
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMutually antagonistic molecular clips: symmetry-breaking non-covalent bonds at the chiral–nonchiral interface†
Sungryul
Bae
 ,
Younjae
Jeong
and
Dongwhan
Lee
*
,
Younjae
Jeong
and
Dongwhan
Lee
*
Department of Chemistry, Seoul National University, 1 Gwanak-ro, Gwanak-gu, Seoul 08826, Korea. E-mail: dongwhan@snu.ac.kr
First published on 7th January 2025
Abstract
The homochirality of life remains an unresolved scientific question. Prevailing models postulate that homochirality arose through mutual antagonism. In this mechanism, molecules of opposite handedness deactivate each other, amplifying even a small enantiomeric excess into a larger proportion. In this paper, we present chiral molecular clips that replicate this process. Through π–π stacking and complementary hydrogen bonds, shape-persistent clips of opposite chirality bind to each other more strongly than those of the same chirality, resulting in chiral amplification. This process was studied quantitatively, revealing a remarkably high degree (180-fold) of stereoselection, unmatched by any non-covalent assemblies reported to date. We demonstrate how this symmetry-breaking, in conjunction with the chiral composition of the host, impacts the binding of nonchiral molecules. Our findings illustrate how chirality transfer and amplification occur non-covalently from hosts to guests, offering insights into the evolutionary origins of homochirality in life's molecular building blocks.
Introduction
Life on Earth predominantly exists in single-handed forms, with fundamental building blocks such as amino acids and sugars found exclusively in one chiral form.1 This homochirality is considered essential for information storage and transfer,2,3 as well as the evolution of complex structures in biological systems.4,5 Despite its importance, the origin of homochirality still remains a profound mystery.6–10 Prevailing theories conjecture that uniform handedness has emerged in stages through the interaction between pre-existing chirality and nonchiral entities.7–11 Autocatalysis is a prominent model by which chiral molecules self-replicate from nonchiral precursors and amplify the asymmetry.6,9–11 This process involves mutual antagonism between opposite chiralities, where mirror-image molecules deactivate each other via strong heterochiral association (Scheme 1). As a consequence, the enantiomer initially present in even a slight excess (blue in Scheme 1d) survives, while its mirror image (gray in Scheme 1d) is effectively removed from the solution population. The result is a significant increase in host enantiomeric excess (ee) for subsequent guest binding. By definition, the nonchiral guest (green in Scheme 1d) binds both mirror-image hosts with equal strength. However, the depletion of one enantiomer from the solution through mutual antagonism ensures that the resulting host–guest complex carries only the chiral information of the surviving enantiomer.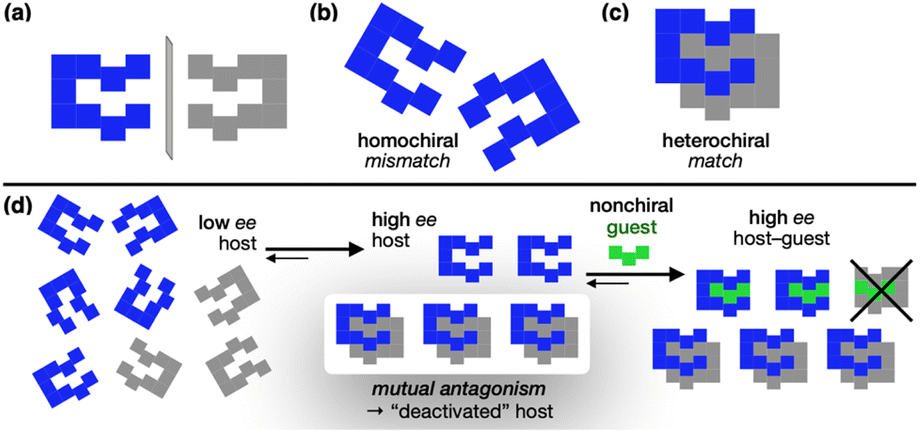 | ||
| Scheme 1 Mutual antagonism and chirality transfer. | ||
Supramolecular chemistry often employs reversible secondary interactions to replicate structure-specific biological processes.12–14 Chirality is a central theme in these efforts,15,16 drawing inspiration from how living systems differentiate enantiomeric substrates. In typical approaches, a chiral host is deployed to preferentially recognize a chiral guest over its mirror image. In this work, however, we show how chiral hosts can exploit mutual antagonism to dramatically enhance net binding affinity and relay chiral information to a nonchiral guest.
Although the concept of mutual antagonism was proposed several decades ago, its experimental demonstration has been limited to only a handful of metal-based catalysts,17–21 phosphoric acids,22,23 and biomolecules.24,25 While the non-linear effects in asymmetric catalysis are often attributed to mutual antagonism, the specific mode of action remains poorly understood. Structural information on the active form of the catalyst and how the chiral assembly disrupts interaction with the substrates remain elusive,26–29 with no definitive theoretical models.30 Even for the Soai reaction,31–33 the most ideal manifestation of homochirality through autocatalysis, these questions still remain unanswered.
In light of renewed interest in mutual antagonism, we present the supramolecular chemistry of molecular clips C* (Scheme 2) as a functional model. With its shape-persistent nature, particularly advantageous for structure-based studies, chiral C* operates solely through weak non-covalent bonds, such as π–π stacking and hydrogen bonding (HB), with unparalleled stereoselectivity (vide infra).
 | ||
| Scheme 2 Design principles of chiral molecular clips, and preferred assembly modes depending on the chiral composition. | ||
Background and design principles
To maximize mutual antagonism, we need strong heterochiral but weak homochiral association.34–38 Molecular clips, comprising a C-shaped cavity defined by a pair of arms supported by a spine, are an ideal structural platform to implement this idea, since they can engage in inter-host dimerization39 as well as host–guest interactions.40,41 For a handful of chiral clips known,34,42–47 homochiral association has been the primary focus of investigation.42–44 The preference for heterochiral association has not been established, with the exception of only one case.34 Asymmetry in known chiral clips typically resides in the spine rather than the arm units,43–47 so that the binding site remains largely unaffected by chirality. As a result, the geometric differences between the structure and its mirror image are minimal, making it difficult for the system to exhibit a strong preference for the assembly mode, either homochiral or heterochiral.We thus aimed to deliberately create chirality in the arm units by extending them differently on either side of the spine.34 This structural variation creates pronounced asymmetry between the two mirror images, providing a structural basis for the system to preferentially favor a specific mode of chiral assembly. Chirality in ring-fused, aromatic-rich π-system is often implemented using helical symmetry, as seen in helicene derivatives.48,49 This topology, however, tends to favor homochiral over heterochiral association.38,50 We thus decided to employ point chirality rather than helical chirality to build mirror-image binders.
We recently reported a series of molecular clips, C, which self-assemble to form quadruple π-stacks (Scheme 2a).51 Comprised of two parallel-oriented π-surfaces supported by a rigid tetrahydrocarbazole spine, these shape-persistent molecules exhibit strong self-association through complementary N–H⋯N hydrogen bonds between the indole N–H group of one molecule and the pyridine N atom of the other molecule. We noted that the mirror plane bisecting the parent C clip renders its two pyridyl groups equivalent, but at any given moment, only one of them participates in HB for dimer formation. As such, even after removing the redundant non-HB pyridine ring (Scheme 2b), we envisioned that the dimeric structure would still be maintained, which is comprised of two chiral clips of opposite handedness, (R)-C* and (S)-C* (Scheme 2c). Here, the stereogenic center is manifested when the two equivalent substituents at the quaternary center of the spiro-junction in C become distinct after the removal of one pyridine ring.
A simple steric model predicted that heterochiral dimerization of C* would produce stable dimers with two HB pairs, reinforced by the perfect shape complementarity between two mirror images (Movie S1†). On the other hand, homochiral dimerization is likely to be weak, as a severe twisting of the clip backbone is needed to form two HB pairs between two identical molecules (Movie S2†). As a consequence, enantiopure clips would outperform racemic clips in the complexation of flat aromatic guests, in the competitive equilibrium depicted in Scheme 2d. Depending on the chiral composition of the population, the clips adjust their preference between self-dimerization and host–guest binding to achieve chiral amplification and transfer. To test the feasibility of this idea, we set out to prepare C*.
