2e), indicating particles with a highly ordered internal structure. We hypothesize that the surface morphology results from the rapid growth mechanism of these particles. Accordingly, we observed that increasing the concentration of acetic acid modulator in the synthesis slowed the rate of crystallization and facilitated the growth of larger, faceted particles approximately 25 μm in diameter (Fig. S4b†). Through this modulation strategy, we synthesized diffraction-quality single crystals of NU-2500 (Fig. S5†).
Single-crystal X-ray diffraction (SCXRD) analysis revealed NU-2500 crystallized in the cubic Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group. In NU-2500, the BTC linkers are highly disordered throughout the crystal; the oxygen atoms are fully deprotonated and coordinated with mononuclear Th(IV) ions. Because the rotation of the disordered BTC linker enables two possible orientations for the carboxylate groups to coordinate to adjacent Th ions, the mononuclear Th ion nodes of NU-2500 also show a highly disordered connection to the oxygen of the carboxylate groups. To avoid the short contact of O⋯O on the Th node and linker steric hindrance, the final occupancy of the BTC linker was determined as 0.75, resulting in the densely packed, negatively charged 3D framework [Th2(BTC)3][H3O] with 9-coordinated Th(IV) ion nodes (Table S1†). Based on the synthetic conditions, a [H3O]+ ion is expected to be the counterion for charge-balancing. However, this species is difficult to resolve as its locations can overlap with the missing linkers that are distributed throughout the framework. To provide a clear visualization of NU-2500 without the presence of all possible linker orientations and disorder, we depict one possible arrangement of the BTC linkers within the crystal in Fig. 2c. This model is consistent with the average coordination environment of each Th(IV) ion and the chemical formula of the overall structure, though it is idealized as we expect the linkers to be randomly arranged throughout the framework. Thermogravimetric analysis (TGA) of NU-2500 (Fig. S6†) demonstrates the NU-2500 framework decomposes at approximately 400 °C. To probe the porosity of this material, we performed N2 adsorption isotherm measurements at 77 K on activated NU-2500 powder (Fig. S7†) and observed limited uptake of 35 cm3 g−1 at P/P0 = 0.8. The calculated Brunauer–Emmett–Teller (BET) area is 100 m2 g−1, which is consistent with the low porosity based on the densely packed crystal structure. The DFT pore size distribution shows broad peaks centered at approximately 12 and 24 Å (Fig. S8†), consistent with the disordered internal pore structure of NU-2500 caused by the different potential orientations of the linkers. Despite the disorder present within the framework, the rapid formation of NU-2500 is highly favorable for a precipitation-based separation strategy. Following selective crystallization, the framework can be disassembled to recover the Th(IV) ions for later use.
m space group. In NU-2500, the BTC linkers are highly disordered throughout the crystal; the oxygen atoms are fully deprotonated and coordinated with mononuclear Th(IV) ions. Because the rotation of the disordered BTC linker enables two possible orientations for the carboxylate groups to coordinate to adjacent Th ions, the mononuclear Th ion nodes of NU-2500 also show a highly disordered connection to the oxygen of the carboxylate groups. To avoid the short contact of O⋯O on the Th node and linker steric hindrance, the final occupancy of the BTC linker was determined as 0.75, resulting in the densely packed, negatively charged 3D framework [Th2(BTC)3][H3O] with 9-coordinated Th(IV) ion nodes (Table S1†). Based on the synthetic conditions, a [H3O]+ ion is expected to be the counterion for charge-balancing. However, this species is difficult to resolve as its locations can overlap with the missing linkers that are distributed throughout the framework. To provide a clear visualization of NU-2500 without the presence of all possible linker orientations and disorder, we depict one possible arrangement of the BTC linkers within the crystal in Fig. 2c. This model is consistent with the average coordination environment of each Th(IV) ion and the chemical formula of the overall structure, though it is idealized as we expect the linkers to be randomly arranged throughout the framework. Thermogravimetric analysis (TGA) of NU-2500 (Fig. S6†) demonstrates the NU-2500 framework decomposes at approximately 400 °C. To probe the porosity of this material, we performed N2 adsorption isotherm measurements at 77 K on activated NU-2500 powder (Fig. S7†) and observed limited uptake of 35 cm3 g−1 at P/P0 = 0.8. The calculated Brunauer–Emmett–Teller (BET) area is 100 m2 g−1, which is consistent with the low porosity based on the densely packed crystal structure. The DFT pore size distribution shows broad peaks centered at approximately 12 and 24 Å (Fig. S8†), consistent with the disordered internal pore structure of NU-2500 caused by the different potential orientations of the linkers. Despite the disorder present within the framework, the rapid formation of NU-2500 is highly favorable for a precipitation-based separation strategy. Following selective crystallization, the framework can be disassembled to recover the Th(IV) ions for later use.
Contrary to what is commonly reported with d-block metals such as zirconium, we did not observe variation in framework structure resulting from coordination modulation in our synthetic screening with La(III) and Th(IV). Moving beyond our initial hypothesis of leveraging coordination modulation, we decided to target metal separations based on different crystallization thermodynamics and kinetics. We noted that both [La(BTC)(H2O)6] and NU-2500 could be synthesized rapidly under mild reaction conditions: within 30 minutes of heating at 80 °C, white powder precipitated from solution for both the La and Th syntheses. PXRD measurements demonstrated the high crystallinity of these materials and their agreement with simulated patterns generated from the crystal structures of [La(BTC)(H2O)6] and NU-2500 (Fig. S9 and S10†). While both individual frameworks can form rapidly, we predicted that the crystallization of NU-2500 would be thermodynamically preferred from a mixture of Th(IV) and La(III), due to the increased oxophilicity of Th(IV) compared to trivalent REs. This preferential crystallization of NU-2500 could then be used to efficiently separate Th from the initial mixed metal solution.
