
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesigning photolabile ruthenium polypyridyl crosslinkers for hydrogel formation and multiplexed, visible-light degradation†
Teresa L. Rappa,
Yanfei Wangb,
Maegan A. Delessioa,
Michael R. Gaua and
Ivan J. Dmochowski *a
*a
aDepartment of Chemistry, University of Pennsylvania, 231 S 34th St., Philadelphia, PA, USA. E-mail: ivandmo@sas.upenn.edu
bDepartment of Anesthesiology, Division of Critical Care Medicine Boston Children's Hospital, Harvard Medical School, 300 Longwood Avenue, Boston, MA, USA
First published on 8th February 2019
Abstract
Photoresponsive materials afford spatiotemporal control over desirable physical, chemical and biological properties. For advanced applications, there is need for molecular phototriggers that are readily incorporated within larger structures, and spatially-sequentially addressable with different wavelengths of visible light, enabling multiplexing. Here we describe spectrally tunable (λmax = 420–530 nm) ruthenium polypyridyl complexes functionalized with two photolabile nitrile ligands that present terminal alkynes for subsequent crosslinking reactions, including hydrogel formation. Two Ru crosslinkers were incorporated within a PEG–hydrogel matrix, and sequentially degraded by irradiation with 592 nm and 410 nm light.
Introduction
Photoresponsive molecules and materials are transforming multiple areas of research, from drug delivery,1–6 to materials engineering,7–15 and biology.16–26 Many natural biological processes are not photoresponsive, making light a versatile trigger for controlling complex biological systems.27 The incorporation of photoactive moieties within biomolecules,24 small-molecule drugs,28 and materials7 provides a method for modulating their activity. Likewise, photoactive moieties incorporated within soft materials, e.g., polymers, hydrogels, and elastomers, enable spatiotemporally precise, light-guided modulation of structure–function properties. Photoresponsive hydrogels in particular have long been used as platforms for cell growth and delivery, for small and large molecule drug delivery,29,30 and for basic materials applications.31 To expand methods for tuning soft material properties, e.g., shape and viscosity, we developed differentially photoresponsive ruthenium moieties suitable for hydrogel formation and subsequent multiplexed ligand dissociation.A drawback to most current photoresponsive molecules is the high-energy light required for bond dissociation. Common photoresponsive organic chromophores, e.g., o-nitrobenzyl,32 azobenzene,14 and coumarin,30,33 respond to near-UV and blue light, which barely penetrates most biomaterials or live tissue. Attempts to red-shift the activation wavelength have focused on multiphoton excitation,10,34–37 coupling with upconverting nanoparticles36,38 or chemically modified chromophores.39 Some limiting factors include the small activation volume of multiphoton processes, the potential toxicity of embedded nanoparticles, low quantum yields (leading to sample heating and photodamage during repeated illumination), and synthetic complexity.
To address these challenges, we have worked to develop inorganic photoactive molecules that absorb orange-red light, which has greater penetration depth and is less prone to photodamage in clinical applications.37 Our laboratory has expanded the use of photolabile ruthenium crosslinkers for applications in biology and soft materials. The first Ru-based crosslinker, (Ru(bipyridine)2(3-ethynylpyridine)2) (Ru-BEP), presented two alkynes for the circularization of antisense bis-azide-modified oligonucleotides for light-activated gene knockdown in zebrafish embryos.21 A related compound, Ru(bipyridine)2(3-pyridinaldehyde)2 (RuAldehyde), provided a light-responsive crosslinker for hydrogel formation, site-selective degradation and protein release.29
These Ru polypyridyl complexes share the unique ability to exchange a monodentate pyridine ligand with solvent upon irradiation with visible light. Single-site photo-substitution has been observed for other [Ru(polypyridyl)2X2]2+ complexes, where X = pyridine-2,40–42 or sulphur-containing43 ligands. Alternatively, two nitrile ligands44,45 can both undergo rapid photo-substitution (Fig. 1A). Excitation into the singlet metal-to-ligand charge transfer (1MLCT) band initiates intersystem crossing to a low-lying triplet state (Fig. 1B). In most photo-responsive Ru-polypyridyl complexes this triplet state is primarily 3MLCT in character, with another triplet metal-centred (3MC) state close enough in energy to be thermally populated.
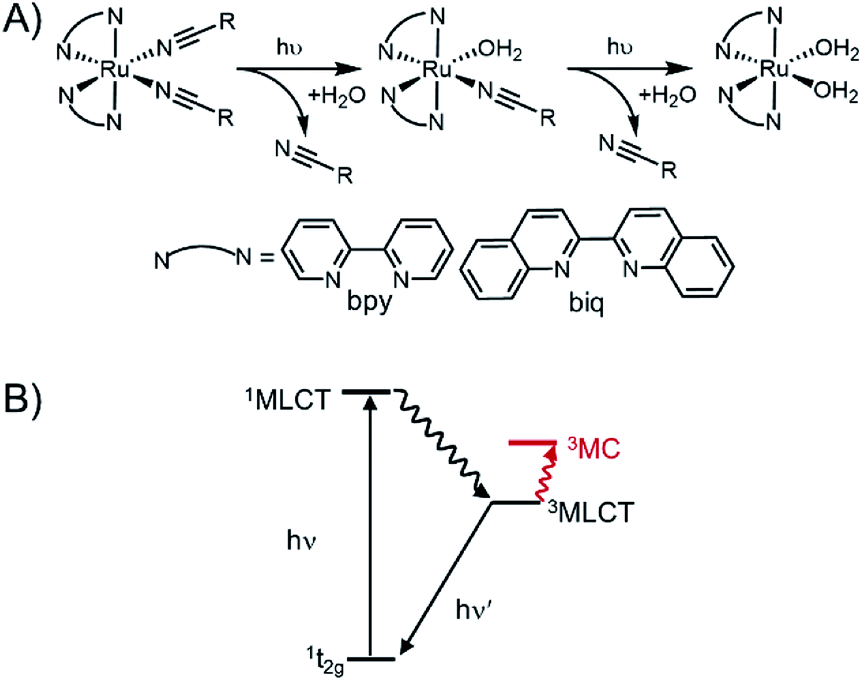 | ||
| Fig. 1 Photoinitiated ligand exchange in ruthenium polypyridyl complexes. (A) Photolysis observed for Ru(II)–nitrile complexes is a two-step process in which both ligands are exchanged with coordinating solvent. (B) Jablonski diagram showing excited states responsible for ligand. | ||
In the current study, our goal was to red-shift the absorption of Ru crosslinkers for multiplexing applications, while incorporating two photolabile nitrile-based ligands for maximum photodissociation within a hydrogel.46 Inspired by previous work from Turro and coworkers, we designed a series of Ru crosslinkers incorporating biquinoline ligands that red-shift the maximum absorption wavelength, λmax.46 The biquinoline also increased the steric strain around the Ru center, increasing the quantum yield of photorelease, Φpr.47 This technique has led to several applications of red-light-absorbing, photoresponsive materials incorporating polypyridyl ruthenium compounds.48,49 Here, we present the first examples of red-shifted Ru compounds that incorporate crosslinking functionality and achieve hydrogel formation, while enabling wavelength-selective degradation with visible light.
