
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProtonated glycine supramolecular systems: the need for quantum dynamics†
Fabio
Gabas
 ,
Giovanni
Di Liberto
,
Riccardo
Conte
* and
Michele
Ceotto
*
,
Giovanni
Di Liberto
,
Riccardo
Conte
* and
Michele
Ceotto
*
Dipartimento di Chimica, Università degli Studi di Milano, via Golgi 19, 20133 Milano, Italy. E-mail: riccardo.conte1@unimi.it; michele.ceotto@unimi.it
First published on 17th September 2018
Abstract
IR spectroscopy is one of the most commonly employed techniques to study molecular vibrations and interactions. However, characterization of experimental IR spectra is not always straightforward. This is the case of protonated glycine supramolecular systems like Gly2H+ and (GlyH + nH2), whose IR spectra raise questions which have still to find definitive answers even after theoretical spectroscopy investigations. Specifically, the assignment of the conformer responsible for the spectrum of the protonated glycine dimer (Gly2H+) has led to much controversy and it is still debated, while structural hypotheses formulated to explain the main experimental spectral features of (GlyH + nH2) systems have not been theoretically confirmed. We demonstrate that simulations must account for quantum dynamical effects in order to resolve these open issues. This is achieved by means of our divide-and-conquer semiclassical initial value representation technique, which approximates the quantum dynamics of high dimensional systems with remarkable accuracy and outperforms not only the commonly employed but unfit scaled-harmonic approaches, but also pure classical dynamics simulations. Besides the specific insights concerning the two particular cases presented here, the general conclusion is that, due to the widespread presence of protonated systems in chemistry, quantum dynamics may play a prominent role and should not be totally overlooked even when dealing with large systems including biological structures.
Introduction
Protonated systems are ubiquitous in chemistry.1 They are involved in many different processes and activities, ranging from organic reactions and intermediates to biological and interstellar-medium events. Furthermore, protonation is determinant for the chemical properties of heteroatomic compounds, such as amino acids. For instance, hydrolysis of amides, peptides, and proteins at biological pH is initiated and driven by the process of protonation. In general, the electronic and conformational structure of proteins as well as their dynamics are strongly influenced by protonation with the resulting three-dimensional structure playing a key role in their biological activity.Protonated glycine compounds are pivotal examples of protonated systems because they are the smallest building blocks of more complex biological entities. A full comprehension of their dynamics is indeed essential for a correct understanding of the stability and reactivity of many other protonated systems. For this reason, in the past, protonated glycine compounds have been the subject of extensive experimental and computational studies.2–9 However, there are some fundamental questions about these systems which are still open: specifically, to what extent is the proton shared between the amide and the carboxylic group? Is it a static or a dynamical effect? Do nuclear quantum mechanical contributions play a major or a minor role in determining the properties of these protonated compounds? One should expect very peculiar quantum mechanical effects when the proton is shared between nucleophilic groups either belonging to different molecules, like in the case of the glycine proton-bound dimer, or when they are part of the same molecule, as in protonated glycine. The main reason for this expectation is that the proton is the only ion with basically zero ionic radius and it has the lightest mass. These peculiarities are at the origin of proton mobility and reactivity, and one would expect quantum mechanical contributions to be determinant. This paper aims at providing answers to the open questions illustrated above and at estimating the impact of quantum mechanical effects by comparing quantum and classical simulations versus available experimental results.
One popular experimental spectroscopic technique to study the vibrational features of protonated compounds is represented by infrared multiple photon dissociation (IRMPD). IRMPD provides enhanced signals of gaseous molecular ions in the infrared region once they have been trapped in the high vacuum cells of mass spectrometers.2,10–13 However, when applied to the glycine proton-bound dimer, Gly2H+, IRMPD does not permit undisputed identification of which Gly2H+ conformer is more representative of the IRMPD spectrum.
This open issue, which has to be resolved in order to properly characterize the structural properties not only of the dimer but also of complex peptide chains, has been the topic of previous joint experimental and computational studies in 2005 by McLafferty et al.14 and in 2007 by McMahon et al.15 with conclusions clearly at odds. Specifically, Gly2H+ has two low energy gas-phase conformers, named CS01 and CS02, plus a zwitterionic form ZW01 (see the ESI† for more information) which rapidly interconverts to CS01 during the dynamics. In the CS01 conformer, the two moieties making up the dimer are bound by means of an O⋯H+N interaction, while a N⋯H+N interaction is peculiar of CS02. According to some studies, including McLafferty's one,9,14 CS02 is the most representative conformer in the experimental IRMPD spectrum, while for other studies, with McMahon's paper among them,4,5,15 it is CS01 that deserves recognition, so a definitive conclusion has not been reached yet.
In both studies14,15 the authors employed a scaled-harmonic approach to interpret the main features of the IRMPD spectrum. In the scaled-harmonic technique,16 first the normal mode frequencies (i.e. the purely harmonic frequencies of vibration) at the minimum geometry are calculated by diagonalizing the equilibrium nuclear Hessian matrix and taking the square roots of the Hessian eigenvalues. Then, they are scaled to account for anharmonicity and coupling between modes. Such an approach is widely employed since it is easily doable even for large size molecules. It requires calculation of just a single Hessian matrix. However, the approach remarkably neglects any dynamical and anharmonic effects that may become crucial when interactions such as hydrogen bonds dominate the interaction picture.17 Even if several research groups have provided full sets of scaling constants for the different levels of theory and electronic basis sets employed16,18 as well as different scaling constants for calculations of different observables (frequency, zero point energy, enthalpy, entropy, etc.), the scaled harmonic approach is misleading for the interpretation of the glycine proton-bound dimer spectrum. Furthermore, it is generally classified as an ab initio method in an improper way, since an empirical tuning parameter is enforced.
