
Upgrading biomass-derived furans via acid-catalysis/hydrogenation: the remarkable difference between water and methanol as the solvent†
Xun
Hu
,
Roel J. M.
Westerhof
,
Liping
Wu
,
Dehua
Dong
and
Chun-Zhu
Li
*
Fuels and Energy Technology Institute, Curtin University of Technology, GPO Box U1987, Perth, WA 6845, Australia. E-mail: chun-zhu.li@curtin.edu.au; Fax: (+) 61 8 9266 1138; Tel: (+) 61 8 9266 1131
First published on 7th October 2014
Abstract
In methanol 5-hydroxymethylfurfural (HMF) and furfuryl alcohol (FA) can be selectively converted into methyl levulinate via acid-catalysis, whereas in water polymerization dominates. The hydrogenation of HMF, furan and furfural with the exception of FA is much more selective in methanol than in water.
Introduction
Furans (e.g. furfural and 5-hydroxylmethylfurfural) can be produced from abundantly available biomass via hydrolysis.1 They are the platform chemicals which can be upgraded to value-added chemicals such as pharmaceutical intermediates2 and fuel additives3via acid-catalysis or hydrogenation or both. For example, 5-hydroxylmethylfurfural (HMF) and furfuryl alcohol (FA, the hydrogenation product of furfural) can be converted into levulinic acid/ester via acid-catalyzed conversion4 or to 2-methyltetrahydrofuran (MTHF)5 and valerate esters6via hydrogenation. Furfural can also be converted to alkyl levulinates (fuel additives)7 or to MTHF8via acid-catalysis coupled with hydrogenation.During either acid-catalysis or hydrogenation, the solvent and the catalyst are the critical factors for determining overall efficiency of the processes. The solvent not only provides a reaction environment but also may react directly with reactants, modifying their behaviors. For example, during the acid-catalyzed conversion of levoglucosan and glucose, water as a solvent promotes polymerization and diminishes production of levulinic acid, while methanol reacts with the sugars, stabilizes the reactive intermediates and promotes methyl levulinate production.9 During the one-step conversion of xylose to levulinic acid, using water and methanol as solvents also makes remarkable difference: much higher yields of levulinic acid/ester produced in methanol than in water.10 Numerous efforts have been devoted to the development of acid catalysts or hydrogenation catalysts for the production of these furans.11 However, much less attention was given to roles of solvent, which, in many cases, is almost equally important to a catalyst in both acid-catalyzed reactions and hydrogenation reactions.
In this study, the acid-treatment (catalyst: Amberlyst 70) and hydrogenation (catalyst: Pd/C) of the biomass-derived furans (FA, HMF, furan and furfural) were performed in water and methanol to demonstrate the importance of methanol in the catalytic reactions. Our results indicate that: (1) the furans with distinct molecular structures demonstrate very different levels of interaction with methanol and water; (2) in methanol both FA and HMF can be selectively converted into methyl levulinate. In water both FA and HMF polymerized but to very different extents (FA ≫ HMF); (3) the hydrogenation efficiency and selectivity of HMF, furan and furfural with the exception of FA is much more efficient in methanol than in water.