We present a series of alkyne-bearing Ru(II) compounds with nitrile-based photolabile ligands (compounds 1–3, Fig. 2). Starting from Ru(bipyridine)2(5-hexynenitrile)2, λmax was sequentially red-shifted by incorporating 1 or 2 biquinoline ligands (Fig. 2). A crystallographic analysis confirmed that 5-hexynenitrile appropriately positions the pendant alkyne for subsequent reaction with an azide-modified branched polyethylene glycol (PEG) polymer (10 kDa) via copper(I)-catalyzed alkyne–azide cycloaddition (CuAAC).50,51 The resulting hydrogels, formed with Ru crosslinkers 1 and 3, allowed spatially selective degradation via two different wavelengths of visible light (592 and 410 nm).
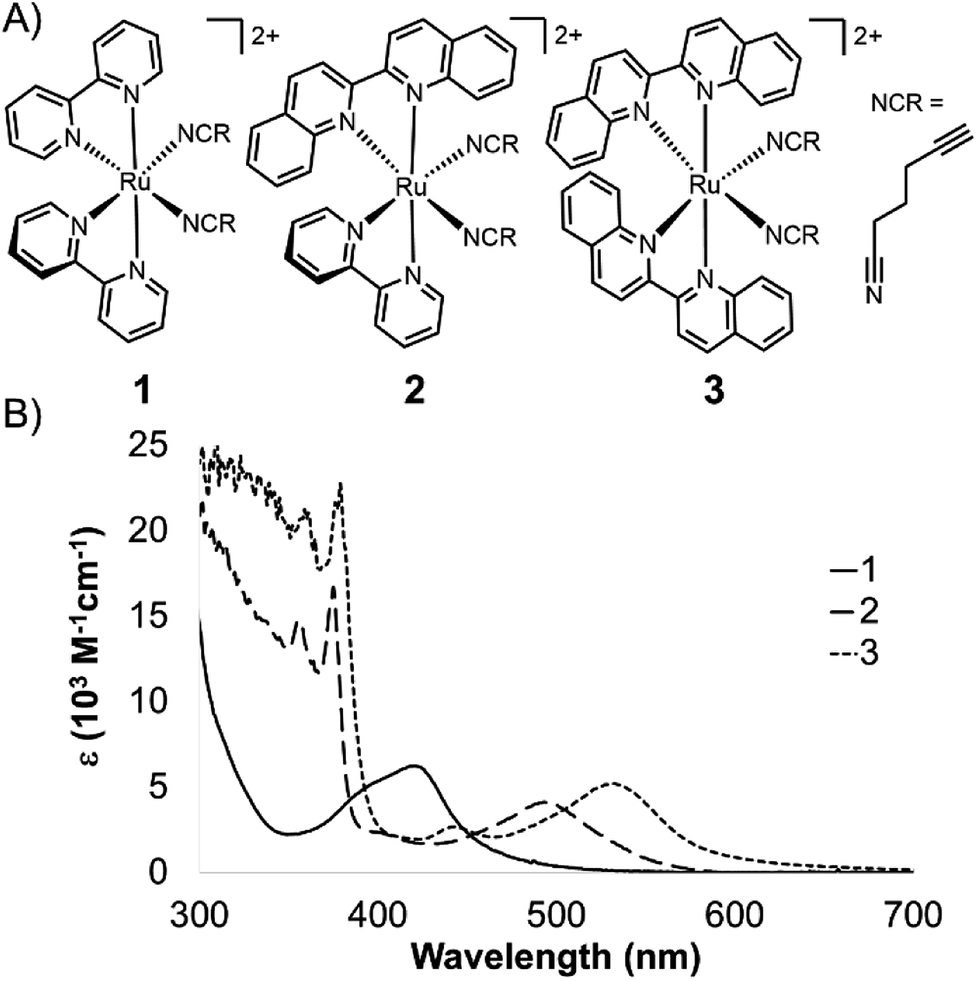 | ||
| Fig. 2 Ru crosslinkers with two photolabile nitrile ligands. (A) Three compounds synthesized in this study. (B) Molar absorption spectra for 1–3. | ||
Results and discussion
Compound 1 was synthesized from commercially available Rubpy2Cl2 and 5-hexynenitrile through the Ru(bpy)2(H2O)2 intermediate generated by the addition of AgPF6 to form AgCl precipitate. 1–3 were purified as the PF6− salt via silica column flash chromatography (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 acetonitrile:methylene chloride mobile phase), and isolated as the nitrate salt using an Amberlite© IRA-410 column in good yield (54%) (see ESI† for synthetic details). The nitrate counterion gave Ru2+ polypyridyl complexes with excellent solubility and stability in water (Fig. S1†).