Results and discussion
Synthesis, resolution, and structural characterization
Chiral clips C*-NI and C*-P2 share the same chiral skeleton but differ in the chemical structure of the upper canopy units (Scheme 3). As outlined in Scheme 3, the upper π-surface was installed onto the rigid tricyclic spine through C–C or C–N cross-coupling reactions as we previously reported.51 Each of the ketone intermediates (1 or 2) was subjected to a condensation reaction with the common diamine precursor 3 to afford the corresponding aminals 4. This key step introduces the chirality by transforming the flat carbonyl group into the quaternary stereogenic center. Subsequent oxidation converted 4 to the final isobenzimidazole products, as racemic clips rac-C*-NI or rac-C*-P2 (see ESI† for details on synthesis and characterization).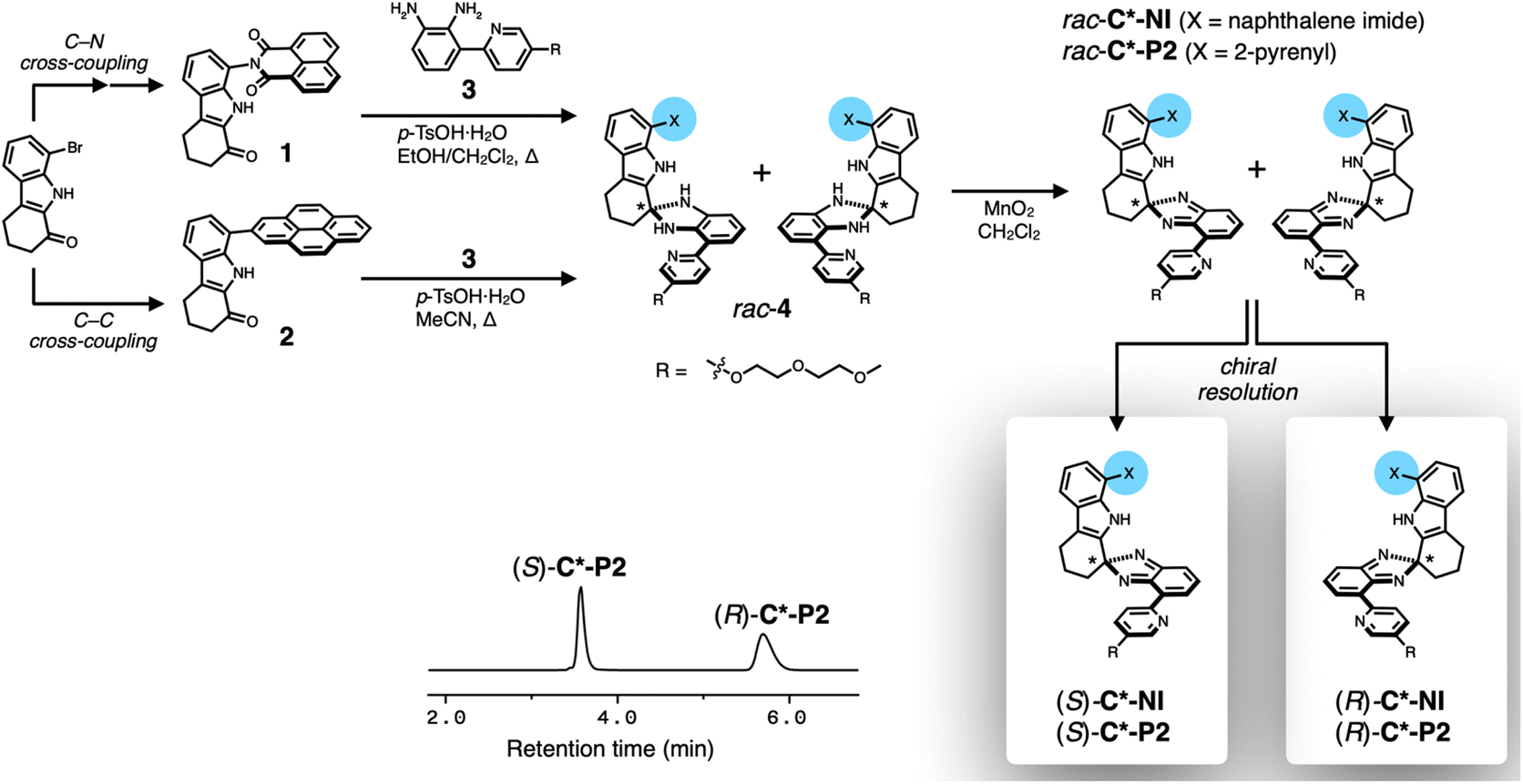 | ||
| Scheme 3 Synthesis and chiral resolution of chiral clips. | ||
For each racemic pair, enantiopure samples were obtained using preparative chiral HPLC (Scheme 3 and Fig. S3†). The absolute configuration of each enantiomer was assigned by circular dichroism (CD) spectroscopy in conjunction with time-dependent density functional theory (TD-DFT) computational studies (Fig. S4†). Structural characterization by single crystal X-ray diffraction (SC-XRD, Fig. S1†) on the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 adduct, prepared deliberately by combining two different enantiopure clips (R)-C*-P2 and (S)-C*-NI (Fig. 1i and j), unambiguously validated our stereochemical assignments (vide infra).52 The SC-XRD analysis on another cross-over 1:1 adduct of (R)-C*-NI and (S)-BrC*-NI, a mono-brominated analogue of (S)-C*-NI prepared to enhance anomalous scattering, confirmed the consistency in the absolute configuration, although heavy disorder in the chain units prevented full refinement (Fig. S2†). In the HPLC chromatograms, the earlier eluting fraction corresponds to the (S)-form, and the latter to the (R)-form (Scheme 3 and Fig. S3†).
:1 adduct, prepared deliberately by combining two different enantiopure clips (R)-C*-P2 and (S)-C*-NI (Fig. 1i and j), unambiguously validated our stereochemical assignments (vide infra).52 The SC-XRD analysis on another cross-over 1:1 adduct of (R)-C*-NI and (S)-BrC*-NI, a mono-brominated analogue of (S)-C*-NI prepared to enhance anomalous scattering, confirmed the consistency in the absolute configuration, although heavy disorder in the chain units prevented full refinement (Fig. S2†). In the HPLC chromatograms, the earlier eluting fraction corresponds to the (S)-form, and the latter to the (R)-form (Scheme 3 and Fig. S3†).
 | ||
| Fig. 1 X-ray structure of C*-NI as (a) ORTEP diagram with thermal ellipsoids at the 50% probability level, (b) capped-stick representation built with crystallographically determined atomic coordinates of meso-[C*-NI]2, and (c) brick-like schematic representation. (d) Intermolecular Nindole–H⋯Npyridyl hydrogen bond of meso-[C*-NI]2. Capped-stick representations of the X-ray structures of (e and f) meso-[C*-NI]2, (g and h) meso-[C*-P2]2, and (i and j) quasi-racemate (R)-C*-P2@(S)-C*-NI with metric parameters for π–π stacking and hydrogen bonding. | ||
To test whether the mirror-image clips indeed form heterochiral dimers to fulfill mutual antagonism (Scheme 2d), single crystals of the racemic clips were grown from chlorinated solvents in combination with ethers or toluene. For C*-NI, SC-XRD analysis revealed the formation of Ci symmetric meso-[C*-NI]2 as a 1:1 adduct of (R)-C*-NI and (S)-C*-NI (Fig. 1b and e), a simplified schematic representation of which is shown in Fig. 1c. Here, heterochiral dimerization resulted in centrosymmetric arrangements of two Nindole–H⋯Npyridyl hydrogen bonds (dN⋯N = 2.887 (4) Å) supporting a quadruple π-stack (Fig. 1d and f). When viewed along the vertical direction (Fig. 1e and f), the quadruple π-stack adopts an a–b–b–a type arrangement with interplanar distances of 3.3225(17) Å and 3.373(2) Å for the naphthalene imide⋯isobenzimidazole (= a⋯b) and isobenzimidazole⋯isobenzimidazole (= b⋯b) pairs, respectively, which maximizes van der Waals (vdW) contacts.51,53 A structurally related heterochiral meso-[C*-P2]2 (Fig. 1g and h) was also obtained from rac-C*-P2.
Notably, heterochiral dimerization occurs even between clips of different canopies. A 1:1 mixture of (S)-C*-NI and (R)-C*-P2 produced single crystals of (R)-C*-P2@(S)-C*-NI quasi-racemate (Fig. 1i and j) resembling the genuine heterochiral dimers meso-[C*-NI]2 and meso-[C*-P2]2.52,54,55 This quasi-racemate assisted in unambiguously assigning the absolute stereochemistry of individual chiral clips.52 Even with repeated attempts using numerous combinations of solvents and temperatures, however, we were not successful in obtaining diffraction-quality crystals of enantiopure clips. Unlike racemic samples, enantiopure clips are highly soluble even in ethers and toluene, suggesting their inefficient packing due to shape mismatching between individual molecules (vide infra).