Results and discussion
Acid-catalyzed conversion of the furans in water and methanol
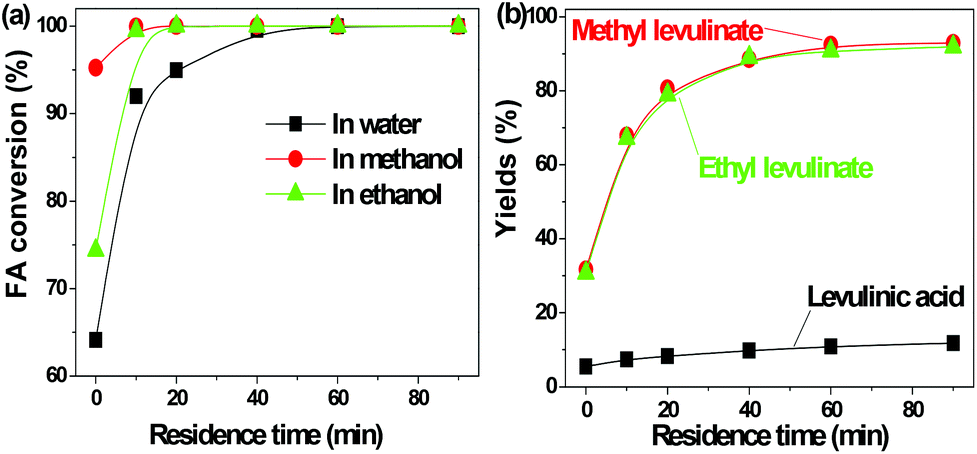 | ||
| Fig. 1 Acid-catalyzed conversion of furfuryl alcohol in water, methanol and ethanol. Reaction conditions: furfuryl alcohol: 3 g, Amberlyst 70: 3 g, solvent: 100 ml, T = 130 °C, residence time: 90 min. | ||
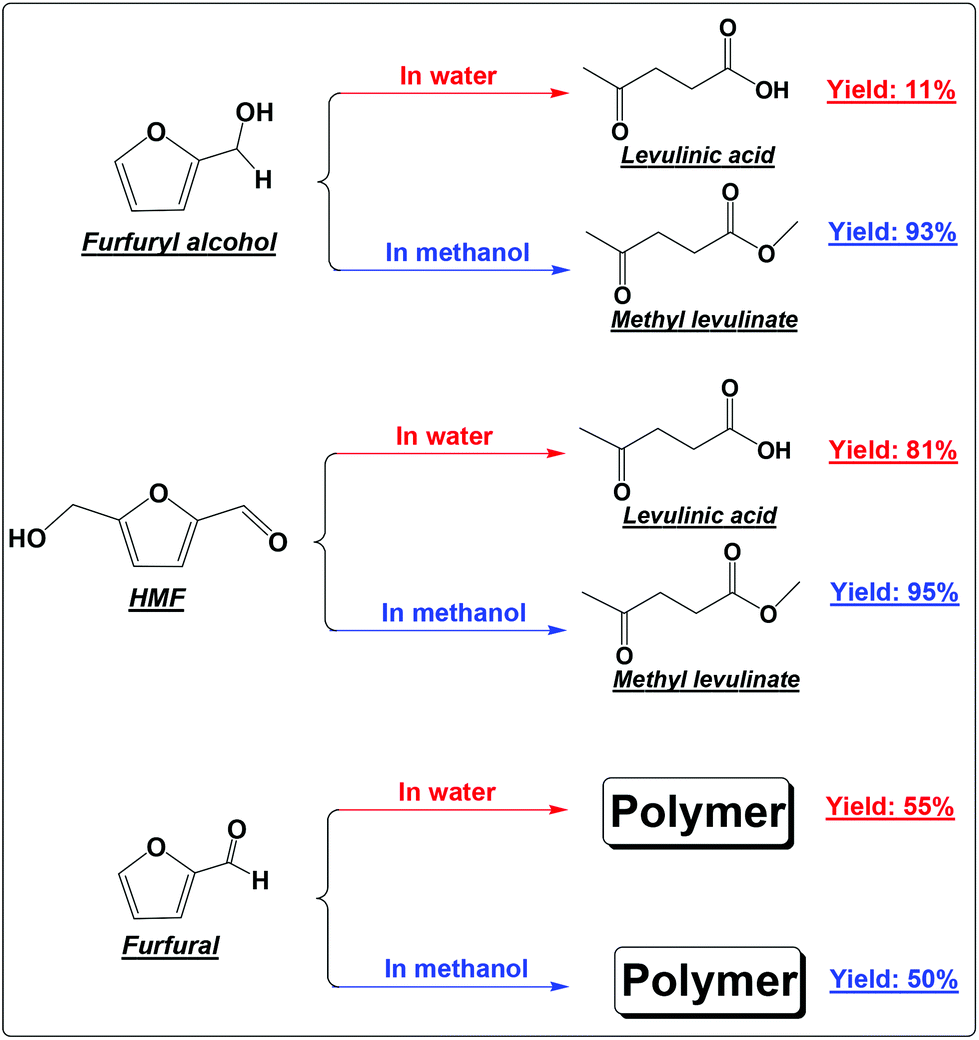 | ||
| Scheme 1 Acid-catalyzed conversion of FA, HMF and furfural in water and methanol. | ||
FA was selectively converted into methyl levulinate in methanol or ethyl levulinate in ethanol, but in water the opposite was observed. Polymerization of FA in the acidic environment is believed to be the main reason for low selectivity of levulinic acid in water. In the acid-treatment of FA in water, the used catalyst became very sticky, which was difficult to be scrubbed from the reactor wall and stirrer. In methanol, the polymerization is suppressed and the production of methyl levulinate from FA is the preferred reaction pathway. The formation of acetal in the alcohols possibly suppresses the aldol condensation which leads to polymer formation.