:4 acetonitrile:methylene chloride mobile phase), and isolated as the nitrate salt using an Amberlite© IRA-410 column in good yield (54%) (see ESI† for synthetic details). The nitrate counterion gave Ru2+ polypyridyl complexes with excellent solubility and stability in water (Fig. S1†).
To generate Ru(bpy)(biq)Cl2 for 2 we found it necessary to use the benzene ruthenium dimer [(benzene)RuCl2]2 to ensure conversion to the mixed ligand product. Bipyridine was coordinated first to generate Ru(bpy)Cl42−, which was purified by filtration, followed by addition of biquinoline and heating to give Ru(bpy)(biq)Cl2, which was purified by precipitation into diethyl ether, in 55% yield. Subsequent coordination of two 5-hexynenitrile ligands gave 2 in a final overall yield of 13.5%. Compound 3 was synthesized starting with RuCl3; 2.2 equivalents of biquinoline were added with hydroquinone as the reducing agent and excess LiCl to generate the intermediate Rubiq2Cl2, which was isolable by precipitation into ether in 33% yield. Coordination of 5-hexynenitrile proceeded by the same procedure as for 1 and 2, giving 3 as nitrate salt in overall 24% yield. All compounds were characterized by 1H NMR spectroscopy, high-resolution ESI mass spectrometry, and UV-Vis absorption spectroscopy (see ESI†).
Ruthenium polypyridyl complexes exhibit strong absorbance in the visible region due to the low-lying metal-to-ligand charge transfer (MLCT) band. In this state, electrons are excited from the ground state orbital located primarily on the metal center to a low-lying excited orbital located on the polypyridyl ligand, at higher energy for bipyridine than biquinoline.46 Ligands with more extended pi bonding tend to lower the energy of the 1MLCT band, and red shift the absorbance.
The 1MLCT absorption maxima for 1, 2, and 3 were 419, 491, and 529 nm, respectively (ε reported in Table 1). A shift of over 70 nm was observed with the first substitution of a bipyridine for biquinoline ligand, from 1 to 2 (Fig. 2A), followed by a nearly 40 nm red-shift from 2 to 3. This shows good agreement with previously published spectra for Ru(phen)2(MeCN)2 (λmax = 420 nm), Ru(phen)(biq)(MeCN)2 (λmax = 497 nm), and Ru(biq)2(MeCN)2 (λmax = 535 nm).46
| ε (M−1 cm−1) | Φpr | |
|---|---|---|
| 1 | 6140 ± 100 | 0.16 ± 0.02@450 nm |
| 2 | 1900 ± 100 | 0.19 ± 0.005@532 nm |
| 3 | 7400 ± 400 | 0.07 ± 0.01@532 nm |
The photolysis of ruthenium polypyridyl compounds can be observed directly using UV-Vis spectroscopy. As the compound undergoes ligand exchange of a coordinated ligand for a solvent molecule, a significant red shift is observed in the MLCT band. Under continuous irradiation, compounds 1–3 sequentially exchanged both nitrile ligands (Fig. 3A, S2†). The UV-Vis photolysis curve for 3 is shown in Fig. 3B, where peaks at 560 and 590 nm indicated a stepwise process, with a monoaquated intermediate. The clear isosbestic points at 550 and 570 nm also indicated the stepwise transition from 3 to monoaquated 3′ to bisaquated 3′′, although the first transition point at 550 nm included early formation of 3′′ under continuous irradiation.
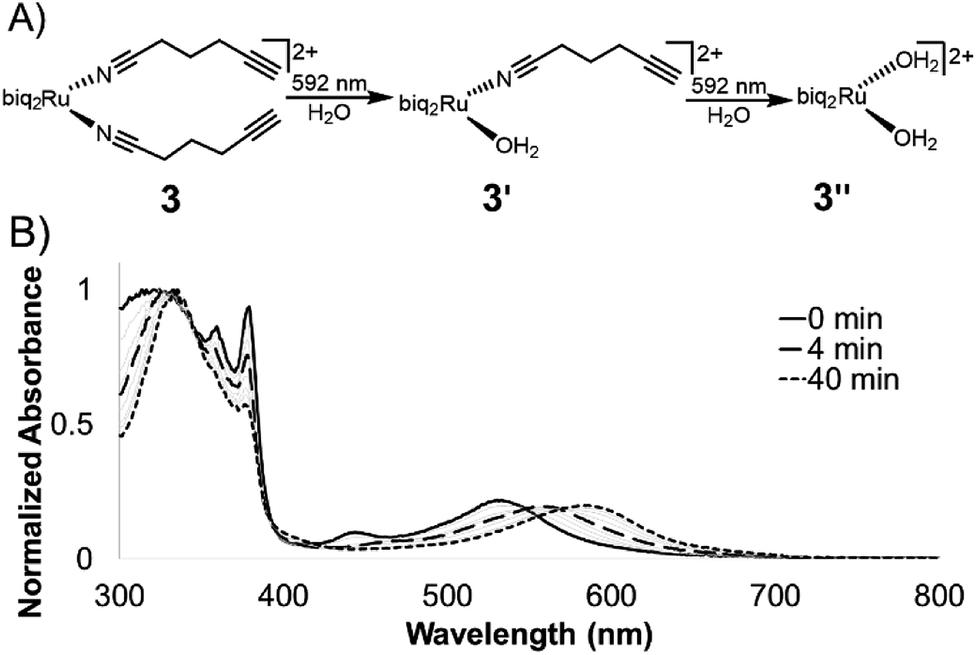 | ||
| Fig. 3 Photolysis of 3 in water. (A) Compounds 1–3 undergo a stepwise ligand exchange of both nitrile ligands when irradiated in water. The second step takes much longer than the first. (B) Photolysis trace of 3 in water under irradiation from 592 nm LED (25 mW cm−2). | ||
The loss of the second nitrile ligand in 3 was slower, occurring on the order of 40 min (Fig. 3B), compared to the first ligand exchange event, which was completed within 4 min of constant irradiation in the bulk sample. This trend was observed for 1 and 2 as well (Fig. S2†). The Ru MLCT band extends well beyond the λmax, which can be used to induce ligand exchange at longer wavelengths of light; irradiation at 600–700 nm (red incandescent light bulb, 5 mW) was less efficient but led to complete photolysis of 3 in 4 h (Fig. S3†).