Chirality-dependent clip association in solution
Comparative 1H NMR spectroscopic studies (Fig. 2) revealed distinctively different tendencies of homochiral vs. heterochiral association. The aromatic proton resonances of the N-methylated rac-C*-P2Me and the substructure model 5 are similar (Fig. 2c and d), whereas (R)-C*-P2 shows slight upfield shifts (Δδ = 0.03–0.17 ppm) in the pyridyl and isobenzimidazolyl C–H protons (Fig. 2b). The most pronounced upfield shifts (Δδ = 0.41–0.71 ppm) were observed for rac-C*-P2 (Fig. 2a). This spectral behavior is consistent with the X-ray structure of meso-[C*-P2]2 (Fig. 1g), with the protons sandwiched between the π-faces upon dimer formation experiencing significant shielding. The pyrenyl protons show similar shielding effects but are less noticeable because they belong to the outermost layers of the π-stack (blue circles, Fig. 2a–c).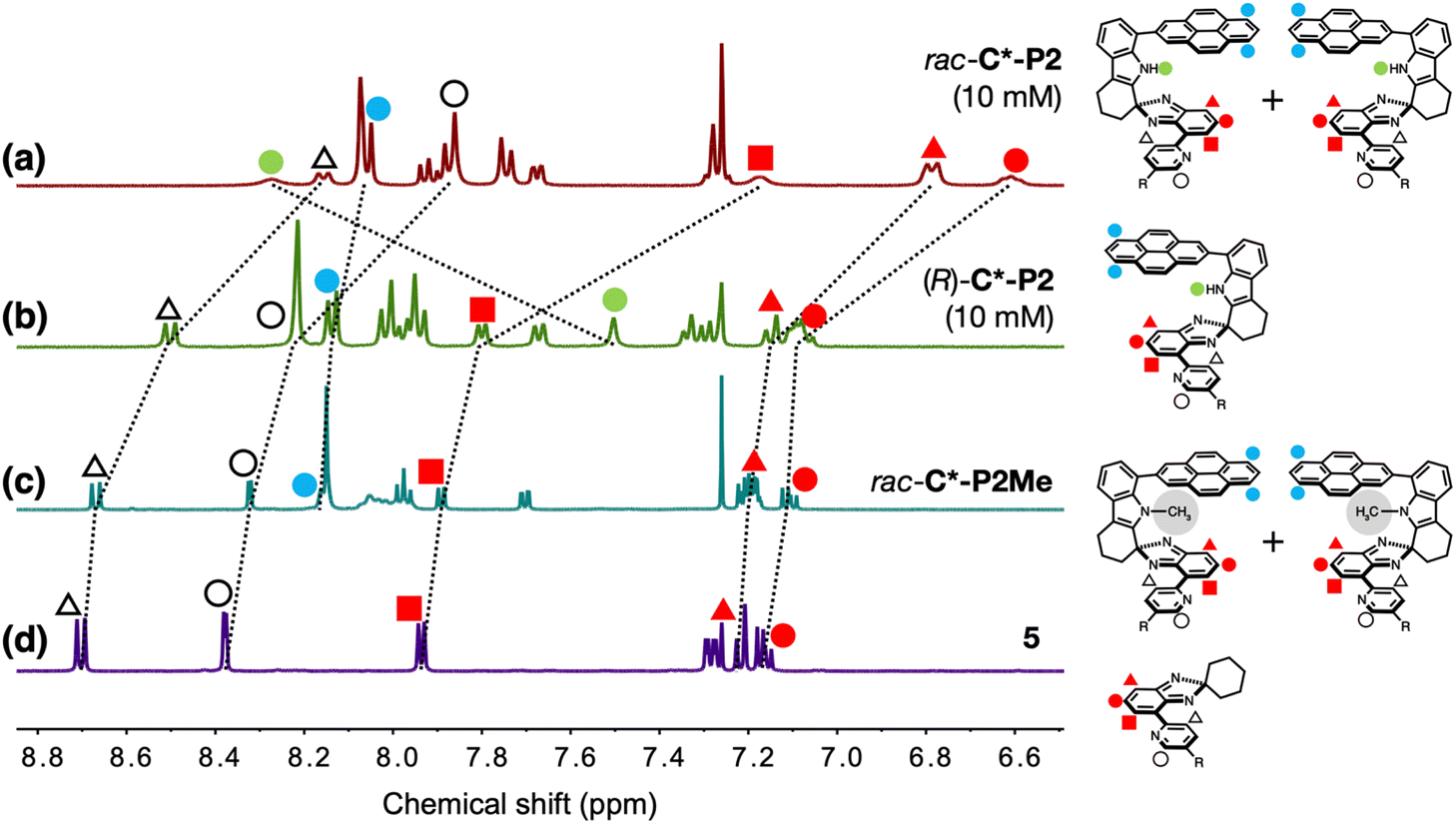 | ||
| Fig. 2 Partial 1H NMR spectra of (a) rac-C*-P2 (10 mM), (b) (R)-C*-P2 (10 mM), (c) rac-C*-P2Me (6.1 mM), and (d) 5 (23 mM) in CDCl3 at T = 298 K. Proton resonances are labeled with the symbols denoted in the chemical structures. | ||
Enantiopure (R)-C*-P2 can only form homochiral dimers, whereas rac-C*-P2 can form both homochiral and heterochiral dimers. As such, the larger upfield shifts for the C–H protons of rac-C*-P2 suggest that heterochiral association is stronger than homochiral association. Supporting this interpretation, the N–H proton of rac-C*-P2 appears further downfield (δ = 8.27 ppm) compared to (R)-C*-P2 (δ = 7.50 ppm) (green circles, Fig. 2a and b), indicating its stronger involvement in HB during dimerization.
To gain a quantitative understanding of this chirality-dependent association behavior, the binding constants for homochiral and heterochiral dimerization were determined and compared. Fig. 3a shows the 1H NMR spectra of enantiopure (R)-C*-P2 at sample concentrations of 20 mM and 2 mM in CDCl3 at r.t. Upon dilution, downfield shifts in the π–π stacked region (Fig. 3a) were observed, which is consistent with monomer–dimer equilibrium. Additional 1H NMR spectra were obtained by varying the sample concentration from 20 mM to 0.1 mM (Fig. S5†). Using the pyridine resonances as a spectroscopic handle, a monomer–homochiral dimer isotherm (ESI Note 1†) was constructed with a very small homochiral dimerization constant (Khomo) of only 6.2 (±0.7) M−1, indicating essentially negligible self-association of (R)-C*-P2 (Fig. 3e, g, and S5†). Global fitting with additional proton resonances displaying concentration dependence yielded a comparable Khomo value (Fig. S7†).
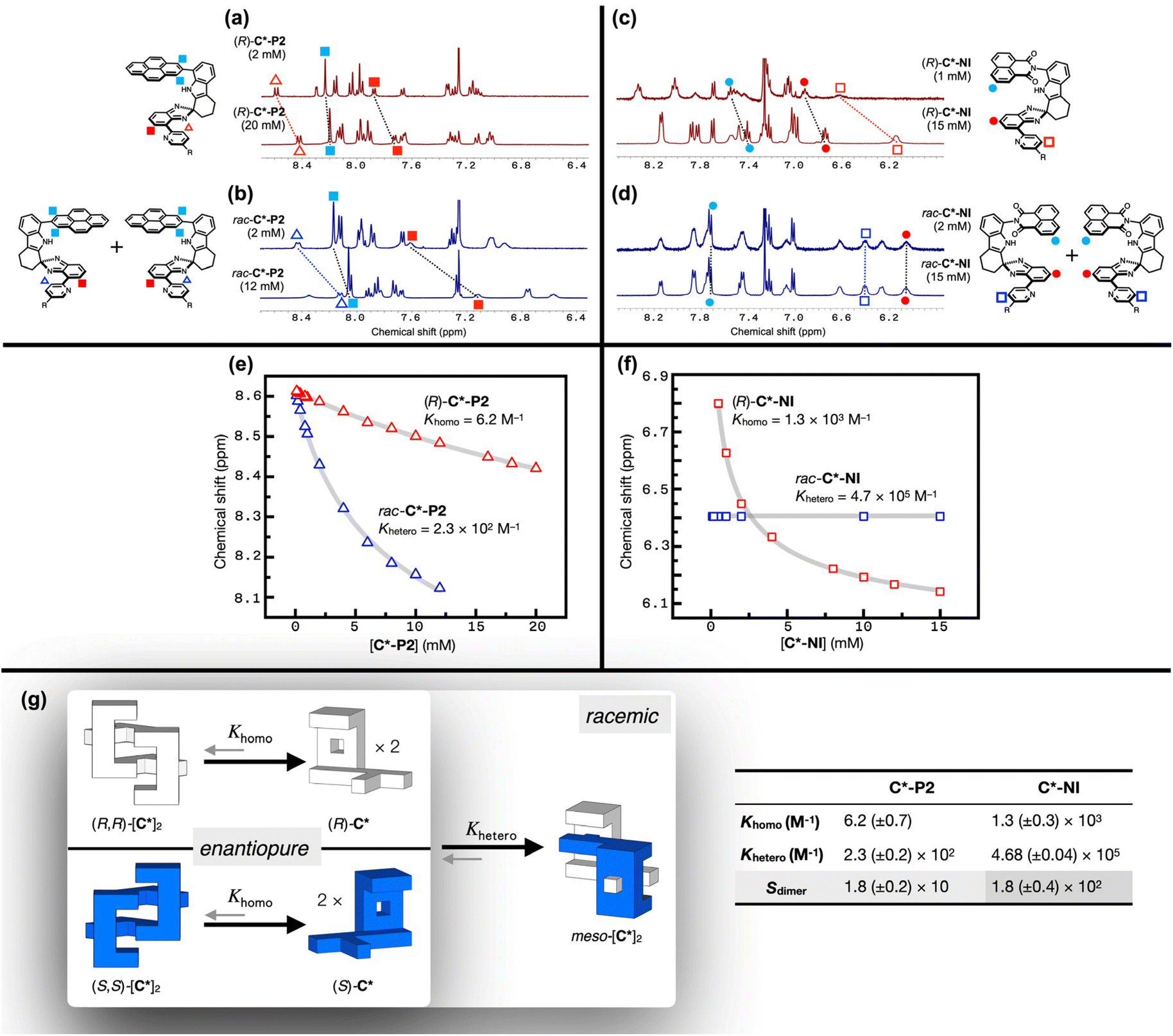 | ||
| Fig. 3 1H NMR (400 MHz, T = 298 K) spectra of (a) (R)-C*-P2, (b) rac-C*-P2, (c) (R)-C*-NI, and (d) rac-C*-NI obtained at two different concentrations are shown for qualitative comparison. For a full analysis of the concentration-dependence, changes in the chemical shifts of the pyridyl proton resonances (annotated by blank triangles or squares in the chemical structures) are plotted as a function of the concentration of (e) (R)-C*-P2 and rac-C*-P2, or (f) (R)-C*-NI and rac-C*-NI in CDCl3 at T = 298 K. The overlaid gray lines in (e) and (f) are theoretical fits from non-linear least-squares regression, taking into account both homochiral dimerization (Khomo) and heterochiral dimerization (Khetero), as described in (g). The solution equilibrium, schematically shown in (g), is dictated by the dimerization constants Khomo and Khetero in the table on the right, which were obtained by numerical global fitting. The selectivity coefficient Sdimer is defined as Khetero/(2Khomo). | ||
Under similar conditions, however, rac-C*-P2 (Fig. 3b) showed dramatically different concentration-dependent spectral shifts compared to enantiopure (R)-C*-P2 (Fig. 3a). As a qualitative measure, the 1H NMR spectrum of a concentrated (20 mM) (R)-C*-P2 solution closely resembles that of a dilute (2 mM) rac-C*-P2 solution, indicating a greater proportion of the dimeric forms in the latter. For a quantitative analysis, the 1H NMR spectra of rac-C*-P2 were acquired in a broader concentration range from 12 mM to 0.1 mM (Fig. S8†) and fitted to an isotherm that accounts for the equilibrium among monomer, homochiral dimer, and heterochiral dimer (Fig. 3g and ESI Note 2†). The heterochiral dimerization constant (Khetero) is 2.3 (±0.2) × 102 M−1 (Fig. 3e, g, and S8†). Without enthalpic preference for either type of dimerization, the equilibrium constant for heterochiral dimerization is twice that of homochiral dimerization by the configurational entropic contribution (ESI Note 3 and Fig. S9†).31 For the selectivity factor defined as Sdimer = Khetero/(2Khomo), the Sdimer value is 18 (±2) for C*-P2 (Fig. 3g), underscoring the predominance of heterochiral clip association (Fig. 4b) (vide infra).