MOF crystallization from a binary metal mixture
We proceeded by replicating the synthesis conditions described above for individual metals, but instead employing an equimolar mixture of La(III) and Th(IV) (Fig. 3a). With both metals in solution, multiple framework products are potentially accessible: NU-2500, [La(BTC)(H2O)6], a mixture of both individual MOFs, or a mixed phase in which both La and Th metals are incorporated into a single lattice. Following solvothermal synthesis, we observed the rapid precipitation of a white powder from solution. By PXRD, we saw that the resulting precipitate consisted of a single phase for all modulators studied, with the exception of 2-fluorobenzoic acid. This phase is consistent with the simulated powder pattern of NU-2500 (Fig. 3b). For 2-fluorobenzoic acid, we note the formation of a mixture of NU-2500 and Th-MOF-808, as observed with the Th-only synthesis. In all cases, we did not observe PXRD peaks corresponding to [La(BTC)(H2O)6], indicating that crystallization of the Th phase is preferred over La under these synthetic conditions.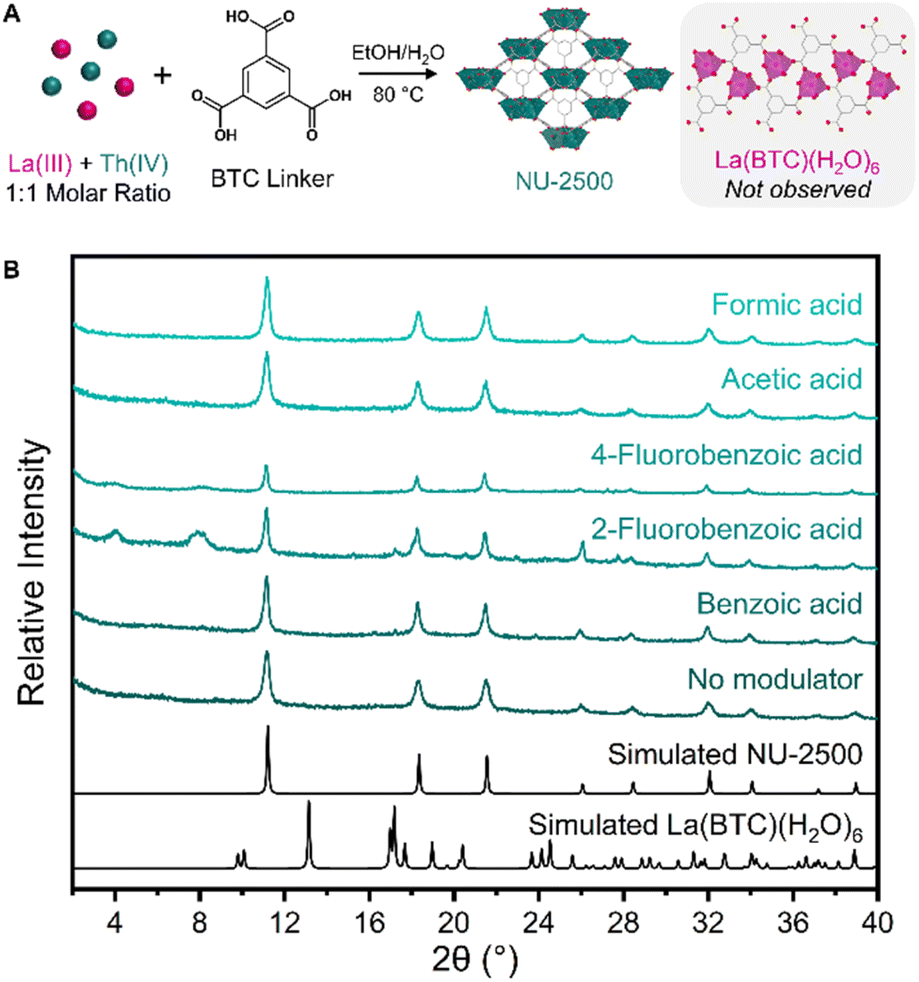 | ||
| Fig. 3 (A) Selective crystallization of Th-MOF product, NU-2500, from an initial equimolar mixture of La(III) and Th(IV) ions. (B) PXRD patterns of crystalline products resulting from syntheses with equimolar La + Th, BTC linker, and specified modulators. We note the formation of a mixture of NU-2500 and Th-MOF-808 when 2-fluorobenzoic acid is used as the modulator, though we do not observe the formation of La(BTC)(H2O)6 for any of the modulators studied. Simulated patterns of potential products were generated from single crystal structures.63 | ||
While we could not detect the presence of [La(BTC)(H2O)6] in our bulk products by PXRD, we next attempted to determine whether La(III) ions could potentially be co-crystallizing with Th(IV) into the NU-2500 lattice. To this end, we turned to inductively coupled plasma-optical emission spectroscopy (ICP-OES) to precisely quantify the metal contents of the crystalline products. Following synthesis, the crystalline powders were isolated by centrifugation, washed, and microwave-digested before ICP-OES analysis to determine the relative concentrations of La and Th (see ESI† for complete sample preparation details). We found that all crystalline products were highly enriched in Th (Table S2†), supporting our finding of exclusive crystallization of the NU-2500 phase by PXRD, regardless of modulator identity (Fig. 3b). However, we noted differences in the degree of separation of Th and La present in the crystalline products across the different modulators studied. The aromatic modulators studied here (benzoic acid, 2-fluorobenzoic acid, and 4-fluorobenzoic acid) all yielded crystalline products with comparable Th![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :La molar ratios (22:1, 23:1, and 18:1, respectively). In comparison, selectivity for Th crystallization was increased with the smaller organic acid modulators; with a Th:La molar ratio of 39:1 for formic acid and 45:1 for acetic acid (Table S2†). Given the density and limited porosity of the framework, we hypothesize that the relatively larger modulators may create additional steric hindrance around the Th(IV) ions while the NU-2500 crystallites are forming, limiting the “error correction” process of reversible linker coordination during framework crystallization. However, the smaller formic and acetic acids can modulate the rate of crystallization without obstructing linker binding, facilitating the reversible correction process and minimizing erroneous incorporation of La sites within the framework.