The transformation of FA to levulinic acid initiates from the hydration and opening of the furan rings.14 Many reactive compounds with the carbonyl group and carbon–carbon double bonds were produced as intermediates.14 In the acidic environment they can easily polymerize via aldol condensation. The UV florescence characterization clearly showed the formation of the extensive conjugated carbon–carbon double bonds (Fig. S1†). In methanol the polymerization of these reactive intermediates were suppressed while the formation of methyl levulinate were promoted. This result is in line with our previous study that methanol plays a crucial role in suppression of polymerization during the acid-catalyzed conversion of C6 sugars like levoglucosan and glucose.9
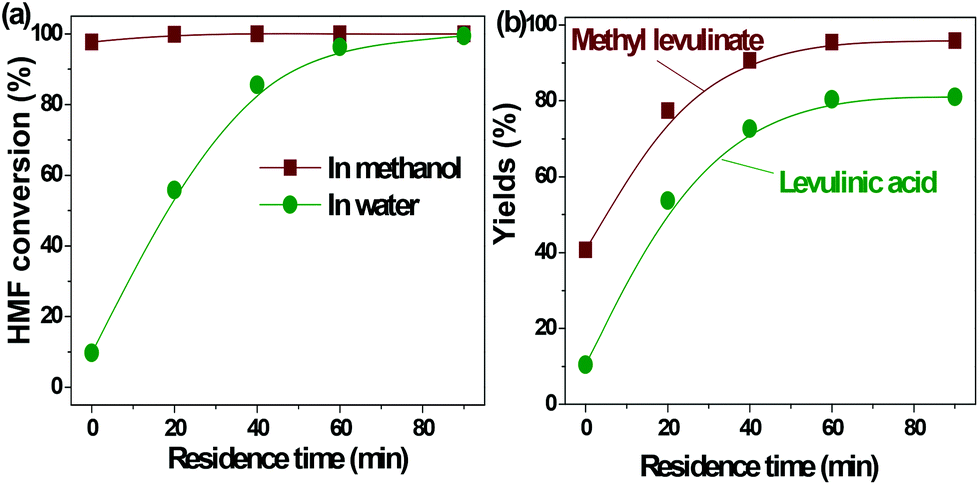 | ||
| Fig. 2 Acid-catalyzed conversion of HMF in water and methanol. Reaction conditions: HMF loaded: 3 g, Amberlyst 70: 3 g, solvent: 100 ml, T = 170 °C, residence time: 90 min. | ||
In our previous study about the conversion of levoglucosan in methanol, we found that HMF reacted with methanol, forming 5-(methoxymethyl)-2-furancarboxadhyde and 2-(dimethoxymethyl)-5-(methoxymethyl)furan.9 It was believed that the conversion of the hydroxyl group to the ether functionality and the carbonyl group to the acetal functionality suppressed the polymerization of HMF. This leads to selective production of methyl levulinate from HMF in methanol. However, the polymerization of HMF in both methanol and water did occur, as evidenced by the formation of a soluble polymer with the progress of the reaction (Fig. S2†), but to a very limited level. In addition, the result here also proves that in water the decomposition of HMF to levulinic acid is the preferred reaction pathway, but not the polymerization.
