Palladium-catalyzed Suzuki reaction in aqueous solvents applied to unprotected nucleosides and nucleotides
Gwénaëlle Hervéa,
Guillaume Sartoria,
Gérald Enderlina,
Grahame Mackenzieb and
Christophe Len*a
aTransformations Intégrées de la Matière Renouvelable, UTC-ESCOM, Centre de Recherche Royallieu, BP 20529, F-60200 Compiègne, France. E-mail: christophe.len@utc.fr; Fax: +33 (0)3 44 97 15 91; Tel: +33 (0)3 44 23 88 28
bDepartment of Chemistry, University of Hull, Hull, HU6 7RX, England
First published on 13th March 2014
Abstract
Nucleoside analogues have attracted much attention due to their potential biological activities. Amongst all synthetic nucleosides, C5-modified pyrimidines and C2- or C8-modified purines have been particularly studied. A large variety of palladium cross-coupling reactions, with a majority of them based on the Suzuki–Miyaura reaction, have been developed for preparing the desired nucleoside derivatives. Our objective is to focus this review on the Suzuki–Miyaura cross-coupling of nucleosides using methodologies compatible with green chemistry and sustainable development for one part and bioorthogonality for the other part, which means using aqueous medium and no protection/deprotection steps.
This work is dedicated to Professor Gordon Shaw (1922–1997)
Gordon Shaw obtained a first class honours degree in chemistry in 1942 from Imperial College of Science and Technology and a PhD (supervised by Sir Ian Heilbron and A. H. Cook on the synthesis of penicillin and analogues) in 1945. Following a spell in the pharmaceutical industry, he was a Senior Lecturer in Organic Chemistry at the University of New South Wales between 1948 and 1959. He won a Nuffield Research Fellowship to work at the University of Cambridge with Lord Todd, the Nobel Prize Laureate, from 1955 and 1956. In 1960 he took up the post of Reader in Organic Chemistry at the University of Bradford, and in 1989 he became a Professor at the University of Bradford. He continued his teaching and research until his death on 25 June 1997.He published work in the field of nucleosides and nucleotides but also focused on the origins of life on Earth largely through the study of the polymer sporopollenin, which is the extremely stable polymer that coats pollen grains and is found in some of Earth's oldest sedimentary rocks. He wrote a book with J. Brooks entitled ‘Origin and Development of Living Systems’. He showed that sporopollenin could be adapted as a solid phase support for such applications as peptide synthesis. Most of his nucleoside and nucleotide work was reported in his 67-part series in the Journal of the Chemical Society on the ‘synthesis of purines, pyrimidines and imidazoles including nucleosides’. His approach was notable by being the first to employ an acyclic intermediate to obtain regiospecificity and in many cases stereoselectivity. His work shone much light on the de novo biosynthesis of purines and included such achievements as the first synthesis of the naturally occurring cytokinin zeatin. In the latter part of his work, he used an innovative route to prepare anthracyclinones and their derivatives via a modified Marschalk reaction using a carbohydrate as a chiral template. He published this work in a five-part series in the J. Chem. Soc.
Gordon Shaw will be well remembered by a wide circle of collaborators and friends for his modesty, humility and capacity for original thought.
1. Introduction
Natural nucleosides are of great biological importance in metabolic pathways and as building blocks of nucleic acids. Their common structural characteristic is the presence of two molecular moieties: D-ribo or D-2′-deoxyribopentofuranose as the glycone fragments and purine or pyrimidine as the aglycone moiety. These two moieties are covalently bonded at the anomeric site (C1′) of the glycone to either the N1 of the pyrimidine (uracil, thymine, and cytosine) or N9 of the purine (adenine and guanine) in a β-D configuration to form, correspondingly, the nucleosides thimidine (1), uridine (2), cytidine (3), adenosine (4) and guanosine (5) (Fig. 1).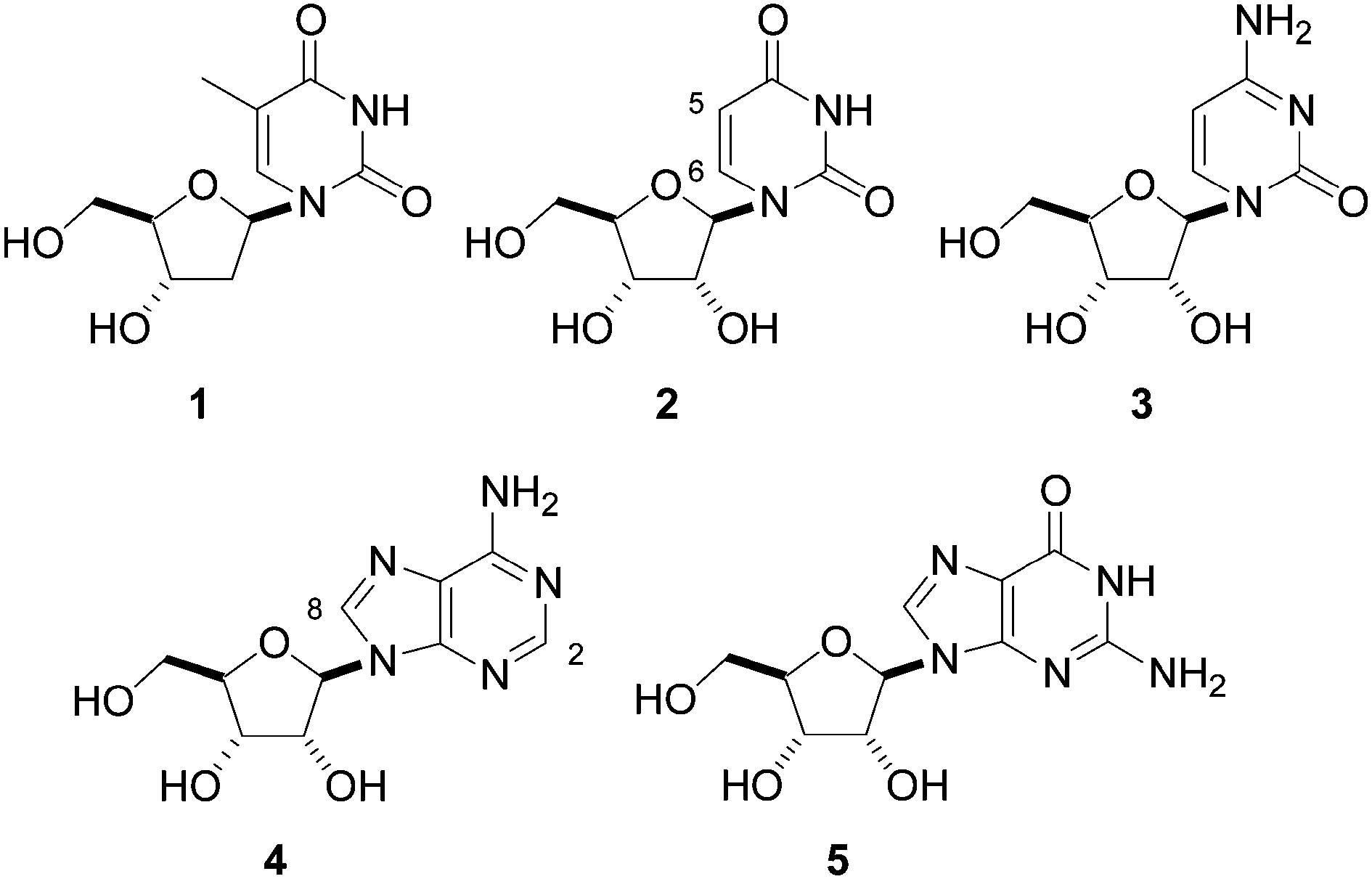 | ||
| Fig. 1 Nucleosides 1–5. | ||
Since the discovery that nucleoside analogues have shown high effectiveness as antiviral and antitumor agents,1 the search for new agents with a higher therapeutic index has started. In this regard, many modifications to the glycone moiety and/or the nucleobase have been reported. A variety of functionalities2 have been introduced into the ribose glycone moiety. Examples of simple substituents include halogeno, N3, CF3, CN, alkyl, alkenyl, alkynyl, aryl, thio, and seleno groups, while more complicated modifications are required to form acyclic nucleosides, L-isomers, thio or amino analogues, C-nucleosides and nucleosides having restricted conformations2k,3 such as bicyclonucleosides, cyclonucleosides and cyclic phosphoesters. Major modifications to the nucleobase have included the introduction of aryl, polyaryl, heteroaryl and heteropolyaryl groups via C–C bond coupling. These were synthesized for the study of biological environments such as protein–DNA complexes, DNA damage, mutation and cancers.4 These new tools such as DNA and RNA structural probes are aimed at gaining a better understanding of various diseases and establishing methods for diagnosis and high-throughput screening. Starting from the natural nucleosides 1–5, the synthesis of the aryl nucleoside analogues, having a C–C bond coupling, permitted selective modification at the C-5 and C-6 positions of the pyrimidine moiety and the C-2 and C-8 positions of the purine ring system. Of the main strategies used for the preparation of aryl, polyaryl, heteroaryl and heteropolyaryl nucleoside analogues (C sp2–C sp2 bond formation), the palladium-catalyzed Suzuki–Miyaura, Stille, Negishi and Hiyama reactions appear to be the most prefered.5 In general, the C–C cross-coupling reactions have been effected in organic solvents using totally protected nucleosides.
The aims of green chemistry6 are to (i) develop less hazardous chemical syntheses; (ii) use safer solvents and auxiliaries; (iii) use renewable feedstocks; (iv) reduce intermediates; and (v) develop or discover better catalysts. Even if the Stille, Negishi and Hiyama reactions comply with some of the green chemistry principles, only the Suzuki–Miyaura reaction offers the most attractive potential for development. An important feature is that the Suzuki–Miyaura reaction can be carried out in green solvents, such as water, without the need for protection and deprotection steps. The general Suzuki–Miyaura catalytic cycle occurs by oxidative addition, transmetallation and reductive elimination.7 After formation of the catalytic species Pd(0), generated in situ starting from palladium Pd(II) or directly from Pd(0) derivatives, the oxidative addition of the aryl halide ArX leads to the palladium complex [ArPdXLn]. The transmetallation step occurs by conversion of the palladium halide [ArPdXLn] in the presence of base RO− to a nucleophilic palladium alkoxy complex [ArPdORLn] that subsequently reacts with a neutral organoboron compound Ar′B(OH)2 to afford the diaryl complex [ArPdAr′Ln] in a cis–trans equilibrium. Reductive elimination of the cis form gives the biaryl derivative Ar–Ar′ and Pd(0) (Scheme 1).7e
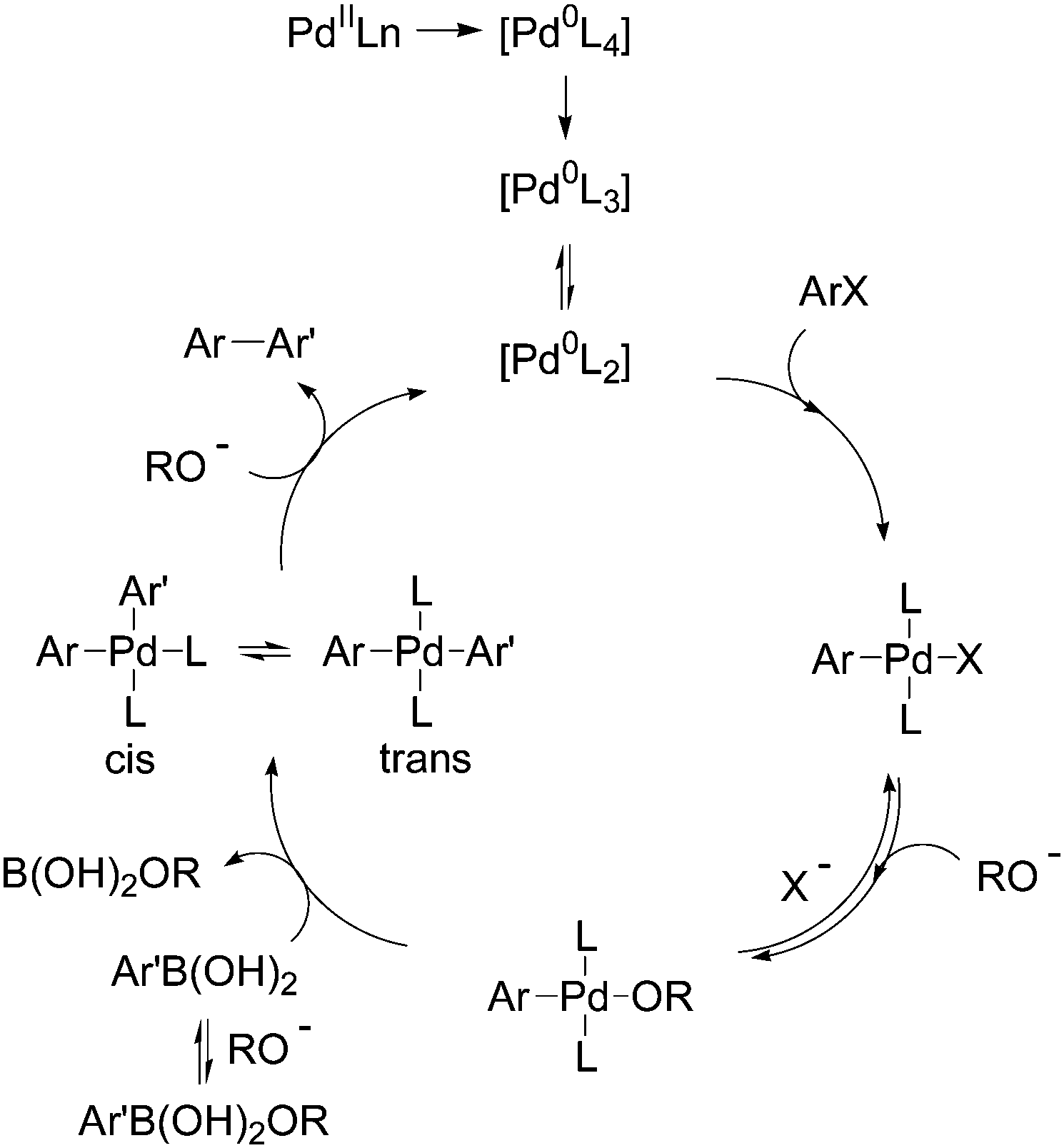 | ||
| Scheme 1 Mechanism for the Suzuki–Miyaura reaction. | ||
This review is concerned with green chemistry and sustainable development in the field of modified nucleoside analogues with particular focus on the Suzuki–Miyaura C–C cross-coupling reaction applied to unprotected nucleosides and nucleotides in aqueous media or water as the sole solvent. The particular C–C bond formations in this review are those in positions 5 and 6 of the pyrimidine nucleoside analogues and in position 8 of the purine nucleoside analogues. Examples of Suzuki–Miyaura coupling in position 6 of purine and using 7-deazapurine are also covered along with postsynthetic arylation of DNA by Suzuki–Miyaura cross-coupling. For the sake of clarity, this review has been arranged to describe the different methodologies by varying (i) the nature of the solvent THF–MeOH–H2O, DME–H2O, DMF–H2O, CH3CN–H2O, MeOH–H2O, and H2O; (ii) the palladium source and nature with respect to Pd(II) and Pd(0); and (iii) the nature of the base. One special chapter is dedicated to the postsynthetic strategy.
2. Suzuki–Miyaura in THF–MeOH–H2O
The Suzuki–Miyaura cross-coupling reaction in THF–MeOH–H2O was developed using both Pd(0) as the active catalyst and Pd(II) as a pre-catalyst when used in a large amount (10–11 mol%).2.1. Pd(PPh3)4 as Pd(0) and NaOH
In 2002, Wagenknecht and co-workers reported the first direct synthesis of unprotected arylated pyrimidine derivatives using a palladium-catalyzed Suzuki–Miyaura cross-coupling reaction.8 Starting from the 5-iodo-2′-deoxyuridine (6), the modified nucleoside 5-(pyren-1-yl)-2′-deoxyuridine (8) was obtained in 70% yield using pyren-1-yl boronic acid (7) (1.0 eq) and a large amount of Pd(PPh3)4 as the catalyst (10 mol%) in a mixture of THF–MeOH–H2O (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2) as the solvent (Scheme 2) for 20 hours under reflux. NaOH (20 eq) as a strong base was required to obtain the desired sterically hindered coupling product 8 in sufficient yield. The authors reported a slightly higher yield of nucleoside analogue 8 (79% vs. 70% yield) using the same experimental conditions a few months later.9
:1:2) as the solvent (Scheme 2) for 20 hours under reflux. NaOH (20 eq) as a strong base was required to obtain the desired sterically hindered coupling product 8 in sufficient yield. The authors reported a slightly higher yield of nucleoside analogue 8 (79% vs. 70% yield) using the same experimental conditions a few months later.9
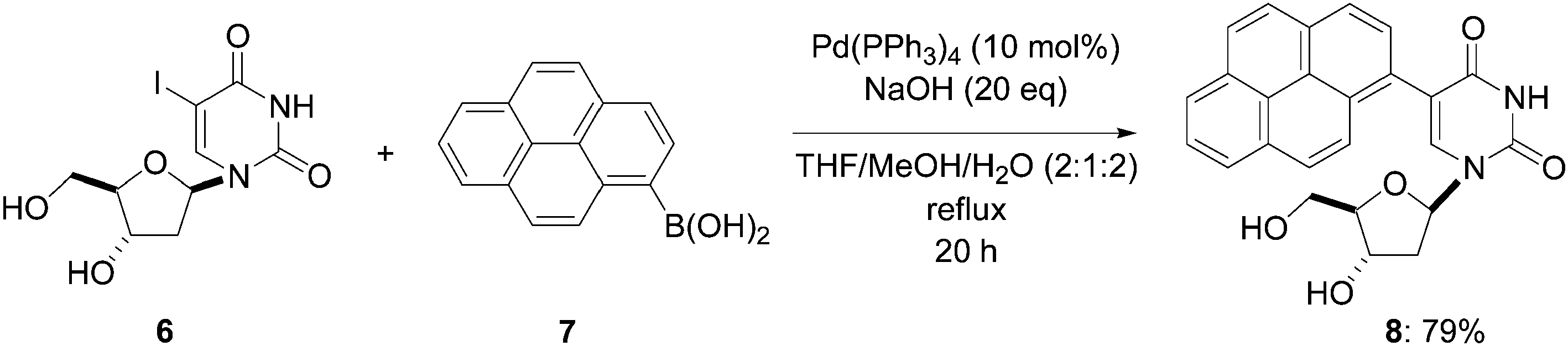 | ||
| Scheme 2 Synthesis of pyren-1-yl-modified nucleoside 8. | ||
The authors showed that protection of the two hydroxyl groups of the 2′-deoxyribose moiety of compound 6 was not necessary. In order to validate their new pathway, Wagenknecht and co-workers prepared compound 8 starting from the 3′,5′-di-O-acetyl-2′-deoxy-5-iodouridine (9) and boronic acid 7 (1.0 eq). The use of the protected nucleoside analogue 9 required modifications to the protocol. In this case, dry THF was used as solvent and Et3N (4.2 eq) was used as base.8 The cross-coupling furnished the pyrene analogue 10, and subsequent classical deprotection of the nucleoside analogue 10 afforded the target pyrene derivative 8 in 55% yield (two steps) (Scheme 3).
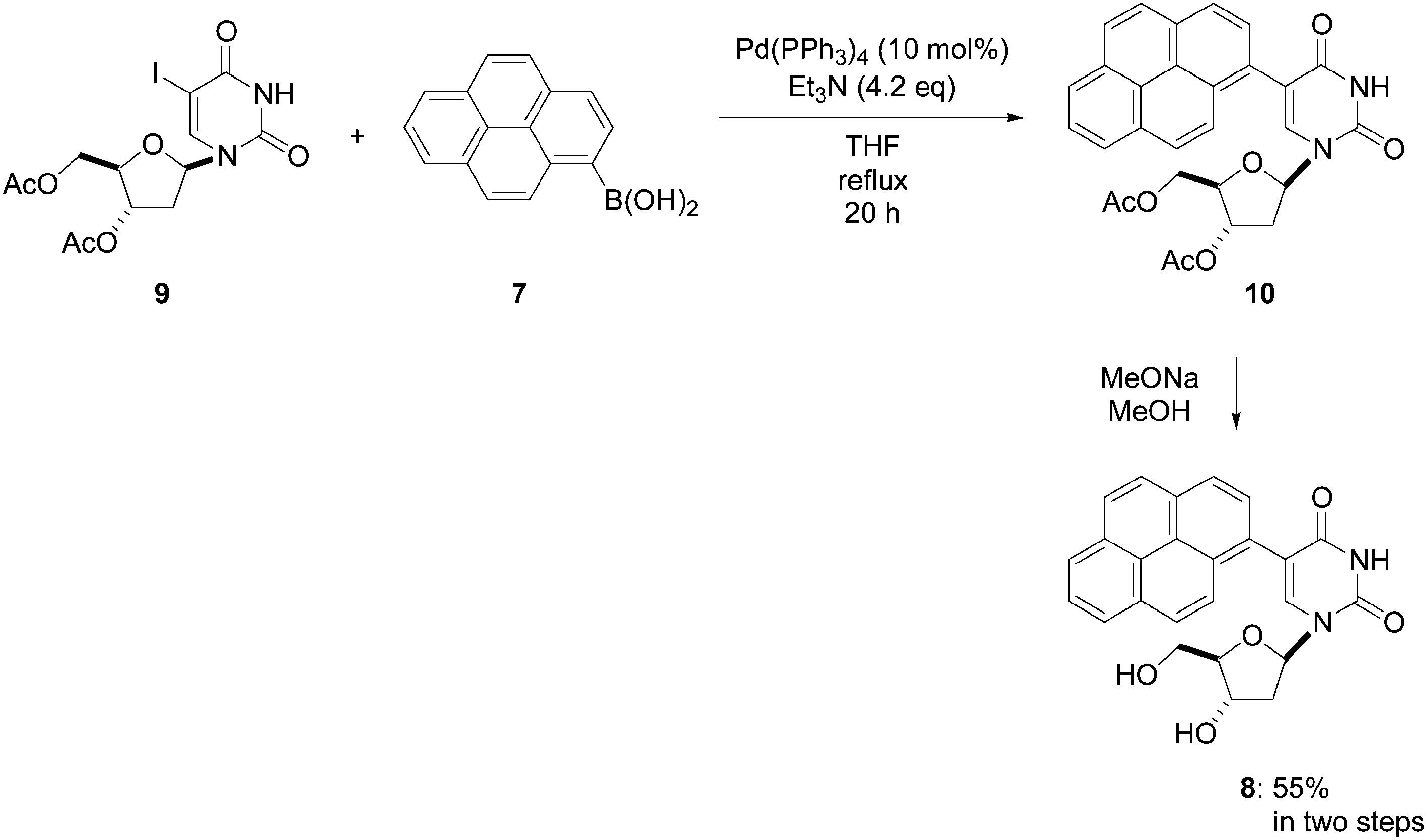 | ||
| Scheme 3 Synthesis of pyren-1-yl-modified nucleoside 8 starting from the protected nucleoside analogue 9. | ||
A second 8-(pyren-1-yl)-nucleoside (12) was prepared in a similar way (Scheme 4) starting from 8-bromo-2′-deoxyguanosine (11) having an unprotected hydroxyl group on the ribose moiety and an exocyclic amino group on the nucleobase and boronic acid 7 (1.0 eq). After purification of the crude product, the pyrene-modified nucleoside 12 was obtained in 65% yield.8
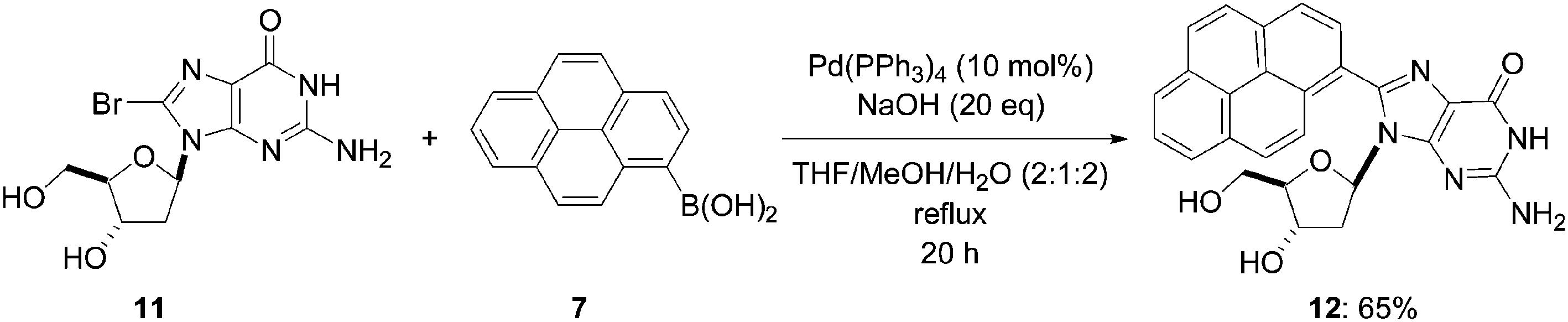 | ||
| Scheme 4 Synthesis of pyren-1-yl-modified nucleoside 12. | ||
Subsequently, Wagenknecht and co-workers applied their protocol to the preparation of two others pyrene-modified derivatives, 14 and 16, starting from the corresponding 5-iodo and 8-bromo derivatives 13 and 15, respectively. Each of the starting materials 13 and 15 had two hydroxyl groups and one exocyclic amino group (Scheme 5).10 Starting from the nucleoside analogues 11, 13 and 15, the Suzuki–Miyaura cross-couplings were performed with an excess of boronic acid (1.2 eq) using the same previously described catalyst and base in THF–MeOH–H2O (2:2:1) as in their earlier study. Even with a little excess of boronic acid 7 (1.2 eq vs. 1.0 eq) and modulation of the water ratio (20% vs. 40%), the guanosine analogue 12 was obtained in similar yield (Schemes 4 and 5). The target compounds 14 and 16 were prepared in 21% and 10% yields, respectively, with a longer reaction time required for the cytosine analogue 14.
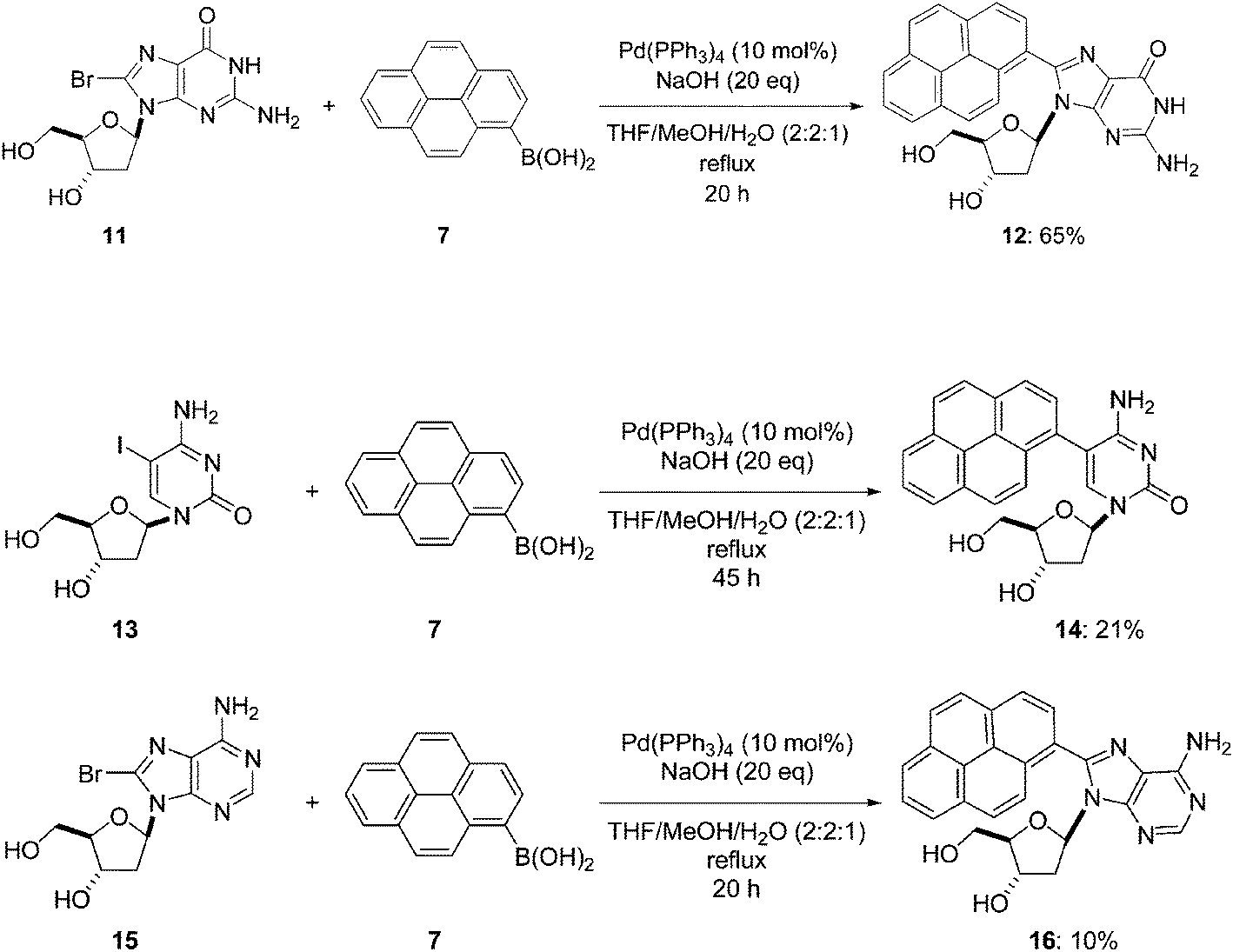 | ||
| Scheme 5 Synthesis of pyren-1-yl-modified nucleoside analogues 12, 14 and 16. | ||
Compared with preceding results, compounds 14 and 16 were obtained in lower yields. Since the authors have consistently used unprotected starting nucleosides, the difference in reactivity is likely to be attributed to the structural differences in the four nucleobases but more particularly to the exocyclic amino function present in the cytidine and adenosine scaffold. Furthermore, the exocyclic amino function could be involved in palladium-complex formation, leading to side-products. Therefore, to improve the yields of compound 14, further attempts were performed such as (i) changing the catalyst Pd(PPh3)4 to PdCl2(dppf); (ii) substituting the boronic acid with the propanediol ester derivative; and (iii) extending the reaction time. However, these modifications were not successful. Finally, Wagenknecht and co-workers decided to protect their starting cytidine analogue and to proceed with the cross-coupling in dry THF as a solvent in the presence of Et3N as a base using a similar strategy to that outlined in Scheme 3. As expected, the yield of the cross-coupling adduct 14 was higher (49%).10
Two years later, the same team directed their efforts to synthesize chromophore-modified nucleosides, by applying the palladium Pd(0)-catalyzed Suzuki cross-coupling reaction to the 8-bromo-2′-deoxyguanosine (11), employing an excess of the pinacol ester 17 of boronic acid (1.2 eq). After 24 hours of reflux, the target C-8 adduct 18 was obtained in 25% yield (Scheme 6).11 It is noteworthy that the benzo[a]pyren-6-yl boronic acid was difficult to isolate and the synthesis of the corresponding pinacol ester 17 was the preferred product.
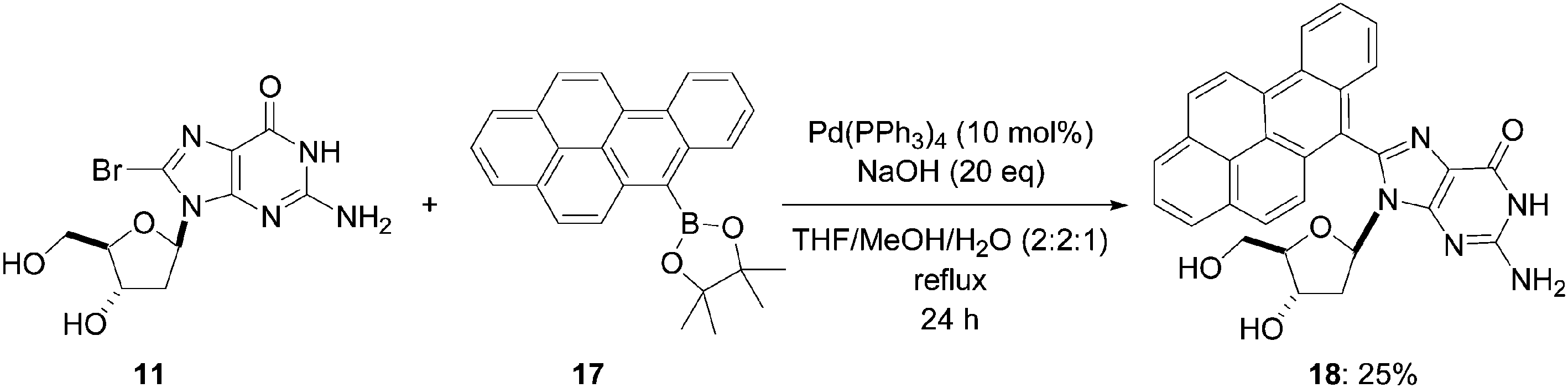 | ||
| Scheme 6 Synthesis of modified nucleoside 18. | ||
The synthesis of the anthraquinone-modified nucleoside 20 (Scheme 7) was described by Gothelf and co-workers in 2009 using the methodology developed by Wagenknecht.12 Starting from the 5-iodo derivative 6 and the corresponding pinacolate 19, the authors modified the solvent ratio THF–MeOH–H2O (20:12:15) but used Pd(PPh3)4 (10 mol%) as a catalyst in keeping with their previous work except that they employed NaOH (20 eq) as base. In their hands, the target nucleoside analogue 20 was obtained in 52% yield.
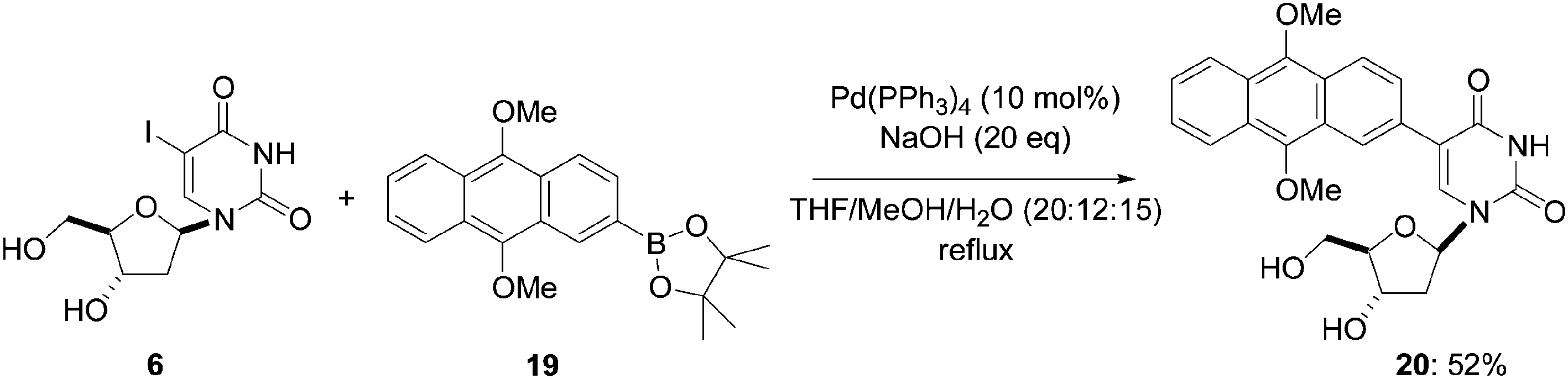 | ||
| Scheme 7 Synthesis of anthraquinone-conjugated nucleoside analogue 20. | ||
2.2. PdCl2(dppf) as Pd(II) and NaOH
Wagenknecht and co-workers also reported the synthesis of the phenothiazine derivative of 2′-deoxyuridine 22.13 Starting from compound 6 and the corresponding pinacolate 21 (1.2 eq), they changed their source of palladium Pd(PPh3)4 to PdCl2(dppf) [Pd(II) vs. Pd(0)] and modified the ratio of the solvent mixture (THF–MeOH–H2O, 2:2:1 vs. THF–MeOH–H2O, 1:1:0.83). After 44 hours under reflux, compound 22 was obtained in 34% yield (Scheme 8).
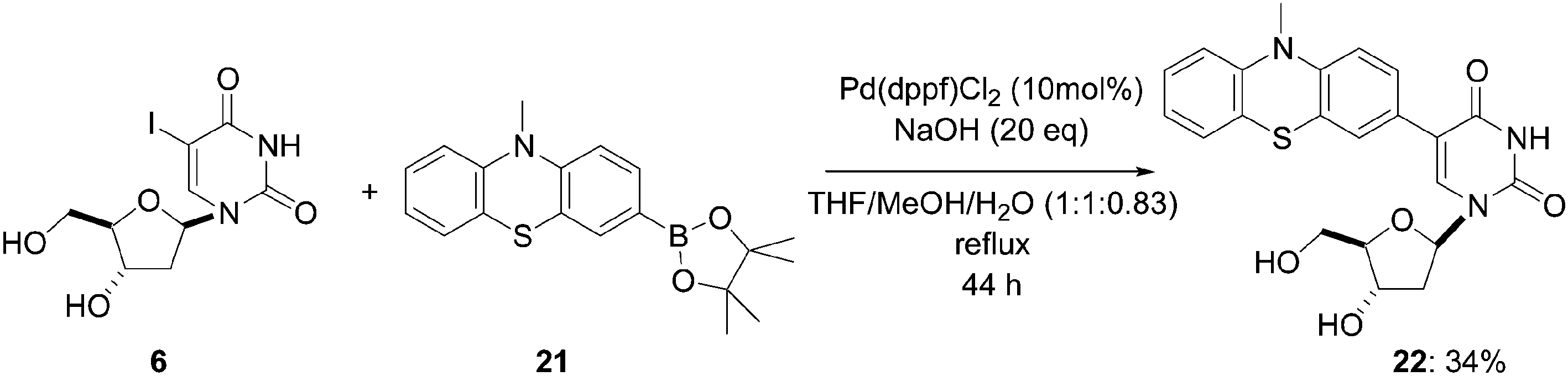 | ||
| Scheme 8 Synthesis of phenothiazine-modified nucleoside 22. | ||
2.3. PdCl2(dppf)2 as Pd(II) and NaOH
In the continuity of their work on pyrene-modified nucleosides, Wagenknecht and co-workers described the synthesis of 5-(pyren-2-yl)-2′-deoxyuridine (24) in 2008 (Scheme 9).14 In this case, the uridine moiety was linked to the 2-position of the pyrene chromophore instead of the 1-position as on previously described compounds. For this purpose, the main modifications were the palladium source PdCl2(dppf)2 (11 mol%) and the ratio of the solvent mixture (THF–MeOH–H2O, 2:1:1 vs. THF–MeOH–H2O, 2:2:1). Starting from the uridine analogue 6 and the boronic acid 23, the pyren-2-yl derivative 24 was prepared in 62% yield after 60 hours treatment at 65 °C. Using this catalyst system, the yield of compound 24 was lower than that obtained for the preparation of the regioisomer 8 (79% vs. 62%, Scheme 2).
