
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhysicochemical properties and application of concentrated KN(SO2F)2/sulfolane solution in high-voltage high-power K-ion batteries†
Daisuke
Igarashi
 a,
Ryoichi
Tatara‡
a,
Satoshi
Yasuno
b and
Shinichi
Komaba
*a
a,
Ryoichi
Tatara‡
a,
Satoshi
Yasuno
b and
Shinichi
Komaba
*a
aDepartment of Applied Chemistry, Tokyo University of Science, Shinjuku, Tokyo, 162-8601, Japan. E-mail: komaba@rs.tus.ac.jp
bJapan Synchrotron Radiation Research Institute (JASRI), Sayo-gun, Hyogo 679-5198, Japan
First published on 26th October 2024
Abstract
To achieve improved performance and practical applications of K-ion batteries (KIBs), which are considered next-generation batteries without resource constraints, electrolytes that can maximize the electrochemical performance of K-insertable electrode materials are crucial. In this study, a potassium bis(fluorosulfonyl)amide (KFSA)/sulfolane (SL) system was investigated as a potential electrolyte candidate for KIBs. The KFSA/SL solutions were found to be in the liquid state at room temperature, even at a highly concentrated KFSA/SL molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (corresponding to 5.1 mol dm−3). A systematic spectroscopic analysis revealed that the KFSA/SL solution has a K+–SL–K+ bridge-type structure unique to SL-based electrolytes. In addition, the solutions remained in the liquid state at high concentrations by suppressing the crystallization of solvates, because the interaction between K+ and SL was weaker than that between Li+ or Na+ and SL systems. The concentrated KFSA/SL solution (1:1) exhibited superior electrochemical stability, which enabled stable cycling of the graphite negative electrodes and a high-voltage operation of the K2Mn[Fe(CN)6] and KVPO4F positive electrodes. Furthermore, the superior cation-transport properties of the electrolyte owing to the bridge-type structure improved the rate capability of the electrode-active materials. This study advances the possibility of using novel electrolytes for realizing high-power, high-voltage KIBs.
:1 (corresponding to 5.1 mol dm−3). A systematic spectroscopic analysis revealed that the KFSA/SL solution has a K+–SL–K+ bridge-type structure unique to SL-based electrolytes. In addition, the solutions remained in the liquid state at high concentrations by suppressing the crystallization of solvates, because the interaction between K+ and SL was weaker than that between Li+ or Na+ and SL systems. The concentrated KFSA/SL solution (1:1) exhibited superior electrochemical stability, which enabled stable cycling of the graphite negative electrodes and a high-voltage operation of the K2Mn[Fe(CN)6] and KVPO4F positive electrodes. Furthermore, the superior cation-transport properties of the electrolyte owing to the bridge-type structure improved the rate capability of the electrode-active materials. This study advances the possibility of using novel electrolytes for realizing high-power, high-voltage KIBs.
Introduction
K-Ion batteries (KIBs) have emerged as sustainable next-generation batteries that do not require minor elements, and are promising alternatives to Li-ion batteries (LIBs).1 Compared with Na-ion batteries (NIBs),2 KIBs are advantageous because the standard electrode potential of K+/K is as low as that of Li+/Li in non-aqueous electrolytes. They also benefit from the maturity of graphite negative electrodes that are widely used in conventional LIBs.3–5 In recent years, extensive research has been conducted on highly concentrated electrolytes for LIBs, NIBs, and KIBs6–8 for developing electrolytes with a wide potential window, which are compatible with both the high 4 V-class positive electrodes9,10 and the negative electrodes operated close to the metal-plating potential.11,12 The specific solution structure of concentrated electrolytes, with contact ion pairs (CIPs) and ionic aggregates (AGGs), improves the electrochemical stability of the electrolyte in terms of both thermodynamics and kinetics. This enhancement is due to the increased oxidation stability of the solvents owing to the minimized free solvent molecules and the protection of the electrode surface by the anion-derived solid electrolyte interphase (SEI).13,14Sulfolane (SL) or sulfone-based concentrated electrolytes have been widely studied as LIB electrolytes because of their desirable characteristics: (1) they generally demonstrate good oxidation stability to enable the operation of 5 V-class positive electrodes.15–17 (2) A bridge structure is formed in which one SL molecule coordinates to two different Li+ ions at a high Li salt concentration, thereby resulting in a remarkably high Li+ transference number.18 Because of these favorable properties, SL-based solutions have been reported as electrolytes not only for LIBs,17 Li metal batteries,19 and Li–S batteries20 but also for NIBs.21,22 The use of SL-based solutions as KIB electrolytes is also of immense interest. However, few studies have applied SL-based electrolytes to K systems. Li et al. reported a concentrated electrolyte comprising potassium bis(fluorosulfonyl)amide (KFSA) and SL in a molar ratio of approximately 1:1.6.23 However, their study focused only on dual graphite batteries and not on typical KIBs, and hence, the K+ transport properties of the SL-based electrolytes were not examined.
In this study, we prepared and investigated KFSA/SL solutions (see Fig. S1† for the chemical structures) as KIB electrolytes in terms of their fundamental physical properties, cathodic and anodic electrochemical stabilities, and impact on the battery performance of the active materials. In addition, we systematically examined the solution chemistry of the KFSA/sulfone electrolyte and compared it with those of the lithium bis(fluorosulfonyl)amide (LiFSA)/sulfone and sodium bis(fluorosulfonyl)amide (NaFSA)/sulfone systems to understand the effect of the cationic species in sulfone-based electrolytic systems.
Experimental section
Materials for preparing the electrolytes
KFSA (>99.9%, extra dry) was purchased from Solvionic, France. KPF6 (battery grade, H2O < 110 ppm) and battery-grade electrolyte solvents (H2O < 30 ppm) such as SL, 3-methylsulfolane (MeSL), triglyme (G3), ethylene carbonate (EC), propylene carbonate (PC) and diethyl carbonate (DEC) were purchased from Kishida Chemicals Co., Ltd, Japan. LiFSA (>99.0%, H2O < 200 ppm) was purchased from Kishida Chemicals Co., Ltd, Japan, while NaFSA (>99%) was purchased from Solvionic, France. All reagents were used as received without any further purification and stored in an Ar-filled glovebox with a dew point of approximately −80 °C.Synthesis
A Prussian blue analogue, K2Mn[Fe(CN)6], was synthesized via a chelate-assisted precipitation method reported in our previous study.24 In particular, 40 mmol of K4Fe(CN)6 trihydrate (Kanto Chemical Co., Inc., Japan) and 40 mmol of MnCl2 tetrahydrate (Sigma-Aldrich, USA) were separately dissolved in 1 L of a 0.2 mol dm−3 aqueous solution of tripotassium citrate (Kanto Chemical Co., Inc., Japan). The MnCl2 solution was then added dropwise to the K4Fe(CN)6 solution at a rate of 5 mL min−1 under constant N2 bubbling. The as-obtained white precipitate was aged for 15 h, collected via centrifugation, washed several times with water and once with ethanol, and finally, vacuum dried at 100 °C overnight. The chemical composition of the final product was determined to be K1.95Mn[Fe(CN)6]0.95 using inductively coupled plasma-optical emission spectrometry (ICP-OES).KVPO4F was synthesized via a two-step solid-state reaction according to a previously reported method, with some modifications.25 First, a VPO4 precursor was prepared using the carbothermal method. In particular, V2O5 (Kanto Chemical Co., Inc., Japan), NH4H2PO4 (Wako Pure Chemical Industries, Japan), and acetylene black (Li-100, Denka Co., Ltd, Japan) were mixed in a molar ratio of 1:2:2.1 via planetary ball milling using an acetone dispersion medium and dried at room temperature for several hours and then at 80 °C in an oven. The mixed powder was pelletized, heated to 750 °C at a ramp rate of 5 °C min−1 under Ar gas flow (200 mL min−1), and finally sintered at 750 °C for 4 h. The as-obtained VPO4 powder was mixed with KF (Wako Pure Chemical Industries, Japan) via planetary ball milling without a dispersant. The KF/VPO4 mixture was pelletized, wrapped in Au foil, and heated at 700 °C for 4 h under an Ar gas flow (50 mL min−1). Wrapping with Au foil was essential to suppress the evaporation of F during the heating process.