It has been well documented that the yield of levulinic acid from glucose in water is typically ca. 50% (mol basis).16 The loss of levulinic acid production is due to the polymerization during the dehydration of glucose or HMF or both.16 Here based on the yields of levulinic acid from HMF (ca. 81%) and from glucose (ca. 50%, another experiment under similar conditions), we concluded that: (1) the polymerization of HMF itself contributes to ca. 20% of the loss of levulinic acid production; (2) the polymerization incurred from glucose contributes another ca. 30% to the loss of levulinic acid production, which was calculated by the loss of levulinic acid production directly from glucose (ca. 50%) minus that directly from HMF (ca. 20%); (3) polymerization takes place in both the dehydration of glucose to HMF and the degradation of HMF to levulinic acid, but more significant in the initial dehydration of glucose. Effort should be made to suppress both polymerization in the up-stream dehydration of the sugars and the down-stream degradation of HMF, which are the keys for enhancing levulinic acid production from sugars in water.
Both HMF and FA in water can be converted into levulinic acid, but their tendencies towards polymerization are very different. Although the molecular structures of HMF and FA are different, the intermediates involved during their transformation to levulinic acid have similar structures.14 It has not been understood why FA has a much higher tendency towards polymerization than HMF in water, while methanol or ethanol can stabilize FA and promote the selective conversion of FA to methyl levulinate or ethyl levulinate.
Furfural is the main product from the dehydration of xylose. It is so reactive towards polymerization that methanol and water make almost no difference.18 Polymer is the only main product in either methanol or water (Scheme 1). Furfural itself contains the reactive carbonyl group and the furan ring. Opening of the furan ring of furfural under acidic conditions would produce more reactive intermediates. These intermediates, unlike those from the degradation of HMF, cannot be further converted into the relatively stable levulinic acid. With the prolonged residence time, they have no other way to go but polymerizes as their main destiny.19
Hydrogenation of the furans in water and methanol
 | ||
| Scheme 2 Hydrogenation of furfuryl alcohol in water and methanol. Reaction conditions: furfuryl alcohol loaded: 2 g, Pd/C loaded: 2 g, solvent: 40 ml, T = 170 °C, residence time: 120 min, P = 70 bar. TFA: tetrahydrofurfuryl alcohol; TFME: tetrahydrofurfuryl methyl ether. | ||
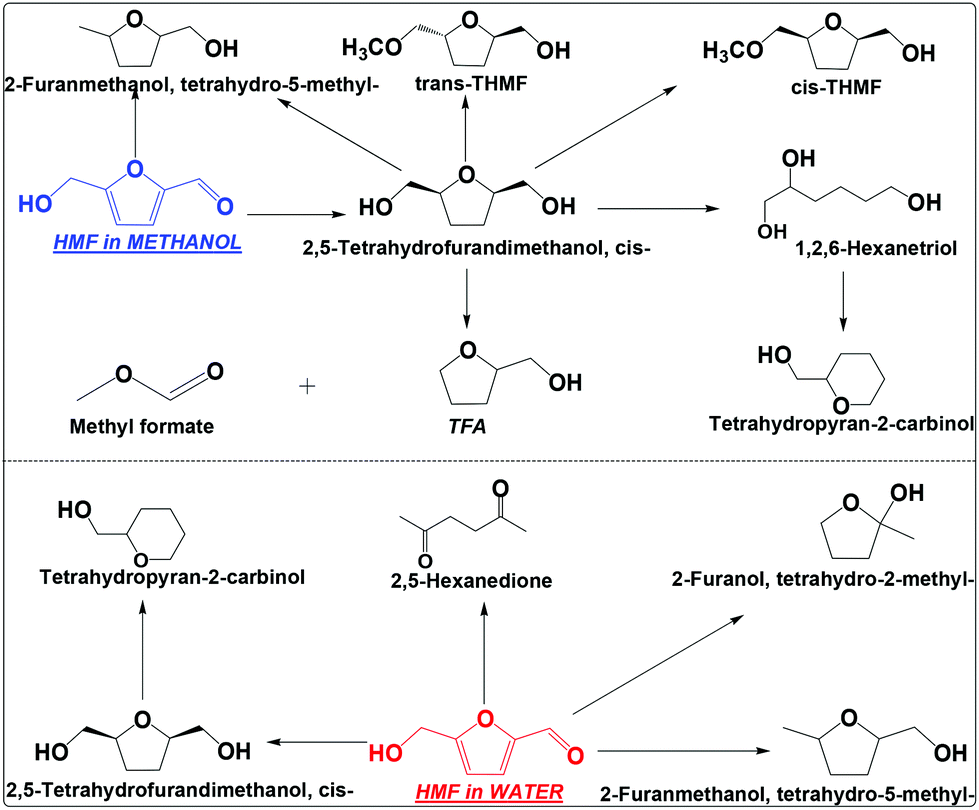 | ||
| Scheme 3 Hydrogenation of HMF in water and methanol. Reaction conditions are the same as that in Scheme 2. THMF: tetrahydro-5-(methoxymethyl)furan-2-yl)methanol; TFA: tetrahydrofurfuryl alcohol. | ||
TFM has at least four ways to go under the conditions employed (Scheme 3), which includes cracking of the hydroxymethyl group to TFA and methyl formate, hydrodeoxygenation of the hydroxyl group to tetrahydro-5-methyl-2-furanmethanol, opening of the furan ring and then dehydrate to tetrahydropyran-2-carbinol, and etherification to tetrahydro-5-(methoxymethyl)furan-2-yl)methanol (THFM, its identification was based on the analysis of its mass spectrum, see Fig. S7 and S8†). Interestingly, THFM has both the trans and cis isomers. During the etherification of cis-TFM, methanol can attack from both sides of the furan ring, forming the isomers. This is different from the hydrogenation of HMF that the molecule accesses the metallic sites from only one side of the furan ring.