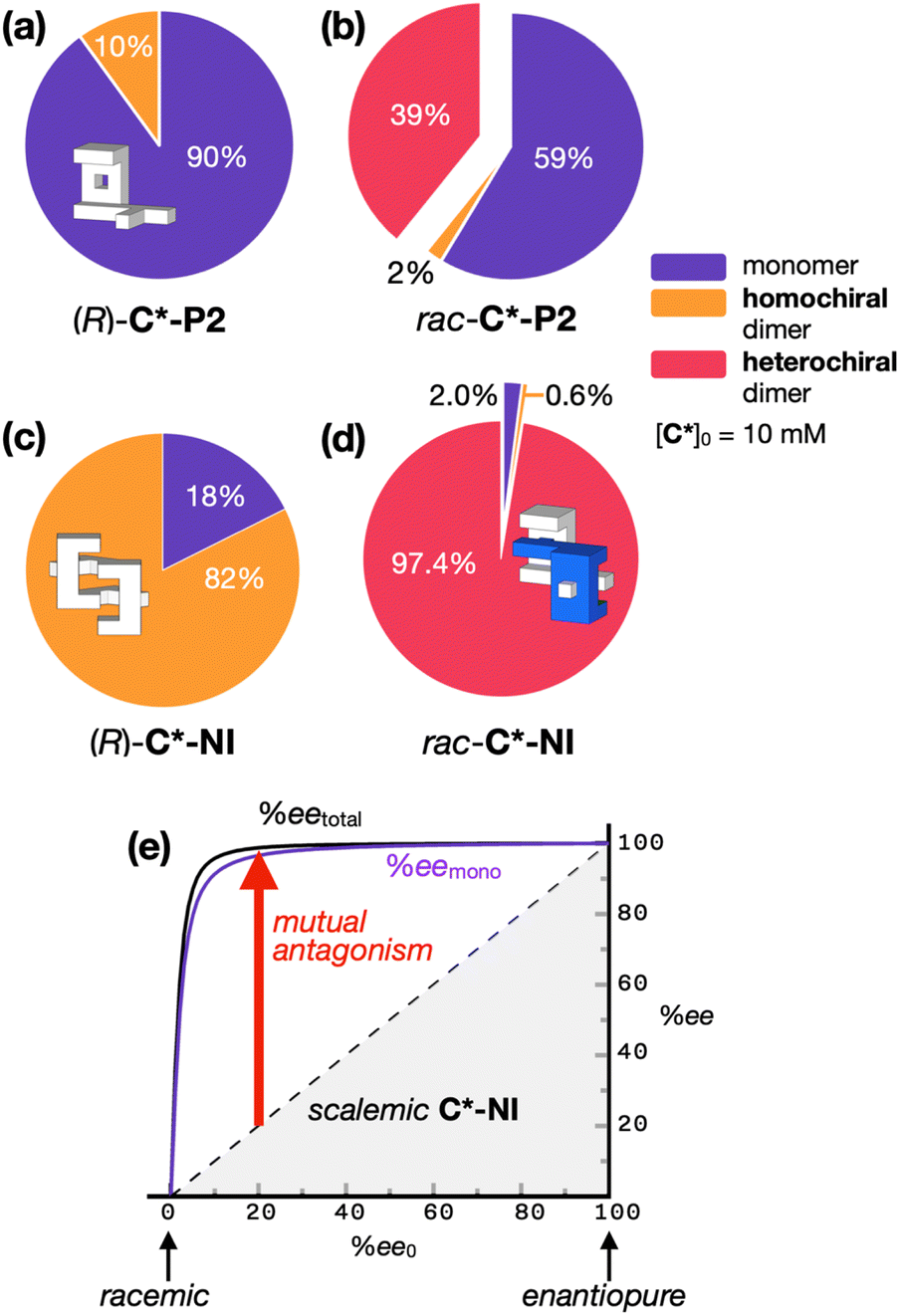 | ||
| Fig. 4 Speciation pie charts showing percent fractions of individual species for (a) (R)-C*-P2, (b) rac-C*-P2, (c) (R)-C*-NI, and (d) rac-C*-NI with [C*]0 = 10 mM. Color code: monomeric (R)-C* or (S)-C*, violet; homochiral dimeric (R,R)-[C*]2 or (S,S)-[C*]2, orange; heterochiral dimeric meso-[C*]2, magenta. (e) Plot of redistributed enantiomeric excess (ee) of C*-NI as a function of the initial enantiomeric excess (ee0). The purple line represents the percent enantiomeric excess of the monomer only (eemono); black line indicates the percent enantiomeric excess of both the monomer and homochiral dimer (eetotal); dashed lines denote the relationship between ee0 and ee in the absence of dimerization. See Fig. 3 for the Khomo and Khetero values used in the numerical simulation. | ||
With a larger dipole moment associated with the naphthalene imide canopy promoting donor–acceptor (D–A) type π–π stacking, C*-NI is anticipated to show a stronger dimerization propensity than C*-P2.51 Indeed, enantiopure (R)-C*-NI displayed much more pronounced concentration-dependent spectral shifts compared to (R)-C*-P2 (Fig. 3a), with Khomo = 1.3 (±0.3) × 103 M−1 determined by isotherm fitting (Fig. 3c, f, g, and S6†). With a better shape match between mirror-image clips, the heterochiral dimerization of C*-NI becomes even stronger. Unlike all other cases, no spectral change was observed for rac-C*-NI upon dilution from 15 mM to 20 μM (Fig. 3d, f, and S10†), indicating the predominance of a single chemical species within this concentration range.
As summarized in Fig. 3g, five different species can co-exist in rac-C*-NI solution: enantiomeric pair of monomers, homochiral dimers, and the heterochiral meso-dimer. For the enantiopure (R)-C*-NI, neither the monomer nor the homochiral dimer predominates in this concentration range (Fig. 3c, f, and S6†). Therefore, the seemingly invariant spectra of rac-C*-NI can only be explained by the overwhelming presence of the heterochiral dimer. In support of this notion, the 2D-NOESY 1H NMR spectrum of rac-C*-NI in CDCl3 (Fig. S11†) revealed strong cross-peaks originating from through-space interactions of isobenzimidazole with indole, pyridine with indole, and naphthalene imide with ether chain. Such spatial proximities are achieved only by heterochiral dimerization, which maximizes vdW contacts and the number of intermolecular HB.
