
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective cleavage of lignin and lignin model compounds without external hydrogen, catalyzed by heterogeneous nickel catalysts†
Liang
Jiang‡
 a,
Haiwei
Guo‡
bc,
Changzhi
Li
*b,
Peng
Zhou
a and
Zehui
Zhang
*a
a,
Haiwei
Guo‡
bc,
Changzhi
Li
*b,
Peng
Zhou
a and
Zehui
Zhang
*a
aKey Laboratory of Catalysis and Materials Sciences of the Ministry of Education, South-Central University for Nationalities, Wuhan, 430074, China. E-mail: zehuizh@mail.ustc.edu.cn; Fax: +86-27-67842572; Tel: +86-27-67842572
bState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China. E-mail: licz@dicp.ac.cn
cUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 21st March 2019
Abstract
Selective hydrogenolysis of the Caryl–O bonds in lignin is a key strategy for the generation of fuels and chemical feedstocks from biomass. Currently, hydrogenolysis has been mainly conducted using hydrogen, which is flammable and not sustainable or economical. Herein, an external hydrogen-free process for aryl ethers hydrogenolysis in lignin models and dioxasolv lignin over nickel nanoparticles supported on Al2O3, is reported. Kinetic studies reveal that the transfer hydrogenolysis activity of the three model compounds decreased in the following order: benzyl phenyl ether (α-O-4), 2-phenylethyl phenyl ether (β-O-4) and diphenyl ether (4-O-5), which linearly corresponds to their binding energies and the activation energies. The main reaction route for the three model compounds was the cleavage of the ether bonds to produce aromatic alkanes and phenol, and the latter was further reduced to cyclohexanol. Dioxasolv lignin depolymerization results exhibit a significant Caryl–O decrease over the Ni nanoparticles supported on Al2O3 with iso-propanol as the hydrogen source through 2D-HSQC-NMR analysis, which confirmed the transfer hydrogenolysis conclusion in the model study. This work provides an economical and environmentally-friendly method for the selective cleavage of lignin and lignin model compounds into value-added chemicals.
Introduction
With the increasing crises of shrinking fossil fuel supplies and global climate change, great effort has been devoted to the development of new routes for producing chemicals and liquid fuels from renewable sources.1,2 Biomass, the only carbon containing renewable resource, is undoubtedly regarded as the best candidate to provide sustainable chemicals and energy for our society. Lignin is the second most abundant component in lignocellulose with 20–30 weight percentage.3 It is a three-dimensional bio-polymer composed of various C–O linkages connected with methoxylated phenyl-propane units.4 Therefore, the depolymerization of lignin could produce diverse aryl ether fragments (CAr–O bond) either in the form of α-O-4 or β-O-4 linkages.5–8 However, it is a great challenge to cleave the CAr–O bond in lignin due to the high C–O bond energy (218–314 kJ mol−1).9Both homogeneous and heterogeneous catalysts have been explored for the cleavage of aryl ethers in liquid phase using hydrogen. Homogeneous catalysts including Ru, V, and Ni complexes were investigated for the cleavage of the CAr–O bond,10–12 which were performed under relatively mild conditions (80–135 °C, 1 bar H2) due to the flexibility of the homogeneous catalysts, allowing them to contact the CAr–O bonds freely without high steric limitation. However, these catalytic systems demonstrated several drawbacks such as: difficulty in recycling the homogeneous catalysts, the purification of products, the requirement of complex ligands, and the use of air-sensitive organic bases.10 In addition, the use of air-sensitive bases such as t-BuOK increases the production cost water has to be removed from raw biomass or the solvents.