Results and discussion
Protonated glycine dimer
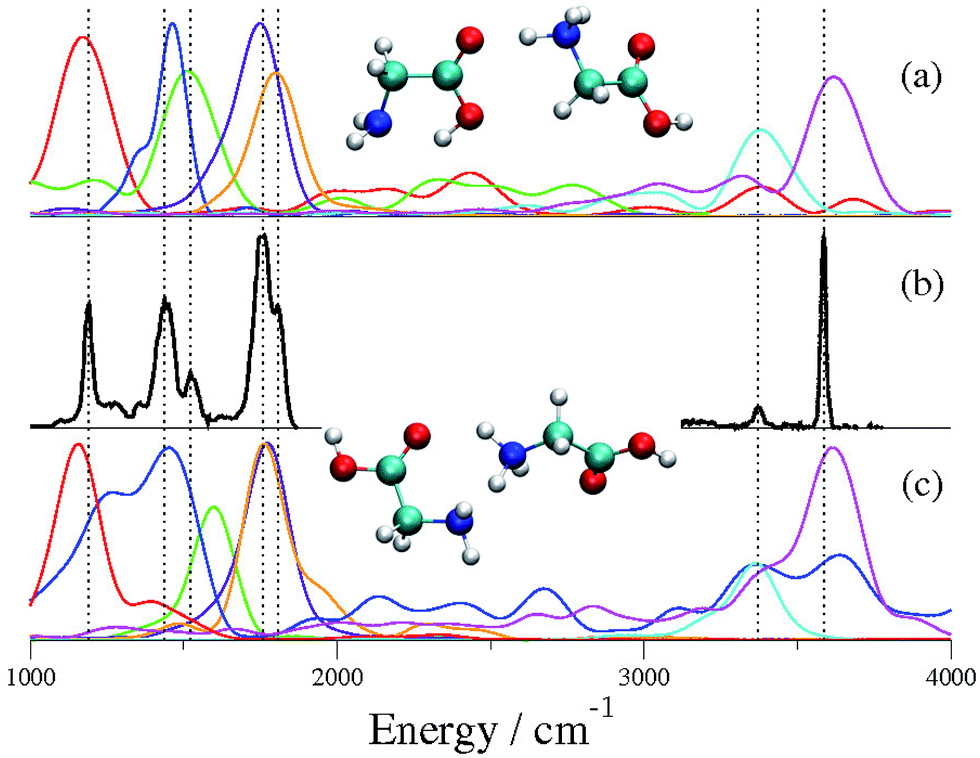
To prove the inaccuracy and ineffectiveness of scaled-harmonic frequency estimates, following ref. 15, we performed geometry optimizations of Gly2H+ at the DFT-B3LYP level of theory with the 6-311+G(d,p) basis set, followed by a scaled-harmonic analysis. In agreement with the previous studies, we found that conformer CS01 is the lowest energy conformer, while CS02 is just 2.1 kcal mol−1 higher in energy. Given this small difference in energy, the determination of the conformer responsible for the IRMPD spectrum in panel (a) of Fig. 1 implies the assignment and the interpretation of the spectrum in full detail. By scaling all the harmonic frequencies at the CS01 geometry by a factor equal to 0.96, panel (b), the OH and NH stretching region is well reproduced while the mid-range one is not. Likewise, simulations based on the 0.97 factor suggested in ref. 14 would follow a similar destiny. Conversely, if the scaling factor equal to 0.985 suggested in ref. 15 is applied, then the stick spectrum of panel (c) is found. This scaled-harmonic spectrum mimics reasonably well the experimental peaks in the mid-range region between 1000 cm−1 and 2000 cm−1. However, the same scaling factor performs poorly in the H-stretching region above 3000 cm−1. Since the anharmonicity parameters for each vibration are not known a priori, we conclude that it is impossible to model IR spectra on the basis of harmonic calculations, at least when different conformers are present. For these reasons, we deem that previous investigations cannot be recognized as conclusive ones. | ||
| Fig. 1 Vibrational spectrum of Gly2H+. Panel (a) is the IRMPD spectrum that has been obtained by combining figures from ref. 14 and 15. Panels (b) and (c) report the stick harmonic spectra at the DFT-B3LYP level with the 6-311+G(d,p) basis set, scaled respectively by a factor of 0.96 and 0.985 (ref. 15). | ||
A computational technique able to account for conformational and dynamical effects should be conveniently based on (quantum) molecular dynamics, since the dynamics allows exploration of the actual Potential Energy Surface (PES) even far from the harmonic region.19–21 Such a non-local approach may be crucial for a correct interpretation of hydrogen bonding.
The quantum dynamical way to spectroscopy and frequency computation (i.e. the power spectrum) is given by the Fourier transform of the autocorrelation of the time-evolved nuclear wavepacket averaged over the quantum density matrix of vibrational states22
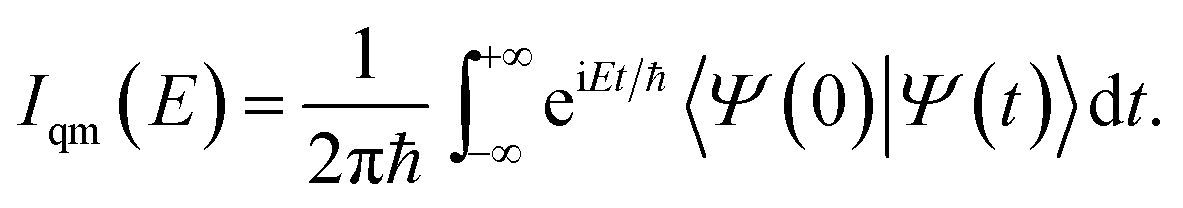 | (1) |
Eqn (1) includes all quantum mechanical spectroscopic information like zero point energy (ZPE), fundamental and overtone frequencies, anharmonicities and couplings, tunneling effects and quantum resonances between overtones and fundamentals. The classical equivalent of eqn (1) is the Fourier transform of the velocity autocorrelation function
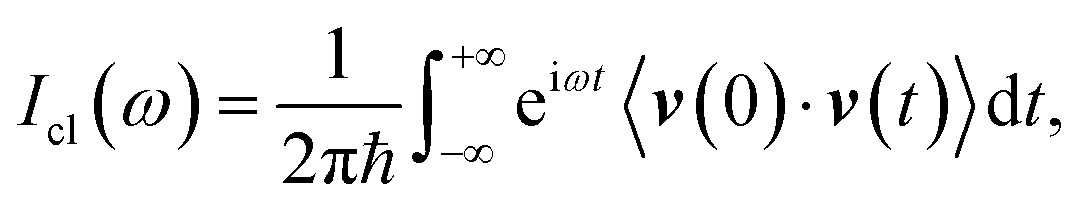 | (2) |
Unfortunately, when dealing with high dimensional systems, purely quantum mechanical simulations based on eqn (1) are out of reach because of the so-called curse of the dimensionality problem. Furthermore, accurate and fast-to-evaluate analytical PESs are usually not available and must be replaced by more computationally expensive ab initio “on-the-fly” calculations, whereby the dynamics can be performed (even if at a lower level of electronic theory), and which demand for a theoretical formalism that permits a convenient interface to them. Semiclassical (SC) dynamics can be interfaced to ab initio “on-the-fly” calculations straightforwardly, so we adopted it to calculate Iqm(E). In a SC simulation, quantum mechanical effects are reproduced by employing many entangled coherent states, which are time-evolved on top of classical trajectories. The semiclassical initial value representation (SC-IVR) approximation27–30 of quantum dynamics has been proved to be reliable and robust.31–47 Recently, we developed an SC-IVR method based on a “divide-and-conquer” strategy (DC SCIVR) to undertake the spectroscopic calculations of eqn (1).48 The method can deal with very high dimensional molecular and supra-molecular systems, and it is very accurate when compared to available exact vibrational quantum mechanical calculations.49,50 Specifically, our DC-SCIVR method has been tested successfully against systems with up to hundreds of degrees of freedom,48 and in particular it has been employed to study the vibrational features of the protonated water dimer, the Zundel cation. The results are very accurate (within a few wavenumbers) even for the vibrational bands of the proton doublet in the region of the O–H–O stretching frequency and associated with the proton transfer (∼1000 cm−1), when compared to exact grid-based quantum dynamics results on the same PES. The DC-SCIVR idea is to calculate the power spectrum as a sum of partial reduced-dimensionality spectra. First, full dimensional ab initio on-the-fly classical trajectories are calculated. Then, the normal modes are divided into vibrational subspaces according to their mutual coupling,48,49,51 and the partial spectra are calculated by projecting the classical trajectory information according to the following formula48
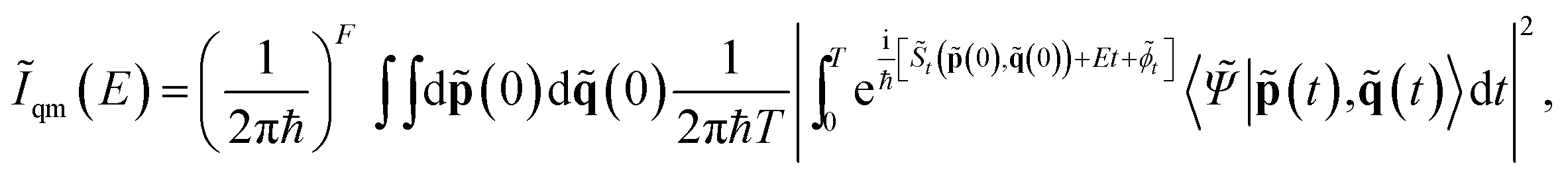 | (3) |