Electrode preparation and electrochemical measurements
:5 using deionized water as the dispersion medium to form a homogeneous slurry, followed by application onto an Al foil and drying at 80 °C.
:9.8:9.7 using N-methylpyrrolidone (NMP, Kanto Chemical Co., Inc., Japan) to form a homogeneous slurry. A single-walled carbon nanotube (SWCNT) dispersant consisting of 0.4% SWCNT, 0.6% PVDF, and 99% NMP was added to the slurry such that the resulting composition ratio of the active material, KB, SWCNT, and PVdF was 80:9.8:0.2:10. The mixed slurry was applied onto an Al foil and dried at 80 °C.
:10:10 using dehydrated NMP as the dispersion medium. The subsequent preparation process was identical to that described above for the graphite electrode.
The active material//K half cells, KVPO4F//graphite full cells, and K//K symmetric cells were assembled into 2032-type coin cells (Hohsen Corp., Japan). A glass fiber filter disc (GB-100R, ADVANTEC Inc., Japan) was used as the separator. The galvanostatic charge and discharge tests were conducted at 25 °C using a TOSCAT-3100 charge–discharge test system (TOYO SYSTEM, Japan). All other electrochemical measurements were conducted using a VMP-3 potentiostat (BioLogic, France) at 25 °C. Rate capability test of K2Mn[Fe(CN)6], cyclic voltammetry (CV) and linear sweep voltammetry (LSV) of the metallic substrates were performed using a three-electrode cell (SB9 for the rate capability test and SB1A for CV and LSV, EC FRONTIER, Japan) with activated carbon as the counter electrode and K metal wire as the reference electrode. The detailed cell configuration is described in our previous study.26 The potential sweep rate was set to 1 mV s−1 for both CV and LSV.
Characterization
The ionic conductivities of the electrolytes were measured using an ionic conductivity meter (CM-41X, TOA DKK, Japan) in an incubator set at different temperatures. The viscosities and densities of the electrolytes were evaluated using a rolling ball viscometer (Lovis 2000 M/ME, Anton Paar, Austria) equipped with a density meter. The thermal phase transition behaviors of the electrolytes were analyzed using differential scanning calorimetry (DSC 3500 Sirius, NETZSCH, Germany). The electrolyte samples were sealed in aluminum pans in an Ar-filled glovebox. The sample temperature was first increased to 110 °C and held for 10 min, decreased to −100 or −130 °C and again held for 10 min, and finally increased to 110 °C at a heating/cooling rate of 5 °C min−1. The thermograms were recorded during the final heating process. Raman spectra of the electrolyte solutions were recorded using a 532 nm laser Raman spectrometer (Raman 11i, Nanophoton, Japan), and were then deconvoluted using the pseudo-Voigt function as required. Hard X-ray photoelectron spectroscopy (HAXPES) was conducted at the BL46XU beamline of SPring-8 at an X-ray excitation energy of 7939 eV. The photoelectron detection angle was set at 80° and the pass energy of the analyzer was set to 100 eV. The obtained spectra were deconvoluted using the pseudo-Voigt function. The binding energy of the obtained spectrum was calibrated with the binding energy of the sp2 carbon of graphite (284.6 eV), and the peak intensity was normalized to that of sp2 carbon. Ex situ X-ray diffraction (XRD) measurements of the graphite electrode were performed using an X-ray diffractometer with Ni-filtered Cu Kα radiation (SmartLab, Rigaku Corporation, Japan). A custom-made airtight sample holder was used to prevent the electrode samples from being exposed to air.Results and discussion
Photographs of the XFSA/SL mixtures (X = Li, Na, or K) with various mixing ratios are displayed in Fig. 1a. Henceforth, samples with a salt/solvent mixing ratio of 1/n will be denoted as “salt/solvent = 1/n.” According to the phase diagrams reported by Dokko et al., the LiFSA/SL and NaFSA/SL systems exhibited stable solid solvate crystals at room temperature when the LiFSA/SL ratio was 1/5 or 1/1 and the NaFSA/SL ratio was 1/3 or 1/0.5.21,27 Therefore, the LiFSA/SL and NaFSA/SL systems with highly concentrated mixing ratios cannot be used as battery electrolytes at room temperature. In contrast, the KFSA/SL solutions prepared in this study were homogeneous at room temperature, even at a concentrated mixing ratio of KFSA/SL > 1/1. Although SL is a solid at room temperature with a melting point of approximately 28 °C, the mixture with even a small amount of KFSA salt added (KFSA/SL = 1/20) was liquid at room temperature. The molar concentration of KFSA/SL = 1/1 was calculated to be 5.1 mol dm−3 from the density measured at room temperature (see Table S1†), which is higher than those of previously reported KFSA/SL solutions.23,28 The phase diagrams of the KFSA/SL = 1/n binary system electrolytes, based on DSC measurements (see Fig. S2†), is shown in Fig. 1b. In the region where the mole fraction of KFSA was low (xKFSA < 0.29, n < 2.5), the KFSA/SL mixtures exhibited melting points. The melting point reached a maximum for n = 4 (xKFSA = 0.2), and the melting point depression appeared to be caused by the eutectic effect between KFSA/SL = 1/4 and the solvates of other compositions before and after xKFSA = 0.2. However, the phase diagram for xKFSA < 0.4 is complicated, and no stable compositions other than that with n = 4 can be determined. In the high-concentration region (xKFSA > 0.29), only glass-transition points were observed, and these mixtures did not exhibit crystallization or subsequent melting behavior. Therefore, the concentrated KFSA/SL solutions were found to be in a metastable liquid state; however, this state remained for a very long period, and no signs of crystallization were observed. The KFSA/SL = 1/1 electrolyte remained in the liquid state without any precipitate, even after storing in a freezer for more than one year at approximately −18 °C (see Fig. S3†). Therefore, the concentrated KFSA/SL solution can be concluded to have more favorable physicochemical properties as a battery electrolyte than the analogous Li or Na systems. | ||
| Fig. 1 (a) Photographs of the XFSA/SL = 1/n mixtures (X = Li, Na, or K). Numbers on the bottle caps indicate the value of n for each solution. (b) Phase diagram of the KFSA/SL = 1/n binary system. The red circles, blue squares, and black crosses indicate the melting points (Tm), glass-transition points (Tg), and phase transition points that are not specifically attributed (Tx), respectively. (c) Walden plots of the KIB electrolytes. Raman spectra corresponding to (d) SO2 vibration of SL and (e) S–N–S vibration of the FSA− anion in KFSA/SL = 1/1. | ||
As shown in Fig. S4,† the dependence of the ionic conductivity and viscosity on the KFSA concentration is similar to that of many other non-aqueous electrolytes. The ionic conductivity reached its maximum value when the KFSA concentration was approximately 1 mol dm−3. The relationship between the molar conductivity, Λ, and viscosity, η, at 25 °C, of several KIB electrolytes, including KFSA/SL = 1/1, is shown in Fig. 1c as a Walden plot (see Table S2† for the physicochemical properties of KFSA/G3 = 1/1 and 1 mol dm−3 KPF6/EC:PC (1:1 v/v%)). The plot of KFSA/SL = 1/1 appeared immediately below the KCl ideal line, and it was located considerably closer to the ideal line than those of KFSA/G3 = 1/1 or 1 mol dm−3 KPF6/EC:PC, indicating that the KFSA/SL = 1/1 electrolyte exhibited better ionic dissociation than the other electrolytes. The KFSA/SL = 1/1 electrolyte demonstrated better ionic dissociation owing to its higher KFSA concentration compared to the KFSA/SL = 1/10 electrolyte (see Fig. S5a† for a more detailed analysis of the concentration dependence). The values of KFSA/SL = 1/1 on this plot were also substantially affected by the temperature, as shown in Fig. S5b;† they deviated from the ideal line at higher temperatures.