:La molar ratios (22:1, 23:1, and 18:1, respectively). In comparison, selectivity for Th crystallization was increased with the smaller organic acid modulators; with a Th:La molar ratio of 39:1 for formic acid and 45:1 for acetic acid (Table S2†). Given the density and limited porosity of the framework, we hypothesize that the relatively larger modulators may create additional steric hindrance around the Th(IV) ions while the NU-2500 crystallites are forming, limiting the “error correction” process of reversible linker coordination during framework crystallization. However, the smaller formic and acetic acids can modulate the rate of crystallization without obstructing linker binding, facilitating the reversible correction process and minimizing erroneous incorporation of La sites within the framework.
To probe the effect of modulator quantity on Th selectivity, we repeated our synthesis trial with an equimolar La(III) and Th(IV) mixture, BTC linker, and five concentrations of acetic acid ranging from 1–10 mmol. At all concentrations, the PXRD patterns of the crystalline products were consistent with NU-2500 (Fig. S11†). As the acetic acid concentration is increased from 1–5 mmol, the products are increasingly enriched in Th, with the greatest Th:La molar ratio of 71:1 resulting from 5 mmol of acetic acid (Table S3†). Increasing acetic acid further beyond this concentration did not result in increased selectivity for Th in the products, and we also observed slower crystallization rates and decreased product formation. Therefore, we opted to continue our crystallization trials with 5 mmol of acetic acid to target the greatest Th selectivity in the product frameworks.
We also tested the effect of varying the relative concentrations of metal and linker present in the synthesis. Controlling the stoichiometry of Th and BTC is necessary to ensure the crystallization is both efficient and selective. Without enough linker present to ensure complete crystallization of the NU-2500 product based on the Th ions available, there would be leftover Th(IV) ions remaining in solution. Conversely, we sought to test if increasing the relative concentration of the linker would result in improved separation efficiency. We repeated our binary crystallization trial with an equimolar La(III) and Th(IV) mixture (0.065 mmol each), and increased quantities of BTC (0.13 and 0.26 mmol, representing twice and four times the concentration of each individual metal, respectively). We observed rapid crystallization and confirmed the formation of NU-2500 by PXRD (Fig. S12†). However, ICP-OES analysis of the crystalline products showed that as the relative concentration of BTC increases, the Th:La selectivity decreases. Compared to a product Th:La ratio of 71:1 from an initial equimolar Th:BTC mixture, we observed Th:La ratios of 63:1 and 24:1 resulting from trials with BTC quantities of 0.13 and 0.26 mmol, respectively (Table S4†). While we did not detect any bulk phases other than NU-2500 in the PXRD patterns, we hypothesize that the increased BTC presence may be providing additional opportunities for La to incorporate within the NU-2500 framework. For instance, once the Th(IV) ions have been consumed in the crystallization, remaining open carboxylate groups on excess BTC ligands at the periphery of the crystallites could coordinate La(III) ions, thus decreasing the Th:La selectivity of our crystallization scheme. To target the most effective separation of Th(IV), we did not further pursue increased BTC concentrations.
Kinetics of NU-2500 crystallization from a La/Th mixture
Next, we investigated the selectivity of Th crystallization from a binary mixture over time. We set up a series of identical crystallization trials with an equimolar initial mixture of La(III) and Th(IV), BTC linker, and 5 mmol acetic acid modulator. Samples were placed in an 80 °C oven and removed from heat after specified time intervals to stop the crystallization process. Within 30 minutes of heating, we observed the formation of white powder, indicating rapid framework crystallization. We sampled the solids from each timepoint and analyzed them with PXRD (Fig. S13†). At each time interval between 30 minutes and 48 hours, we observe PXRD patterns consistent with the exclusive formation of crystalline NU-2500. Preferential crystallization of the Th framework is in line with our prediction of the more favorable thermodynamics of Th(IV) crystallization compared to La(III). Interestingly, at no point throughout the reaction do we observe the formation of [La(BTC)(H2O)6], meaning it is not necessary to stop the crystallization reaction at a specific time interval to capture the synthesis of phase-pure NU-2500. Instead, La framework crystallization is effectively prevented in favor of the formation of the thermodynamically preferred NU-2500 product.As a control experiment, we soaked a sample of [La(BTC)(H2O)6] in an aqueous solution of Th(NO3)4·4H2O at 80 °C. After washing, the PXRD pattern remained consistent with [La(BTC)(H2O)6] (Fig. S14†) and ICP-OES analysis demonstrated that there was no significant exchange of Th(IV) ions into the framework, which contained a La molar fraction of 97% (Table S5†). This indicates that the exclusive synthesis of NU-2500 is due to preferential crystallization kinetics, not a structural transformation from [La(BTC)(H2O)6].
