
Asymmetric ring opening of racemic epoxides for enantioselective synthesis of (S)-β-amino alcohols by a cofactor self-sufficient cascade biocatalysis system†
Jian-Dong
Zhang
 *a,
Xiao-Xiao
Yang
a,
Qiao
Jia
a,
Jian-Wei
Zhao
a,
Li-Li
Gao
b,
When-Chao
Gao
a,
Hong-Hong
Chang
a,
Wen-Long
Wei
a and
Jian-He
Xu
c
*a,
Xiao-Xiao
Yang
a,
Qiao
Jia
a,
Jian-Wei
Zhao
a,
Li-Li
Gao
b,
When-Chao
Gao
a,
Hong-Hong
Chang
a,
Wen-Long
Wei
a and
Jian-He
Xu
c
aDepartment of Biological and Pharmaceutical Engineering, College of Biomedical Engineering, Taiyuan University of Technology, Taiyuan, Shanxi, China. E-mail: zhangjiandong@tyut.edu.cn
bCollege of Environmental Science and Engineering, Taiyuan University of Technology, Taiyuan, Shanxi, China
cLaboratory of Biocatalysis and Bioprocessing, State Key Laboratory of Bioreactor Engineering, East China University of Science and Technology, Shanghai, China
First published on 4th December 2018
Abstract
A novel one-pot epoxide hydrolase/alcohol dehydrogenase/transaminase cascade process for the asymmetric ring opening of racemic epoxides to enantiopure β-amino alcohols is reported. The product (S)-β-amino alcohols were obtained in 97–99% ee and 79–99% conversion from readily available racemic epoxides.
Chiral β-amino alcohols are very important chiral molecules that are used as building block and structural motifs in pharmaceutically-active molecules and natural products, and serve as main sources of chirality in asymmetric synthesis.1 For instance, (S)-(+)-2-amino-1-butanol 4a is utilized as an intermediate for the synthesis of ethambutol (EMB), which is an important anti-tubercular drug,1a (S)-2-amino-2-phenylethanol 4c is used as a building block for the preparation of indolin-2-one derivatives as potent p21-activated kinase (PAK4) inhibitors,1c and (S)-2-amino-2-(4-bromophenyl) ethanol 4f is used as an intermediate for the synthesis of 4, 4′-bis(biphenyl) substituted bis(oxazoline) (BOX) ligands, which are among the most widely studied ligands in asymmetric catalysis.1e
To date, a wealth of methods have been developed for the synthesis of chiral β-amino alcohols.2 Due to the low cost and commercial availability of epoxides, the asymmetric ring opening (ARO) of epoxides provides efficient and straightforward access to chiral β-amino alcohols.3 In the past few decades, considerable efforts have been devoted to asymmetric ring opening reactions of epoxides using amines as nucleophiles that have shown moderate to good selectivity.4 However, most of these methodologies, which have to date predominantly been achieved using chemical methods, have suffered from one or more disadvantages, such as a limited scope of substrates, unsatisfactory product ee, moderate yields and regioselectivities, and the need for extreme conditions. Due to their exquisite chemo-, regio- and stereoselectivity, enzymes have been widely used as green catalysts in organic synthesis. Biocatalytic methods have been developed for the synthesis of chiral β-amino alcohols using lipase,5 transaminase,6 and ketone reductase.7 However, the high cost or commercial nonavailability of the substrates and their low yield (50% theoretical yield) of kinetic resolution has rendered some of these methods inconvenient and uneconomic. Recently, as a green and useful tool, cascade biocatalysis reactions catalyzed by multienzymatic systems have strongly moved into the focus of researchers because of their unique potential for the environmentally benign synthesis of chemicals,8 and several types of cascade biocatalysis systems have been developed for the synthesis of chiral amino alcohols,9 for instance in the transformation of glycolaldehyde and β-hydroxypyruvate to 2-amino-1,3,4-butanetriol (ABT),9b and the conversion of benzaldehyde and pyruvate to nor(pseudo)ephedrine N(P)E.9c However, to date, no reports have appeared in the literature detailing the asymmetric ring opening of racemic epoxides for the enantioselective synthesis of chiral β-amino alcohols via enzymatic catalysis, probably as a consequence of the difficulties in finding robust enzymes with excellent activity and enantioselectivity.10 Besides that, how to solve the problem of cofactor regeneration in some cascade biocatalysis reactions is still a challenge.