The solvation structures of the KFSA/SL solutions were analyzed using Raman spectroscopy. Fig. 1d shows the Raman spectra of the neat SL and KFSA/SL = 1/n solutions, wherein the Raman scattering of the SO2 scissoring vibration of neat SL is observed at approximately 567 cm−1. This peak not only shifted toward higher wavenumbers with increasing KFSA concentrations because of an increase in the solvent–cation interactions, but also broadened and exhibited an asymmetric tail. This is a typical behavior observed for concentrated sulfone-based solutions,18,21 and the peak asymmetrization is due to the appearance of a new peak at approximately 578 cm−1, originating from a bridge-type SL, where one SL molecule simultaneously coordinates to two cations, as shown in Fig. S6.† The peak of KFSA/SL = 1/1 was observed at a lower wavenumber than that of KFSA/SL = 1/1.5, which is probably due to the presence of fewer SL molecules in the highly concentrated KFSA/SL = 1/1 solution (see Fig. S7† for a clearer depiction). Assuming that the bridge-type fraction is maximized and that no free SL molecules are present in the KFSA/SL = 1/1.5 solution, the peak positions of the monodentate and bridge type SL were estimated to be 571.3 and 578.1 cm−1, respectively, based on the peak deconvolution of the spectrum of KFSA/SL = 1/1.5 shown in Fig. S8.† Notably, the shift and asymmetrization of the SO2 scissoring peak are typical phenomena of concentrated SL solutions;18 however, the degree of asymmetrization depends on the cationic species. Fig. S9† shows the Raman spectra of neat SL, KFSA/SL = 1/3, and LiFSA/SL = 1/3. The spectrum of LiFSA/SL = 1/3 exhibited more pronounced tailing on the side of higher wavenumbers than that of KFSA/SL = 1/3.
The Raman spectra of the KFSA/SL = 1/n solutions corresponding to the S–N–S vibration of FSA− anions are shown in Fig. 1e. The peak position shifted to a higher wavenumber with increasing KFSA concentration owing to an increase in the cation–anion interactions caused by the formation of CIPs and AGGs, as observed in the case of other FSA salt-based concentrated electrolytes. The formation of CIPs and AGGs in such concentrated electrolytes is considered beneficial in many studies because it facilitates the decomposition of anions and subsequent SEI formation on the surface of the metal or carbon negative electrodes.11,29,30
Although the effect of cationic species on the solution structure of the XFSA/SL system is of immense interest, the large difference in the melting points of stable solvates depending on the cationic species makes it difficult to compare the physical properties of XFSA/SL systems for the same salt/SL mixing ratio. Therefore, instead of the XFSA/SL system, we compared the physical properties of mixtures of the XFSA (X = Li, Na, or K) and MeSL (which has a methyl group added to the five-membered ring of SL, Fig. S10†) system. In the LiFSA/MeSL and NaFSA/MeSL systems, the crystallization of the XFSA·xMeSL solvate at highly concentrated compositions is prevented by the steric hindrance caused by the methyl group, which makes the LiFSA/MeSL = 1/n and NaFSA/MeSL = 1/n (1 < n ≤ 7) solutions glass-forming liquids at room temperature.22,27
The phase diagram of the KFSA/MeSL system in Fig. 2a shows that the concentrated KFSA/MeSL solutions exhibit a glass-forming liquid state at ambient temperature over a wide range of mixing ratios, similar to the LiFSA/MeSL and NaFSA/MeSL systems. The corresponding DSC curves are presented in Fig. S11.† In our study, uniform liquids were obtained at room temperature for XFSA/MeSL = 1/1.5, for all the LiFSA, NaFSA, and KFSA/MeSL systems. Solid precipitates were observed in solutions that were more concentrated than NaFSA/MeSL = 1/1.4, whereas the LiFSA/MeSL and KFSA/MeSL solutions remained as liquids even for n = 1.
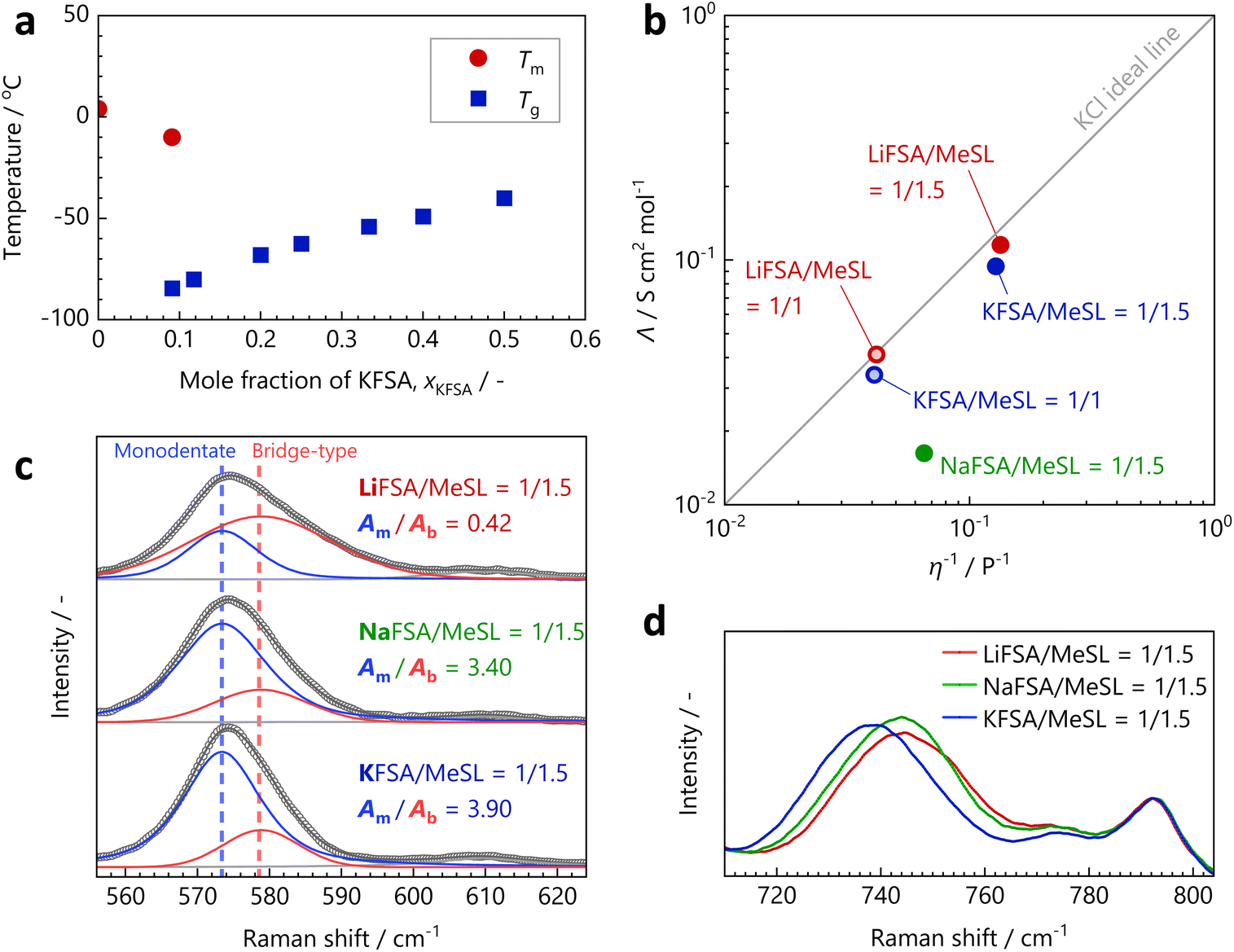 | ||
| Fig. 2 (a) Phase diagram of the KFSA/MeSL binary system. (b) Walden plot of the concentrated XFSA/MeSL (X = Li, Na, or K) solutions at room temperature. (c) Raman spectra of XFSA/MeSL = 1/1.5 corresponding to the SO2 scissoring vibration of MeSL. The open circles, and blue, red, and black solid lines indicate the experimental values, deconvoluted peaks attributable to monodentate MeSL, deconvoluted peaks corresponding to bridge-type MeSL, and the fitting total, respectively. The minor peak observed at approximately 610 cm−1 is attributed to the alkyl chains of MeSL. (d) Raman spectra of XFSA/MeSL = 1/1.5 corresponding to the S–N–S vibration of the FSA− anions. The minor peaks observed at approximately 774 and 792 cm−1 are attributed to the alkyl chains of MeSL. | ||
The Walden plot of the concentrated XFSA/MeSL solutions at room temperature is shown in Fig. 2b. The ionic conductivity, viscosity, and density of each electrolyte are listed in Tables S3 and S4.† The plots of the LiFSA/MeSL and KFSA/MeSL solutions were observed near the KCl ideal line, and the plot of the Li system was closer to this line than that of the K system for the same salt/MeSL mixing ratio. By contrast, the plot of NaFSA/MeSL = 1/1.5 was located far from the ideal line. This suggests that the approximate estimated degree of ionic dissociation for the Li, Na, and K systems is in the order of Li > K ≫ Na. A pronounced increase in the viscosity and decrease in the conductivity of concentrated solutions of NaFSA have been generally observed for other solvent systems, such as 1,2-dimethoxyethane and propylene carbonate.31,32
The Raman spectra of the XFSA/MeSL = 1/1.5 solutions corresponding to the SO2 scissoring vibration of MeSL are shown in Fig. 2c. Each experimental spectrum was deconvoluted into peaks for two components, monodentate and bridge-type MeSL, with peak areas of Am and Ab, respectively. The Am/Ab ratios for the Li, Na, and K systems were calculated to be 0.42, 3.40, and 3.90, respectively, and the integral intensity of Ab followed the order of Li ≫ Na > K. The bridge-type solvation structure was considerably more developed in the Li system than in the other systems, probably because of its strong Lewis acidity. The Raman spectra of the XFSA/MeSL = 1/1.5 solutions corresponding to the S–N–S vibration of the FSA− anions are shown in Fig. 2d, wherein a more pronounced Raman shift can be observed for smaller cationic systems. However, the Raman shift values for the Li and Na systems were almost the same. This indicates that K+ interacts more weakly with the anions than Na+ and Li+, or that Na+ has a stronger interaction than what is intuitively estimated based on the order of the ionic radius and Lewis acidity. Briefly, the Raman spectral analysis results indicate that Li+ interacts strongly with both the solvents and anions, whereas Na+ interacts strongly only with the anions.