From the viewpoint of green and suitable chemistry, heterogeneous catalysts are a better choice for the aqueous-phase conversion of aryl ethers. Either unsupported nanoparticles Fe,13 RuNi,14 AuNi,15 or supported metal catalysts16–24 have been used for the cleavage of lignin derived aryl ethers to a mixture of aromatics and phenolics/cycloalcohols (Scheme 1a). Particularly, nickel catalysts have recently received great interest for this transformation because they are readily available and low cost.18–20 Due to the inherent lower catalytic activity in comparison with noble-metal catalysts, the heterogeneous Ni catalyst systems were previously performed at high temperature and high hydrogen pressure.21–23 Subsequently, Ni/SiO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 18 and Ni/TiN19 catalysts were observed to promote the cleavage of lignin derived aryl ethers under relatively mild conditions (120–150 °C, and 6–12 bar H2) at the expense of high Ni loading (Scheme 1b and c). Recently, a heterogeneous Ni catalyst formed by the in situ reduction of a homogeneous Ni complex, was found to be effective for this transformation with low content of Ni (0.005–0.25 equiv.).24 However, the excessive use of a moisture-sensitive base (2.5–10 equiv. t-BuONa), the low availability of the Ni complex, a long reaction time (up to 24–96 h), and high reaction temperature (up to 180 °C) limit its practical application (Scheme 1d).
18 and Ni/TiN19 catalysts were observed to promote the cleavage of lignin derived aryl ethers under relatively mild conditions (120–150 °C, and 6–12 bar H2) at the expense of high Ni loading (Scheme 1b and c). Recently, a heterogeneous Ni catalyst formed by the in situ reduction of a homogeneous Ni complex, was found to be effective for this transformation with low content of Ni (0.005–0.25 equiv.).24 However, the excessive use of a moisture-sensitive base (2.5–10 equiv. t-BuONa), the low availability of the Ni complex, a long reaction time (up to 24–96 h), and high reaction temperature (up to 180 °C) limit its practical application (Scheme 1d).
 | ||
| Scheme 1 Methods for the cleavage of lignin model compound diphenyl ether. | ||
Methods for the cleavage of the CAr–O bond in aryl ethers usually require hydrogen. While the use of hydrogen for the cleavage produces water as the oxidation product, the use of hydrogen at high temperature and high pressure requires specialist equipment, and has potential safety issues.25 In addition, caution is needed to transport and store hydrogen gas which increases production costs. Therefore, developing new catalytic systems for the cleavage of CAr–O bonds selectively and environmentally is highly desirable. Compared with the hydrogenation reactions involving hydrogen, transfer hydrogenation using hydrogen donors such as alcohols and formic acid are safer and easier to handle.26 In fact, transfer hydrogenation of the lignin model compounds, even lignin, has been reported in recent years.27 However, the reactions were performed under harsh conditions (supercritical methanol, 300–320 °C).28 RANEY®-Ni could promote transfer hydrogenation of the lignin model compounds as well as wood biomass under mild conditions.29 However, the process of the preparation of RANEY®-Ni is non-environmentally friendly and RANEY®-Ni is very sensitive to air, requiring special treatment for both storage and application. Therefore, it is highly desirable to develop supported non-noble metal catalysts for the transfer hydrogenation of lignin model compounds as well as lignin to produce value added chemicals.
Inspired by the fact that some supported nickel catalysts are capable of promoting the transfer hydrogenation and the release of hydrogen from hydrogen donors,30,31 we report the transfer hydrogenation of CAr–O bonds in lignin model compounds using biomass-derived iso-propanol over supported nickel catalysts (Ni/Al2O3). The Ni/Al2O3 catalysts were prepared by the calcination of Ni–Al layered double hydroxide compounds, followed by reduction under a hydrogen atmosphere. High activity and selectivity for the hydrogenation of CAr–O bonds was observed during transfer hydrogenation with iso-propanol under mild conditions. More importantly, the developed method is also effective for the transfer hydrogenation cleavage of actual lignin to produce valuable chemicals. Our developed methods could overcome the drawbacks as mentioned above. Not only does our method not require the use of high-pressure H2, but the catalysts are stable and do not require cautious treatment. More importantly, our transfer hydrogenation processes were performed under mild conditions with comparable or even lower temperatures in comparison with reported methods using H2 and other hydrogen donors. Thus, this work provides an economical and environmentally-friendly method for the selective cleavage of lignin model compounds, as well as lignin, into value-added chemicals.