In this study, we envisioned a one-pot three-step enzymatic cascade reaction for the asymmetric ring opening of racemic epoxides to enantiopure β-amino alcohols (Scheme 1). The process includes the complete hydrolysis of a racemic epoxide to a vicinal diol by an epoxide hydrolase (EH), the oxidation of the vicinal diol intermediate to an α-hydroxy ketone by two stereocomplementary alcohol dehydrogenases (ADH) and the asymmetric reductive amination of an α-hydroxy ketone intermediate to an enantiopure β-amino alcohol by a transaminase (TA). ADH/(R)-(+)-1-phenylethylamine (R-MBA) was also utilized in the cascade system to drive the equilibrium towards the product and recycle the NAD+ cofactor.
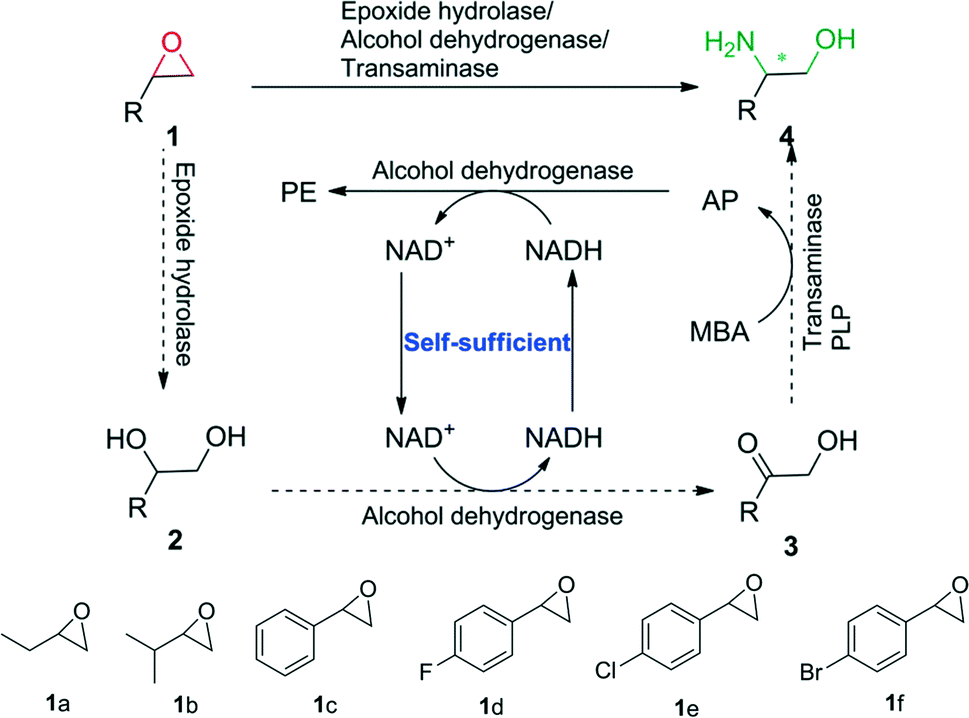 | ||
| Scheme 1 One-pot asymmetric ring opening of racemic epoxides to enantiopure β-amino alcohols via epoxide hydrolase/alcohol dehydrogenase/transaminase cascade biocatalysis. | ||
In order to realize the novel cascade biocatalysis system, the transaminases for the conversion of α-hydroxy ketones to enantiopure β-amino alcohols in the third step of the cascade biocatalysis system were selected. Based on our previously constructed high-throughput method for the screening of highly enantioselective amino alcohol specific transaminases,11 (R)-ω-transaminase (MVTA) from Mycobacterium vanbaalenii12 was identified as a robust enzyme with high activity and excellent enantioselectivity toward β-amino alcohols, and the enzyme was utilized to synthesise chiral β-amino alcohols from the corresponding α-hydroxy ketones 3a–f for the first time. After 2 to 3 h of reaction time, the conversion reached 99% with the product in 97–99% ee (see the ESI,† Table S1). Due to (R)-MBA being used as an efficient amine donor of MVTA, the co-product acetophenone (AP) was produced in the asymmetric reductive amination process. In order to recycle the NAD+ in the second step of the cascade biocatalysis system, we designed an in situ self-sufficient cofactor regeneration system (Scheme 1) that involves the alcohol dehydrogenase catalyzed oxidation of vicinal diols and the same alcohol dehydrogenase catalyzed AP reduction. Then, the alcohol dehydrogenase (ADH)/(R)-MBA in the cascade system can be utilized to recycle the NAD+ cofactor and simultaneously drive the equilibrium towards the product. In order to realize this process, sixteen alcohol dehydrogenases were tested for diol 2c oxidation activity and AP reduction activity (see the ESI,† Table S2). BDHA from Bacillus subtilis and GoSCR from Gluconobacter oxydans13 showed the highest activity toward the diols (R)-2c (0.80 