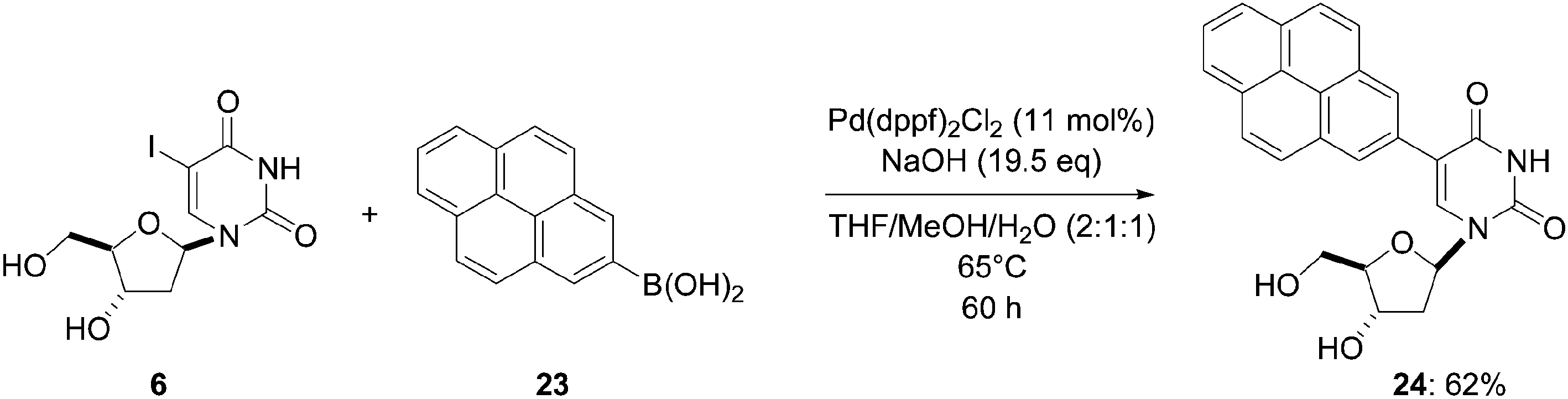 | ||
| Scheme 9 Synthesis of pyren-2-yl-modified nucleoside analogue 24. | ||
3. Suzuki–Miyaura in DME–H2O
Only Pd(0) as catalyst in a large quantity (10 mol%) and Pd(II) as precatalyst (2.5 mol%) were used for the development of a Suzuki–Miyaura cross-coupling reaction in DME–H2O.3.1. Pd(PPh3)4 as Pd(0) and K2CO3
In 2004, Yamamoto and co-workers described the Suzuki–Miyaura cross-coupling reaction starting from 8-bromoadenosine (25) and phenyl boronic acid (1.5 eq) using Pd(PPh3)4 (10 mol%) as the catalyst and K2CO3 (6 eq) as the base in DME–H2O (2:1) at 90 °C for 16 hours (Table 1, entry 1).15 The 8-phenyl derivative 26 was obtained in 81% yield (Table 1, entry 1).
|
|||
|---|---|---|---|
| Entry | R | Product | Yield (%) |
| 1 | 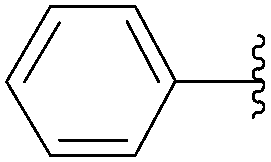 |
26 | 81 |
| 2 | 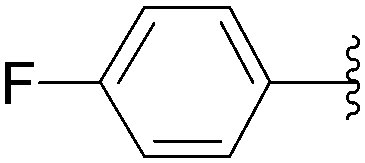 |
27 | 75 |
| 3 | 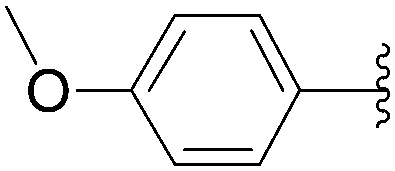 |
28 | 73 |
| 4 | 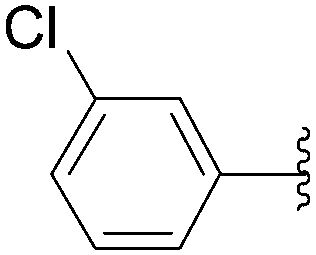 |
29 | 63 |
| 5 | 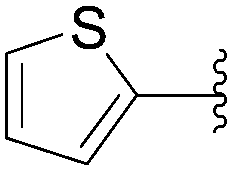 |
30 | 40 |
| 6 | 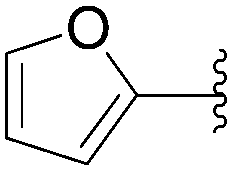 |
31 | 25 |
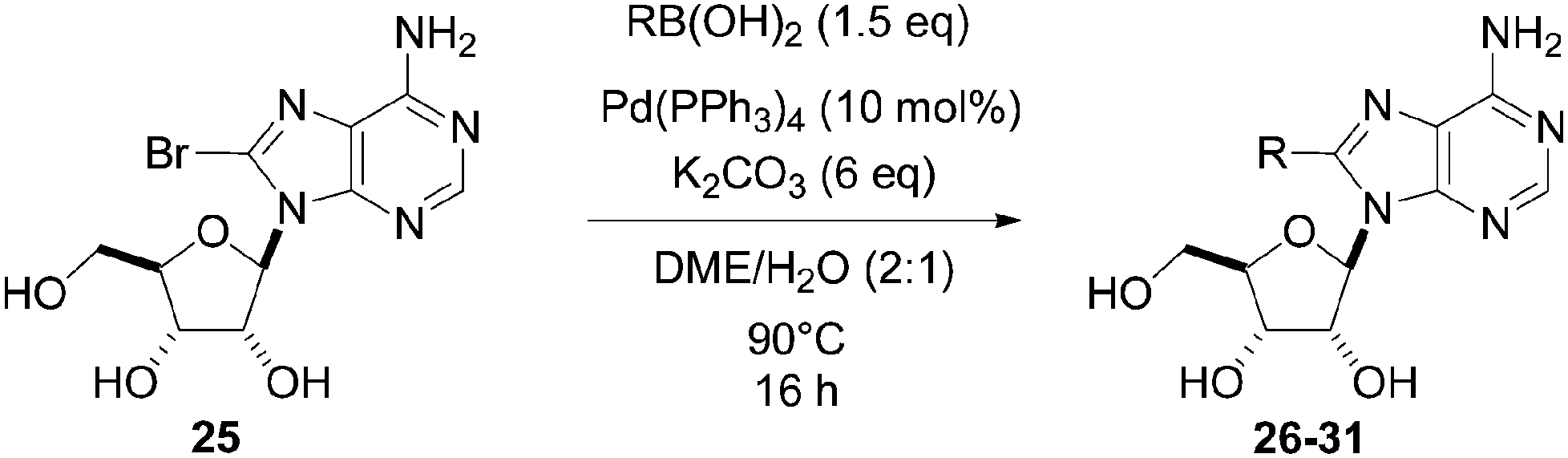
The methodology was applied to different boronic acids, and the adducts 27–31 were obtained (Table 1, entries 2–6). One can note that the yields were dependent on the boronic acid used. In the case of para-substituted phenyl boronic acids, the electron-rich substituents (fluorine and methoxy group) afforded the cross-coupling adducts 27 and 28 in 75% and 73% yields, respectively (Table 1, entries 2 and 3). In the case of the meta-substituted phenyl boronic acid, the electron-deficient substituent (chloride atom) furnished the target compound 29 in lower yield (63%) (Table 1, entry 4). The heterocyclic boronic acids having thien-2-yl and furan-2-yl moieties gave the compounds 30 and 31 in 40% and 26% yields, respectively (Table 1, entries 5 and 6). In these cases, the lower yields might be explained by the presence of a heteroatom, steric hindrance and the lack of solubility in chloroform and methanol eluents used in the chromatography step.15
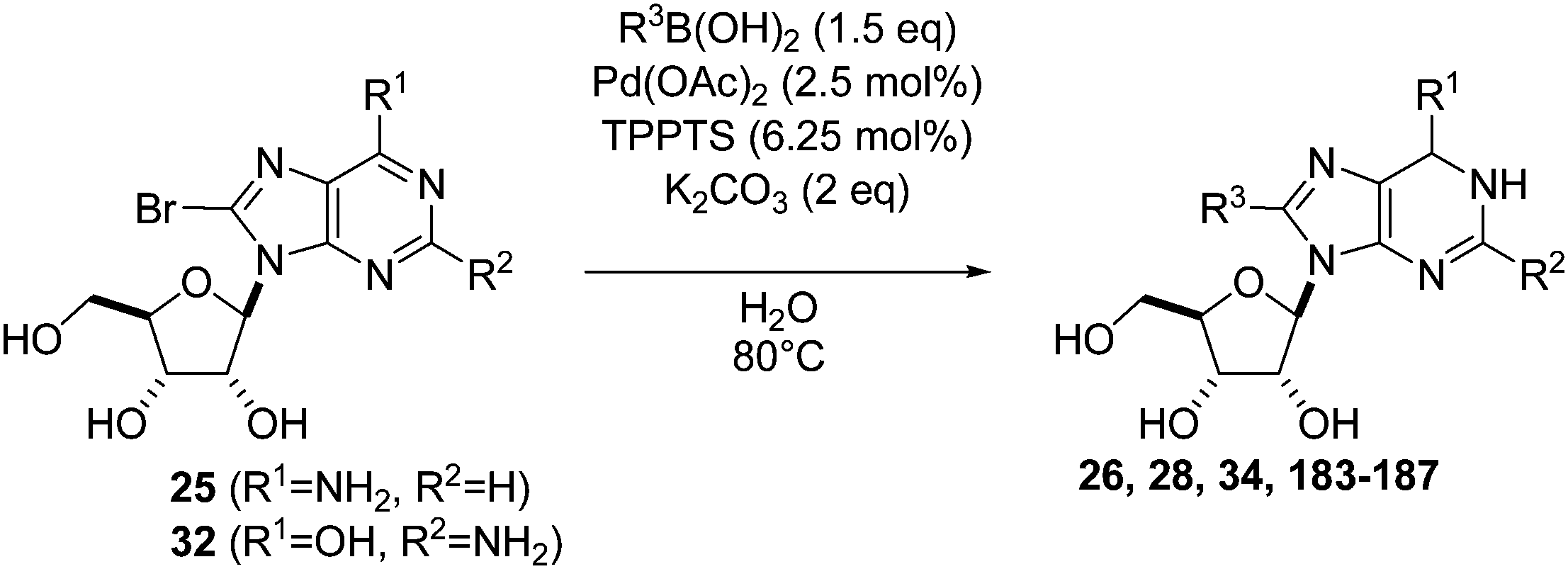
Two years later, Wagner and co-workers used a slightly different methodology to conduct the Suzuki–Miyaura cross couplings of 8-bromoadenosine (25) and 8-bromoguanosine (32) in the presence of phenyl boronic acid (33) (1.5 eq).16 In the cases where Pd(PPh3)4 (10 mol%) and K2CO3 (6 eq) continued to be used by the authors, reactions were conducted at 80 °C for 24 hours in DME–H2O (2:1). Using these conditions, 8-phenyladenosine (26) was obtained in 60% yield, but the authors were unsuccessful in isolating 8-phenyl guanosine (34). However, in the case of adenosine analogue, it could have been that the increase in temperature (90 °C vs. 80 °C) contributed to an increase in the yield (81% vs. 60% yield) (Table 1, entry 1 and Scheme 10).
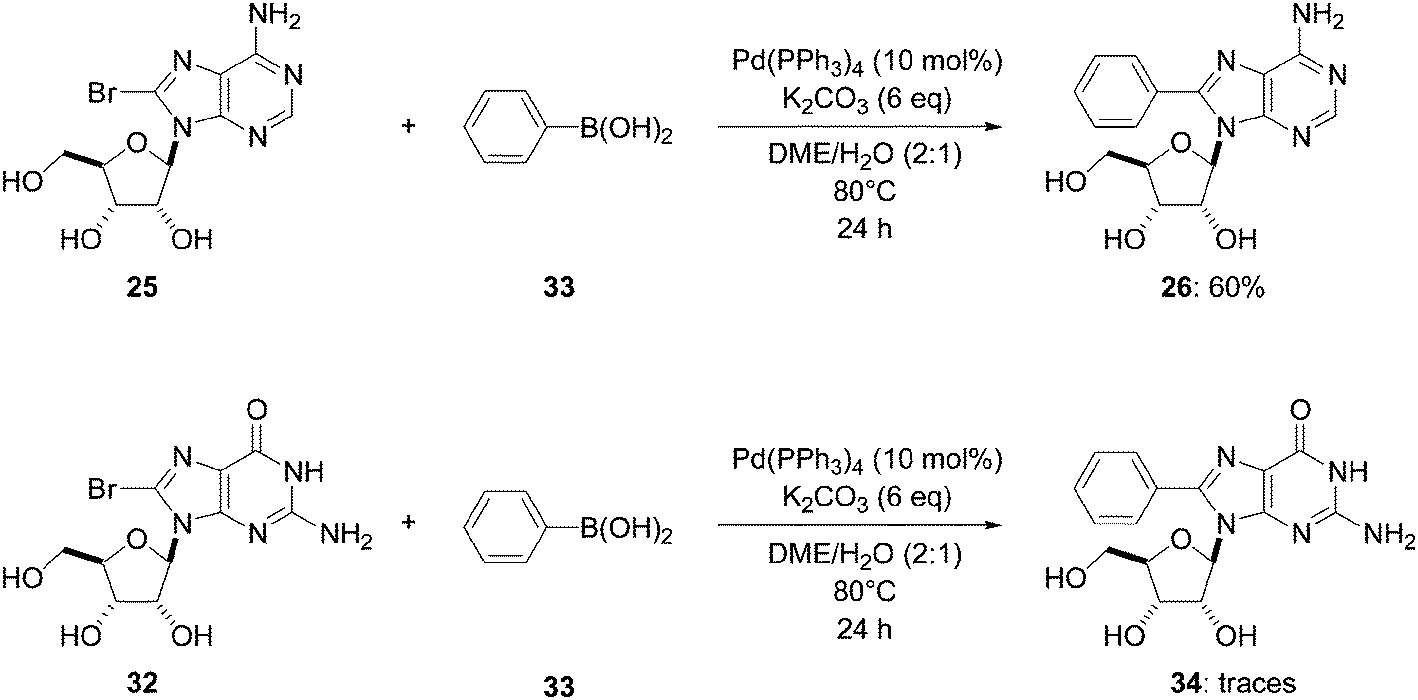 | ||
| Scheme 10 Synthesis of 8-phenyladenosine and guanosine analogues 26 and 34. | ||
Ealick and co-workers used Yamamoto's procedure to obtain 8-phenyladenosine (26) starting from deprotected 8-bromoadenosine (25) in the course of their studies towards new inhibitors of human S-adenosylmethionine decarboxylase.17 No yield was reported but this research group claimed that the use of the unprotected starting material was superior both in time and yield to that using a blocked nucleoside.
3.2. Pd(OAc)2 as Pd(II), TPPTS and K2CO3
When Wagner and co-workers used the catalyst system Pd(OAc)2 (2.5 mol%), TPPTS (6.25 mol%) and a lesser amount of K2CO3 as base (2 eq) in a DME–H2O (2:1) mixture at 80 °C, the target compound 26 was obtained in 69% yield starting from the bromine derivative 25 (Scheme 11).16 As described, when using Pd(PPh3)4 and the guanosine derivative 32, only traces of the coupling adduct could be detected.
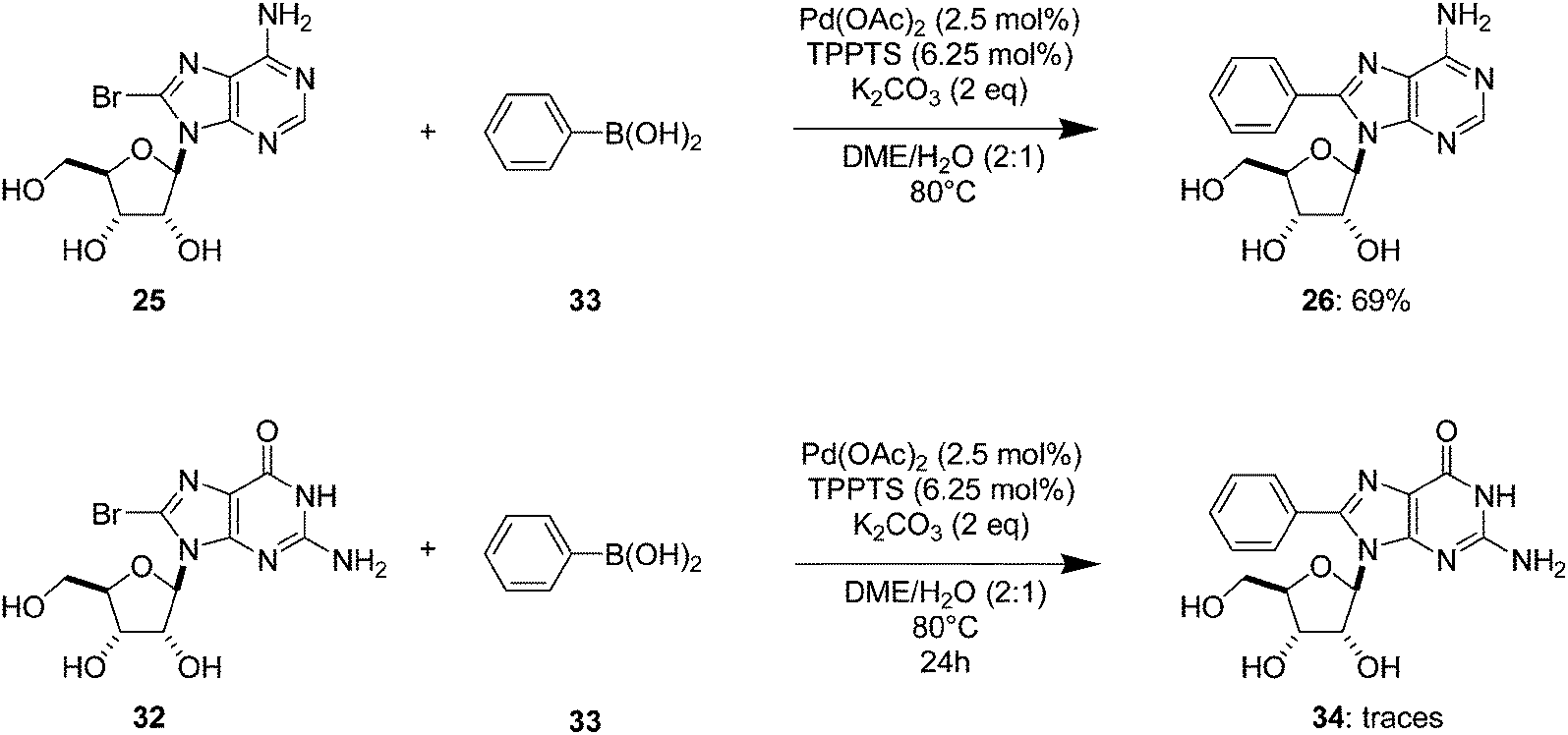 | ||
| Scheme 11 Synthesis of 8-phenyladenosine and guanosine analogues 26 and 34. | ||
4. Suzuki–Miyaura in DMF–H2O
The Suzuki–Miyaura cross-coupling reaction in DMF–H2O was described with Pd(0) as catalyst (10 mol%).4.1. Pd(PPh3)4 as Pd(0) and CsF
In 2011, Herdewijn's group reported a study on the synthesis and evaluation of 5-substituted-2′-deoxyuridine monophosphate analogues against Mycobacterium tuberculosis.18 In this work, the authors decided to use the unprotected 5-iodo-2′-deoxyuridine (6) as the starting material and the chosen boronic acids. They conducted a systematic study and showed that the varying nature of the base (K2CO3, Na2CO3, NaOH, or KF) in organic solvent–H2O mixtures led predominantly to dehalogenation. Herdewijn and co-workers successfully achieved the cross-coupling by reacting 4-fluorophenyl boronic acid (1.2 eq) with CsF (2.5 eq) as base in a mixture of DMF–H2O (2:1) as solvent and Pd(PPh3)4 (10 mol%) as the catalyst at 60 °C. Under these reaction conditions, only a small amount of 2′-deoxyuridine was detected, and the target nucleoside analogue 35 was obtained in 78% yield (Scheme 12). Application of this protocol afforded the 3-methoxy analogue 36 in 95% yield (Scheme 12).18
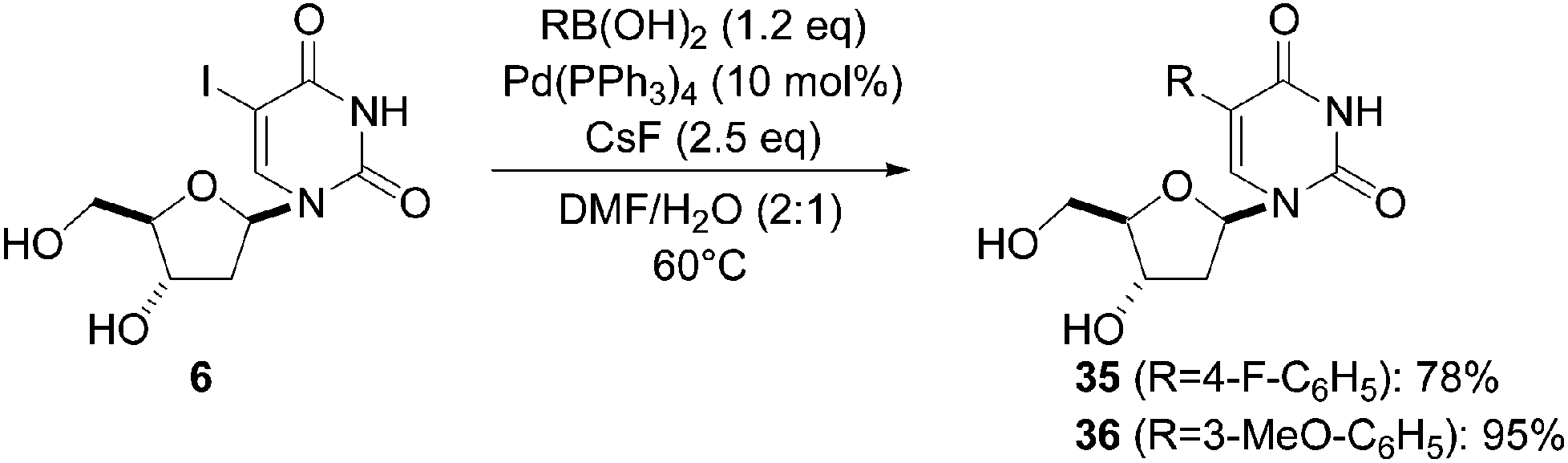 | ||
| Scheme 12 Synthesis of 5-aryl uridine analogues 35 and 36. | ||
Phosphorylation of compounds 35 and 36 afforded the desired 5-aryl-2′-deoxyuridine monophosphate analogues 37 and 38, respectively (Fig. 2). The 2-(4-fluorophenyl) analogue 37 exhibited good activity against a novel flavin-dependent thymidylate synthase in Mycobacterium tuberculosis (ThyX), whereas no activity could be detected against the classical mycobacterial thymidylate synthase (ThyA). On the other hand, the 5-(3-methoxyphenyl) analogue 38 showed no activity against either enzyme. This indicates that the nature of the substitution pattern on the phenyl ring has a large influence on the biological activity.
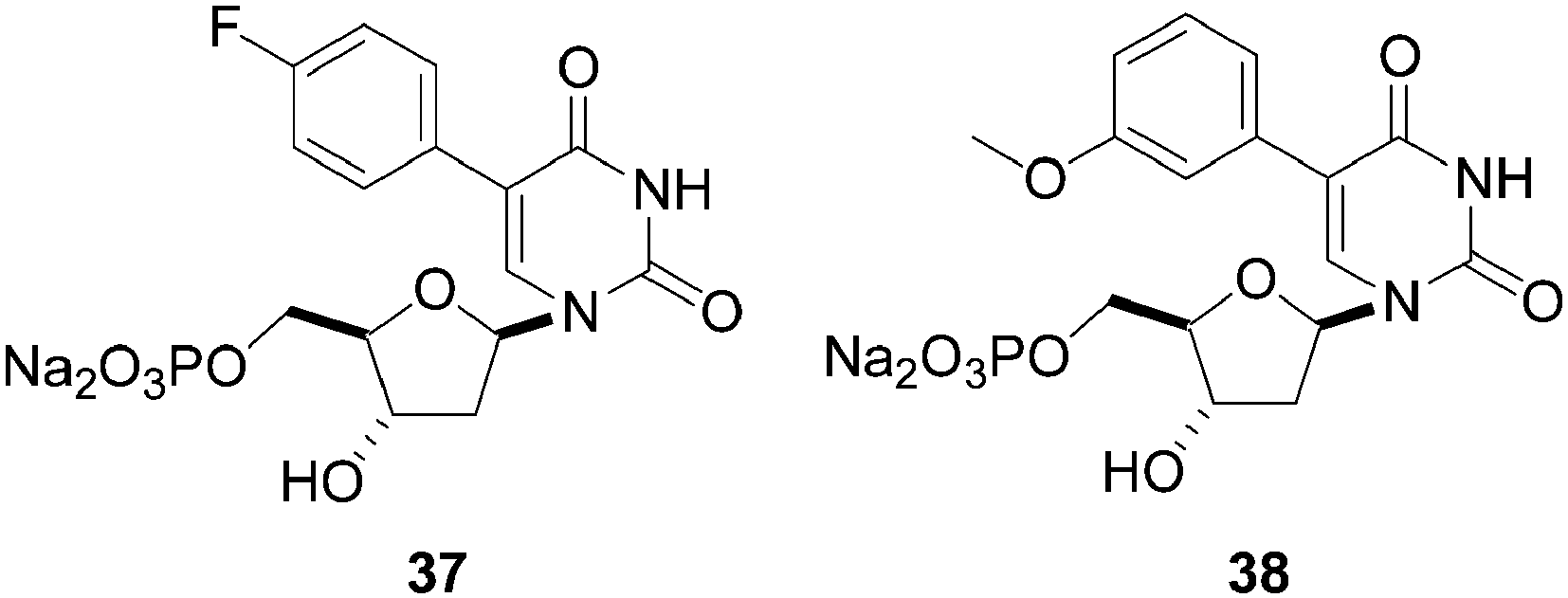 | ||
| Fig. 2 5-Phenyl uridine monophosphate analogues 37 and 38. | ||
5. Suzuki–Miyaura in CH3CN–H2O
In contrast to the Suzuki–Miyaura cross-coupling reaction developed in aqueous solvents, Pd(0) and Pd(II) were used as catalyst and pre-catalyst, respectively, in different excess (1.8–10 mol%) in CH3CN–H2O.5.1. PdCl2(dppf)2 as Pd(II) and Na2CO3
In 2008, Wagenknecht's group used Pd(dppf)2Cl2 as source of palladium for their Suzuki–Miyaura cross-coupling to furnish the nucleoside analogue 40 as a potential fluorescent probe for nucleic acids. Reacting 2′-deoxyuridine 6, 4-formylphenylboronic acid (39) (1.2 eq) and Na2CO3 (2 eq) as base in the presence of Pd(dppf)2Cl2 (6.5 mol%) as catalyst in a CH3CN–H2O (1:2) mixture for 5 hours at 80 °C afforded the target aldehyde 40 in 76% yield (Scheme 13).19 The presence of a formyl group, which has an electron-withdrawing function, in the para position of the phenyl core was influential in obtaining a good yield.
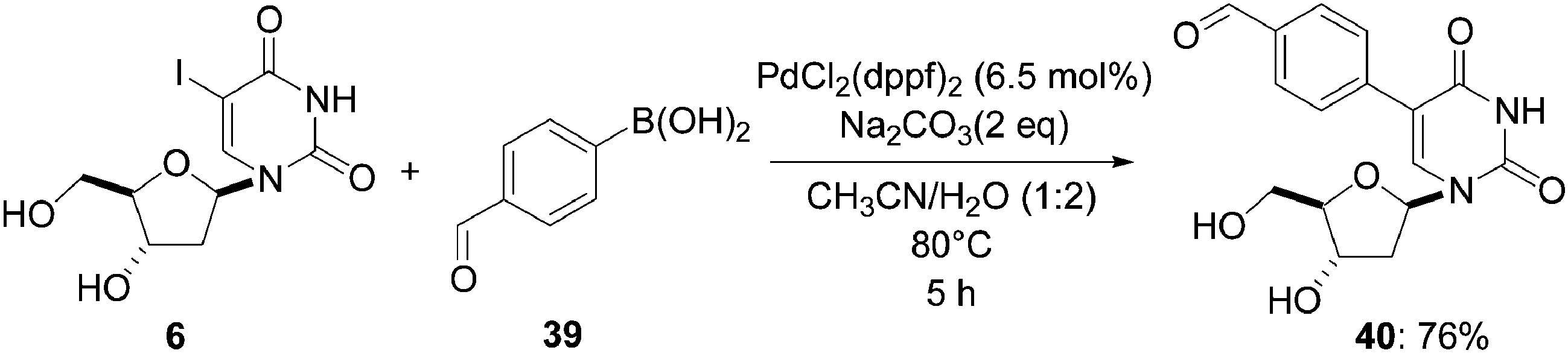 | ||
| Scheme 13 Synthesis of 5-(4-formylphenyl)uridine analogue 40. | ||
5.2. Pd(PPh3)4 as Pd(0) and K2CO3
In 2006, Wagner and co-workers described the synthesis of the 8-phenyladenosine analogue 26 in 90% yield.16 The reaction was undertaken at 80 °C, using the 8-bromo derivative 25, boronic acid 33 (1.5 eq) and a palladium catalyst based on a hydrophobic triphenylphosphine ligand (10 mol%) in the presence of K2CO3 as base (6 eq) in a CH3CN–H2O (1:2) mixture as solvent and furnished the target nucleoside analogue 26 (Scheme 14). This method afforded the targeted compound 26 in a higher yield than that using DME–H2O (Table 1, entry 1). With 8-bromoguanosine (32), no reaction was observed as previously described using DME–H2O as solvent (Scheme 10).16
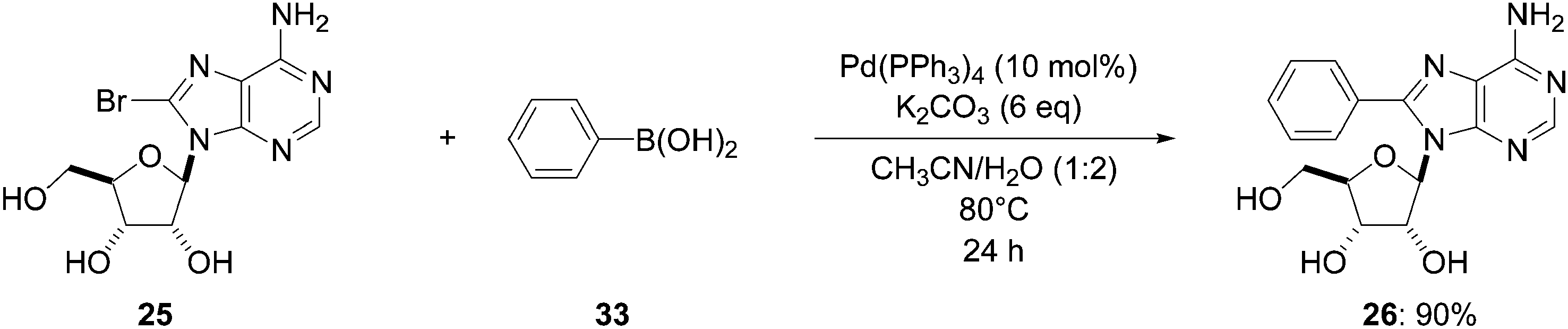 | ||
| Scheme 14 Synthesis of 5-phenyladenosine analogue 26. | ||
5.3. Pd(OAc)2 as Pd(II), TPPTS and Na2CO3
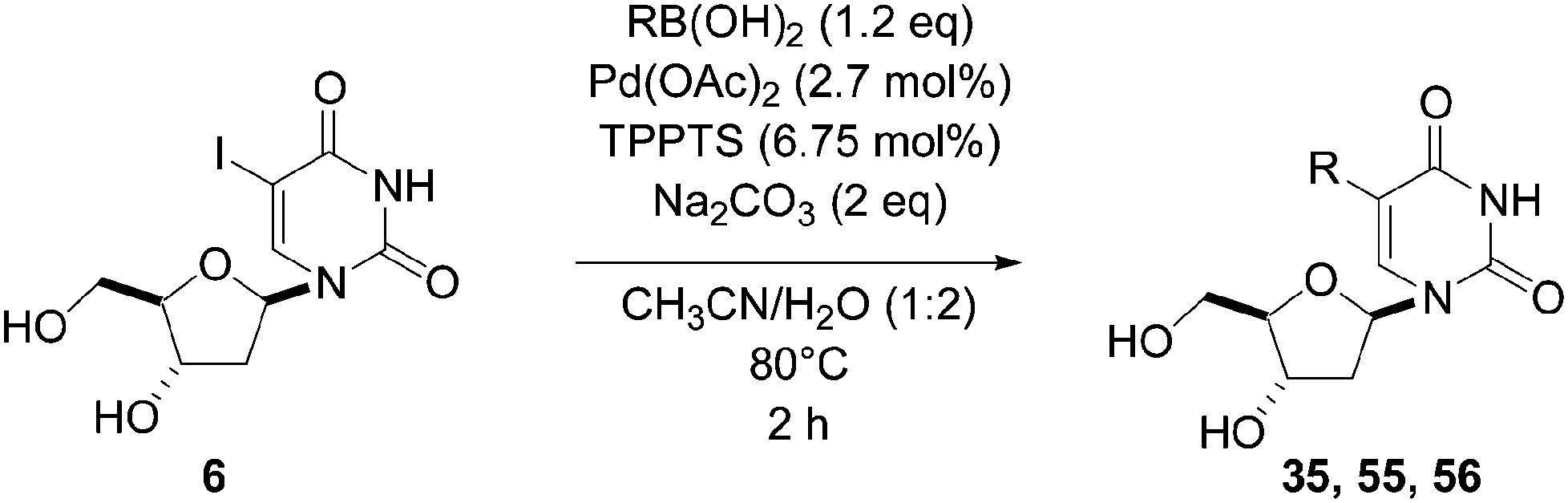
In 2003, Shaughnessy and co-workers reported the first methodology using commercially available, or easily prepared, water-soluble phosphine ligands and their application to the Suzuki coupling of different halonucleoside analogues.20 The methodology started from 8-bromo-2′-deoxyguanosine (11) and phenyl boronic acid (33). The ligands explored were water-soluble phosphines such as TPPTS, t-Bu-Pip-phos and DCPES and hydrophobic phosphines such as tri-tert-butylphosphine (41) and tri-o-tolylphosphine (42) (Fig. 3). Screening reactions were carried out in the presence of Pd(OAc)2 (5 mol%), the ligand (5–12.5 mol%) and Na2CO3 (2 eq) in a CH3CN–H2O (1:2) mixture as solvent. Shaughnessy and co-workers found that the best catalytic system was TPPTS–Pd(OAc)2. After optimization, the catalyst loading was lowered to 2.5–2.7 mol% without any change in reactivity.
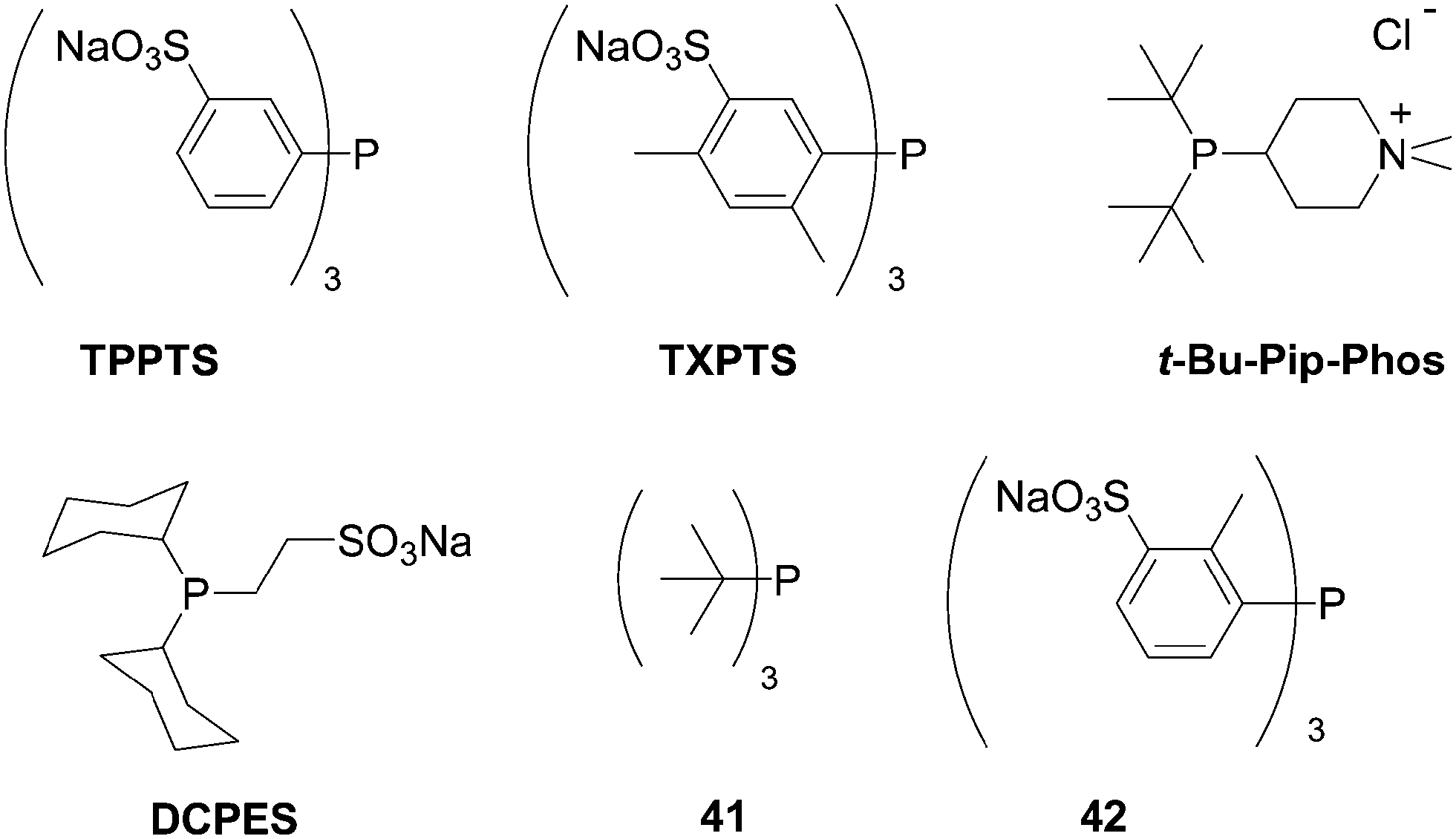 | ||
| Fig. 3 Phosphine derivatives TPPTS, TXPTS, t-Bu-Pip-Phos, DCPES, 41 and 42. | ||
The reaction of compound 11 and boronic acid 33 in the presence of Pd(OAc)2 (2.5 mol%) and TPPTS (6.25 mol%) at 80 °C over 2 hours resulted in complete conversion of 8-bromo derivative 11 to the target nucleoside analogue 43 in 82% yield (Table 2, entry 2). The same authors extended their work, using the same reaction conditions, to a series of both aryl boronic acids and halonucleoside analogues 6, 11, 15, 25 and 32 to determine the scope and utility of the reaction (Tables 2–4). Targets were chosen both to probe the electronic effects of the boronic acid on the reaction and to provide nucleoside and 2′-deoxynucleoside adducts of interest 16, 26–28, 34, 35, and 43–56 (Tables 2–4).20 For a given starting material, the nature of the substituent in position 4 of the aryl boronic acid did not modulate the yield. It is noteworthy that starting from the guanosine derivative, the 2′-deoxy derivatives 43, 45 and 47 were always prepared in better yields (+20%) than the corresponding guanosine analogues 34, 44 and 46 (Table 2, entries 1–6). The presence or not of a hydroxyl group in 2′ position of adenosine compounds had no such impact on the yield of the cross-coupling adducts (Table 2). Other halonucleoside analogues 6, 15 and 25 were explored (Tables 3 and 4), and the methodology was efficient for the tested starting materials even if the coupling of 8-bromoguanosine (32) furnished lower yield.