In water, TFM is also the main product, but its abundance is much lower than that in methanol (Scheme 3 and Fig. S9†). The selectivity for hydrogenation of HMF is much lower in water than in methanol. This is also evidenced by the formation of 2,5-hexanedione in water, but was not detected in methanol. Clearly, hydrogenation of HMF in methanol is much more efficient in methanol than in water.
 | ||
| Scheme 4 Hydrogenation of furfural in water and methanol. Reaction conditions are the same as that in Scheme 2. | ||
The furan ring and the carbonyl group can be hydrogenated in either water or methanol, producing TFA as the main product. TFA yields are much higher in methanol than in water. This is not even counted in the ether, TFME, formed via etherification of TFA. The polymerization of furfural and hydrogenation of furfural at the elevated temperatures are competitive reactions. Furfural is very reactive towards polymerization due to its reactive carbonyl group and furan ring, while saturation of the carbonyl group and the furan ring eliminates the reactive functional groups and prevent further polymerization. The soluble polymer was not detected in water (Fig. S10†). The low selectivity towards hydrogenation of the furan ring in furfural in water is responsible for the low yields of TFA production. For example, cyclopentanone was formed in water but not in methanol. Its formation in water was probably promoted by the synergistic effect of metal and the protons generated by water at high temperature. In addition, furfural also cracked to butyrolactone in water, while in methanol the products are mainly from the hydrogenation reactions. The selective hydrogenation in methanol favors only the production of the direct hydrogenation product, TFA.
Catalytic conversions of furfural in the presence of both the hydrogenation catalyst (Pd/C) and the acid catalyst (Amberlyst 70) were also performed to understand their competitions (Scheme 5). This is because acid-catalysis and hydrogenation are always coupled during the conversion of furans, sugars and cellulose to the fuel or fuel additives. With only Amberlyst 70, furfural mainly polymerized.18 With only Pd/C, furfural was mainly converted into TFA. With both Amberlyst 70 and Pd/C, the acid-catalysis and hydrogenation were expected to occur in parallel.
 | ||
| Scheme 5 Acid-catalyzed conversion and hydrogenation of furfural in water and methanol. Reaction conditions: furfural loaded: 2 g, Pd/C loaded: 2 g, Amberlyst 70 loaded: 2 g; solvent: 40 ml, T = 170 °C, residence time: 120 min, P = 70 bar. TFA: tetrahydrofurfuryl alcohol. MMHF: 2-(methoxymethyl)tetrahydrofuran. | ||
In methanol, with both Amberlyst 70 and Pd/C as the catalysts it was found that the hydrogenation reactions dominated. TFA is the main product (yield: 44.3%) from the hydrogenation (Scheme 5). Only a very small amount of furfural was partially hydrogenated to 2-(methoxymethyl)furan and further forming trace amounts of methyl levulinate (yield: 0.2%) via acid-catalysis (Scheme 5). The presence of Amberlyst 70 in methanol mainly catalyzes the etherification to form the ethers like 1,5-dimethoxy-pentane.