U mg−1) and (S)-2c (0.12 U mg−1) and moderate activity toward AP (0.428 U mg−1 for BDHA and 1.257 U mg−1 for GoSCR), respectively. Therefore, BDHA and GoSCR were selected as promising candidates to catalyze the diol oxidation step of the cascade reaction. Substrate specific analysis revealed that BDHA and GoSCR have high activity and excellent enantioselectivity toward vicinal diol 2a–f, with an activity from 0.133 to 0.791 U mg−1 protein for BDHA toward (R)-2a–f, and 0.067 to 0.119 U mg−1 protein for GoSCR toward (S)-2a–f (see the ESI,† Table S3). For the first hydrolysis step, the epoxide hydrolase (SpEH) from Sphingomonas sp. HXN-200 was selected as a promising enzyme due to its remarkable activity in the enantioselective hydrolysis of epoxides.14 The substrate specific and enantioselectivity of SpEH was further analyzed with six racemic epoxides 1a–f, with 20 mM of substrate. After reaction for 2 to 3 h, the substrates were completely converted to vicinal diols 2 (99% conversion), forming the product diol 2 in 3–40% ee (see the ESI,† Table S4). As the intermediate product 2 was formed in the first step of the cascade reaction with low ee values, it was then essential to employ two stereocomplementary alcohol dehydrogenases (2,3-butanediol dehydrogenase, BDHA, and polyol dehydrogenase, GoSCR) in the second step of the cascade biocatalysis system to ensure the complete conversion of both enantiomers of the vicinal diol intermediate.
After SpEH, BDHA, GoSCR and MVTA were selected as the best candidates for the new cascade biocatalysis system. The combination of the last two steps into a simultaneous one-pot in the cascade reaction was first tested with BDHA, GoSCR and MVTA (Scheme 2). The reaction conditions for the conversion of diols to chiral β-amino alcohols were optimized using 2c as a model substrate, and the highest conversion of 4c was obtained at pH 7.4 and 30 °C, with 0.5 mM of NAD+ added (see the ESI,† Tables S5–S7). The adjustment of the two enzyme concentrations at a ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (20 mg mL−1 of ADH and 10 mg mL−1 of MVTA) resulted in the highest conversion of 4c (see the ESI,† Table S8). Under the optimized conditions, the enantioselective conversion of racemic diols 2c (50 mM) with the mixture of BDHA, GoSCR and MVTA (2:2:1) reached up to 99% with the product 4c in >99% ee. The generality of this cascade system (BDHA/GoSCR/MVTA) was further investigated using five other racemic diols (2a–b and 2d–f) and 31–99% conversions were obtained in 97–99% ee with a mixture of BDHA, GoSCR and MVTA (2:2:1) (see the ESI,† Table S9).