![[S with combining tilde]](https://www.rsc.org/images/entities/i_char_0053_0303.gif) t(
t(![[p with combining tilde]](https://www.rsc.org/images/entities/b_char_0070_0303.gif) (0),
(0),![[q with combining tilde]](https://www.rsc.org/images/entities/b_char_0071_0303.gif) (0))), the phase of the semiclassical prefactor (
(0))), the phase of the semiclassical prefactor (![[small phi, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e2.gif) t),46 and the overlap between the reference state |
t),46 and the overlap between the reference state |![[capital Psi, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e11c.gif) 〉 and the coherent state |(t),(t)〉. More details can be found in the ESI.†Fig. 2 shows the resulting DC-SCIVR power spectra, where the ZPE has been shifted to zero for better comparison with the experiments, which cannot be used to measure it. The reported peaks are those of the vibrational modes which have the highest oscillator strengths.
〉 and the coherent state |(t),(t)〉. More details can be found in the ESI.†Fig. 2 shows the resulting DC-SCIVR power spectra, where the ZPE has been shifted to zero for better comparison with the experiments, which cannot be used to measure it. The reported peaks are those of the vibrational modes which have the highest oscillator strengths.
 | ||
| Fig. 2 DC-SCIVR power spectra for conformer CS01 in panel (a) and CS02 in panel (c) of Gly2H+. Panel (b) is the experimental IRMPD spectrum. Starting from the left, the red continuous line is for the free OH bending mode, the blue one is for the OH hydrogen-bonded bending, the green line is for the umbrella inversion mode, the violet and orange ones are both for carbonyl stretchings, the cyan line is for the NH symmetric stretching and, finally, the magenta one is for the free OH stretching. | ||
A peak-by-peak comparison between the semiclassical spectra of the two possible conformers shown in panel (a) and (c) in Fig. 2 and the experimental one which appears in the middle panel (b) shows clearly that in panel (a) all the experimental vibrational features are accurately reproduced, differently from the case of panel (c) where the agreement is not as good. By looking at the actual calculated frequencies for each peak, a mean absolute error from the experimental peaks of 14 cm−1 is associated with the spectrum of conformer CS01 in panel (a), while a deviation of 32 cm−1 characterizes the spectrum of conformer CS02 in panel (c). This discrepancy in the mean absolute error values is pretty significant and conclusive even upon weighing in the typical accuracy of semiclassical calculations.49 More specifically, in the fingerprint region, the two carbonyl stretching frequencies (violet and orange lines) are degenerate in the case of the CS02 conformer, while they are not for CS01, in agreement with the experimental spectrum. Furthermore, the two vibrational peaks at around 1500 cm−1, corresponding to the fundamentals for the OH hydrogen-bonded bending (blue profile) and the umbrella inversion mode (green line), are well reproduced in panel (a), while panel (c) shows more elaborate vibrational coupling features. Similar considerations are valid also for the symmetric NH stretching and the free OH stretching in the high frequency region, with the spectrum in panel (a) better resembling the experimental profile. We stress that our assignment of the experimental spectrum to conformer CS01 has been obtained without any tuning parameter and in a fully ab initio way. Furthermore, no ad hoc frequency scaling factor nor fitting procedures have been applied to the calculated spectra reported here. From our simulations we note that there are numerous peaks in the frequency domain between the fingerprint peaks and the NH and OH stretching region which have not been detected by the experiments. These peaks belong to mode overtones and combinations of them, and are consequently experimentally much less intense. Finally, energetics calculations for both conformers have been performed at a quantum level by adding ZPE values to the classical minimum, i.e. the bottom of the well, and CS01 has been found to be about 2.5 kcal mol−1 more stable than CS02, revealing that the conformer that we identified as the major contributor to the experimental IR spectrum is also the (quantum mechanical) global minimum.
Despite the success of the semiclassical simulations presented above, one key methodological question remains open. In fact, if on the one hand quantum effects are hallmarks of spectroscopy, on the other hand for systems of dimensionality similar or bigger than Gly2H+ it could be argued that a classical picture is enough to describe with sufficient accuracy the spectral features of at least fundamental transitions. This would require much less effort since a semiclassical simulation is significantly more computationally intense than a classical one. Eqn (2) requires calculating at each time step the cartesian velocities v(t) of the nuclei only. Instead, in eqn (3), the calculation of Iqm(E) in semiclassical approximation implies the evaluation of not only the position and the velocities of the nuclei at each time step, but also the nuclear Hessian (for evolution of the phase term). This is about an order of magnitude more expensive in terms of computational efforts.
