
Intravenous injection of L-BMAA induces a rat model with comprehensive characteristics of amyotrophic lateral sclerosis/Parkinson–dementia complex†
Ke-Wei
Tian
a,
Hong
Jiang
b,
Bei-Bei
Wang
c,
Fan
Zhang
a and
Shu
Han
*a
aInstitute of Anatomy and Cell Biology, Medical College, Zhejiang University, Hangzhou 310058, China. E-mail: han00shu@zju.edu.cn; Fax: +86-571-88208094; Tel: +86-571-88208160
bDepartment of Electrophysiology, Sir Run Run Shaw Hospital, Medical College, Zhejiang University, Hangzhou 310058, China
cCore Facilities, Zhejiang University School of Medicine, Hangzhou 310058, China
First published on 10th November 2015
Abstract
Non-protein amino acid beta-N-methylamino-L-alanine (L-BMAA) is a neurotoxin that was associated with the high incidence of Amyotrophic Lateral Sclerosis/Parkinson–Dementia Complex (ALS/PDC) in Guam. This neurotoxin has been implicated as a potential environmental factor in amyotrophic lateral sclerosis, Alzheimer's disease and other neurodegenerative diseases, and was found to accumulate in brain tissues of ALS/PDC patients. It is extremely important to establish a reliable animal model that has the comprehensive characteristics of ALS/PDC for studying mechanisms underlying neurodegeneration, and exploring effective therapies. However, very few good animal models that mimic ALS/PDC have been established. In this study, an ideal rat model that mimicked most characteristics of ALS/PDC was established by administering continuous intravenous (i.v.) injections of neurotoxic L-BMAA. Based on the data obtained, it was demonstrated that continuous i.v. injections of L-BMAA induced mitochondrial morphology and structural changes, astrogliosis, motor neuronal death, and other relative functional changes, which led to the overexpression of pro-inflammatory cytokines cyclooxygenase-2 (COX-2), nuclear factor kappa B (NF-κB) and tumor necrosis factor-alpha (TNF-α), and resulted in the upregulation of glycogen synthase kinase-3 (GSK3), downregulation of astrocytic glutamate transporter-1 (GLT-1), accumulation of microtubule-associated protein tau and cytosolic aggregates of TAR DNA-binding protein-43 (TDP-43) in degenerating motor neurons. These results suggest that this model could be used as a useful tool for the mechanistic and therapeutic study of ALS/PDC.
Introduction
Amyotrophic lateral sclerosis (ALS) is the most common motor neuronal disease characterized by upper and lower motor neuron degeneration.1 Previous studies have revealed that approximately 8–10% of all cases are familial cases, and approximately half of these cases are characterized by superoxide dismutase mutation (SOD-1), while the other 90–92% of all ALS cases occur sporadically with no known familial history.2 Previous studies have examined the relationship between genetic and environmental risk factors, and proposed that ALS can be considered to be the result of environmental risks and time acting on a pre-existing genetic load, followed by an automatic, self-perpetuating decline to death.3 At present, there is no known exact cause of sporadic ALS. Smoking, electromagnetic radiation, pesticides and heavy metals such as lead and mercury, possible exposure to formaldehyde, and head trauma are all suspected to be possible reasons.2,3 The cyanobacteria-derived neurotoxin L-BMAA continues to attract attention due to its ability to cause neurodegeneration, which provides support for its potential role in the etiology of sporadic ALS.2 Cox and colleagues have demonstrated that L-BMAA in cycad flour is mainly concentrated in proteins.2L-BMAA is a neurotoxic amino acid produced by cyanobacteria and diatoms. Since its discovery in 1967 by Vega and Bell, its toxicity has been associated with Amyotrophic Lateral Sclerosis/Parkinson–Dementia Complex (ALS/PDC) in the Isle of Guam.4 The consumption of cycad flour, flying foxes, and other animals that fed on cycad seeds by the indigenous Chamorro people led to the bio-concentration of protein-bound L-BMAA up the food chain, which resulted in the accumulation of L-BMAA in the brains of Chamorro patients with ALS/PDC (mean concentration of L-BMAA was 627 μg g−1).2–7 Notably, several studies have supported the relationship between L-BMAA and sporadic ALS.8,9 Beyond this, the research in Guam suggests that ALS/PDC complex was also due to L-BMAA contained in the seeds of the Cycas micronesica and consumed by the Chamorro there.10 Both Guamanian ALS and sporadic ALS could be caused by the oral,11 sc,12 I.V.,5 I.P.13 or intrathecal14 administration of L-BMAA.However, despite similar causes and clinical similarities, Guamanian ALS lacks the glutamate deficiency and taurine excess characteristic of sporadic ALS, which may lead to some histological difference between Guamanian ALS and typical sporadic ALS.15
In ALS studies, mutant SOD1 transgenic mice with overexpressed human mutant SOD1 have been used to study familial ALS, because they could develop adult-onset progressive muscle weakness and atrophy caused by prominent motor neuron degeneration.1,5 Previous studies have attempted to produce pathologies of ALS/PDC by consecutive intraperitoneal injection or intrathecal infusion of L-BMAA,13,14 which has partially produced ALS/PDC characteristic disorders.
In the present study, we chose to carry out continuous intravenous (i.v.) injections of L-BMAA to establish a rat model with ALS/PDC-like phenotypes. Intravenous injection of L-BMAA can deliver neurotoxins directly and quickly compared to intraperitoneal injections, and is more affordable compared to intrathecal infusion via osmotic mini-pumps. Multiple behavioral and neurological evaluations, histological and immunohistochemical staining, western blotting, electron microscopy observation, and electrophysiological tests were adopted to assess endoplasmic reticulum stress, neurodegeneration, changes of relative expression factors, astrogliosis, and functional neuronal loss related to injured and degenerating motor neurons (MNs) in motor cortices and the anterior horn of the spinal cord. We hope that this animal model may provide a useful tool for studying sporadic ALS/PDC in the future.
Results
L-BMAA injections led to functional disabilities and electrophysiological changes
Two or three weeks after L-BMAA injections, treated rats began to exhibit functional disabilities compared with normal controls. In the rotarod test (Fig. 1A), the length of time that L-BMAA-treated rats stayed on the device was significantly shorter than in controls (P < 0.05). Tilt plane angles, on which L-BMAA-treated rats stood on, also remarkably declined (P < 0.05, Fig. 1B). Meanwhile, the average grip strength of forepaws of L-BMAA-treated rats significantly decreased (P < 0.05, Fig. 1C), and L-BMAA-treated rats had much more limb loss in the beam walking test compared with normal controls at each time point (P < 0.05, Fig. 1D). These results suggest the evident loss of muscle force both in forepaws and hind paws. Moreover, fear conditioning test results revealed that L-BMAA treatment could significantly reduce freezing behavior compared with normal controls at each trial time point (P < 0.05, Fig. 1E), suggesting memory deterioration induced by L-BMAA toxicity.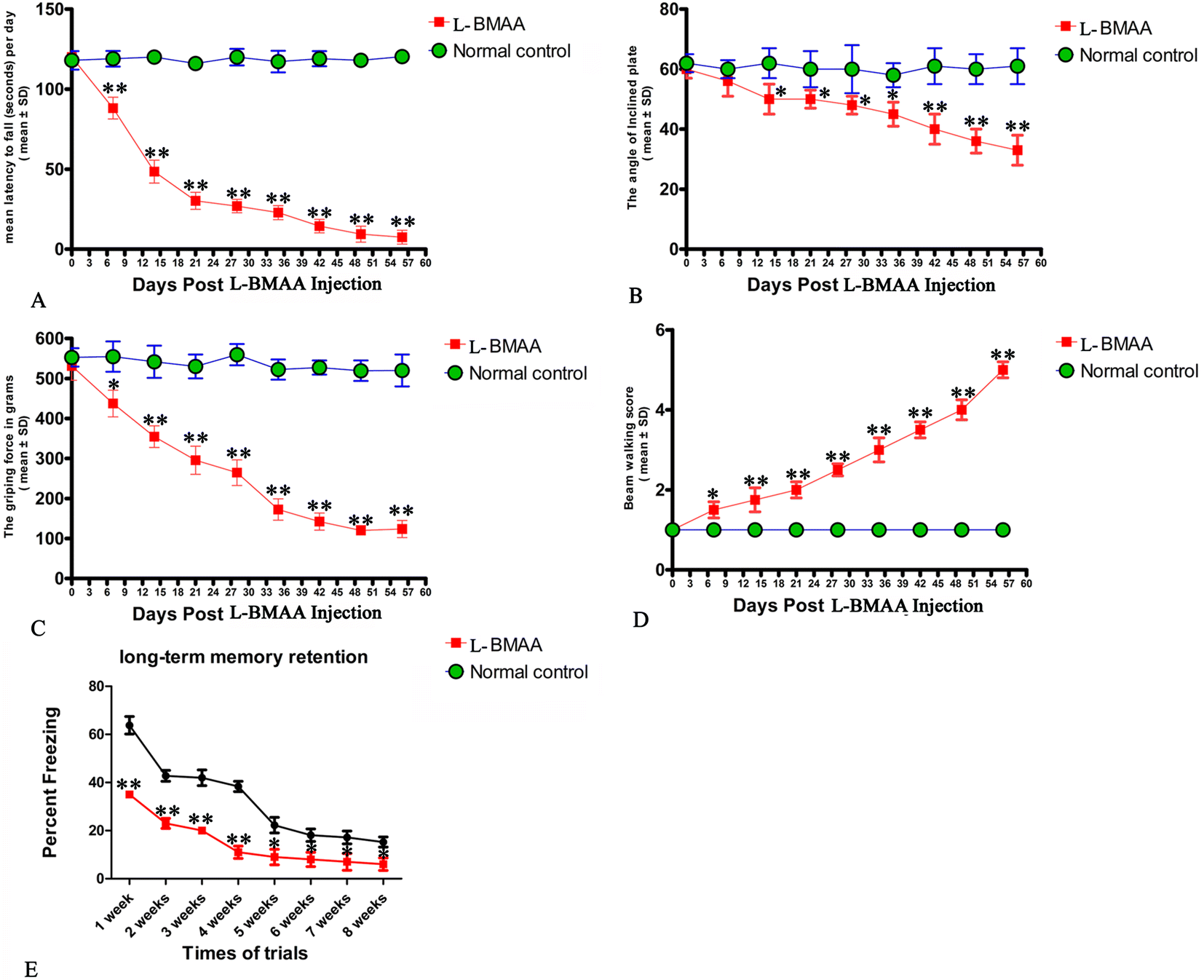 | ||
| Fig. 1 Motor functional assessment revealed neurological disabilities in L-BMAA-treated rats. (A) Rotarod test: the length of time L-BMAA-treated rats stayed on the device was significantly shorter compared to their respective controls. (B) Inclined plane test: tilt plane angles, on which L-BMAA-treated rats stood on, remarkably declined from the second week after L-BMAA injection. (C) Average grip strength of forepaws of L-BMAA-treated rats obviously decreased compared with controls at each time point. (D) Beam-walking test: average beam walking score has shown that L-BMAA-treated rats had more limb losses in the beam walking test compared with controls at each time point. (E) Fear conditioning: L-BMAA treatment obviously reduced freezing behaviors compared with controls at each trial time point. *P < 0.05 and **P < 0.01 vs. the control group. | ||
In terms of clinical severity revealed by the behavior tests above, electrophysiological examinations revealed pathological changes in nerve conduction. At each time point, MEP amplitude (related to the number of surviving fibers) became significantly lower and latency (related to the speed of conduction) was significantly prolonged, compared with normal controls; especially within 4–8 weeks post L-BMAA injections (P < 0.05, ESI Fig. 1† and Table 1). Furthermore, the mean amplitude of MUPs increased, the mean duration was prolonged, and the percentage of polyphasic potentials increased much more than in the control group (>120%; ESI Fig. 2† and Table 2), all of which indicate the destruction of muscular innervation.