ICP-OES analysis of samples from each time interval confirms exclusive crystallization of the Th framework from an equimolar La(III) and Th(IV) mixture, as all of the crystalline solids recovered from solvothermal synthesis are highly enriched in Th (Fig. 4a and Table S6†). After 30 minutes of heating, the recovered solid products have an average molar fraction of Th greater than 98%, corresponding to a Th:La molar ratio of 78:1 (Table S6†). This high selectivity for Th(IV) crystallization is maintained over the course of 48 hours, indicating that co-crystallization of La(III) into the NU-2500 lattice does not occur with additional reaction time. This demonstrates that our crystallization strategy is highly effective in producing phase-pure Th frameworks without La contamination.
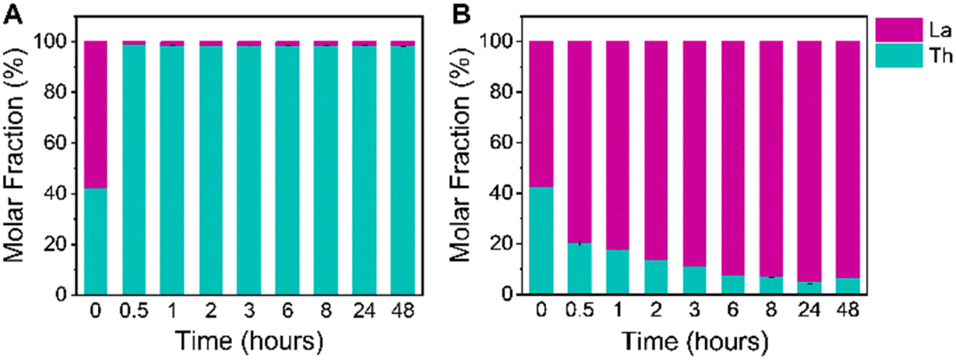 | ||
| Fig. 4 Molar fractions of metals present in (A) crystalline products and (B) supernatants at specified timepoints during binary crystallization trials with an initial mixture of La(III) and Th(IV) as determined by ICP-OES. The time = 0 bar represents the initial concentration of metals in solution before selective crystallization. Error bars shown represent the standard deviation of three replicate trials. | ||
While our NU-2500 products themselves had high selectivity for Th crystallization, we were also interested in the efficiency of our platform for removal of Th from the reaction mixture. The ability to remove a greater quantity of Th(IV) ions from the initial mixed metal solution would be highly advantageous for a separation scheme, as it would decrease the number of crystallization cycles necessary to isolate rare earth ions of a desired purity. To this end, we employed an equimolar initial mixture of La(III) and Th(IV) to perform our selective crystallization and quantified the relative concentrations of each metal remaining in solution throughout the reaction as NU-2500 was crystallized. Samples of the supernatants from each time interval were analyzed with ICP-OES (Fig. 4b and Table S7†). We found that after 30 minutes of heating time for NU-2500 to crystallize, the molar fraction of Th remaining in solution had decreased to approximately 20%, corresponding to a Th(IV)/La(III) separation factor of 302; after 24 hours of crystallization time, the Th molar fraction had been reduced further to 5%, corresponding to a Th(IV)/La(III) separation factor of 1279 (Table S6†). This indicates that our selective crystallization platform is capable of rapidly and efficiently recovering Th(IV) ions from an initial equimolar mixture in a single crystallization step. The Th/La selectivity we observe is comparable to other reported crystallization-based strategies to separate actinides from lanthanides: for instance, the polyoxometalate crystallization strategy employed by Zhang et al.42 reported an Am(VI)/Eu(III) separation factor of 780. Additionally, we note the impressive selectivities achieved by Wang et al. in their study on selenite crystallization,43 which demonstrated a Th(IV)/La(III) separation factor of 2.1 × 105. While the selectivity for Th-MOF crystallization reported here is lower, we emphasize the mild conditions used in our crystallization platform, which produce pure NU-2500 within 30 minutes of mild heating at 80 °C, compared to 3 days of heating at 230 °C for the selenite crystallization study.
Generally, the strategy of selective crystallization for the separation of Th from RE elements represents a significant improvement over the numerous sequential extraction steps that are often necessary to perform a complete extraction with conventional homogeneous extraction techniques. The high efficiency of Th(IV) ion crystallization eliminates the need for additional consumption of organic solvents and extractants that would be required for multiple-cycle separations. By sequestering the Th(IV) ions within a solid framework material that can be easily isolated from the solution phase, radioactive contamination of the solvent phase is minimized, resulting in less hazardous waste streams from this separation process.