Protonated glycine tagged by hydrogen molecules
To point out clearly the importance of quantum mechanical effects in vibrational spectra influenced by proton dynamics, we consider that, recently, Masson, Williams and Rizzo published a series of very interesting IRMPD spectra, where protonated glycine, GlyH+, was tagged by an increasing and controlled number of hydrogen molecules.3 We focus particularly on two of these investigations. The first one regards protonated glycine solvated by a single hydrogen molecule (GlyH + H2)+, while in the other instance three H2 molecules are involved (GlyH + 3H2)+. The minimum geometries of these systems are reported in Fig. 3.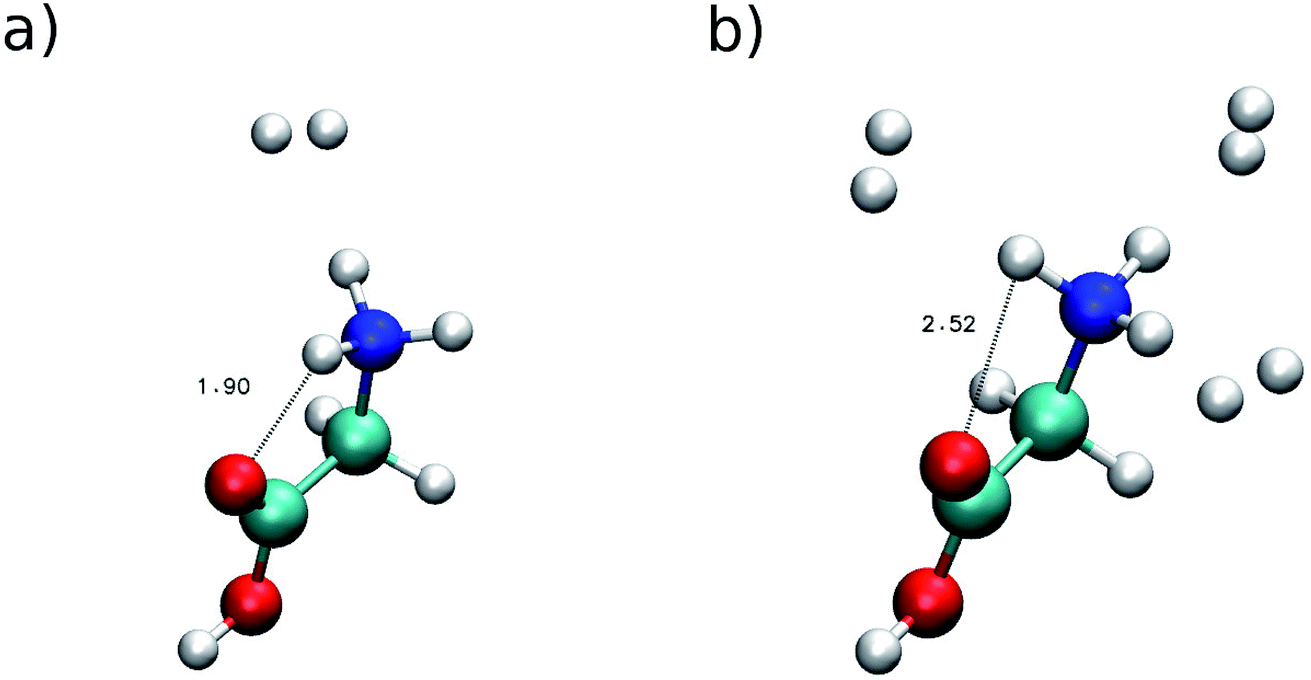 | ||
| Fig. 3 Minimum configuration geometries. Panel (a) for (GlyH + H2)+ and (b) for (GlyH + 3H2)+. Distances are in Å. | ||
This figure suggests that panel (a) is characterized by a strong hydrogen bond interaction between one of the amide hydrogens and the carbonylic oxygen atom of the carboxylic acid group, while in panel (b) the presence of the three hydrogen molecules may suppress the hydrogen bond interaction by inducing a reorientation of the amide group. In panel (a) the H⋯O distance at the minimum geometry is about 1.90 Å, while in panel (b) the distance is equal to 2.52 Å. This last distance is still shorter than the sum of hydrogen and oxygen van der Waals radii (2.72 Å), which is considered, as a rule of thumb, the limit for hydrogen bonding.
To check whether the hydrogen bond is lifted or not by virtue of the H2 tagging process and if quantum mechanical effects play any relevant role in this kind of interaction, we first performed ab initio “on-the-fly” DC-SCIVR simulations using the DFT-B3LYP level of theory and employing the aVDZ basis set, and then compared the results with the experimental spectra. Fig. 4 reports the IRMPD and simulated spectra for (GlyH + H2)+.
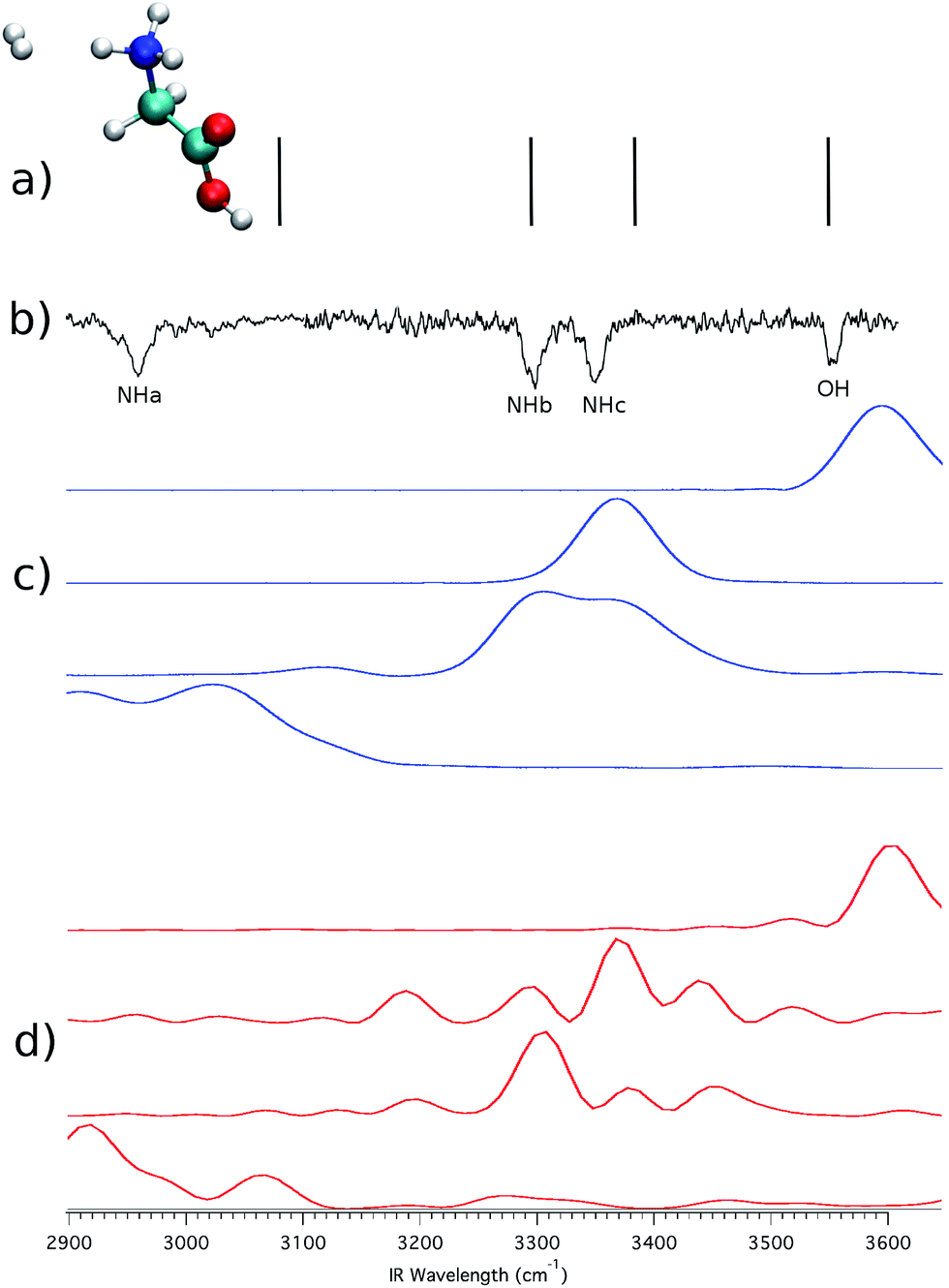 | ||
| Fig. 4 (GlyH + H2)+ spectra. In (a) the scaled-harmonic stick spectrum is presented; panel (b) shows the experimental IRMPD spectrum;3 (c) is the Icl classical spectrum according to eqn (2), and (d) is the Iqm semiclassical spectrum from eqn (1). In the experimental spectrum, the label NHa is for the hydrogen bonded NH stretching frequencies, while NHb and NHc indicate the unbound ones. OH labels the homonymous stretching frequency. Numerical values of the frequencies are reported in the ESI.† | ||
The experimental results are reproduced in spectrum (b), where the amide N–H stretching involved in the intramolecular hydrogen bond is located between 2950 and 3000 cm−1 and labeled as NHa, while NHb and NHc indicate the free NH stretching peaks. The signal at 3546 cm−1 corresponds to the free OH stretching. Upon adoption of a scaling coefficient equal to 0.96 to match the harmonic OH frequency (at the MP2 level of theory with the aVDZ basis set) with the experimental OH band, the NHb and NHc peaks are also reproduced quite well, while NHa is off by about 117 cm−1, as shown by the stick spectrum on the top panel of Fig. 4.3 Moving to classical simulations, we