| Group | Latency (ms) | Wave amplitude (μV) |
|---|---|---|
| Data are expressed as mean ± SD. *P < 0.05 and **P < 0.01 vs. the control group. | ||
| Control | 4.95 ± 1.91 | 10.52 ± 1.83 |
| L-BMAA 0 day | 5.64 ± 1.54 | 5.72 ± 0.88* |
| L-BMAA 1 week | 6.98 ± 0.55* | 1.1 ± 0.35** |
| L-BMAA 2 weeks | 6.58 ± 0.5* | 0.9 ± 0.12** |
| L-BMAA 3 weeks | 6.55 ± 0.69* | 0.23 ± 0.11** |
| L-BMAA 4 weeks | 7.41 ± 0.35** | 0.45 ± 0.13** |
| L-BMAA 5 weeks | 7.5 ± 0.52** | 0.4 ± 0.1** |
| L-BMAA 6 weeks | 7.65 ± 0.15** | 0.3 ± 0.12** |
| L-BMAA 7 weeks | 7.8 ± 0.83** | 0.2 ± 0.1** |
| L-BMAA 8 weeks | 14.59 ± 1.3** | 0.12 ± 0.1** |
| Group | Wave amplitude (μV) | Duration (ms) | Rate of polyphasic action potential (%) |
|---|---|---|---|
| Data are expressed as mean ± SD. *P < 0.05 and **P < 0.01 vs. the control group. | |||
| Control | 252 ± 19.63 | 8.9 ± 1.24 | 15 ± 3.2% |
| L-BMAA 0 day | 263 ± 24.56 | 8.3 ± 1.51 | 14 ± 2.75% |
| L-BMAA 1 week | 287 ± 28.79 | 10 ± 1.98* | 18 ± 3.42% |
| L-BMAA 2 weeks | 319 ± 35.58* | 9.8 ± 2.63* | 22 ± 3.69%* |
| L-BMAA 3 weeks | 343 ± 28.64* | 9.7 ± 3.15* | 25 ± 3.91%* |
| L-BMAA 4 weeks | 358 ± 21.84** | 9.92 ± 1.86* | 23 ± 3.67%* |
| L-BMAA 5 weeks | 409 ± 33.72** | 12.7 ± 2.83** | 30 ± 3.82%* |
| L-BMAA 6 weeks | 462 ± 41.26** | 11.5 ± 3.91** | 39 ± 5.35%** |
| L-BMAA 7 weeks | 473 ± 39.88** | 12.9 ± 4.31** | 41 ± 4.23%** |
| L-BMAA 8 weeks | 548 ± 30.16** | 15.8 ± 2.57** | 58 ± 3.59%** |
L-BMAA injections induced the expression of pro-inflammatory factors, microglia activation and reactive gliosis
Expression levels of pro-inflammatory factors (NF-κB, COX-2 and TNF-α) and CD68, a marker for activated microglia and extravasated macrophages, were detected by western blotting (Fig. 2) and immunostaining (Fig. 3). In L-BMAA-treated rats, we found that COX-2, NF-κB and TNF-α pro-inflammatory cytokine expression significantly increased (P < 0.05); and CD68 expression was also upregulated by L-BMAA injections at early stages, which reached a peak at four weeks and significantly decreased at eight weeks post injection (P < 0.05, Fig. 2).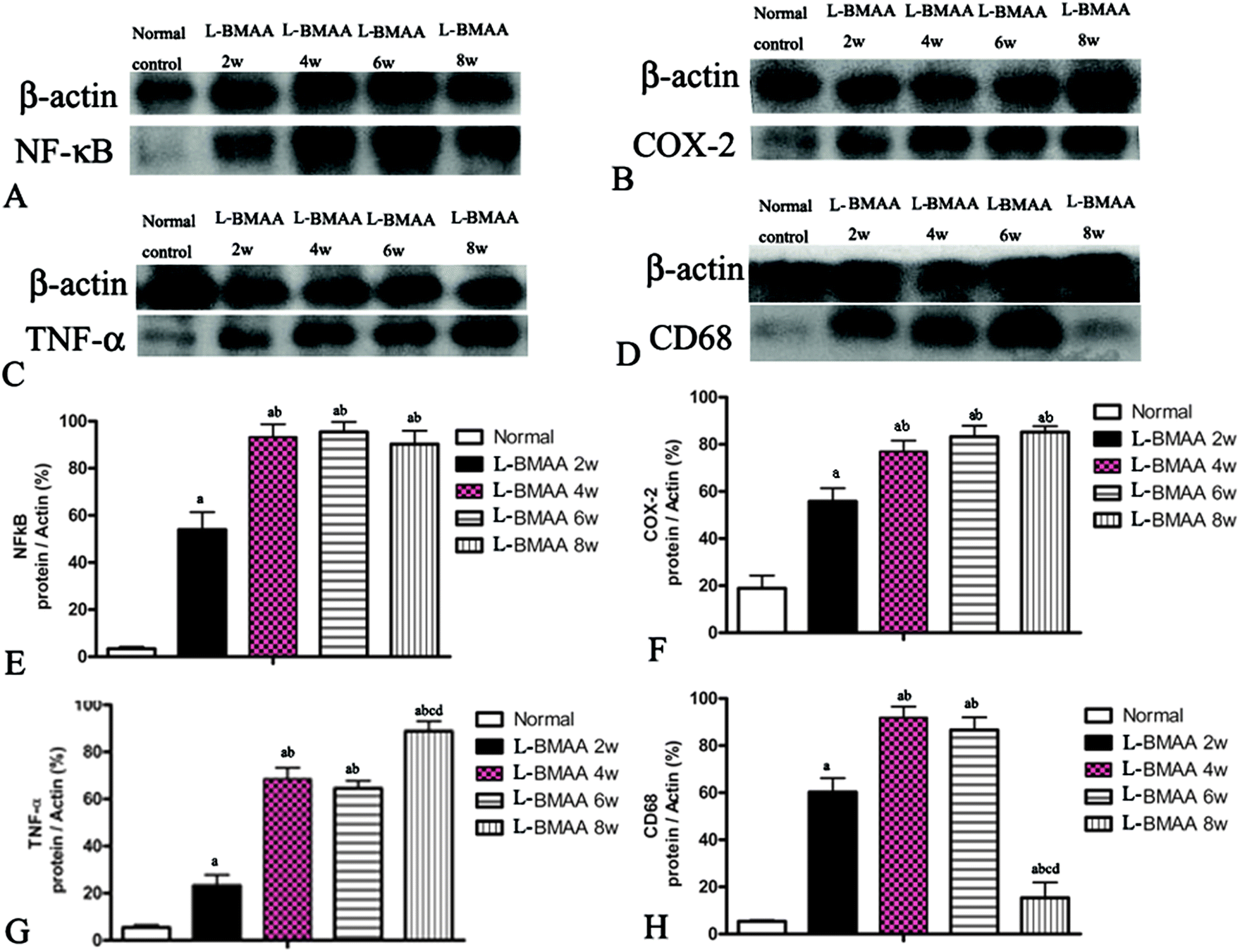 | ||
| Fig. 2 NF-κB, COX-2, TNF-α and CD68 expression levels were altered by L-BMAA treatment, as detected by western blotting. (A and B) NF-κB and COX-2 expression levels were upregulated by L-BMAA injection, especially at 4–8 weeks post treatment. (C) TNF-α expression was also upregulated by L-BMAA injection, and reached a peak at eight weeks post treatment. (D) CD68 expression was upregulated by L-BMAA injection, which reached a peak at four weeks and evidently decreased at eight weeks post injection. (E–H) A–D images were quantitated as described in the Experiments section: (a) P < 0.05 vs. the control group, (b) P < 0.05 vs.L-BMAA-treated rats at two weeks following injections, (c) P < 0.05 vs.L-BMAA-treated rats at four weeks following injections, (d) P < 0.05 vs.L-BMAA-treated rats at six weeks following injections. | ||
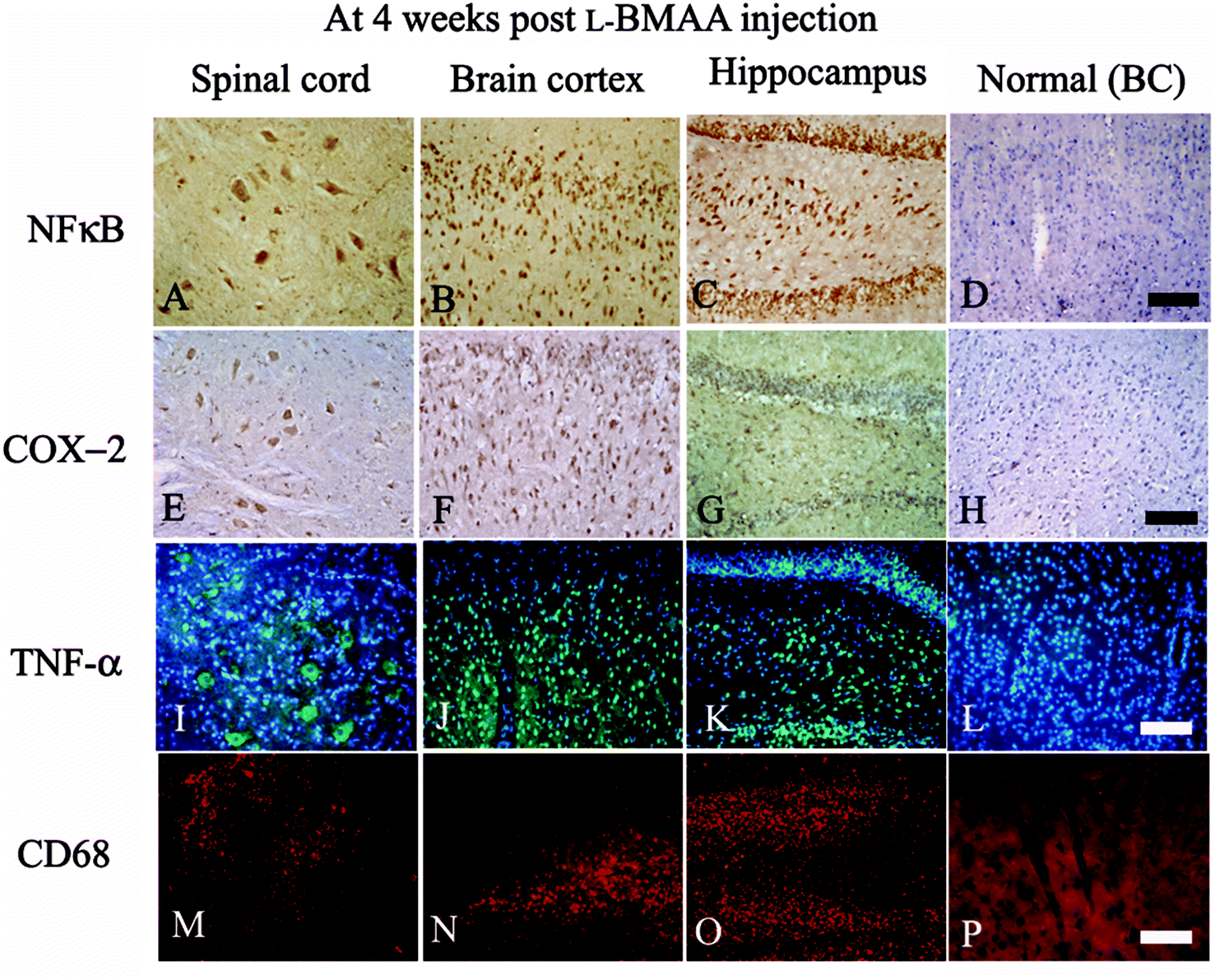 | ||
| Fig. 3 At four weeks following L-BMAA injections, NF-κB (A–C, immunostaining), COX-2 (E–G, immunostaining), TNF-α (I–K, green color immunofluorescence staining, nuclei of all cells were stained with blue color Hoechst 33342) and CD68 (M–O, red color immunofluorescence staining) exhibited visible expression levels in the lumbar spinal cord, brain motor cortex and hippocampus, when compared with the normal control (D, H, L and P). BC: brain cortex. Scale bar = 100 μm. | ||
To assess whether L-BMAA injections could promote reactive gliosis, we examined the expression of GFAP, a specific marker for astrocytes. Abnormal proliferated astrocytes were detected at 2–4 weeks post injections of L-BMAA (Fig. 4C–F), and reactive astrogliosis formed notable glial scarring at 6–8 weeks post injections (Fig. 4G–J). Western blotting results also revealed that GFAP expression was upregulated in L-BMAA-treated rats (Fig. 4K and L).