Experimental
Materials
All chemicals were purchased from Aladdin Chemicals Co. Ltd. (Beijing, China). All solvents were purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China), and used directly.Catalyst preparation
NiAl-LDH with the formula [Ni2Al(OH)6](NO3)·0.6H2O was prepared according to the published procedure.30 Typically, 0.20 mol of Ni(NO3)2·6H2O, 0.10 mol of Al(NO3)3·9H2O and 0.70 mol of urea were added into 100 mL of deionized water, and then the mixture was stirred at room temperature until a clear solution was observed. After that, the mixture was stirred at 140 °C for 9 h in an autoclave. After cooling to room temperature, the solid slurry was filtered, washed several times with deionized water and finally dried at 60 °C under vacuum.Preparation of Ni/Al2O3-T catalysts
Ni/Al mixed oxide (NiO–Al2O3) was first prepared by calcining the NiAl-LDH precursor at 500 °C for 5 h under static air. Then, the NiO–Al2O3 sample was reduced under a H2 flow at 400 °C for 30 min with a heating rate of 2 °C min−1, and increased the reduction temperature to 500, 550, 600, 650 or 700 °C for 30 min with a heating rate of 2 °C min−1 (denoted as Ni/Al2O3-T, T refers to the reduction temperature). After being cooling to room temperature in a continuous H2 flow, the Ni/Al2O3-T was passivated in a 1.0 mol% O2/N2 mixture for 1 h, and stored under air for subsequent catalytic evaluation.Catalyst characterization
Transmission electron microscopy (TEM) images of the samples were obtained on an FEI Tecnai G2-20 instrument. The samples were first dispersed in ethanol under ultrasonication and dropped onto copper grids for observation. Mesopore surface area and pore size measurements were performed with N2 adsorption/desorption isotherms at 77 K on a V-Sorb 2800P instrument. Before the measurements, the samples were degassed at 100 °C for 12 h. X-ray powder diffraction (XRD) patterns of samples were determined with a Bruker advanced D8 powder diffractometer (Cu Kα). All XRD patterns were collected with a scanning rate of 0.016° s−1. The nickel content in the Ni/Al2O3-T catalysts was quantitatively determined by inductively coupled-atomic emission spectrometer (ICP-AES) on an IRIS Intrepid II XSP instrument (Thermo Electron Corporation).General procedure for the hydrogenation of aryl ethers
The catalytic reactions were carried out in a 50 mL autoclave reactor. In a typical experiment, the lignin model compound of aryl ethers (1.0 mmol), Ni/Al2O3-600 (20 mg), and iso-propanol (10 mL) were added into the autoclave reactor. The air in the reactor was removed by N2, and finally the autoclave was pressurised with 10 bar N2. Then the reactor was heated at room temperature to 150 °C within 10 min, the reaction started at 150 °C and was left for the desired time with a stirring speed of 1000 rpm. The reaction was quenched at ambient temperature using ice-water.The organic products were analyzed with a 7890F gas chromatograph system (GC) with a flame ionization detector, both equipped with HP-5 capillary columns (30 m × 0.32 mm × 0.4 mm). The temperature of the column was initially kept at 80 °C for 3 min, and then increased at a rate of 20 °C min−1 to 220 °C. Products were identified by comparison of the retention time of the unknown compounds with those of standard compounds, and quantified based on the internal standard method. Ethylbenzene and diphenyl ether were used as the internal standard to calibrate the liquid product concentrations when the reaction substrate was benzyl phenyl ether; ethylbenzene and benzyl alcohol were used as the internal standard when the reaction substrate was diphenyl ether; toluene and diphenyl ether were used as the internal standard when the reaction substrate was 2-phenylethyl phenyl ether. These internal standards were added after the reaction had finished. Conversion is defined as the amount of change in raw materials during the reaction, divided by the total amount of starting material, multiplied by 100%. The selectivity is defined as the mole percentage of the target products with the total amount of starting material.