:1 (20 mg mL−1 of ADH and 10 mg mL−1 of MVTA) resulted in the highest conversion of 4c (see the ESI,† Table S8). Under the optimized conditions, the enantioselective conversion of racemic diols 2c (50 mM) with the mixture of BDHA, GoSCR and MVTA (2:2:1) reached up to 99% with the product 4c in >99% ee. The generality of this cascade system (BDHA/GoSCR/MVTA) was further investigated using five other racemic diols (2a–b and 2d–f) and 31–99% conversions were obtained in 97–99% ee with a mixture of BDHA, GoSCR and MVTA (2:2:1) (see the ESI,† Table S9).
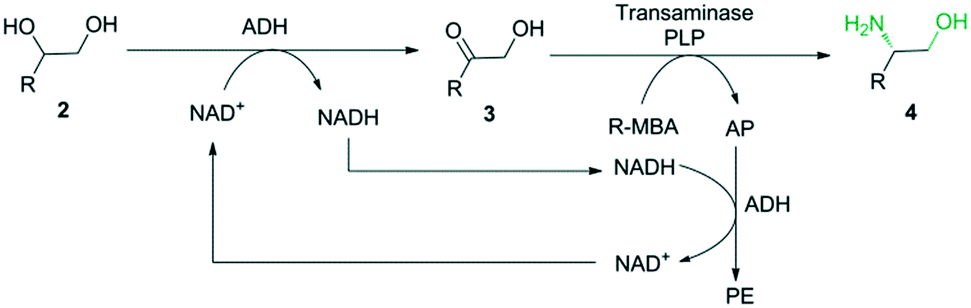 | ||
| Scheme 2 Enantioselective conversion of racemic vicinal diol 2 to enantiopure β-amino alcohol 4 using a mixture of BDHA, GoSCR and MVTA. | ||
We next sought to combine the SpEH, BDHA, GoSCR and MVTA biocatalysts in a one-pot cascade to convert 1 to 4. Racemic 1c was exposed to SpEH followed by BDHA/GoSCR and MVTA, and the target (S)-4c was obtained in 86.5%–99% conversion and >99% ee (Table 1, entries 3–4). The generality of the cascade biocatalysis system was investigated by examining five other racemic epoxides (1a–b and 1d–f) using SpEH, BDHA/GoSCR and MVTA (1:2:2:1) in the presence of 0.5 mM of NAD+, and the (S)-β-amino alcohols 4a–b were obtained in 97–99% ee and 47.3%–61.2% conversion from the racemic epoxides 1a–b (20 mM) after 24 h of reaction time (Table 1, entries 1–2). For the halogen para substituted styrene oxide substrates 1d–f, good to excellent conversions (34%–96.9%) of the (S)-β-amino alcohols 4d–f were obtained in >99% ee (Table 1, entries 4–6).