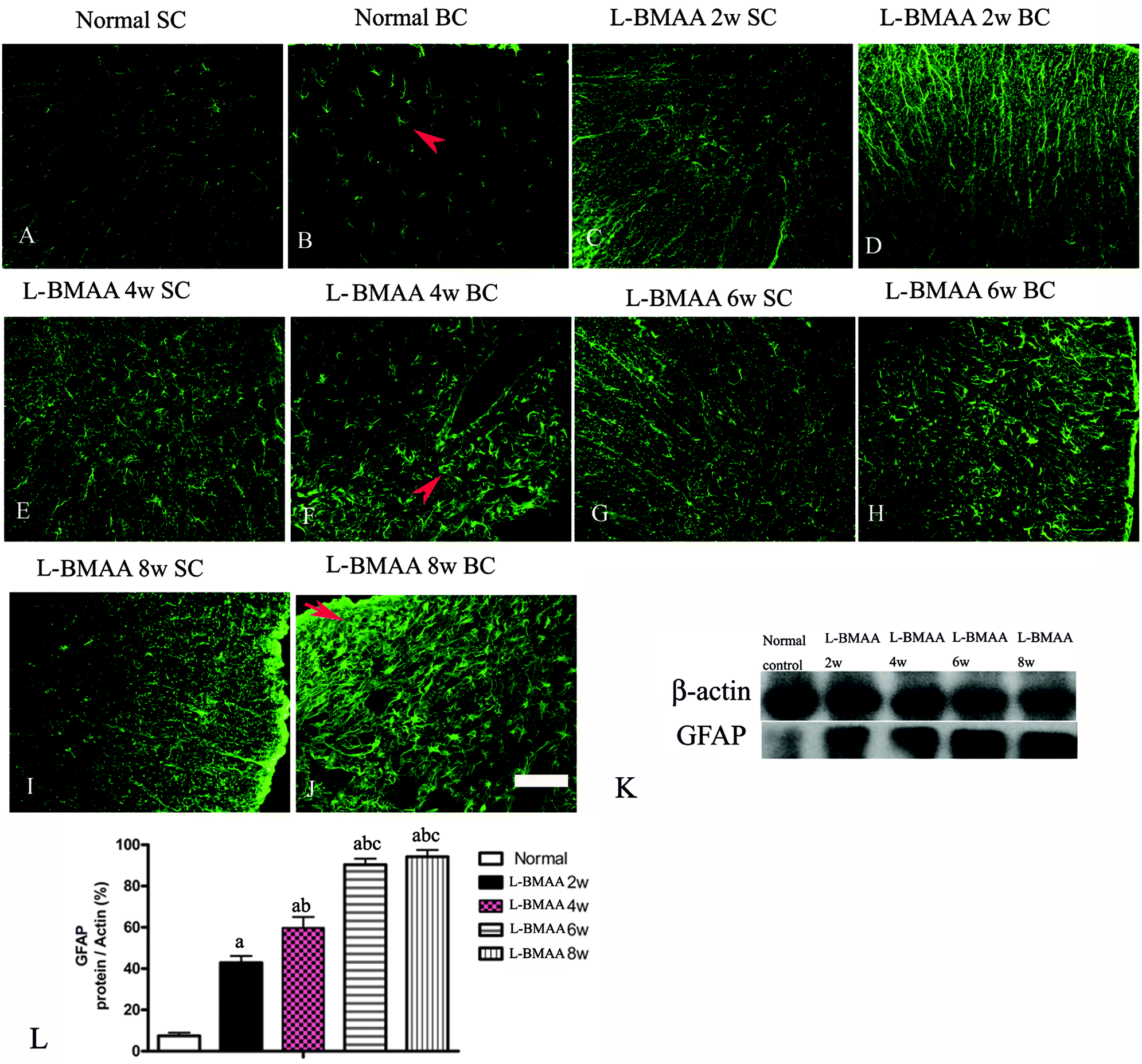 | ||
| Fig. 4 (A–J) L-BMAA-treated rats revealed evident reactive gliosis by FITC-conjugated GFAP immunofluorescence staining. SC: spinal cord; BC: brain cortex. Scale bar = 100 μm. Arrows in (B) show normal astrocytes, (F) show abnormal proliferated astrocytes, and (J) show that reactive astrogliosis forms glial scarring. (K–L) GFAP expression levels were upregulated in L-BMAA-treated rats as shown by western blotting; (a) P < 0.05 vs. the control group; (b) P < 0.05 vs.L-BMAA-treated rats at two weeks following injections; (c) P < 0.05 vs.L-BMAA-treated rats at four weeks following injections. | ||
L-BMAA injections increased GSK3β and tau expression, promoted the translocation of the cytosolic TDP-43 protein, and reduced the expression of GLT-1
Since glycogen synthase kinase-3β (GSK3β) could play a role in some neuropathological processes,13 we analyzed the expression levels of GSK3β in L-BMAA-treated rats. Our results indicate an increase in total GSK3β levels in the lumbar spinal cord, motor cerebral cortex and hippocampus, especially at the late stage of ALS/PDC induction (Fig. 5A and B; ESI Fig. 3A–D†).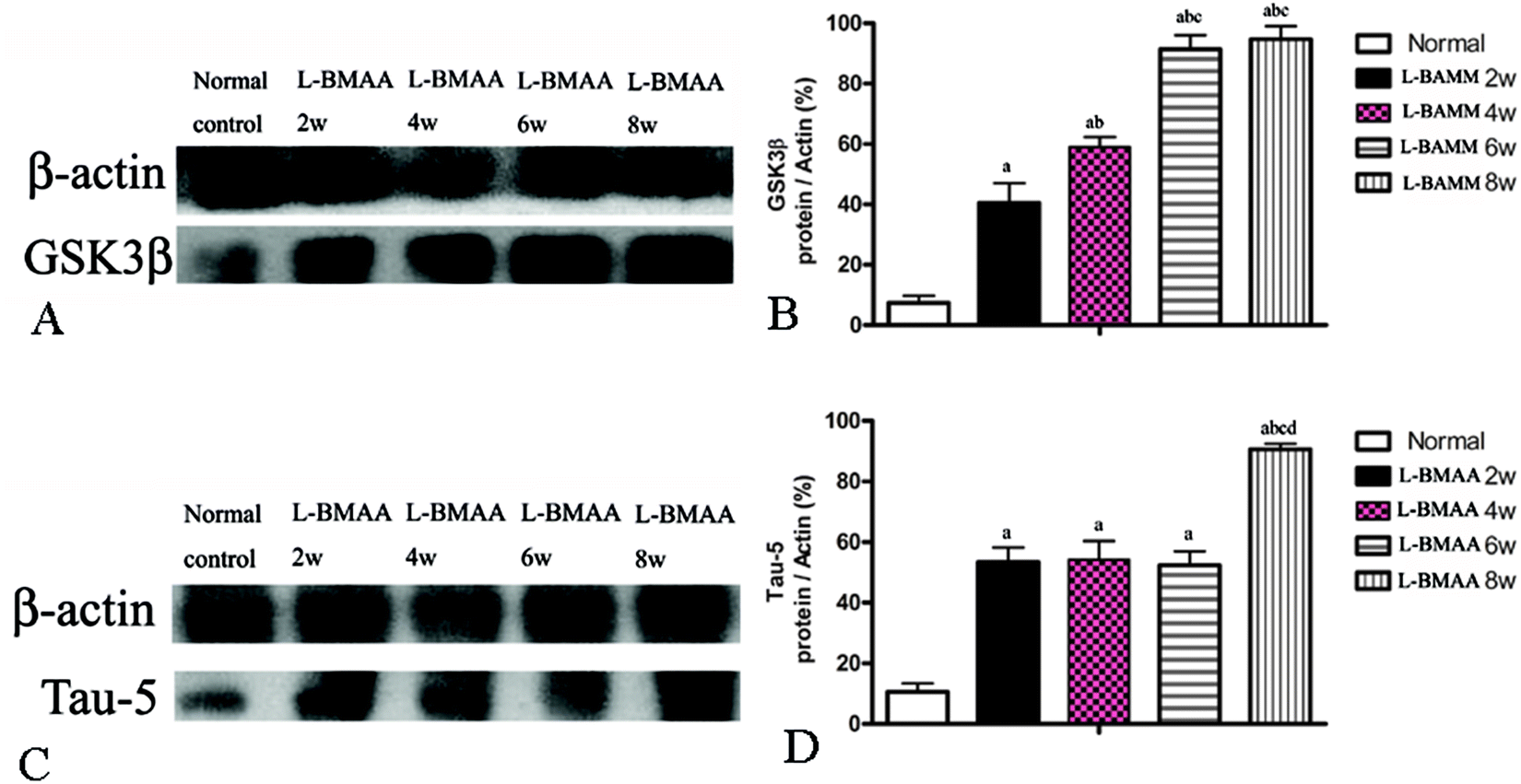 | ||
| Fig. 5 (A and B) GSK3β expression was upregulated as revealed by western blotting. (C and D) The expression of tau-5 was upregulated as revealed by western blotting; (a) P < 0.05 vs. the control group; (b) P < 0.05 vs.L-BMAA-treated rats at two weeks following injections; (c) P < 0.05 vs.L-BMAA-treated rats at four weeks following injections; (d) P < 0.05 vs.L-BMAA-treated rats at six weeks following injections. | ||
The accumulation of microtubule-associated protein tau (MAPT) aggregates correlates well with neurodegeneration in many diseases. Our results revealed that L-BMAA injection could increase tau-5 expression in neurons of the anterior horn of the spinal cord (ESI Fig. 3E–H†). Western blotting also confirmed the increased expression of tau proteins (Fig. 5C and D).
Current ALS research has focused on the role of TAR DNA-binding protein 43 (TDP-43) in neurodegeneration.13,14 TDP-43 expression is normally restricted to the nucleus, as shown in MNs in the control group (Fig. 6B and D). At 2–4 weeks post L-BMAA injection, loss of the distinct nuclear expression of TDP-43 accompanied by the appearance of some cytosolic staining was noted in a fraction of degenerating MNs (Fig. 6E–H). A significant loss of TDP-43 in the nucleus of damaged MNs was revealed at 6–8 weeks post L-BMAA injection, and the nucleus exhibited a bulge and vacuolar appearance (shown by arrows in Fig. 6I–L). In the hippocampus, neurons also displayed visible disruptions of homogeneous nuclear labeling and accumulation of cytosolic aggregates of TDP-43 in L-BMAA-treated rats (arrows in Fig. 6O and P), compared with normal controls (Fig. 6M).
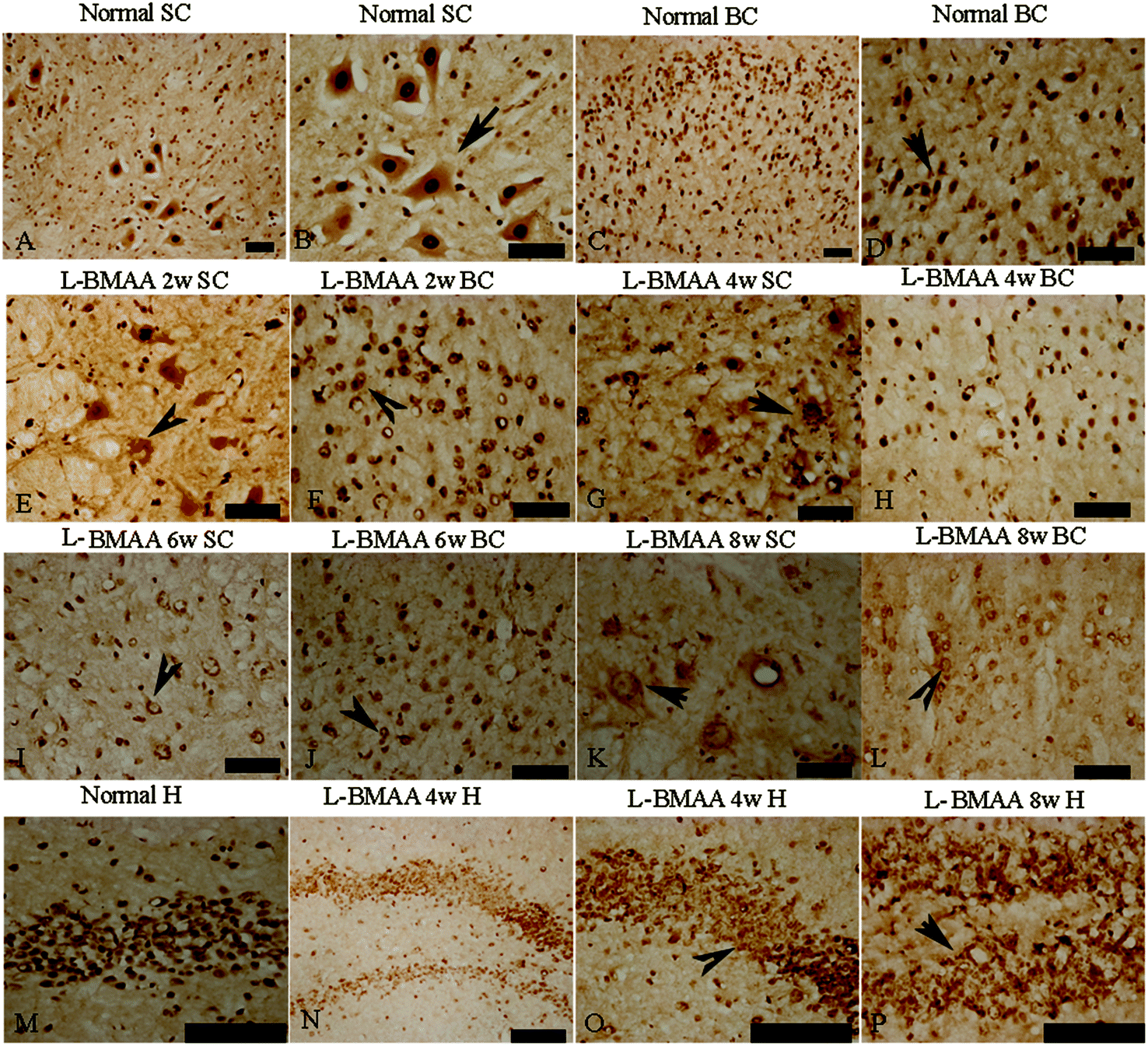 | ||
| Fig. 6 L-BMAA injections induced the appearance of cytosolic TDP-43 immunoreactive aggregates in motor neurons (MNs) of the spinal cord and brain motor cortex (A–L), and neurons in the hippocampus (M–P); scale bar = 100 μm. (A–D) TDP-43 expression is normally quite strongly restricted in the nucleus (shown by the arrow), as in the MNs of rats in the control group; (E–H) at 2–4 weeks post L-BMAA injections, loss of distinct nuclear expression of TDP-43 accompanied by the appearance of some cytosolic staining was noted in a fraction of degenerating MNs; (I–L) obvious loss of TDP-43 in the nucleus of damaged MNs was revealed at 6–8 weeks post L-BMAA injections (shown by the arrow), and the nucleus exhibited a bulge and vacuolar appearance. (M–P) In the hippocampus, neurons also displayed a clear disruption of homogeneous nuclear labeling and accumulation of cytosolic aggregate signs (arrows in O and P), compared with the control group (M). | ||
GLT-1 is abundantly distributed in the central nervous system (CNS). Downregulation of GLT-1 was reported in various neurological diseases.14 In our study, an obvious decrease in GLT-1 expression at eight weeks post injections was revealed by western blotting (Fig. 7).