General procedures for the transfer cleavage of lignin
The catalytic conversion of lignin was conducted in a stainless steel autoclave (50 mL) with an initial N2 pressure of 1 MPa. In a typical process, the catalyst (30 mg), lignin (100 mg), and iso-propanol (20 mL) were charged in the autoclave and the reactor was stirred at 800 rpm for 12 h at 170 °C. After reaction, the mixture was cooled to room temperature and was filtered. The filtrate was distilled at 45 °C under reduced pressure to obtain light-yellow liquid oil. The liquid oil was weighed and was then diluted with methanol to 1.5 mL, the monomer products in the liquid oil were qualitatively determined with a HP 5973 GC-MS (HP-5 column, 30 m × 0.32 mm × 0.25 μm) and were quantified with GC-FID (HP-5 column, 30 m × 0.32 mm × 0.25 μm) by an internal standard method using mesitylene as the standard. NMR spectra were recorded on a Bruker AVANCE III HD 700 MHz spectrometer at 25 °C. The Bruker standard pulse program hsqcetgpsi was used for HSQC acquisition. Fifty milligrams of lignin was dissolved in 0.5 mL d6-DMSO and the solvent peak was used as the internal reference. HSQC cross-signals were analyzed and assigned by comparing with the published literature.41Results and discussion
Catalyst preparation and characterization
Ni–Al layered double hydroxide compound (NiAl-LDH) is a class of two dimensional (2D) anion-intercalated materials, which was prepared by traditional co-precipitation.32 Powder X-ray diffraction patterns (Fig. S1†) illustrated the crystalline nature of NiAl-LDH, which can be indexed to a characteristic feature of the layered NiAl-LDH phase.33 The XRD pattern of NiAl-LDH presents the basal spacing (003), which is characteristic of the layered structures.33 After calcination at 500 °C in air, the basal reflections of NiAl-LDH disappear, and 3 new peaks with 2θ = 37.3°, 43.5° and 63.2° appear in the XRD patterns of NiO–Al2O3, corresponding to (111), (200), and (220) reflections of the NiO (Fig. 1).34 However, no reflection peaks of Al2O3 in the XRD patterns of NiO–Al2O3 were detected, indicating that an amorphous phase of Al2O3 is present. These results suggest that the calcination of NiAl-LDH in air at 500 °C generates NiO and an amorphous Al2O3 phase. The