The above discussion on the compositions with stoichiometric solvates and melting points of the XFSA/SL mixtures, and the solution structure of the XFSA/MeSL solutions can be summarized as follows: Li+ tends to form CIPs and AGGs easily with solvents, with anions and bridge-type solvation structures. This facilitates ion dissociation and cation transport, and is therefore favorable for use in LIB electrolytes. However, strong Li+–solvent and Li+–anion interactions enhance the thermodynamic stability of the solvate crystal phase and induce solvate precipitation in concentrated compositions such as LiFSA/SL = 1/1. Na+ has a significantly weaker interaction with solvents to form a bridge-type structure than Li+; however, it exhibits unusually strong CIP- and AGG-forming interactions with anions, which make concentrated NaFSA solutions viscous and poorly ion-dissociable, thereby making their use as NIB electrolytes difficult, unless they are in a high-temperature environment.21,22 The weak Lewis acidity of K+ results in weak interactions with both solvents and anions and ensures a wide liquid-phase range, where even equimolar mixtures of salts and solvents can remain as liquids. The less rigid bridge solvation structure is disadvantageous compared with that of the Li analog system. The plot of the LiFSA/SL = 1/3 solution was located closer to the ideal line of the Walden plot than that of the KFSA/SL = 1/3 solution. However, the K system enabled the formation of concentrated compositions with better dissociation, such as KFSA/SL = 1/1, without solvate precipitation, because of its weaker K+–solvent and K+–anion interactions (see Fig. S12†).
Because one of the advantages of KIBs is the applicability of a graphite negative electrode, compatibility with graphite, such as cathodic stability and SEI formation ability, is an important aspect of KIB electrolytes. Prior to the electrochemical test with K metal electrodes, the K metal foils were soaked overnight in KFSA/SL = 1/10 or KFSA/SL = 1/1 mixtures to confirm their compatibility for K-half cell tests, as shown in Fig. 3a. The metallic luster was retained in the case of KFSA/SL = 1/1, whereas the surface was dull and black in the case of KFSA/SL = 1/10. The cathodic stability of KFSA/SL = 1/1 was also confirmed from the reversible K-plating/stripping reaction on the Cu substrate, as shown in the cyclic voltammogram in Fig. S13.†
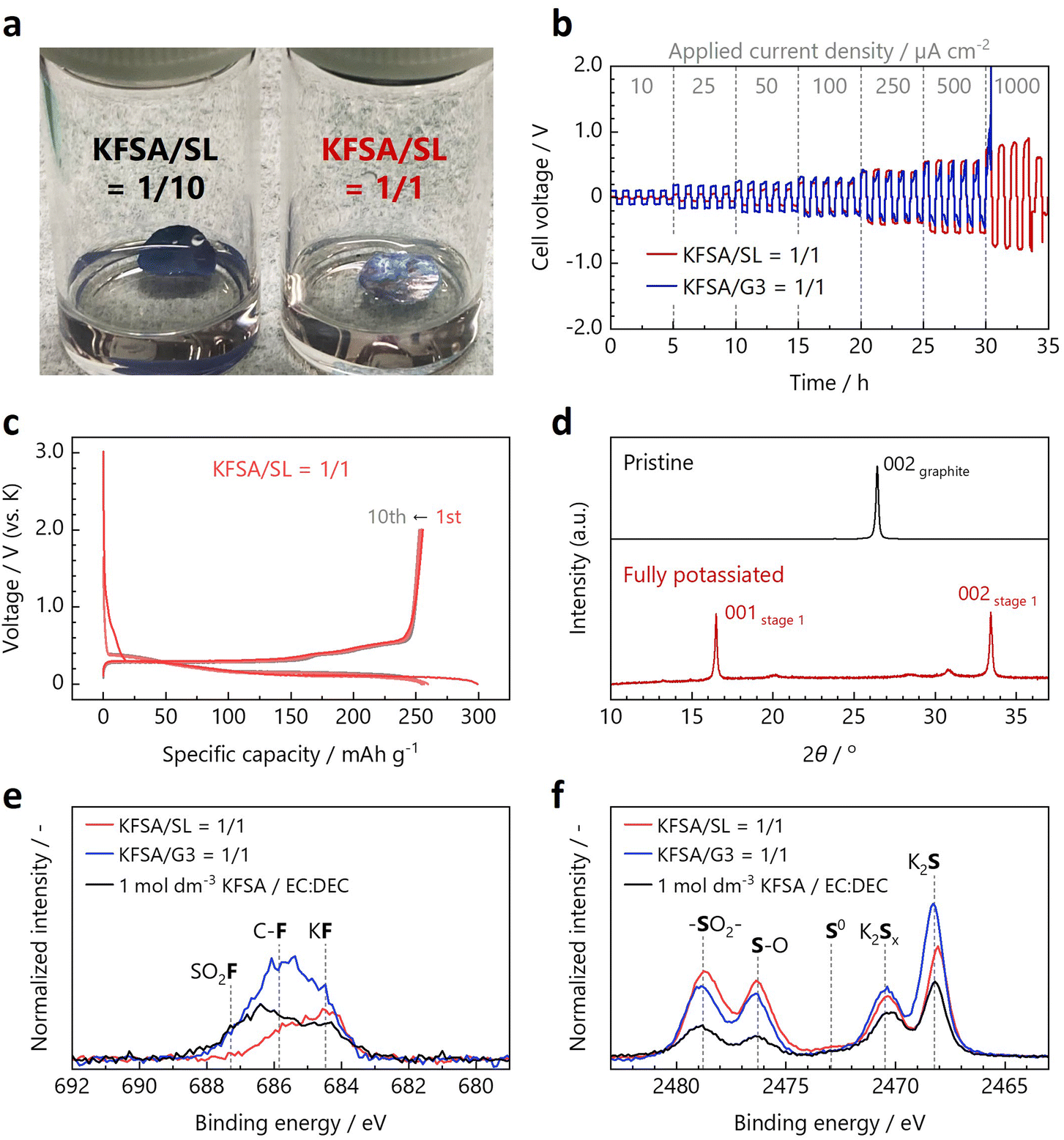 | ||
| Fig. 3 (a) Photographs of K metal disks soaked in the KFSA/SL = 1/10 (left) and KFSA/SL = 1/1 solutions (right). (b) Voltage profiles of K//K symmetric cells with the KFSA/SL = 1/1 and KFSA/G3 = 1/1 electrolytes. (c) Charge and discharge curves of the graphite//K cell with the KFSA/SL = 1/1 electrolyte. (d) Ex situ XRD patterns of pristine and fully potassiated graphite electrodes. (e) F 1s and (f) S 1s HAXPES profiles of graphite electrodes cycled in the KFSA/SL = 1/1, KFSA/G3 = 1/1, and 1 mol dm−3 KFSA/EC:DEC electrolyte. | ||
The current-reversal voltage profiles of the K//K symmetric cells with the KFSA/SL = 1/1 electrolyte and a previously reported ether-based concentrated electrolyte, KFSA/G3 = 1/1, are shown in Fig. 3b. The cell voltage of the K//K cells was smaller when KFSA/SL = 1/1 was used, especially when the applied current density was less than 250 μA cm−2, which is in good agreement with the electrochemical impedance spectroscopy results of the K//K symmetric cells (see Fig. S14†). Although this finding was partially unexpected, considering the cathodically unstable nature of SL at lower potentials,16,33 it could be related to the large desolvation energy in the concentrated G3 electrolyte, which is a long-chain glyme, and the SEI composition on the electrode surface, as discussed later. The key difference between the two electrolytes is their K-plating/stripping behaviors under high current density conditions. The plating/stripping reaction did not proceed properly, while the cell voltage diverged above 2 V at 1000 μA cm−2 in the case of KFSA/G3 = 1/1, whereas it proceeded smoothly under the high current density in the case of KFSA/SL = 1/1. This indicated that the diffusion-limited current density in KFSA/SL = 1/1 was larger than that in KFSA/G3 = 1/1, under this anion-blocking condition. Unlike in the cases of the Li salt/SL and NaFSA/SL systems, the absence of a nuclear magnetic resonance (NMR)-sensitive nucleus in the case of K and the large and unstable impedance of the K-plating/stripping reaction make it difficult to quantify the self-diffusion coefficient of K+ using pulse-field gradient NMR27 and the K+ transference number using the DC polarization technique.21 However, we believe that the large gap in the limiting current density is sufficient to demonstrate fast K+ transport in the KFSA/SL = 1/1 electrolyte, as expected from the solution structure inferred from the Raman spectra. The variations in the limiting current density for the KFSA/SL = 1/1 and KFSA/G3 = 1/1 electrolytes are also in good agreement with the Walden plot shown in Fig. 1c.