Photolysis data were fit to an equation derived from a pseudo-first order kinetics process, and the time constants were determined (Fig. S4†). The value of Φpr was found for the first exchange event from the rate constant coupled with the laser power (Fig. S4†). As expected, Φpr decreased roughly 2-fold as the MLCT band was shifted further to the red, from 0.16 (in 1) to 0.07 (in 3), Table 1.52,53 Similarly, the efficiency of photolysis for 3 (ε × Φpr = 520 M−1 cm−1) was lower than 1 at 980 M−1 cm−1. Although these values were lower than other published ruthenium caging groups, with efficiencies that range from 2000 (ref. 46) to 4000 M−1 cm−1,29 they are significantly improved over other green-light-sensitive caging groups like BODIPY, with Φpr on the order of 100 M−1 cm−1.54,55
Diffraction-quality crystals of 3 as PF6− salt were grown via vapour diffusion from 3–5 mg dissolved in acetonitrile/methanol/THF (0.1 mL each) with diethyl ether, stored at −20 °C for 2 weeks (Fig. 4). Bond lengths between Ru2+ and ligands were within expected ranges, with variations due to the steric strain in the system. The angle between the nitrile ligands is stretched significantly to >95° perhaps due to the strain caused by bulkier biq ligands coordinated to Ru2+. In the less crowded Ru(bpy)(biq)(5-hexynenitrile)2 compound, the nitrile–Ru–nitrile angle is ∼90° (Fig. S5†). Crystal structure of 3 shows alkynes positioned 4.3 Å and 4.9 Å from biquinolines, angled such that they are accessible for cycloaddition with azide and copper catalyst (Table 2).
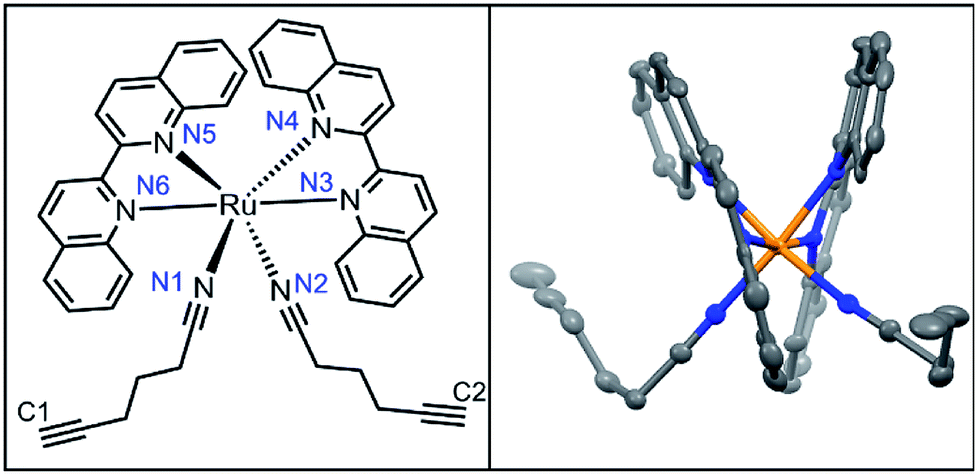 | ||
| Fig. 4 Crystal structure of 3. | ||
| Bond | 3 (Å) | |
|---|---|---|
| Ru–biq | Ru–N4 | 2.084(6) |
| Ru–N3 | 2.093(6) | |
Ru–N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C C |
Ru–N1 | 2.025(6) |
| Ru–N2 | 2.024(6) | |
| CC to biq |
C1–biq | 4.267 |
| C2–biq | 4.926 |
The conformational flexibility of the nitrile-alkyl ligands required synthesis and testing of several Ru compounds to identify competent crosslinkers. Initially, Ru compounds employing a shorter 4-pentynenitrile ligand were synthesized and found to be incapable of Cu(I)-mediated PEG gelation (Fig. S6†). Incorporation of longer 5-hexynenitrile ligands led to functional Ru crosslinkers, but only after mild synthetic conditions for nitrile coordination were employed. Ru-(5-hexynenitrile) coordination performed at elevated temperatures and longer reaction times resulted in Ru compounds found to be incapable of Cu(I)-mediated PEG gelation. X-ray crystal structure analysis of one such example shows alkyne positioned much closer to the biquinoline, only 3.7 Å (Fig. S5†). The cis-alkane conformation should disfavour Cu(I)-mediated alkyne–azide cycloaddition chemistry. Ru compounds 1–3 were synthesized using the mild conditions detailed in the Synthetic Procedure and confirmed to be excellent crosslinkers in gelation studies.
CuAACs have been widely used for materials design, with several studies showing the generation of hydrogel materials. Hyaluronic acid,56,57 polyethylene glycol (PEG),58 dextran,59 poly(vinyl) alcohol (PVA),60 along with several other polymers have been modified with azides and terminal alkynes to facilitate hydrogel formation. The need for a Cu(I) catalyst has limited some bio-applications as it can be toxic to cells,61 but can also provide spatiotemporal control. In one example Bowman and co-workers used a photocatalyst to reduce Cu(II) for the formation of a hydrogel with precise control.62 Copper can be dialysed away from preformed hydrogels, which is acceptable for many drug delivery platforms.