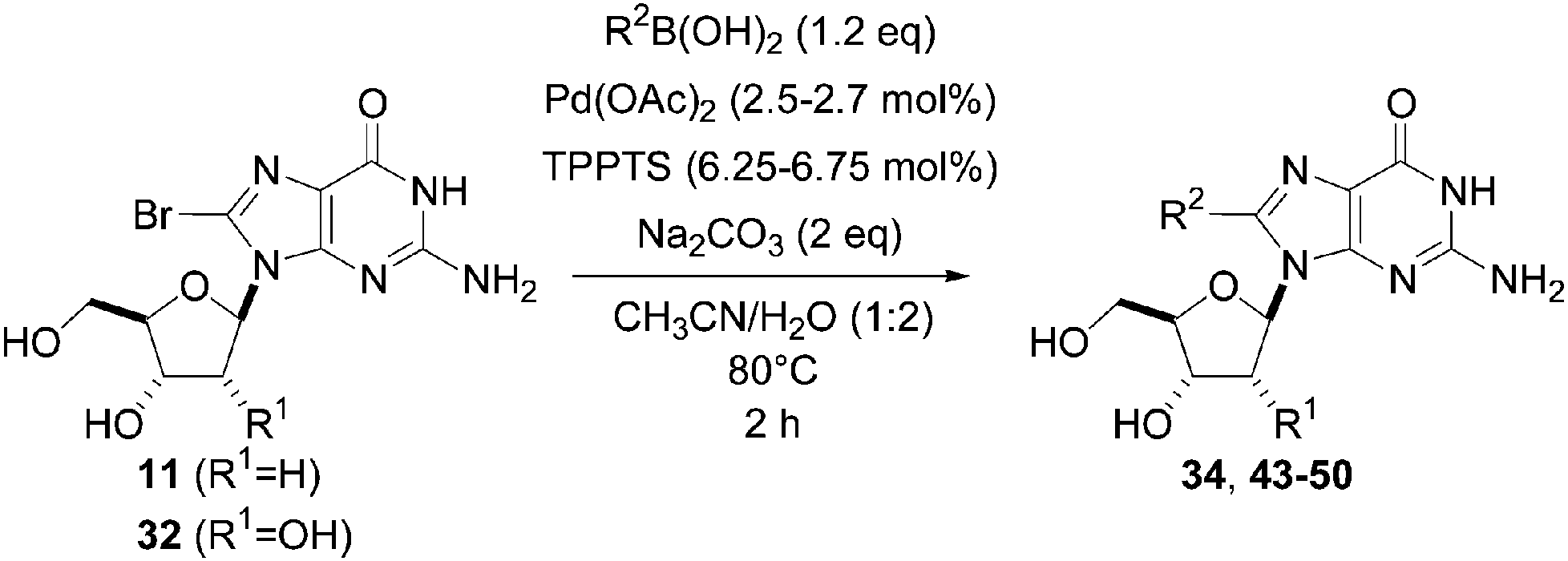
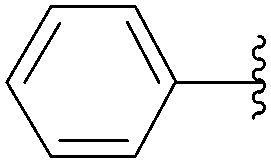
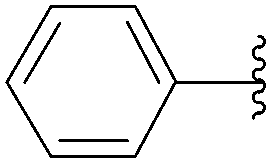
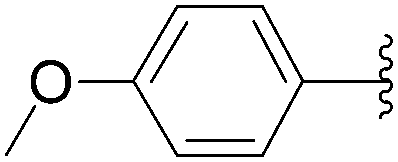
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | Starting material | R2 | Product | Yield (%) |
| 1 | OH | 25 |  |
26 | 88 |
| 2 | H | 15 |  |
51 | 87 |
| 3 | OH | 25 |  |
28 | 85 |
| 4 | H | 15 |  |
52 | 77 |
| 5 | OH | 25 |  |
27 | 99 |
| 6 | H | 15 |  |
53 | 90 |
| 7 | H | 15 | 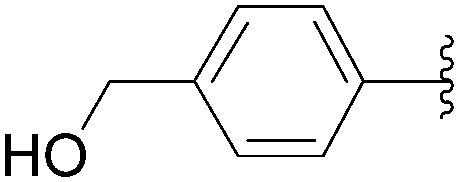 |
54 | 91 |
| 8 | H | 15 | 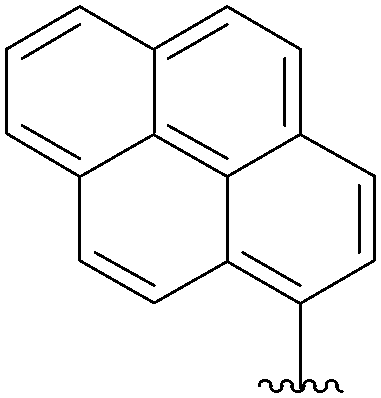 |
16 | 68 |
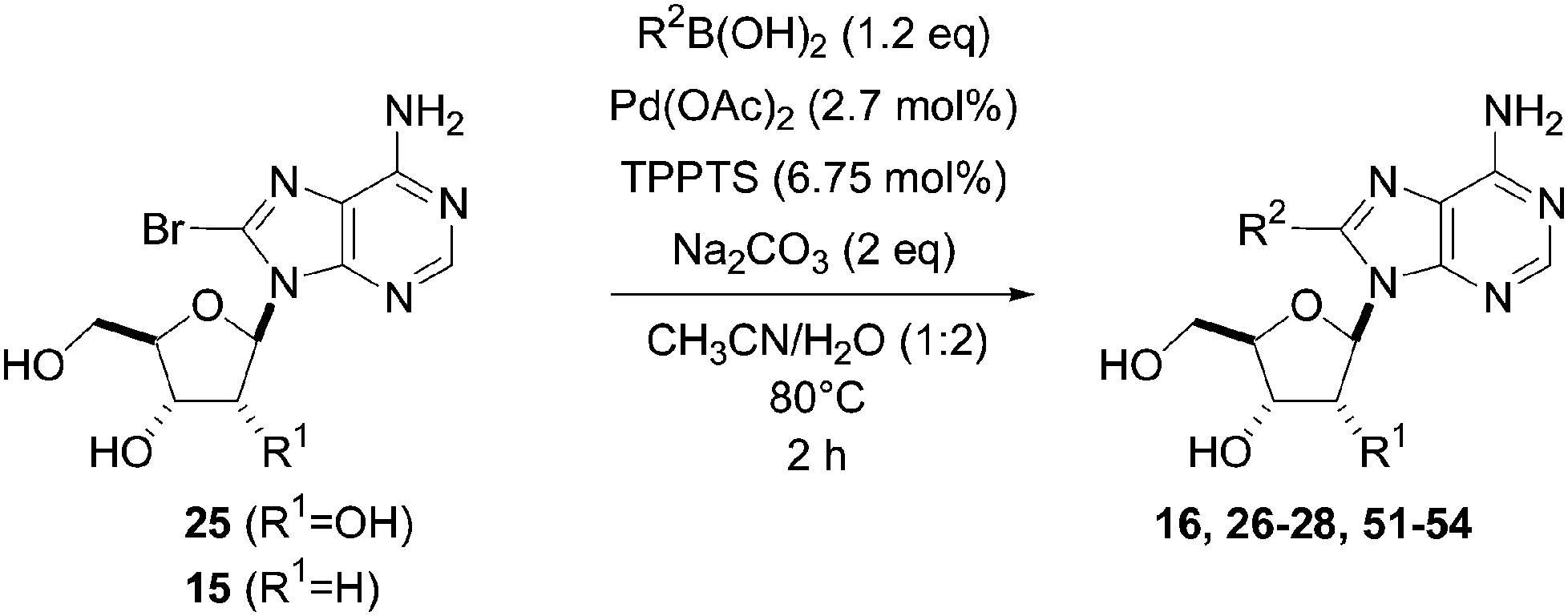
|
|||
|---|---|---|---|
| Entry | R | Product | Yield (%) |
| 1 |  |
55 | 83 |
| 2 | 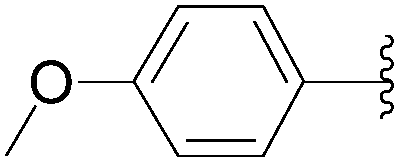 |
56 | 82 |
| 3 | 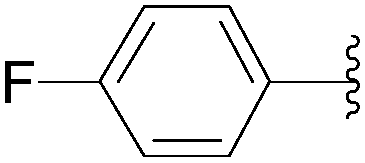 |
35 | 92 |
In 2008, Shaughnessy and co-workers reported the development of a general strategy for the synthesis of C8-arylpurine phosphoramidites.21 C8-Arylation of both the 8-bromo-2′-deoxyguanosine (11) and the corresponding adenosine analogue 15 was the key step of the process and was achieved using a Suzuki–Miyaura reaction. This cross-coupling reaction was conducted in a CH3CN–H2O (1:2) mixture containing Na2CO3 (2 eq) and Pd(OAc)2 (2.6 mol%) at 80 °C for 2.5 hours. The ligand TPPTS (6.84 mol%) was used as the catalyst in all cases. Reactions were readily followed by TLC and run until the C8-bromopurine starting material had been consumed (Table 5). The yields obtained using this method were similar to those obtained in 2003 by the same authors except for the tolyl derivative 53 (90% vs. 80%) (Table 3, entry 6 and Table 5, entry 7).21
|
||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | Starting material | R3 | Product | Yield (%) |
| 1 | OH | NH2 | 11 | 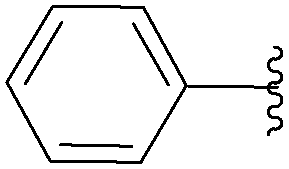 |
43 | 83 |
| 2 | OH | NH2 | 11 |  |
48 | 99 |
| 3 | OH | NH2 | 11 |  |
57 | 83 |
| 4 | OH | NH2 | 11 |  |
58 | 72 |
| 5 | OH | NH2 | 11 |  |
59 | 67 |
| 6 | NH2 | H | 15 | 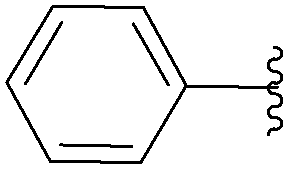 |
51 | 85 |
| 7 | NH2 | H | 15 | 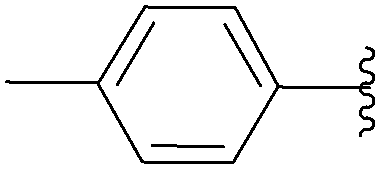 |
53 | 80 |
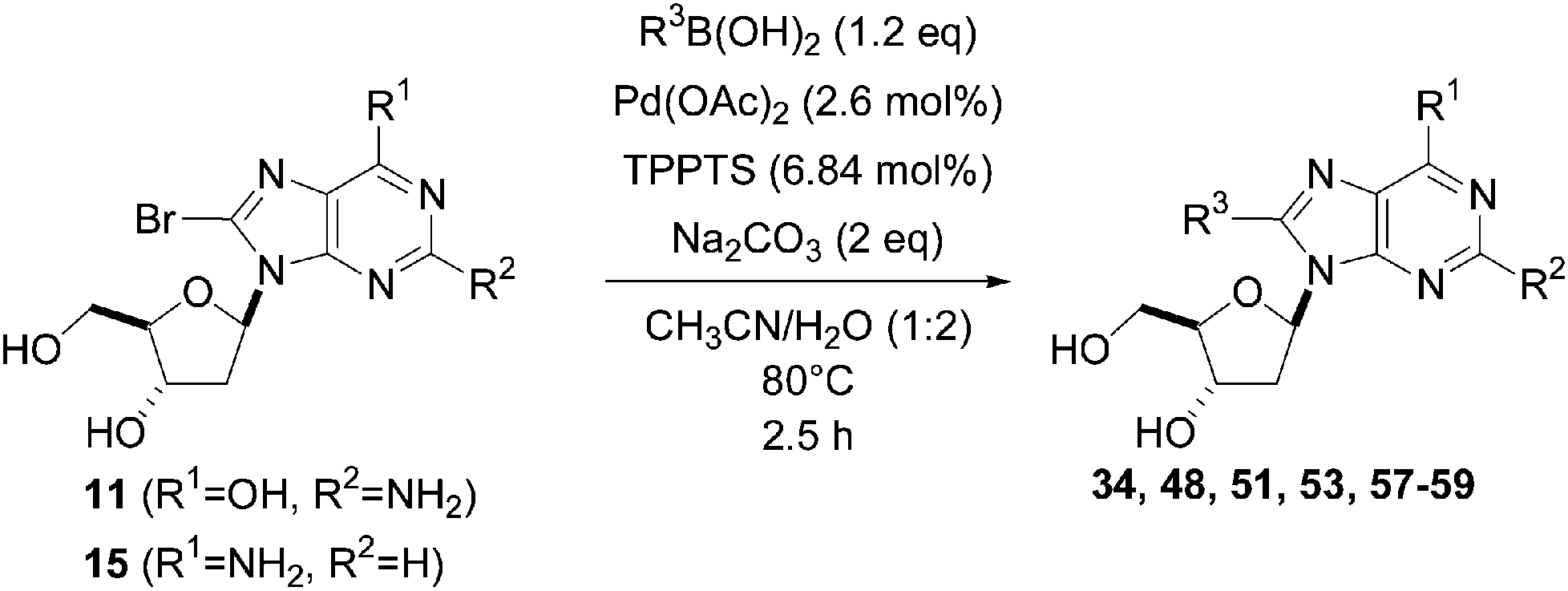
Shaughnessy and co-workers showed that the reactivity was dependant on the nature of the halonucleoside analogue.20 After optimization by varying the nature of the solvent, the palladium source and the ligand, the authors found two new methods starting from 2′-deoxyadenosine derivative 15 and 2′-deoxyuridine (6), respectively. These new methodologies were followed by HPLC; only relative HPLC peaks areas were reported. The starting substrate 15 and the boronic acid 33 (1.2 eq) were reacted in the presence of Pd(OAc)2 (10 mol%), TXPTS (25 mol%) and Na2CO3 (2 eq) in CH3CN–H2O (1:2) at room temperature for 30 minutes. The target nucleoside analogue 51 was obtained in yield higher than 95% (Scheme 15). It was the first reporting of a room temperature reaction giving a cross-coupling adduct in good yield.
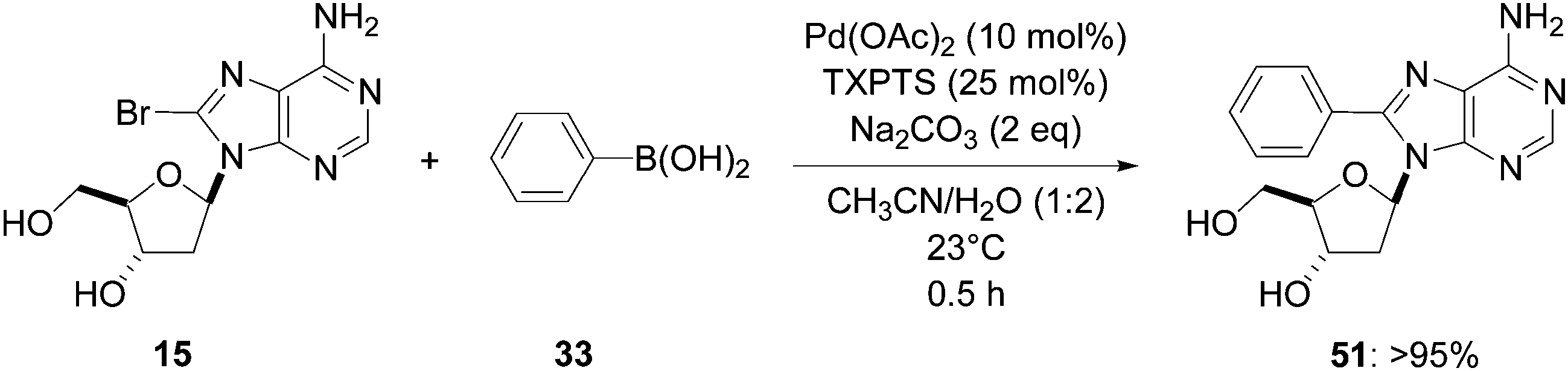 | ||
| Scheme 15 Synthesis of 8-phenyl-2′-deoxyadenosine (51). | ||
Starting from substrate 6, a similar reaction over 1 hour at room temperature but using a larger amount of Na2CO3 (7 eq) gave the uridine analogue 55 in a yield higher than 95% (Scheme 16).
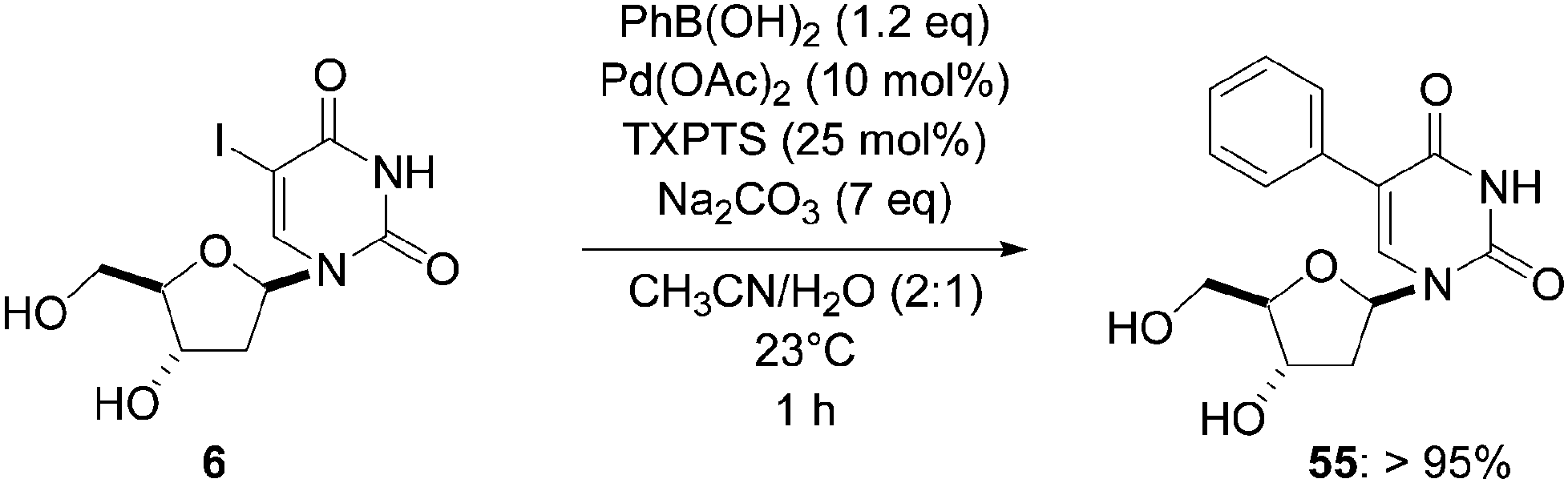 | ||
| Scheme 16 Synthesis of 8-phenyl-2′-deoxyuridine (55). | ||
With the aim to design a preparatory scale reaction, Shaughnessy and co-workers used the TXPTS–Pd(OAc)2 (2.5:1) catalyst system with a lower amount of palladium (2.7 mol%). This protocol afforded the nucleoside analogues 51 and 52 in 94% and 99% yields, respectively (Scheme 17). Application to the uridine and guanosine derivatives 6 and 11 furnished the corresponding nucleoside analogues 61 and 43 in 45% and 50% yields, respectively. It is clear that at room temperature, the use of TXPTS improved the method for the adenosine derivatives 51 and 52 (for compound 51: 94% vs. 87%; for compound 52: 99% vs. 77%, Scheme 17 and Table 3) but not for the uridine and guanosine nucleoside analogues 61 and 43.20
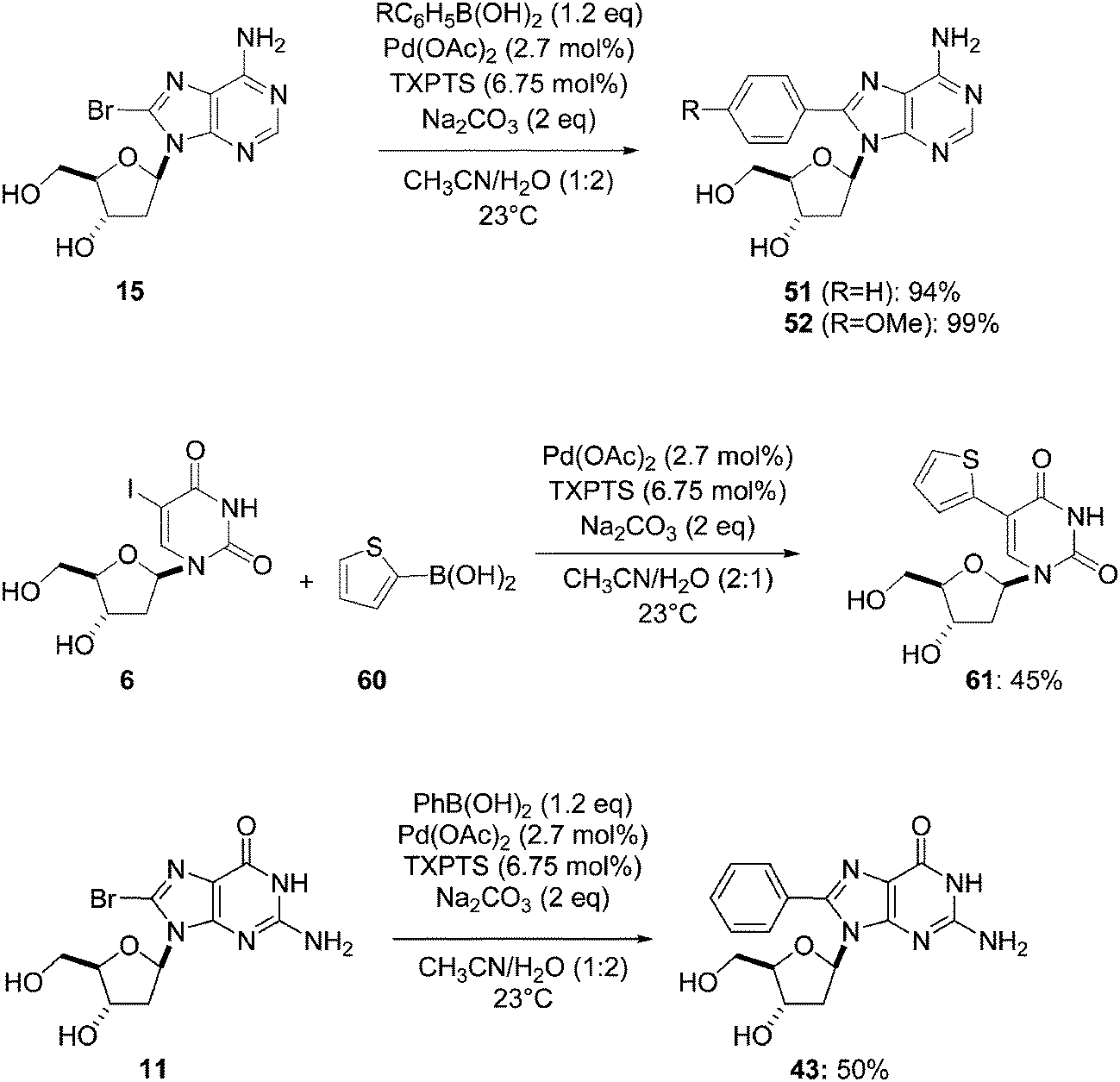 | ||
| Scheme 17 Synthesis of 8-aryl nucleoside analogues 43, 51, 52 and 61. | ||
Shaughnessy and co-workers noted that there was an unexpected ligand dependence on catalytic activity starting from 8-bromo derivative 11. In order to investigate this phenomenon, the authors carried out a series of reactivity and NMR spectroscopic studies, which showed that the guanine nucleoside 11 coordinated to Pd(II), thus inhibiting reduction to the Pd(0) active species.22 The authors showed that this competitive coordination could be avoided by replacement of the acidic N-1 proton of the nucleoside (Fig. 4).
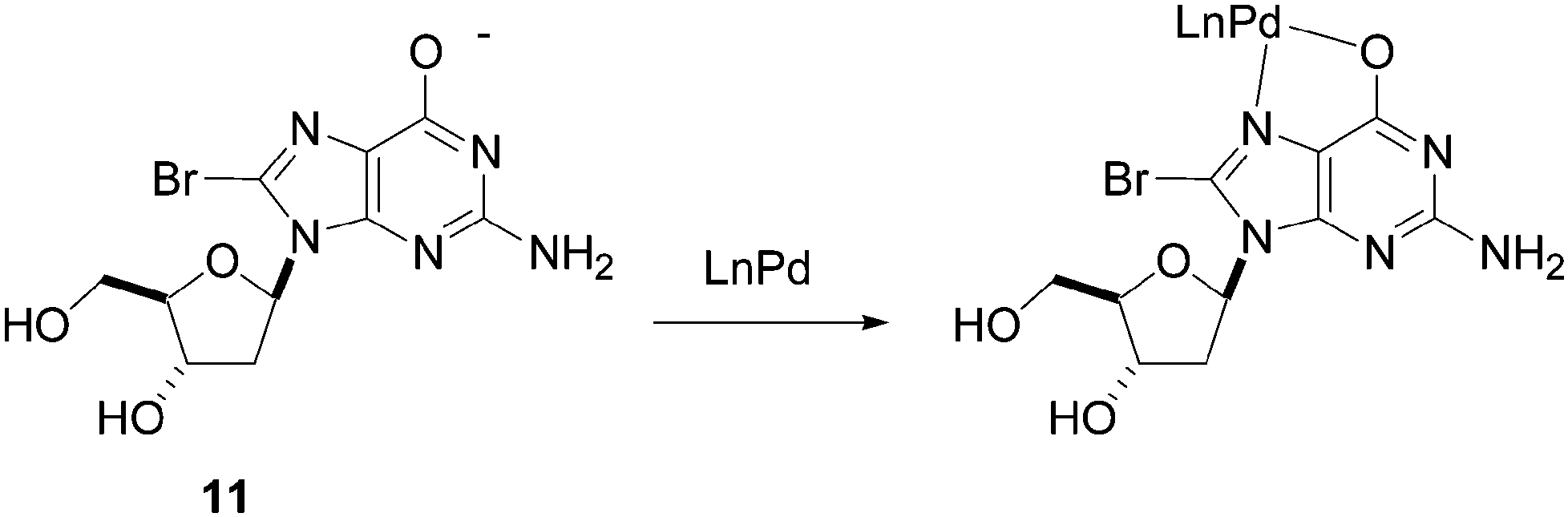 | ||
| Fig. 4 Palladium coordination to nucleoside analogue 11. | ||
The previously described Shaughnessy methodology20 was used in 2008 by Blaauwen and co-workers.23 After a longer reaction time (3 h vs. 2 h), they were able to isolate compound (34) in 75% yield instead of 64% yield.
In 2007, Harvey and co-workers tried to extend the work of Wagenknecht and Shaughnessy to obtain new C8-pyrenyl adducts starting from 8-halogenated purines.24 For this purpose, they synthesized different boronic acids and engaged them in a Suzuki cross-coupling reaction with 8-bromo-2′-deoxyguanosine (11) and 8-bromo-2′-deoxyadenosine (15) (Table 6). Their catalytic system consisted of Pd(OAc)2 (1.8 mol%) and TPPTS (4.72 mol%), maintaining the choice of CH3CN–H2O (1:2) in the presence of the corresponding boronic acid (1.0 eq) and Na2CO3 (1.4 eq). Reactions were conducted in a sealed tube heated at 80 °C overnight. In these conditions, the nucleoside analogues 12, 62 and 63 were obtained in yields higher than 58%. Although a range of polycyclic aromatic hydrocarbon boronic acids were tested, only the three adducts 12, 62 and 63 were obtained. The other boronic acids failed to engage in coupling with both 8-halogenated purines 11 and 15, and afforded instead the corresponding hydrogenated polycyclic aromatic molecules as the major product. The authors concluded that the failure of coupling to take place appeared to be the net consequence of steric retardation of the rate of coupling and acceleration of the rate of hydrolysis of the boronic acid group.24
|
||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | Starting material | R3 | Product | Yield (%) |
| 1 | H | NH2 | 15 |  |
62 | 58 |
| 2 | H | NH2 | 15 | 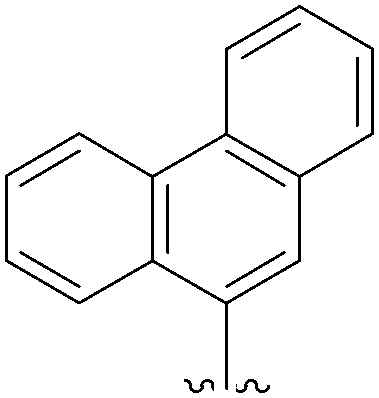 |
63 | 80 |
| 3 | NH2 | OH | 11 | 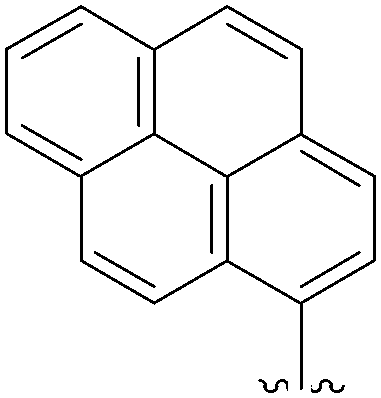 |
12 | 65 |
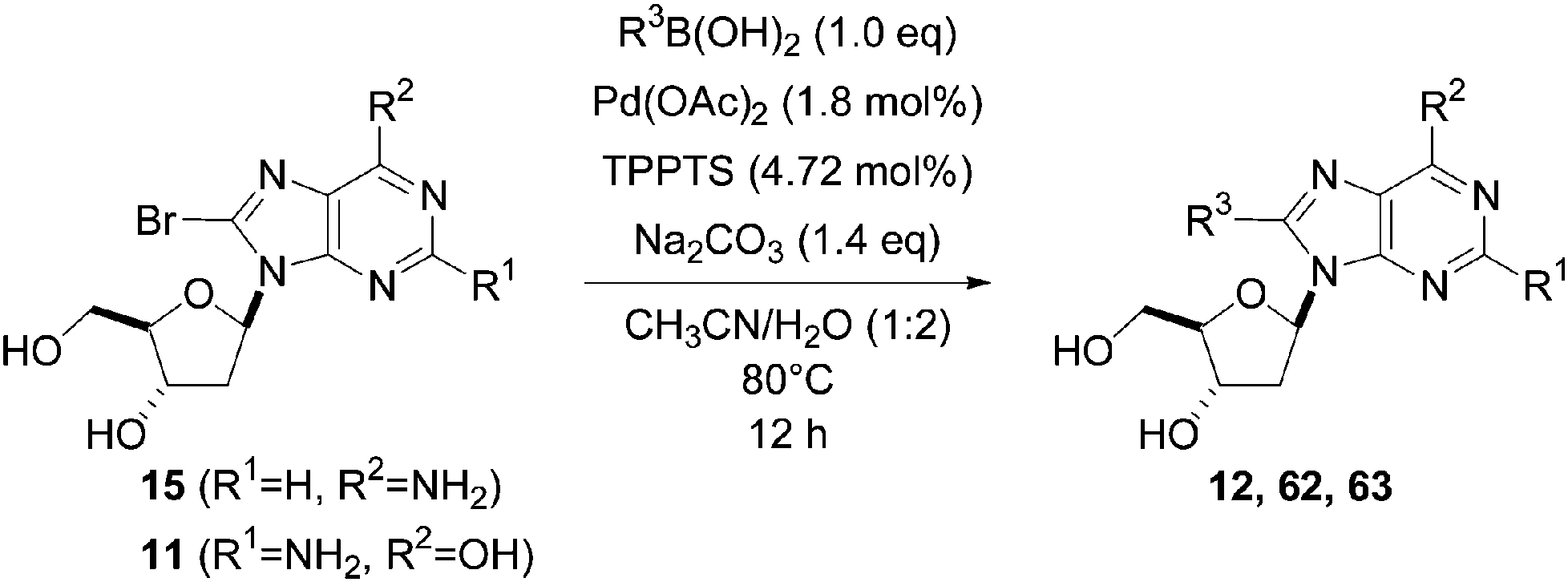
Still in 2007, Sekine's group published an attempt to use 5-aryl cytosine derivatives 64–66 as an artificial base pair to form a DNA triplex.25 The very efficient synthesis of such derivatives was achieved by reacting a mixture of 5-iodo-deoxycytidine (13), the water-soluble phosphine ligand TPPTS (8 mol%), Pd(OAc)2 (3 mol%), CH3CN–H2O (2:1) and Na2CO3 (2.2 eq) at 45 °C (Table 7). It is important to note that on this occasion, H2O was not the main part of the solvent mixture but represented only 1/3 of the solvent mixture.
|
|||
|---|---|---|---|
| Entry | R | Product | Yield (%) |
| 1 | 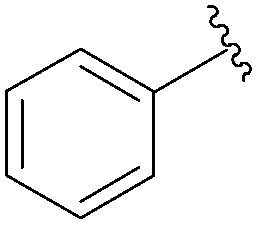 |
64 | 85 |
| 2 | 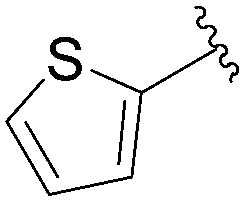 |
65 | 99 |
| 3 | 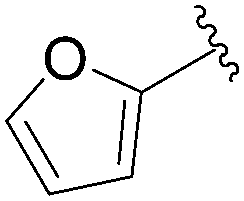 |
66 | 100 |
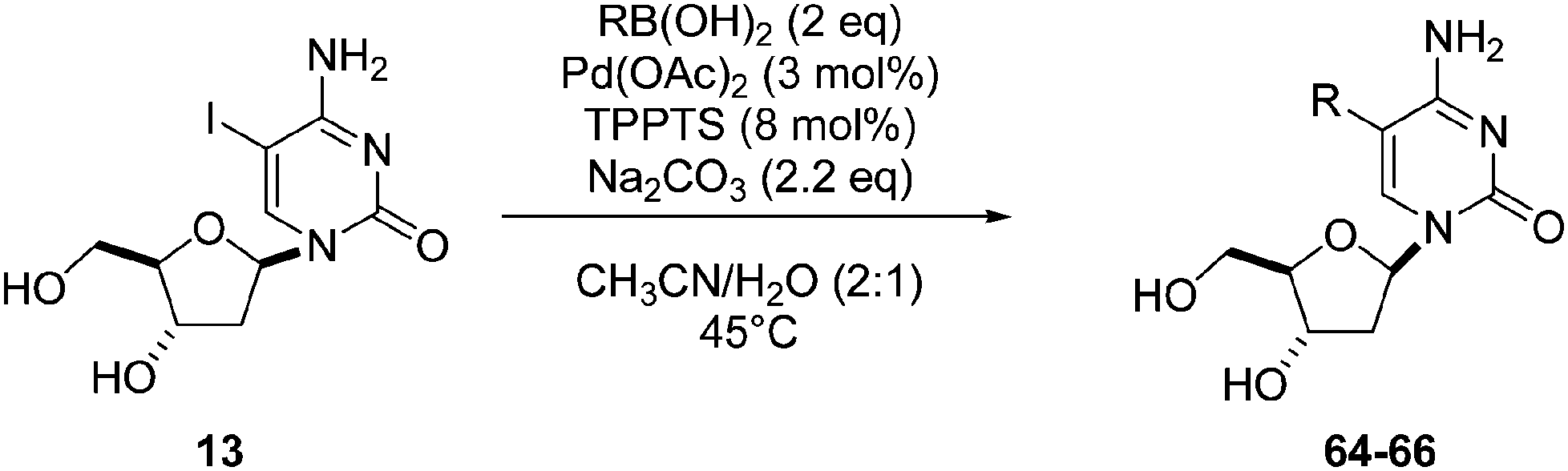
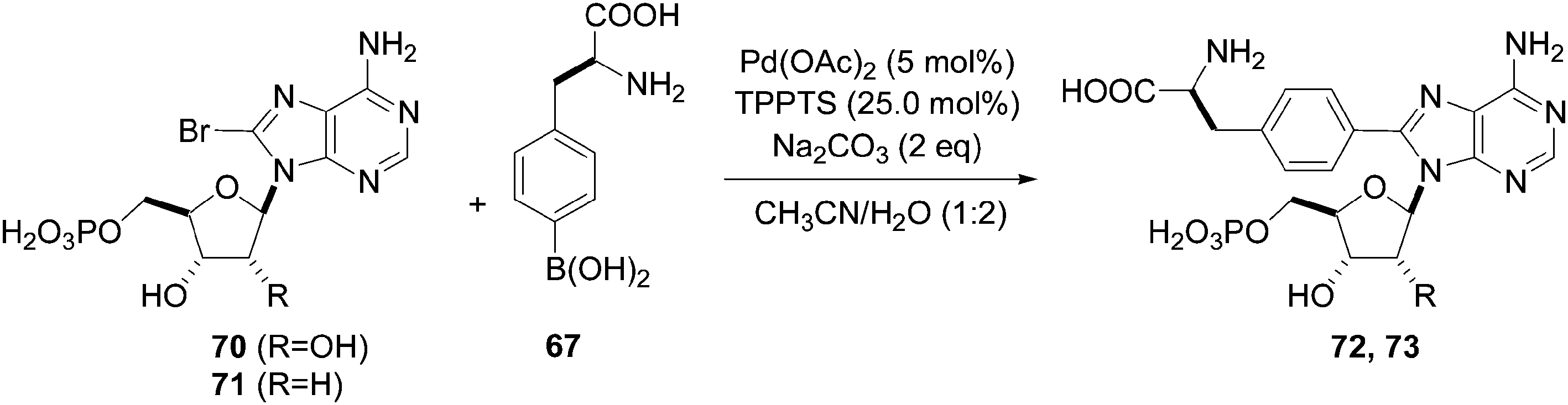
In 2006, Hocek and co-workers reported an application of Shaughnessy's conditions for the synthesis of 4-(adenosine-8-yl)alanine derivatives.26 After screening different ligands, bases and solvent mixtures, the authors chose to use a large amount of catalyst Pd(OAc)2 (5 mol%), TPPTS (12.5 mol%), boronic acid 67 (1.25 eq) and Na2CO3 (2 eq) in a mixture of CH3CN–H2O (1:2) for their coupling reactions as well as two ways of heating: conventional heating (90 °C) and microwave irradiation assistance (150 °C). The reactions of unprotected (S)-4-boronophenylalanine 67 with 8-bromoadenine nucleosides 15 and 25 gave the desired compounds 68 and 69, respectively, in a single step (Table 8). Microwave irradiation assistance permitted reduction of the reaction time but the preparative yields were somewhat lower. Additional studies of microwave-mediated reactions showed that yields could be further improved (>68%) (Table 8, entries 5 and 6) by increasing the ratio of ligand to Pd(OAc)2 to 5:1 instead of 2.5:1.