NiO–Al2O3 sample was then reduced at 5 representative temperatures of 500, 550, 600, 650 and 700 °C. NiO was the dominant phase for the Ni–Al-500 catalyst, while new peaks corresponding to metallic Ni were clearly observed in the XRD patterns of the Ni–Al-550 catalyst. Further increasing the reduction temperature to 600 °C, metallic Ni became the main phase for the NiO–Al2O3-600 catalyst. Three peaks with 2θ = 44.2°, 51.6° and 76.2° clearly appeared in the XRD patterns of the NiO–Al2O3-600, NiO–Al2O3-650 and NiO–Al2O3-700 catalysts, which were assigned to (111), (200), and (220) reflections of metallic Ni (PDF number 01-087-0712). The XRD results indicate that the NiO phase gradually transformed into metallic Ni with the increase of the reduction temperature. The particle size of metallic Ni calculated by the Scherrer equations (Table S1†) increased from 5.5 nm for the Ni/Al2O3-500 catalyst to 10.2 nm for the Ni/Al2O3-700 catalyst, suggesting the growth of Ni nanoparticles with the increase of reduction temperature. Ni loadings in the Ni/Al2O3-500, Ni/Al2O3-550 and Ni/Al2O3-600 catalysts determined by ICP-AES slightly increased with the increase of the reduction temperature (Table S1†). The ICP-AES results were consistent with the XRD results. XRD results reveal that NiO in the NiO–Al2O3 sample were gradually reduced to metallic Ni by increasing the reduction temperature from 500 °C to 600 °C, and thus the Ni loading in the Ni/Al2O3-T catalysts increased in the order of Ni/Al2O3-500, Ni/Al2O3-550 and Ni/Al2O3-600. | ||
| Fig. 1 XRD patterns of NiAl-MMO and Ni/Al2O3-T catalysts. | ||
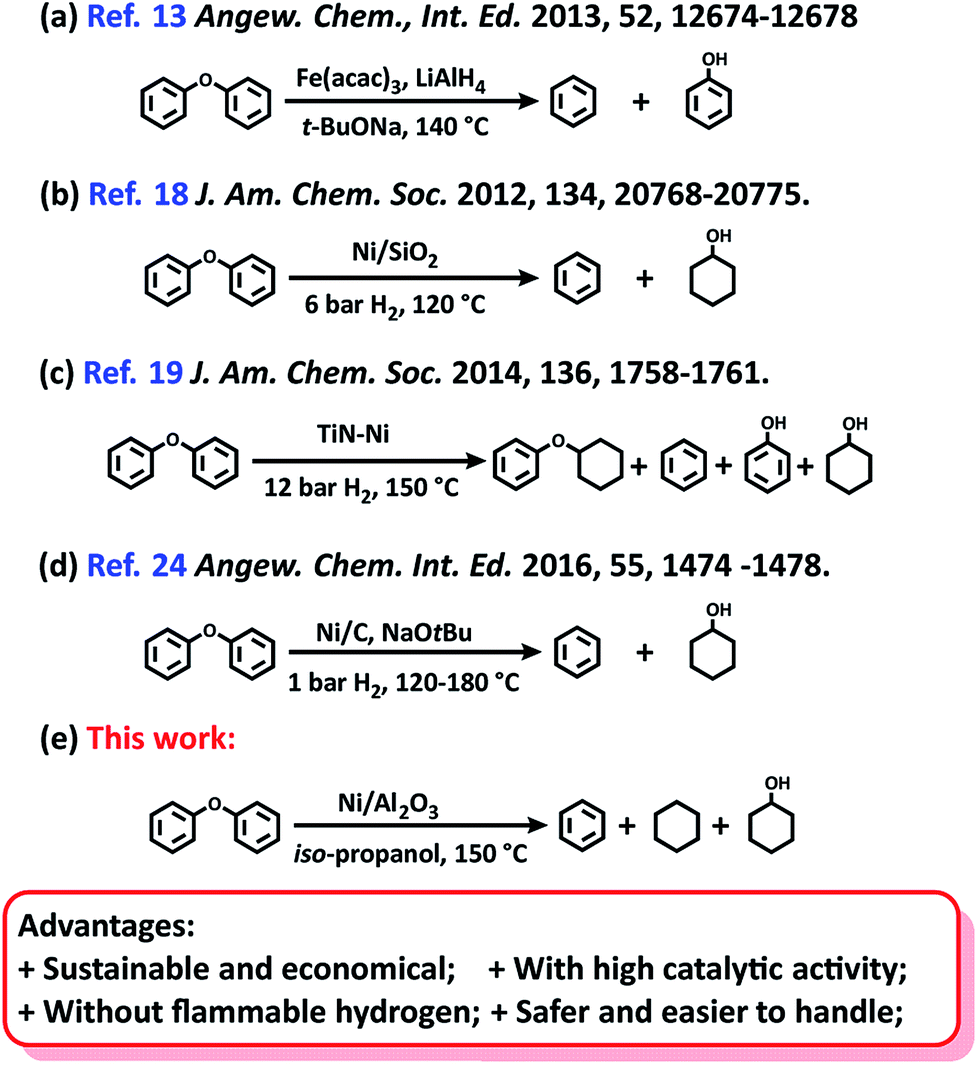
The BET surface areas and average pore diameter of the as-prepared Ni/Al2O3-T catalysts are listed in Table S1.† All the Ni/Al2O3-T catalysts display a type IV isotherm shape (Fig. 2a), characteristic of the mesoporous structure and wide mesopore size distribution (Fig. 2b). Interestingly, the surface area of the Ni/Al2O3-T catalysts gradually decreased with the increase of the reduction temperature (Table S1†). The average size of the Ni/Al2O3-T catalysts increased with the increase of the reduction temperature, which is contrary to the effect on the surface area of the Ni/Al2O3-T catalysts (Table S1†).