Compounds 1–3 were tested for crosslinking reaction with azido-PEG (MW 10000 Da) in the presence of CuSO4, THPTA ligand, and sodium ascorbate reducing agent (Scheme 1), forming a strong hydrogel within 30 s (results shown for 3, Fig. 5). Hydrogels formed at a final weight percent of 7.5 wt% with stoichiometric ruthenium crosslinker, generating elasticity nearing 1 kPa (Fig. 5). As expected, when exposed to visible light (400–500 nm) the hydrogel rapidly lost its elastic properties, becoming a viscous liquid within 5 min (Fig. 5).
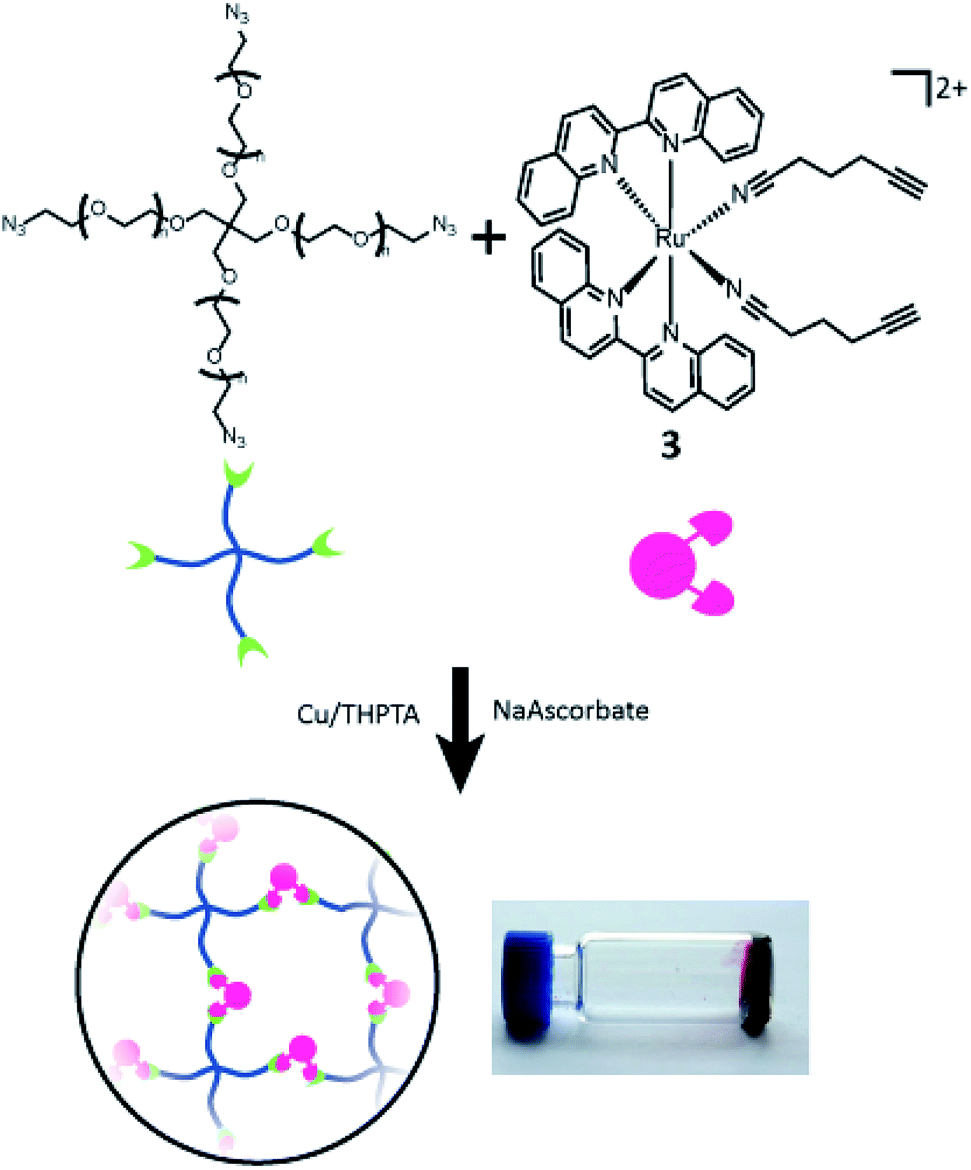 | ||
| Scheme 1 Gelation of branched PEG via crosslinking reaction with 3. | ||
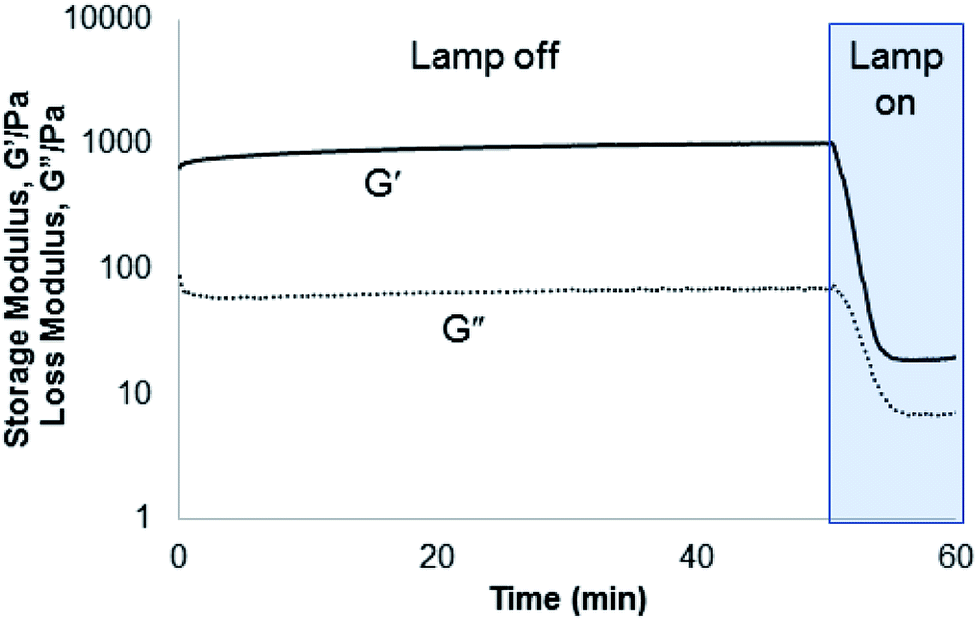 | ||
| Fig. 5 Rheometry demonstrating gelation formed from the incorporation of 3 into a PEG hydrogel. The hydrogel was rapidly degraded under irradiation with 400–500 nm light (25 mW cm−2). | ||
Next, a striped hydrogel was formed for multiplexing experiment via “layer-by-layer” reaction of azido-PEG with crosslinker 1, alternating with crosslinker 3 (Fig. 6). Orange light (592 nm) was used to degrade 3 selectively while leaving 1 intact, as demonstrated both in a solution experiment with equal parts 1 and 3 (Fig. 6A) and in the gel (Fig. 6B). A significant increase in absorbance at 590 nm confirmed the formation of the bisaquated product Ru(biq)2(H2O)2, 3′′ (Fig. 6A). Finally, irradiation at 410 nm led to a significant decrease in absorbance at 423 nm and small increase at 590 nm due to formation of Ru(bpy)2(H2O)2, 1′′ (Fig. 6A, S1†), and rapidly degraded the remaining hydrogel sections crosslinked by 1 (Fig. 6B). The sequence of irradiation is important in this case, as compound 3 absorbs both 410 and 592 nm light and will be degraded by both wavelengths.