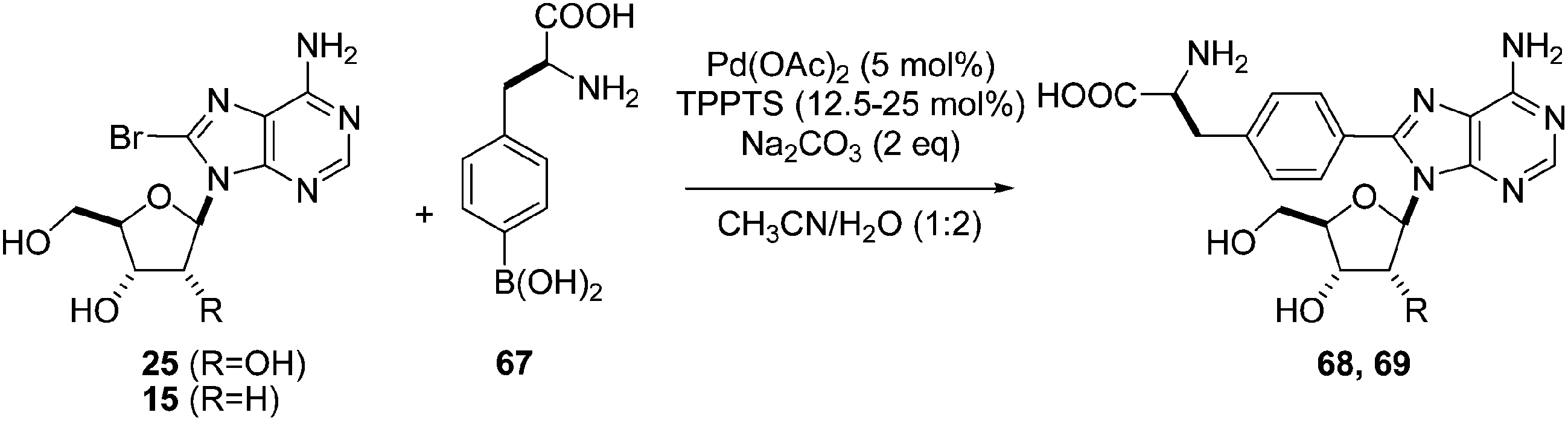
In the study to obtain the monophosphate derivatives 72 and 73, it was found that compound 72 could not be obtained by conventional heating at 90 °C (Table 9, entry 1). However, application of microwave irradiation at 150 °C was more successful and showed the possibility of cross-coupling (Table 9, entry 2).26 Finally, classical heating at the optimum temperature of 125 °C provided the desired nucleotide 72 in 72% yield (Table 9, entry 3). In contrast to the nucleoside analogue 70, complete decomposition of the nucleotide monophosphate 71 was observed in the case of microwave-mediated conditions to obtain 73 (Table 9, entry 4). In this case, the nucleotide analogue 73 was furnished under classical heating (125 °C) in 71% yield (Table 9, entry 5). These results confirmed that, in the Suzuki–Miyaura conditions, the 2′-deoxy derivatives of adenosine were less stable than the corresponding adenosine analogue using alternative technologies such as microwave mediation.
|
||||||
|---|---|---|---|---|---|---|
| Entry | R1 | Starting material | T (°C) | Time | Product | Yield (%) |
| 1 | OH | 70 | 90 | 4 h | 72 | 0 |
| 2 | OH | 70 | 150 (MW) | 5 min | 72 | 51 |
| 3 | OH | 70 | 125 | 1.5 h | 72 | 72 |
| 4 | H | 71 | 150 (MW) | 5 min | 73 | <1 |
| 5 | H | 71 | 125 | 20 min | 73 | 71 |
In contrast to the successful preparation of the monophosphate derivatives 72 and 73 using conventional heating, complete decomposition of the nucleotide triphosphates was observed when using microwave-mediated activation.
In continuity of their work on nucleosides and nucleotides chemistry, Hocek and co-workers reported new results on aqueous cross-coupling reactions of halogenated 2′-deoxynucleosides.27 They applied the above optimized catalytic system, namely, Pd(OAc)2 (5 mol%), TPPTS (25 mol%) in reaction with 5-iodo-2′-deoxyuridine (6) in the presence of boronic acid 67 (1.3 eq) and a large excess of Na2CO3 (3 eq) as base. Reactions were run for 1.5 hours at 100 °C to give compound 74 in 78% yield (Scheme 18).
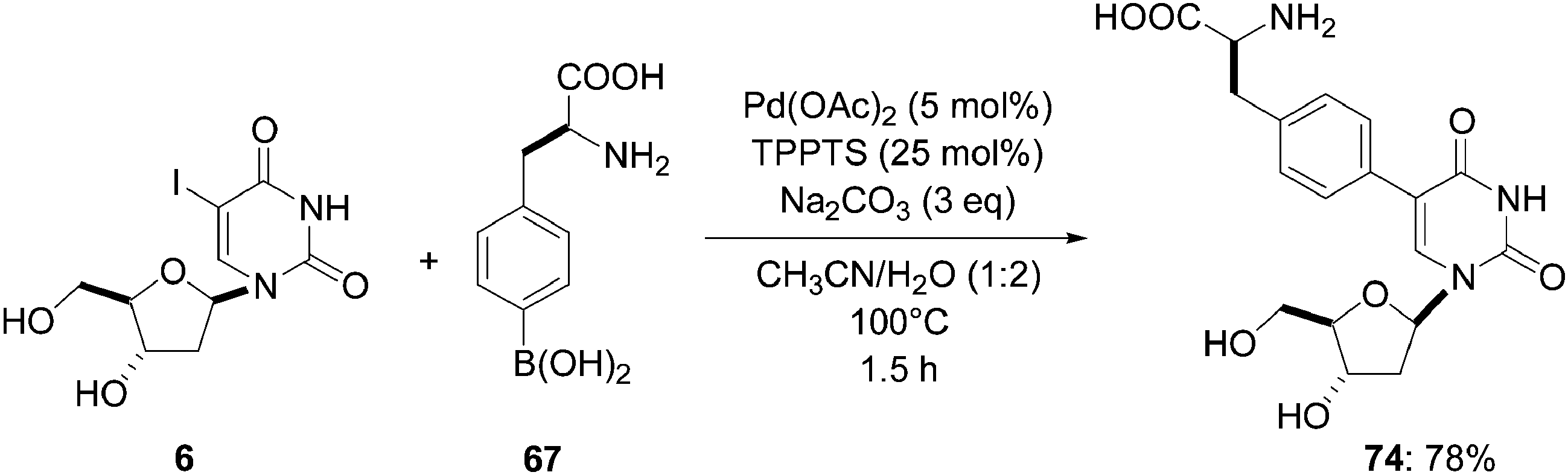 | ||
| Scheme 18 Synthesis of 5-aryl uridine analogue 74. | ||
In the continuity of this work, Hocek's group reported, in 2010, a series of 6-hetearyl-7-deaazapurine riboside analogues 76–81. Those compounds were prepared directly from the corresponding unprotected 6-chloro-7-deazapurine (75) and boronic acid (1.25 eq) using Pd(OAc)2 (5 mol%), TPPTS (12.5 mol%) as the catalyst and Na2CO3 (3 eq) as the base at 100 °C for 3 hours (Table 10).28 In this work the concentration of ligand was lower (12.5 mol% vs. 25 mol%) than for the synthesis of analogues 72–73 (Table 9).
|
|||
|---|---|---|---|
| Entry | R | Product | Yield (%) |
| 1 | 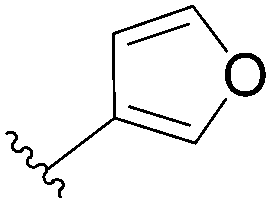 |
76 | 69 |
| 2 | 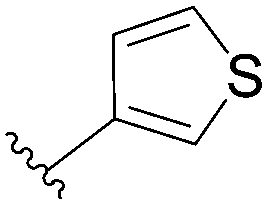 |
77 | 67 |
| 3 | 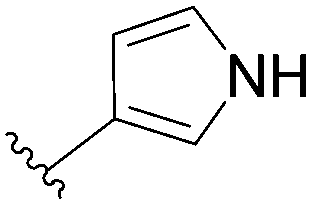 |
78 | 55 |
| 4 | 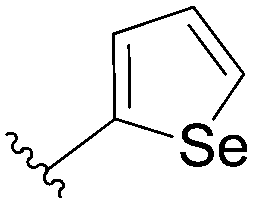 |
79 | 64 |
| 5 | 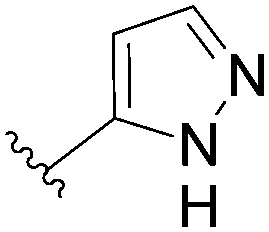 |
80 | 64 |
| 6 | 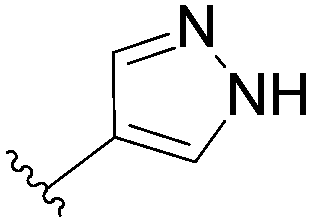 |
81 | 12 |
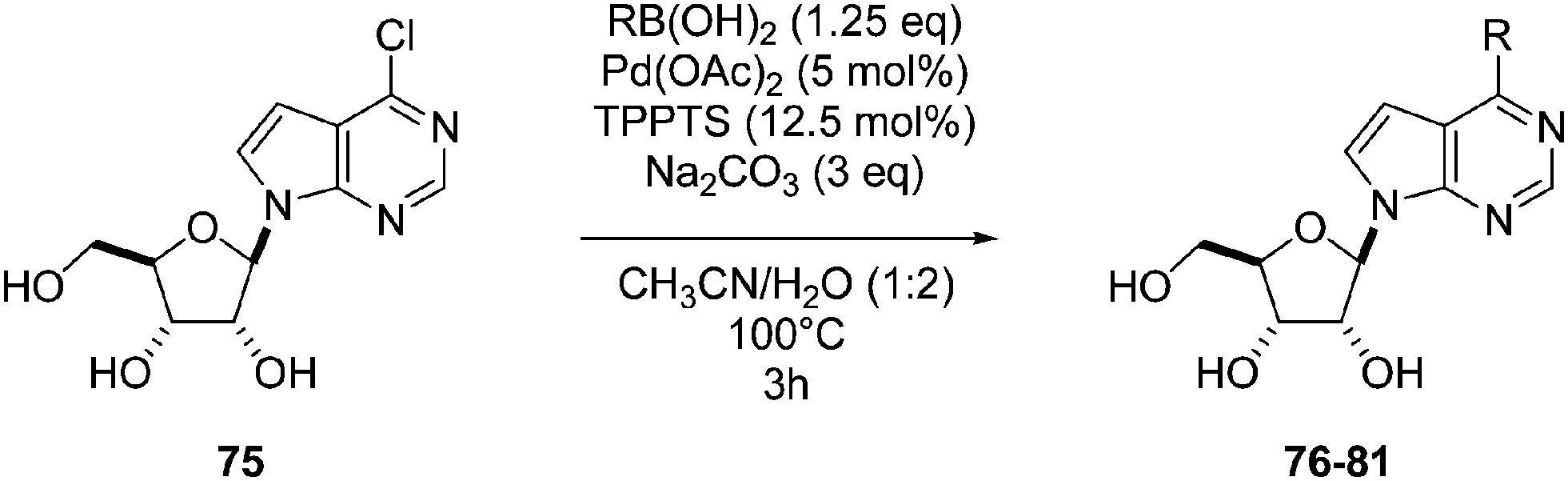
In the case of 3-pyrrolyl derivative, the boronic acid used was an N-protected triisopropylsilyl derivative (Table 10, entry 3). Authors reported that the protecting group was removed simultaneously with the Suzuki coupling due to the strongly basic conditions of the reaction. It has been noted that in case of NH containing boronic acid, a N-arylated product was formed by coupling of the 5-membered heterocycle with chlorinated starting material.
Application of previously described methodology26 with a larger amount of catalyst and base allowed Hocek and co-workers to obtain, in 2011, fourteen 7-aryl- and 7-hetearayltubercidin derivatives 83–96.29 The desired products were prepared in a single step mostly in good yields (Table 11).
|
|||
|---|---|---|---|
| Entry | R | Product | Yield (%) |
| 1 |  |
83 | 54 |
| 2 |  |
84 | 36 |
| 3 |  |
85 | 74 |
| 4 | 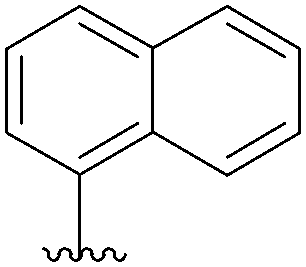 |
86 | 47 |
| 5 | 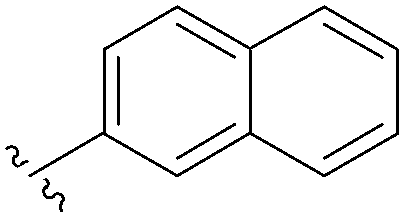 |
87 | 56 |
| 6 | 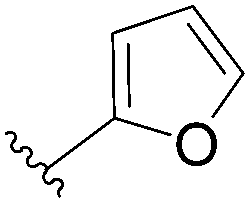 |
88 | 48 |
| 7 | 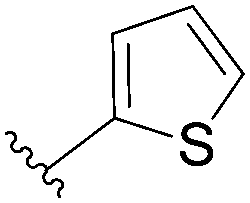 |
89 | 82 |
| 8 | 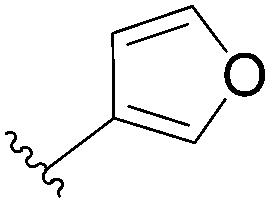 |
90 | 72 |
| 9 | 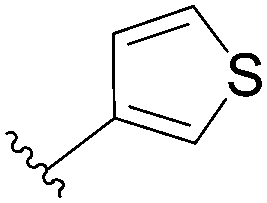 |
91 | 77 |
| 10 | 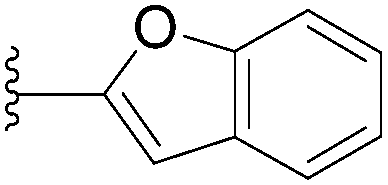 |
92 | 38 |
| 11 | 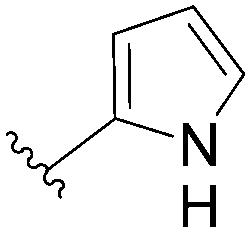 |
93 | 77 |
| 12 | 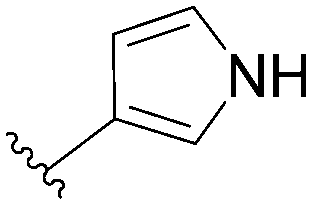 |
94 | 63 |
| 13 | 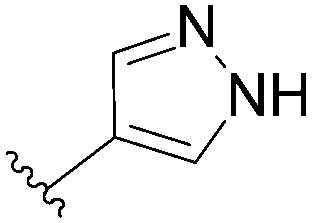 |
95 | 9 |
| 14 | 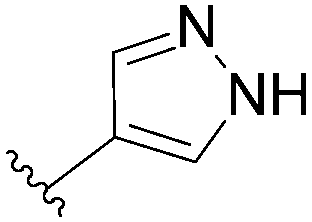 |
96 | 82 |
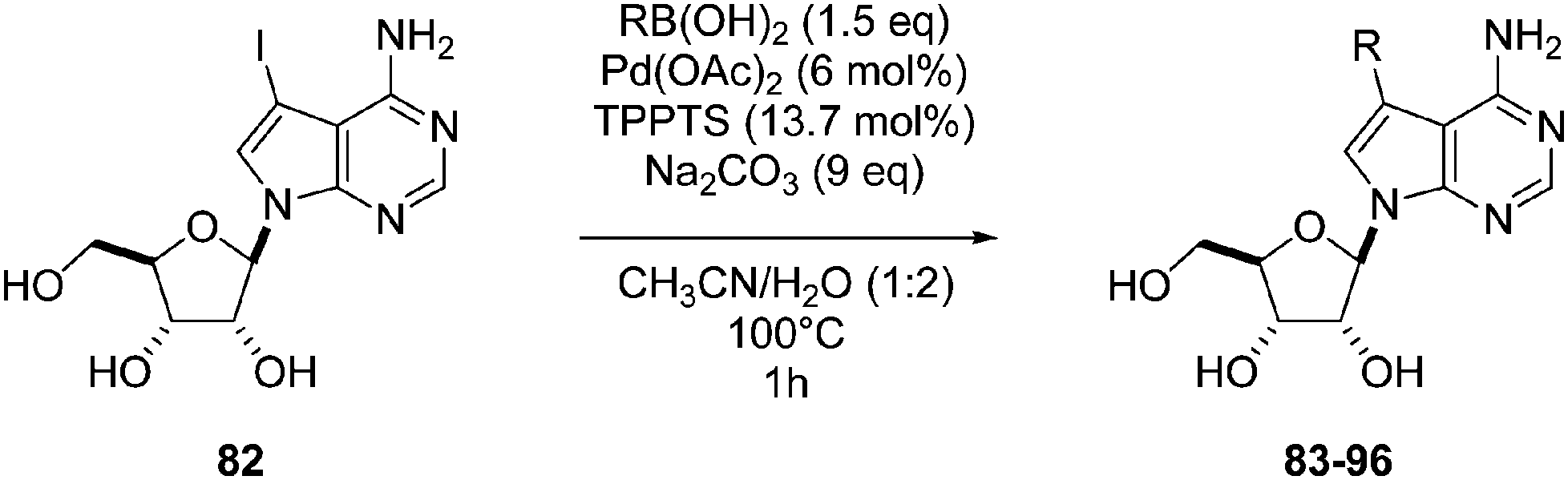
In addition to the compounds 83–96 (Table 11), the authors prepared several nucleotides, e.g., 5-O-monophosphate and 5′-O-triphosphate derivatives, by the aqueous Suzuki–Miyaura reaction (Table 12).29
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | Starting material | R2 | Product | Yield (%) |
| 1 | PO3H2 | 97 |  |
99 | 46 |
| 2 | P3O9H4 | 98 |  |
100 | 94 |
| 2 | PO3H2 | 97 |  |
101 | 27 |
| 3 | P3O9H4 | 98 |  |
102 | 34 |
| 4 | PO3H2 | 97 |  |
103 | 53 |
| 5 | P3O9H4 | 98 |  |
104 | 51 |
| 6 | PO3H2 | 97 |  |
105 | 37 |
| 7 | P3O9H4 | 98 |  |
106 | 45 |
| 8 | PO3H2 | 97 | 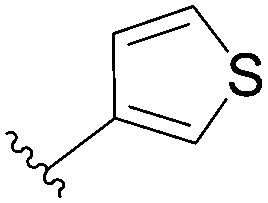 |
107 | 67 |
| 9 | P3O9H4 | 98 | 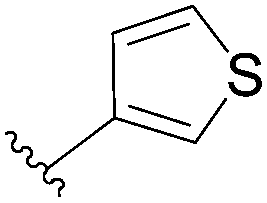 |
108 | 47 |
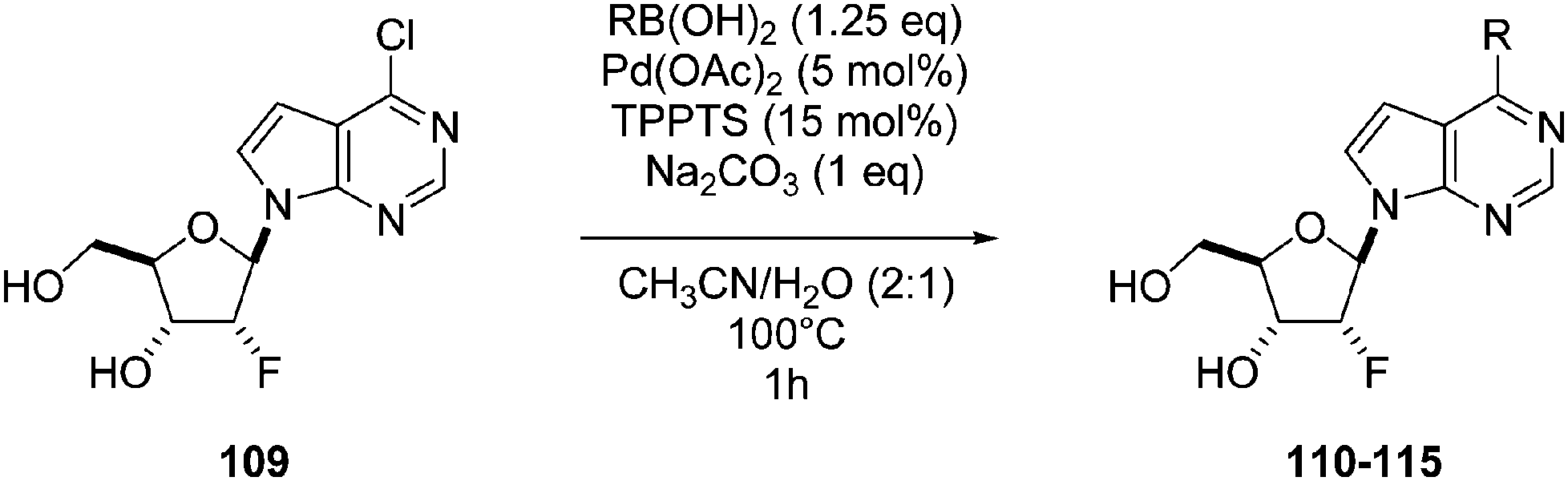
In their continuous research on the synthesis and biological activity of modified purine nucleoside derivatives and analogues, Hocek and co-workers reported the preparation of 2′-deoxy-2′-fluororibonucleosides derived from 6-chloro-7-deazapurines.30 This time, the cross-coupling reactions were done in a H2O–CH3CN (1:2) medium, still in the presence of Pd(OAc)2 (5 mol%), TPPTS (15 mol%) and a smaller amount of Na2CO3 (1 eq) at 100 °C for 1 hour. Different boronic acids were used, and desired products 110–115 were isolated in moderate-to-good yields (Table 13).
|
|||
|---|---|---|---|
| Entry | R | Product | Yield (%) |
| 1 | 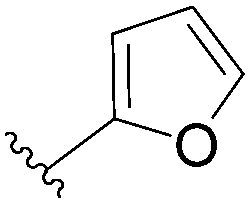 |
110 | 47 |
| 2 | 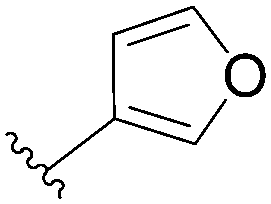 |
111 | 39 |
| 3 | 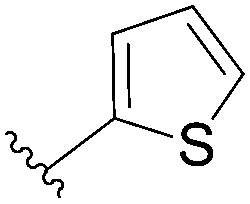 |
112 | 65 |
| 4 |  |
113 | 37 |
| 5 |  |
114 | 89 |
| 6 |  |
115 | 89 |
In 2012, application of Hocek's method was described by Fisher and co-workers as part of their work to produce fluorescence studies on modified nucleosides.31 This team studied the contribution of both the type and position of the aromatic substituents on the fluorescence of the resulting uridine derivatives. For this purpose, they synthesized and characterized different uridine and 2′-deoxyuridine analogues, which have an aromatic system directly linked to the C5 positions of the unprotected starting nucleoside (Table 14). Once again, the catalytic system was composed of Pd(OAc)2 (3.7 mol%), TPPTS (22 mol%) and Na2CO3 (3 eq) as base in a mixture H2O–CH3CN (2:1). In this protocol, the authors used a large excess of TPPTS as ligand.
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | Starting material | R2 | Product | Yield (%) |
| 1 | OH | 116 | 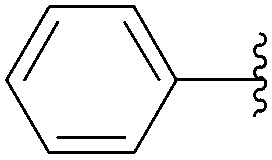 |
117 | 50 |
| 2 | OH | 116 | 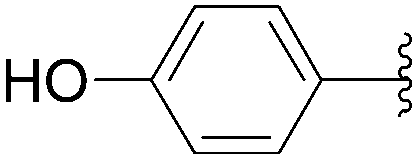 |
118 | 64 |
| 3 | OH | 116 | 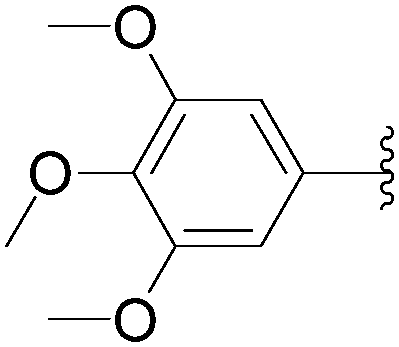 |
119 | 42 |
| 4 | OH | 116 | 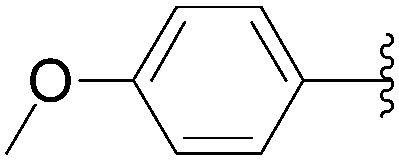 |
120 | 13 |
| 5 | H | 6 | 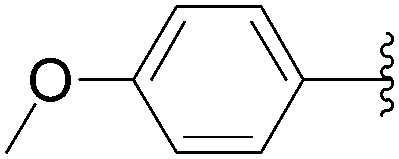 |
56 | 50 |
Recently, Hocek and co-workers reported the preparation of five 6-hetearyl-7-deazapurine ribonucleosides 121–125 starting from deprotected 6-chloro-7-deazapurine analogues using a direct aqueous Suzuki coupling in the presence of Pd(OAc)2 (5 mol%), TPPTS (15 mol%) and Na2CO3 (3 eq) (Table 15).32
|
|||
|---|---|---|---|
| Entry | R | Product | Yield (%) |
| 1 | 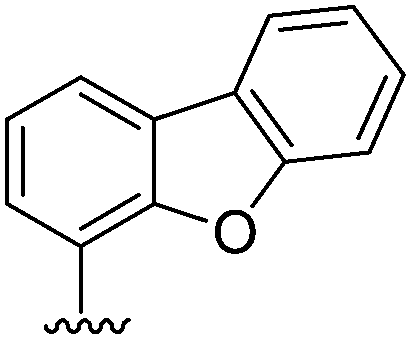 |
121 | 76 |
| 2 | 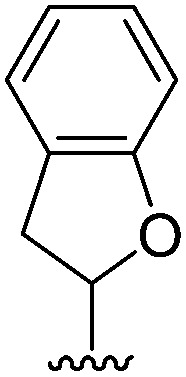 |
122 | 75 |
| 3 |  |
123 | 71 |
| 4 | 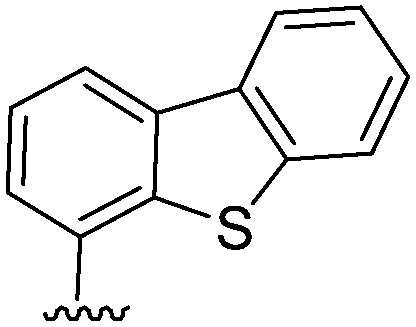 |
124 | 73 |
| 5 | 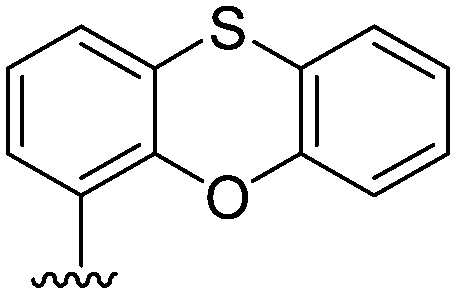 |
125 | 71 |
5.4. Pd(OAc)2 as Pd(II), TPPTS and K2CO3
In 2006, Wagner and co-workers used part of the methodology described by Shaughnessy and co-worker20 for the synthesis of 8-phenyladenosine analogue 26.16 They reacted the mixture of 8-bromo derivative 25, boronic acid 33 (1.5 eq), Pd(OAc)2 (2.5 mol%) and TPPTS (6.25 mol%) in the presence of K2CO3 as base (2 eq) in CH3CN–H2O (1:2) to give the target nucleoside analogue 26 in 94% yield (Scheme 19). Using a similar method but with K2CO3 as base instead of Na2CO3 permitted them to increase the yield of compound 26 (94% vs. 88%, Table 3, entry 1). In contrast with the use of Pd(PPh3)4 catalyst, this method permitted 8-phenyl guanosine (34) to be obtained in 75% yield. It is notable that none of the reagents Pd(PPh3)4 or Pd(OAc)2, TPPTS in a DME–H2O mixture or Pd(PPh3)4 in CH3CN–H2O allowed the target guanosine derivative 34 to be obtained (Scheme 19).
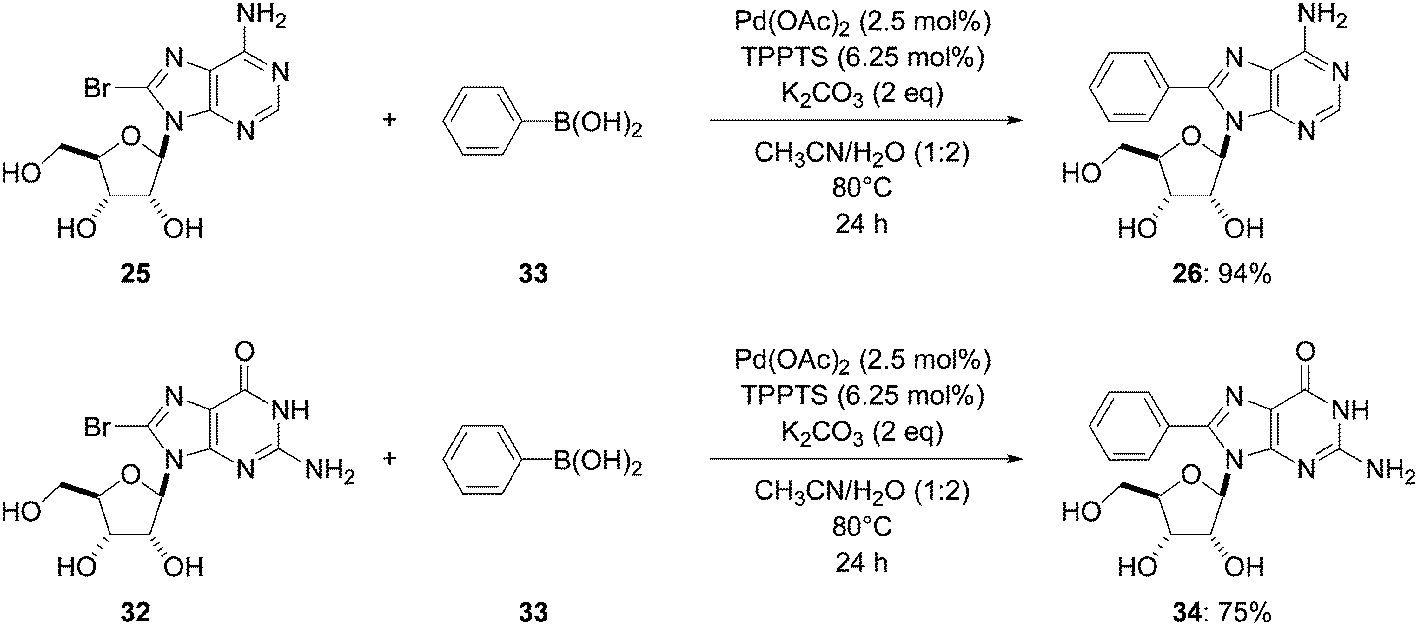 | ||
| Scheme 19 Synthesis of 8-phenyladenosine and 8-phenyl guanosine analogues 26 and 34. | ||
5.5. Pd(OAc)2 as Pd(II), TPPTS and Cs2CO3
In order to prepare new nucleotide analogues having an amino acid moiety, different studies were reported by Hocek's group.26 In 2003, the authors described an efficient methodology for cross-coupling reactions starting from nucleotide triphosphate analogues. As those particular nucleosides are known to be rather labile compounds, different mild methodologies such as lower temperature or microwaves irradiation assistance were tested but without any success, because a complex mixture of by-products was obtained including hydrolysis products of the starting nucleotide. The use of conventional base most often afforded the degradation of the triphosphate analogue. To limit this step, other bases were studied. Thus, the triphosphate derivatives 126 and 127, boronic acid 67 (1.5 eq), Pd(OAc)2 (10 mol%), TPPTS (50 mol%) as catalyst and Cs2CO3 (5 eq) as base in a mixture of CH3CN–H2O (1:2) at 125 °C for 20–30 minutes furnished the target nucleotide analogues 128 and 129 in 51% and 55% yields, respectively (Scheme 20).
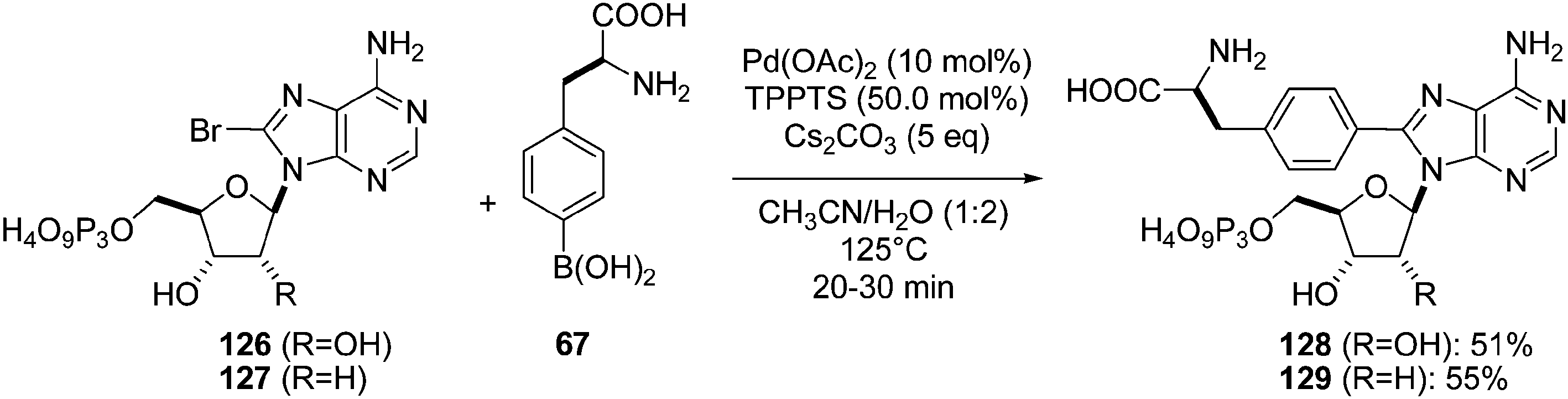 | ||
| Scheme 20 Synthesis of (adenine-8-yl)phenylalanine analogues 128 and 129. | ||
Starting from the 5-iodo derivative 130 and modifying the above methodology allowed Hocek and co-workers to obtain the cytosine analogue 131. The authors used the unchanged catalytic system but preferred to modulate the temperature (110 °C vs. 125 °C). The mixture of 5-iodo derivative 130 and boronic acid 67 (2.0 eq) in the presence of Pd(OAc)2 (10 mol%), TPPTS (50 mol%) and Cs2CO3 (5 eq) in a CH3CN–H2O (1:2) afforded the amino acid derivative 131 in 56% yield (Scheme 21) in 30 minutes.27
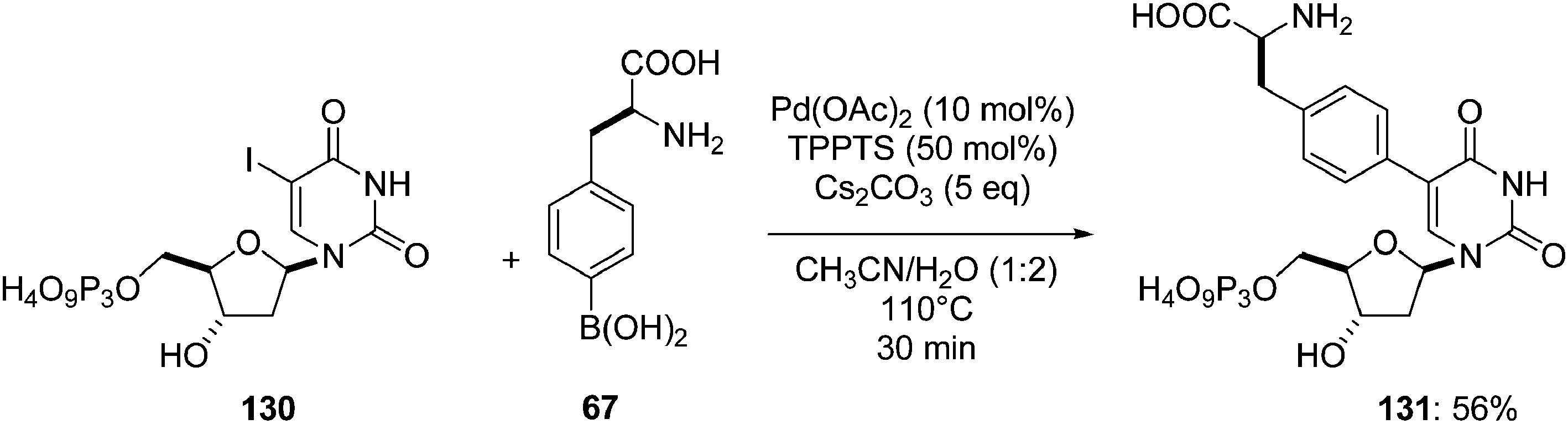 | ||
| Scheme 21 Synthesis of (uridin-5-yl)phenylalanine analogue 131. | ||
In 2008, Hocek and co-workers described the preparation of 7-deaza-2′-deoxyadenosine derivatives bearing bipyridine and phenanthroline ligands linked via acetylene or phenylene to position 7 of the nucleoside, as well as their Ru(II) complexes.33 The aim of this work was to study the electrochemical and photophysical properties of the resulting labeled nucleosides. In order to attach the phenylene linker to position 7 of the 7-deaza-2′-deoxyadenosine, the authors used an aqueous-phase Suzuki–Miyaura cross-coupling reaction, and the phenylene-bridged conjugates 133–135 were prepared in excellent yields. Starting from 7-iodo-7-deaza-2′-deoxyadenosine (132), pinacol boronate ester (1.2 eq), Pd(OAc)2 (5 mol%) and TPPTS (12.5 mol%) in the presence of Cs2CO3 (3 eq) in a CH3CN–H2O (1:2) mixture as solvent furnished the target compounds 133–135 in 78–96% yields (Table 16).
|
|||
|---|---|---|---|
| Entry | R | Product | Yield (%) |
| 1 |  |
133 | 95 |
| 2 |  |
134 | 96 |
| 3 |  |
135 | 78 |
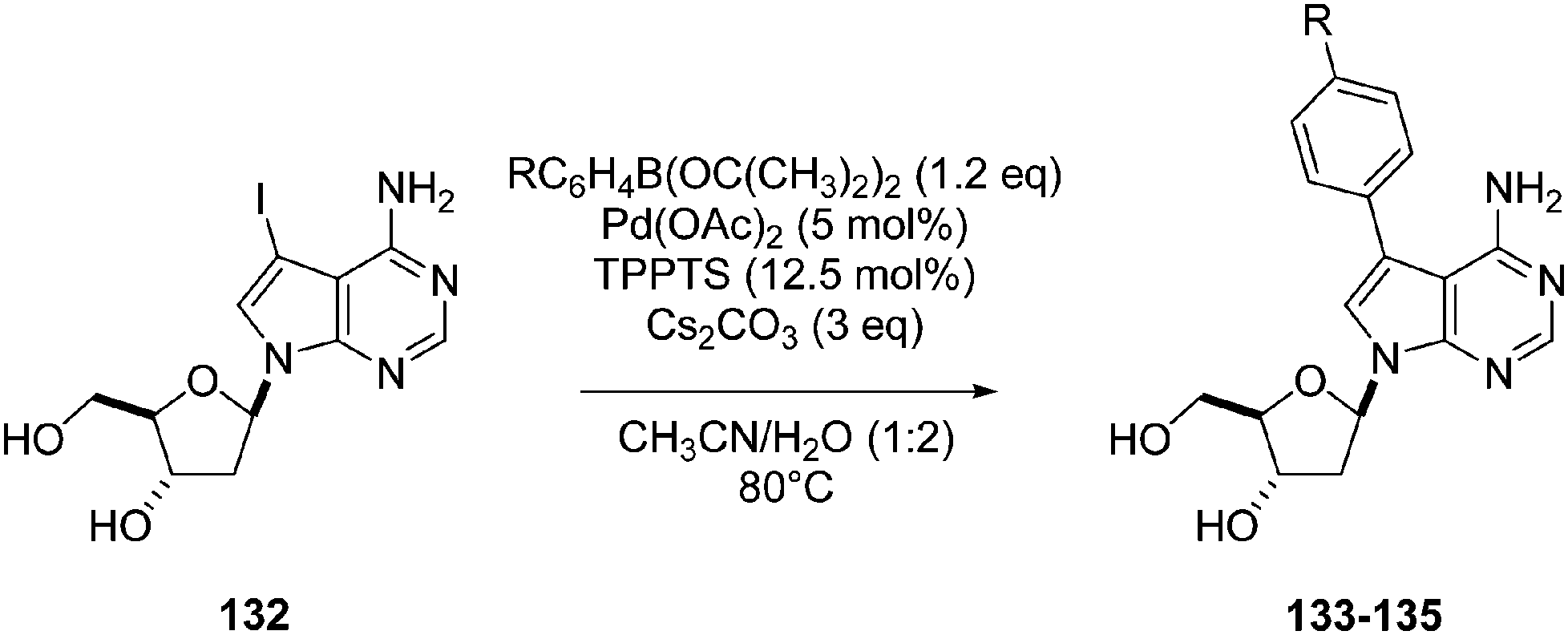
Using exactly the same methodology, the authors also obtained three corresponding Ru(II) complexes 136–138 of the 7-deazapurine nucleoside in good yields (Table 17). The authors themselves prepared the boronate–Ru(II) building blocks necessary for the Suzuki–Miyaura cross couplings.
|
|||
|---|---|---|---|
| Entry | R | Product | Yield (%) |
| 1 |  |
136 | 95 |
| 2 |  |
137 | 73 |
| 3 | 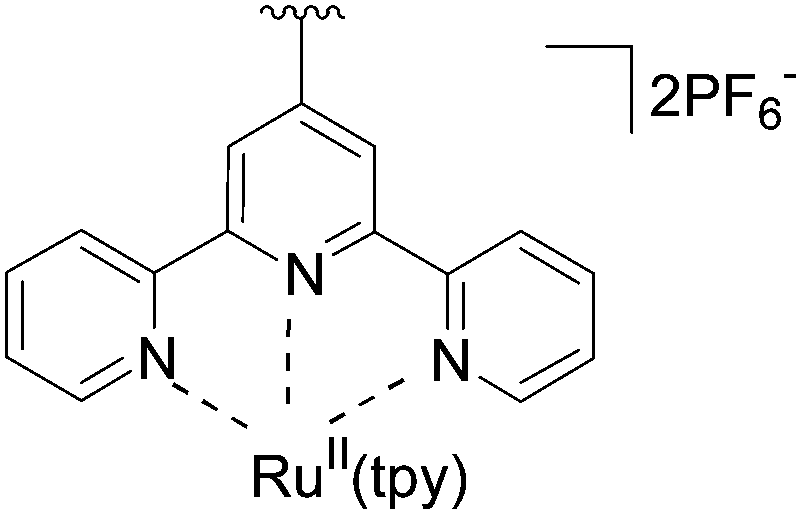 |
138 | 76 |
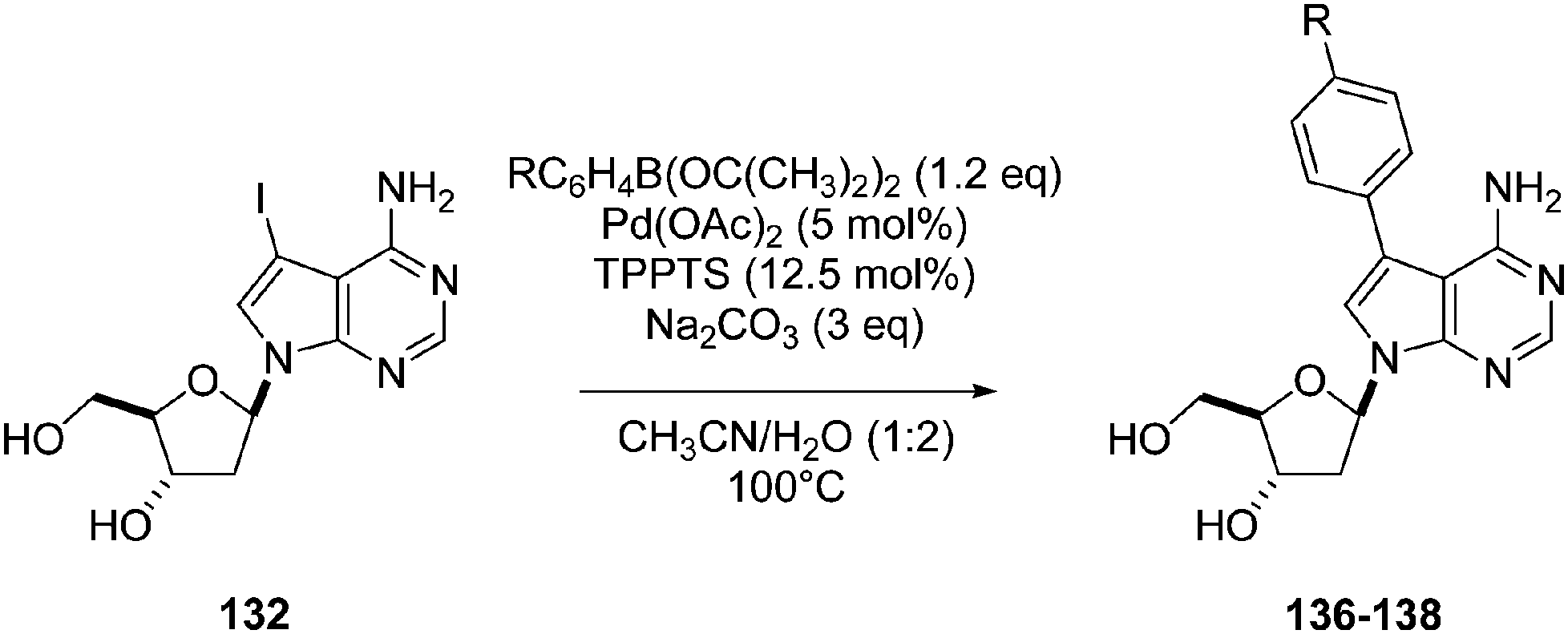
Still in 2008, Hocek's group reported the synthesis of 8-phenyl-dATP 139 and its use as substrate for DNA polymerases.34 The Suzuki–Miyaura cross-coupling reaction of 8-Br-dATP 127 with phenyl boronic acid 33 (2 eq) was performed in the presence of Pd(OAc)2 (10 mol%) and TPPTS (50 mol%) in a CH3CN–H2O (1:2) mixture for 30 minutes at 120 °C (Scheme 22).