Fig. 3c shows the charge–discharge curves of a natural graphite//K cell with the KFSA/SL = 1/1 electrolyte in the voltage range of 0–2 V (vs. K) at a current density of 27.9 mA g−1. The reversible capacity was 256 mA h g−1 in the first cycle, which is sufficiently close to the theoretical capacity of 279 mA h g−1, and the graphite electrode showed acceptable cyclability for 50 cycles (see Fig. S15†). The irreversible capacity of 44 mA h g−1 at the first cycle can be attributed to the electrolyte decomposition and SEI formation.3 The initial coulombic efficiency was calculated to be 85.4%. The obtained curves were almost identical to those obtained for the graphite//K cells with the conventional 1 mol dm−3 KFSA/EC:DEC (1:1 v/v%) and KFSA/G3 = 1/1 electrolytes shown in Fig. S16.† Despite the inferior reduction resistance of SL compared to ether solvents, the graphite electrode with KFSA/SL = 1/1 electrolyte showed a similar performance to that using KFSA/G3 = 1/1. This may be because the KFSA concentration is higher for KFSA/SL = 1/1 (5.1 mol dm−3) than KFSA/G3 = 1/1 (3.5 mol dm−3), where the effect of the improved SEI formation compensates for the poor reduction stability of the SL solvent. Fig. 3d shows the ex situ XRD patterns of the pristine and fully potassiated graphite electrodes. The stage 1 potassium–graphite intercalation compound, KC8, was detected as the main phase in the fully potassiated electrode, which was consistent with the reversible capacity determined from the galvanostatic charge–discharge measurements. Therefore, the KFSA/SL = 1/1 electrolyte is suitable for graphite negative electrodes for KIBs.
The surface conditions of the cycled graphite electrodes were analyzed using HAXPES. The C 1s, O 1s, F 1s, and S 1s HAXPES spectra are presented in Fig. S17, S18† and 3e, f, respectively. It is well known that a KF-rich SEI is formed in electrolytes containing a large amount of KFSA.34–36 The graphite electrodes cycled in the concentrated electrolytes in this study also showed a larger KF signal than that cycled in the conventional electrolyte. In addition, in the case of KFSA/SL = 1/1, the intensity of the peak obtained at approximately 686 eV, which originates from the F-containing organic compounds, is smaller than that obtained in the case of KFSA/G3 = 1/1.
The intensities of the S 1s peaks were also higher for the electrodes cycled in the concentrated electrolytes than that in the conventional electrolyte. Considering that a recent study suggested that not only a KF-rich SEI but also a K2S-rich SEI is ionically conductive and improves the negative electrode properties in K metal battery systems,37 the FSA−-derived S-rich SEI may also contribute toward enhancing the electrochemical properties of graphite electrodes in concentrated electrolytes with respect to those in conventional electrolytes. S–O bonding was detected at a higher binding energy predominantly for the graphite electrode cycled in KFSA/SL = 1/1, whereas the K2Sx and K2S peaks were observed at lower binding energies with higher intensities for the electrode cycled in KFSA/G3 = 1/1. This may imply that the S–O bond-rich SEI components deposited in KFSA/SL = 1/1 are formed via the decomposition of both FSA− anions and SL solvent. Although it is practically difficult to determine which chemical species contributes more to the SEI layer, since sulfur can be supplied from both FSA− and SL, the contribution of SL to the SEI formation has been suggested in studies of graphite electrodes in LIBs by Cai et al. and Zheng et al.38,39 and hard carbon electrodes in NIBs by Kuang et al.40
The compatibility of the KFSA/SL = 1/1 electrolyte with the positive electrode materials was evaluated using LSV. Fig. 4a shows the LSV curves of Pt foil in the KFSA/G3 = 1/1 and KFSA/SL = 1/1 electrolytes. The detected anodic current in KFSA/SL = 1/1 was smaller than that in KFSA/G3 = 1/1 in the potential region of 4 V vs. K+/K. Because the KFSA/G3 = 1/1 electrolyte was previously reported to be compatible with the K2Mn[Fe(CN)6] electrodes, the KFSA/SL = 1/1 electrolyte was expected to exhibit the same or better performance for positive electrodes compared with KFSA/G3 = 1/1.34
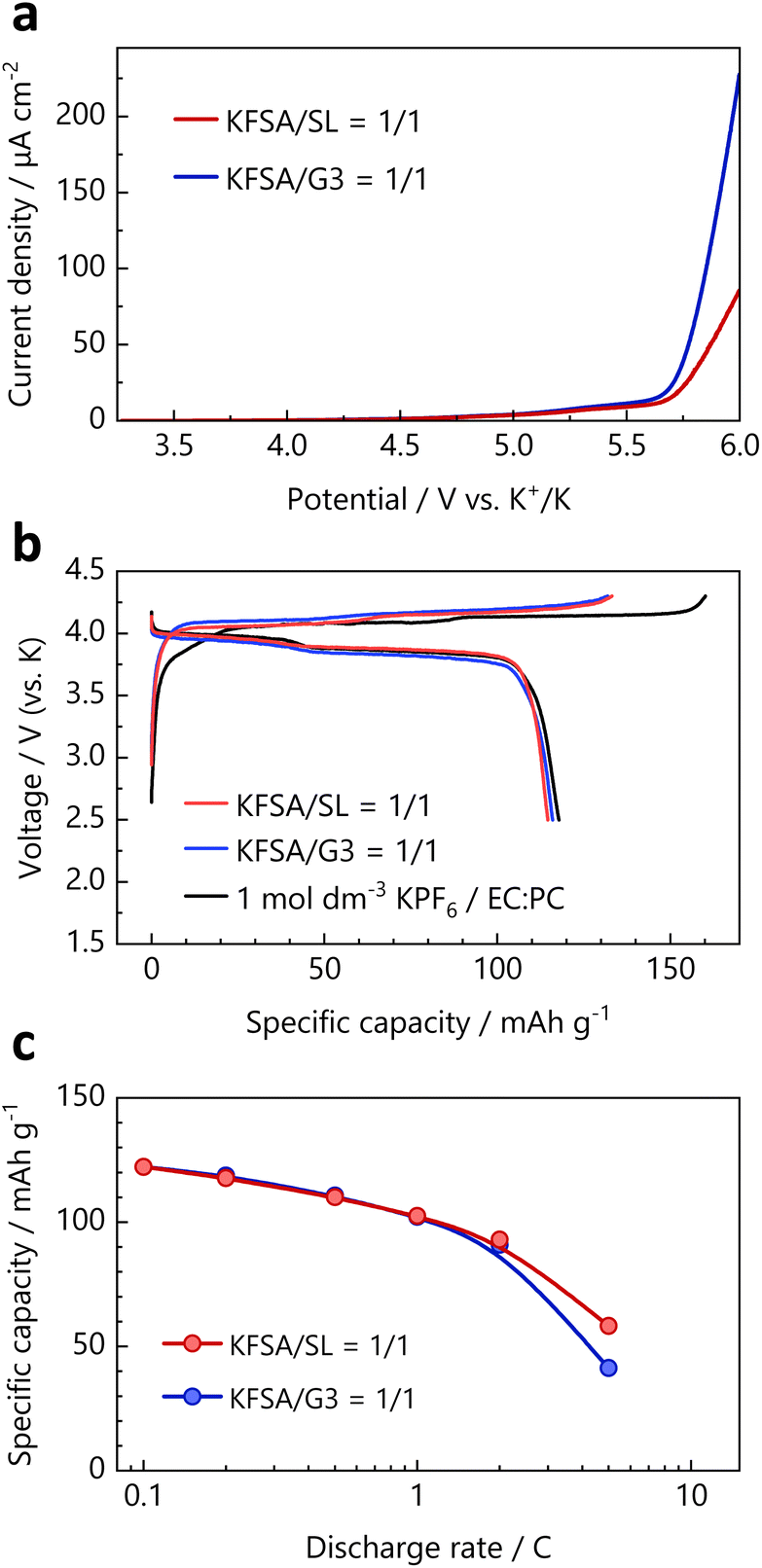 | ||
| Fig. 4 (a) LSV of Pt foil in the KFSA/SL = 1/1 and KFSA/G3 = 1/1 electrolytes. (b) Initial charge–discharge curves of the K2Mn[Fe(CN)6] electrodes in different electrolytes. The current density was 15.5 mA g−1 (corresponding to 0.1C). (c) Discharge rate dependence of the discharge capacity of the K2Mn[Fe(CN)6] electrodes in highly concentrated electrolytes. | ||