Using the competition between clip dimerization and guest encapsulation (vide infra), the heterochiral dimerization constant of C*-NI was determined as Khetero = 4.68 (±0.04) × 105 M−1 (Fig. 3g, 6c, and S20; ESI Note 6†).56–59 This remarkably large Khetero value reproduces the negligible 1H NMR spectral changes in the concentration range of Fig. 3f (gray solid line overlaid on the experimental data points). The selectivity factor of C*-NI is Sdimer = 1.8 (±0.4) × 102 (Fig. 3g). In other words, clips of opposite handedness bind each other 180 times more tightly than those of the same handedness, indicating a strong mutual antagonism. For stereoselection between homochiral and heterochiral dimerizations, this Sdimer value is the highest. The only known example of Sdimer exceeding 180 is the Soai reaction catalysts,60 but recent studies suggest that these catalysts form tetramer or higher-order aggregates, rather than dimers, making the previously reported Sdimer values less relevant.33,61 Remarkably, C* has achieved a record-high level of stereoselectivity solely through π–π stacking and hydrogen bonding.
Chirality-dependent speciation and chiral amplification
The monomer–dimer equilibrium constants determined above (Fig. 3g) allowed us to construct speciation pie charts (Fig. 4a–d) to analyze the solution population. While the equilibrium lies toward the homochiral dimer for enantiopure C*-NI (Fig. 4c), guest species can still effectively compete (Scheme 2d; vide infra). For racemic C*-NI, however, the dominant heterochiral dimer is unbeatably robust against disassembly (Fig. 4d). As a result, mutual antagonism becomes particularly pronounced in the scalemic population, i.e., a mixture of mirror-image isomers at a ratio other than 1:1. For example, when [(R)-C*-NI]0 outnumbers [(S)-C*-NI]0, nearly all (S)-C*-NI exists as 1:1 complex with (R)-C*-NI, with the free monomers and weakly-bound homochiral dimers almost exclusively comprised of (R)-C*-NI. This situation dramatically skews the enantiomeric excess of the monomer (eemono), as well as the total ee (= eetotal; taking into account both the monomer and homochiral dimer) from the initial ee value (ee0), resulting in a distinct non-linearity (Fig. 4e).
The strong mutual antagonism of C*-NI drives the eemono and eetotal towards 100%. As modeled in Fig. 4e, even when the ee0 is 10%, the eetotal value reaches 96%. For ee0 = 20%, the eetotal rises to 99%. In other words, a mixture of (R)- and (S)-C*-NI with an ee > 20% behaves like an enantiopure sample due to mutual antagonism and the non-linear effect (see ESI Notes 1, 2, and 4† for details on the numerical simulations), which effectively amplifies the available chirality.
Molecular basis of stereoselectivity: computational modeling
To gain structural insights into the remarkable stereoselectivity of C* in dimerization (Fig. 4), density functional theory (DFT) computational studies were carried out. While X-ray crystallography unambiguously established the three-dimensional structures of heterochiral dimers (Fig. 1), structural evidence for the corresponding homochiral dimers remains elusive. We thus turned to computational modeling of all potential homochiral dimers.The downfield shifts of the indole N–H proton (Fig. S5 and S6†) implicate the involvement of Nindole–H⋯Npyridyl hydrogen bonds in the putative homochiral dimerization, which helped us narrow down the models to three candidates: two with a single HB (I and II in Fig. 5a and S12a†) and one with two HBs requiring severe structural distortion (III in Fig. 5a and S12a†). Steric hindrance precluded trimers or higher-order aggregates.
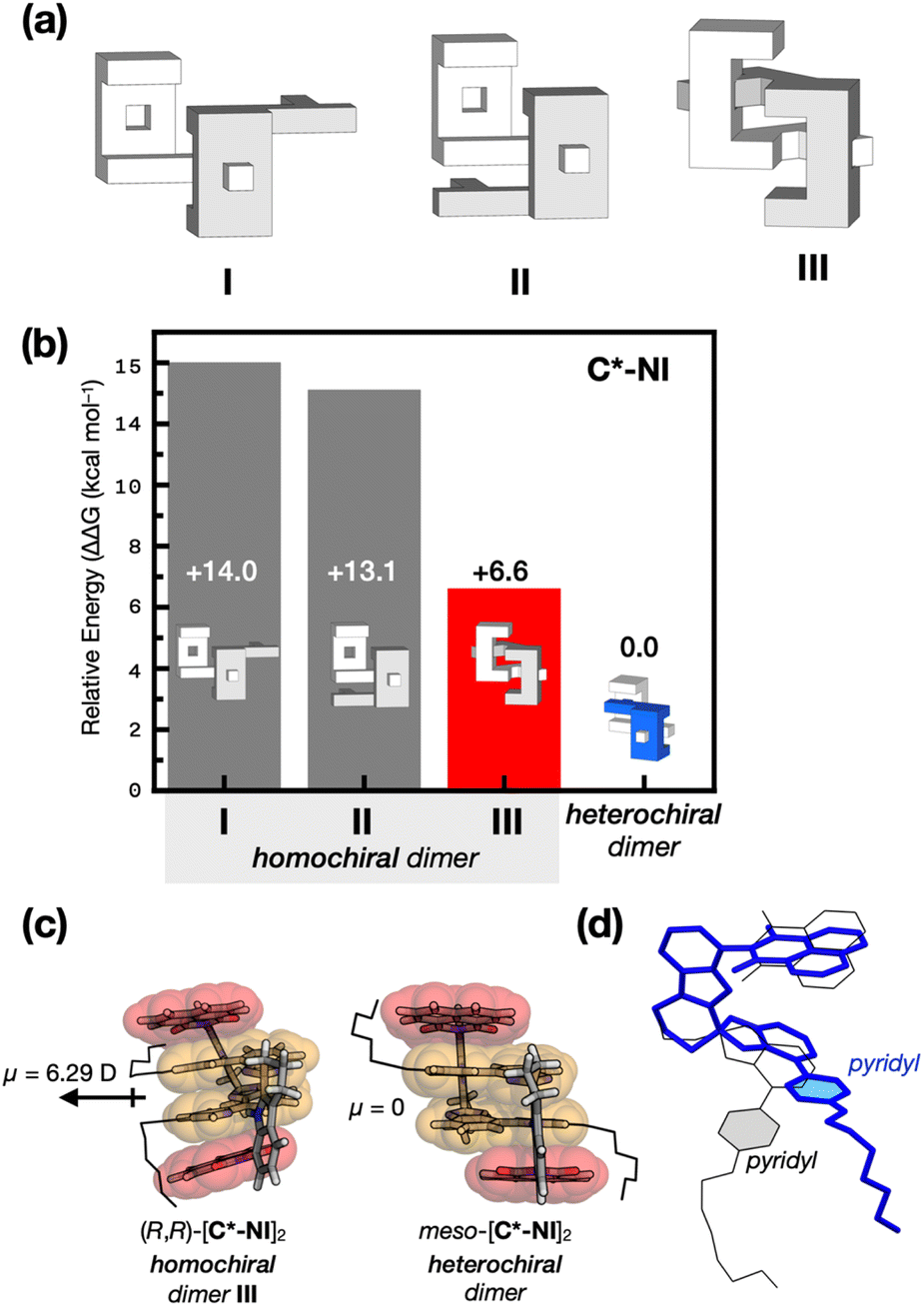 | ||
| Fig. 5 (a) Schematic representations of possible structures of the homochiral dimer (R,R)-[C*]2. (b) Bar graphs comparing the free energies computed at the B3LYP-D3/6-31++G** level, relative to the energy of the heterochiral dimer as the reference point. (c) Dipole moments of energy-minimized DFT models of the homochiral (R,R)-[C*-NI]2 (III in (a)) and meso-[C*-NI]2. (d) Overlaid DFT energy-minimized structures of C*-NI comprising the homochiral (blue capped-stick) vs. heterochiral (black wireframe) dimer. | ||
The dimeric DFT models I–III form the quadruple π-stack and Nindole–H⋯Npyridyl HB, similar to the structurally characterized heterochiral dimers (Fig. 1), although they differ in the number of HB and degree of structural distortion (Fig. S12†). The energy-minimized DFT models predicted that, for both C*-P2 and C*-NI, III is preferred over I or II by significant energy differences (>3.7 kcal mol−1; Fig. 5b and S12b†). The computed π–π stacking pattern of III is consistent with more pronounced upfield shifts of the proton resonances (Fig. 2b, c, S13b and c†) observed for the buried pyridine–isobenzimidazole π-surfaces (yellow, Fig. 5c) compared with the exposed canopy units (red, Fig. 5c), either naphthalene imide (for (R)-C*-NI) or pyrenyl (for (R)-C*-P2) constituting the quadruple π-stack. With the pseudo-C2 symmetry, the dipole moments contributed by each monomer cannot be canceled out upon homochiral association. Indeed, DFT computational studies revealed a substantial build-up of the dipole moments: 6.29 D for (R,R)-[C*-NI]2 (Fig. 5c and S14a†) and 5.04 D for (R,R)-[C*-P2]2 (Fig. S14b†). This result indicates that the centrosymmetric orientation of the π-clips within the heterochiral meso-adducts benefits from stronger dipole–dipole interactions, which enhance their stability.