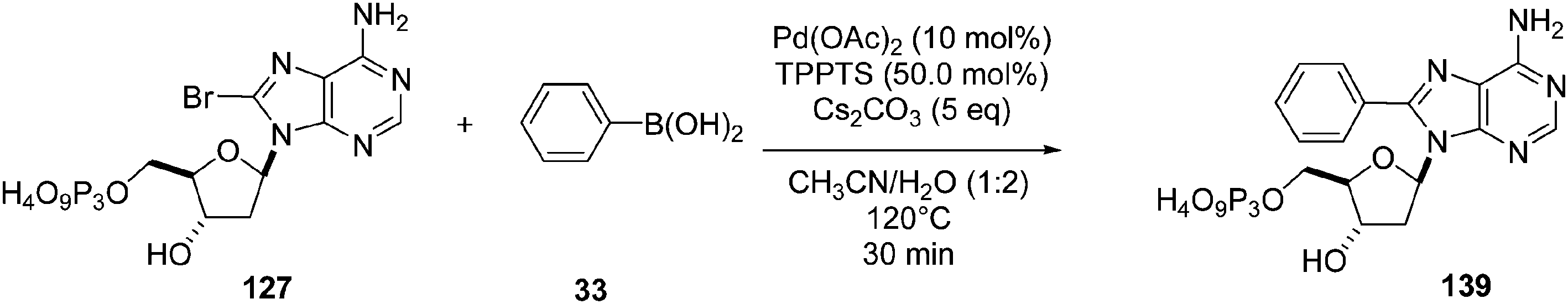 | ||
| Scheme 22 Synthesis of 8-phenyl-dATP 139. | ||
The coupling of more conventional aromatic boronic acids was described by the Hocek group using a similar method.35,36 By application of the first methodology, starting from 5-iodo nucleotide derivatives 130 and 140, Hocek and co-workers obtained the uridine and cytosine analogues 141–145. The reactions were performed at 110 °C for 30 minutes using a mixture of 130 or 140 and boronic acid (2 eq) in the presence of Pd(OAc)2 (10 mol%), TPPTS (50 mol%) and Cs2CO3 (5 eq) in CH3CN–H2O (1:2). The large amount of palladium resulted in furnishing the target uridine analogues 141 and 142 and the cytosine derivatives 143–145 in 26–65% yields (Table 18).
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | Starting material | R2 | Product | Yield (%) |
| 1 | OH | 130 | 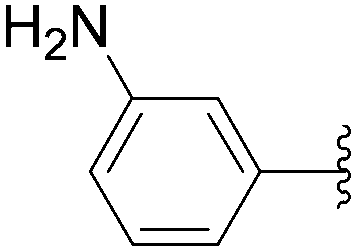 |
141 | 43 |
| 2 | OH | 130 | 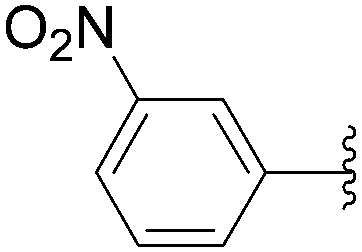 |
142 | 26 |
| 3 | NH2 | 140 | 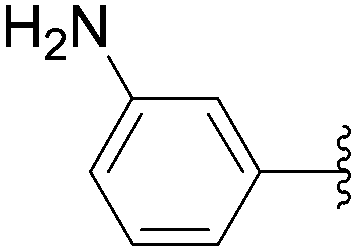 |
143 | 43 |
| 4 | NH2 | 140 | 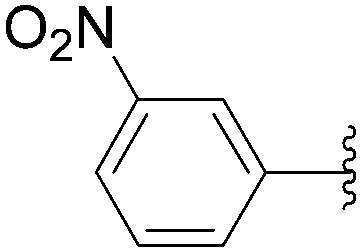 |
144 | 26 |
| 5 | NH2 | 140 | 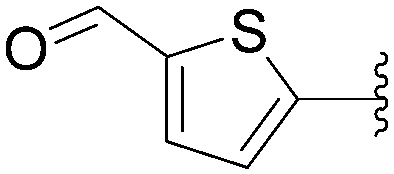 |
145 | 65 |

Application of this cross-coupling protocol in the reactions of the iodinated nucleotide monophosphate 146 and 5-formylthiophene boronic acid (147) gave the corresponding triphosphate 148 in 50% yield (Scheme 23).36
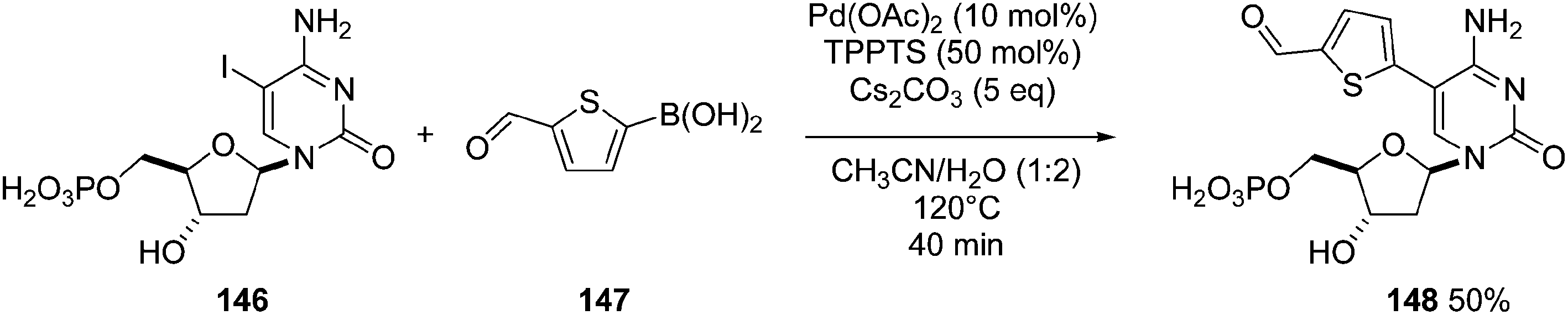 | ||
| Scheme 23 Synthesis of 5-aryl cytosine monophosphate analogue 148. | ||
Recently, Hocek and co-workers targeted nucleoside derivatives functionalized with alkylsulfanylphenyl groups at the 5-position of the pyrimidine.37 Their project started with a model study of cross-couplings of different halogenated nucleosides with alkylsulfanylphenylboronic acid. Because the free-sulfanyl phenyl boronic acid was presumed to be potentially unreactive due to Pd-catalyst poisoning, they selected stable methylsulfanyl derivatives, as well as more labile benzyl- and tritylsulfanyl derivatives with potentially cleavable protecting groups at the sulfur atom. The iodinated nucleosides 13, 140 and 146 were selected, and the previously described conditions [Pd(OAc)2 (10 mol%), TPPTS (50 mol%), Cs2CO3 (5 eq) in CH3CN–H2O (1:2)] were applied. All the reactions were performed at 100 °C for 30 minutes. The final nucleoside analogue products 150–154 were obtained in 78–82% yields (Table 19, entries 2–6). The corresponding nucleotide analogues 155–158 were prepared in lower yields, with the worst one possessing the trityl protecting group (Table 19, entries 7 and 10). As expected by the authors, reactions involving free-sulfanyl phenyl boronic acid gave no positive results, which supported the anticipated Pd-catalyst poisoning (Table 19, entry 1). In all cases, the desired triphosphates were isolated by semi-preparative HPLC and were accompanied by substantial amounts (15–25%) of the corresponding diphosphates resulting from hydrolysis.37
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | Starting material | R2 | Product | Yield (%) |
| 1 | H | 13 | 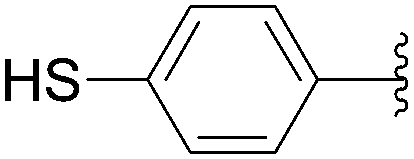 |
149 | 0 |
| 2 | H | 13 | 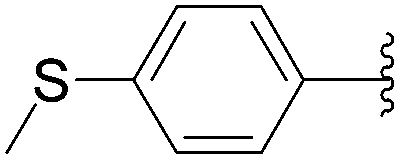 |
150 | 82 |
| 3 | H | 13 | 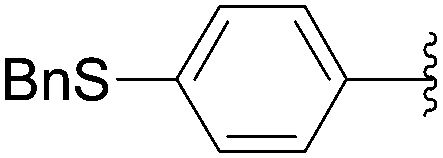 |
151 | 79 |
| 4 | H | 13 | 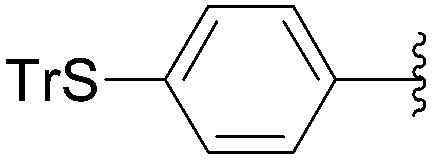 |
152 | 78 |
| 5 | PO3H2 | 146 | 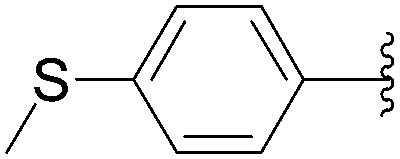 |
153 | 78 |
| 6 | PO3H2 | 146 | 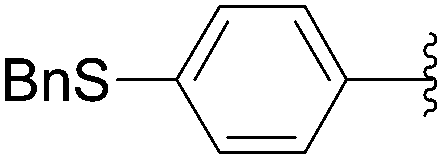 |
154 | 96 |
| 7 | PO3H2 | 146 | 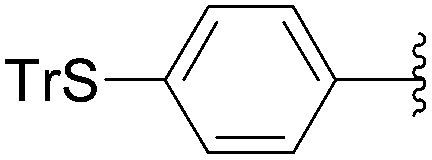 |
155 | 14 |
| 8 | P3O9H4 | 140 | 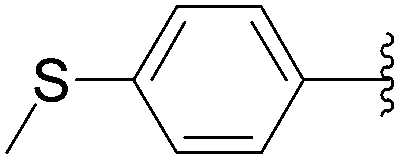 |
156 | 50 |
| 9 | P3OH4 | 140 | 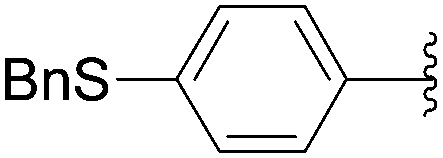 |
157 | 34 |
| 10 | P3O9H4 | 140 | 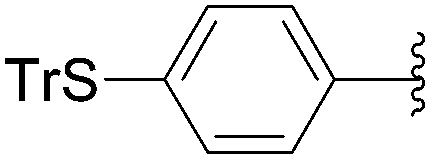 |
158 | 10 |
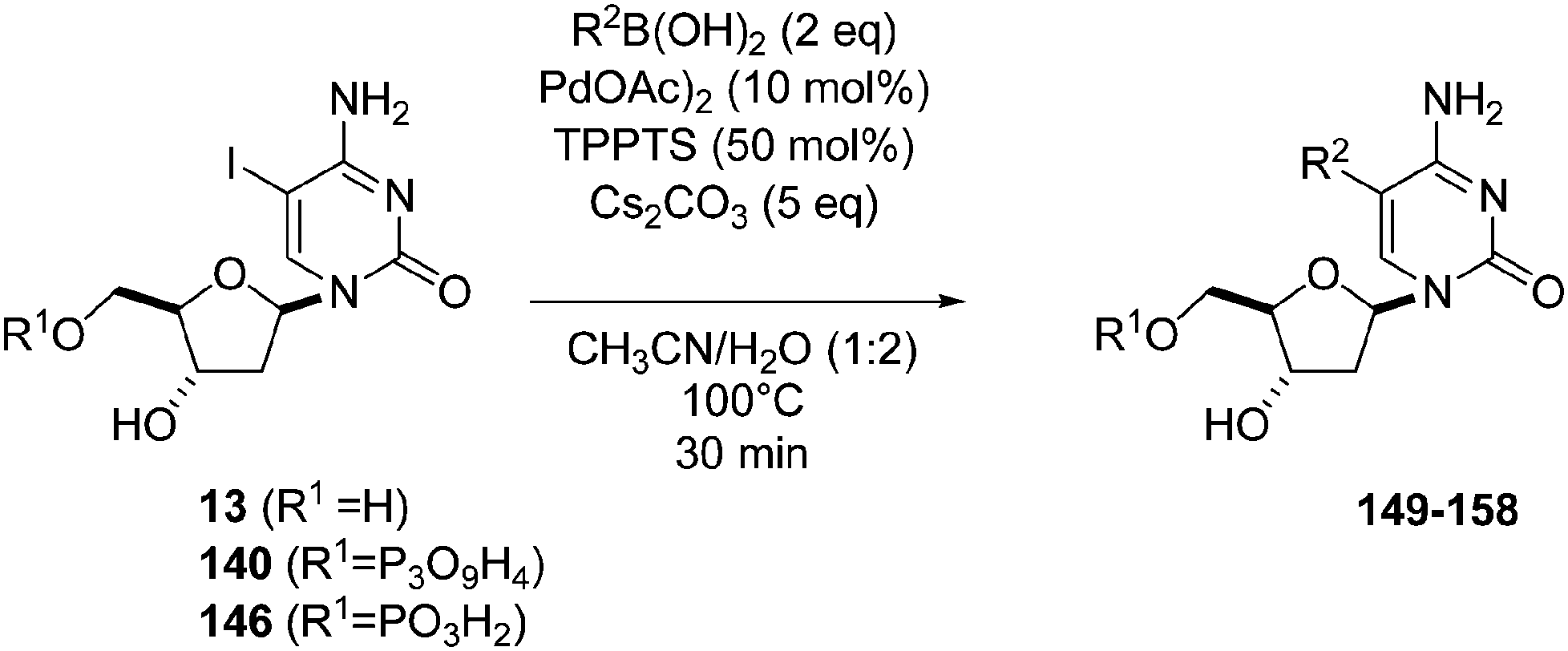
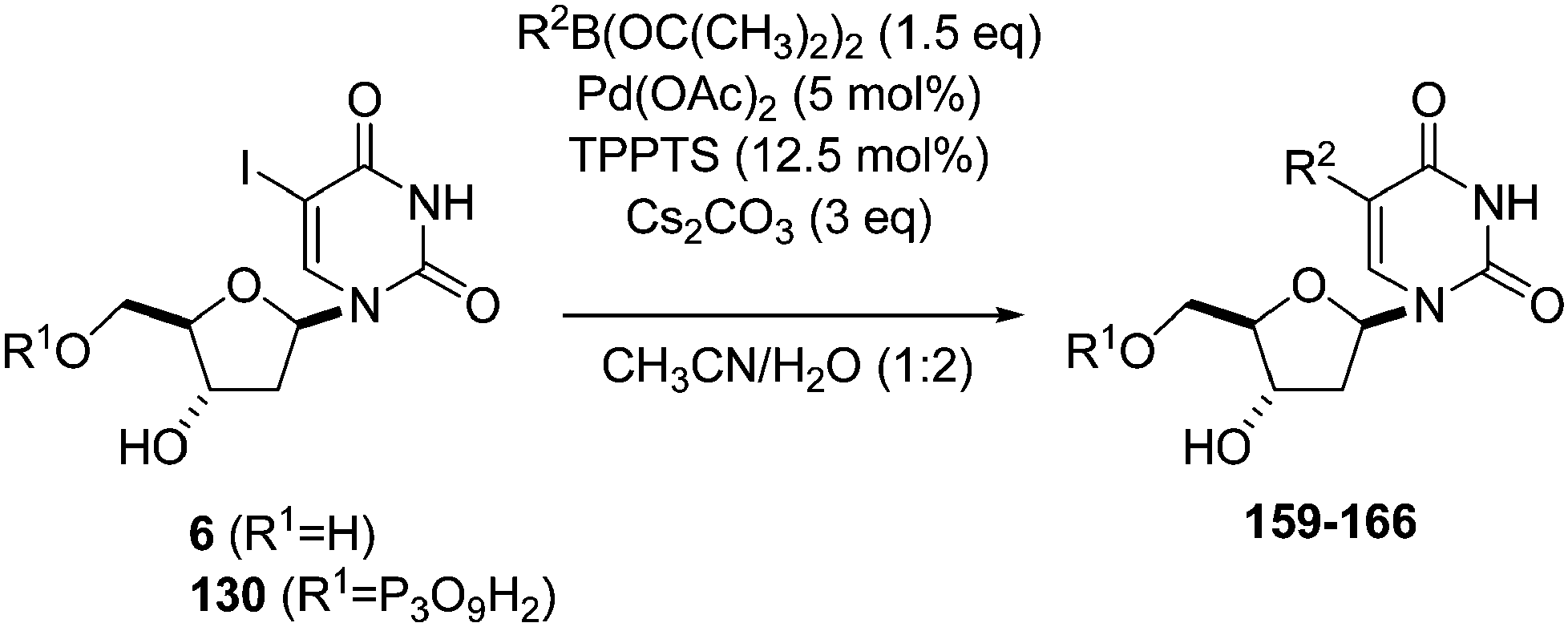
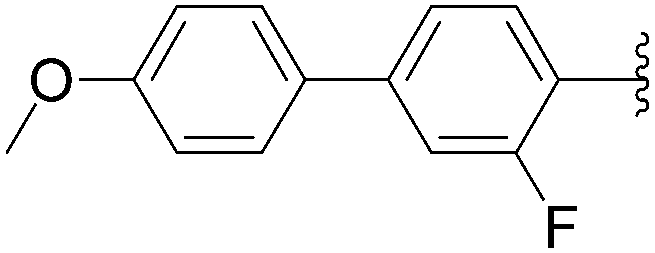
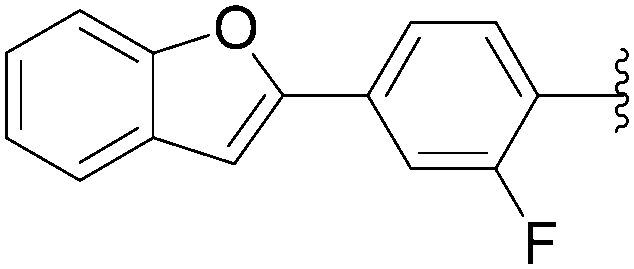
In 2012, Hocek's group reported four new fluorinated biaryl fluorescent labels and their attachment to nucleosides and nucleosides triphosphate by aqueous cross-coupling reactions of biarylboronates.38 For this purpose, four different types of biaryl groups were designed in which the following were attached directly at the 5-position of pyrimidines: 4-methoxybiphenyl (BIF); 2-phenylbenzofuryl (BFU); 2-phenylbenzoxazole (BOX); and 2-phenyl-5-aminobenzoxazole (ABOX). The frequently described cross-coupling protocol involving pinacol ester boronate acid (1.5 eq), Pd(OAc)2 (5 mol%), TPPTS (12.5 mol%), Cs2CO3 (3 eq) and CH3CN–H2O (1:2) was used for the two reactions conducted at 80 °C for 2 hours starting from iodinated nucleoside 6 and 90 °C for 45 minutes starting from iodinated nucleotides triphosphates 130 (Table 20). The desired biaryl-substituted nucleosides 159–162 were obtained in good-to-moderate yields (59–71%). Analogously, the halogenated nucleotide triphosphate 130 reacted with the four boronates to give directly the biaryl compounds 163–166 in somewhat lower yields (17–25%), due to the hydrolysis of the triphosphates during the coupling reaction.
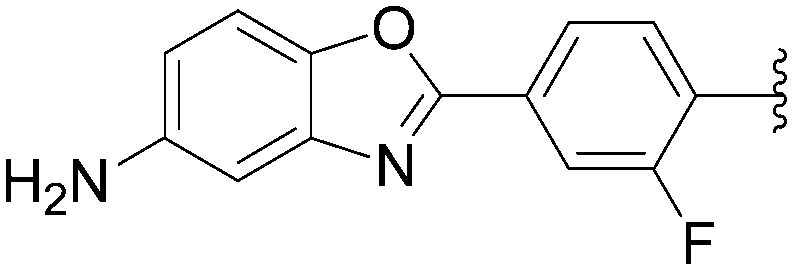
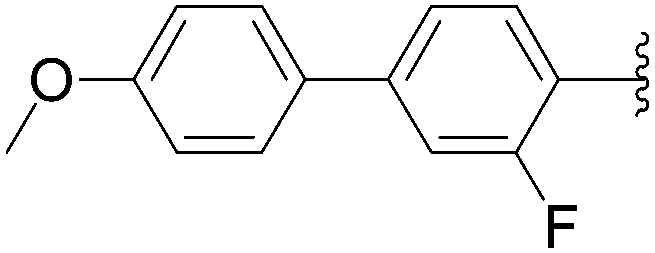
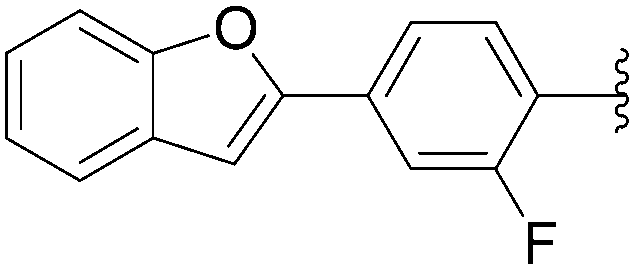
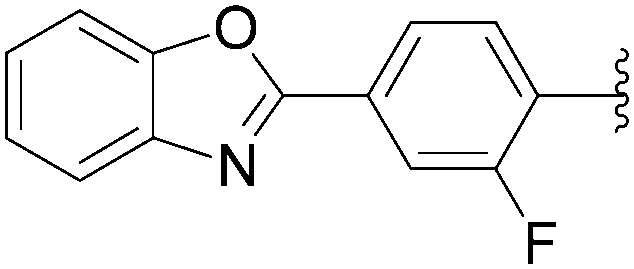
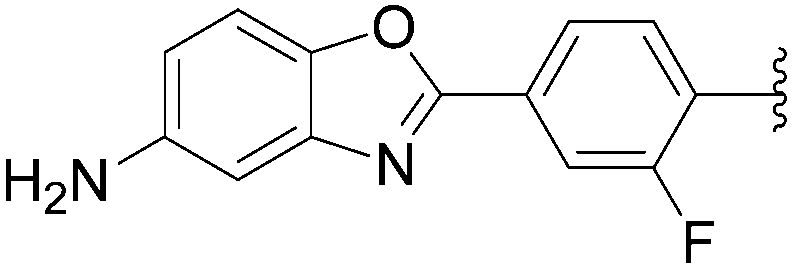
In 2009, the same laboratory reported the synthesis of 2′-deoxyuridine and 2′-deoxycytidine nucleoside analogues 167–172 bearing bipyridine (bpy) or terpyridine (tpy) ligands linked via phenylene linkers.39 For the first time, the Suzuki–Miyaura couplings were achieved using 4-(pinacolboronato)phenyl derivatives (1.2 eq) in the presence of Cs2CO3 (3eq), Pd(OAc)2 (5 mol%) and TPPTS (12.5 mol%) in a mixture of CH3CN–H2O (1:2) at 80 °C (Table 21, entries 1–3 and 7–9). Under these conditions, the yields of the target compounds 167 and 170 were acceptable at 60% and 65%, respectively, but using the other boronate esters the coupling adducts were not obtained in satisfactory yields. In a second phase of the study, with variations in the source of palladium, the nature of ligand revealed that the best results were obtained when Pd(OAc)2 was present at 10 mol% in association with TPPTS (50 mol%) in a mixture of CH3CN–H2O (2:1) at 90 °C. All the target nucleoside analogues 167–172 were obtained in better yields (35–75% vs. 7–65%) (Table 21, entries 4–6 and 10–12). In the work of Sekine and co-workers published in 2007,25 water was not the main solvent.
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | Starting material | R2 | Product | Yield (%) |
| a Pd(OAc)2 (5 mol%), TPPTS (12.5 mol%), CH3CN–H2O (1:2), 80 °C.b Pd(OAc)2 (10 mol%), TPPTS (50 mol%), CH3CN–H2O (2:1), 90 °C. |
|||||
| 1 | OH | 6 | 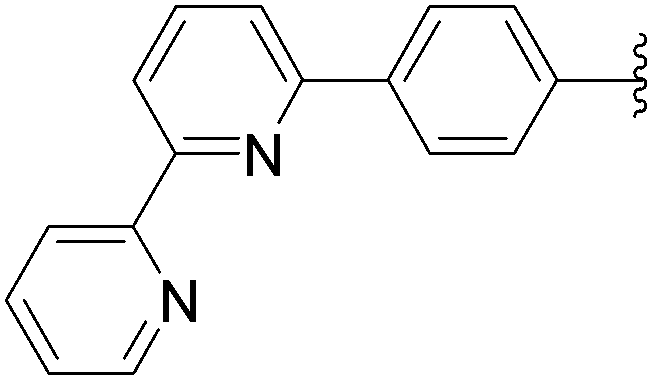 |
167 | 60a |
| 2 | OH | 6 | 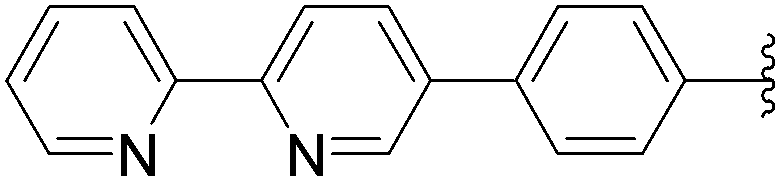 |
168 | 7a |
| 3 | OH | 6 | 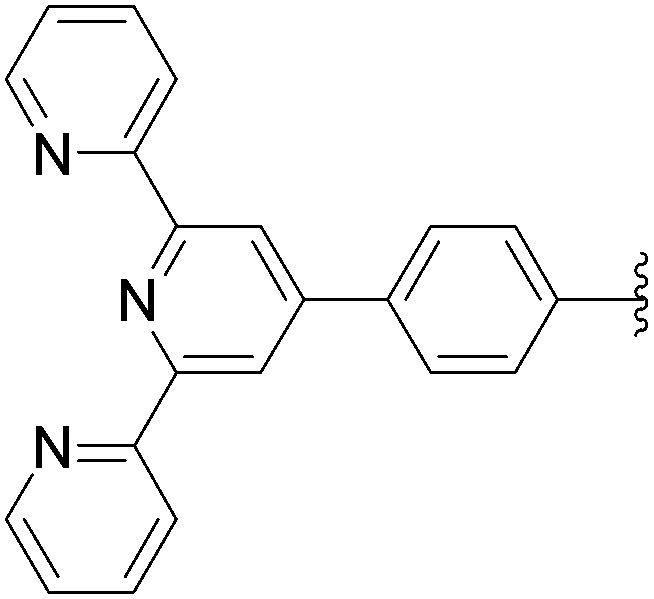 |
169 | 24a |
| 4 | OH | 6 | 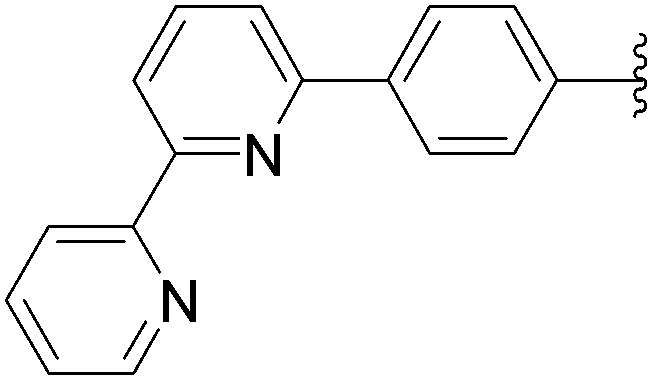 |
167 | 75b |
| 5 | OH | 6 | 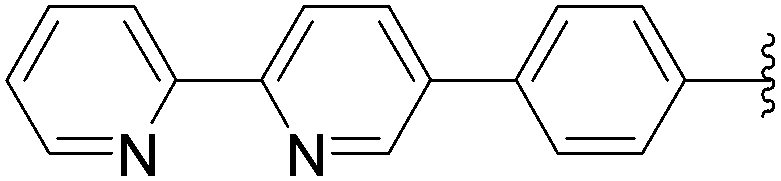 |
168 | 35b |
| 6 | OH | 6 | 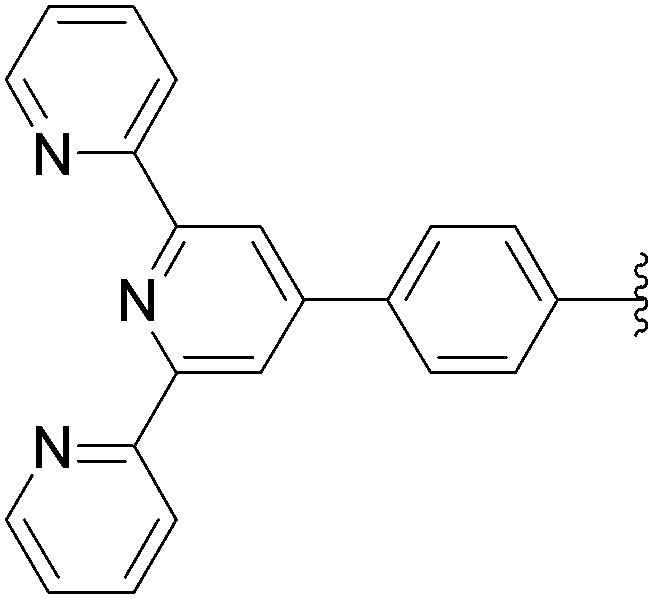 |
169 | 70b |
| 7 | NH2 | 13 | 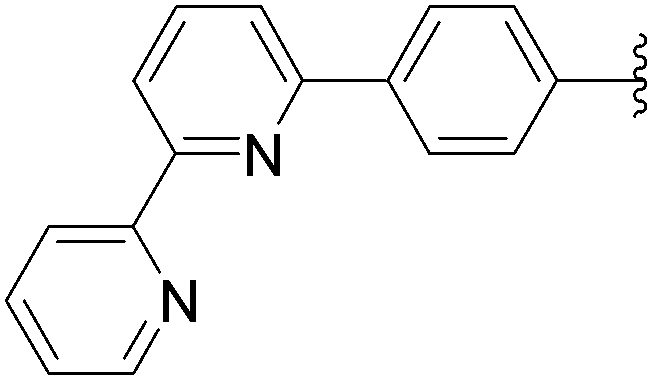 |
170 | 65a |
| 8 | NH2 | 13 | 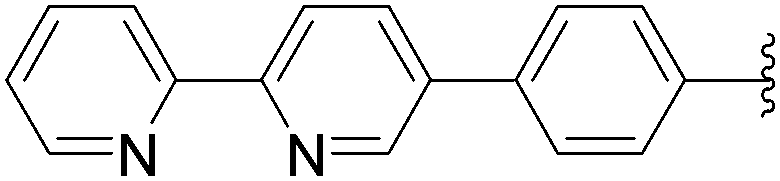 |
171 | 12a |
| 9 | NH2 | 13 | 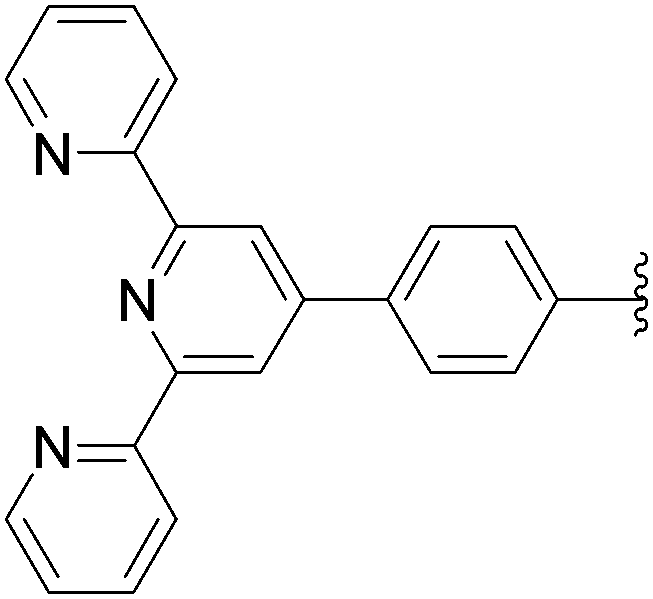 |
172 | 28a |
| 10 | NH2 | 13 | 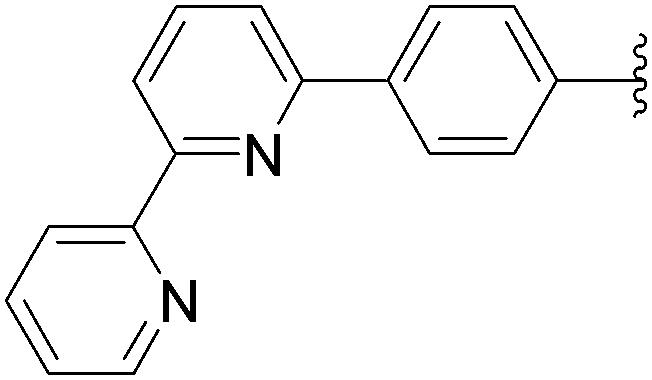 |
170 | 75b |
| 11 | NH2 | 13 | 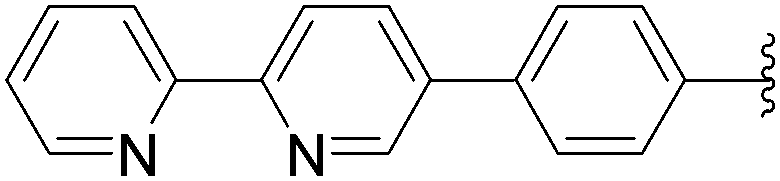 |
171 | 55b |
| 12 | NH2 | 13 | 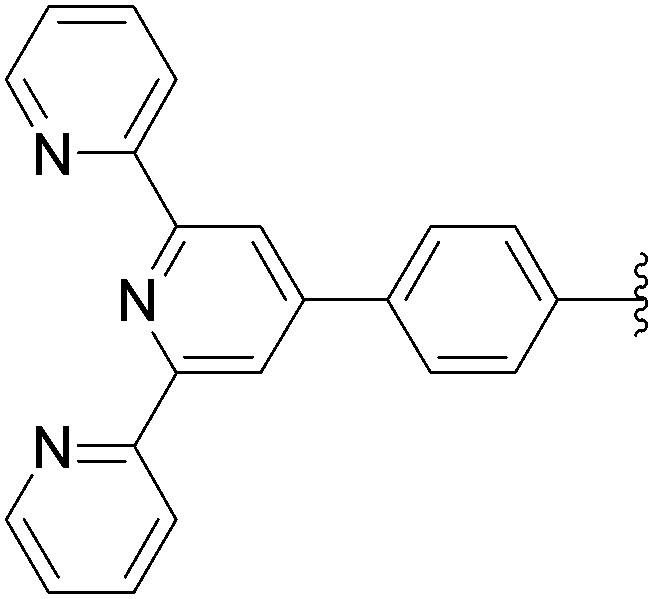 |
172 | 70b |
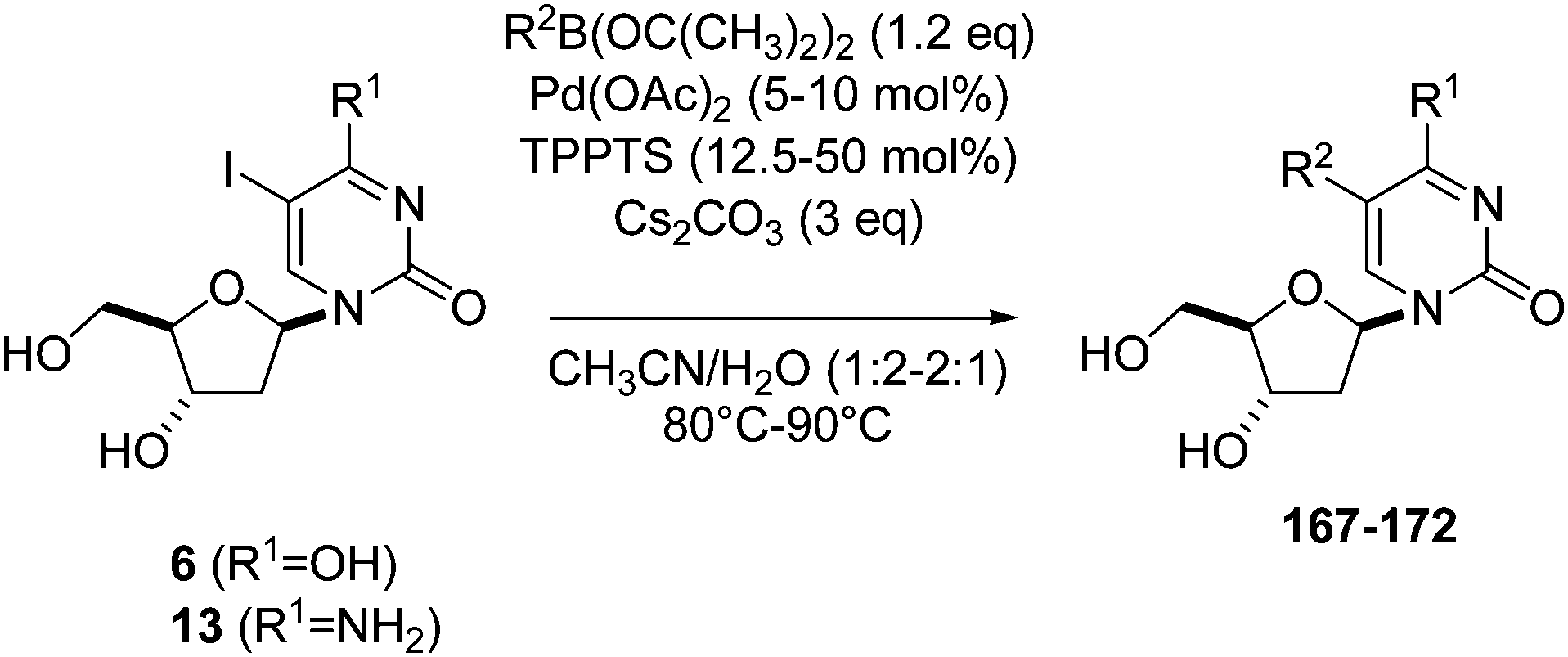
In 2013, Hocek and co-workers used their previously described methodology33 in order to obtain four benzofurazane labeled derivatives of 5-iodo-2′-deoxycytidine (13) and 7-deaza-7-iodo-2′-deoxyadenosine (132) and their respective triphosphate forms 173 and 174.40 The Suzuki–Miyaura coupling was done in the presence of benzo[c][1,2,5]oxadiazole-5-boronic acid (175) (1.2 eq) and a large excess of catalyst and base at 75 °C for 45 minutes to 1 hour. The resulting compounds 176–179 were isolated in good-to-moderate yields (Scheme 24).