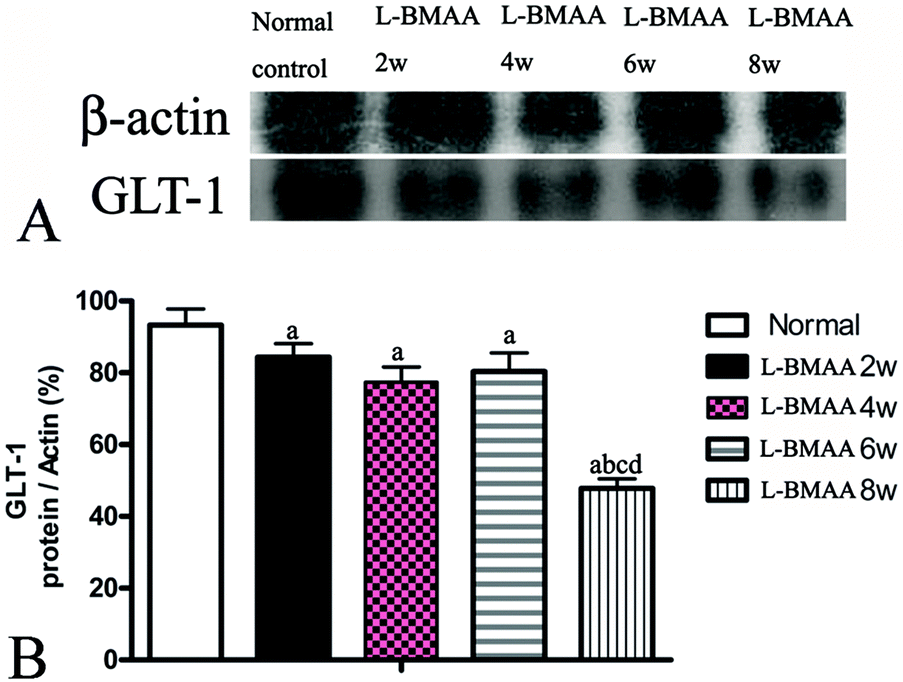 | ||
| Fig. 7 L-BMAA injections reduced GLT-1 expression, especially at eight weeks post treatment, as detected by western blotting; (a) P < 0.05 vs. the control group; (b) P < 0.05 vs.L-BMAA-treated rats at two weeks following injections; (c) P < 0.05 vs.L-BMAA-treated rats at four weeks following injections. | ||
L-BMAA injections induced fragmentation of neurofilaments and axonal loss
Bielschowsky's silver staining revealed the sparseness of neurofilament density in L-BMAA-treated rats (Fig. 8), and a significant loss of axons was detected four weeks post L-BMAA treatment (Fig. 8G–I). Loss of dendrites in neurons at six weeks post L-BMAA injections (Fig. 8J–L) displayed deformed ovoid formations with a swelling nucleus (arrows in Fig. 8J and K). Finally, at eight weeks post-injections (Fig. 8M–O), the normal structure of neurons, especially in the anterior horn of the spinal cord, completely disappeared; while neurons presented a bulge and necrotic appearance with only a few remaining neurofilaments (arrow in Fig. 8N). Moreover, SMI-32 immunolabeling, which could recognize non-phosphorylated epitopes of neurofilament proteins necessary for maintaining large neurons with highly myelinated processes, revealed widespread fragmentation of dendritic processes and the absence of SMI-32 staining in both somatic and dendrites of MNs in L-BMAA-treated rats (Fig. 9). Visibly swollen MNs (arrows in Fig. 9B and C), atrophy (arrow in Fig. 9D), and significant loss of SMI-32 staining (arrow in Fig. 9E) were also observed.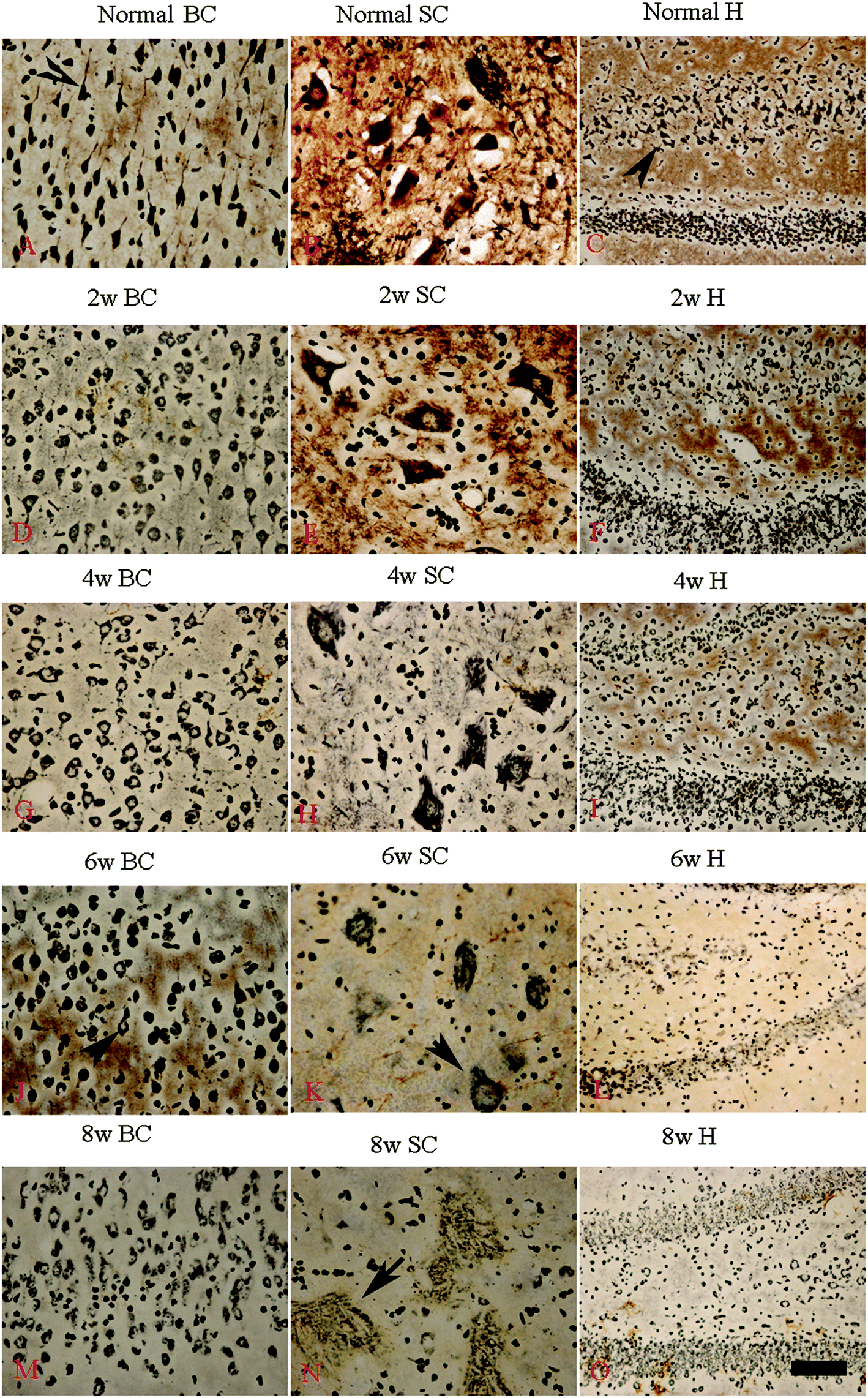 | ||
| Fig. 8 Bielschowsky's silver impregnation of axons was used to measure axonal density in neurons of the spinal cord, brain motor cortex and hippocampus. SC: spinal cord; BC: brain cortex; H: hippocampus; scale bar = 100 μm. (A–C) Bielschowsky's silver staining method non-specifically stains normal neurons including pyramidal cells in the brain motor cortex and hippocampus, showing a typical regular and triangular shape (arrows in A and C); and large motor neurons (MNs) in the anterior horn of the spinal cord (B). Density of neurofilaments began to become sparse from the second week after L-BMAA injections (D–F), and significant loss of axons was detected at four weeks post L-BMAA treatment (G–I). Loss of neurites in neurons at six weeks post L-BMAA injections (J–L) displayed deformed ovoid-shaped forms with a swelling nucleus (arrows in J and K); and finally, at eight weeks post injection (M–O), the normal structure of neurons especially in the anterior horn of the spinal cord completely disappeared, and neurons presented a dissolving necrotic appearance with only a few remaining neurofilaments (arrow in N). | ||
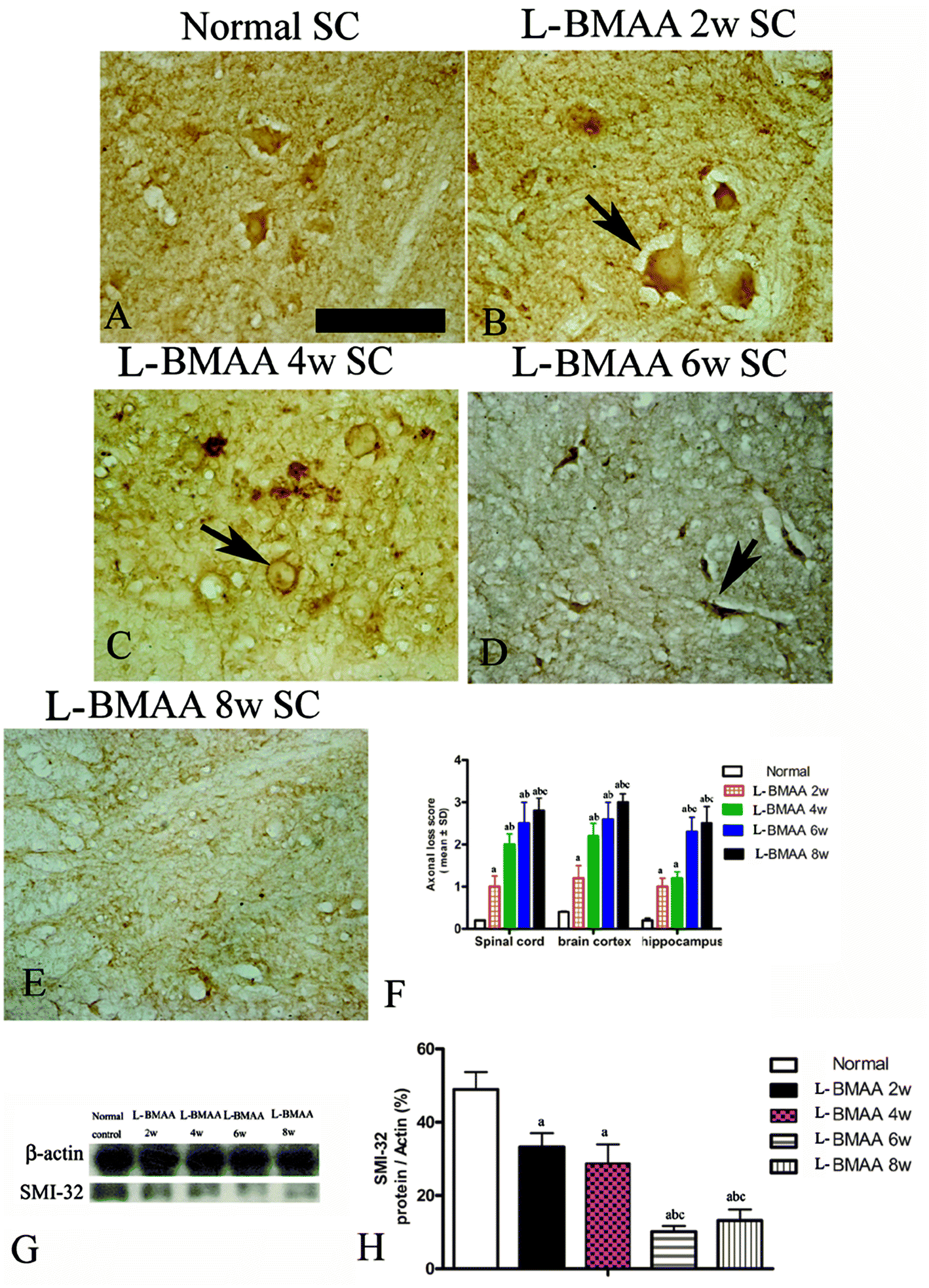 | ||
| Fig. 9 Morphological changes were detected in lumbar ventral horn motor neurons (MNs). SMI-32 immunolabeling, which recognizes non-phosphorylated epitopes of neurofilament proteins necessary for maintaining large neurons with highly myelinated processes, could provide visualization of changes in somatic and dendritic morphology, and show structural damage. Scale bar = 100 μm. (A) MNs of control rats; (B) arrow indicates a MN with swelling appearance; (C) widespread fragmentation of dendritic processes and the absence of SMI-32 staining both in somatic and dendrites of MNs as indicated by an arrow; (D) MN atrophy (arrow) was found; (E) significant loss of SMI-32 staining was revealed at eight weeks post L-BMAA injections; (F) the condition of axonal loss was determined by axonal loss scoring; (G–H) SMI-32 expression was downregulated in L-BMAA-treated rats, especially at 6–8 weeks post injections, as shown by western blotting; (a) P < 0.05 vs. the control group; (b) P < 0.05 vs.L-BMAA-treated rats at two weeks following injections; (c) P < 0.05 vs.L-BMAA-treated rats at four weeks following injections. | ||
L-BMAA injections induced mitochondrial degeneration, demyelination, as well as neuronal apoptosis and loss
Electron micrograph examination results revealed disorganized mitochondrial changes such as vacuolization and swollen cristae (arrows in Fig. 10F, H and I); and the myelin sheath of neurofibers displayed splitting, vacuoles, and loose and fused changes (Fig. 10E, G and I). At the late stage, some mitochondria were totally degenerated, and their cristae completely disappeared (Fig. 10J). More severe demyelination changes and axonal loss were also detected (Fig. 10K). Neurons showed apoptotic signs with a shrunken nucleus; and a condensed, fragmented and marginated nuclear chromatin at the late stage of disease induction (Fig. 10L).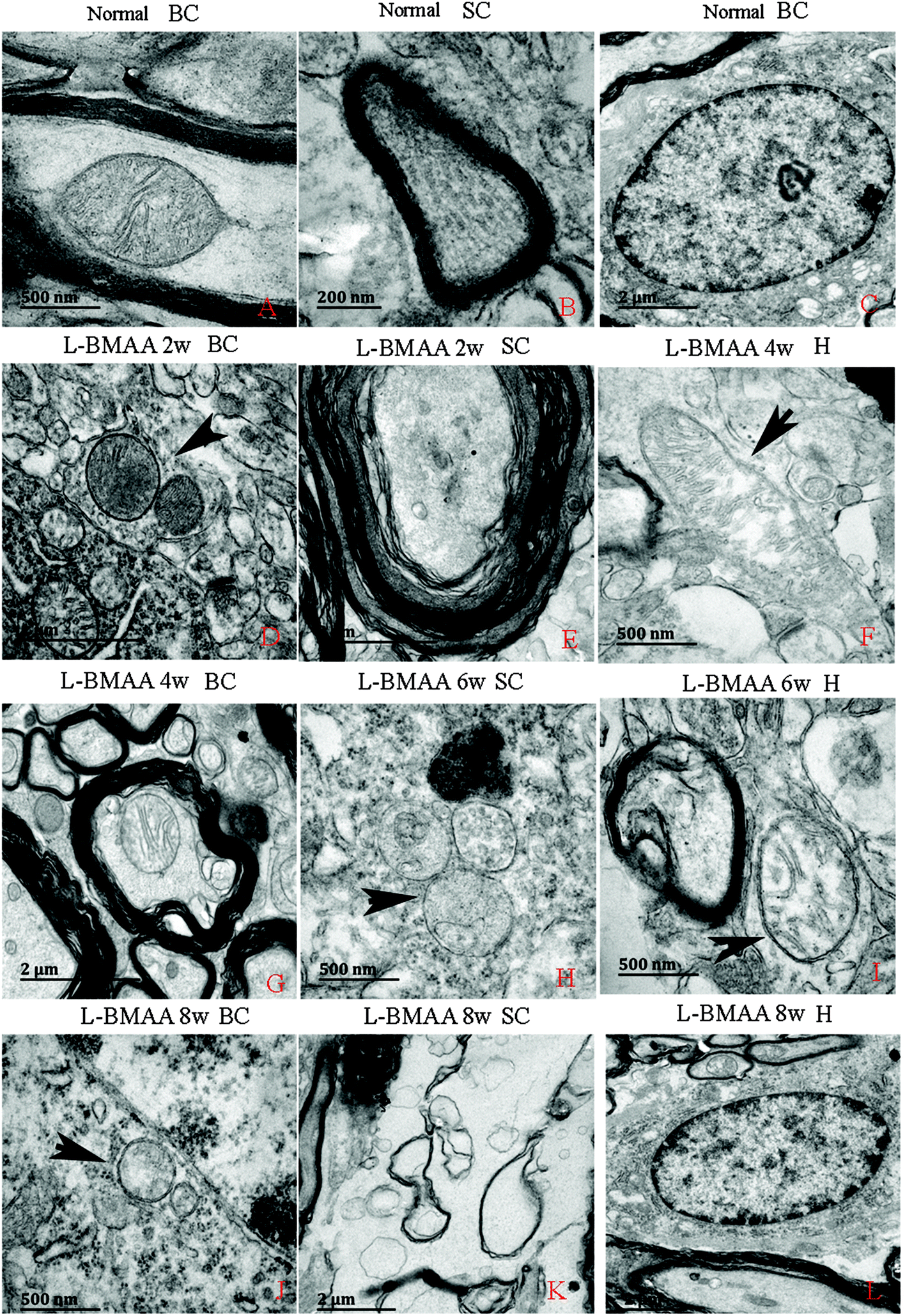 | ||