In water, the hydrogenation and acid-catalysis occurred in parallel. TFA is one of the main products, but the yield (10.6%) is much lower than that in methanol. Levulinic acid yield (8.4%) in water is much higher than the yield of its counterpart (methyl levulinate) in methanol (Scheme 5). This clearly indicates that in water furfural has much more chance to be partially hydrogenated to furfuryl alcohol. In methanol the furan ring and the carbonyl group were hydrogenated very quickly. Furfural has little chance to go to FA and methyl levulinate. Obviously, hydrogenation proceeds very efficiently in methanol even in the presence of a strong solid acid catalyst. In water the hydrogenation is much less efficient. The hydrogenation and the acid-catalysis occur in parallel.
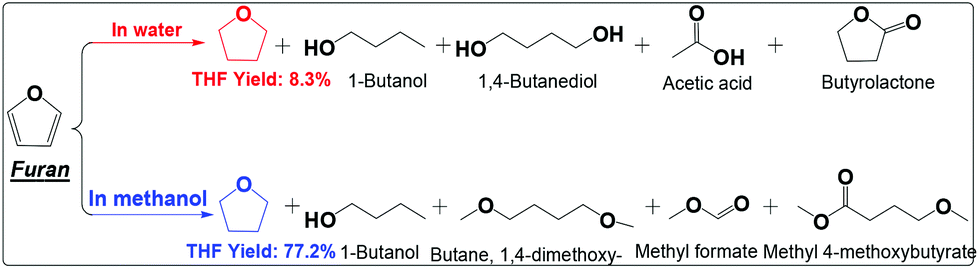 | ||
| Scheme 6 Hydrogenation of furan in water and alcohol. Reaction conditions are the same as that in Scheme 2. | ||
Conclusions
The biomass derived furans behave very differently during their acid-catalyzed conversion in water and in methanol. FA and HMF can be selectively converted into methyl levulinate in methanol. In water they both polymerize but to different extents. Furfuryl alcohol mostly (>80%) polymerizes in water while a much smaller portion of HMF (ca. 20%) polymerizes. Furfural, in comparison, polymerizes either in water or methanol almost completely.For furan, HMF and furfural the hydrogenation is much more efficient in methanol than in water. The acid-catalyzed reactions can even compete with the hydrogenation of furfural in water but not in methanol. The efficiency for the acid-catalysis and hydrogenation of the furans in methanol is much higher or at least equal (for hydrogenation of furfuryl alcohol) to that in water. Hence, methanol or other alcohols are the preferred solvents for the upgrading of furans via both acid-catalysis and hydrogenation.
Experimental section
All the chemicals used in this study are of analytical grade. Commercially available solid acid catalysts, Amberlyst 70 (obtained from Dow Chemical), and Pd/C (obtained from Sigma-Aldrich) were used directly without any further treatment. Acid-catalysis and hydrogenation of the furans were performed in a Hastalloy autoclave reactor (Autoclave Engineers, Division of Snap-Tite Inc.). The procedures for handling the autoclave are similar to that in our previous work.19 The experiments for acid-catalysed conversion of the furans were performed under autonomous vapour pressure generated by the solvent with a stirring rate of 500 rpm. Sampling was performed at certain time intervals to understand progress of the reactions. Mass balance between reactants and collected liquid products was calculated and used to calibrate the yields of the products. Hydrogenation of the furans was performed at 170 °C with the same stirring rate but no sampling was performed. Water or methanol (40 ml) was used as the solvent and loading of the catalyst and furans was specified in the schemes. The hydrogen pressure before heating-up the reactor is 40 bar and was maintained at 70 bar at 170 °C by feeding additional hydrogen, if needed.The products were analysed with an Agilent GC/MS (6890 series GC with a 5973 MS detector) equipped with a HP-INNOWax capillary column. Acetone was used