 | ||
| Fig. 2 (a) N2 adsorption/desorption isotherms of Ni/Al2O3-T catalyst and, (b) corresponding Barrett–Joyner–Halenda (BJH) pore-size distribution curve. | ||
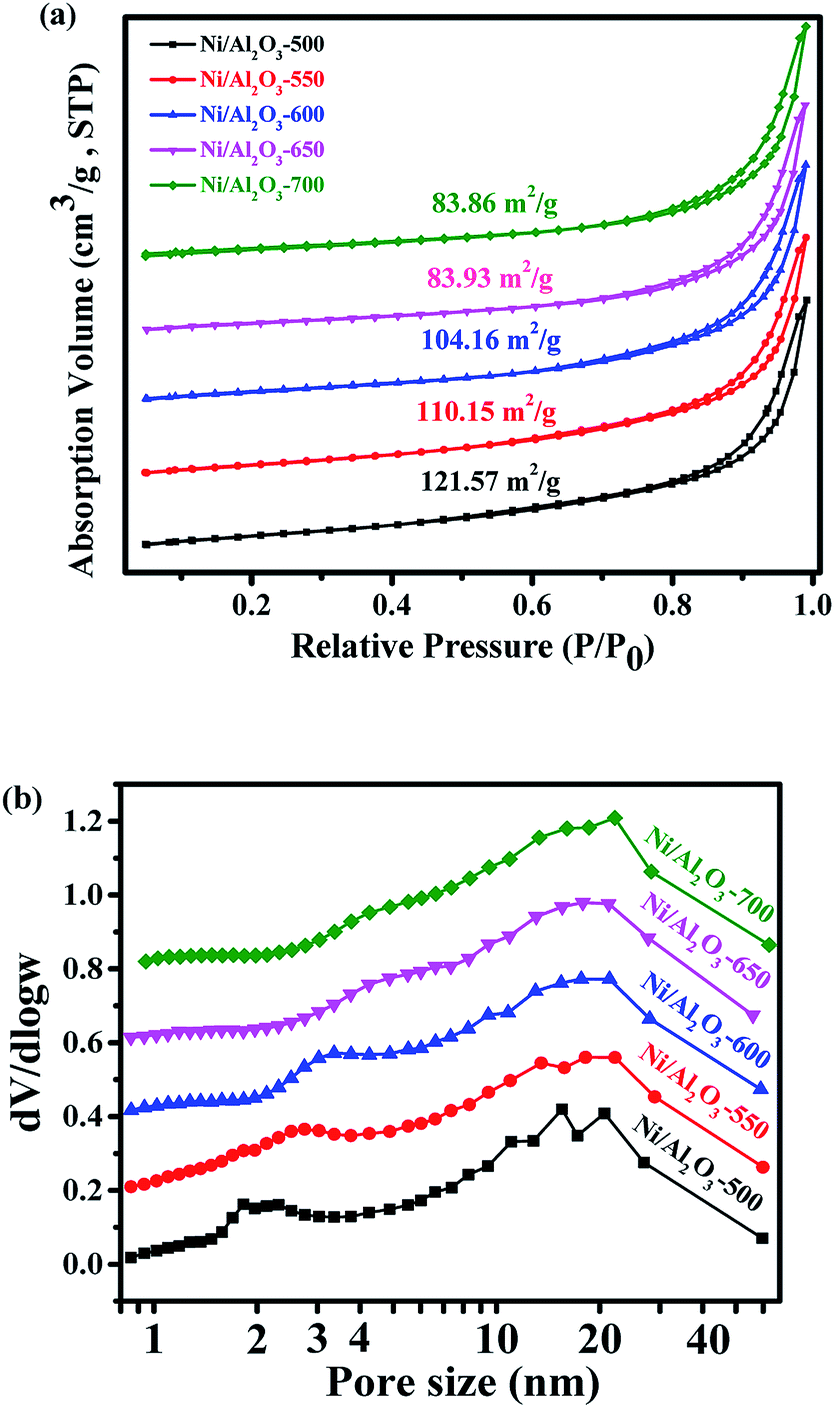
X-ray photoelectron spectroscopy (XPS) was carried out to detect the valence states of surface Ni of the representative Ni/Al2O3-600 catalyst (Fig. S2†). The peaks of binding energy at ∼851.7 (Ni 2p3/2), ∼854.3/872.1 (Ni 2p1/2), and ∼860.1/878.7 eV can be assigned to metallic Ni, divalent valence state Ni2+ and the satellite peak, respectively.35 Obviously, most of the nickel nanoparticles were present in the oxidation state as detected by XPS, albeit the XRD results of the Ni/Al2O3-600 catalyst reveal that the nickel species presented mainly in the metallic state. The crystal phase detected by XRD was metallic state, while the surface of the metallic nickel nanoparticle was highly oxidized into its oxidation state by oxygen either from the passivated process or air storage. Nickel nanoparticles were clearly observed and well dispersed on the surface of the Ni/Al2O3-T catalysts (Fig. 3). The average size of the nickel nanoparticle slightly increased to 5.6, 6.9 and 7.2 nm for the Ni/Al2O3-500, Ni/Al2O3-550 and Ni/Al2O3-600 catalysts, respectively. However, its size greatly increased to 14.9 and 16.5 nm for the Ni/Al2O3-650 catalyst and Ni/Al2O3-700 catalyst with a large size distribution. These results suggest that the nickel nanoparticles grow into large-sized nanoparticles at high reduction temperatures beyond 600 °C.
 | ||
| Fig. 3 TEM images of the as-prepared Ni/Al2O3-T catalysts and particle size distribution of the Ni nanoparticles ((a) Ni/Al2O3-500; (b) Ni/Al2O3-550; (c) Ni/Al2O3-600; (d) Ni/Al2O3-650; (e) Ni/Al2O3-700). | ||
Catalyst screen
Firstly, the activity of the Ni/Al2O3-T catalysts was evaluated by the transfer hydrogenation of benzyl phenyl ether (α-O-4) as the model reaction with iso-propanol as the hydrogen donor as well as the solvent. The hydrogenation of benzyl phenyl ether did not occur without the catalyst (Table 1, entry 1). Similarly, no conversion of benzyl phenyl ether was observed in the presence of Al2O3, suggesting that Al2O3 was inactive for this transformation (Table 1, entry 2). To our delight, all the Ni/Al2O3-T catalysts were active for the transfer hydrogenation of benzyl phenyl ether, with iso-propanol