Selectivity of Th crystallization from mixtures with decreased initial Th concentration
Beyond equimolar mixtures of Th and rare earths, we attempted to challenge our selective crystallization platform with initial solutions containing decreased concentrations of Th relative to REs. We prepared mixed metal stock solutions containing initial La:Th molar ratios of 3:1, 6:1, and 15:1. With increased concentrations of La(III) ions available in solution to coordinate with the linker, we intended to determine whether our synthesis would remain selective for NU-2500 crystallization. Through visual analysis of these vials during solvothermal synthesis at 80 °C with BTC linker and acetic acid modulator, we noted slower precipitation of product when the initial relative Th(IV) concentration is lower, indicating reduced crystallization kinetics compared to trials without an excess of La(III) ions available in solution. However, after 2 hours we had synthesized white crystalline powders consistent with the exclusive formation of NU-2500 by PXRD (Fig. S15†). We observe no peaks corresponding to the competing [La(BTC)(H2O)6] lattice, demonstrating the strong thermodynamic preference for NU-2500 crystallization even in the presence of excess La(III) ions available in solution. ICP-OES analysis of the crystalline products from these syntheses demonstrates the high selectivity for Th(IV) in the NU-2500 powders (Fig. 5a and Table S8†). From an initial mixture of 3:1 La:Th, the recovered products contain a molar fraction of Th greater than 97%. Even in products crystallized from an initial 15:1 La:Th solution, the molar fraction of Th in the products exceeds 93% (Table S8†). Despite the vastly increased presence of La(III) ions, our crystallization strategy remains highly selective towards the exclusive synthesis of the thermodynamically preferred NU-2500 framework. This indicates that our platform can be utilized to efficiently extract Th(IV) from RE ores with variable Th concentrations. Further, the ability to selectively crystallize Th from solutions at low relative Th concentrations represents an effective strategy to remove trace radioactive contaminants from RE processing streams.
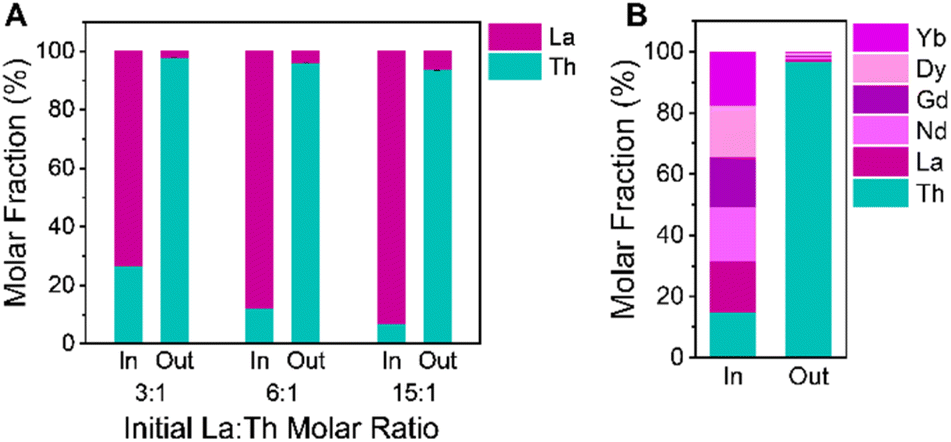 | ||
| Fig. 5 Molar fractions of metals present in selective crystallization trials with (A) increased initial La:Th ratios and (B) mixture of five different trivalent REs and Th(IV). The “in” bar represents the initial concentration of metals in solution while the “out” bar represents the contents of the solid products following selective crystallization. Error bars shown represent the standard deviation of three replicate trials. | ||
Th crystallization against additional REs
To ensure our Th removal strategy was feasible for REs beyond La, we expanded our separation scheme to include additional lanthanides. In addition to La, we selected Ce, Nd, Gd, Dy, and Yb to test their crystallization against Th in equimolar initial metal mixtures. These REs are all trivalent, with slight variations in their Lewis acidities and ionic radii, which range from 103.2 (La) to 86.8 pm (Yb).69 In solvothermal crystallization trials with binary, equimolar RE(III) + Th(IV) mixtures, we observe rapid precipitation of white powders from solution for all of the REs studied. Regardless of RE metal identity, we observe the exclusive formation of NU-2500 by PXRD (Fig. S16†). Consistent with our findings from binary trials of La + Th mixtures, we note minimal co-crystallization of the REs into the NU-2500 lattice, as determined by ICP-OES of the solid products (Fig. S17 and Table S9†). High selectivity for Th crystallization is evidenced by average Th molar fractions greater than 98% for all products synthesized from equimolar mixtures of Th(IV) with Ce(III), Nd(III), Gd(III), Dy(III), and Yb(III) (Table S9†). For the case of Ce(III), we note that our crystallization conditions do not result in oxidation to Ce(IV), preserving the efficacy of our valence-selective scheme. Finally, we tested an equimolar, six component mixture of Th(IV), La(III), Nd(III), Gd(III), Dy(III), and Yb(III) with our crystallization platform. Following solvothermal synthesis, we confirmed the formation of phase-pure NU-2500 by PXRD (Fig. S18†). ICP-OES demonstrated that the molar fraction of Th in the solid products exceeds 96% (Fig. 5b and Table S10†), indicating that our selective crystallization strategy is highly effective at producing pure samples of Th frameworks in the presence of multiple other competing REs in solution.Conclusions
We have developed a valence-selective crystallization strategy to distinguish Th(IV) from RE(III) ions through metal–organic framework synthesis. Using simple carboxylate-based linkers and coordination modulators, we have established synthetic conditions to selectively crystallize a Th-MOF from an initial mixture containing both Th and multiple REs. When individual RE or Th salts are used separately, we report the formation of two distinct MOFs: the previously reported [La(BTC)(H2O)6] or a novel Th framework, NU-2500. However, from a mixture of La(III) and Th(IV), we observe the exclusive formation of the thermodynamically preferred NU-2500 product. In a single one-pot reaction from an equimolar La(III) and Th(IV) mixture, we can synthesize phase-pure NU-2500 with a Th molar fraction exceeding 98%. Through this process, we can remove 95% of Th(IV) ions from solution in a single crystallization step. This high selectivity for Th crystallization is retained even at low initial concentrations of Th(IV) and in the presence of multiple other competing RE(III) metals.By sequestering Th(IV) ions within a solid MOF product, radioactive species can be easily separated from RE(III) ions left behind in solution through simple filtration. Our heterogeneous approach also reduces the solvent demands that are associated with currently employed homogeneous extraction methods, which generate substantial hazardous and radioactive chemical waste. Notably, our selective crystallization strategy utilizes heating at low temperatures and an environmentally-friendly solvent system consisting of water and ethanol, conditions that are far milder than typically used in industrial RE processing. Given the strategic importance of REs and particularly their relevance to clean energy technologies, the development of efficient, selective, and eco-friendly RE processing strategies is vital. Our selective crystallization strategy provides an alternative, efficient approach to remove Th impurities from REs by confining the radioactive Th(IV) ions within a stable MOF, which can be deconstructed downstream to reuse these Th ions. Considering their synthetic tunability and vast range of accessible structures, MOFs are a promising target for diverse selective crystallization applications. We anticipate future work to leverage the versatility of MOF synthesis to broaden the scope of this selective crystallization method and address further challenges related to RE purification.