Moreover, significant torsional strain disfavors homochiral dimerization. In particular, the pyridine units have to undergo substantial dislocation to accommodate two HBs in the homochiral dimer (Fig. 5d and S15†), compared to the fully relaxed geometry of the heterochiral dimer. In the DFT model, the two monomers constituting the homochiral dimer (R,R)-[C*-NI]2 are calculated as 4.15 kcal mol−1 and 2.91 kcal mol−1 higher in electronic energy than those in the heterochiral dimer. For (R,R)-[C*-P2]2, they are 2.55 kcal mol−1 and 0.34 kcal mol−1. These considerable energy differences preferentially stabilize the heterochiral clips.
Chiral–nonchiral interface: chirality-dependent host assembly and guest encapsulation
To investigate whether the chirality amplified by mutual antagonism can be effectively transmitted to nonchiral molecules, we studied the interaction between the clips C* and a nonchiral guest. Enantiopure clips are more effective at guest binding than racemic clips, even toward nonchiral guests. Studies on this intriguing phenomenon were prompted by our previous work on the nonchiral molecular clip C, which can recognize 1,10-phenanthroline-5,6-dione (PHD) to form a 1:1 host–guest complex (Scheme 2a).51 As a flat, polyheterocyclic molecule, PHD is capable of forming hydrogen bonds with the indole N–H group buried inside the clip. With rac-C*-P2, the three-dimensional structure of the inclusion complex, rac-[C*-P2 ⊃ PHD], was confirmed by SC-XRD, which revealed tight π–π stacking with interplanar distances of 3.241(2) Å and 3.242(2) Å (Fig. 6a). The addition of PHD to (R)-C*-NI also elicited changes in the CD spectrum, with a new transition appearing at around 260 nm, distinct from (R)-C*-NI alone (Fig. S16†). This optical signal lies within the absorption range of PHD, indicating that PHD is positioned within the chiral environment provided by the host.
 | ||
| Fig. 6 (a) Chemical structure of PHD and capped-stick representation of the X-ray structure of (R)-[C*-P2 ⊃ PHD] and (S)-[C*-P2 ⊃ PHD] with vdW surfaces overlaid on the stacked aromatic regions, and ether chains simplified as wireframes. Changes in the chemical shifts of PHD proton resonances (denoted by green circles in the chemical structure; [PHD]0 = 0.45 mM; T = 298 K) in CDCl3 during titrations with (b) C*-P2, and (c) C*-NI. Experiments conducted with enantiopure clips are denoted by green filled circles, and those with racemic clips by green hollow circles. (d) Schematic representation of equilibrium between monomeric and dimeric hosts and host–guest complexes. The overlaid gray lines on (b) and (c) are theoretical fits that take into account homochiral dimerization (Khomo), host–guest complexation (KHG), and, if applicable, heterochiral dimerization (Khetero) of the hosts, as described in (d). Dashed lines in the plots (b) and (c) denote expected titration curves in the absence of host dimerization. | ||
To gain a quantitative understanding of this chemistry, particularly the effects of mutual antagonism of chiral hosts on guest recognition, we conducted 1H NMR spectroscopic studies on (R)-C*-P2 and PHD in CDCl3. The proton resonances of the host (R)-C*-P2 reflect at least three different species in solution: (R)-C*-P2, (R,R)-[C*-P2]2, and (R)-[C*-P2 ⊃ PHD]. The rapid exchange among these species complicates the situation, making the interpretation of spectral changes less straightforward. We thus focused on the simpler 1H NMR patterns of the guest PHD, which exists either as host-entrapped (R)-[C*-P2 ⊃ PHD] or free PHD. Specifically, the C–H proton resonance at the 2-position of PHD was followed as a function of host concentration. For (R)-C*-P2, we observed an upfield shift of Δδ = 0.36 ppm with 25 equivalents of the host in a 0.45 mM guest solution (Fig. 6b and S17†). Under similar conditions, the upfield shift was much less pronounced for rac-C*-P2 with Δδ = 0.20 ppm (Fig. 6b and S18†). This difference suggests that the heterochiral dimerization of the racemic host depletes the free host available to capture the guest, whereas a much lower fraction of homochiral dimers of the enantiomeric host enhances the availability of the host for guest complexation (Fig. 4a and b).
The 1H NMR titration data was fitted to a non-linear regression model (Fig. 6d and ESI Note 5†) to determine the host–guest complexation constant of KHG = 1.22 (±0.08) × 102 M−1 (Fig. 6b and S17†). In the absence of any self-association of the host, whether homochiral or heterochiral, the titration curve should resemble the dashed lines in Fig. 6b (see ESI Note 5† for details on the numerical simulation). The minor deviation of the experimental data (green filled circles, Fig. 6b) from this theoretical prediction signifies that host dimerization has essentially no effect on its ability to recognize the guest. Weak self-association between hosts of the same handedness dramatically enhances the guest complexation ability of the clip, even toward the nonchiral guest, which contrasts with the behavior of rac-C*-P2 (green hollow circles, Fig. 6b). For the latter, strong self-association necessitates a higher concentration of the host to achieve the same degree of guest complexation.
Similar trends were observed for the intercalation of other nonchiral guests. The 1H NMR spectra of (S)-C*-P2vs. rac-C*-P2, obtained in the presence of various flat aromatic molecules, consistently showed more pronounced spectral shifts for the enantiopure clip. Notably, phenanthroline and diazafluorenone, which underwent negligible changes with the racemic clip, displayed significant upfield shifts with the enantiopure clip (Fig. S21†). As shown in Fig. 6a, the host–guest complexes of chiral C*-P2 and nonchiral PHD are mirror images of each other. Therefore, the host chirality should not affect the intrinsic binding affinity. Mutual antagonism of the host breaks this symmetry in the binding isotherm, by which enantiopurity enhances the net binding affinity toward the nonchiral guest.
A similar behavior was observed for C*-NI. Titration with the enantiopure (R)-C*-NI resulted in a systematic upfield shift of the PHD proton resonance, with a host–guest complexation constant of KHG = 4.9 (±0.1) × 102 M−1 (Fig. 6c and S19†). Under similar conditions, the spectral shifts induced by the racemic clips were notably smaller, which could be attributed to strong heterochiral dimerization (Fig. 4d and 6c). By analyzing the effects of this heterochiral dimerization on the host–guest complexation, we were able to back-calculate the heterochiral dimerization constant of Khetero = 4.68 (±0.04) × 105 M−1 for C*-NI, which reproduced the data shown in Fig. 3d, f, and g (see also Fig. 6d and S20; ESI Note 6†).56–59
For a side-by-side comparison with C*-P2, a theoretical titration curve was also constructed for C*-NI (Fig. 6c) without assuming host dimerization. Although the homochiral dimerization of C*-NI is moderately strong and attenuates its guest intercalating ability, it still performs much better than the racemic clips. Individual C*-NI monomers have a strong propensity for guest uptake (KHG = 4.9 × 102 M−1), but this potential is not fully realized due to strong heterochiral dimerization. With 6 equiv. of the PHD guest, the enantiopure clip predominantly forms the triple π-stack (R)-[C*-NI ⊃ PHD] (93%), along with minor fractions of the homochiral dimer (R,R)-[C*-NI]2 (4%) and unbound (R)-C*-NI (4%) (Fig. 7a). Under similar conditions, rac-C*-NI is more or less equally distributed between the heterochiral dimeric meso-[C*-NI]2 (56%) and the triple π-stack rac-[C*-NI ⊃ PHD] host–guest complex (42%) (Fig. 7b).