| Fig. 10 Electron micrograph demonstrating the following: mitochondrial degeneration; splitting; vacuoles; loose and fused changes of the myelin; and apoptotic signs of the nucleus. SC: spinal cord; BC: brain cortex; H: hippocampus. (A, F, and H–J) Scale bar = 500 nm; (B) scale bar = 200 nm; (C, E, G, K and L) scale bar = 2 μm; (D) scale bar = 20 μm. (A–C) Rats in the control group: (A) mitochondria with normal shape; (B) normal myelinated axons exhibiting dark, ring-shaped myelin sheaths surrounding axons; (C) normal neuronal nuclei with uncondensed chromatin. (D–L) Rats treated with L-BMAA. At two weeks post injections (D–E), normal mitochondrial structure could still be retained (D); while splitting, vacuoles, loose and fused changes on the myelin sheath were found (E). From the 4th week post L-BMAA injections, disorganized mitochondrial changes such as vacuolization and swollen cristae (arrows in F, H and I) were found. At the 8th week post L-BMAA injections (J–L), some mitochondria were totally degenerated, and their cristae completely disappeared (J). Meanwhile, more severe changes in demyelination and axonal loss were detected (K); and neurons also showed apoptotic signs with a shrunken nucleus and condensed, fragmented, and marginated nuclear chromatin (L). | ||
Corresponding to the above changes, immunofluorescence staining and western blotting of caspase-3, an enzyme involved in the execution of the mammalian apoptotic programme, revealed its upregulation following L-BMAA treatment (Fig. 11). The upregulation of caspase-3 expression reached a peak at six weeks post injections, and declined with neuronal loss (Fig. 12A and B). Meanwhile, gradual neuronal loss in the anterior horn of the spinal cord, motor brain cortex and hippocampi of L-BMAA-treated rats was observed by nissl staining (Fig. 12C and D; ESI Fig. 4†).
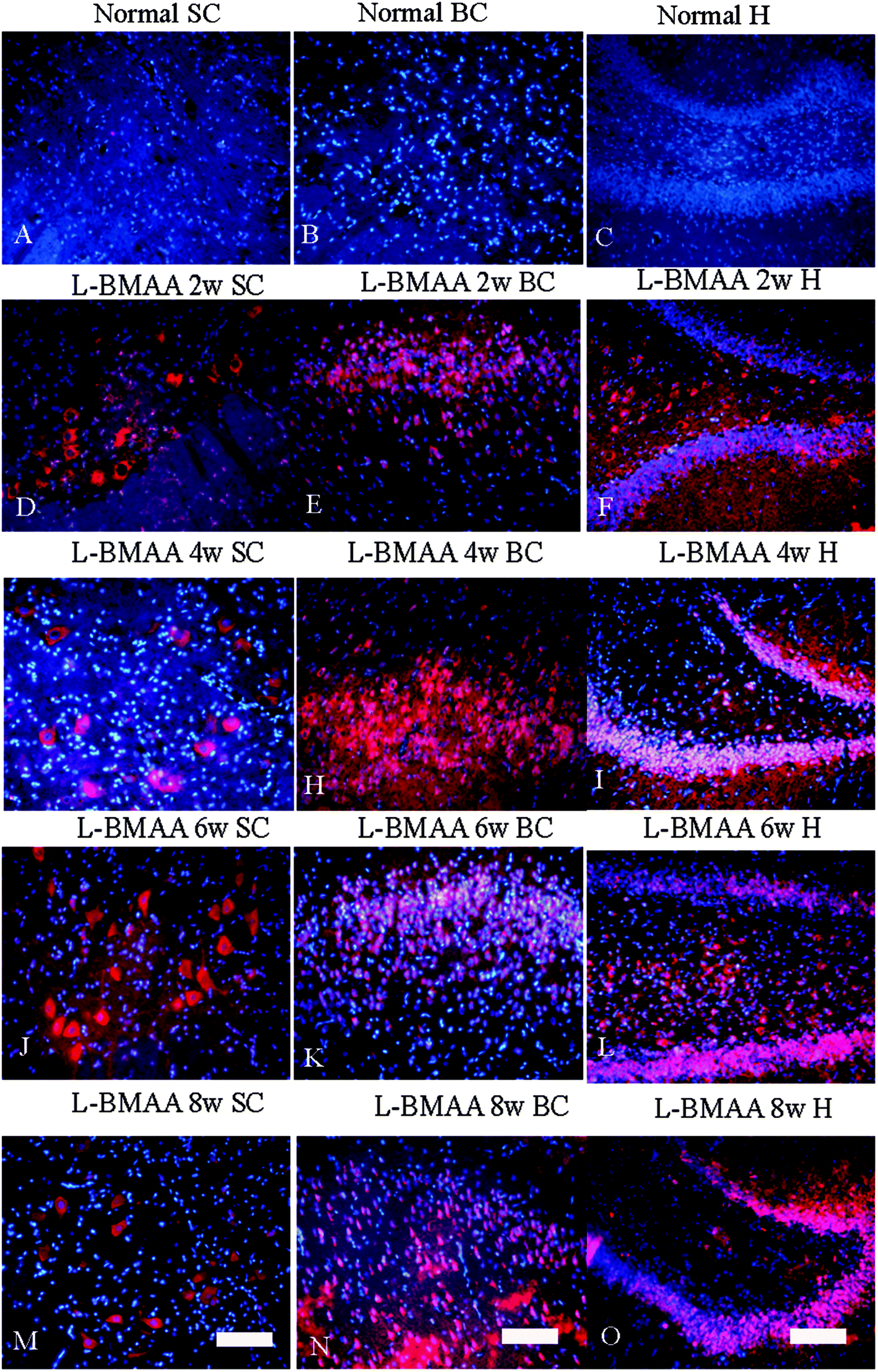 | ||
| Fig. 11 The expression of caspase-3, an enzyme involved in the execution of the mammalian apoptotic programme, increased after L-BMAA treatment, which reached a peak at six weeks post injections, and declined relatively. The expression of caspase-3 was detected by immunofluorescence staining. The red color shows caspase-3-labeled neurons, and the nuclei of all cells were stained with Hoechst 33342 (blue). Scale bar = 100 μm; SC: spinal cord; BC: brain cortex; H: hippocampus. | ||
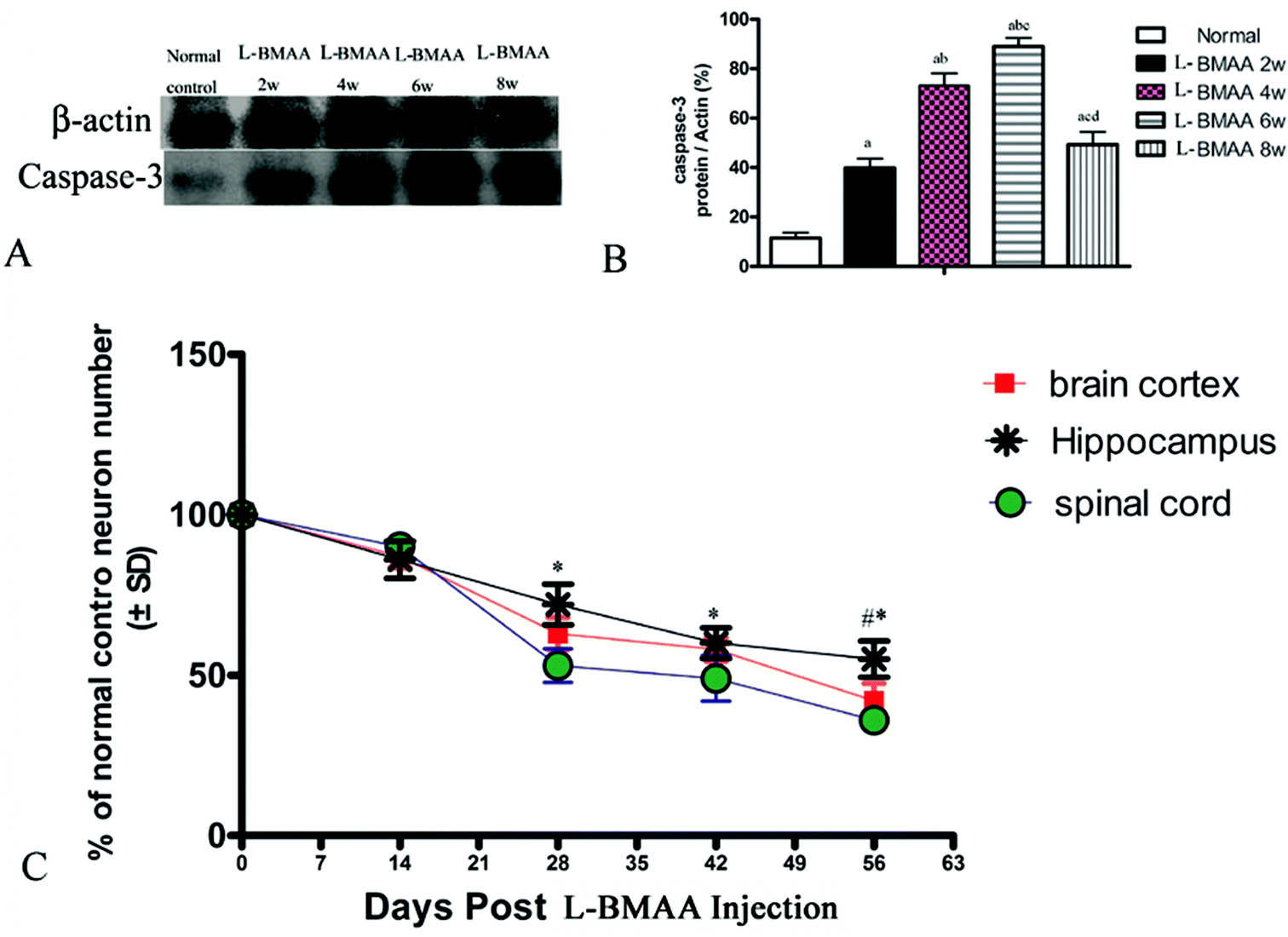 | ||
| Fig. 12 (A and B) Changes in caspase-3 expression levels are shown by western blotting; (a) P < 0.05 vs. the control group; (b) P < 0.05 vs.L-BMAA-treated rats at two weeks following injections; (c) P < 0.05 vs.L-BMAA-treated rats at four weeks following injections; (d) P < 0.05 vs.L-BMAA-treated rats at six weeks following injections. (C) The rate of neuronal survival (percentage of normal control) was exhibited based on neuronal counting. *P < 0.05 (hippocampus vs. spinal cord); #P < 0.05 (hippocampus vs. brain cortex). | ||
Discussion
ALS/PDC among the indigenous Chamorro people of Guam and other Mariana islands has been known for decades.5 In recent years, the rapid increase in the number of genes in which mutations have been associated with ALS has led to in vitro and in vivo models. All these models were able to exhibit signs including protein misfolding, mRNA splicing, oxidative stress, as well as proteasome and mitochondrial dysfunction. A number of therapeutic treatments for ALS have been tested in the widely used TgN(SOD1-G93A)1Gur transgenic mouse model. Moreover, culture models, which are also ideal systems for ALS, could identify the specific features of MNs at a single cell level. Neuron–astrocyte metabolic interactions, which have a critical role in MNs through glutamatergic toxicity, have been mostly defined in vitro. Ca2+ metabolism, which appears to play a critical role in inducing MN loss in ALS, has also been successfully studied using in vitro cell models.16Recently, toxic effects of L-BMAA were observed in animal studies.13,14 An ALS-PDC mouse model of the Western Pacific has been established by feeding mice with washed cycad flour, which exhibited significant and progressive motor, cognitive and sensory behavioral deficits (a major epidemiological link to ALS-PDC).17 Another study suggested that chronic oral administration is not neurotoxic to mice, in which a total of 15.5 g kg−1 of beta-N-methylamino-L-alanine was administered by gavage to mice over 11 weeks without observing any behavioral abnormalities.18 Contrarily, L-BMAA (25–400 mg kg−1) was acutely administered to rats by i.v. injection into the femoral vein, or chronically infused into the brain with osmotic mini-pumps that could induce neuronal and behavioral changes.5L-BMAA influx across the blood–brain barrier was found to be saturable with a maximal transfer rate (Vmax) of 1.6 ± 0.3 × 10(−3) μmol s−1 g−1 and a half-saturation constant (Km) of 2.9 ± 0.7 mM based on total perfusate L-BMAA concentration.19 Brain L-BMAA levels peaked within eight hours after injection, and declined with a t1/2 similar to that of plasma.20 After two weeks of continuous infusion (100 mg per kg per day), steady-state brain concentrations equaled 10–30 μg g−1 and only moderately exceeded those in plasma.20 These results suggest that L-BMAA may reach potentially toxic levels in the brain (i.e. >250 μM) after large doses (>100 mg kg−1) or a prolonged time of infusion.20 In addition, another study has reported behavioral changes (cognitive impairment) following fetal and neonatal exposure to repeated L-BMAA treatments (200 or 600 mg per kg sc).21 In the present study, we adopted a medium dose of 300 mg kg−1 to observe possible morphologic and functional changes.