calculated the classical spectrum, Icl, by means of eqn (2) and report it in panel (c) separately for each mode for a better comparison with the experiment. The main spectroscopic features are reproduced, even if the signal corresponding to the NHa and NHb bands is quite broad. Finally, in the semiclassical spectrum of panel (d), calculated with eqn (3), the fundamental bands are accurately reproduced, with the addition of overtones that are too weak to be detected in the experiment and that are missing in the classical and scaled-harmonic simulations. In general, the simulated peaks are broader than the experimental ones because, on the one hand, experiments are performed at very low temperature (a few K) and rotations are hindered or even blocked, while, on the other hand, in the simulations the dynamics is propagated only for a short time (less than 1 ps) before the Fourier transform is undertaken, and every mode (including internal rotators) is given an amount of energy according to its contribution to the ZPE and left free to evolve without any artificial constraints. Furthermore, the dynamics of the hydrogen bonding may contribute to the broadening of the NH stretching bands as shown by Gaigeot and coworkers by applying finite temperature classical molecular dynamics to small protonated peptides, such as Ala2H+ and Ala3H+.6,7,25
We now turn to the other system, i.e. (GlyH + 3H2)+, reported in panel (b) of Fig. 3. Fig. 5 shows the spectra corresponding to those presented in Fig. 4 but this time for this bigger system. One may think that hydrogen molecules do not interact significantly with GlyH+ and that it is possible to obtain in good approximation the IRMPD signal of the isolated molecule. However, there are clear differences between the experimental spectra of Fig. 4 and 5. One of them is represented by the blue-shifted NHa peak. As usual, scaled-harmonic calculations are shown as a stick spectrum in panel (a) of Fig. 5 and they fail to account correctly for the anharmonicity of the NHa stretching motion. Once more, the scaling of harmonic frequencies brings us to a dead end. A classical approach based on eqn (2) is not helpful in this circumstance as demonstrated by the set of spectra in panel (c) of Fig. 5 that clearly show classical mechanics overestimating the NHa stretching frequency. We believe that this is due to the fact that the intramolecular hydrogen bond and the dynamics of the involved proton have a prevalent quantum nature. In other words, a scaled-harmonic or classical dynamics approach leads to the wrong conclusion that the intramolecular hydrogen bond is broken in the presence of 3H2 molecules interacting with GlyH+. Conversely, a semiclassical simulation based on eqn (1), reported in panel (d), reproduces accurately all the vibrational features of the IRMPD spectrum also in this case, including the strong anharmonicity of the NHa stretching and the consequent red shift, thus confirming that the hydrogen bond interaction is only weakened and not completely broken, even in the presence of three H2 molecules coordinated to the amide group.