Data availability
Crystallographic data for NU-2500 has been deposited in the Cambridge Crystallographic Data Centre (CCDC) under deposition number 2378389.Author contributions
M. A. G. and O. K. F. designed the project and led the investigation, with the help of K. O. K. M. A. G. performed materials synthesis and characterization, crystallization trials, and interpreted the results. C. S. S. performed additional characterization experiments. Z.-M. Y., C. L. S., and C. D. M. contributed to the interpretation of single crystal diffraction data. M. A. G. wrote the manuscript. O. K. F. supervised the project, and all authors contributed to editing and revision of the manuscript.Conflicts of interest
O. K. F. has a financial interest in NuMat Technologies, a startup company that is seeking to commercialize metal–organic frameworks. All other authors declare no competing interests.Acknowledgements
The authors gratefully acknowledge financial support from the Army Research Office (award number W911NF2020136) and the Defense Threat Reduction Agency (HDTRA1-19-1-0007). M. A. G. and C. S. S. gratefully acknowledge support from the Ryan Fellowship and the International Institute for Nanotechnology (IIN) at Northwestern University. This work made use of the IMSERC Crystallography facility at Northwestern University, which has received support from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS-2025633), and Northwestern University. Metal analysis was performed at the Northwestern University Quantitative Bio-element Imaging Center (QBIC). M. A. G. would like to thank Rebecca Sponenburg from QBIC for her guidance with ICP-OES measurements. This work also made use of the EPIC facility of Northwestern University's NUANCE Center, which has received support from the SHyNE Resource (NSF ECCS-2025633), the IIN, and Northwestern's MRSEC program (NSF DMR-2308691).Notes and references
- T. Cheisson and E. J. Schelter, Science, 2019, 363, 489–493 CrossRef CAS PubMed.
- V. Balaram, Geosci. Front., 2019, 10, 1285–1303 CrossRef CAS.
- B. S. Van Gosen, P. L. Verplanck, K. R. Long, J. Gambogi and R. R. I. Seal, The rare-earth elements—Vital to modern technologies and lifestyles, U.S Geological Survey, 2014, report number: 2014-3078, DOI:10.3133/fs20143078.
- J. Lucas, P. Lucas, T. Le Mercier, A. Rollat and W. Davenport, in Rare Earths, ed. J. Lucas, P. Lucas, T. Le Mercier, A. Rollat and W. Davenport, Elsevier, Amsterdam, 2015, ch. 13, pp. 213–230, DOI:10.1016/B978-0-444-62735-3.00013-9.
- R. J. Gorte, AIChE J., 2010, 56, 1126–1135 CrossRef CAS.
- C. R. Ronda, T. Jüstel and H. Nikol, J. Alloys Compd., 1998, 275–277, 669–676 CrossRef CAS.
- A. Elshkaki and T. E. Graedel, Appl. Energy, 2014, 136, 548–559 CrossRef.
- R. Lacal-Arántegui, J. Cleaner Prod., 2015, 87, 275–283 CrossRef.
- J.-C. G. Bünzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048–1077 RSC.
- T. E. Lipman and P. Maier, MRS Bull., 2021, 46, 1164–1175 CrossRef.
- E. Alonso, A. M. Sherman, T. J. Wallington, M. P. Everson, F. R. Field, R. Roth and R. E. Kirchain, Environ. Sci. Technol., 2012, 46, 3406–3414 CrossRef CAS PubMed.
- T. Watari, K. Nansai and K. Nakajima, Resour., Conserv. Recycl., 2020, 155, 104669 CrossRef.
- S. Hoenderdaal, L. Tercero Espinoza, F. Marscheider-Weidemann and W. Graus, Energy, 2013, 49, 344–355 CrossRef.
- K. Habib and H. Wenzel, J. Cleaner Prod., 2014, 84, 348–359 CrossRef CAS.