Previous studies have demonstrated that intracerebroventricular (icv) 500 μg injections of L-BMAA could lead to behavioral changes including splay (flat posture with spreading of all limbs), clonic convulsions, rigidity, forepaw and hind paw grooming, wet-dog shakes and deep breathing in 60% of treated rats.22 In our study, motor functional declines were measured by rotarod test, inclined plane test, beam-walking and grip strength assessment. In general, dyskinesia in L-BMAA-treated rats became gradually serious with time, and was accompanied by the gradual degeneration and loss of MNs in the anterior horn of the spinal cord and brain motor cortex, as shown by nissl and immunochemical staining of SMI-32, which strongly labels the cell bodies of MNs and their neuritic processes.
In the MN degeneration process, the MN cell body swelled with obvious vacuolar changes at the early stage, displayed signs of atrophy, and finally exhibited a dissolving and necrotic appearance with a few remaining neurofilaments, as demonstrated by SMI-32 immunostaining and Bielschowsky's silver impregnation. Loss of neurites, axons and splitting, vacuoles, loose and fused changes of the myelin sheath, and apoptotic signs of the nucleus were all consistent with the gradual death of MNs and the decrease of motor function. This confirms that our model effectively and comprehensively obtained the characteristics of ALS.
Some complex genetic and environmental interactions may contribute to sporadic ALS. Approximately two-third of familial cases are triggered by mutations of four genes including chromosome 9 open reading frame 72 (C9ORF72), Cu/Zn superoxide dismutase (SOD1), fused in sarcoma/translocated in liposarcoma (FUS/TLS), and TAR-DNA binding protein 43 (TDP43).23 Loss of nuclear TDP-43 characterizes sporadic and most familial forms of ALS.24 A distinct mechanism that is strongly suspected of contributing to MN degeneration in ALS is the accumulation of cytosolic TDP-43 proteins. TDP-43 is a protein primarily located in the nucleus, whereas only small amounts exist in the cytosol under normal conditions.13,14 It has been found that TDP-43 has a critical role in diverse neurodegenerative diseases, and TDP-43 was observed to leave the nucleus and accumulate in cytosolic aggregates.13,14,25 This phenomenon has been investigated in human ALS, as well as in damaged MNs of a rodent ALS model with mutant SOD1.15,26L-BMAA can also bind to serine tRNA and is incorporated into the polypeptide chain, which causes the synthesized protein not to fold properly. Clumps of misfolded proteins may form aggregates that characterize neurodegenerative diseases.13,25 With a good prototype of human ALS,26 this phenomenon was also discovered in our L-BMAA injected rodent model (i.e. in neurons of the anterior horn of the spinal cord, brain motor cortex and hippocampus); especially at the late stage post L-BMAA treatment. Moreover, another index that was seen in human ALS is the loss of GLT-1 expression, which is a finding that initially lent its strong support to the idea that excitotoxic activation of glutamate receptors might contribute to MN damage in the disease.15,27 We obtained similar results in our rodent model, which could confirm the previous research on L-BMAA toxicity.
Elevated GSK3β levels are related to neurodegenerative diseases, especially to Alzheimer's disease (AD),13 and increased GSK3β levels were sometimes found in ALS. Previous studies have shown that GSK3β levels increased in the frontal and temporal cortex of ALS patients with cognitive impairments,28 and the active form of GSK3β was also elevated in the lumbar spinal cord and motor cerebral cortex of L-BMAA-treated rats.13 In our study, GSK3β upregulation was observed, which corresponded to the loss of muscular force associated with the degeneration of MNs in the spinal cord and motor cortex, as well as to the impairment of memory associated with neuronal loss and active caspase-3 overexpression in the hippocampus. In addition, recent ALS/PDC neuropathological studies suggest this disorder as a ‘tangle’ disease.6 The elevated expression of tau-5 in our L-BMAA-induced model showed that tauopathy, which has been identified in Guamanian ALS/PDC, shares some identical biochemical characteristics with classical AD.6
Neurodegenerative diseases such as AD, Parkinson's disease and ALS are all characterized by the appearance of reactive microglial and astroglial cells, which is referred to as neuro-inflammation; and this can contribute to MN death.29 In our L-BMAA-induced model, inflammation-associated CD68-positive microglia30 diffusely infiltrated within the parenchyma of the spinal cord, brain motor cortex and hippocampus; and was sustained until the late stage when most of the MNs have undergone apoptosis/necrosis. This is consistent with an opinion that the accumulation of reactive microglia in degenerating areas of ALS tissue is a key cellular event creating a chronic inflammatory environment that results in MN death.31 The expression of active caspase-3 (a specific marker of apoptosis) and CD68 (a specific marker of activated microglia and macrophage) reached a peak at 4–6 weeks following L-BMAA injection, and decreased with the gradual death and loss of neural cells, which is similar to our findings at eight weeks following L-BMAA injection. This is consistent with the neuronal loss phenomenon revealed by nissl staining in Fig. 12C.
Meanwhile, abnormal GFAP expression was detected at two weeks post injections of L-BMAA. Reactive astrogliosis formed glial scarring, and the glial scar became more and more augmented with the increase of the treatment time of L-BMAA, which is disadvantageous to remyelination and functional recovery.29,32,33
In a previous study, TNF-α and interferon gamma (IFN-γ) proinflammatory cytokines have been proposed to be involved in ALS-linked microglial activation.34 The exposure to these cytokines results in the increased expression of pro-oxidative enzymes including inducible nitric oxide synthase (iNOS), the catalytic subunit of the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and COX-2.34 Their cooperation could activate NF-κB and induce apoptotic MN death, as measured by immunoreactivity assays of NF-κB, TNF-α, COX-2 and active caspase-3.31,35,36
Electron microscopy images revealed a disorganization of mitochondria such as vacuolization, swollen cristae, or cristae that has completely disappeared in L-BMAA-treated rats. Since redox-sensitive targets such as intact membranes are highly dependent on mitochondrial function, mitochondrial damage is considered to be both a major contributor to oxidative stress (OS, a condition arising from unbalanced production and removal of reactive oxygen species) and a major cause of OS.37 The NF-κB pathway has also been found to promote these processes.37,38 Mitochondrial damage and induced OS contributed to several neurodegenerative diseases including ALS, at least in most studied SOD1-linked genetic variants.37 Our study suggests that L-BMAA injections could also lead to mitochondrial injury, followed by a series of OS and neuronal damage.
Detailed knowledge of electromyography (EMG) is critical for evaluating muscular atrophy and reinnervation in the earliest changes of motor units in ALS.39,40 Altered synaptic transmission accompanied by loss of MNs has been reported in a SOD1G93A mouse model of ALS.41 Instability of MUPs, consistent with a very early phase of increased axonal excitability, also appeared in the early stage ALS.39 Afterwards, widespread neuronal dysfunction and atrophy of muscles causes widespread loss of MUPs.40 Both the duration and amplitude of MUPs depend on the number of innervated muscle fibers; and polyphasic potentials are a sign that impulses from corresponding muscle fibers of a motor unit recorded by a needle occurred at slightly different times.40 Polyphasic potentials can arise from the presence of fine and slowly conducting branches of nerve fibers that sprout to innervate nearby denervated muscle fibers.40 Our nerve electrophysiology test displayed a prolonged MEP latency associated with the delay of conduction speed and a declined amplitude associated with the decrease of surviving fiber numbers. However, the test also displayed irregular MUPs characterized by a high amplitude, an increased rate of polyphasia, prolonged duration, similarities to those recorded in the case of neurogenic lesions, and muscular dystrophy.41 All these signs were consistent with pathological EMG changes in ALS.42
Conclusion
The work of establishing an animal model of ALS by L-BMAA administration has been applied for more than 50 years. At present, many groups continue to study the neurotoxic effects of L-BMAA in animals. Using different doses (from 1.5 μg to 28 mg kg−1) and different species (chick, rats, mice, monkey and apes), they have developed animal models with significant clinical symptoms such as splay, rigidity, jerking movements, body shakes, dragging gait, weakness, convulsions and complete inability to extend the legs and stand.5 In addition to cognitive disturbances and morphological brain changes in adult rodents exposed to L-BMAA during their development, L-BMAA exposure in neonatal rats has also caused significant biochemical and behavioral effects.43 Previous studies have supported the relationship between L-BMAA and the sporadic occurrence of ALS and other neurodegenerative diseases. Therefore, studying the underlying mechanisms of ALS through an L-BMAA-induced animal model has assumed great relevance in recent years. In 2015, de Munck E. et al. adopted the dose of L-BMAA (300 mg kg−1, i.p. for five consecutive days) and produced ALS characteristics found in patients such as decreased muscle volume, thinner motor cortex, enlarged lateral ventricles of the brain, and alterations in both bulbar nuclei and neurotransmitter levels. They also concluded that L-BMAA-treated rats could be a good model that mimics degenerative features in ALS patients.9 Moreover, they carried out an ultrastructural cell analysis of spinal motor neurons and revealed alterations in both the endoplasmic reticulum and mitochondria in 2013.13 These upregulated alterations in the active form of GSK3 and caspase-3 levels in the lumbar spinal cord and motor cerebral cortex of L-BMAA-treated rats have also been shown. On the other hand, they also reported that TDP-43 increased in L-BMAA-treated animals.13 Although parts of our studies overlapped with previous studies, we observed the astrogliosis and motor neuronal death process, overexpression of pro-inflammatory factors (COX-2, NF-κB and TNF-α), downregulation of GLT-1, cytosolic aggregates of TDP-43 and accumulation of microtubule-associated protein tau in degenerating motor neurons following L-BMAA injection. Moreover, multiple functional disabilities and pathologic EMG changes in MEP and MUPs were also seen. All these signs obtained most of the pathological features of ALS/PDC and displayed comprehensive neuropathological, electromyographic and functional impairments that resemble sporadic ALS symptoms, which could provide meaningful supplementary and reference materials for the data of other groups. Since the investigations on our model only lasted for eight weeks, we did not perform any special long-term observations (such as the worsening of the symptoms with age or the thinning of the motor cortex). Further long-term studies on our new model would be carried out in the future.Experiments
Animals
A total of 40 adult male SD rats weighing 250 g were obtained from the Laboratory Animal Center of Zhejiang University. Rats were divided into two groups: eight rats were assigned to the control group and 32 rats were assigned to the ALS/PDC-induced group. Eight rats were sacrificed at two, four, six and eight weeks post L-BMAA injection. Among these rats, five rats were sacrificed for histological assessment, immunohistological and immunofluorescence staining, and transmission electron microscopy observation; while three rats were sacrificed for western blotting assay. All animal procedures used in this study were carried out in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals. The study was approved by the animal ethics committee of Zhejiang University.Intravenous injection
L-BMAA was purchased from Sigma (St. Louis, MO, USA), and was dissolved in 0.5 ml of sterile distilled water. Rats in the ALS/PDC-induced group (n = 32) received continuous L-BMAA (300 mg kg−1) i.v. injections. We dissolved 75 mg of L-BMAA in 0.5 ml of sterile distilled water, in which the concentration of L-BMAA was 150 mg ml−1. Each SD rat, approximately 250 g in body weight, received 75 mg of L-BMAA (300 mg kg−1) via direct tail vein injection daily for three consecutive days. Rats in the control group (vehicle, n = 8) received i.v. injections of distilled water (0.5 ml).Motor function assessments
Previous studies have established a set of motor functional scales to evaluate the neurological disability of ALS/PDC.8 This evaluation was performed by researchers who were blind to the experimental animal groups.Rotarod test
The animal was placed on a rotarod (9 cm in diameter, 8 cm wide; KN-75, Natsume Seisakusho Co., Ltd) rotating at 15 rpm in the forward direction, and the longest time that the animal remained on the rod before falling was measured. Animals were trained two or three times a week before symptoms, and animals that could remain on the cylinder for over two minutes were chosen for this experiment.13 This test was carried out once a week for eight weeks.Inclined plane test
Animals were placed on an inclined board that was set at 60°, 55°, 50°, 45°, 40°, 35° or 30° angles. The angle at which the animal could remain without slipping for five seconds or more was determined. The test started from a 55° angle. From the second test onwards, measurements started from one angle setting higher than the maximum angle the animal had tolerated from the previous test.44 This test was carried out once a week for eight weeks.Forepaw gripping ability
Each rat was handled for five minutes and trained on a grip strength meter (GSM, designed by TSE-Systems) for two days. Then, rats were held by the tail just over the bar of the GSM; wherein rats reached out and grasped the bar with the un-restrained paw. Rats were then pulled away from the GSM by its tail in one smooth motion until the grip was released. In this way, the GSM measured the force (in grams) at the instant before the bar was released.45Fear conditioning
Fear conditioning trainings and trials were conducted as previously described.46 Long-term memory retention was evaluated in a test session carried out after three days of trainings. Animals were tested twice each week for eight weeks. Freezing was defined as the absence of all movement except that required for respiration, and was measured with instantaneous observations by observers who were blind to the test conditions. Behavior was recorded and video images were transferred into a computer equipped with an analysis program. The percentage of changed pixels between two adjacent 0.5 second images was used as a measure of activity.Beam-walking test
Beam-walking test was performed using a 2.5 cm wide rectangular wooden beam, which was 70 cm long and placed 1 m above the bench top.47 The first training session was done a day before the test, and the second training session was done just before the test. After these two training sessions, animals walked along the beam from one end to the other without any exogenous stimulus or footslip. After injection, beam-walking tests were performed once a week for eight weeks. The performance was graded by a blinded observer on a scale from 1 to 5: 1 = the rat crossed the beam normally without shaking; 2 = the rat crossed the beam normally while shaking; 3 = the rat crossed the beam with more than three footslips (any foot slipped off the top surface of the beam); 4 = the rat stood on the beam, and the affected hind limb hanged and did not aid in forward locomotion; and 5 = the rat was unable to stand on the beam and fell down immediately.48Nerve electrophysiology test
Motor evoked potential (MEP) and indexed motor unit potentials (MUPs) were detected each week following L-BMAA injection. An acupuncture pin electrode was adopted to initiate stimulus. Latency and amplitude of MEP were recorded as previously described.49,50 Electronic signals were collected and transferred into a computer for analysis.MUPs were analyzed for abnormalities in conventional electromyography. A disposable monopolar needle electrode (Dantec 13R1 with a recording surface area of 0.07 mm2) was used to record MUPs. A window discriminator was used to reduce contamination of the index motor unit from other motor units.39,40 MUPs were considered abnormal if it was more than 20% of that in normal controls.