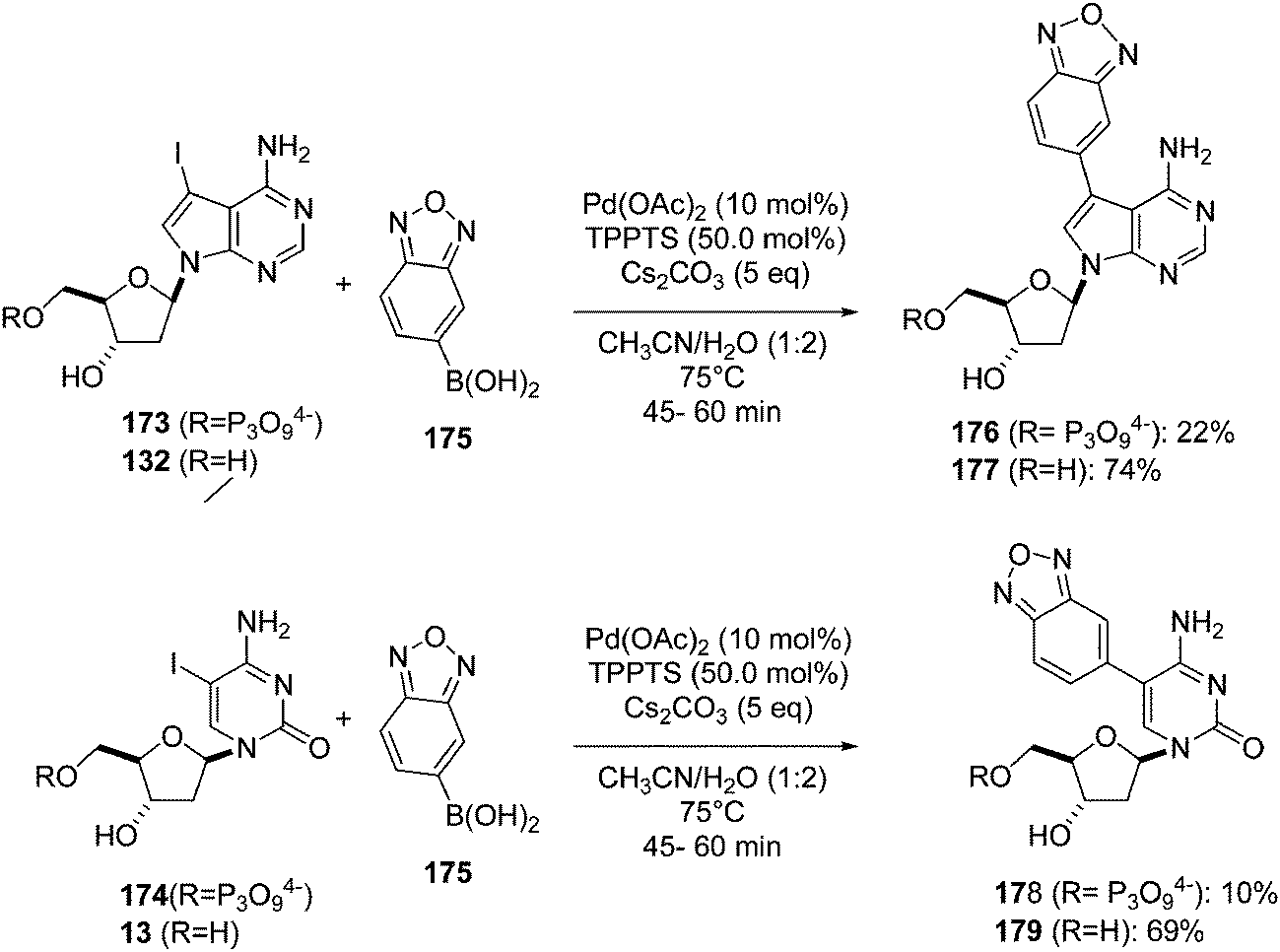 | ||
| Scheme 24 Synthesis of benzofurazane nucleosides 175–178. | ||
6. Suzuki–Miyaura in CH3OH–H2O
The Suzuki–Miyaura cross-coupling reaction in a mixture of CH3OH–H2O was studied only using Pd(0).6.1. Pd(PPh3)4 as Pd(0) and NaOH
In 2004, Saito and co-workers conducted a Suzuki–Miyaura cross coupling of 5-iodo-2′-deoxyuridine (6) with 4-formylboronic acid (39) in MeOH–H2O (5:1) as solvent mixture.41 If it were that the authors used Pd(PPh3)4 and NaOH as palladium source and base, respectively, they gave no indication about the amounts employed. However, it was claimed that the desired cross-coupling product 40 was obtained in 28% yield after refluxing for 24 hours (Scheme 25). The target compound 40 was also obtained by Wagenknecht in 2008 in 76% yield.19 Due to the lack of information in the work of Saito, it is difficult to compare the results.
 | ||
| Scheme 25 Synthesis of 5-(4-formylphenyl)uridine analogue 40. | ||
7. Suzuki–Miyaura in H2O
Surprisingly, only a few publications report the palladium-catalyzed cross-coupling syntheses of modified nucleosides using Pd(0) or Pd(II) and water as the sole solvent. In the case of Pd(II), recent reports describe a ligand-free Suzuki–Miyaura reaction for the first time in nucleoside chemistry.7.1. Pd(PPh3)4 as Pd(0) and Na2CO3
The first reported use of water as the sole solvent for a Suzuki cross-coupling reaction was in 2003 by Williams and co-workers. They studied this reaction involving 5-iodouridine (75), boronic acid 180, Pd(PPh3)4 (3 mol%) and reverse-phase glass beads (Scheme 26).42 The authors described the reaction as fast with a full conversion of the starting material 75 being completed within 4 hours. The cross-coupling product 181 and self-coupling product were obtained in a ratio of roughly 7:3. Further purification of the target carboxy derivative 181 was not mentioned in the report. Relatively low levels of palladium loading were used, and the authors described briefly an investigation into the recycling of the reverse-phase glass beads but not for the nucleoside case. It is noteworthy that palladium leaching of 0.40% was detected.
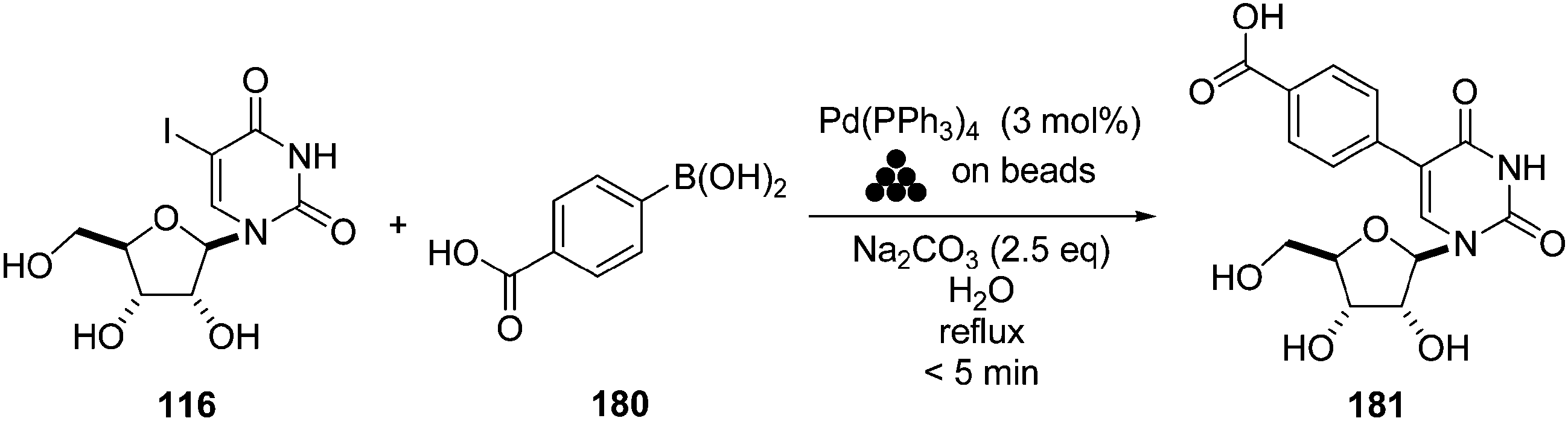 | ||
| Scheme 26 Synthesis of 5-(4-formylphenyl)uridine analogue 181. | ||
7.2. Pd(PPh3)4 as Pd(0) and K2CO3
Wagner and co-workers reported the Suzuki–Miyaura coupling of unprotected compounds 25 and 32 carried out with the traditional catalyst Pd(PPh3)4 in water.16 Starting from adenosine derivative 25, addition of phenyl boronic acid (33) (1.5 eq), Pd(PPh3)4 (10 mol%) and a large excess of K2CO3 (6 eq) in neat water at 80 °C for one day furnished the target derivative 26 in 75% yield (Table 22, entry 1). Application of the methodology to the 8-bromoguanosine (32) did not give a cross-coupling product (Table 22, entry 2). However, the use of 4-hydroxyphenylboronic acid gave the adduct 182 albeit in low yield (Table 22, entry 3).
|
||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | Starting material | R3 | Product | Yield (%) |
| 1 | NH2 | H | 25 |  |
26 | 75 |
| 2 | OH | NH2 | 32 |  |
34 | 0 |
| 3 | NH2 | H | 25 |  |
182 | 26 |
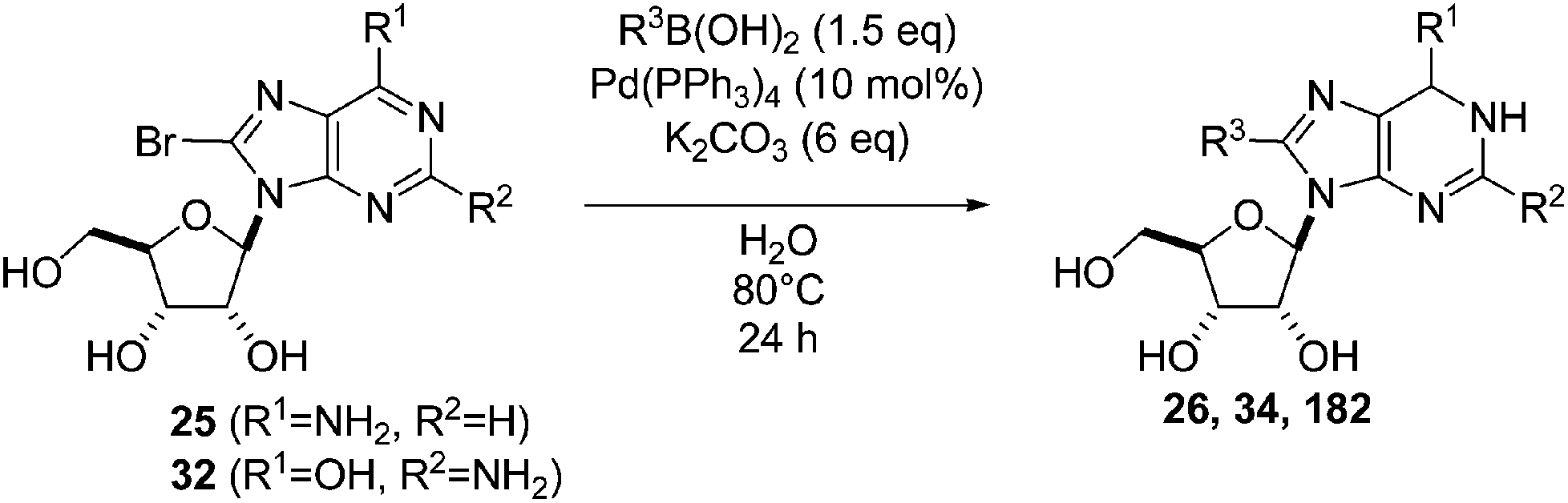
7.3. Pd(OAc)2 as Pd(II), TPPTS and K2CO3
In the same paper, Wagner and co-workers described the use of Pd(OAc)2 with TPPTS as catalytic system (Table 23).16 At 80 °C, a mixture of 8-bromo nucleoside derivative 25 and boronic acid 33 (1.5 eq) in the presence of Pd(OAc)2 (2.5 mol%), TPPTS (6.25 mol%) and K2CO3 (2 eq) in water furnished the phenyl analogue 26 in 75% yield (Table 23, entry 1) as described by the authors with the Pd(PPh3)4 (Table 22, entry 1). In contrast to the former method, the guanosine derivative 34 was prepared in 61% yield (Table 23, entry 2). By varying the boronic acid, the aryl nucleoside analogues 26, 28, 34, 44 and 183–187 were obtained successfully (Table 23). It is noteworthy that the cross-coupling was not achieved using 4-hydroxyphenylboronic acid (Table 23, entry 9). The authors noted that most of the 8-arylguanosine derivatives prepared by this method were obtained as black powders that required further purification by either recrystallization or column chromatography.
|
||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | Starting material | R3 | Product | Yield (%) |
| 1 | NH2 | H | 25 |  |
26 | 75 |
| 2 | OH | NH2 | 32 |  |
34 | 61 |
| 3 | NH2 | H | 25 |  |
183 | 70 |
| 4 | OH | NH2 | 32 |  |
184 | 83 |
| 5 | NH2 | H | 25 | 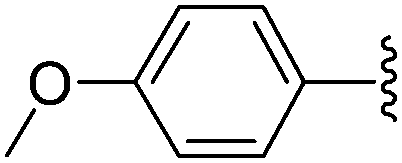 |
28 | 79 |
| 6 | OH | NH2 | 32 | 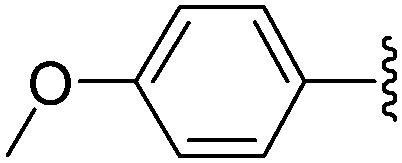 |
44 | 87 |
| 7 | NH2 | H | 25 | 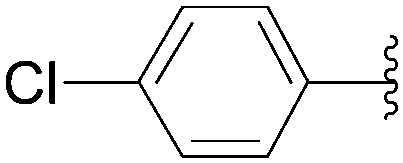 |
185 | 96 |
| 8 | OH | NH2 | 32 | 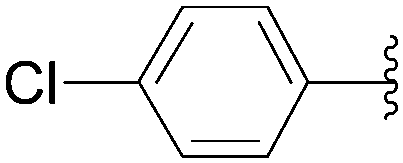 |
186 | 84 |
| 9 | NH2 | H | 25 |  |
187 | 0 |
7.4. Pd(OAc)2 as Pd(II), TPPTS and Na2CO3
Recently, Len's group described the use of the same catalytic system described by Wagner's team16 except for the choice of base.43 Reaction of the uridine analogue 6, with boronic acid 33 (1.3 eq) in the presence of a much smaller amount of palladium Pd(OAc)2 (1.0 mol%), TPPTS (2.5 mol%) and Na2CO3 in water at 80 °C for 4 hours furnished the corresponding adduct 55 in 75% yield (Scheme 27).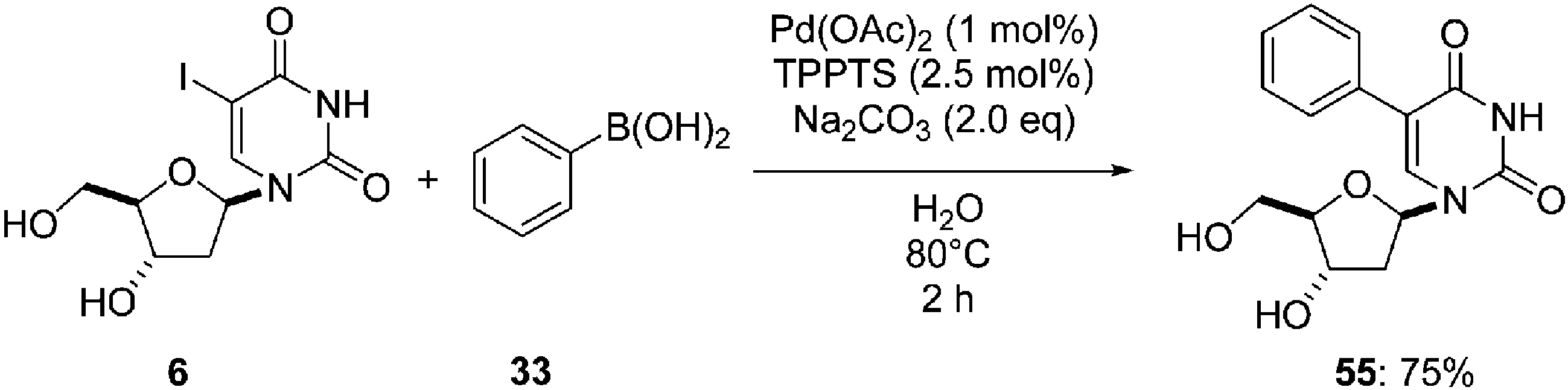 | ||
| Scheme 27 Synthesis of 5-phenyl uridine analogue 55. | ||
7.5. Pd(OAc)2 as Pd(II), PPh3 and Na2CO3
Agrofoglio and co-workers investigated the development of a Suzuki–Miyaura reaction involving 2′-deoxyuridine in a completely aqueous medium using a readily available and inexpensive catalyst–ligand system.44 The conditions of this model reaction were optimized using the unprotected 5-iodo-2′-deoxyuridine (6) and 4-methoxyphenylboronic acid (188). After varying the nature of the ligand (e.g. PPh3, TXTPS, CataXcium F Sulf or tris[bis(N-2-hydroxyethyl)aminomethyl]phosphine) and the amount of either the ligand or the solvent, the best method found was the coupling of compound 6 with the boronic acid (1.5 eq) in the presence of Na2CO3 (1.5 eq), Pd(OAc)2 (3 mol%) and PPh3 (5.4 mol%) in neat water at 80 °C for 4 hours. The authors decided then to use, under such optimised conditions, microwave irradiation assistance at 120 °C, which permitted a significant reduction in the reaction time with the same yield (4 hours vs. 10 minutes) and the furnishing of the desired adduct 56 in 75% yield (Scheme 28).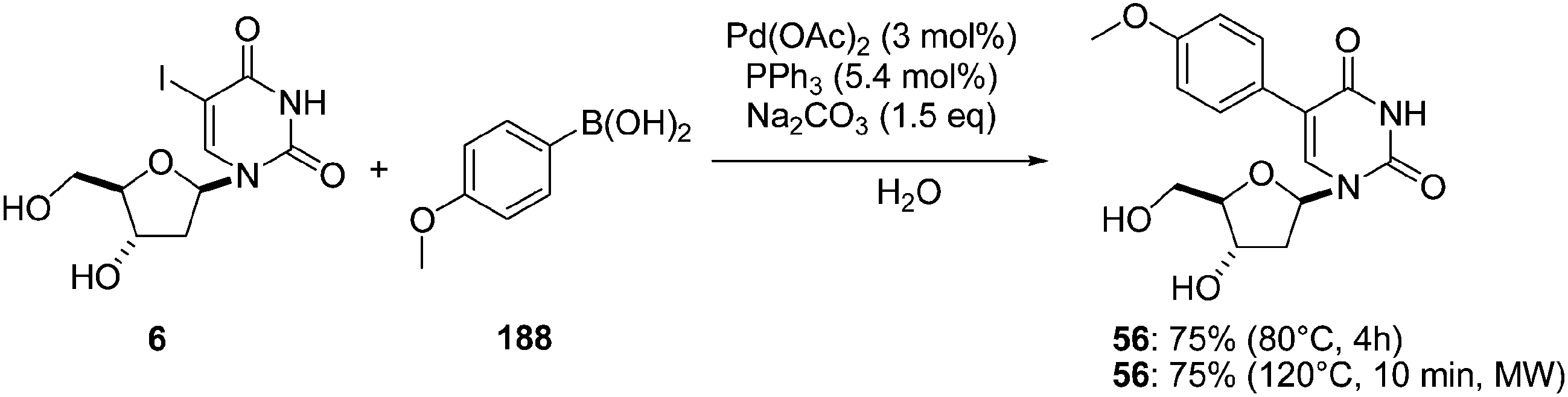 | ||
| Scheme 28 Synthesis of 5-(4-methoxyphenyl)uridine analogue 56. | ||
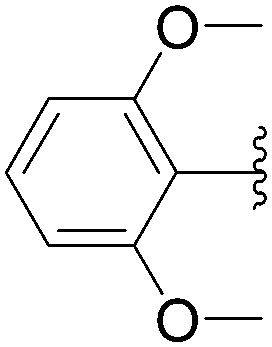
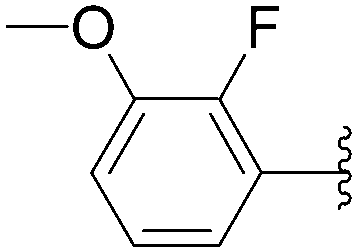
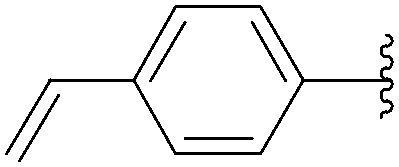
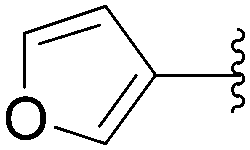
Application of the optimized method to different aryl boronic acids afforded the cross-coupled products 35, 40, 55 and 189–197 in good yield with substrates that contained either electron-donating or -withdrawing groups in the para and meta positions of the aromatic core. Thus, 5-iodo-2′-deoxyuridine (6) was coupled with a variety of heteroarylboronic acids including thiophene-3-, furan-2-, and furan-3-boronic acids (Table 24, entries 11–13).
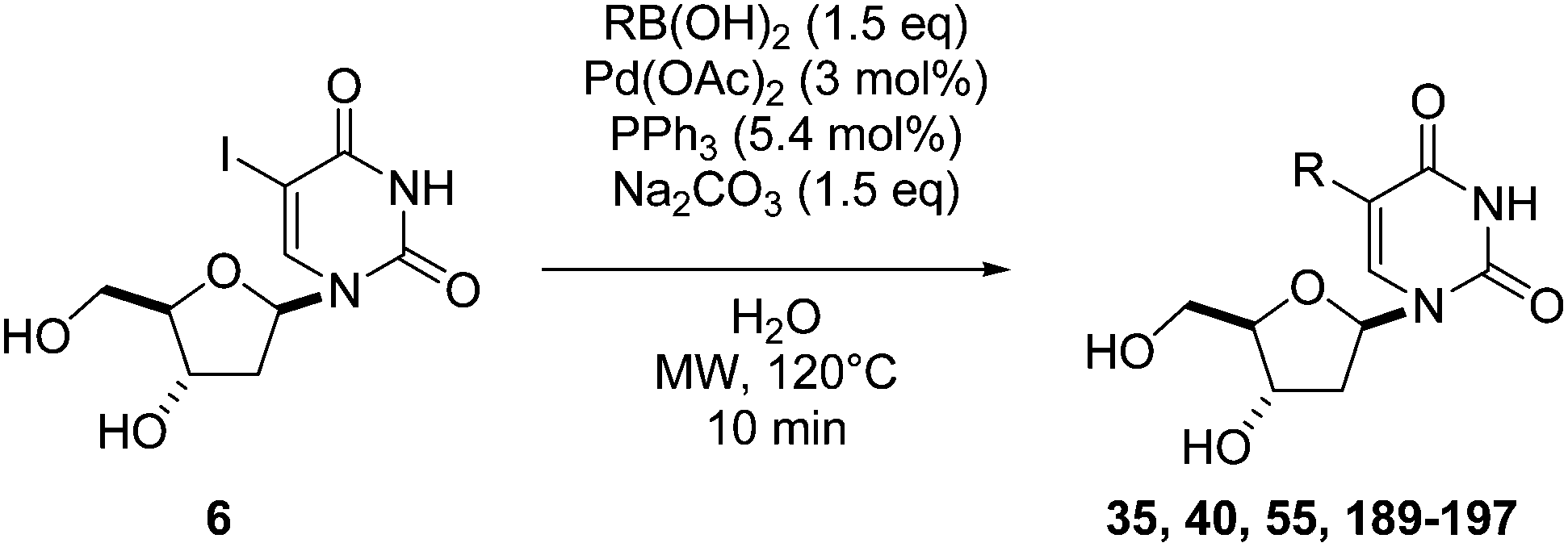
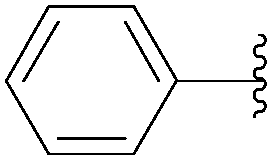
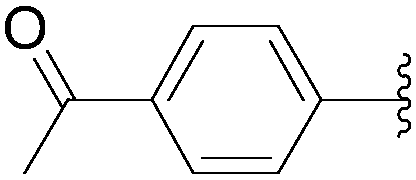
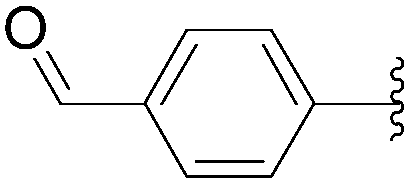
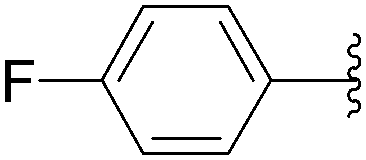
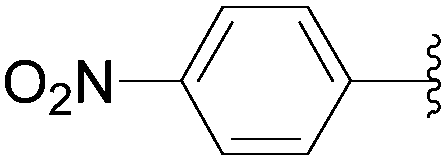
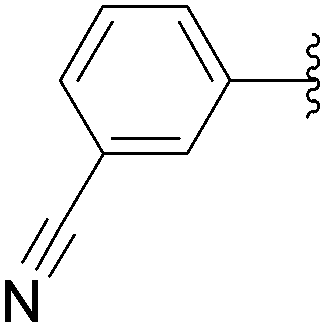
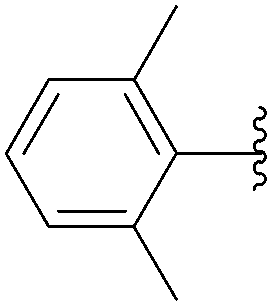
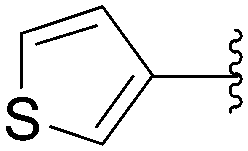
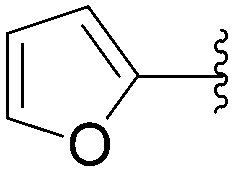
7.6. Na2PdCl4 as Pd(II) and K2CO3
Wagner and co-workers reported a challenging Suzuki–Miyaura coupling of unprotected 8-bromo-5′-mono- and triphosphates guanosine.45 In the same paper, the authors initially chose a model reaction starting from 8-bromoguanosine (32) to identify the optimum conditions in neat water.45 Several catalytic systems were tested including Pd(OAc)2, Pd(NO3)2, Na2PdCl4 and PdCl2 as the source of palladium and TPPTS along with the Buchwald ligand and EDTA as the source of ligand. Out of the catalytic systems screened, only those with a phosphine ligand were found to be active. The role of Pd source was highlighted by the finding that replacement of Pd(OAc)2 with a water-soluble Pd source resulted in a cleaner product. The best results, with regard to yield and reaction times, were achieved with catalytic systems combining either Pd(NO3)2 or Na2PdCl4 with the water-soluble phosphine ligand TPPTS. Because of the lower cost and higher stability of Na2PdCl4 [compared to Pd(NO3)2], the catalytic system Na2PdCl4 (2.5 mol%) and TPPTS (6.25 mol%) was chosen for the reaction in the presence of K2CO3 (2 eq) in neat water at 80 °C (Table 25). Using this method, the nucleoside analogues 34, 44, 184 and 186 were obtained in yields higher than 70%. It is noteworthy that Shaughnessy and co-workers obtained similar results (determination by HPLC analysis) starting from 8-bromo-2′-deoxyadenosine (15) to furnish the corresponding 8-phenyl derivative 51 using different sources of palladium and TPPTS or TXPTS as ligand.20
|
||||
|---|---|---|---|---|
| Entry | R | Product | Time (min) | Yield (%) |
| 1 |  |
34 | 20 | 77 |
| 2 |  |
186 | 30 | 84 |
| 3 | 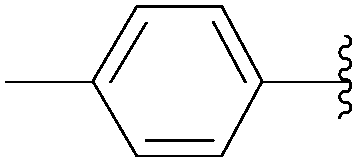 |
184 | 20 | 70 |
| 4 | 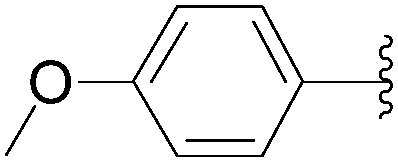 |
44 | 30 | 84 |
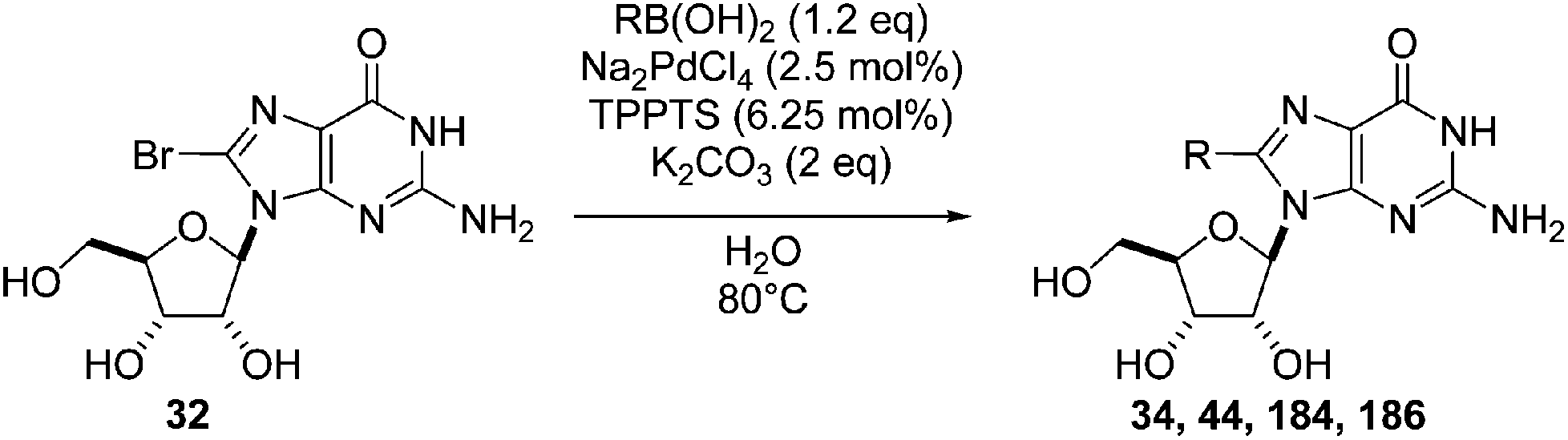
The authors next applied their optimized cross-coupling conditions with a lower excess of base (K2CO3 1.5 eq) to the reaction of the mono- and triphosphate guanosine derivatives 198 and 199.45 All of the 8-aryl monophosphate derivatives 200–203 were obtained in good-to-excellent yields, and most reactions were completed within 2 hours at 80 °C (Table 26, entries 1–4). The cross-coupling reactions of the nucleotides triphosphates 204–206 proceeded smoothly, in short reaction times and with good yields (Table 26, entries 5–7).
|
||||||
|---|---|---|---|---|---|---|
| Entry | R1 | Starting material | R2 | Product | Time (min) | Yield (%) |
| 1 | PO3H2 | 198 | 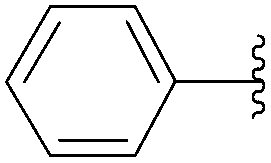 |
200 | 15 | 63 |
| 2 | PO3H2 | 198 | 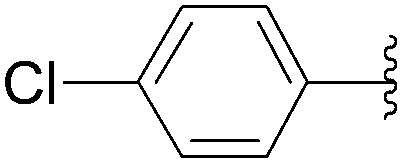 |
201 | 80 | 92 |
| 3 | PO3H2 | 198 | 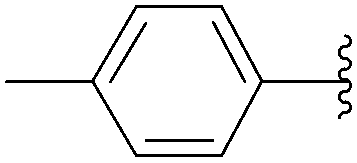 |
202 | 60 | 90 |
| 4 | PO3H2 | 198 | 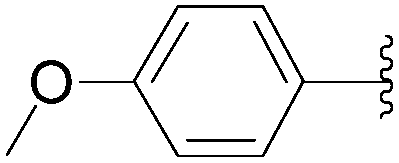 |
203 | 360 | 71 |
| 5 | P3O9H4 | 199 | 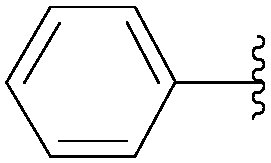 |
204 | 60 | 85 |
| 6 | P3O9H4 | 199 | 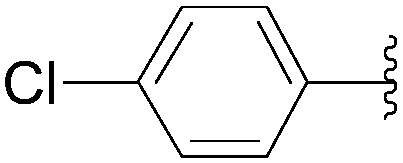 |
205 | 60 | 56 |
| 7 | P3O9H4 | 199 | 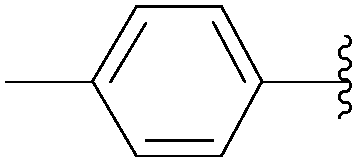 |
206 | 120 | 65 |
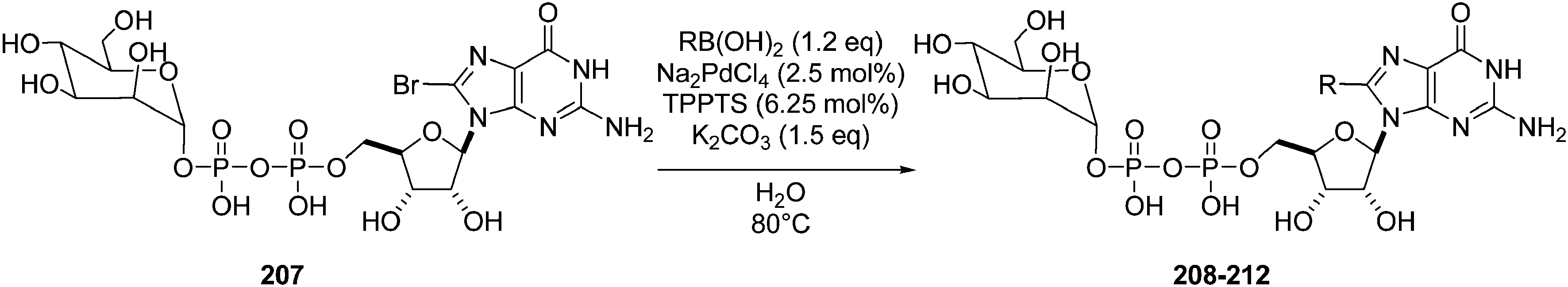
In 2008, the same team extended their methodology to the preparation of 8-aryl-5′-diphosphateguanosine-mannose analogues 208–212 starting from the bromo derivative 207 (Table 27).46 This synthesis is the first example, to date, of the cross coupling of a sugar-nucleotide in aqueous media.