Fig. 4b shows the initial charge–discharge curves for the K2Mn[Fe(CN)6]//K cells with the three types of electrolytes: 1 mol dm−3 KPF6/EC:PC, KFSA/G3 = 1/1, and KFSA/SL = 1/1 in the voltage range of 2.5–4.3 V (vs. K) at a current density of 15.5 mA g−1. Both the highly concentrated electrolytes improved the initial coulombic efficiency of the K2Mn[Fe(CN)6] electrodes. In addition to the better oxidation stability of the concentrated electrolytes, we believe the primary reason for this phenomenon to be the favorable anion-derived SEI that forms on the K metal surface in the concentrated electrolytes, which suppresses solvent decomposition on the K metal surface.41,42 This inhibits the so-called “cross-talk” effect between the positive electrode and K metal, which has recently been identified as the dominant factor responsible for the irreversible capacity of the positive electrode in KIB electrolytes.24,43,44 The improvement in the coulombic efficiency by using the KFSA/SL = 1/1 electrolyte also indicates that the corrosion of the Al current collector was suppressed in it, as well as in most highly concentrated FSA-salt-based electrolytes.31,34,45 Fig. S19† shows the coulombic efficiency and long-term cyclability of the K2Mn[Fe(CN)6] electrodes. The concentrated electrolytes improved the coulombic efficiency not only in the first cycle but also during and after the second cycle.
The rate capability of the K2Mn[Fe(CN)6] electrodes in each electrolyte was evaluated using a three-electrode setup. Unlike two-electrode systems, such as coin-type cells, the three-electrode system allows the correct evaluation of the rate capability of the working electrodes without the effect of the large polarization of the K metal-based counter electrodes.24,46 Fig. S20† shows the discharge rate curves of the K2Mn[Fe(CN)6] electrodes at different current densities, and the observed discharge capacity versus the discharge C-rate is plotted in Fig. 4c. The discharge capacities were almost the same at lower C-rates; however, larger discharge capacities were obtained in the KFSA/SL = 1/1 electrolyte than in the KFSA/G3 = 1/1 electrolyte at a high discharge rate of 5C, despite its very low ionic conductivity and high viscosity. This could be because the KFSA/SL = 1/1 electrolyte exhibited a larger diffusion-limited current density than the KFSA/G3 = 1/1 electrolyte, as shown in Fig. 3b. The above discussion demonstrates for the first time the superior cation-transport properties of SL-based concentrated electrolytes for the KFSA/SL system, as well as for the Li and Na analogs.20,21
A schematic of the solution structure of the KFSA/SL = 1/1 electrolyte based on the above experimental results is presented in Fig. 5. The concentrated KFSA/sulfone-based electrolytes not only enlarged the potential window owing to the increased CIPs and AGGs, as observed in other concentrated electrolytes, but also enabled fast K+ hopping transport via the K+–SL–K+ bridge-type solvation structure. It should be emphasized that an electrolyte system with the same ionic concentration cannot be realized for the Li and Na analogs at room temperature.
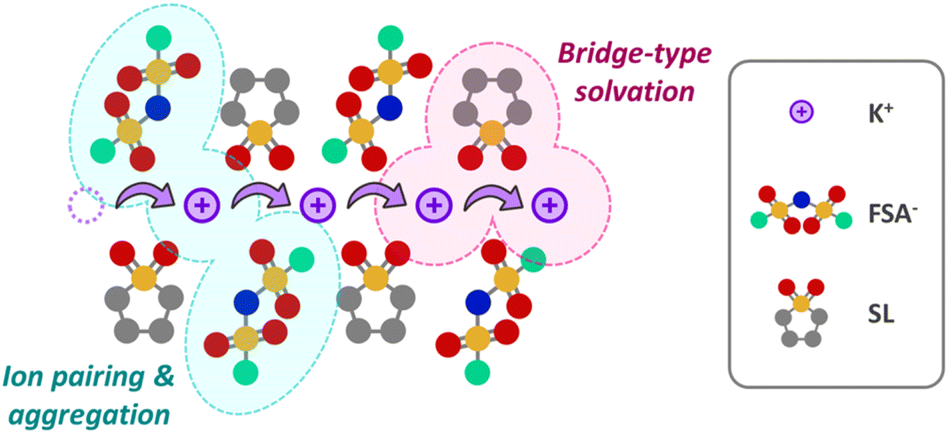 | ||
| Fig. 5 Schematic illustration of the solution structure of the KFSA/SL = 1/1 electrolyte. | ||
The KFSA/SL = 1/1 electrolyte, which exhibits durable anodic stability, is expected to be appropriate for high-voltage KIB materials. KVPO4F is an outstanding positive electrode material, which exhibits sufficient practical capacity (with a maximum of approximately 100 mA h g−1) and has a high working potential of over 4 V vs. K+/K.25,47,48 However, in most cases, a large irreversible capacity is observed in the electrochemical tests on KVPO4F composite//K cells because of its very high working potential, which causes a large amount of electrolyte decomposition. Fig. 6a displays the charge–discharge curves of KVPO4F in the conventional 1 mol dm−3 KPF6/EC:PC electrolyte in the voltage range of 2–4.9 V (vs. K) at a current density of 13.1 mA g−1. Reversible cycling was observed; however, a large irreversible reaction occurred during each charging process, as demonstrated in previous studies.25,48,49 The KFSA/G3 = 1/1 electrolyte continuously decomposed at approximately 4.7 V vs. K+/K during the first charging process (Fig. 6b), and thus, the electrode could not be fully charged to the upper voltage limit of 4.9 V. This was possibly due to the anodic decomposition of G3, an ether-based solvent with relatively poor anodic stability, even in a highly concentrated state.9,34 The reversibility of the charge–discharge reactions was highly improved in the KFSA/SL = 1/1 electrolyte (Fig. 6c) compared with that in the two above-mentioned electrolytes. This can be explained on the basis of two factors: (1) suppression of the cross-talk effect between the KVPO4F and K-metal electrodes owing to the formation of the SEI layer on the K metal surface and (2) the inherently strong anodic stability of SL solvents.15,17 In addition, several studies suggested that SL can be decomposed on the surface of the electrode to produce a cathode electrolyte interphase (CEI).38,40
 | ||
| Fig. 6 Charge–discharge curves of the KVPO4F electrodes in the (a) 1 mol dm−3 KPF6/EC:PC, (b) KFSA/G3 = 1/1, and (c) KFSA/SL = 1/1 electrolytes. (d) Cycling performance and coulombic efficiency of the KVPO4F electrodes. | ||
Therefore, such passivation layer may also contribute to the improvement of the apparent oxidation resistance of KFSA/SL.