- L. Grandell, A. Lehtilä, M. Kivinen, T. Koljonen, S. Kihlman and L. S. Lauri, Renewable Energy, 2016, 95, 53–62 CrossRef CAS.
- K. Smith Stegen, Energy Policy, 2015, 79, 1–8 CrossRef CAS.
- A. Jordens, Y. P. Cheng and K. E. Waters, Miner. Eng., 2013, 41, 97–114 CrossRef CAS.
- W. D. Judge and G. Azimi, Hydrometallurgy, 2020, 196, 105435 CrossRef CAS.
- Z. Zhu, Y. Pranolo and C. Y. Cheng, Miner. Eng., 2015, 77, 185–196 CrossRef CAS.
- T. Xu, X. Zheng, B. Ji, Z. Xu, S. Bao, X. Zhang, G. Li, J. Mei and Z. Li, Sep. Purif. Technol., 2024, 330, 125501 CrossRef CAS.
- K. T. Rim, K. H. Koo and J. S. Park, Saf. Health Work, 2013, 4, 12–26 CrossRef CAS PubMed.
- F. Xie, T. A. Zhang, D. Dreisinger and F. Doyle, Miner. Eng., 2014, 56, 10–28 CrossRef CAS.
- N. V. Thakur, Miner. Process. Extr. Metall. Rev., 2000, 21, 277–306 CrossRef CAS.
- T. Liu and J. Chen, Sep. Purif. Technol., 2021, 276, 119263 CrossRef CAS.
- B. Weaver, F. A. Kappelmann and A. C. Topp, J. Am. Chem. Soc., 1953, 75, 3943–3945 CrossRef CAS.
- D. F. Peppard, G. W. Mason, J. L. Maier and W. J. Driscoll, J. Inorg. Nucl. Chem., 1957, 4, 334–343 CrossRef CAS.
- Z. Weng, S. M. Jowitt, G. M. Mudd and N. Haque, Econ. Geol., 2015, 110, 1925–1952 CrossRef.
- X.-A. Chen, Y.-E. Cheng and Z. Rong, J. Radiol. Prot., 2005, 25, 451 CrossRef CAS PubMed.
- S. H. Ali, Resources, 2014, 3, 123–134 CrossRef.
- D. S. Sholl and R. P. Lively, Nature, 2016, 532, 435–437 CrossRef PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- H. Ghasempour, K.-Y. Wang, J. A. Powell, F. ZareKarizi, X.-L. Lv, A. Morsali and H.-C. Zhou, Coord. Chem. Rev., 2021, 426, 213542 CrossRef CAS.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS PubMed.
- P. A. Kobielska, A. J. Howarth, O. K. Farha and S. Nayak, Coord. Chem. Rev., 2018, 358, 92–107 CrossRef CAS.
- Z. Hasan and S. H. Jhung, J. Hazard. Mater., 2015, 283, 329–339 CrossRef CAS PubMed.
- J. de Decker, J. de Clercq, P. Vermeir and P. van der Voort, J. Mater. Sci., 2016, 51, 5019–5026 CrossRef CAS.
- L. Jiang, W. Zhang, C. Luo, D. Cheng and J. Zhu, Ind. Eng. Chem. Res., 2016, 55, 6365–6372 CrossRef CAS.
- Y.-R. Lee, K. Yu, S. Ravi and W.-S. Ahn, ACS Appl. Mater. Interfaces, 2018, 10, 23918–23927 CrossRef CAS PubMed.
- W. Yang, Z.-Q. Bai, W.-Q. Shi, L.-Y. Yuan, T. Tian, Z.-F. Chai, H. Wang and Z.-M. Sun, Chem. Commun., 2013, 49, 10415–10417 RSC.
- Z.-Q. Bai, L.-Y. Yuan, L. Zhu, Z.-R. Liu, S.-Q. Chu, L.-R. Zheng, J. Zhang, Z.-F. Chai and W.-Q. Shi, J. Mater. Chem. A, 2015, 3, 525–534 RSC.
- N. Zhang, L.-Y. Yuan, W.-L. Guo, S.-Z. Luo, Z.-F. Chai and W.-Q. Shi, ACS Appl. Mater. Interfaces, 2017, 9, 25216–25224 CrossRef CAS PubMed.
- H. Zhang, A. Li, K. Li, Z. Wang, X. Xu, Y. Wang, M. V. Sheridan, H.-S. Hu, C. Xu, E. V. Alekseev, Z. Zhang, P. Yan, K. Cao, Z. Chai, T. E. Albrecht-Schönzart and S. Wang, Nature, 2023, 616, 482–487 CrossRef CAS PubMed.
- Y. Wang, H. Lu, X. Dai, T. Duan, X. Bai, Y. Cai, X. Yin, L. Chen, J. Diwu, S. Du, R. Zhou, Z. Chai, T. E. Albrecht-Schmitt, N. Liu and S. Wang, Inorg. Chem., 2018, 57, 1880–1887 CrossRef CAS PubMed.
- X. Yin, Y. Wang, X. Bai, Y. Wang, L. Chen, C. Xiao, J. Diwu, S. Du, Z. Chai, T. E. Albrecht-Schmitt and S. Wang, Nat. Commun., 2017, 8, 14438 CrossRef CAS PubMed.
- X. Zhao, M. Wong, C. Mao, T. X. Trieu, J. Zhang, P. Feng and X. Bu, J. Am. Chem. Soc., 2014, 136, 12572–12575 CrossRef CAS PubMed.