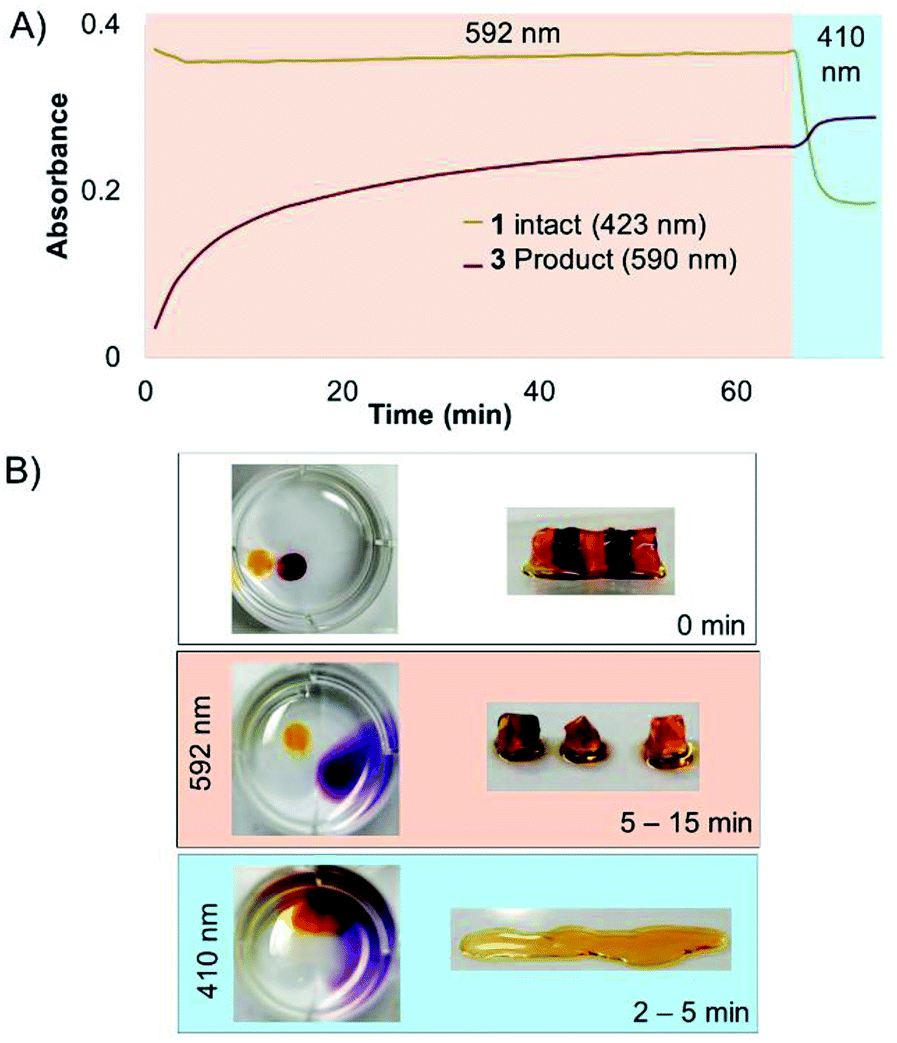 | ||
| Fig. 6 Selective degradation of 1 and 3 in solution and hydrogel. (A) Irradiation at 592 nm photolyzed 3 in solution while leaving 1 intact, until irradiation at 410 nm. (B) Striped hydrogel incorporating alternating sections of 1 and 3 was selectively degraded at 592 nm, leaving the orange gel regions intact. The remaining sections crosslinked by 1 were degraded by 410 nm light. | ||
We developed spectrally tuneable ruthenium polypyridyl crosslinkers with two pendant alkynes for hydrogel formation, and high photolysis efficiency for multiplexed, visible-light gel degradation. Replacing bipyridine with more pi-conjugated biquinoline ligands red-shifted the absorbance, generating a series of sequentially red-shifted compounds. Incorporation of two flexible 5-hexynenitrile ligands at the Ru2+ center enabled CuAAC crosslinking reactions, while also facilitating subsequent photodegradation of gels incorporating these Ru crosslinkers. This represents the first example to our knowledge of a two-color hydrogel system that can be selectively activated by two different visible wavelengths. Such Ru crosslinkers may be applied broadly in materials chemistry, or alternately employed for generating photoactive versions of circular oligos,21 peptides, or other bis-azide containing molecules.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Many thanks to Jonathan Gallarga for his assistance with the rheometry experiments, and Pat Carroll for his work in solving the crystal structures. The authors gratefully acknowledge financial support from the NIH (R01 GM-083030), the NSF through the University of Pennsylvania, Materials Research Science and Engineering Center (MRSEC, DMR-1720530), and the UPenn Chemistry Department.References
- N. A. Smith, P. Zhang, S. E. Greenough, M. D. Horbury, G. J. Clarkson, D. McFeely, A. Habtemariam, L. Salassa, V. G. Stavros and C. G. Dowson, et al., Chem. Sci., 2017, 8, 395–404 RSC.