| Entry | Sub. | Sub. conc [mM] | SpEH [mg mL−1] | BDHA [mg mL−1] | GoSCR [mg mL−1] | MVTA [mg mL−1] | Time [h] | Conv. to 4a [%] | (S)-4 eeb [%] |
|---|---|---|---|---|---|---|---|---|---|
| The reactions were conducted using 25–55 mM of the amine donor (R-MBA) in 100 mM sodium phosphate buffer (pH 7.5) containing 0.1 mM pyridoxal-5′-phosphate (PLP), 0.5 mM NAD+, 20–50 mM substrates, and 10% DMSO at 30 °C. a Conversion was determined by GC, error limit: <2% of the stated values. b ee values were determined by chiral GC. | |||||||||
| 1 | 1a | 20 | 10 | 20 | 20 | 10 | 24 | 47.3 | 97 |
| 2 | 1b | 20 | 10 | 20 | 20 | 10 | 24 | 61.2 | 98 |
| 3 | 1c | 20 | 10 | 20 | 20 | 10 | 16 | 99.0 | >99 |
| 4 | 1c | 30 | 20 | 40 | 40 | 20 | 16 | 86.5 | >99 |
| 5 | 1d | 20 | 10 | 20 | 20 | 10 | 24 | 96.9 | >99 |
| 6 | 1e | 20 | 10 | 20 | 20 | 10 | 24 | 70.5 | >99 |
| 7 | 1f | 20 | 10 | 20 | 20 | 10 | 24 | 34.0 | >99 |
Compared to enzymatic catalysis, whole-cell biocatalysts provide unique properties,15 such as that the intact cells can protect the enzyme from rapid deactivation, and the cofactor NAD+/NADH in cells can be used for targeted reactions with co-factor recycling. Mixed resting cells of E. coli (SpEH–MVTA) and E. coli (BDHA–GoSCR) (Fig. S1†) were first examined for the conversion of epoxides 1a–f to (S)-β-amino alcohols 4a–f. Combining E. coli (SpEH–MVTA) cells at 10 g cdw L−1 and E. coli (BDHA–GoSCR) cells at 16 g cdw L−1 led to the production of (S)-4a–c in 97–99% ee with 42.5%–68.1% conversion from 10 mM of 1a and 1b and 20 mM of 1c, respectively. For the conversion of 20 mM of the epoxides 1d–f, resting cells of E. coli (SpEH–MVTA) and E. coli (BDHA–GoSCR) were mixed in a 14:22 ratio, and after 24 h of reaction time, the products (S)-4d–f were obtained in >99% ee and 61.8–77.7% conversion (see the ESI,† Table S10).
A recombinant E. coli strain that co-expresses all of the necessary enzymes is often used for cascade biocatalysis since this avoids having to cultivate cells of multiple strains.16 Resting cells of E. coli (SpEH–BDHA–GoSCR–MVTA) co-expressing four enzymes were thus used for the one-pot conversion of the epoxides 1a–f to the (S)-amino alcohols 4a–f. The E. coli (SpEH–BDHA–GoSCR–MVTA) cells showed specific activities of 14.8–40.6 U g−1 cdw for the conversion of 1c to 4c after growth for 2–16 h (see the ESI,† Fig. S3). The SDS-PAGE of the cell-free extracts of the E. coli cells harvested at 4–16 h is shown in Fig. S2.† Reactions of 10–20 mM of the racemic epoxide 1c with E. coli (SpEH–BDHA–GoSCR–MVTA) at 10–20 g cdw L−1 afforded (S)-4c in >99% ee and 99% conversion. When the concentration of the racemic epoxide 1c was increased to 50 mM, only a trace amount of the desired product (S)-4c was formed (5.6% conversion) (see the ESI,† Table S11). We attribute this to the low solubility and high toxicity of the epoxide substrate in aqueous phase. To circumvent the solubility and toxicity problem of the epoxides, an aqueous–organic two-phase system consisting of sodium phosphate buffer (100 mM, pH 7.5) and hexadecane at 1:1 (v/v) was used as the reaction medium. Reactions of 20 mM of epoxides 1a and 1b with E. coli (SpEH–BDHA–GoSCR–MVTA) at 18 g cdw L−1 afforded (S)-4a and (S)-4b in 97%–98% ee and 80.4–99% conversion within 5 h of reaction time. The reaction of epoxide 1c (50 mM) with the same catalysts at 20 g cdw L−1 gave (S)-4c in >99% ee and 92% conversion (Table 2). The time course of this reaction is shown in Fig. 1. At 2 h, epoxide 1c was totally converted to 1-phenyl-1, 2-ethanediol 2c due to the high activity of SpEH, 33.5 mM diol 2c remained in the reaction mixture, and the remainder of diol 2c was converted to 15.5 mM of (S)-2-amino-2-phenylethanol 4c in >99% ee by BDHA/GoSCR and MVTA. From 2 to 16 h, the concentration of (S)-4c increased linearly upon a linear decrease in the concentration of 2c. At 16 h, 46 mM of (S)-4c was produced in >99% ee, and a trace amount of 2c remained in the reaction mixture. In the whole reaction process, almost no 2-hydroxyacetophenone 3c was detected. These results show that the α-hydroxy ketone produced in the second step was quickly converted to an β-amino alcohol by MVTA in the third step of the cascade reaction. Further conversion of epoxides 1d–f (20 mM) with E. coli (SpEH–BDHA–GoSCR–MVTA) at 18 g cdw L−1 gave (S)-4d–f in >99% ee and 79.6–99% conversion.