|
|||
|---|---|---|---|
| Entry | R2 | Product | Yield (%) |
| 1 | 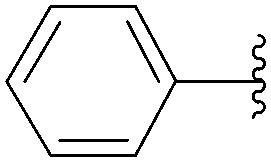 |
208 | 48 |
| 2 | 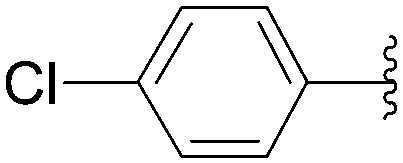 |
209 | 82 |
| 3 | 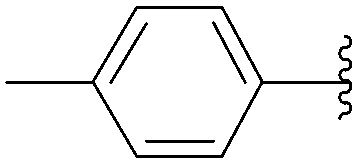 |
210 | 82 |
| 4 | 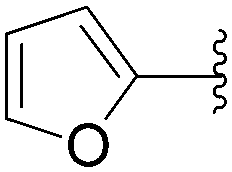 |
211 | 57 |
| 5 | 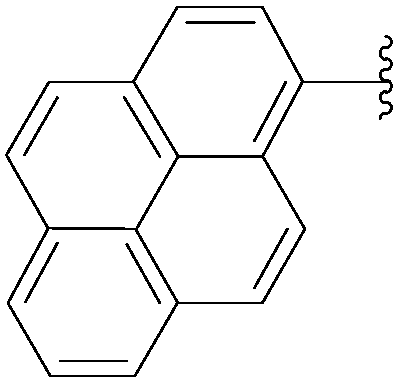 |
212 | 73 |
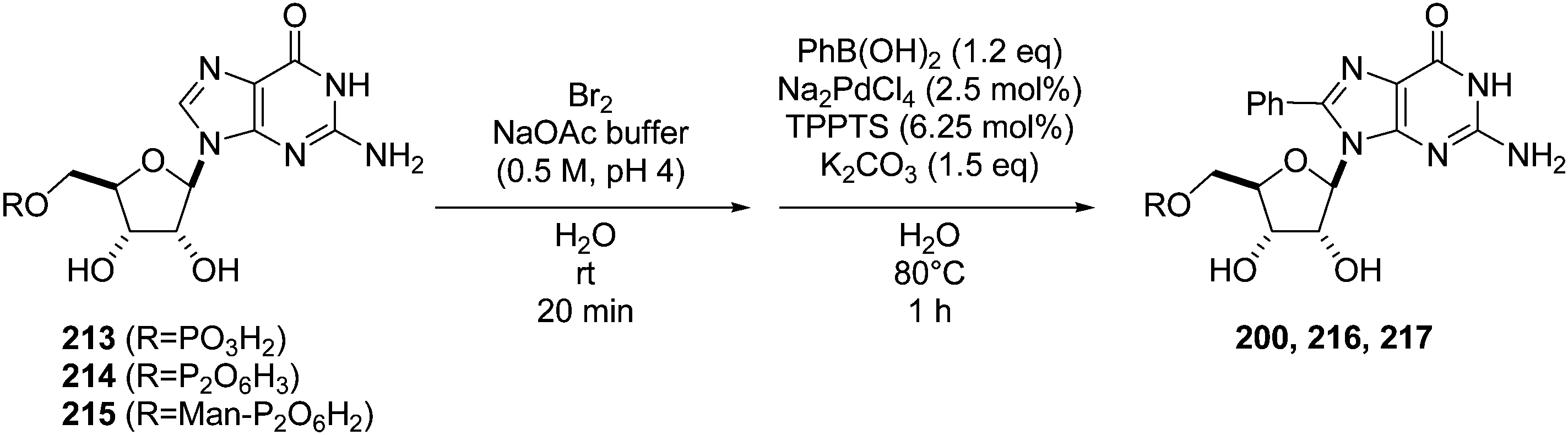
To minimize the number of lengthy purification steps in the sequence, the one-pot, two-step procedure (bromination and cross coupling) starting from the guanosine derivatives 213–215 was studied. The corresponding monophosphate, diphosphate and sugar-nucleotide furnished the target 8-phenyl derivatives 200, 216 and 217 in 75%, 50% and 44% yields (two steps), respectively (Table 28).46
|
||||
|---|---|---|---|---|
| Entry | R | Starting material | Product | Yield (%) |
| 1 | PO3H2 | 213 | 200 | 75 |
| 2 | P2O6H3 | 214 | 216 | 50 |
| 3 | Man-(PO3)2− | 215 | 217 | 44 |
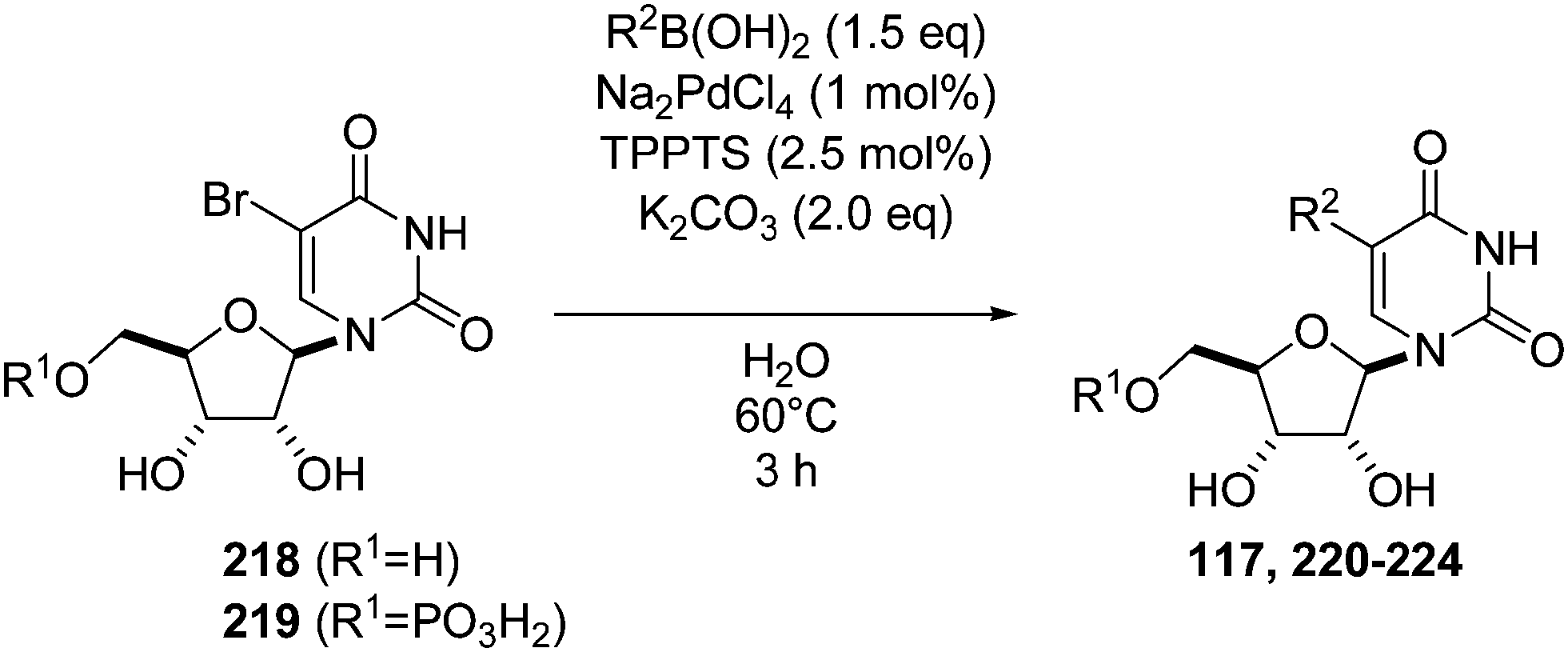
Wagner and co-workers in 2008, reported the first successful Suzuki–Miyaura reaction of an unprotected 5-halo-5′-mono and diphosphate uridine-sugar.47 The authors found an optimized catalytic system for the coupling of 5-bromouridine (218) and 5-bromouridine monophosphate (219) in neat water involved treating Na2PdCl4 (1 mol%), TPPTS (2.5 mol%) and K2CO3 (2 eq) as base at 60 °C for 3 hours. Six adducts 117 and 220–224 were obtained using this methodology (Table 29). It was the first time that a bromo derivative was used as the starting material in the case of pyrimidine analogues.
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | Starting material | R2 | Product | Yield (%) |
| 1 | H | 218 |  |
117 | 45 |
| 2 | H | 218 |  |
220 | 43 |
| 3 | H | 218 | 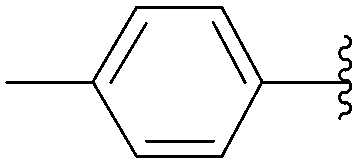 |
221 | 46 |
| 4 | PO3H2 | 219 | 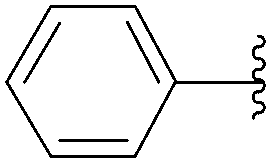 |
222 | 57 |
| 5 | PO3H2 | 219 | 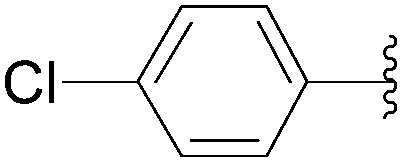 |
223 | 26 |
| 6 | PO3H2 | 219 | 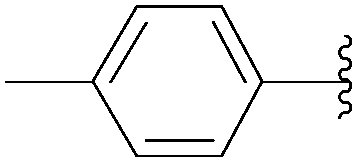 |
224 | 27 |
The authors advised that neither an increase in the amount of base (K2CO3) nor use of a stronger base (NaOH) improved cross-coupling efficiency. However, replacement of K2CO3 (2 eq) with Cs2CO3 (2 eq) for 2 hours resulted in a substantially higher yield of the nucleotide 222 (71% vs. 57%) (Scheme 29).
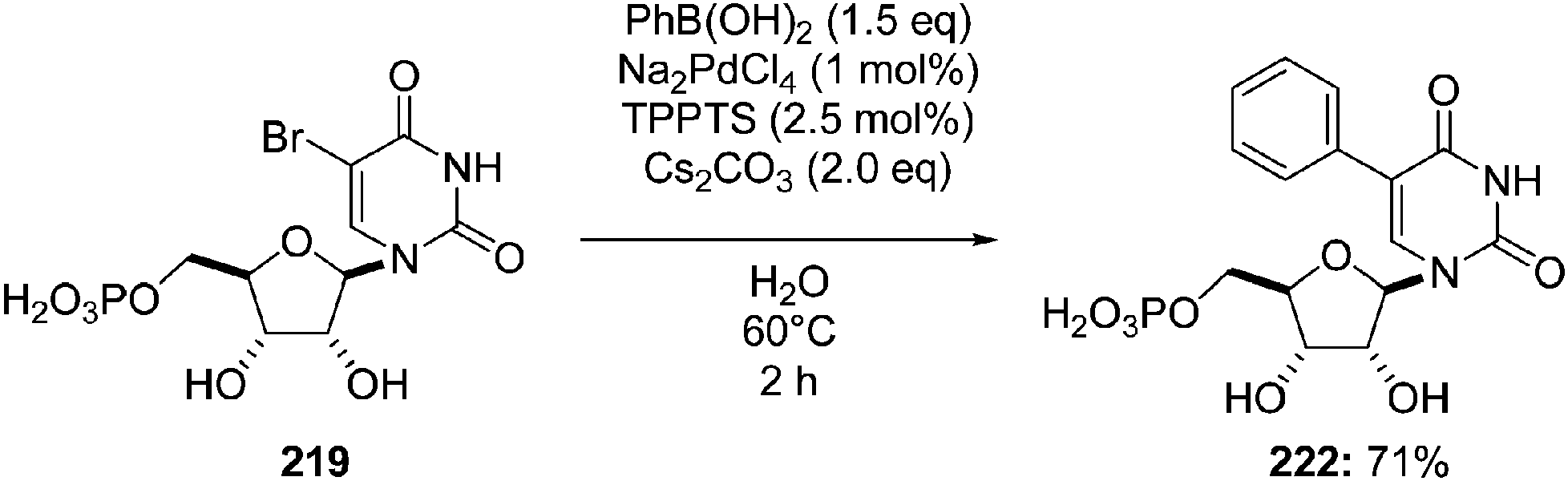 | ||
| Scheme 29 Synthesis of monophosphate 5-(phenyl)uridine analogue 222. | ||
It is of note that the aforementioned optimized Suzuki–Miyaura conditions were unsuccessfully applied to 5-bromouridine diphosphate.47 The authors attempted a similar conversion involving the iodinated derivatives 225 and 226 instead of the brominated analogues, since the iodides are generally more reactive in cross-coupling reactions than the corresponding bromide analogues. In this case, six different adducts 117, 221 and 227–230 were prepared by addition of boronic acid (1.5 eq) in the presence of Cs2CO3 (2.0 eq) as base, Na2PdCl4 (2.4 mol%) and TPPTS (6.21 mol%) in neat water at 50 °C for 1 hour (Table 30). Using this method, compounds 117 and 221 were obtained in better yields than those previously mentioned here (for 117 71% vs. 45%; for 221 66% vs. 46%).
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | Starting material | R2 | Product | Yield (%) |
| 1 | PO3H2 | 225 |  |
117 | 71 |
| 2 | PO3H2 | 225 |  |
221 | 66 |
| 3 | Glu-(P2O6H2) | 226 | 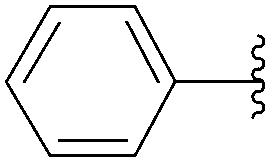 |
227 | 64 |
| 4 | Glu-(P2O6H2) | 226 | 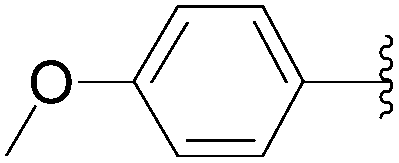 |
228 | 58 |
| 5 | Glu-(P2O6H2) | 226 | 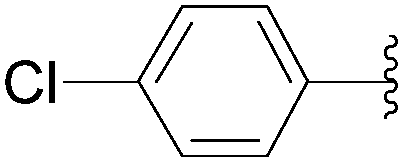 |
229 | 57 |
| 6 | Glu-(P2O6H2) | 226 | 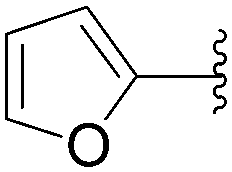 |
230 | 40 |
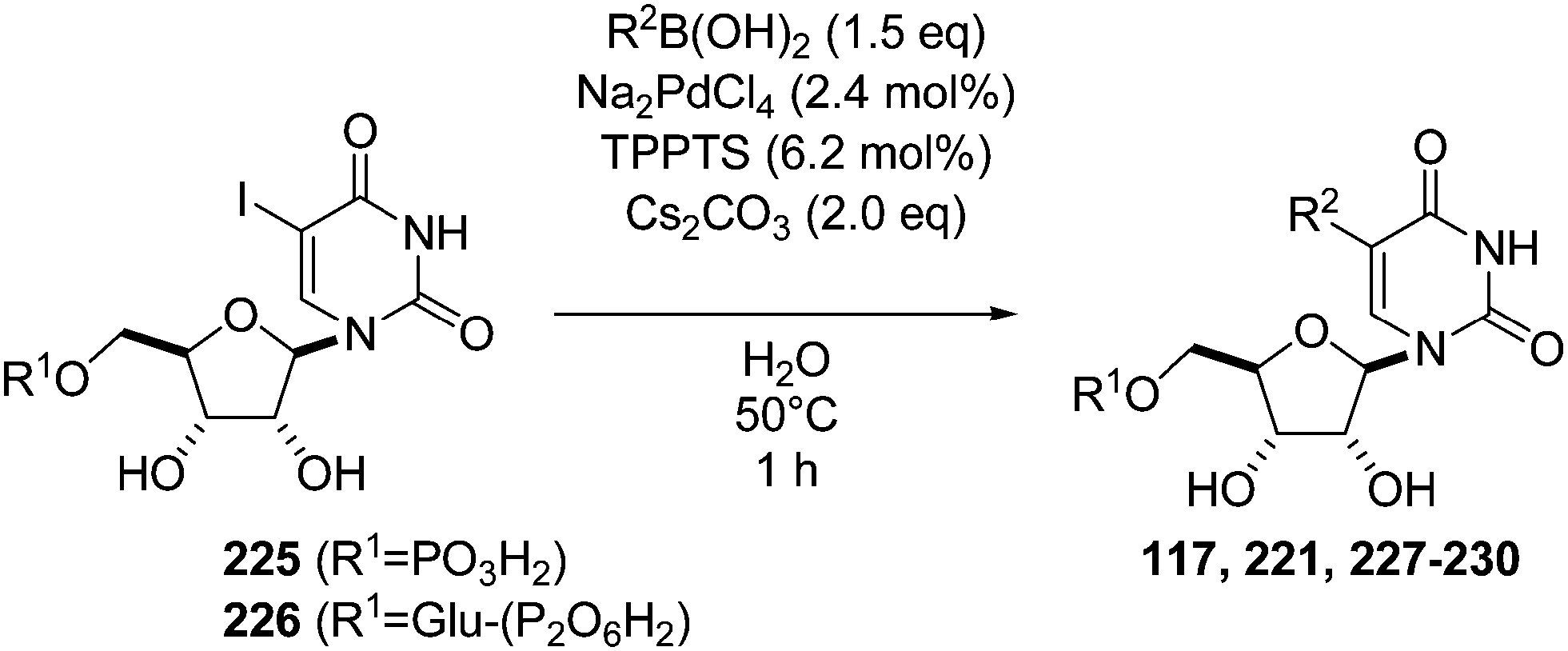
To investigate the possibility that the 5-brominated derivative 231 may, in solution, adopt a conformation, which restricts access of the palladium-ligand catalytic complex, the authors carried out a comparative conformational analysis of both sugar-nucleotides 226 and 231 (Fig. 5). It appears that the iodinated derivative 226 adopts a syn conformation in which the 5-iodo substituent is pointing away from the glucose diphosphate group, and the 5 position is more easily accessible by the Pd catalyst. The brominated derivative 231 was found to prefer the anti conformation where the 5-bromo substituent is facing towards the bulky glucose diphosphate moiety. Therefore, authors concluded that the superior cross-coupling reactivity of the iodinated substrate may be attributed, as least in part, to the capacity of the bulky iodo-substituent to induce a conformation, which is favorable for the insertion of the reactive Pd species during the oxidative addition step.47
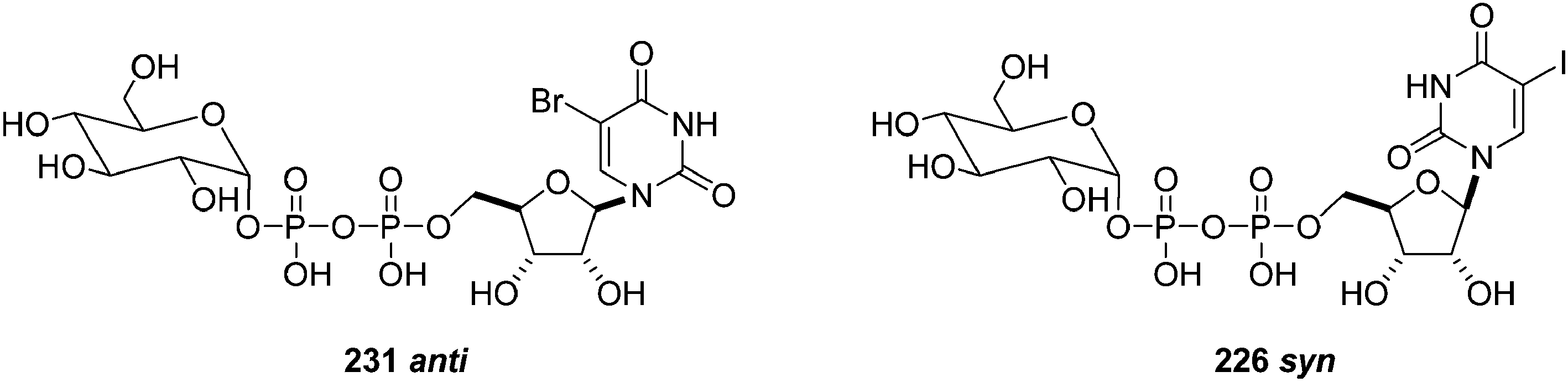 | ||
| Fig. 5 Anti and syn conformation of nucleotides 231 and 226. | ||
7.7. Na2PdCl4 as Pd(II) and KOH
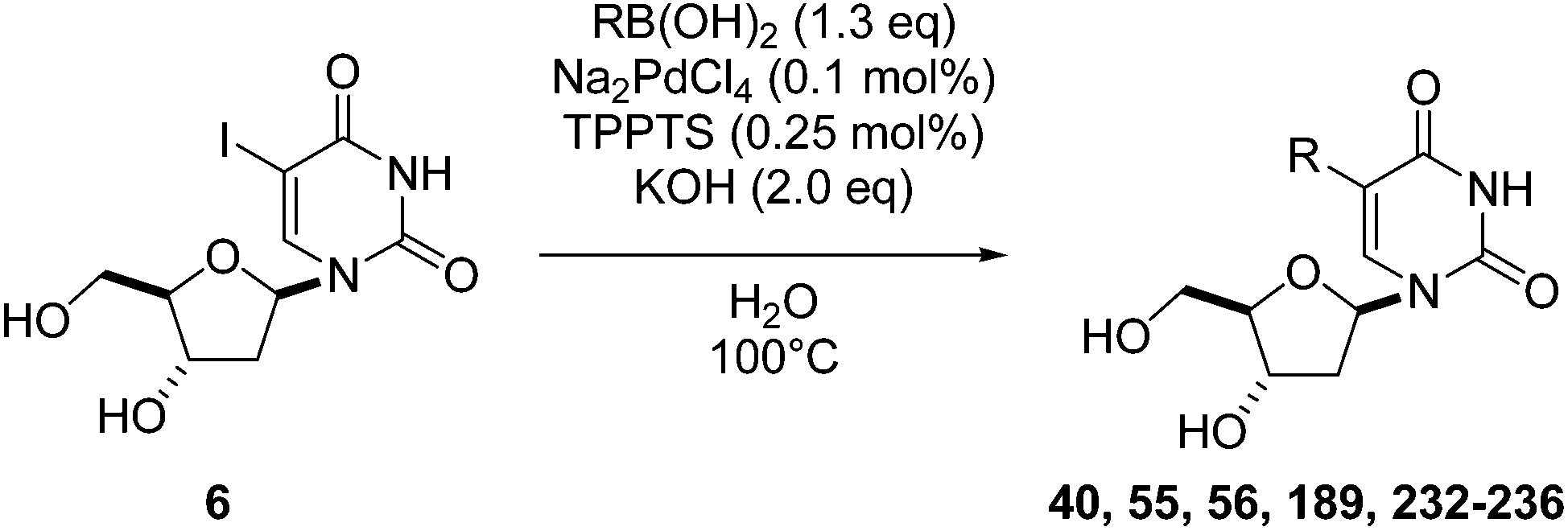
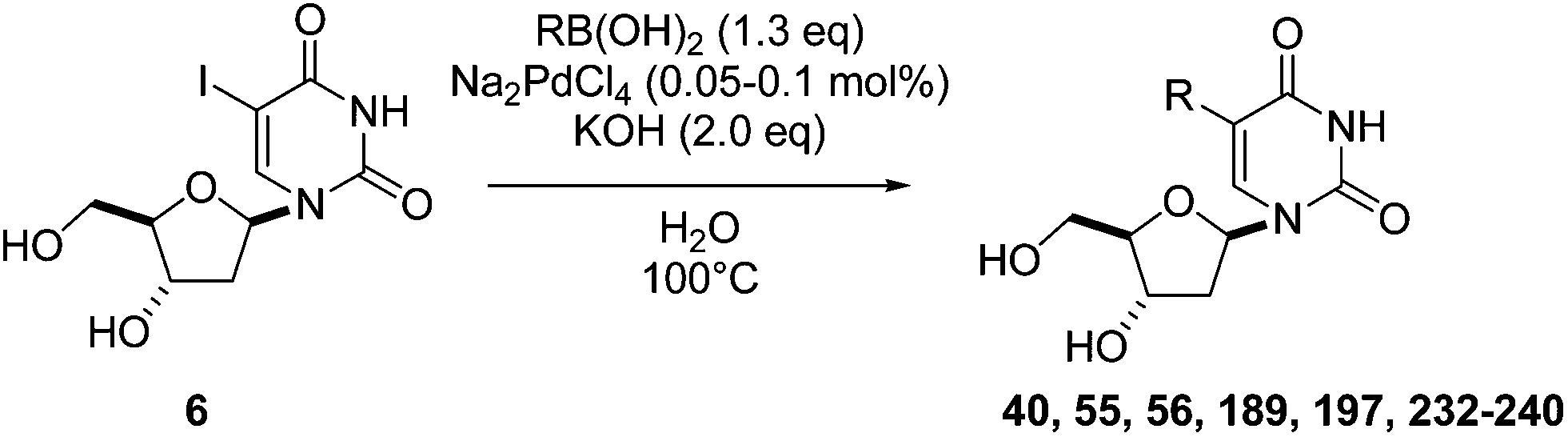
In an attempt to expand on the sustainable aspect of the Suzuki–Miyaura reaction, our group recently reported the development of three successful water-based methods starting from the commercially available unprotected 5-iodo-2′-deoxyuridine (6).43,48–50 Using a very low loading of catalyst (Na2PdCl4 0.1 mol% and TPPTS 0.25 mol%) in the presence of KOH (2 eq) as base, various 5-aryl-2′-deoxyuridine analogues 40, 55, 56, 189 and 232–236 were obtained in good-to-very good yields (Table 31).43 Substituent effects in the aryl boronic acids did not appear to be significant, with the exception of compound 233, which has a nitrile substituent in the para position (Table 31, entry 6). No hydrolysis was detected under our aqueous basic conditions used for the synthesis of nucleoside analogue 233; hence, the reaction was stopped after no more conversion was detected. All the aryl boronic acids with diverse electron-donating and electron-withdrawing substituents delivered the cross-coupled products in good yields (Table 31). The sterically demanding 2-methyl- and 2-methoxyphenyl boronic acids (Table 31, entries 7 and 8) proved to be difficult substrates for the Suzuki–Miyaura reaction. However, after extending reaction times, the target nucleoside analogues 234 and 235 were isolated in high (94%) and modest (36%) yields, respectively. A good conversion using 2-naphthylboronic acid furnished the nucleoside analogue 236 in 77% yields after 5 hours (Table 31, entry 9).
|
||||
|---|---|---|---|---|
| Entry | Time (h) | R | Product | Yield (%) |
| 1 | 0.5 |  |
55 | 85 |
| 2 | 1 |  |
232 | 78 |
| 3 | 6 | 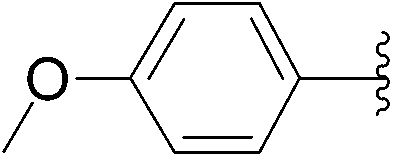 |
56 | 66 |
| 4 | 1 | 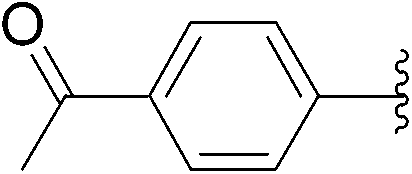 |
189 | 70 |
| 5 | 4 | 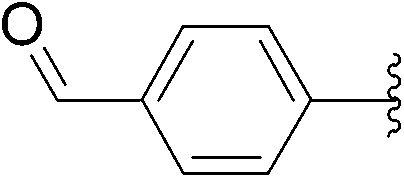 |
40 | 84 |
| 6 | 0.5 | 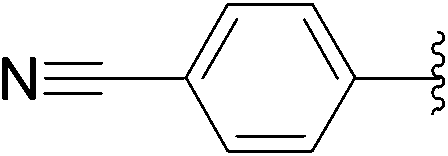 |
233 | 26 |
| 7 | 24 | 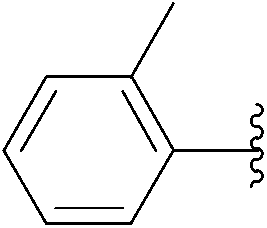 |
234 | 94 |
| 8 | 24 | 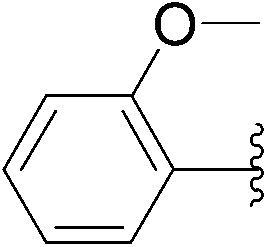 |
235 | 36 |
| 9 | 5 | 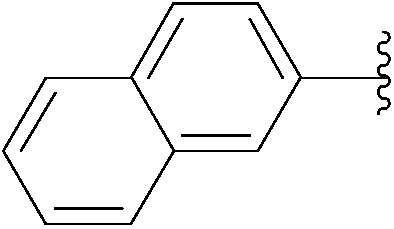 |
236 | 77 |
With the objective of designing a “greener” protocol, attention was drawn to improving atom economy and reducing the number of derivatives. Our group reported an efficient extension of the previous work to establish whether the presence of the ligand may or not influence cross-coupling efficiency.48 For this purpose, 5-iodo-2′-deoxyuridine (6) and phenyl boronic acid (33) were engaged in Suzuki–Miyaura cross-coupling reactions with low amounts of palladium (0.5–0.01 mol%), either in the presence or absence of TPPTS (2.5 eq to Pd) at 100 °C in neat water (Table 32). Our results showed clearly that the presence of TPPTS in the mixture was not necessary at 100 °C. Whatever the amount of palladium (0.5–0.01 mol%) employed, the absence of TPPTS had no inhibitory effect on the reaction course. In our hands, no significant differences in reaction time and/or yields were observed between 0.5 mol%, 0.1 mol% and 0.05 mol% of Pd(II). Indeed using such catalytic conditions (Table 32, entries 1–6), the desired cross-coupling product 55 was obtained in very good yields (79–86%) within a maximum of 30 minutes. One can also see that by dramatically decreasing the amount of palladium there is an expected increasing of the reaction time. However, even after 24 hours 5-phenyl-2′-deoxyuridine (55) was obtained in moderate yields even with only 0.01 mol% of Pd(OAc)2 used (Table 32, entries 9 and 10).
|
||||
|---|---|---|---|---|
| Entry | Na2PdCl4 (mol%) | TPPTS (mol%) | Time (h) | Yield (%) |
| 1 | 0.5 | 1.25 | 0.25 | 79 |
| 2 | 0.5 | 0 | 0.08 | 82 |
| 3 | 0.1 | 0.25 | 0.25 | 80 |
| 4 | 0.1 | 0 | 0.25 | 80 |
| 5 | 0.05 | 0.125 | 0.50 | 84 |
| 6 | 0.05 | 0 | 0.50 | 86 |
| 7 | 0.02 | 0.05 | 4 | 73 |
| 8 | 0.02 | 0 | 4 | 72 |
| 9 | 0.01 | 0.025 | 24 | 60 |
| 10 | 0.01 | 0 | 24 | 58 |
These encouraging experiments (Table 32, entries 4 and 6) led us to define two ligand-free methods to: (i) establish a different optimized reaction; and (ii) produce a consistent library of nucleoside analogues. For this purpose different aryl boronic acids were engaged in the cross-coupling reactions. Various substrates were examined with electron-donating and -withdrawing groups in the para or ortho positions accordingly. Also different means of heating were studied: classical and microwave irradiation assisted.49
The reaction times and yields in the presence of Na2PdCl4 (0.1 mol%) were always shorter and higher, respectively, than those observed in the presence of a lower loading of Na2PdCl4 (0.05 mol%) under thermal as well as microwave irradiation conditions.49 In addition, the cross-coupling reactions were more efficient when the microwave irradiation was employed. Aryl boronic acids with an electron-donating substituent in the para position gave the cross-coupling products 56 and 232 in good yields (Table 33, entries 3–6) either with 0.1 mol% or 0.05 mol% Na2PdCl4. Compared with compound 55, the presence of a methyl group in the para position of the aromatic ring does not change significantly the reaction result since compound 232 was isolated in the same range of yields and times (Table 33, entries 1–4). On the contrary, when a heteroatom was directly bound to the aryl boronic acid in the para position, an extended time reaction was needed to reach completion (Table 33, entries 5 and 6). Due to a lower nucleophilicity of aryl boronic acids with electron-withdrawing groups, particularly with a nitrile and formaldehyde groups, reactions were less efficient (Table 33, entries 9–12). The sterically demanding 2-methyl- and 2-methoxyphenyl boronic acids proved to be difficult substrates for Suzuki–Miyaura cross coupling of nucleosides even using “standard” conditions. It is of note that both methods proved successful albeit in giving modest yields (Table 33, entries 15–18). However, whatever the atom that was directly attached to the aromatic moiety of the boronic (methyl and methoxy groups) acid, no difference was observed in reaction time. It is proposed that steric restrictions may take over from electronic effects in such cases. In the cases of boronic acids which involved both steric effect and electron-withdrawing substituents such as 2-acetylphenylboronic acid and 2-formylphenylboronic acid, as might be expected no cross-coupling reaction was observed when using conventional heating, and only in one case a product was observed when microwave irradiation (Table 33, entries 19–22) was applied. To expand on the array of substrates for study, 5-iodo-2′-deoxyuridine (6) was coupled with a variety of heteroarylboronic acids (Table 33, entries 23–26) and with the (E) styrylboronic acid (Table 33, entries 27 and 28). It was found that thiophen-2-boronic acid was the only heteroarylboronic acid reactive enough to give the desired cross-coupling product 239 albeit in modest yield using either conventional heating or microwave irradiation (Table 33, entries 23–24). Also furan-2-yl boronic acid was only sufficiently reactive when the alternative technology was used (Table 33, entries 25 and 26). Interestingly, compound 240 was obtained using our methodology via the Suzuki–Miyaura reaction under heating with either condition (Table 33, entries 27 and 28). To the best of our knowledge, this is the first time in nucleoside chemistry that the Suzuki–Miyaura reaction has been shown to be successful in respect of the use of green and economic conditions.
|
|||||
|---|---|---|---|---|---|
| Entry | Na2PdCl4 (mol%) | Time (h) | R | Product | Yield (%) |
| a Isolated yields.b Under microwave irradiation.c Under conventional heating.d Reaction did not reach completion. | |||||
| 1 | 0.1 | 0.08b (0.25)c |  |
55 | 85b (80)c |
| 2 | 0.05 | 0.25b (0.50)c |  |
55 | 82b (86)c |
| 3 | 0.1 | 0.08b (0.50)c |  |
232 | 79b (79)c |
| 4 | 0.05 | 0.50b (2.00)c | 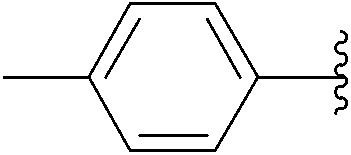 |
232 | 79b (80)c |
| 5 | 0.1 | 1.00b (7.00)c | 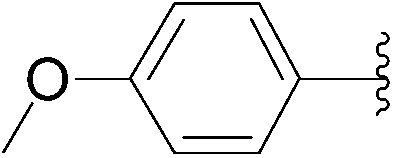 |
56 | 84b (64)c |
| 6 | 0.05 | 1.00b (24.00)c | 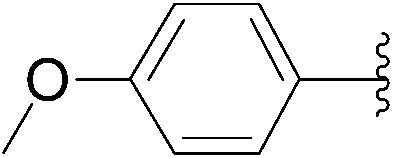 |
56 | 67b (57)c |
| 7 | 0.1 | 0.50b (2.00)c | 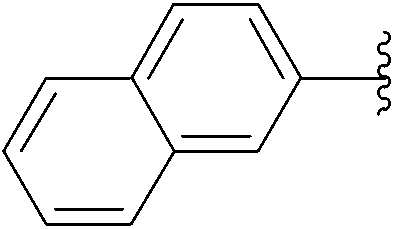 |
236 | 84b (80)c |
| 8 | 0.05 | 1.00b,d (24.00)c | 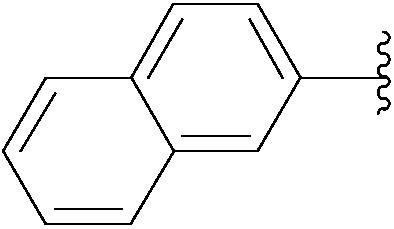 |
236 | 36b (70)c |
| 9 | 0.1 | 0.25b (1.00)c | 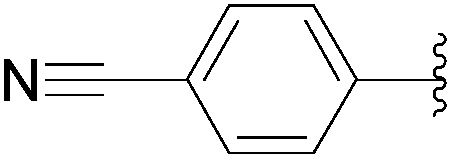 |
233 | 33b (39)c |
| 10 | 0.05 | 0.50b (5.00)c | 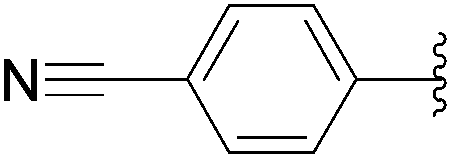 |
233 | 10b (13)c |
| 11 | 0.1 | 0.50b (6.00)c | 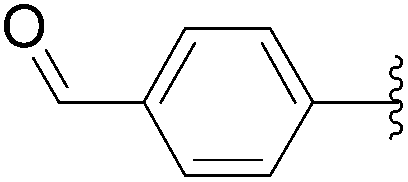 |
40 | 66b (17)c |
| 12 | 0.05 | 1.00b,d (5.00)c | 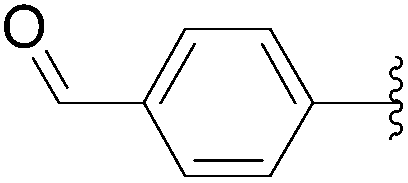 |
40 | 56b (13)c |
| 13 | 0.1 | 0.25b (0.50)c | 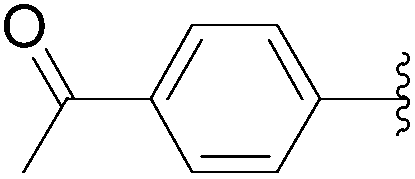 |
189 | 78b (69)c |
| 14 | 0.05 | 0.50b (24.00)c | 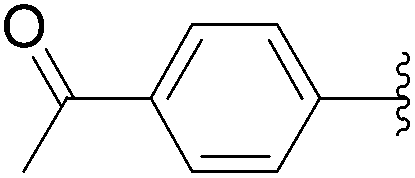 |
189 | 57b (49)c |
| 15 | 0.1 | 1.00b,d (24.00)c | 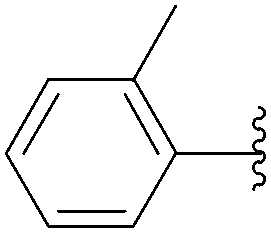 |
234 | 30b (22)c |
| 16 | 0.05 | 1.00b,d (24.00)c | 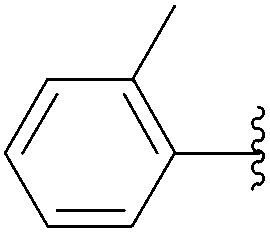 |
234 | 21b (19)c |
| 17 | 0.1 | 1.00b,d (24.00)c | 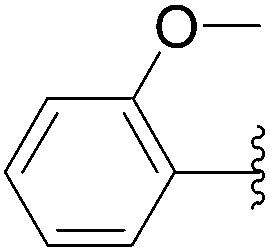 |
235 | 45b (23)c |
| 18 | 0.05 | 1.00b,d (24.00)c | 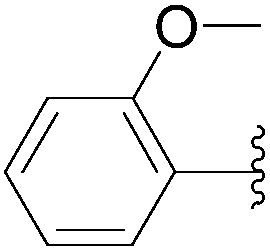 |
235 | 23b (18)c |
| 19 | 0.1 | 1.00b,d (24.00)c | 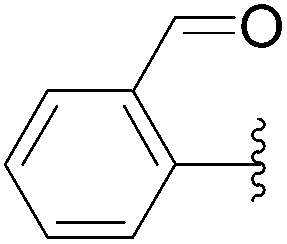 |
237 | 0b (0)c |
| 20 | 0.05 | 1.00b,d (24.00)c | 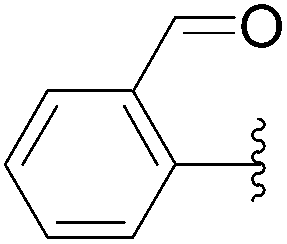 |
237 | 0b (0)c |
| 21 | 0.1 | 1.00b (24.00)c | 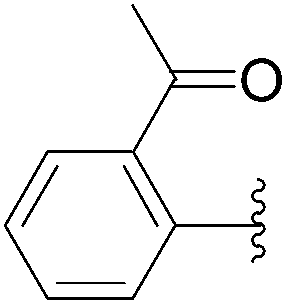 |
238 | 23b (0)c |
| 22 | 0.05 | 1.00b,d (24.00)c | 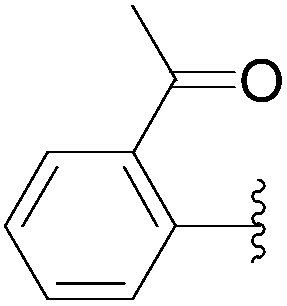 |
238 | 0b (0)c |
| 23 | 0.1 | 1.00b,d (6.00)c | 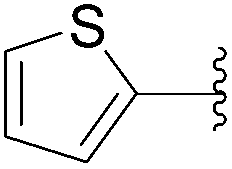 |
239 | 40b (32)c |
| 24 | 0.05 | 1.00b,d (24.00)c | 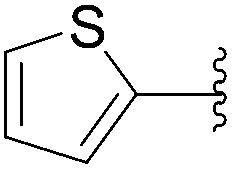 |
239 | 45b (0)c |
| 25 | 0.1 | 1.00b,d (24.00)c | 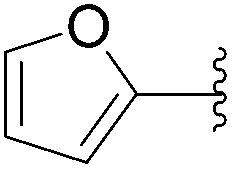 |
197 | 75b (0)c |
| 26 | 0.05 | 1.00b,d (24.00)c | 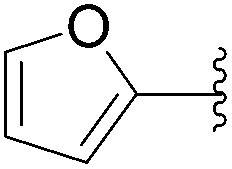 |
197 | 18b (0)c |
| 27 | 0.1 | 0.50b (2.00)c |  |
240 | 85b (58)c |
| 28 | 0.05 | 1.00b (4.00)c |  |
240 | 66b (33)c |
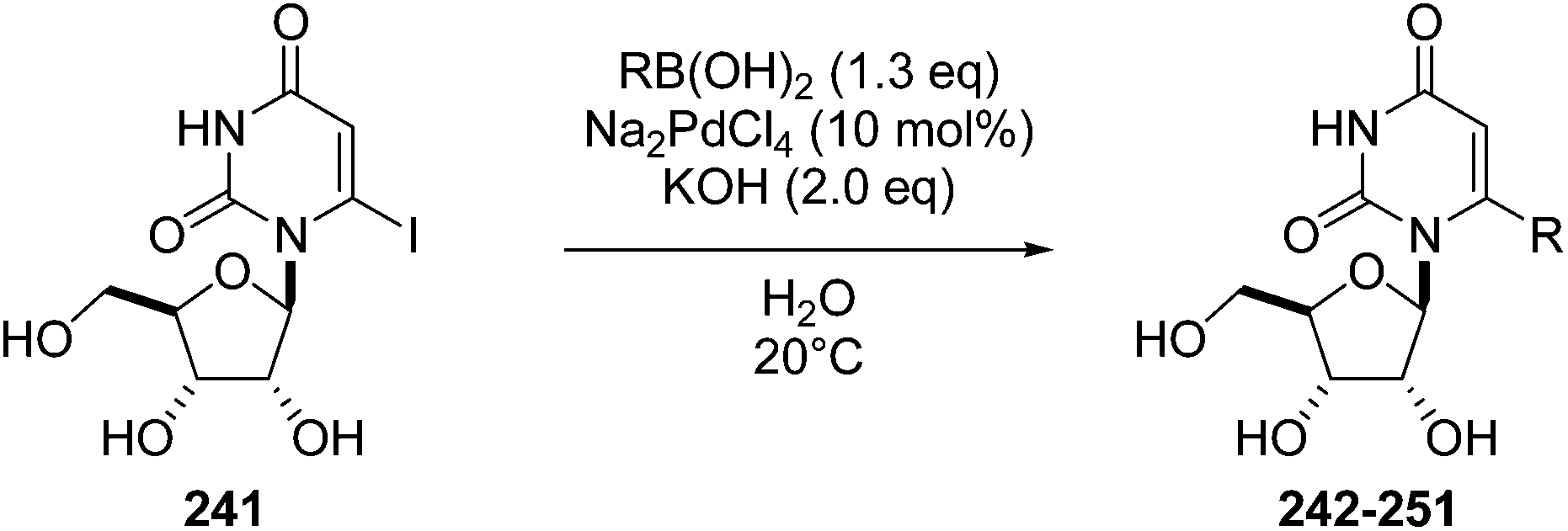
In 2013, Len's group described the first synthesis of the 6-aryl nucleoside analogues 242–251 using Suzuki–Miyaura cross-coupling reaction in water without the use of a ligand.50 After a systematic variation of different parameters, the optimized method found was that described above, which involved an enhancement of the amount of palladium Na2PdCl4 (10 mol%) in water without ligand. Due to the instability of the starting material 241 having an iodine atom in position 6, the cross coupling was developed further to enable it to be conducted effectively at room temperature. Thus, a series of aryl boronic acids with different electronic and steric factors were successfully screened leading to the target compounds 242, 243, 245–247 and 251 (Table 34).