- A. Li, C. Turro and J. J. Kodanko, Chem. Commun., 2018, 54, 1280–1290 RSC.
- F. Reeßing and W. Szymanski, Curr. Med. Chem., 2018, 24, 4905–4950 CrossRef.
- A. Döbber, A. F. Phoa, R. H. Abbassi, B. W. Stringer, B. W. Day, T. G. Johns, M. Abadleh, C. Peifer and L. Munoz, ACS Med. Chem. Lett., 2017, 8, 395–400 CrossRef.
- M. M. Dcona, D. Mitra, R. W. Goehe, D. A. Gewirtz, D. A. Lebman and M. C. T. Hartman, Chem. Commun., 2012, 48, 4755 RSC.
- S. Ki Choi, T. Thomas, M.-H. Li, A. Kotlyar, A. Desai and J. R. Baker Jr, Chem. Commun., 2010, 46, 2632 RSC.
- A. M. Kloxin, A. M. Kasko, C. N. Salinas and K. S. Anseth, Science, 2009, 324, 59–63 CrossRef CAS PubMed.
- A. Ovsianikov, A. Deiwick, S. Van Vlierberghe, P. Dubruel, L. Möller, G. Dräger and B. Chichkov, Biomacromolecules, 2011, 12, 851–858 CrossRef CAS PubMed.
- D.-S. Shin, J. You, A. Rahimian, T. Vu, C. Siltanen, A. Ehsanipour, G. Stybayeva, J. Sutcliffe and A. Revzin, Angew. Chem., Int. Ed., 2014, 53, 8221–8224 CrossRef CAS PubMed.
- K. Peng, I. Tomatsu, B. van den Broek, C. Cui, A. V. Korobko, J. van Noort, A. H. Meijer, H. P. Spaink and A. Kros, Soft Matter, 2011, 7, 4881 RSC.
- C. M. Kirschner, D. L. Alge, S. T. Gould and K. S. Anseth, Adv. Healthcare Mater., 2014, 3, 649–657 CrossRef CAS PubMed.
- C. K. Arakawa, B. A. Badeau, Y. Zheng and C. A. DeForest, Adv. Mater., 2017, 29, 1703156 CrossRef PubMed.
- D. R. Griffin and A. M. Kasko, J. Am. Chem. Soc., 2012, 134, 13103–13107 CrossRef CAS PubMed.
- A. M. Rosales, K. M. Mabry, E. M. Nehls and K. S. Anseth, Biomacromolecules, 2015, 16, 798–806 CrossRef CAS PubMed.
- T. E. Brown, I. A. Marozas and K. S. Anseth, Adv. Mater., 2017, 29, 1605001 CrossRef PubMed.
- L. Wu, Y. Wang, J. Wu, C. Lv, J. Wang and X. Tang, Nucleic Acids Res., 2013, 41, 677–686 CrossRef CAS.
- L. Kröck and A. Heckel, Angew. Chem., Int. Ed., 2005, 44, 471–473 CrossRef PubMed.
- D. D. Young, M. O. Lively and A. Deiters, J. Am. Chem. Soc., 2010, 132, 6183–6193 CrossRef CAS.
- S. Yamazoe, Q. Liu, L. E. McQuade, A. Deiters and J. K. Chen, Angew. Chem., Int. Ed., 2014, 53, 10114–10118 CrossRef CAS PubMed.
- S. Yamazoe, I. A. Shestopalov, E. Provost, S. D. Leach and J. K. Chen, Angew. Chem., Int. Ed., 2012, 51, 6908–6911 CrossRef CAS PubMed.
- J. C. Griepenburg, T. L. Rapp, P. J. Carroll, J. Eberwine and I. J. Dmochowski, Chem. Sci., 2015, 6, 2342–2346 RSC.
- J. C. Griepenburg, B. K. Ruble and I. J. Dmochowski, Bioorg. Med. Chem., 2013, 21, 6198–6204 CrossRef CAS PubMed.
- B. N. Goguen, B. D. Hoffman, J. R. Sellers, M. A. Schwartz and B. Imperiali, Angew. Chem., Int. Ed., 2011, 123, 5785–5788 CrossRef.
- C. W. Riggsbee and A. Deiters, Trends Biotechnol., 2010, 28, 468–475 CrossRef CAS PubMed.
- N. Ankenbruck, T. Courtney, Y. Naro and A. Deiters, Angew. Chem., Int. Ed., 2018, 57, 2768–2798 CrossRef CAS.
- J. Hemphill, E. K. Borchardt, K. Brown, A. Asokan and A. Deiters, J. Am. Chem. Soc., 2015, 137, 5642–5645 CrossRef CAS PubMed.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- M. Huisman, J. K. White, V. G. Lewalski, I. Podgorski, C. Turro and J. J. Kodanko, Chem. Commun., 2016, 52, 12590–12593 RSC.
- T. L. Rapp, C. B. Highley, B. C. Manor, J. A. Burdick and I. J. Dmochowski, Chem.–Eur. J., 2018, 24, 2328–2333 CrossRef CAS PubMed.
- M. A. Azagarsamy and K. S. Anseth, Angew. Chem., Int. Ed., 2013, 52, 13803–13807 CrossRef CAS.