Perfusion and tissue processing
Animals in each group were sacrificed at two, four, six and eight weeks (five/per time point in each group) of post L-BMAA injection. Rats were anesthetized with sodium pentobarbital and intracardially perfused with cold saline, followed by 4% paraformaldehyde in 0.1 M of phosphate buffer solution (PBS, pH 7.4). The whole spinal cord and brain tissues were carefully harvested and dissected. The lumbar spinal cord (1 cm) and half of the brain of each animal were fixed in the same fixative for four hours, and transferred in 30% sucrose solution in PBS until the tissue sank to the bottom of the container. Twenty μm-thick sections were cut on freezing microtome through the coronal plane of the brain and transverse plane of the spinal cord using a Leica cryostat, and the sections were mounted onto 0.02% poly-L-lysine-coated slides. All sections were collected for histological assessment, and for immunohistological and immunofluorescence staining. The remaining central nervous tissues were fixed in 10% glutaraldehyde solution and examined by using a transmission electron microscope.51,52Histology assessment
Cresyl violet (nissl) staining was employed to assess inflammation and neuron survival. Five sections from the cortex, hippocampi and anterior horn of the spinal cord of each animal were randomly selected; and images were photographed under 200× magnification in three vision fields per section. Neuron counts were performed, which was restricted to neurons with a well-defined nucleolus and a cell body with enrichment of the endoplasmic reticulum.Bielschowsky silver staining was performed to estimate the nerve axonal loss situation. Sections were placed in 20% silver nitrate at room temperature in the dark for 15 minutes. The solution was washed off with distilled water and replaced by ammoniacal silver solution for 10 minutes in the dark. Then, ammonium hydroxide/formaldehyde was added to stain slides until the tissue turned brown with a gold background, followed by treatment with gold chloride and sodium thiosulphate. Axonal loss was assessed using the following scale:53 0, no axonal loss; 1, few foci of superficial axonal loss involving less than 25% of tissues; 2, foci of deep axonal loss that encompassed over 25% of tissues; and 3, diffuse and widespread axonal loss.
Immunohistochemical staining
Slides were warmed for 20 minutes on a slide warmer, and a ring of wax was applied around the sections with a PAP pen (Invitrogen, Carlsbad, CA). After rinsing in 0.01 M of tris-buffered saline (TBS) for 10 minutes, the sections were permeabilized and blocked with 0.3% Triton X-100/10% normal goat serum in 0.01 M of PBS for 30 minutes; then, incubated with polyclonal rabbit antibodies anti-SMI-32 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :500; Sternberger Monoclonals, Berkeley, CA), anti-GLT-1 (1:1000; Chemicon, Temecula, CA), anti-tau-5 (1:500; Invitrogen Corp., Carlsbad, CA), anti-TDP-43 (1:1000; Proteintech Group, Chicago, IL), anti-caspase-3 (1:500; Cayman Chemical, Ann Arbor, MI), anti-GSK3β (1:500; Thermo Fisher Scientific, Waltham, MA), anti-NF-κB p65 (1:500; Abcam, MA), and anti-TNF-α (1:1000; ProSci Inc., CA) overnight at 4 °C. Sections were incubated with secondary biotinylated goat anti-rabbit/mouse IgG antibody (1:400; Vector Laboratories, CA) for one hour at room temperature, followed by avidin–biotin–peroxidase complex (ABC kit; Thermo Fisher Scientific, CA). The sections were counterstained with hematoxylin after five minutes of incubation with 0.02% DAB and 0.003% H2O2 in 0.005 Tris-HCl. Primary antibody omission controls were used in order to further confirm the specificity of immunohistochemical labeling.
:500; Sternberger Monoclonals, Berkeley, CA), anti-GLT-1 (1:1000; Chemicon, Temecula, CA), anti-tau-5 (1:500; Invitrogen Corp., Carlsbad, CA), anti-TDP-43 (1:1000; Proteintech Group, Chicago, IL), anti-caspase-3 (1:500; Cayman Chemical, Ann Arbor, MI), anti-GSK3β (1:500; Thermo Fisher Scientific, Waltham, MA), anti-NF-κB p65 (1:500; Abcam, MA), and anti-TNF-α (1:1000; ProSci Inc., CA) overnight at 4 °C. Sections were incubated with secondary biotinylated goat anti-rabbit/mouse IgG antibody (1:400; Vector Laboratories, CA) for one hour at room temperature, followed by avidin–biotin–peroxidase complex (ABC kit; Thermo Fisher Scientific, CA). The sections were counterstained with hematoxylin after five minutes of incubation with 0.02% DAB and 0.003% H2O2 in 0.005 Tris-HCl. Primary antibody omission controls were used in order to further confirm the specificity of immunohistochemical labeling.
Immunofluorescence staining
The sections were pretreated with the same method described above, and incubated with primary monoclonal mouse antibodies CD68/ED1 (1:100; Santa Cruz Biotechnology, Santa Cruz, CA), anti-cyclooxygenase-2 (COX-2, 1:1000; BioVision, Milpitas, CA), and polyclonal rabbit anti-GFAP (1:200; Thermo Fisher Scientific, Waltham, MA) overnight at 4 °C; then, washed with PBS and incubated with 1:200 TRITC (Rhodamine)-conjugated goat anti-mouse or FITC-conjugated goat anti-rabbit IgG secondary antibodies for one hour at 37 °C (Invitrogen, CA). The sections were finally coverslipped with antifade Gel/Mount aqueous mounting media (Southern Biotech, AL). All control sections were incubated in PBS without primary antibodies.
Western blotting
Rats in each group were sacrificed by decapitation at two, four, six and eight weeks (three rats per time point in each group) post L-BMAA injection. Spinal cords and the cerebral cortex (150 mg) were homogenized in lysis buffer (1% Nonidet P-40, 10% glycerol, 137 mM of NaCl, 20 mM of Tris-HCl, pH 7.5) including an EDTA-free protease inhibitor cocktail (Roche Diagnostics, Mannheim, Germany), followed by centrifugation at 800g for 15 minutes. Protein concentrations of all samples were determined by the Bradford assay (Sigma-Aldrich Inc., St. Louis, MO, USA). Proteins (150 μg) from these supernatants were resolved by SDS-PAGE and transferred to Immobilon-P membranes (Millipore Corp.). Membranes were incubated with rabbit anti-GSK3β (1:1000; Thermo Fisher Scientific, Waltham, MA), tau-5 (1:1000; Invitrogen Corp., Carlsbad, CA), SMI-32 (1:1000; Sternberger Monoclonals, Berkeley, CA), anti-GLT-1 (1:1000; Chemicon, Temecula, CA), caspase-3 (1:800; Cayman Chemical, Ann Arbor, MI), CD68/ED1 (1:500; Santa Cruz Biotechnology, Santa Cruz, CA), COX-2 (1:1000; BioVision, Milpitas, CA), GFAP (1:500; Thermo Fisher Scientific, Waltham, MA), anti-TNF-α (1:2000; ProSci Inc., CA) and anti-NF-κB p65 (1:500; Abcam, MA). After washing, blots were incubated with HRP-conjugated secondary antibody. Proteins were detected by enhanced chemiluminescence, as described in the technical manual of Amersham Pharmacia Biotech; then, imaged and quantified by Bio-Rad Quantity One 1D software. To normalize protein bands to a gel loading control, membranes were washed in TBST and re-probed with rabbit anti-β-actin (1:5000; Abcam, MA); followed by incubation with peroxidase-conjugated goat anti-rabbit (1:5000; Santa Cruz, CA) and ECL detection. For negative controls, the primary antibody was omitted.
Electron microscopy
The 10% glutaraldehyde fixed tissues were washed three times with 0.1 M of PBS. Post-fixed tissues were placed in 1% osmium tetroxide at 4 °C overnight, and washed three times with 0.1 M of PBS. After post-fixation, the following steps were done in an EM processor with agitation at room temperature: dehydrated tissues in graded ethanol (30%, 50%, 70%, 80%, 90% and 95% ethanol) for five minutes each, followed by three changes of absolute ethanol for 10 minutes each; after two changes in 1,2-propylene oxide (PO) for 15 minutes each, tissues were immersed in a 1:1 PO:Epon mixture for one hour, overnight in pure Epon, and finally embedding tissues in pure Epon at 60 °C for three days. Epon-embedded tissues were cut into 90 nm sections with a diamond knife on an Ultracut microtome, and collected on a 200 mesh copper grid. Lead citrate (approximately 3%) and 8% uranyl acetate were filtered before use. Grids were stained with lead citrate droplets for 20 minutes in a Petri dish, and washed three times with distilled water. Then, the sections were stained with droplets of 8% uranyl acetate inside a Petri dish at room temperature for 20 minutes, washed three times with distilled water, and the grids were ready for electron microscopic analysis. Images were first captured at low resolution, and imaged at higher magnifications in the spinal cord and brain tissues.
Statistical analysis
Overall treatment effects were examined with SPSS 13.0 (SPSS, Chicago, IL). A one-way analysis of variance (ANOVA) was performed on each dataset to identify whether significant differences existed between treatment groups, followed by the Newman–Keuls post hoc test and Tukey's honest significant difference test. A P-value <0.05 was considered statistically significant. Kruskal–Wallis nonparametric one-way ANOVA was used for data presented as percentages. Rotarod test results were analyzed with Dunnett's test, and inclined plate test results were analyzed with Steel's test. Data are presented as mean ± standard deviation (SD). Graphs were created using GraphPad Prism 4 software (GraphPad Prism Software, Inc., CA, USA).Acknowledgements
This work was funded by the Zhejiang Provincial Natural Science Foundation of China (Grant No. R2110025) and the National Natural Science Foundation of China (Grant No. 81271333).References
- M. Watanabe, M. Dykes-Hoberg, V. C. Culotta, D. L. Price, P. C. Wong and J. D. Rothstein, Histological evidence of protein aggregation in mutant SOD1 transgenic mice and in amyotrophic lateral sclerosis neural tissues, Neurobiol. Dis., 2001, 8, 933–941 CrossRef CAS PubMed
.