as the hydrogen donor (Table 1, entries 3–7). The catalytic activity of Ni/Al2O3-T greatly increased with the increase of reduction temperature from 500 to 600 °C. The conversions of benzyl phenyl ether were 15.4%, 22.0% and 55.3% for the Ni/Al2O3-500, Ni/Al2O3-550 and Ni/Al2O3-600, respectively (Table 1, entries 3–5). Comparing the results in entries 3–5, shows that the active phase for the transfer hydrogen from iso-propanol was metallic Ni. The highest conversion of benzyl phenyl ether was attained in 58.9% over the Ni/Al2O3-650 catalyst (Table 1, entry 6). However, further increasing the pyrolysis temperature to 700 °C, the conversion of benzyl phenyl ether significantly decreased to 33.1% (Table 1, entry 7). Although Ni/Al2O3-700 has the largest content of metallic nickel, its lower catalytic activity should be due to its large particle size, as metal nanoparticles with smaller size have been generally considered to provide more active sites for chemical reactions.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Con (%) | Yield (%) | |||
| 1 | 2 | 3 | 4 | |||
| a Reaction conditions: benzyl phenyl ether (185 mg, 1 mmol), 130 °C, 3 h, catalyst (20 mg), iso-propanol (10 mL), stirring at 1000 rpm. | ||||||
| 1 | — | 0 | — | — | — | — |
| 2 | Al2O3 | 0 | — | — | — | — |
| 3 | Ni/Al2O3-500 | 15.4 | 15.4 | 14.4 | — | 1.0 |
| 4 | Ni/Al2O3-550 | 22.0 | 22.0 | 20.9 | — | 1.1 |
| 5 | Ni/Al2O3-600 | 55.3 | 55.3 | 50.5 | 0.5 | 4.3 |
| 6 | Ni/Al2O3-650 | 58.9 | 58.9 | 57.8 | 0.2 | 0.9 |
| 7 | Ni/Al2O3-700 | 33.1 | 33.1 | 32.6 | — | 0.5 |

For all the reactions, the C–O–C bond was selectively cleaved at the position of the aliphatic carbon, producing phenol and toluene. In addition, the gas phase was also investigated with gas chromatography, and hydrogen was not detected. These results suggest that the cleavage of benzyl phenyl ether was a real hydrogen-transfer process by iso-propanol over the Ni/Al2O3-T catalysts. Although the Ni/Al2O3-600 catalyst showed a similar catalytic activity as the Ni/Al2O3-650 catalyst, the former demonstrated stronger ability to promote the subsequent hydrogenation of phenol into cyclohexanol (Table 1, entries 5 vs. 6). Therefore, the Ni/Al2O3-600 catalyst was used for the following studies.
Transfer hydrogenation of benzyl phenyl ether at different temperatures