- D. Y. Wong, D. R. Griffin, J. Reed and A. M. Kasko, Macromolecules, 2010, 43, 2824–2831 CrossRef CAS.
- J. W. Walker, J. A. Mccray and G. P. Hess, Biochemistry, 1986, 25, 1799–1805 CrossRef CAS PubMed.
- M. A. Azagarsamy, D. D. McKinnon, D. L. Alge and K. S. Anseth, ACS Macro Lett., 2014, 3, 515–519 CrossRef CAS.
- I. Aujard, C. Benbrahim, M. Gouget, O. Ruel, J.-B. Baudin, P. Neveu and L. Jullien, Chem.–Eur. J., 2006, 12, 6865–6879 CrossRef CAS PubMed.
- R. G. Wylie and M. S. Shoichet, J. Mater. Chem., 2008, 18, 2716 RSC.
- B. Yan, J.-C. Boyer, N. R. Branda and Y. Zhao, J. Am. Chem. Soc., 2011, 133, 19714–19717 CrossRef CAS.
- X. Zeng, X. Zhou and S. Wu, Macromol. Rapid Commun., 2018, 1800034 CrossRef.
- B. Yan, J.-C. Boyer, D. Habault, N. R. Branda and Y. Zhao, J. Am. Chem. Soc., 2012, 134, 16558–16561 CrossRef CAS.
- D. Wang, M. Wagner, H.-J. Butt and S. Wu, Soft Matter, 2015, 11, 7656–7662 RSC.
- Z. Leonardo, C. Cecilia, A. Pablo, L. Baraldo and R. Etchenique, J. Am. Chem. Soc., 2003, 125, 882–883 CrossRef.
- M. Huisman, J. K. White, V. G. Lewalski, I. Podgorski, C. Turro and J. J. Kodanko, Chem. Commun., 2016, 52, 12590–12593 RSC.
- L. N. Lameijer, D. Ernst, S. L. Hopkins, M. S. Meijer, S. H. C. Askes, S. E. LeDévédec and S. Bonnet, Angew. Chem., Int. Ed., 2017, 56, 11549–11553 CrossRef CAS PubMed.
- R. N. Garner, L. E. Joyce and C. Turro, Inorg. Chem., 2011, 50, 4384–4391 CrossRef CAS PubMed.
- R. Sharma, J. D. Knoll, P. D. Martin, I. Podgorski, C. Turro and J. J. Kodanko, Inorg. Chem., 2014, 53, 3272–3274 CrossRef CAS.
- T. Respondek, R. Sharma, M. K. Herroon, R. N. Garner, J. D. Knoll, E. Cueny, C. Turro, I. Podgorski and J. J. Kodanko, ChemMedChem, 2014, 9, 1306–1315 CrossRef CAS.
- B. A. Albani, C. B. Durr and C. Turro, J. Phys. Chem. A, 2013, 117, 13885–13892 CrossRef CAS PubMed.
- J. D. Knoll, B. A. Albani, C. B. Durr and C. Turro, J. Phys. Chem. A, 2014, 118, 10603–10610 CrossRef CAS PubMed.
- X. Zeng, X. Zhou and S. Wu, Macromol. Rapid Commun., 2018, 1800034 CrossRef.
- C. Xie, W. Sun, H. Lu, A. Kretzschmann, J. Liu, M. Wagner, H.-J. Butt, X. Deng and S. Wu, Nat. Commun., 2018, 9, 3842 CrossRef PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- C. Tornoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef CAS.
- L. N. Lameijer, D. Ernst, S. L. Hopkins, M. S. Meijer, S. H. C. Askes, S. E. LeDévédec and S. Bonnet, Angew. Chem., Int. Ed., 2017, 56, 11549–11553 CrossRef CAS.
- L. M. Loftus, A. Li, K. L. Fillman, P. D. Martin, J. J. Kodanko and C. Turro, J. Am. Chem. Soc., 2017, 139, 18295–18306 CrossRef CAS.
- P. P. Goswami, A. Syed, C. L. Beck, T. R. Albright, K. M. Mahoney, R. Unash, E. A. Smith and A. H. Winter, J. Am. Chem. Soc., 2015, 137, 3783–3786 CrossRef CAS PubMed.
- N. Umeda, H. Takahashi, M. Kamiya, T. Ueno, T. Komatsu, T. Terai, K. Hanaoka, T. Nagano and Y. Urano, ACS Chem. Biol., 2014, 9, 2242–2246 CrossRef CAS PubMed.
- V. Crescenzi, L. Cornelio, C. Di Meo, S. Nardecchia and R. Lamanna, Biomacromolecules, 2007, 8, 1844–1850 CrossRef CAS PubMed.
- X. Hu, D. Li, F. Zhou and C. Gao, Acta Biomater., 2011, 7, 1618–1626 CrossRef CAS PubMed.
- S. Q. Liu, P. L. Rachel Ee, C. Y. Ke, J. L. Hedrick and Y. Y. Yang, Biomaterials, 2009, 30, 1453–1461 CrossRef CAS PubMed.
- D. A. Heller, Y. Levi, J. M. Pelet, J. C. Doloff, J. Wallas, G. W. Pratt, S. Jiang, G. Sahay, A. Schroeder and J. E. Schroeder, et al., Adv. Mater., 2013, 25, 1449–1454 CrossRef CAS PubMed.
- D. A. Ossipov and J. Hilborn, Macromolecules, 2006, 39, 1709–1718 CrossRef CAS.
- Y. Jiang, J. Chen, C. Deng, E. J. Suuronen and Z. Zhong, Biomaterials, 2014, 35, 4969–4985 CrossRef CAS PubMed.
- B. J. Adzima, Y. Tao, C. J. Kloxin, C. A. DeForest, K. S. Anseth and C. N. Bowman, Nat. Chem., 2011, 3, 256–259 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1875400–1875402. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra09764j |
| This journal is © The Royal Society of Chemistry 2019 |