| Sub. | Sub. conc. [mM] | R(+)-MBA [mM] | Catalyst 3 [g cdw L−1] | T [h] | Conv. to 4a [%] | (S)-4 eeb [%] |
|---|---|---|---|---|---|---|
|
The reactions were conducted using 25–55 mM of the amine donor (R-MBA) in a two-liquid phase system consisting of sodium phosphate buffer (100 mM, pH 7.5) and hexadecane (1:1). The reactions were carried out using 0.1 mM PLP and 10–50 mM of the substrates at 30 °C.
a Conversion was determined by GC, error limit: <2% of the stated values.
b ee values were determined by chiral GC.
|
||||||
| 1a | 20 | 25 | 18 | 5 | 99.0 | 97 |
| 1b | 20 | 25 | 18 | 5 | 80.4 | 98 |
| 1c | 50 | 55 | 20 | 16 | 92.0 | >99 |
| 1d | 20 | 25 | 18 | 5 | 99.0 | >99 |
| 1e | 20 | 25 | 18 | 5 | 97.1 | >99 |
| 1f | 20 | 25 | 18 | 5 | 79.6 | >99 |
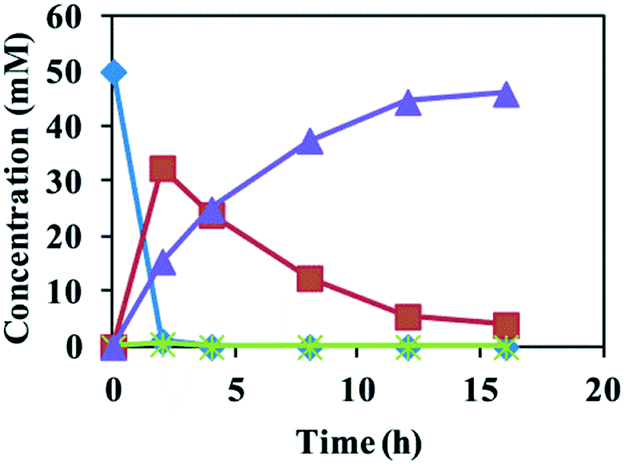 | ||
Fig. 1 Time course of the conversion of styrene oxide 1c (50 mM) to (S)-4c with resting cells of E. coli (SpEH–BDHA–GoSCR–MVTA) (20 g cdw L−1) in a two-liquid phase system.  = the concentration of 1c; = the concentration of 1c;  = the concentration of 2c; = the concentration of 2c;  = the concentration of 3c; and = the concentration of 3c; and  = the concentration of (S)-4c. = the concentration of (S)-4c. | ||
The applicability of this new type of cascade biocatalysis was tested in a 100 mL scale preparative experiment. After 24 h of reaction times, pure (S)-4c was isolated in >99% ee and 80% isolated yield after extraction with ethyl acetate and flash chromatography (see the ESI,† Fig. S17 and S18). This example demonstrates the facile and green application of this new type of cascade biocatalysis for the asymmetric ring opening of epoxides to enantiopure β-amino alcohols in high-yield.