The cycling performance and variation trend of the coulombic efficiency of the KVPO4F electrodes in 1 mol dm−3 KPF6/EC:PC and KFSA/SL = 1/1 are shown in Fig. 6d. The coulombic efficiency and cyclability were improved in the KFSA/SL = 1/1 electrolyte. Consistent with this study, Kim et al. suggested that the origin of the capacity degradation of KVPO4F can be attributed to an increase in the surface resistance owing to the deposition of electrolyte decomposition products.50 Parasitic reactions between the charged KVPO4F and electrolyte components in the high-potential region have also been recently reported by Wernert et al.51 The robust anodic stability of KFSA/SL = 1/1 contributes toward improving the battery properties of such high-voltage electrode materials, and it can thus be a remarkable option as an electrolyte for high-voltage KIBs. Based on the above results, a KIB full cell was fabricated using a KVPO4F positive electrode, graphite negative electrode, and KFSA/SL = 1/1 electrolyte. The charge–discharge curves of the full cell are shown in Fig. S21.† The cell exhibited reversible operation with an average discharge voltage of 3.9 V at the initial cycle. Although the further optimization of electrode manufacturing and cell design to improve the cycle performance remains a challenge, the results indicate that the KFSA/SL electrolyte may contribute to the realization of 4 V-class KIBs with high-voltage electrode materials. In addition, Fig. S22† shows the charge–discharge curves of the full cell operated at lower temperature (5 °C) in comparison with that at 25 °C. Despite the high melting point of the SL solvent, the full cell exhibited the reversible operation at 5 °C, although the capacity decreased due to the increased cell resistance at low temperature.
Conclusions
In this study, the solution chemistry of KFSA/SL and XFSA/MeSL mixtures (X = Li, Na, or K) was systematically investigated, and the compatibility of KFSA/SL = 1/1 as a battery electrolyte was evaluated. In contrast to the Li and Na analogs, the KFSA/SL system maintained its liquid state at room temperature over a wide composition range, including the highly concentrated region. The calorimetric analysis revealed that the concentrated KFSA/SL solutions formed glass-forming metastable phases. According to the Raman spectroscopy analysis results of the XFSA/MeSL solutions, the weak interactions between K+ and both the solvent and FSA− anions could enable the application of both liquid states in battery electrolytes and also result in desirable cation-transport properties owing to the weaker Lewis acidity of K+ than Li+ and Na+. Fast K+ transport in the KFSA/SL = 1/1 electrolyte, as evidenced by the limiting current density under anion-blocking conditions, improved the high-power operation of the K2Mn[Fe(CN)6] positive electrodes. The CIP- and AGG-rich solution structure of KFSA/SL = 1/1, which is typical for highly concentrated solutions, achieved a desirable SEI, resulting in the effective passivation of the K metal surface and reversible operation of the graphite negative electrode. Furthermore, the superior oxidative stability of the SL and K+–SL–K+ bridge-type solvation structure enabled the operation of 5 V-class positive electrodes and improved the rate capability of the electrode-active materials, which was hardly achieved using conventional ether-based concentrated electrolytes. We believe that this study will contribute toward the exploration of novel alkali metal ion chemistry and the development of a new family of electrolytes for next-generation batteries.Data availability
The datasets used in this study are available from the corresponding author upon request.Author contributions
D. I. performed the experiments, analyzed the data, and wrote the first draft of the manuscript. R. T. proposed the study concept and contributed to writing and revising the manuscript. S. Y. contributed to the experiments and analysis of the HAXPES data. S. K. supervised the study. All authors reviewed and revised the manuscript in several steps and approved the final version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was partially funded by the MEXT Program, Data Creation and Utilization Type Materials Research and Development Project (grant no. JPMXP1122712807), JST-CREST (grant no. JPMJCR21O6), JST-PRESTO (grant no. JPMJPR2374), JST-GteX (grant no. JPMJGX23S4), and JSPS KAKENHI (grant no. JP20H02849, JP21K20561, JP22K14772, and JP23K13829). The HAXPES measurements were performed at the BL46XU beamline of SPring-8 with the approval of JASRI (proposal no. 2023B1630). The authors thank Mizuki Kawabe, Nanako Ito, and Ryusei Fujimoto for helping with DSC measurements and Dr Akio Iwanade for helping with ICP-OES measurements at NIMS under ARIM-MEXT (grant no. JPMXP1223NM0077). The authors would like to thank Editage (http://www.editage.jp) for English language editing. D. I. thanks JST for establishing university fellowships for the Creation of Science and Technology Innovation (grant no. JPMJFS2144), and the academic crowdfunding platform “Academist” and its supporters. R. T. thanks the TEPCO Memorial Foundation for a basic research grant and the Takahashi Industrial and Economic Research Foundation.References
- T. Hosaka, K. Kubota, A. S. Hameed and S. Komaba, Chem. Rev., 2020, 120, 6358–6466 CrossRef CAS.
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636–11682 CrossRef CAS PubMed.
- S. Komaba, T. Hasegawa, M. Dahbi and K. Kubota, Electrochem. Commun., 2015, 60, 172–175 CrossRef CAS.
- Z. Jian, W. Luo and X. Ji, J. Am. Chem. Soc., 2015, 137, 11566–11569 CrossRef CAS PubMed.
- W. Luo, J. Wan, B. Ozdemir, W. Bao, Y. Chen, J. Dai, H. Lin, Y. Xu, F. Gu, V. Barone and L. Hu, Nano Lett., 2015, 15, 7671–7677 CrossRef CAS PubMed.
- Y. Yamada, J. Wang, S. Ko, E. Watanabe and A. Yamada, Nat. Energy, 2019, 4, 269–280 CrossRef CAS.
- M. Zhou, P. Bai, X. Ji, J. Yang, C. Wang and Y. Xu, Adv. Mater., 2021, 33, 2003741 CrossRef CAS PubMed.
- T. Hosaka and S. Komaba, Bull. Chem. Soc. Jpn., 2022, 95, 569–581 CrossRef CAS.
- K. Yoshida, M. Nakamura, Y. Kazue, N. Tachikawa, S. Tsuzuki, S. Seki, K. Dokko and M. Watanabe, J. Am. Chem. Soc., 2011, 133, 13121–13129 CrossRef CAS PubMed.
- J. Wang, Y. Yamada, K. Sodeyama, C. H. Chiang, Y. Tateyama and A. Yamada, Nat. Commun., 2016, 7, 12032 CrossRef CAS PubMed.
- Y. Yamada, K. Furukawa, K. Sodeyama, K. Kikuchi, M. Yaegashi, Y. Tateyama and A. Yamada, J. Am. Chem. Soc., 2014, 136, 5039–5046 CrossRef CAS PubMed.