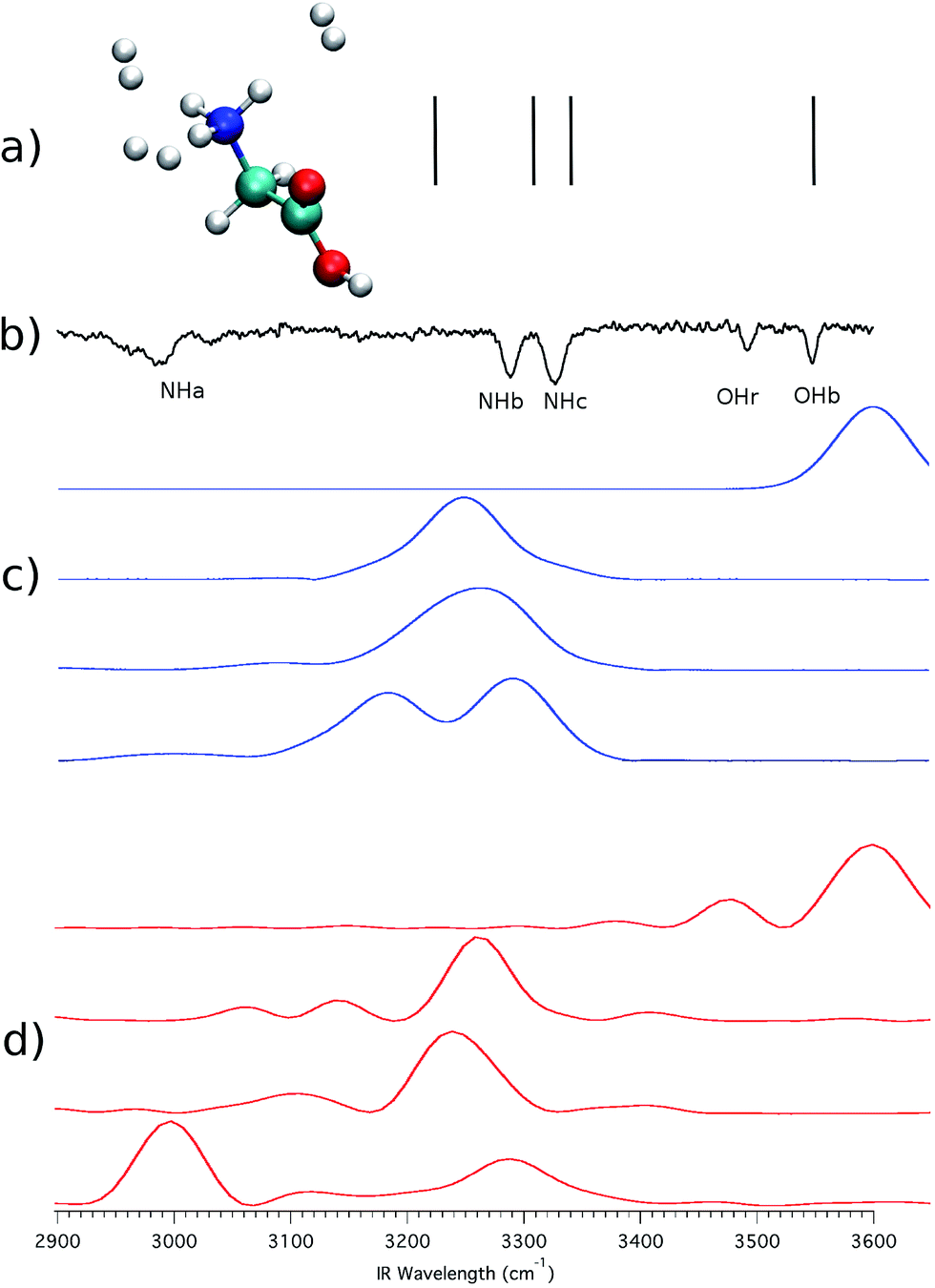 | ||
| Fig. 5 The same as in Fig. 4 but for (GlyH + 3H2)+. Numerical values of the frequencies are reported in the ESI.† | ||
Another key difference between Fig. 4 and 5 lies in the appearance of a second OH stretching band, located at 3491 cm−1, and labeled as OHr. Masson et al.3 suggested that this band is the result of a configuration where one of the three H2 molecules interacts with the carboxylic group. Indeed, the peak is red-shifted by about 55 cm−1 with respect to the free OH stretching (the OHb band). This would mean that the experimental spectrum (b) of Fig. 5 actually originates from two different conformers. To validate the previous conformational hypothesis we consider a configuration with a single H2 molecule tagging the carboxylic group. This geometry is not stable experimentally (in fact the OHr peak appears only when 3 or more H2 molecules are involved), but it can be investigated theoretically. The system is reported in panel (a) of Fig. 6, and we focus on the OH stretching.
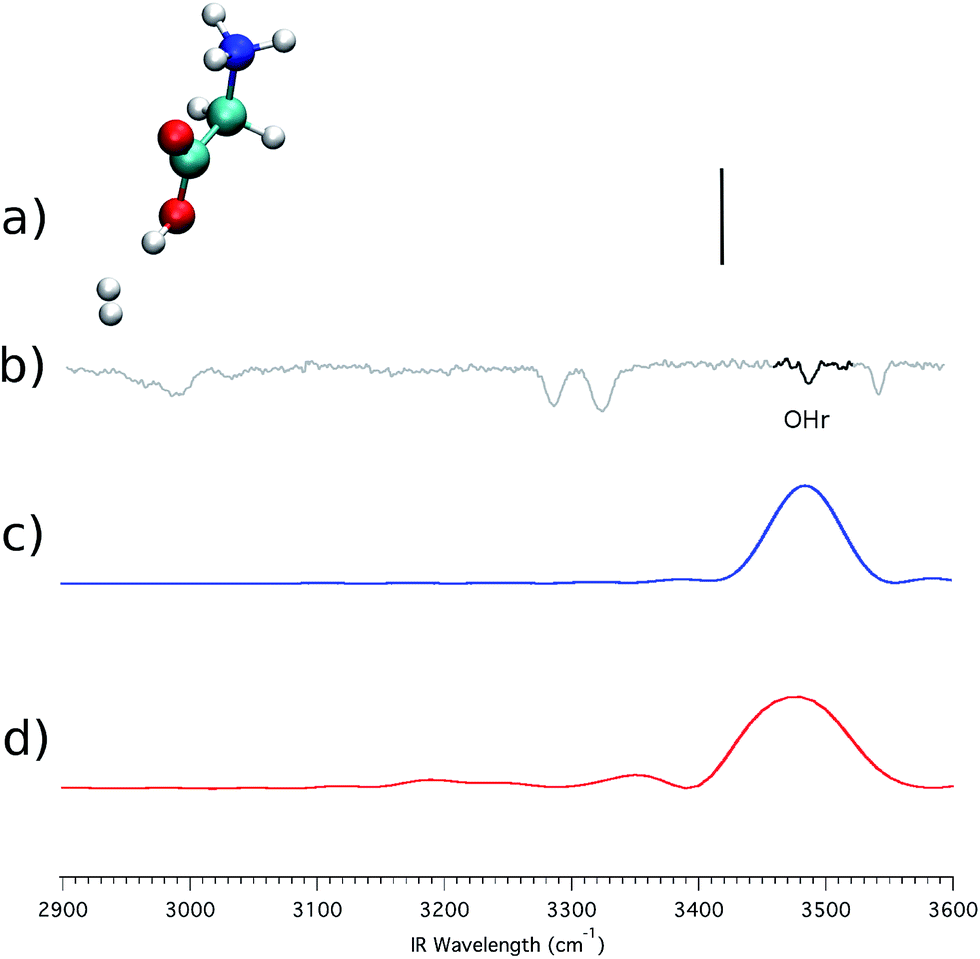 | ||
| Fig. 6 (GlyH + H2)+ spectra, with the H2 molecule coordinated to the carboxylic group. Labels (a)–(d) are as in Fig. 4. | ||
Still in panel (a) the harmonic stick spectrum (at the DFT-B3LYP level of theory with the aVDZ basis set) for the OH stretching is presented after scaling by a factor of 0.96, which is the same coefficient employed in the previous simulations. This estimate is definitely off the mark. In contrast, both the classical (panel c) and the semiclassical (panel d) peaks are quite accurate for the OH stretching, confirming that the OHr band is indeed due to the interaction between a H2 molecule and the carboxylic group. The presence of the H2 molecule weakens the OH bond, leading to the observed red shift.