In summary, a new type of cascade biocatalysis reaction was successfully developed for the asymmetric ring opening of racemic epoxides to useful and valuable chiral β-amino alcohols in high ee via epoxide hydrolysis, diol oxidation and α-hydroxy ketone asymmetric reductive amination. In vitro cascade biocatalysis with mixtures of recombinant SpEH, BDHA, GoSCR and MVTA was conducted in the presence of a trace amount of NAD+ to convert the epoxides 1a–f to the corresponding (S)-β-amino alcohols 4a–f in 97–99% ee and 34–99% conversion. The alcohol dehydrogenase (ADH)/(R)-(+)-1-phenylethylamine (R-MBA) in the cascade biocatalysis system was utilized to drive the equilibrium towards the product and recycle the NAD+ cofactor. Whole-cell based cascade biocatalysis was also achieved using the engineered recombinant E. coli cells without any additional NAD+ cofactor, and the produced (S)-β-amino alcohols were obtained in good to excellent conversion (79–99%) and 97–99% ee. Preparative biotransformation was also demonstrated on a 100 mL scale with cells of E. coli (SpEH–BDHA–GoSCR–MVTA). The designed cascade process might be generally applicable to access a variety of different chiral β-amino alcohols starting from the corresponding inexpensive racemic epoxides by combining appropriate enzymes.
This study was supported by the National Natural Science Foundation of China (Grant No. 21772141), the Shanxi Province Science Foundation for Youths (grant No. 201701D221042) and the Open Funding Project of the State Key Laboratory of Bioreactor Engineering.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) R. E. Lee, M. Protopopova, E. Crooks, R. A. Slayden, M. Terrot and C. E. Barry III, J. Comb. Chem., 2003, 5, 172–187 CrossRef CAS PubMed; (b) A. V. Malkov, M. A. Kabeshov, M. Bella, O. Kysilka, D. A. Malyshev, K. Pluháčková and P. Kočovský, Org. Lett., 2007, 9, 5473–5476 CrossRef CAS PubMed; (c) J. Guo, M. Zhu, T. Wu, C. Hao, K. Wang, Z. Yan, W. Huang, J. Wang, D. Zhao and M. Cheng, Bioorg. Med. Chem., 2017, 25, 3500–3511 CrossRef CAS; (d) Y. Hamada, T. Nakanishi, K. Suzuki, R. Yamaguchi, T. Hamada, K. Hidaka, S. Ishiura and Y. Kiso, Bioorg. Med. Chem. Lett., 2012, 22, 4640–4644 CrossRef CAS PubMed; (e) X. Cattoën and M. A. Pericàs, Tetrahedron, 2009, 65, 8199–8205 CrossRef.
- (a) K. B. Sharpless, D. W. Patrick, L. K. Truesdale and S. A. Biller, A new reaction, J. Am. Chem. Soc., 1975, 97, 2305–2307 CrossRef CAS; (b) G. Li, H. Chang and K. B. Sharpless, Angew. Chem., Int. Ed. Engl., 1996, 35, 451–454 CrossRef CAS; (c) J. A. Birrell and E. N. Jacobsen, Org. Lett., 2013, 15, 2895–2897 CrossRef CAS PubMed; (d) C. X. Ye, Y. Y. Melcamu, H. H. Li, J. T. Cheng, T. T. Zhang, Y. P. Ruan, X. Zheng, X. Lu and P. Q. Huang, Nat. Commun., 2018, 9, 410 CrossRef PubMed.
- (a) E. J. Jacobsen, Acc. Chem. Res., 2000, 33, 421–431 CrossRef CAS; (b) C. Wang, L. Luo and H. Yamamoto, Acc. Chem. Res., 2016, 49, 193–204 CrossRef CAS.
- (a) A. K. Chakraborti, S. Rudrawar and A. Kondaskar, Org. Biomol. Chem., 2004, 2, 1277–1280 RSC; (b) S. Regati, Y. He, M. Thimmaiah, P. Li, S. Xiang, B. Chen and J. C. G. Zhao, Chem. Commun., 2013, 49, 9836–9838 RSC; (c) M. Kumar, R. I. Kureshy, S. Saravanan, S. Verma, A. Jakhar, N. H. Khan, S. H. R. Abdi and H. C. Bajaj, Org. Lett., 2014, 16, 2798–2801 CrossRef CAS PubMed; (d) Z. Sun, J. Chen, Y. Liu and T. Tu, Adv. Synth. Catal., 2017, 359, 494–505 CrossRef CAS.