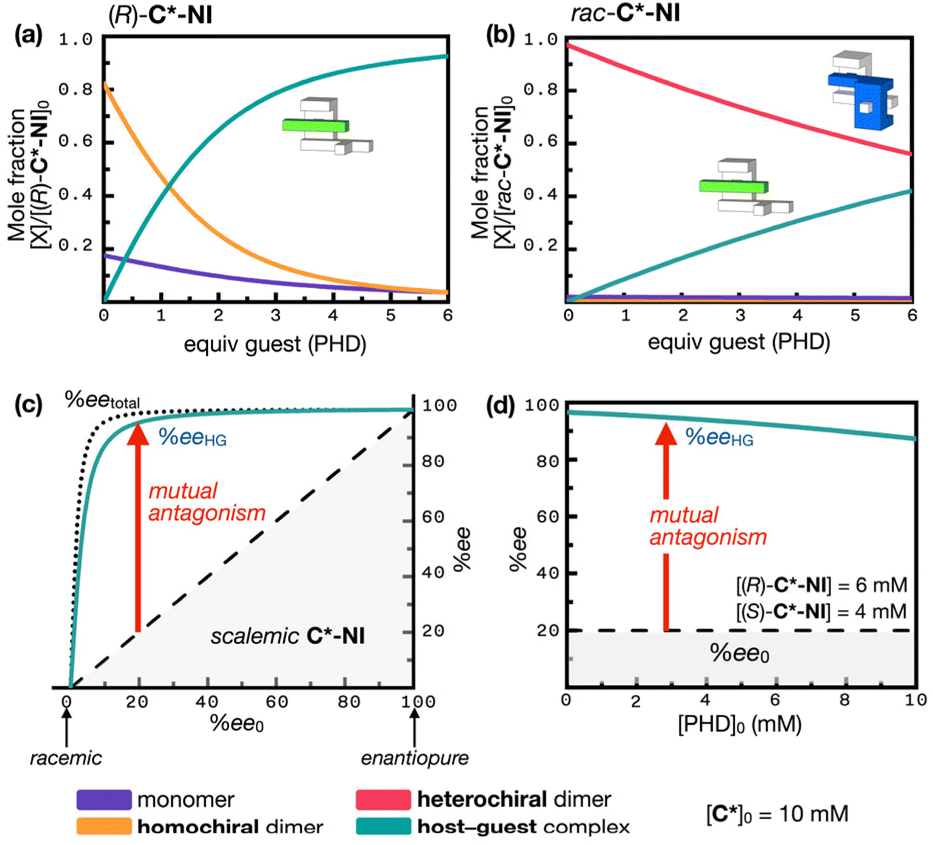 | ||
| Fig. 7 Simulated speciation plots showing changes in the fractions of individual species originating from (a) (R)-C*-NI, and (b) rac-C*-NI clip (unbound (R)-C*-NI and (S)-C*-NI, violet line; homochiral dimeric (R,R)-[C*-NI]2 and (S,S)-[C*-NI]2, orange line; host–guest complex (R)-[C*-NI ⊃ PHD] and (S)-[C*-NI ⊃ PHD], cyan line; heterochiral dimeric meso-[C*-NI]2, magenta line) with the supply of the guest PHD into [C*-NI]0 = 10 mM. (c) Plot of the enantiomeric excess of the host–guest complex (eeHG) C*-NI ⊃ PHD (cyan line) as a function of the initial enantiomeric excess (ee0) of C*-NI in a mixture of [PHD]0 (2 mM) and [C*-NI]0 (10 mM). The black dotted lines represent the relationship between ee0 and eetotal (enantiomeric excess of both the monomer and homochiral dimer) in the absence of PHD; the black dashed lines indicate the relationship between ee0 and eeHG in the absence of host dimerization. (d) Simulated plot illustrating the eeHG (cyan line) of a mixture of [(R)-C*-NI] and [(S)-C*-NI] as a function of the guest (PHD) concentration. Dashed lines denote the expected relationship in the absence of host dimerization; see Fig. 3 and 6 for the Khomo, Khetero, and KHG values used in the numerical simulations. | ||
To assess the effects of host mutual antagonism on molecular recognition, the enantiomeric excess of the host–guest complex (eeHG) was simulated as a function of the initial enantiomeric excess of C*-NI (ee0). With strong heterochiral dimerization, a scalemic mixture of C*-NI behaves similarly to enantiopure C*-NI (10 mM, > 20% ee0) (Fig. 4e). The more abundant enantiomer, which survives mutual antagonism in the scalemic mixture, binds almost exclusively to the PHD guest, while the less abundant enantiomer is subjected to strong heterochiral dimerization and thus excluded from host–guest complexation (Fig. 7b). This situation results in a remarkably high eeHG of C*-NI ⊃ PHD relative to the ee0 of C*-NI. As shown in Fig. 4e, even when the ee0 of C*-NI is as low as 10%, the eetotal value reaches 96%. With ee0 = 20%, eetotal approaches 99%. Under these conditions, in which the active hosts are essentially enantiopure, the addition of a guest ([PHD]0 = 2 mM) results in eeHG values of 87% and 95% for initial ee0 levels of C*-NI of 10% and 20%, respectively (Fig. 7c) (see ESI Notes 5, 6, and 7† for details on the numerical simulation). In this way, the amplified chirality of the host is transmitted to the guest with minimal loss, creating a nearly enantiopure environment for the guest.
To assess the robustness of this chiral amplification, we simulated how the eeHG value changes at a low host ee0 of 20% as the guest concentration increases. As shown in Fig. 7d, even with a 5-fold increase in guest concentration (from 2 mM to 10 mM) depleting the available host, the eeHG value decreased only slightly, from 95% to 85%. The resilience of the system is attributed to the strong heterochiral dimerization, which tolerates perturbation by the host–guest complexation event. Without mutual antagonism, the eeHG value would directly reflect the host's ee0 of 20% under all conditions. With mutual antagonism, however, it is elevated and maintained above 85%. This transfer and amplification of chiral information is effected solely by mutual antagonism of the mirror-image host, occurring at the interface between chiral and nonchiral molecular domains (see ESI Notes 5, 6, and 7† for details on the numerical simulations). The mutual antagonism inevitably depends on host concentration yet remains remarkably resilient. At lower concentrations, the host's reduced tendency to form dimers attenuates the effect, leading to less pronounced chirality amplification and transfer (Fig. S22†). Nevertheless, the exceptionally strong heterochiral dimerization of C*-NI ensures that mutual antagonism persists, even at μM concentrations.
By definition, transfer and amplification require pre-existing chirality and incoming pro-chirality.6,9–11 Evolutionary models propose that the transfer of specific chirality from the primordial soup, which was nearly a racemic mixture, into planar, nonchiral polyaromatic hydrocarbons is a crucial entry point to the RNA world.62–65 The conceptual relevance of our chiral clips, which operate solely through weak π–π interactions and hydrogen bonds, as a model for this chiral–nonchiral juncture is an intriguing question that invites further study.
Conclusions
We have developed molecular clips with remarkable chiral discrimination, regulating the interdependent monomer–dimer exchange and host–guest complexation. A robust heterochiral dimerization between mirror-image clips suppresses guest binding, while weakly associating enantiopure clips better recognize the guests. This property originates from strong, shape-complementary hydrogen bonding between clips of opposite handedness, contrasting with weaker interactions between clips of the same handedness. The behavior mirrors the principle of mutual antagonism, a key hypothesis for the origin of homochirality in nature.In our system, chirality serves as a clever mechanism of controlling the availability of the active host, while not compromising its inherent affinity toward the guest. We demonstrated how nonchiral guests can be recruited into such chiral environments to carry on the chiral information. Future work aims to leverage this host–guest chemistry to integrate chiral amplification by chemical reactions.
Data availability
Experimental procedures, additional data (1H and 13C NMR spectra, FT-IR spectra, HR-MS) of the newly synthesized compounds, and supplementary figures are available in the ESI.† The X-ray crystallographic coordinates for structures reported in this study have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition numbers 2369723 (meso-[C*-NI]2), 2369724 (meso-[C*-P2]2), 2369725 ((R)-C*-P2@(S)-C*-NI), and 2369726 (rac-[C*-P2 ⊃ PHD]). Short video clips of conceptual animations illustrating heterochiral and homochiral dimerization are provided as ESI Movies.† For ESI, crystallographic data in CIF, and ESI Movies,† see DOI: https://doi.org/10.1039/d4sc07655a.Author contributions
All authors have given approval to the final version of the manuscript. Conceptualization: S. B. and D. L.; data curation: S. B. and Y. J.; formal analysis: S. B. and Y. J.; funding acquisition: D. L.; methodology: S. B. and D. L.; visualization: S. B. and D. L.; supervision: D. L.; writing – original draft: S. B. and D. L.; writing – review & editing: S. B., Y. J., and D. L.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (RS-2024-00342767 and 2021R1A5A1030054).References
- F. Devínsky, Symmetry, 2021, 13, 2277 CrossRef.
- J. D. Carroll, Chirality, 2009, 21, 354–358 CrossRef CAS PubMed.
- H. I. Cruz-Rosas, F. Riquelme, A. Ramírez-Padrón, T. Buhse, G. Cocho and P. Miramontes, J. Theor. Biol., 2020, 499, 110316 CrossRef CAS PubMed.
- G. F. Joyce, A. W. Schwartz, S. L. Miller and L. E. Orgel, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 4398–4402 CrossRef CAS PubMed.
- M. Li, X. He, J. Chen, B. Wang, S. Liu and C. Rong, J. Phys. Chem. A, 2021, 125, 1269–1278 CrossRef CAS PubMed.
- D. G. Blackmond, Cold Spring Harbor Perspect. Biol., 2010, 2, a002147 Search PubMed.
- T. Buhse, J.-M. Cruz, M. E. Noble-Terán, D. Hochberg, J. M. Ribó, J. Crusats and J.-C. Micheau, Chem. Rev., 2021, 121, 2147–2229 CrossRef CAS PubMed.
- Q. Sallembien, L. Bouteiller, J. Crassous and M. Raynal, Chem. Soc. Rev., 2022, 51, 3436–3476 RSC.
- K. Soai, T. Kawasaki and A. Matsumoto, Acc. Chem. Res., 2014, 47, 3643–3654 CrossRef CAS PubMed.
- J. M. Ribó and D. Hochberg, Symmetry, 2019, 11, 814 CrossRef.
- F. C. Frank, Biochim. Biophys. Acta, 1953, 11, 459–463 CrossRef CAS PubMed.
- C. M. Hong, R. G. Bergman, K. N. Raymond and F. D. Toste, Acc. Chem. Res., 2018, 51, 2447–2455 CrossRef CAS PubMed.
- S. Wang and M. W. Urban, Nat. Rev. Mater., 2020, 5, 562–583 CrossRef CAS.
- K. A. Podolsky and N. K. Devaraj, Nat. Rev. Chem, 2021, 5, 676–694 CrossRef CAS PubMed.