- H. Ya Gao, W. Li Peng, P. Pan Meng, X. Feng Feng, J. Qiang Li, H. Qiong Wu, C. Sheng Yan, Y. Yang Xiong and F. Luo, Chem. Commun., 2017, 53, 5737–5739 RSC.
- H. Yang, F. Peng, D. E. Schier, S. A. Markotic, X. Zhao, A. N. Hong, Y. Wang, P. Feng and X. Bu, Angew. Chem., Int. Ed., 2021, 60, 11148–11152 CrossRef CAS PubMed.
- P. Yang, Q. Zhuang, Y. Li and J. Gu, Chem. Commun., 2019, 55, 14902–14905 RSC.
- N. Stock and S. Biswas, Chem. Rev., 2012, 112, 933–969 CrossRef CAS PubMed.
- R. S. Forgan, Chem. Sci., 2020, 11, 4546–4562 RSC.
- C. R. Marshall, S. A. Staudhammer and C. K. Brozek, Chem. Sci., 2019, 10, 9396–9408 RSC.
- L. Yang, T. Zhao, I. Boldog, C. Janiak, X.-Y. Yang, Q. Li, Y.-J. Zhou, Y. Xia, D.-W. Lai and Y.-J. Liu, Dalton Trans., 2019, 48, 989–996 RSC.
- T. Tsuruoka, S. Furukawa, Y. Takashima, K. Yoshida, S. Isoda and S. Kitagawa, Angew. Chem., Int. Ed., 2009, 48, 4739–4743 CrossRef CAS PubMed.
- A. Schaate, P. Roy, A. Godt, J. Lippke, F. Waltz, M. Wiebcke and P. Behrens, Chem.–Eur. J., 2011, 17, 6643–6651 CrossRef CAS PubMed.
- Y. Bai, Y. Dou, L.-H. Xie, W. Rutledge, J.-R. Li and H.-C. Zhou, Chem. Soc. Rev., 2016, 45, 2327–2367 RSC.
- M. Perfecto-Irigaray, G. Beobide, O. Castillo, I. da Silva, D. García-Lojo, A. Luque, A. Mendia and S. Pérez-Yáñez, Chem. Commun., 2019, 55, 5954–5957 RSC.
- J.-H. Wang, M.-N. Li, S. Yan, Y. Zhang, C.-C. Liang, X.-M. Zhang and Y.-B. Zhang, Inorg. Chem., 2020, 59, 2961–2968 CrossRef CAS PubMed.
- Y. Chen, X. Zhang, M. R. Mian, F. A. Son, K. Zhang, R. Cao, Z. Chen, S.-J. Lee, K. B. Idrees, T. A. Goetjen, J. Lyu, P. Li, Q. Xia, Z. Li, J. T. Hupp, T. Islamoglu, A. Napolitano, G. W. Peterson and O. K. Farha, J. Am. Chem. Soc., 2020, 142, 21428–21438 CrossRef CAS PubMed.
- S. M. Shaikh, P. M. Usov, J. Zhu, M. Cai, J. Alatis and A. J. Morris, Inorg. Chem., 2019, 58, 5145–5153 CrossRef CAS PubMed.
- X. Gong, H. Noh, N. C. Gianneschi and O. K. Farha, J. Am. Chem. Soc., 2019, 141, 6146–6151 CrossRef CAS PubMed.
- J. Florek, S. Giret, E. Juère, D. Larivière and F. Kleitz, Dalton Trans., 2016, 45, 14832–14854 RSC.
- K. O. Kirlikovali, S. L. Hanna, F. A. Son and O. K. Farha, ACS Nanosci. Au, 2023, 3, 37–45 CrossRef CAS PubMed.
- Y.-H. Wen, J.-K. Cheng, Y.-L. Feng, J. Zhang, Z.-J. Li and Y.-G. Yao, Chin. J. Struct. Chem., 2005, 24, 5 Search PubMed.
- K. Liu, H. You, Y. Zheng, G. Jia, Y. Huang, M. Yang, Y. Song, L. Zhang and H. Zhang, Cryst. Growth Des., 2010, 10, 16–19 CrossRef CAS.
- H. Furukawa, F. Gándara, Y.-B. Zhang, J. Jiang, W. L. Queen, M. R. Hudson and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 4369–4381 CrossRef CAS PubMed.
- T. Islamoglu, D. Ray, P. Li, M. B. Majewski, I. Akpinar, X. Zhang, C. J. Cramer, L. Gagliardi and O. K. Farha, Inorg. Chem., 2018, 57, 13246–13251 CrossRef CAS PubMed.
- J. P. Vizuet, M. L. Mortensen, A. L. Lewis, M. A. Wunch, H. R. Firouzi, G. T. McCandless and K. J. Balkus Jr, J. Am. Chem. Soc., 2021, 143, 17995–18000 CrossRef CAS PubMed.
- D.-X. Xue, A. J. Cairns, Y. Belmabkhout, L. Wojtas, Y. Liu, M. H. Alkordi and M. Eddaoudi, J. Am. Chem. Soc., 2013, 135, 7660–7667 CrossRef CAS PubMed.
- S. Cotton, in Lanthanide and Actinide Chemistry, John Wiley & Sons, Ltd, 2006, pp. 9–22, DOI:10.1002/0470010088.ch2.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and instrumental details, NU-2500 characterization data, and separation trials data. CCDC 2378389. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc07652d |
| This journal is © The Royal Society of Chemistry 2025 |