|
||||
|---|---|---|---|---|
| Entry | Time (h) | R | Product | Yield (%) |
| 1 | 0.5 | 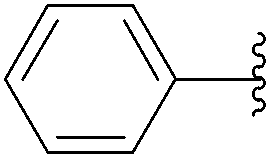 |
242 | 81 |
| 2 | 1.0 | 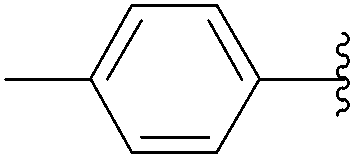 |
243 | 71 |
| 3 | 24 | 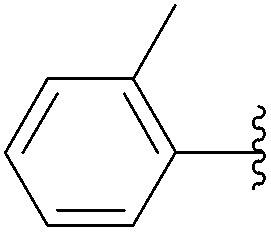 |
244 | 0 |
| 4 | 1.5 | 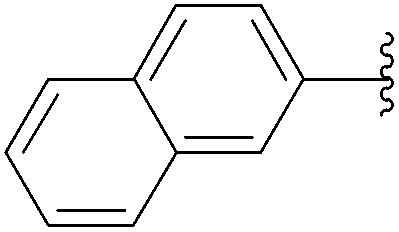 |
245 | 84 |
| 5 | 1.5 | 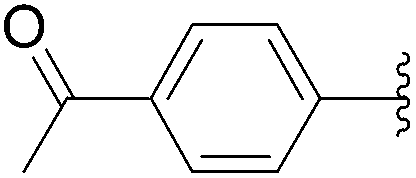 |
246 | 61 |
| 6 | 1.5 | 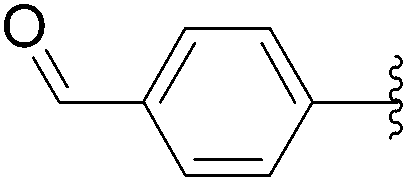 |
247 | 80 |
| 7 | 24 | 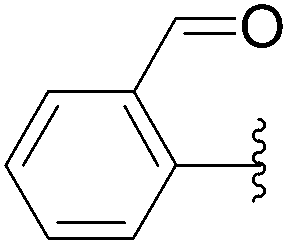 |
248 | 0 |
| 8 | 4 | 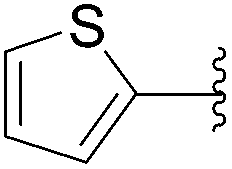 |
249 | 0 |
| 9 | 24 | 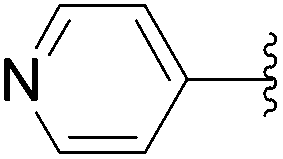 |
250 | 0 |
| 10 | 2 | 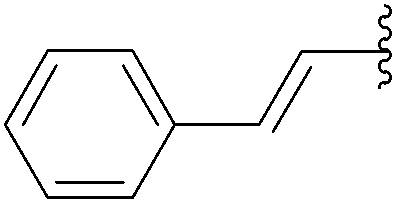 |
251 | 72 |
8. Post-synthetic modification of oligodeoxynucleotides (ODNs)
Multiples methods for the incorporation of modified building blocks with desired properties in oligodeoxynucleotides (ODNs) have been developed.51,52 Each of those methods presents its advantages but also its difficulties and/or drawbacks. An alternative to those methods may exist in the post-synthetic modification of oligodeoxynucleotides. Concerning such Suzuki–Miyaura reactions, the first results were published by Manderville and co-workers in 2011.53 The Suzuki–Miyaura cross-coupling reaction allowed this team to synthesize a number of C8-Ar-G-modified oligonucleotides (dimmers, trimers, decamers and a 15-mer) using a range of aryl boronic acids. In order to optimize the Pd-postsynthetic reaction, the shortest DNA substrates (0.1 μmol) having a bromide group were utilized first and reacted with boronic acid (1.2–10.0 eq) in the presence of Pd(OAc)2 (0.3 mol%), TPPTS (6.7 mol%) and Na2CO3 (2 eq) in H2O–CH3CN (2:1) mixture as solvent at 70 °C for 24 hours (Table 35). The corresponding oligomers were obtained in a large range of yields (15–97%) that was not dependent on the nature of the oligomers. It is notable that the protection of the 4-hydroxy derivatives (Table 35, entries 1–4) did not induce an increase in the yield of the coupling compared with the use of the free hydroxyl derivatives (Table 35, entries 6–8).
|
||||
|---|---|---|---|---|
| Entry | Oligo | R | Product | Yield (%) |
| a 1.2 eq of boronic were used.b 10 eq of boronic acid were used. | ||||
| 1 | ODN1 |  |
252 | 60a |
| 2 | ODN2 |  |
253 | 41a |
| 3 | ODN3 |  |
254 | 15a |
| 4 | ODN3 |  |
255 | 45b |
| 5 | ODN4 |  |
256 | 71b |
| 6 | ODN2 |  |
257 | 60b |
| 7 | ODN3 |  |
258 | 79b |
| 8 | ODN4 |  |
259 | 77b |
| 9 | ODN2 |  |
260 | 39b |
| 10 | ODN3 | 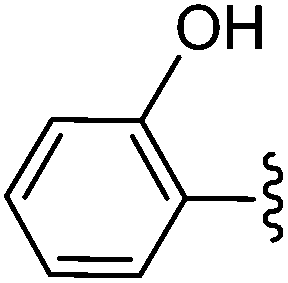 |
261 | 83b |
| 11 | ODN4 | 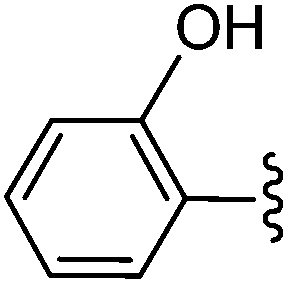 |
262 | 87b |
| 12 | ODN2 |  |
263 | 49b |
| 13 | ODN3 |  |
264 | 55b |
| 14 | ODN4 |  |
265 | 97b |
| 15 | ODN5 |  |
266 | 83b |
| 16 | ODN2 |  |
267 | 47b |
| 17 | ODN3 |  |
268 | 78b |
| 18 | ODN4 |  |
269 | 39b |
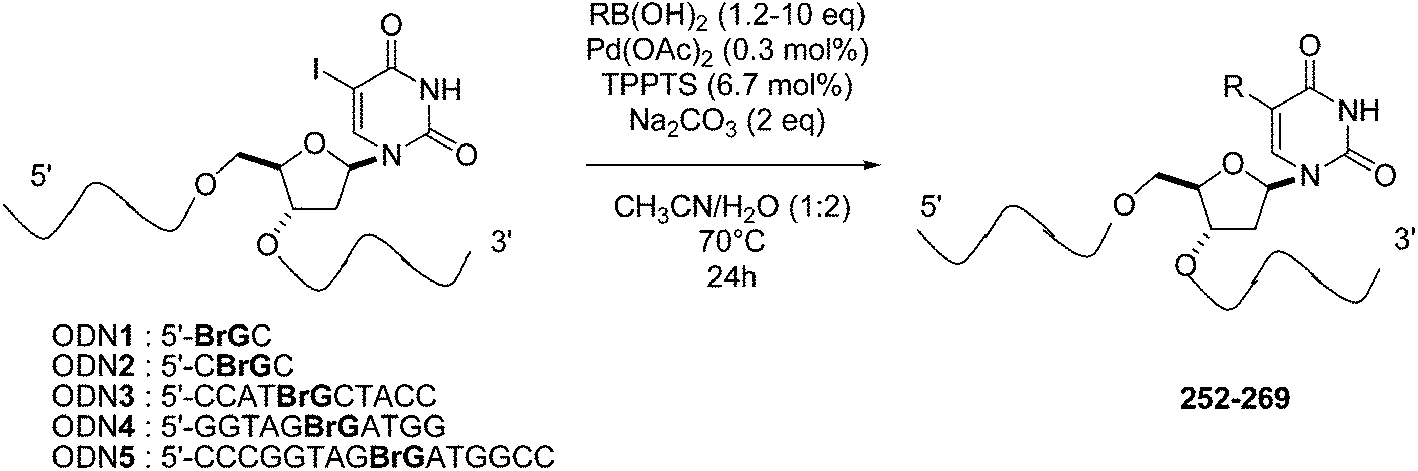
Very recently Jäschke and co-workers reported the preparation of nucleoside-based diarylethene photoswitches starting from 5-iodo-2′-deoxyuridine (6) and 5-iodo-2′-deoxycytidine (13) as well as the preparation of nine photoswitch-modified oligonucleotides.54 Reaction of compound 6 and compound 13 with 2-[2-methyl-5-phenylthien-3-yl]cyclopent-1-ene boronic acid pinacol ester (270) under Suzuki–Miyaura cross-coupling conditions previously described27 gave the corresponding products in yields of 44–61% (Scheme 30).
 | ||
| Scheme 30 Synthesis of nucleoside-base diarylethene photoswitches 271 and 272. | ||
Following this work, the authors reported Suzuki–Miyaura cross couplings of iodinated oligonucleotide and sterically demanding boronic acid.54 First, the authors tested the conditions elaborated by Manderville and co-workers.53 However, no detectable amounts of product were obtained. Therefore, they opted for the conditions developed for sensitive nucleosides triphosphate.35–38 Nine different modified nucleotides bearing one or two photoswitchable groups were then isolated in 16–35% yield (Table 36), starting from halogenated nucleotide (100 μmol), boronic acid pinacol ester (200 eq), Cs2CO3 (220 eq), Pd(OAc)2 (22.5 mol%) and TPPTS (57 mol%) at 120 °C for 1 hour. The authors reported the presence of dehalogenated starting material in addition to the cross-coupled compounds 273–281.
|
||||
|---|---|---|---|---|
| Entry | Oligo | Substrate | Product | Yield (%) |
| 1 | 15mer-dUPS1 | 5′-AGCAACA![[I with combining low line]](https://www.rsc.org/images/entities/char_0049_0332.gif) ![[U with combining low line]](https://www.rsc.org/images/entities/char_0055_0332.gif) CGATCGG-3′ CGATCGG-3′ |
273 | 25 |
| 2 | 15mer-dCPS1 | 5′-AGCAACA![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) CGATCGG-3′ CGATCGG-3′ |
274 | 22 |
| 3 | 15mer-dUPS2 | 5′-GCAACATCGATCGG-3′ |
275 | 26 |
| 4 | 15mer-dCPS2 | 5′-GCAACATCGATCGG-3′ |
276 | 34 |
| 5 | 15mer-dUPS3 | 5′-AGCAACATCGATCG-3′ |
277 | 25 |
| 6 | 15mer-dCPS3 | 5′-AGCAACATCGATCG-3′ |
278 | 35 |
| 7 | 19mer-dUPS1 | 5′-TCTAATACGACTCACATA-3′ |
279 | 20 |
| 8 | 19mer-dUPS2 | 5′-TCTAATACGACTCACTAA-3′ |
280 | 19 |
| 9 | 19mer-dUPS3 | 5′-TCTAATACGACTCACAA-3′ |
281 | 16 |
The authors noticed that a cross coupling done at terminal positions (3′ or 5′) was generally more efficient than an internal one.
Still in 2013, Davis and coworkers reported the generation of functional probes by DNA modification under mild conditions by Suzuki–Miyaura cross coupling.55 For this purpose, the authors started from commercially halogenated ODNs (100 μm) and used several pinacol boronic esters, a catalytic system composed of Pd(OAc)2 and 2-aminopyrimidine-4,6-diol, at biological temperature (37 °C) in a mixture of H2O–CH3CN (3:1) as solvent. Several biologically compatible buffer systems were tested (Na2PO4, NH4OAc and TRIS) as well as a range of basic pH values (8 and 8.5). The authors found that tris(hydroxymethyl)aminomethane (TRIS) buffer at pH 8.5 provided the most robust cross-coupling conditions, meaning full conversion of the starting material with non-noticeable side products, except traces of deiodinated ODN. Further optimization led the authors to obtain a complete conversion in 4 hours at 37 °C using boronic ester (100 eq) and 50 mol% [Pd]. Using these optimized conditions, the authors isolated several modified ODNs 282–288 containing representative functionalities such as diazirine, benzophenone or azobenzene (Table 37).
|
||||
|---|---|---|---|---|
| Entry | Oligo | R | Product | Yield (%) |
| 1 | 3′-AT[-IdU]GC-5′ |  |
282 | 86 |
| 2 | 3′-AT[-IdU]GC-5′ | 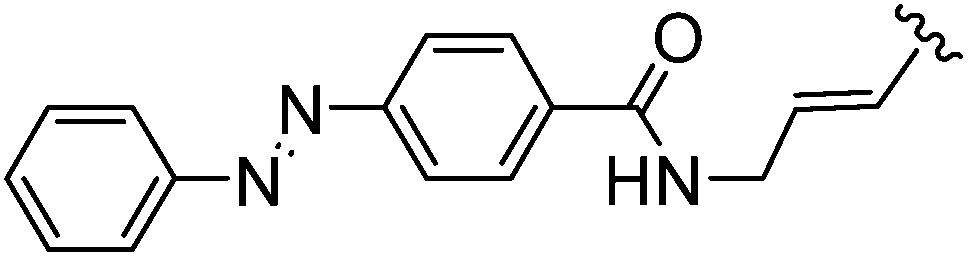 |
283 | 71 |
| 3 | 3′-AT[-IdU]GC-5′ | 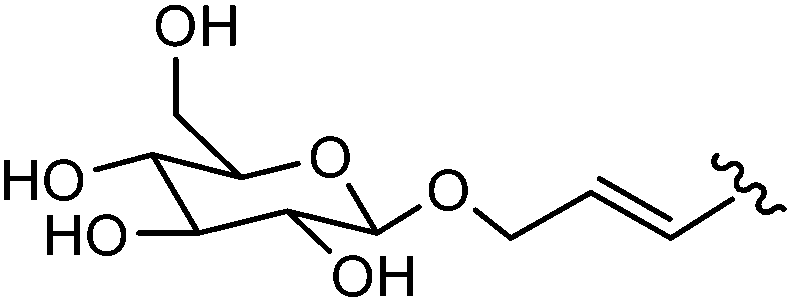 |
284 | 88 |
| 4 | 3′-AT[-IdU]GC-5′ |  |
285 | 93 |
| 5 | 3′-AT[-IdU]GC-5′ |  |
286 | 78 |
| 6 | 3′-AT[-IdU]GC-5′ | 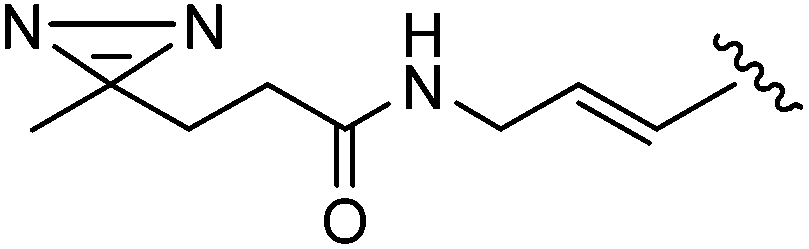 |
287 | 90 |
| 7 | 3′-AT[-IdU]GC-5′ | 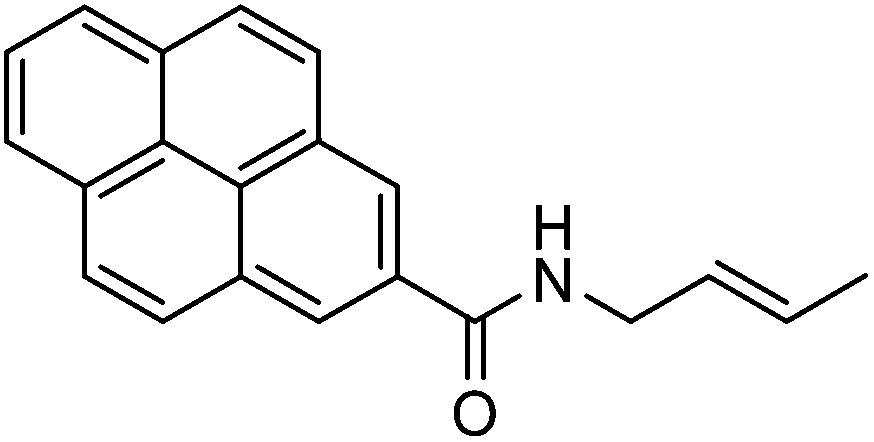 |
288 | 61 |
The authors pointed out that the amount of [Pd] could be reduced to 10 mol% over longer reaction times. The reaction could also be done at room temperature but with a longer full conversion time of starting material (16 hours). The authors tried their reaction conditions on another ODN that did not contain a 5-iodo-2′-deoxyuridine residue in order to rule out nonspecific reaction of other bases and then confirmed site selectivity.
9. Conclusion
A focus of this review has been observance of the 12 principles of green chemistry, with respect to the Suzuki–Miyaura cross-coupling reaction applied to the synthesis of nucleoside and nucleotide analogues having biological activities. To date, the majority of work has been directed towards 8-arylpurine (adenosin-9-yl and guanosin-9-yl) and 5-arylpyrimidine (uracil-1-yl and cytosin-1-yl) analogues having a D-ribose and 2′-deoxy-D-ribose moiety. The normal starting materials used have been 8-bromo, 5-iodo analogues, and in some cases, it has been the 5-bromo derivatives.46 Although most examples in this field have been aryl adducts, some heterocyclic examples have also received attention. The appropriated boronic acids have been of common use but esters have found applications applied for different cross couplings.11–13,38,39 The review encompasses variations of the starting materials, boronic acids, nature of the solvent, palladium source, ligand, base and reaction temperature (often higher than 80 °C).The use of THF–MeOH–H2O (mainly 2:1:2 and 2:2:1)8–14 afforded different 8-aryl-2′-deoxyadenosine, 8-aryl-2′-deoxyguanosine, 5-aryl-2′-deoxyuridine and 5-aryl-2′-deoxycytidine analogues using either Pd(0) (10 mol%) such as Pd(PPh3)4 (ref. 8–10) or Pd(II) (10 mol%) such as PdCl2(dppf)13 and PdCl2(dppf)2 (ref. 14) in the presence of NaOH (20 eq) as base. This solvent mixture was used for the coupling of different boronic acids having 3–5 aromatic cores, with or without heteroatoms. Using these procedures, the yields were low-to-good (10–79%), and starting materials having an exocyclic amino group gave low yields (10–65%).
In DME–H2O (2:1),15–17 the cross coupling was effected by starting from adenosine and guanosine derivatives, which are more polar than the corresponding 2′-deoxy analogues. Of the two nucleoside derivatives, only the adenosine afforded the target aryl adducts in moderate-to-good yield (60–81%). In this solvent mixture, introduction of furan and thiophene cores furnished lower yields (25% and 40%, respectively). Pd(PPh3)4 (10 mol%)15–17 and Pd(OAc)2 (2.5 mol%) with TPPTS (6.25 mol%)16 were used in this reaction media.
The use of DMF–H2O (2:1)18 and Pd(PPh3)4 (10 mol%) was reported only in one study that described the synthesis of the adducts of 2′-deoxyuridine.18
The CH3CN–H2O mixture (1:2)16,19–39 was the most reported solvent for the cross-coupling Suzuki–Miyaura reaction. In this media, Pd(OAc)2 and TPPTS with Na2CO3, K2CO3 or Cs2CO3 were the reagents most studied. Starting materials such as 2′-deoxyadenosine, adenosine, 2′-deoxyguanosine, guanosine, 2′-deoxyuridine, 7-deazapurine and uridine in the presence of Pd(OAc)2–TPPTS (1:2.5) as a catalytic system were transformed into the corresponding target compounds in good-to-excellent yield (>50% yield); however, depending on the steric hindrances, electronic effects and presence or absence of heteroatom, the yields could be lower. The amount of catalyst was often higher than 2.5 mol% with a maximum of 10 mol%. Using Na2CO3, the couple Pd(OAc)2–TPPTS (1:2.5) in CH3CN–H2O enabled different non-canonical adducts such as aniline amino acid derivatives to be obtained.26,27 In this media, the difference between Na2CO3 and K2CO3 was not significant. The use of Cs2CO3 as base led to successful cross-coupling reactions with yields (7–82%), depending on the nature of the boronic acid.38,39 Starting from 2′-deoxycytidine in the presence of Pd(OAc)2 (3 mol%) and TPPTS (8 mol%), a lower temperature (43 °C) was enough to afford the desired amine derivative in good yield (85–100%).25 It is significant that even with the use of room temperature, cross-coupling reactions are still successful. Shaughnessy and co-workers reported the use of TXPTS [Pd(OAc)2 (2.5:1)] with 2.7 mol% of metal at 23 °C to furnish the target nucleoside analogues in 45–99% yields.20 As usually cited in this review, the lower yields (45% and 50%) were obtained starting from 2′-deoxyguanosine or thiophen-2-ylboronic acid. Two publications described the C–C bond formation using either PdCl2(dppf)2 (6.5 mol%)19 or Pd(PPh3)4 (10 mol%),16 respectively, starting from 2′-deoxyuridine and adenosine derivatives. Starting from the nucleotide analogues having 1–3 phosphorus atoms, the CH3CN–H2O mixture (1:2) with Pd(OAc)2–TPPTS (1:2.5) in the presence of Cs2CO3 was found to be the preferred protocol26–38 even if Na2CO3 could afford the cross-coupling conversion.26 Microwave-assisted organic synthesis was applied to the cross-coupling Suzuki–Miyaura reaction starting from adenosine derivatives and the corresponding nucleotide using Pd(OAc)2 (5 mol%), TPPTS (12.5–25 mol%) and Na2CO3 as base. Of course, reduction of the time of reaction was observed but the yields were lower than those obtained with classical thermal reaction.26 There is one report of the mixture MeOH–H2O (5:1) as media for the Suzuki–Miyaura reaction but the yield was low (28%) for the preparation of the 5-formyl-2′-deoxyuridine.41
Recently, water as sole solvent was studied for the Suzuki–Miyaura cross coupling for the preparation of 2′-deoxyuridine, uridine, adenosine, guanosine and diphosphatenucleoside-hexose.16,21,41–50 Suzuki–Miyaura cross coupling using Pd(PPh3)4 (3 mol%) and reverse-phase glass beads was reported for the formation of uridine analogues in 28% yield.42 This process was not fully reported in the literature. The use of Pd(PPh3)4 (10 mol%) in the presence of K2CO3 permitted the synthesis of 8-aryl adenosine derivative in medium-to-high yield (26–75%) but did not furnish the corresponding guanosine derivative.16 Pd(OAc)2 coupled with TPPTS16,43 or PPh3 (ref. 44) was studied as a catalytic system. Using K2CO3 as base, the couple Pd(OAc)2 (2.5 mol%) and TPPTS (6.25 mol%) gave the target adenosine and guanosine derivatives in yields often higher than 70%.16 Microwave-assisted technology using PPh3 instead of TPPTS was described to afford the 5-aryl-2′-deoxyuridine in yields regularly higher than 70%.44 The latest metal to be studied was Na2PdCl4 with or without TPPTS as ligand in the presence of K2CO3 or KOH as base.43–50 Using Na2PdCl4 (2.5 mol%), TPPTS (6.25 mol%) and K2CO3, the cross-coupling reaction afforded 8-arylguanosine derivatives in yields higher than 70%.45 The use of a similar protocol was described to furnish the corresponding mono-, di- and triphosphates frequently in good yields (>50%). To the best of our knowledge, only one report has described of a one-pot, two-step procedure for the preparation of 8-phenyl guanosine derivatives in good yield (44–75%, over two steps). This protocol successively included activation and cross-coupling reactions.45,46
In water, starting from nucleotides, it was not necessary to introduce Cs2CO3 for the preparation of the adduct.45–47 The use of KOH instead of K2CO3 was described by our group.43,48–50 Amongst the different methodologies described, the most significant development of this work has been the low loading of palladium (0.05–0.1 mol%) and the absence of ligand. Starting from 2′-deoxyuridine, the yields of the target 5-aryl nucleoside analogues were at least equal to those already reported in the literature. Microwave-assisted organic synthesis and conventional heating were compared, leading to similar yields whatever the method applied. Typically, the reaction time was faster using the alternative microwave activation. A Suzuki–Miyaura cross-coupling reaction was studied for the first time starting from 6-iodouridine in only water.50 In this case, our group described a novel ligand-free protocol using a more conventional amount of Na2PdCl4 (10 mol%) at room temperature.
In contrast to the published works described in this review, the recent results of Len's group48–50 have revealed that Suzuki–Miyaura cross-coupling reactions applied to 5-iodo-2′-deoxyuridine are feasible: (i) in refluxing pure water; and (ii) with low loading of palladium (iii) without any ligand. Furthermore, when the starting materials are thermally unstable, room temperature and a larger amount of palladium can be used.50
Finally, some very recent papers reported a postsynthetic strategy allowing arylation of DNA on a specific halogenated nucleoside by Suzuki–Miyaura cross-coupling reactions.53–55
Palladium-catalyzed reactions allow access to a wide range of fine chemicals or active pharmaceutical compounds. Nevertheless, they can present a problem in that palladium can often be retained in the isolated product.56
In the future, the Suzuki–Miyaura cross-coupling reaction of canonical or non-canonical nucleoside analogues without a ligand in a green solvent at room temperature with recycling of the catalytic system will represent an interesting challenge for synthetic organic chemists and offer a wider scope of materials for biological investigations.
References
-
(a) E. De Clercq, J. Clin. Virol., 2004, 30, 115–133 CrossRef CAS PubMed
; (b) A. Matsuda and T. Sasaki, Cancer Sci., 2004, 95, 105–111 CrossRef CAS
-
(a) E. R. Samuels, J. McNary, M. Aguilar and A. M. Awad, Nucleosides, Nucleotides Nucleic Acids, 2013, 32, 109–123 CrossRef CAS PubMed
-
(a) J. Lebreton, J. M. Escudier, L. Arzel and C. Len, Chem. Rev., 2010, 110, 3371–3418 CrossRef CAS PubMed
-
(a) D. C. Tahmassebi and D. P. Millar, Biochem. Biophys. Res. Commun., 2009, 380, 277–280 CrossRef CAS PubMed
-
(a) L.-A. Agrofoglio, I. Gillaizeau and Y. Saito, Chem. Rev., 2003, 103, 1875–1916 CrossRef CAS PubMed
- P. T. Anastas and J. C. Warner, Green Chemistry: theory and practice, Oxford University Press, New York, 1998, p. 30 Search PubMed
-
(a) N. Miyaura, T. Yanagi and A. Suzuki, Synth. Commun., 1981, 11, 513–519 CrossRef CAS
-
(a) N. Amann and H.-A. Wagenknecht, Synlett, 2002, 33, 687–691 CrossRef PubMed
- N. Amann, E. Pandurski, T. Fiebig and H.-A. Wagenknecht, Chem.–Eur. J., 2002, 8, 4877–4883 CrossRef CAS
- E. Mayer, L. Valis, R. Huber, N. Amann and H.-A. Wagenknecht, Synthesis, 2003, 15, 2335–2340 Search PubMed
- L. Valis and H.-A. Wagenknecht, Synlett, 2005, 15, 2281–2284 Search PubMed
- M. F. Jacobsen, E. E. Ferapontova and K. V. Gothelf, Org. Biomol. Chem., 2009, 7, 905–908 CAS
- C. Wagner and H.-A. Wagenknecht, Chem.–Eur. J., 2005, 11, 1871–1876 CrossRef CAS PubMed
- C. Wanninger-Weiß and H.-A. Wagenknecht, Eur. J. Org. Chem., 2008, 64–71 CrossRef
- N. Kohyama, T. Katashima and Y. Yamamoto, Synthesis, 2004, 17, 2799–2804 Search PubMed
- A. Collier and G. K. Wagner, Synth. Commun., 2006, 36, 3713–3721 CrossRef CAS
- D. E. McCloskey, S. Bale, J. A. Secrist III, A. Tiwari, T. H. Moss III, J. Valiyaveettil, W. H. W. Brooks, C. Guida, A. E. Pegg and S. E. Ealick, J. Med. Chem., 2009, 52, 1388–1407 CrossRef CAS PubMed
- M. Kogler, B. Vanderhoydonck, S. De Jonghe, J. Rozenski, K. Van Belle, J. Herman, T. Louat, A. Parchina, C. Sibley, E. Lescrinier and P. Herdewijn, J. Med. Chem., 2011, 54, 4847–4862 CrossRef CAS PubMed
- T. Ehrenschwender and H.-A. Wagenknecht, Synthesis, 2008, 3657–3662 CAS
- E. C. Western, J. R. Daft, E. M. Johnson II, P. M. Gannett and K. H. Shaughnessy, J. Org. Chem., 2003, 68, 6767–6774 CrossRef CAS PubMed
- V. Vongsutilers, J. R. Daft, K. H. Shaughnessy and P. M. Gannett, Molecules, 2009, 14, 3339–3352 CrossRef CAS PubMed
- E. C. Western and K. H. Shaughnessy, J. Org. Chem., 2005, 70, 6378–6388 CrossRef CAS PubMed
- T. Läppchen, V. A. Pinas, A. F. Hartog, G.-J. Koomen, C. Schaffner-Barbero, M. Andreu, D. Trambaiolo, J. Löwe, A. Uhem, A. V. Popov and T. den Blaauwen, Chem. Biol., 2008, 15, 189–199 CrossRef PubMed
- Q. Dai, D. Xu, K. Lim and R. G. Harvey, J. Org. Chem., 2007, 72, 4856–4863 CrossRef CAS PubMed
- M. Mizuta, J.-I. Banba, T. Kanamori, A. Ohkubo, M. Sekine and K. Seio, Nucleic Acids Symp. Ser., 2007, 51, 25–26 CrossRef PubMed
- P. Čapek, R. Pohl and M. Hocek, Org. Biomol. Chem., 2006, 4, 2278–2284 Search PubMed
- P. Čapek, H. Cahová, R. Pohl, M. Hocek, C. Gloeckner and A. Marx, Chem.–Eur. J., 2007, 13, 6196–6203 CrossRef PubMed
- P. Nauš, R. Pohl, I. Votruba, P. Džubák, M. Hajdúch, R. Ameral, G. Birkuš, T. Wang, A. S. Ray, R. Mackman, T. Cihlar and M. Hocek, J. Med. Chem., 2010, 53, 460–470 CrossRef PubMed
- A. Bourderioux, P. Nauš, P. Perlíková, R. Pohl, I. Pichová, I. Vortubra, P. Džubák, P. Konečný, M. Hajdúch, K. M. Stray, T. Wang, A. S. Ray, J. Y. Feng, G. Birkus, T. Cihlar and M. Hocek, J. Med. Chem., 2011, 54, 5498–5507 CrossRef CAS PubMed
- P. Perlíková, N. J. Martínez, L. Slavĕtínská and M. Hocek, Tetrahedron, 2012, 68, 8300–8310 CrossRef PubMed
- M. Segal and B. Fischer, Org. Biomol. Chem., 2012, 10, 1571–1580 CAS
- P. Perlíková, P. Konečný, P. Nauš, J. Snášel, I. Votruba, P. Džubák, I. Pichová, M. Hajdúch and M. Hocek, Med. Chem. Commun., 2013, 4, 1497–1500 RSC
- M. Vrábel, R. Pohl, I. Vortruba, M. Sajadi, S. A. Kovalenko, N. P. Ernsting and M. Hocek, Org. Biomol. Chem., 2008, 6, 2852–2860 Search PubMed
- H. Cahová, R. Pohl, L. Bednárová, K. Nováková, J. Cvačka and M. Hocek, Org. Biomol. Chem., 2008, 6, 3657–3660 Search PubMed
- H. Cahová, L. Havran, P. Brázdilová, H. Pivoňková, R. Pohl, M. Fojta and M. Hocek, Angew. Chem., Int. Ed., 2008, 47, 2059–2062 CrossRef PubMed
- V. Raindlová, R. Pohl, M. Šanda and M. Hocek, Angew. Chem., Int. Ed., 2010, 49, 1064–1066 CrossRef PubMed
- H. Macíčková-Cahová, R. Pohl, P. Horáková, L. Havran, J. Špaček, M. Fojta and M. Hocek, Chem.–Eur. J., 2011, 17, 5833–5841 CrossRef PubMed
- J. Riedl, R. Pohl, L. Rulĩšek and M. Hocek, J. Org. Chem., 2012, 77, 1026–1046 CrossRef CAS PubMed
- L. Kalachova, R. Pohl and M. Hocek, Synthesis, 2009, 105, 105–112 Search PubMed
- J. Balintová, M. Plucnara, P. Vidláková, R. Pohl, L. Havran, M. Fojta and M. Hocek, Chem.–Eur. J., 2013, 19, 12720–12731 CrossRef PubMed
- A. Okamoto, T. Inasaki and I. Saito, Tetrahedron Lett., 2005, 46, 791–795 CrossRef CAS PubMed
- K. M. Lawson Daku, R. F. Newton, S. P. Pearce, J. Vile and J. M. J. Williams, Tetrahedron Lett., 2003, 5095–5098 CrossRef CAS
- G. Sartori, G. Enderlin, G. Hervé and C. Len, Synthesis, 2012, 44, 767–772 CrossRef CAS PubMed
- N. Fresneau, M.-A. Hiebel, L. A. Agrofoglio and S. Berteina-Raboin, Molecules, 2012, 17, 14409–14417 CrossRef CAS PubMed
- A. Collier and G. K. Wagner, Org. Biomol. Chem., 2006, 4, 4526–4532 CAS
- A. Collier and G. K. Wagner, Chem. Commun., 2008, 178–180 RSC
- T. Pesnot and G. K. Wagner, Org. Biomol. Chem., 2008, 6, 2884–2891 CAS
- G. Sartori, G. Hervé, G. Enderlin and C. Len, Synthesis, 2013, 45, 330–333 CrossRef CAS PubMed
- S. Gallagher-Duval, G. Hervé, G. Enderlin and C. Len, New J. Chem., 2013, 37, 1989–1995 RSC
- G. Enderlin, G. Sartori, G. Hervé and C. Len, Tetrahedron Lett., 2013, 26, 3374–3377 CrossRef PubMed
- S. L. Beaucage and R. P. Iyer, Tetrahedron, 1993, 49, 6123–6194 CrossRef CAS
- S. Jäger, G. Rashed, H. Kornreich-Leshem, M. Engeser, O. Thum and M. Famulok, J. Am. Chem. Soc., 2005, 127, 15071–15082 CrossRef PubMed
- A. Omumi, D. G. Beach, M. Baker, W. Gabryelski and R. A. Manderville, J. Am. Chem. Soc., 2011, 133, 42–50 CrossRef CAS PubMed
- H. Cahová and A. Jäschke, Angew. Chem., Int. Ed., 2013, 52, 3186–3190 CrossRef PubMed
- L. Lercher, J. F. McGouran, B. M. Kessler, C. J. Schofield and B. G. Davis, Angew. Chem., Int. Ed., 2013, 52, 10553–10558 CrossRef CAS PubMed
- C. E. Garrett and K. Prasad, Adv. Synth. Catal., 2004, 246, 889–900 CrossRef
| This journal is © The Royal Society of Chemistry 2014 |