- X. N. Han, P. F. Li, Y. Han and C.-F. Chen, Angew. Chem., Int. Ed., 2022, 61, e202202527 CrossRef CAS PubMed.
- X. Yang and W. Jiang, J. Am. Chem. Soc., 2024, 146, 3900–3909 CrossRef CAS PubMed.
- K. Soai, T. Shibata, H. Morioka and K. Choji, Nature, 1995, 378, 767–768 CrossRef CAS.
- X. Xu, J. Zhang, S. Dong, L. Lin, X. Lin, X. Liu and X. Feng, Angew. Chem., Int. Ed., 2018, 57, 8734–8738 CrossRef CAS PubMed.
- K. Tanaka, T. Iwashita, E. Yoshida, T. Ishikawa, S. Otuka, Z. Urbanczyk-Lipkowska and H. Takahashi, Chem. Commun., 2015, 51, 7907–7910 RSC.
- B. M. Armstrong, R. I. Sayler, B. H. Shupe, T. A. Stich, R. D. Britt and A. K. Franz, ACS Catal., 2019, 9, 1224–1230 CrossRef CAS.
- T. P. Le, S. Tanaka, M. Yoshimura and M. Kitamura, Bull. Chem. Soc. Jpn., 2020, 93, 1319–1333 CrossRef.
- N. Li, X.-H. Chen, S.-M. Zhou, S.-W. Luo, J. Song, L. Ren and L.-Z. Gong, Angew. Chem., Int. Ed., 2010, 49, 6378–6381 CrossRef CAS PubMed.
- W. Guo, Y. Luo, H. H.-Y. Sung, I. D. Williams, P. Li and J. Sun, J. Am. Chem. Soc., 2020, 142, 14384–14390 CrossRef CAS PubMed.
- M. Klussmann, H. Iwamura, S. P. Mathew, D. H. Wells Jr, U. Pandya, A. Armstrong and D. G. Blackmond, Nature, 2006, 441, 621–623 CrossRef CAS PubMed.
- Y. Hayashi, M. Matsuzawa, J. Yamaguchi, S. Yonehara, Y. Matsumoto, M. Shoji, D. Hashizume and H. Koshino, Angew. Chem., Int. Ed., 2006, 45, 4593–4597 CrossRef CAS PubMed.
- Y. Liu, D. Fiorito and C. Mazet, Chem. Sci., 2018, 9, 5284–5288 RSC.
- L. Ye, Y. Tian, X. Meng, Q.-S. Gu and X.-Y. Liu, Angew. Chem., Int. Ed., 2020, 59, 1129–1133 CrossRef CAS PubMed.
- J. Frey, A. Malekafzali, I. Delso, S. Choppin, F. Colobert and J. Wencel-Delord, Angew. Chem., Int. Ed., 2020, 59, 8844–8848 CrossRef CAS PubMed.
- M. Hatano, H. Okamoto, T. Kawakami, K. Toh, H. Nakatsuji, A. Sakakura and K. Ishihara, Chem. Sci., 2018, 9, 6361–6367 RSC.
- X. Zhu, Y. Li and H. Bao, Chin. J. Chem., 2023, 41, 3097–3114 CrossRef CAS.
- M. Kitamura, S. Suga, H. Oka and R. Noyori, J. Am. Chem. Soc., 1998, 120, 9800–9809 CrossRef CAS.
- D. G. Blackmond, Chem. Rev., 2019, 120, 4831–4847 CrossRef PubMed.
- S. V. Athavale, A. Simon, K. N. Houk and S. E. Denmark, Nat. Chem., 2020, 12, 412–423 CrossRef CAS PubMed.
- A. Wu, A. Chakraborty, J. C. Fettinger, R. A. Flowers II and L. Isaacs, Angew. Chem., Int. Ed., 2002, 41, 4028–4031 CrossRef CAS PubMed.
- M. Chas, G. Gil-Ramírez, E. C. Escudero-Adán, J. Benet-Buchholz and P. Ballester, Org. Lett., 2010, 12, 1740–1743 CrossRef CAS PubMed.
- L.-Y. Yao, T. K.-M. Lee and V. W.-W. Yam, J. Am. Chem. Soc., 2016, 138, 7260–7263 CrossRef CAS PubMed.
- M. Ueda, T. Aoki, T. Akiyama, T. Nakamuro, K. Yamashita, H. Yanagisawa, O. Nureki, M. Kikkawa, E. Nakamura, T. Aida and Y. Itoh, J. Am. Chem. Soc., 2021, 143, 5121–5126 CrossRef CAS PubMed.
- H. Jȩdrzejewska and A. Szumna, Chem. Rev., 2017, 117, 4863–4899 CrossRef PubMed.
- H. Lee and D. Lee, Chem.–Eur. J., 2023, 29, e202302523 CrossRef CAS PubMed.
- F.-G. Klärner and B. Kahlert, Acc. Chem. Res., 2003, 36, 919–932 CrossRef PubMed.
- M. Hardouin-Lerouge, P. Hudhomme and M. Sallé, Chem. Soc. Rev., 2011, 40, 30–43 RSC.
- C.-J. Wallentin, T. Wixe, O. F. Wendt, K.-E. Bergquist and K. Wärnmark, Chem.–Eur. J., 2010, 16, 3994–4002 CrossRef CAS PubMed.
- M. Harmata, Acc. Chem. Res., 2004, 37, 862–873 CrossRef CAS PubMed.
- B. Legouin, P. Uriac, S. Tomasi, L. Toupet, A. Bondon and P. van de Weghe, Org. Lett., 2009, 11, 745–748 CrossRef CAS PubMed.
- V. K. Potluri and U. Maitra, J. Org. Chem., 2000, 65, 7764–7769 CrossRef CAS PubMed.
- B. Saha, S. A. Ikbal, A. G. Petrovic, N. Berova and S. P. Rath, Inorg. Chem., 2017, 56, 3849–3860 CrossRef CAS PubMed.
- Z. Wang, H. Ai, A. Hao and P. Xing, Chem. Mater., 2022, 34, 10162–10171 CrossRef CAS.
- I. G. Stará and I. Starý, Acc. Chem. Res., 2020, 53, 144–158 CrossRef PubMed.
- T. Mori, Chem. Rev., 2021, 121, 2373–2412 CrossRef CAS PubMed.
- E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai and K. Maeda, Chem. Rev., 2016, 116, 13752–13990 CrossRef CAS PubMed.
- H. Lee and D. Lee, Commun. Chem., 2022, 5, 180 CrossRef CAS PubMed.
- X. Wu, J. Malinčík, A. Prescimone and C. Sparr, Helv. Chim. Acta, 2022, 105, e202200117 CrossRef CAS.
- D. Bialas, A. Zitzler-Kunkel, E. Kirchner, D. Schmidt and F. Würthner, Nat. Commun., 2016, 7, 12949 CrossRef CAS PubMed.
- K. A. Wheeler, R. C. Grove, R. E. Davis and W. S. Kassel, Angew. Chem., Int. Ed., 2007, 47, 78–81 CrossRef PubMed.
- M. Kimoto, S. Sugiyama, K. Kumano, S. Inagaki and S. Ito, J. Am. Chem. Soc., 2024, 146, 17559–17565 CrossRef CAS PubMed.
- L. Fielding, Tetrahedron, 2000, 56, 6151–6170 CrossRef CAS.
- L. Cao, M. Šekutor, P. Y. Zavalij, K. Mlinarić-Majerski, R. Glaser and L. Isaacs, Angew. Chem., Int. Ed., 2014, 53, 988–993 CrossRef CAS PubMed.
- M. Yamashina, M. Akita, T. Hasegawa, S. Hayashi and M. Yoshizawa, Sci. Adv., 2017, 3, e1701126 CrossRef PubMed.
- G. Tobajas-Curiel, Q. Sun, J. K. M. Sanders, P. Ballester and C. A. Hunter, Chem. Sci., 2023, 14, 6226–6236 RSC.
- M. Kitamura, S. Suga, M. Niwa, R. Noyori, Z.-X. Zhai and H. Suga, J. Phys. Chem., 1994, 98, 12776–12781 CrossRef CAS.
- A. Matsumoto, T. Abe, A. Hara, T. Tobita, T. Sasagawa, T. Kawasaki and K. Soai, Angew. Chem., Int. Ed., 2015, 54, 15218–15221 CrossRef CAS PubMed.
- R. Shapiro, Origins Life Evol. Biospheres, 1995, 25, 83–98 CrossRef CAS PubMed.
- P. Ehrenfreund, S. Rasmussen, J. Cleaves and L. Chen, Astrobiology, 2006, 6, 490–520 CrossRef CAS PubMed.
- J. Spitzer and B. Poolman, Microbiol. Mol. Biol. Rev., 2009, 73, 371–388 CrossRef CAS PubMed.
- C. D. Bastian and H. Rabitz, Life, 2021, 11, 1419 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and additional data (1H and 13C NMR spectra, FT-IR spectra, HR-MS) of the newly synthesized compounds. CCDC 2369723–2369726. Conceptual animation of heterochiral and homochiral dimerizations as short video clips. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc07655a |
| This journal is © The Royal Society of Chemistry 2025 |