- A. Rouf, G. P. Upta, M. A. Aga, B. Kumar, R. Parshad and S. C. Taneja, Tetrahedron: Asymmetry, 2011, 22, 2134–2143 CrossRef CAS.
- (a) M. S. Malik, E. S. Park and J. S. Shin, Green Chem., 2012, 14, 2137–2140 RSC; (b) H. L. Wu, J. D. Zhang, C. F. Zhang, X. J. Fan, H. H. Chang and W. L. Wei, Appl. Biochem. Biotechnol., 2017, 181, 972–985 CrossRef CAS PubMed; (c) A. Nobili, F. Steffen-Munsberg, H. Kohls, I. Trentin, C. Schulzke, M. Höhne and U. T. Bornscheuer, ChemCatChem, 2015, 7, 757–760 CrossRef CAS.
- R. Patel, A. Banerjee, J. Howell, C. McNamee, D. Brozozowski, D. Mirfakhrae, V. Nanduri, J. Thottathil and L. Szarka, Tetrahedron: Asymmetry, 1993, 4, 2069–2084 CrossRef CAS.
- (a) J. M. Sperl and V. Sieber, ACS Catal., 2018, 8, 2385–2396 CrossRef CAS; (b) J. D. Zhang, S. Wu, J. Wu and Z. Li, ACS Catal., 2015, 5, 51–58 CrossRef CAS; (c) S. Gandomkar, A. Dennig, A. Dordic, L. Hammerer, M. Pickl, T. Haas, M. Hall and K. Faber, Angew. Chem., Int. Ed., 2018, 57, 427–430 CrossRef CAS PubMed.
- (a) P. Gupta and N. Mahajan, New J. Chem., 2018, 42, 12296–12327 RSC; (b) L. Rios-Solis, P. Morris, C. Grant, A. O. O. Odeleye, H. C. Hailes, J. M. Ward, P. A. Dalby, F. Baganz and G. J. Lye, Chem. Eng. Sci., 2015, 122, 360–372 CrossRef CAS; (c) T. Sehl, H. C. Hailes, J. M. Ward, R. Wardenga, E. von Lieres, H. Offermann, R. Westphal, M. Pohl and D. Rother, Angew. Chem., Int. Ed., 2013, 52, 6772–6775 CrossRef CAS.
- I. V. Pavlidis, M. S. Weiβ, M. Genz, P. Spurr, S. P. Hanlon, B. Wirz, H. Iding and U. T. Bornscheuer, Nat. Chem., 2016, 8, 1076–1082 CrossRef CAS PubMed.
- J. D. Zhang, H. L. Wu, T. Meng, C. F. Zhang, X. J. Fan, H. H. Chang and W. L. Wei, Anal. Biochem., 2017, 518, 94–101 CrossRef CAS PubMed.
- M. Höhne, S. Schätzle, H. Jochens, K. Robins and U. T. Bornscheuer, Nat. Chem. Biol., 2010, 6, 807–813 CrossRef PubMed.
- Z. M. Cui, J. D. Zhang, X. J. Fan, G. W. Zheng, H. H. Chang and W. L. Wei, J. Biotechnol., 2017, 243, 1–9 CrossRef CAS PubMed.
- S. Wu, A. Li, Y. S. Chin and Z. Li, ACS Catal., 2013, 3, 752–759 CrossRef CAS.
- (a) F. Garzón-Posse, L. Becerra-Figueroa, J. Hernández-Arias and D. Gamba-Sánchez, Molecules, 2018, 23, 1265 CrossRef PubMed; (b) C. C. C. R. de Carvalho, Microb. Biotechnol., 2017, 10, 250–263 CrossRef PubMed.
- (a) Y. Zhou, S. Wu and Z. Li, Angew. Chem., Int. Ed., 2016, 55, 11647–11650 CrossRef CAS; (b) H. Chen, R. Huang and Y.-H. P. Zhang, Appl. Microbiol. Biotechnol., 2017, 101, 4481–4493 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cy02377h |
| This journal is © The Royal Society of Chemistry 2019 |