as the solvent to dissolve the samples. The temperature of column was maintained at 40 °C for 1.3 min after injection of the sample, and then was increased to 260 °C at a heating rate of 10 °C min−1 and was maintained at this temperature for 10 min. The detailed procedures and parameters of the instrument are described in the literature.20 The soluble polymers were characterized with a Perkin-Elmer LS50B spectrometer by measuring the size and abundance of the conjugated π bonds in the polymer. Details about the parameters are described in the literature.19 Yield of products is defined by the formula:
| Yields (mol%) = (amount of the product produced/theoretical amount of the product from xylose loaded) × 100% |
Acknowledgements
This Project was supported by the Commonwealth of Australia under the Australia–China Science and Research Fund as well as Curtin University of Technology through the Curtin Research Fellowship Scheme.Notes and references
-
(a) M. J. Climent, A. Corma and S. Iborra, Green Chem., 2014, 16, 516–547 RSC
; (b) D. M. Alonso, S. G. Wettstein, M. A. Mellmer, E. I. Gurbuz and J. A. Dumesic, Energy Environ. Sci., 2013, 6, 76–80 RSC
-
(a) A. S. Mamman, J.-M. Lee, Y.-C. Kim, I. T. Hwang, N.-J. Park, Y. K. Hwang, J.-S. Chang and J.-S. Hwang, Biofuels, Bioprod. Biorefin., 2008, 2, 438–454 CrossRef CAS
-
(a) Y.-T. Cheng and G. W. Huber, Green Chem., 2012, 14, 3114–3125 RSC
-
(a) M. Chidambaram and A. T. Bell, Green Chem., 2010, 12, 1253–1262 RSC
-
(a) S. Dutta, S. De, B. Saha and M. I. Alam, Catal. Sci. Technol., 2012, 2, 2025–2036 RSC
-
(a) J.-P. Lange, R. Price, P. M. Ayoub, J. Louis, L. Petrus, L. Clarke and J. Gosselink, Angew. Chem., Int. Ed., 2010, 122, 4581–4585 CrossRef
- B. Chen, F. Li, Z. Huang, T. Lu, Y. Yuan and G. Yuan, ChemSusChem, 2014, 7, 202–209 CrossRef CAS PubMed
- H.-Y. Zheng, Y.-L. Zhu, B.-T. Teng, Z.-Q. Bai, C.-H. Zhang, H.-W. Xiang and Y.-W. Li, J. Mol. Catal. A: Chem., 2006, 246, 18–23 CrossRef CAS
-
(a) X. Hu and C.-Z. Li, Green Chem., 2011, 13, 1676–1679 RSC
- X. Hu, Y. Song, L. Wu, M. Gholizadeh and C.-Z. Li, ACS Sustainable Chem. Eng., 2013, 1, 1593–1599 CrossRef CAS
-
(a) D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493–1513 RSC
- G. M. G. Maldonado, R. S. Assary, J. A. Dumesic and L. A. Curtiss, Energy Environ. Sci., 2012, 5, 6981–6989 Search PubMed
-
(a) G. M. G. Maldonado, R. S. Assary, J. A. Dumesic and L. A. Curtiss, Energy Environ. Sci., 2012, 5, 8990–8997 RSC
- J. Horvat, B. Klaić, B. Metelko and V. Sunjic, Tetrahedron Lett., 1985, 26, 2111–2114 CrossRef CAS
-
(a) J. Song, H. Fan, J. Ma and B. Han, Green Chem., 2013, 15, 2619–2635 RSC
-
(a) H. Heeres, R. Handana, D. Chunai, C. B. Rasrendra, B. Girisuta and H. J. Heeres, Green Chem., 2009, 11, 1247–1255 RSC
- P. Lejemble, A. Gaset and P. Kalck, Biomass, 1984, 4, 263–274 CrossRef CAS
- X. Hu, C. Lievens and C.-Z. Li, ChemSusChem, 2012, 5, 1427–1434 CrossRef CAS PubMed
-
(a) A. S. Dias, S. Lima, M. Pillinger and A. A. Valente, Carbohydr. Res., 2006, 341, 2946–2953 CrossRef CAS PubMed
-
(a) X. Hu, Y. Wang, D. Mourant, R. Gunawan, C. Lievens, W. Chaiwat, M. Gholizadeh, L. Wu, X. Li and C.-Z. Li, AIChE J., 2013, 59, 888–900 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4gc01826e |
| This journal is © The Royal Society of Chemistry 2015 |