- W. G. Bradley, S. A. Banack and P. A. Cox, The ALS/PDC syndrome of Guam and the cycad hypothesis, Neurology, 2009, 72, 473–474 CrossRef PubMed
- A. Al-Chalabi and O. Hardiman, The epidemiology of ALS: a conspiracy of genes, environment and time, Nat. Rev. Neurol., 2013, 9, 617–628 CrossRef CAS PubMed
- A. Vega and A. Bell, a-amino-β-methylaminopropionic acid, a new amino acid from seeds of cycas circinalis, Phytochemistry, 1967, 6, 759–762 CrossRef CAS
- V. T. Karamyan and R. C. Speth, Animal models of BMAA neurotoxicity: a critical review, Life Sci., 2008, 82, 233–246 CrossRef CAS PubMed
- P. G. Ince and G. A. Codd, Return of the cycad hypothesis - does the amyotrophic lateral sclerosis/parkinsonism dementia complex (ALS/PDC) of Guam have new implications for global health?, Neuropathol. Appl. Neurobiol., 2005, 31, 345–353 CrossRef CAS PubMed
- P. S. Spencer, P. B. Nunn, J. Hugon, A. C. Ludolph, S. M. Ross and D. N. Roy,
et al., Guam amyotrophic lateral sclerosis-parkinsonism-dementia linked to a plant excitant neurotoxin, Science, 1987, 237, 517–522 CAS
- A. Delzor, P. Couratier, F. Boumédiène, M. Nicol, M. Druet-Cabanac, F. Paraf, A. Méjean, O. Ploux, J. P. Leleu, L. Brient, M. Lengronne, V. Pichon, A. Combès, S. El Abdellaoui, V. Bonneterre, E. Lagrange, G. Besson, D. J. Bicout, J. Boutonnat, W. Camu, N. Pageot, R. Juntas-Morales, V. Rigau, E. Masseret, E. Abadie, P. M. Preux and B. Marin, Searching for a link between the L-BMAA neurotoxin and amyotrophic lateral sclerosis: a study protocol of the French BMAALS programme, BMJ Open, 2014, 4, e005528 CrossRef PubMed
- E. de Munck, E. Muñoz-Sáez, B. G. Miguel, M. T. Solas, A. Martínez and R. M. Arahuetes, Morphometric and neurochemical alterations found in l-BMAA treated rats, Environ. Toxicol. Pharmacol., 2015, 39, 1232–1245 CrossRef CAS PubMed
- W. G. Bradley and D. C. Mash, Beyond Guam: the cyanobacteria/BMAA hypothesis of the cause of ALS and other neurodegenerative diseases, Amyotrophic Lateral Scler., 2009, 10, 7–20 CrossRef CAS PubMed
- J. M. Wilson, I. Khabazian, M. C. Wong, A. Seyedalikhani, J. S. Bains and B. A. Pasqualotto,
et al., Behavioral and neurological correlates of ALS-parkinsonism dementia complex in adult mice fed washed cycad flour, NeuroMol. Med., 2002, 1, 207–221 CrossRef CAS
- O. Karlsson, E. Roman and E. B. Brittebo, Long-term cognitive impairments in adult rats treated neonatally with beta-N-Methylamino-L-Alanine, Toxicol. Sci., 2009, 112(1), 185–195 CrossRef CAS PubMed
- E. de Munck, E. Munoz-Saez, B. G. Miguel, M. T. Solas, I. Ojeda, A. Martinez, C. Gil and R. M. Arahuetes, beta-N-methylamino-l-alanine causes neurological and pathological phenotypes mimicking Amyotrophic Lateral Sclerosis (ALS): the first step towards an experimental model for sporadic ALS, Environ. Toxicol. Pharmacol., 2013, 36, 243–255 CrossRef CAS PubMed
- H. Z. Yin, S. Yu, C. I. Hsu, J. Liu, A. Acab and R. Wu,
et al., Intrathecal infusion of BMAA induces selective motor neuron damage and astrogliosis in the ventral horn of the spinal cord, Exp. Neurol., 2014, 261, 1–9 CrossRef CAS PubMed
- T. L. Perry, C. Bergeron, J. C. Steele, D. R. McLachlan and S. Hansen, Brain amino acid contents are dissimilar in sporadic and Guamanian amyotrophic lateral sclerosis, J. Neurol. Sci., 1990, 99, 3–8 CrossRef CAS PubMed
- V. Silani, M. Braga, A. Ciammola, V. Cardin and G. Scarlato, Motor neurones in culture as a model to study ALS, J. Neurol., 2000, 247(Suppl 1), I28–I36 CrossRef PubMed
- J. M. Wilson, I. Khabazian, M. C. Wong, A. Seyedalikhani, J. S. Bains and B. A. Pasqualotto,
et al., Behavioral and neurological correlates of ALS-parkinsonism dementia complex in adult mice fed washed cycad flour, NeuroMol. Med., 2002, 1, 207–221 CrossRef CAS
- T. L. Perry, C. Bergeron, A. J. Biro and S. Hansen, Beta-N-methylamino-L-alanine. Chronic oral administration is not neurotoxic to mice, J. Neurol. Sci., 1989, 94, 173–180 CrossRef CAS PubMed
- Q. R. Smith, H. Nagura, Y. Takada and M. W. Duncan, Facilitated transport of the neurotoxin, beta-N-methylamino-L-alanine, across the blood-brain barrier, J. Neurochem., 1992, 58, 1330–1337 CrossRef CAS PubMed
- M. W. Duncan, N. E. Villacreses, P. G. Pearson, L. Wyatt, S. I. Rapoport and I. J. Kopin,
et al., 2-amino-3-(methylamino)-propanoic acid (BMAA) pharmacokinetics and blood-brain barrier permeability in the rat, J. Pharmacol. Exp. Ther., 1991, 258, 27–35 CAS
- O. Karlsson, N. G. Lindquist, E. B. Brittebo and E. Roman, Selective brain uptake and behavioral effects of the cyanobacterial toxin BMAA (beta-N-methylamino-L-alanine) following neonatal administration to rodents, Toxicol. Sci., 2009, 109, 286–295 CrossRef CAS PubMed
- Y. Matsuoka, Z. Rakonczay, E. Giacobini and D. Naritoku, L-beta-methylamino-alanine-induced behavioral changes in rats, Pharmacol., Biochem. Behav., 1993, 44, 727–734 CrossRef CAS
- C. Volonte, S. Apolloni and C. Parisi, MicroRNAs: newcomers into the ALS picture, CNS Neurol. Disord.: Drug Targets, 2015, 14, 194–207 CAS
- J. R. Highley, J. Kirby, J. A. Jansweijer, P. S. Webb, C. A. Hewamadduma and P. R. Heath,
et al., Loss of nuclear TDP-43 in amyotrophic lateral sclerosis (ALS) causes altered expression of splicing machinery and widespread dysregulation of RNA splicing in motor neurones, Neuropathol. Appl. Neurobiol., 2014, 40, 670–685 CrossRef CAS PubMed
- L. M. Igaz, L. K. Kwong, E. B. Lee, A. Chen-Plotkin, E. Swanson and T. Unger,
et al., Dysregulation of the ALS-associated gene TDP-43 leads to neuronal death and degeneration in mice, J. Clin. Invest., 2011, 121, 726–738 CAS
- M. Neumann, D. M. Sampathu, L. K. Kwong, A. C. Truax, M. C. Micsenyi and T. T. Chou,
et al., Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis, Science, 2006, 314, 130–133 CrossRef CAS PubMed
- H. Z. Yin, D. T. Tang and J. H. Weiss, Intrathecal infusion of a Ca(2+)-permeable AMPA channel blocker slows loss of both motor neurons and of the astrocyte glutamate transporter, GLT-1 in a mutant SOD1 rat model of ALS, Exp. Neurol., 2007, 207, 177–185 CrossRef CAS PubMed
- W. Yang, C. Leystra-Lantz and M. J. Strong, Upregulation of GSK3beta expression in frontal and temporal cortex in ALS with cognitive impairment (ALSci), Brain Res., 2008, 1196, 131–139 CrossRef CAS PubMed
- T. Philips and W. Robberecht, Neuroinflammation in amyotrophic lateral sclerosis: role of glial activation in motor neuron disease, Lancet Neurol., 2011, 10, 253–263 CrossRef CAS PubMed
- F. Gonzalez-Scarano and G. Baltuch, Microglia as mediators of inflammatory and degenerative diseases, Annu. Rev. Neurosci., 1999, 22, 219–240 CrossRef CAS PubMed
- M. Mir, V. J. Asensio, L. Tolosa, M. Gou-Fabregas, R. M. Soler and J. Llado,
et al., Tumor necrosis factor alpha and interferon gamma cooperatively induce oxidative stress and motoneuron death in rat spinal cord embryonic explants, Neuroscience, 2009, 162, 959–971 CrossRef CAS PubMed
- M. V. Sofroniew, Molecular dissection of reactive astrogliosis and glial scar formation, Trends Neurosci., 2009, 32, 638–647 CrossRef CAS PubMed
- N. Hibbits, J. Yoshino, T. Q. Le and R. C. Armstrong, Astrogliosis during acute and chronic cuprizone demyelination and implications for remyelination, ASN Neuro, 2012, 4, 393–408 CrossRef CAS PubMed
- T. Prell, J. Lautenschlager, L. Weidemann, J. Ruhmer, O. W. Witte and J. Grosskreutz, Endoplasmic reticulum stress is accompanied by activation of NF-kappaB in amyotrophic lateral sclerosis, J. Neuroimmunol., 2014, 270, 29–36 CrossRef CAS PubMed
- G. Liu, M. Fiala, M. T. Mizwicki, J. Sayre, L. Magpantay and A. Siani,
et al., Neuronal phagocytosis by inflammatory macrophages in ALS spinal cord: inhibition of inflammation by resolvin D1, Am. J. Neurodegener Dis., 2012, 1, 60–74 Search PubMed
- S. Snigdha, E. D. Smith, G. A. Prieto and C. W. Cotman, Caspase-3 activation as a bifurcation point between plasticity and cell death, Neurosci. Bull., 2012, 28, 14–24 CrossRef CAS PubMed
- M. T. Carri, C. Valle, F. Bozzo and M. Cozzolino, Oxidative stress and mitochondrial damage: importance in non-SOD1 ALS, Front. Cell. Neurosci., 2015, 9, 41 Search PubMed
- W. Rui, L. Guan, F. Zhang, W. Zhang and W. Ding, PM -induced oxidative stress increases adhesion molecules expression in human endothelial cells through the ERK/AKT/NF-kappaB-dependent pathway, J. Appl. Toxicol., 2015 DOI:10.1002/jat.3143
- R. B. Stein, B. S. Brucker and D. R. Ayyar, Motor units in incomplete spinal cord injury: electrical activity, contractile properties and the effects of biofeedback, J. Neurol., Neurosurg. Psychiatry, 1990, 53, 880–885 CrossRef CAS
- M. de Carvalho and M. Swash, Fasciculation potentials and earliest changes in motor unit physiology in ALS, J. Neurol., Neurosurg. Psychiatry, 2013, 84, 963–968 CrossRef PubMed
- K. Rowinska-Marcinska, E. Szmidt-Salkowska, A. Fidzianska, E. Zalewska, M. Dorobek and A. Karwanska,
et al., Atypical motor unit potentials in Emery-Dreifuss muscular dystrophy (EDMD), Clin. Neurophysiol., 2005, 116, 2520–2527 CrossRef CAS PubMed
- K. Hokkoku, M. Sonoo, M. Higashihara, E. Stalberg and T. Shimizu, Electromyographs of the flexor digitorum profundus muscle are useful for the diagnosis of inclusion body myositis, Muscle Nerve, 2012, 46, 181–186 CrossRef PubMed
- M. K. Engskog, O. Karlsson, J. Haglöf, A. Elmsjö, E. Brittebo, T. Arvidsson and C. Pettersson, The cyanobacterial amino acid β-N-methylamino-l-alanine perturbs the intermediary metabolism in neonatal rats, Toxicology, 2013, 312, 6–11 CrossRef CAS PubMed
- D. Wang and J. Zhang, Effects of hypothermia combined with neural stem cell transplantation on recovery of neurological function in rats with spinal cord injury, Mol. Med. Rep., 2015, 11, 1759–1767 CAS
- M. K. Strong, J. E. Blanco, K. D. Anderson, G. Lewandowski and O. Steward, An investigation of the cortical control of forepaw gripping after cervical hemisection injuries in rats, Exp. Neurol., 2009, 217, 96–107 CrossRef PubMed
- J. D. Runyan, A. N. Moore and P. K. Dash, A role for prefrontal cortex in memory storage for trace fear conditioning, J. Neurosci., 2004, 24, 1288–1295 CrossRef CAS PubMed
- A. M. Matter, K. A. Folweiler, L. M. Curatolo and A. E. Kline, Temporal effects of environmental enrichment-mediated functional improvement after experimental traumatic brain injury in rats, Neurorehabil Neural Repair, 2011, 25, 558–564 CrossRef PubMed
- V. Beray-Berthat, C. Delifer, V. C. Besson, H. Girgis, B. Coqueran and M. Plotkine,
et al., Long-term histological and behavioural characterisation of a collagenase-induced model of intracerebral haemorrhage in rats, J. Neurosci. Methods, 2010, 191, 180–190 CrossRef PubMed
- H. Bolay, Y. Gursoy-Ozdemir, I. Unal and T. Dalkara, Altered mechanisms of motor-evoked potential generation after transient focal cerebral ischemia in the rat: implications for transcranial magnetic stimulation, Brain Res., 2000, 873, 26–33 CrossRef CAS PubMed
- S. Amadio, S. Pluchino, E. Brini, P. Morana, R. Guerriero and F. Martinelli Boneschi,
et al., Motor evoked potentials in a mouse model of chronic multiple sclerosis, Muscle Nerve, 2006, 33, 265–273 CrossRef PubMed
- S. Han, F. Zhang, Z. Hu, Y. Sun, J. Yang and H. Davies,
et al., Dose-dependent anti-inflammatory and neuroprotective effects of an alphanubeta3 integrin-binding peptide, Mediators Inflammation, 2013, 2013, 268486 Search PubMed
- F. Zhang, J. Yang, H. Jiang and S. Han, An alphanubeta3 integrin-binding peptide ameliorates symptoms of chronic progressive experimental autoimmune encephalomyelitis by alleviating neuroinflammatory responses in mice, J. Neuroimmune Pharmacol., 2014, 9, 399–412 CrossRef PubMed
- L. L. Yin, L. L. Lin, L. Zhang and L. Li, Epimedium flavonoids ameliorate experimental autoimmune encephalomyelitis in rats by modulating neuroinflammatory and neurotrophic responses, Neuropharmacology, 2012, 63, 851–862 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5tx00272a |
| This journal is © The Royal Society of Chemistry 2016 |