Amino group deuteration
To further prove that the differences between the classical and the semiclassical spectra reported respectively in panel (c) and (d) of Fig. 5 are due to quantum mechanical effects only, we quenched them by deuterating all the three hydrogen atoms, pictorially represented by the gray atoms of the molecule in Fig. 7.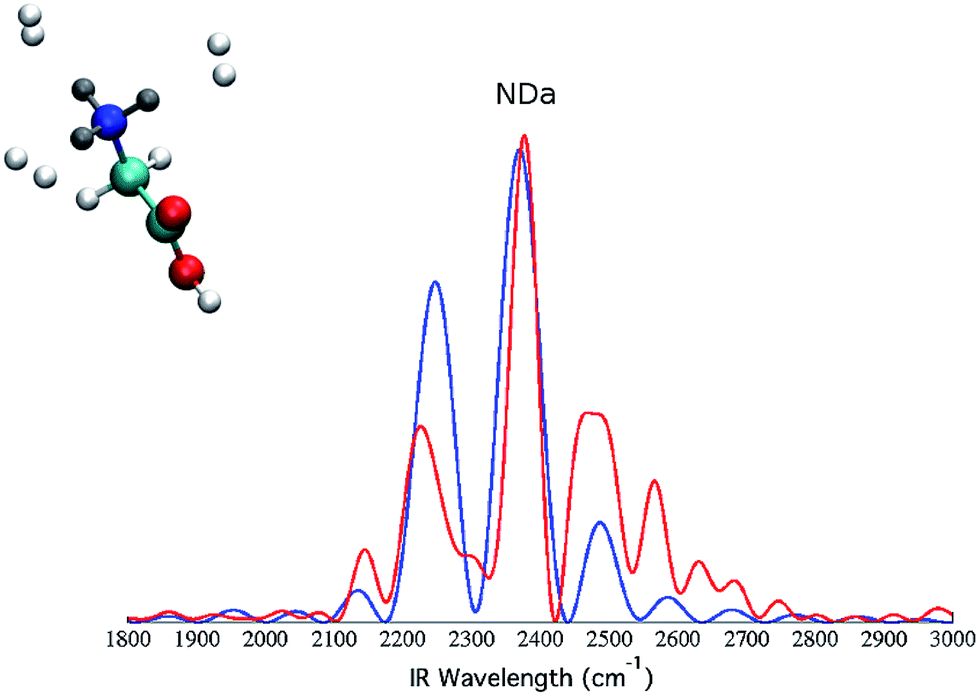 | ||
| Fig. 7 Selectively deuterated (GlyH + 3H2)+ system and the corresponding classical (blue continuous line) and semiclassical (red continuous line) spectra for the amide stretching mode. The deuterium atoms are colored in gray. | ||
Then, we calculated the spectra for the deuterated GlyH+ molecule tagged by the three H2 molecules. We focus on the amino group modes and selectively plot the NDa (previously NHa) band, both using the classical spectrum formulation of eqn (2) and the quantum formulation of eqn (1) and (3). The results are reported in Fig. 7. The NDa band is centered around 2350 cm−1, which is significantly red-shifted with respect to the previous NHa band, because of the heavier deuterium mass. However, we note that the classical and semiclassical peaks are almost identical in this case. This proves that the previous discrepancy of about 150 cm−1 between the classical and the semiclassical NHa band location of Fig. 5 was exclusively due to a quantum mechanical effect of the light hydrogen atom. It is quite surprising that this quantum anharmonic effect is so huge. However, when considering the strong anharmonicity of the NHa potential well and the consequent huge delocalization of the quantum mechanical vibrational eigenfunction (as pictorially represented in the ESI†), the prominent quantum mechanical nature of this hydrogen bond interaction appears fully justified.
Conclusions
We conclude by remarking the importance of employing a quantum dynamical approach in calculating vibrational frequencies and going beyond the scaled-harmonic level. This has been demonstrated by means of divide-and-conquer semiclassical dynamics, which has permitted us to reproduce experimental anharmonicities quite well and to explain some open issues involving the protonated glycine dimer and the tagging of protonated glycine with molecular hydrogen. In particular, for the former, the CS01 conformer has been assigned consistently in the whole frequency range, while, for the latter, some peculiar spectral quantum features due to hydrogen bonding and intermolecular interactions have been rigorously explained, a task that neither scaled-harmonic nor classical approaches were able to accomplish. On this point, we note that the reference experiments were performed at very low temperatures, so we did not run standard thermalized classical simulations (which would have provided just harmonic estimates), but we estimated a classical analog of the quantum mechanical vibrational spectral density. Nevertheless, these classically inspired calculations were not as satisfactory as the semiclassical ones. Interestingly, due to the interaction, vibrational frequency calculations of the tagging H2 molecules display a red shift (∼50 cm−1) comparable to that of the OH stretching of glycine. Remarkably, the adopted DFT-B3LYP level of electronic theory is not only suitable for a realistic description of the entire supramolecular system but is also able to provide frequency estimates in quantitative agreement with the experiments. Finally, we are able to answer the three questions with which we introduced the paper by stating that quantum effects certainly play a very important role in these protonated systems; the intramolecular hydrogen bond interaction has a strong impact on the NH stretching revealing an elevated degree of delocalization of the proton shared with the carboxylic group; the very same interaction is influenced by the dynamics with the hydrogen bond being less and less directional as the number of tagging molecules increases. All these findings point out very clearly the crucial role that quantum dynamics may play, suggesting that it should not be neglected even when dealing with larger systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the European Research Council (ERC) under the European Union's Horizon 2020 Research and Innovation Programme (grant agreement No. [647107] – SEMICOMPLEX – ERC-2014-CoG). We thank Università degli Studi di Milano for further computational time at CINECA (Italian Supercomputing Center), and CINECA under the ISCRA-B (grant QUASP) and LISA (grant GREENTI) initiatives for access to high performance computing resources.References
- T. H. Lowry and K. S. Richardson, Mechanism and theory in organic chemistry, Harper & Row, New York, 1987 Search PubMed.
- U. J. Lorenz and T. R. Rizzo, J. Am. Chem. Soc., 2012, 134, 11053–11055 CrossRef CAS PubMed.
- A. Masson, E. R. Williams and T. R. Rizzo, J. Chem. Phys., 2015, 143, 104313 CrossRef PubMed.
- K. Rajabi and T. D. Fridgen, J. Phys. Chem. A, 2008, 112, 23–30 CrossRef CAS PubMed.
- C. G. Atkins, K. Rajabi, E. A. Gillis and T. D. Fridgen, J. Phys. Chem. A, 2008, 112, 10220–10225 CrossRef CAS PubMed.
- A. Cimas, T. Vaden, T. De Boer, L. Snoek and M.-P. Gaigeot, J. Chem. Theory Comput., 2009, 5, 1068–1078 CrossRef CAS PubMed.