- S. Ko, T. Obukata, T. Shimada, N. Takenaka, M. Nakayama, A. Yamada and Y. Yamada, Nat. Energy, 2022, 7, 1217–1224 CrossRef CAS.
- M. Watanabe, K. Dokko, K. Ueno and M. L. Thomas, Bull. Chem. Soc. Jpn., 2018, 91, 1660–1682 CrossRef CAS.
- Y. Yamada, Bull. Chem. Soc. Jpn., 2020, 93, 109–118 CrossRef CAS.
- A. Abouimrane, I. Belharouak and K. Amine, Electrochem. Commun., 2009, 11, 1073–1076 CrossRef CAS.
- J. Alvarado, M. A. Schroeder, M. Zhang, O. Borodin, E. Gobrogge, M. Olguin, M. S. Ding, M. Gobet, S. Greenbaum, Y. S. Meng and K. Xu, Mater. Today, 2018, 21, 341–353 CrossRef CAS.
- S. Ko, Y. Yamada and A. Yamada, Joule, 2021, 5, 998–1009 CrossRef CAS.
- K. Dokko, D. Watanabe, Y. Ugata, M. L. Thomas, S. Tsuzuki, W. Shinoda, K. Hashimoto, K. Ueno, Y. Umebayashi and M. Watanabe, J. Phys. Chem. B, 2018, 122, 10736–10745 CrossRef CAS.
- Y. Maeyoshi, D. Ding, M. Kubota, H. Ueda, K. Abe, K. Kanamura and H. Abe, ACS Appl. Mater. Interfaces, 2019, 11, 25833–25843 CrossRef CAS PubMed.
- A. Nakanishi, K. Ueno, D. Watanabe, Y. Ugata, Y. Matsumae, J. Liu, M. L. Thomas, K. Dokko and M. Watanabe, J. Phys. Chem. C, 2019, 123, 14229–14238 CrossRef CAS.
- Y. Okamoto, S. Tsuzuki, R. Tatara, K. Ueno, K. Dokko and M. Watanabe, J. Phys. Chem. C, 2020, 124, 4459–4469 CrossRef CAS.
- R. Tatara, Y. Okamoto, Y. Ugata, K. Ueno, M. Watanabe and K. Dokko, Electrochemistry, 2021, 89, 590–596 CrossRef CAS.
- X. Li, X. Ou and Y. Tang, Adv. Energy Mater., 2020, 10, 2002567 CrossRef CAS.
- T. Hosaka, T. Fukabori, H. Kojima, K. Kubota and S. Komaba, ChemSusChem, 2021, 14, 1166–1175 CrossRef CAS PubMed.
- H. Kim, D.-H. Seo, M. Bianchini, R. J. Clément, H. Kim, J. C. Kim, Y. Tian, T. Shi, W.-S. Yoon and G. Ceder, Adv. Energy Mater., 2018, 8, 1801591 CrossRef.
- D. Igarashi, R. Tatara, R. Fujimoto, T. Hosaka and S. Komaba, Chem. Sci., 2023, 14, 11056–11066 RSC.
- Y. Ugata, Y. Chen, S. Sasagawa, K. Ueno, M. Watanabe, H. Mita, J. Shimura, M. Nagamine and K. Dokko, J. Phys. Chem. C, 2022, 126, 10024–10034 CrossRef CAS.
- J. Liao, C. Chen, Q. Hu, Y. Du, Y. He, Y. Xu, Z. Zhang and X. Zhou, Angew. Chem., Int. Ed., 2021, 60, 25575–25582 CrossRef CAS.
- Y. Yamada, K. Usui, C. H. Chiang, K. Kikuchi, K. Furukawa and A. Yamada, ACS Appl. Mater. Interfaces, 2014, 6, 10892–10899 CrossRef CAS.
- K. Takada, Y. Yamada, E. Watanabe, J. Wang, K. Sodeyama, Y. Tateyama, K. Hirata, T. Kawase and A. Yamada, ACS Appl. Mater. Interfaces, 2017, 9, 33802–33809 CrossRef CAS.
- T. Hosaka, K. Kubota, H. Kojima and S. Komaba, Chem. Commun., 2018, 54, 8387–8390 RSC.
- J. Hwang, A. N. Sivasengaran, H. Yang, H. Yamamoto, T. Takeuchi, K. Matsumoto and R. Hagiwara, ACS Appl. Mater. Interfaces, 2021, 13, 2538–2546 CrossRef CAS PubMed.
- B. Kurc, Int. J. Electrochem. Sci., 2018, 13, 5938–5955 CrossRef CAS.
- T. Hosaka, T. Matsuyama, K. Kubota, R. Tatara and S. Komaba, J. Mater. Chem. A, 2020, 8, 23766–23771 RSC.
- G. Zeng, S. Xiong, Y. Qian, L. Ci and J. Feng, J. Electrochem. Soc., 2019, 166, A1217 CrossRef CAS.
- S. Liu, J. Mao, Q. Zhang, Z. Wang, W. K. Pang, L. Zhang, A. Du, V. Sencadas, W. Zhang and Z. Guo, Angew. Chem., Int. Ed., 2020, 59, 3638–3644 CrossRef CAS PubMed.
- J. Park, Y. Jeong, M. H. Alfaruqi, Y. Liu, X. Xu, S. Xiong, M.-G. Jung, H.-G. Jung, J. Kim, J.-Y. Hwang and Y.-K. Sun, ACS Energy Lett., 2022, 7, 401–409 CrossRef CAS.
- H. Cai, H. Jing, X. Zhang, M. Shen and Q. Wang, J. Electrochem. Soc., 2017, 164, A714 CrossRef CAS.
- Q. Zheng, G. Li, X. Zheng, L. Xing, K. Xu and W. Li, Energy Environ. Mater., 2022, 5, 906–911 CrossRef CAS.
- W. Kuang, X. Zhou, Z. Fan, X. Chen, Z. Yang, J. Chen, X. Shi, L. Li, R. Zeng, J.-Z. Wang and S. Chou, ACS Energy Lett., 2024, 9, 4111–4118 CrossRef.
- L. Caracciolo, L. Madec, G. Gachot and H. Martinez, ACS Appl. Mater. Interfaces, 2021, 13, 57505–57513 CrossRef CAS PubMed.
- A. Hofmann, F. Müller, S. Schöner and F. Jeschull, Batteries Supercaps, 2023, 6, e202300325 CrossRef CAS.
- T. Hosaka, T. Fukabori, T. Matsuyama, R. Tatara, K. Kubota and S. Komaba, ACS Energy Lett., 2021, 6, 3643–3649 CrossRef CAS.
- T. Hosaka, T. Matsuyama, R. Tatara, Z. T. Gossage and S. Komaba, Chem. Sci., 2023, 14, 8860–8868 RSC.
- Y. Yamada, C. H. Chiang, K. Sodeyama, J. Wang, Y. Tateyama and A. Yamada, ChemElectroChem, 2015, 2, 1687–1694 CrossRef CAS.
- T. Hosaka, S. Muratsubaki, K. Kubota, H. Onuma and S. Komaba, J. Phys. Chem. Lett., 2019, 10, 3296–3300 CrossRef CAS PubMed.
- T. Hosaka, T. Shimamura, K. Kubota and S. Komaba, Chem. Rec., 2019, 19, 735–745 CrossRef CAS PubMed.
- K. Chihara, A. Katogi, K. Kubota and S. Komaba, Chem. Commun., 2017, 53, 5208–5211 RSC.
- Y. Gao, W. Li, B. Ou, S. Zhang, H. Wang, J. Hu, F. Kang and D. Zhai, Adv. Funct. Mater., 2023, 33, 2305829 CrossRef CAS.
- H. Kim, Y. Tian and G. Ceder, J. Electrochem. Soc., 2020, 167, 110555 CrossRef CAS.
- R. Wernert, L. H. B. Nguyen, A. Iadecola, F. Weill, F. Fauth, L. Monconduit, D. Carlier and L. Croguennec, ACS Appl. Energy Mater., 2022, 5, 14913–14921 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta06029f |
| ‡ Present address: Department of Chemistry and Life Science, Yokohama National University, Yokohama, Kanagawa, 240-8501, Japan. |
| This journal is © The Royal Society of Chemistry 2025 |