- D. Marinica, G. Gregoire, C. Desfrancois, J. Schermann, D. Borgis and M. Gaigeot, J. Phys. Chem. A, 2006, 110, 8802–8810 CrossRef CAS PubMed.
- B. Brauer, G. M. Chaban and R. B. Gerber, Phys. Chem. Chem. Phys., 2004, 6, 2543–2556 RSC.
- A. A. Adesokan and R. Gerber, J. Phys. Chem. A, 2008, 113, 1905–1912 CrossRef PubMed.
- J. R. Eyler, Mass Spectrom. Rev., 2009, 28, 448–467 CrossRef CAS PubMed.
- T. D. Fridgen, Mass Spectrom. Rev., 2009, 28, 586–607 CrossRef CAS PubMed.
- N. C. Polfer, Chem. Soc. Rev., 2011, 40, 2211–2221 RSC.
- L. Jasikova and J. Roithová, Chem.–Eur. J., 2018, 3374–3390 CrossRef CAS PubMed.
- H.-B. Oh, C. Lin, H. Y. Hwang, H. Zhai, K. Breuker, V. Zabrouskov, B. K. Carpenter and F. W. McLafferty, J. Am. Chem. Soc., 2005, 127, 4076–4083 CrossRef CAS PubMed.
- R. Wu and T. B. McMahon, J. Am. Chem. Soc., 2007, 129, 4864–4865 CrossRef CAS PubMed.
- A. P. Scott and L. Radom, J. Phys. Chem., 1996, 100, 16502–16513 CrossRef CAS.
- U. Buck, I. Ettischer, M. Melzer, V. Buch and J. Sadlej, Phys. Rev. Lett., 1998, 80, 2578 CrossRef CAS.
- I. Alecu, J. Zheng, Y. Zhao and D. G. Truhlar, J. Chem. Theory Comput., 2010, 6, 2872–2887 CrossRef CAS PubMed.
- R. Iftimie, P. Minary and M. E. Tuckerman, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 6654–6659 CrossRef CAS PubMed.
- M. Tuckerman, K. Laasonen, M. Sprik and M. Parrinello, J. Chem. Phys., 1995, 103, 150–161 CrossRef CAS.
- S. Pratihar, X. Ma, Z. Homayoon, G. L. Barnes and W. L. Hase, J. Am. Chem. Soc., 2017, 139, 3570–3590 CrossRef CAS PubMed.
- E. J. Heller, Acc. Chem. Res., 1981, 14, 368–375 CrossRef CAS.
- D. Marx and J. Hutter, Ab initio molecular dynamics: basic theory and advanced methods, Cambridge University Press, 2009 Search PubMed.
- G. Mathias, S. D. Ivanov, A. Witt, M. D. Baer and D. Marx, J. Chem. Theory Comput., 2011, 8, 224–234 CrossRef PubMed.
- M.-P. Gaigeot, Phys. Chem. Chem. Phys., 2010, 12, 3336–3359 RSC.
- D. R. Galimberti, A. Milani, M. Tommasini, C. Castiglioni and M.-P. Gaigeot, J. Chem. Theory Comput., 2017, 13, 3802–3813 CrossRef CAS PubMed.
- W. H. Miller, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 6660–6664 CrossRef CAS PubMed.
- W. H. Miller, J. Phys. Chem. A, 2001, 105, 2942–2955 CrossRef CAS.
- K. G. Kay, Annu. Rev. Phys. Chem., 2005, 56, 255–280 CrossRef CAS PubMed.
- E. Pollak, in The Semiclassical Initial Value Series Representation of the Quantum Propagator, ed. D. A. Micha and I. Burghardt, Springer Berlin Heidelberg, Berlin, Heidelberg, 2007, pp. 259–271 Search PubMed.
- R. Gelabert, X. Giménez, M. Thoss, H. Wang and W. H. Miller, J. Phys. Chem. A, 2000, 104, 10321–10327 CrossRef CAS.
- K. G. Kay, J. Chem. Phys., 1994, 100, 4377–4392 CrossRef CAS.
- K. G. Kay, J. Chem. Phys., 1994, 100, 4432–4445 CrossRef CAS.
- K. G. Kay, J. Chem. Phys., 1994, 101, 2250–2260 CrossRef.
- S. Zhang and E. Pollak, J. Chem. Phys., 2004, 121, 3384–3392 CrossRef CAS PubMed.
- S. Zhang and E. Pollak, J. Chem. Theory Comput., 2005, 1, 345–352 CrossRef CAS PubMed.
- F. Grossmann, M. Buchholz, E. Pollak and M. Nest, Phys. Rev. A: At., Mol., Opt. Phys., 2014, 89, 032104 CrossRef.
- E. Zambrano, M. Šulc and J. Vaníček, J. Chem. Phys., 2013, 139, 054109 CrossRef PubMed.
- M. Šulc and J. Vaníček, Mol. Phys., 2012, 110, 945–955 CrossRef.
- M. Ceotto, Y. Zhuang and W. L. Hase, J. Chem. Phys., 2013, 138, 054116 CrossRef PubMed.
- Y. Zhuang, M. R. Siebert, W. L. Hase, K. G. Kay and M. Ceotto, J. Chem. Theory Comput., 2012, 9, 54–64 CrossRef PubMed.
- M. Buchholz, F. Grossmann and M. Ceotto, J. Chem. Phys., 2016, 144, 094102 CrossRef PubMed.
- M. Ceotto, S. Atahan, G. F. Tantardini and A. Aspuru-Guzik, J. Chem. Phys., 2009, 130, 234113 CrossRef PubMed.
- R. Conte, A. Aspuru-Guzik and M. Ceotto, J. Phys. Chem. Lett., 2013, 4, 3407–3412 CrossRef CAS PubMed.
- F. Gabas, R. Conte and M. Ceotto, J. Chem. Theory Comput., 2017, 13, 2378 CrossRef CAS PubMed.
- G. Di Liberto and M. Ceotto, J. Chem. Phys., 2016, 145, 144107 CrossRef PubMed.
- M. Buchholz, F. Grossmann and M. Ceotto, J. Chem. Phys., 2018, 148, 114107 CrossRef PubMed.
- M. Ceotto, G. Di Liberto and R. Conte, Phys. Rev. Lett., 2017, 119, 010401 CrossRef PubMed.
- G. Di Liberto, R. Conte and M. Ceotto, J. Chem. Phys., 2018, 148, 014307 CrossRef PubMed.
- G. Di Liberto, R. Conte and M. Ceotto, J. Chem. Phys., 2018, 148, 104302 CrossRef PubMed.
- M. Wehrle, M. Sulc and J. Vanicek, J. Chem. Phys., 2014, 140, 244114 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Computational setup; geometries, harmonic frequencies and semiclassical vibrational levels of the systems investigated; description of the theory. See DOI: 10.1039/c8sc03041c |
| This journal is © The Royal Society of Chemistry 2018 |