The effect of the reaction temperature on the transfer hydrogenolysis of benzyl phenyl ether was also examined over the Ni/Al2O3-600 catalyst (Table 2). Interestingly, the Ni/Al2O3-600 catalyst was even active for the cleavage of the C–O–C bond at a low temperature of 100 °C (Table 2, entry 1). To the best of our knowledge, there has been no such report on the transfer hydrogenation of benzyl phenyl ether by iso-propanol under such low temperatures over a heterogeneous non-noble metal catalyst. Prolonging the reaction time to 48 h, 100% conversion of benzyl phenyl ether was attained with toluene and phenol as the main product (Table 2, entry 2). The transfer hydrogenolysis of benzyl phenyl ether at low temperature without external hydrogen shows huge potential for the large-scale utilization of lignin to produce valuable chemicals as these processes are environmentally friendly, easy to handle, and economical. It was noted that both the cleavage of the benzyl phenyl ether bond and the subsequent hydrogenation of phenol are sensitive to the reaction temperature. The conversion of benzyl phenyl ether increased with the increase of the reaction temperature from 4.3% at 100 °C to 100% at 150 °C after 3 h (Table 2, entries 1–6). The selectivity of toluene remained 100% at different temperatures, while phenol could be further reduced to cyclohexanol, especially at high temperatures of 140 to 160 °C, (Table 2, entries 5–7). For example, the selectivity of cyclohexanol significantly increased from 13.4% at 140 °C, to 92.3% at 160 °C after 3 h. Unlike the transfer cleavage of the C–O–C bond in benzyl phenyl ether, the selectivity of cyclohexanol was more sensitive to the reaction temperature suggesting that the transfer hydrogenation of phenol into cyclohexanol was more difficult than the transfer cleavage of the C–O–C bond in benzyl phenyl ether. Cyclohexanone, the intermediate of the hydrogenation of phenol into cyclohexanol, remained at low levels irrespective of the reaction temperature, suggesting that cyclohexanone could be readily reduced to cyclohexanol.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Temperature (°C) | Con (%) | Yield (%) | |||
| 1 | 2 | 3 | 4 | |||
| a Reaction conditions: benzyl phenyl ether (185 mg, 1 mmol), Ni/Al2O3-600 (20 mg), iso-propanol (10 mL), 3 h, stirring at 1000 rpm. b The reaction time was 48 h. | ||||||
| 1 | 100 | 4.3 | 4.3 | 4.3 | — | — |
| 2b | 100 | 100 | 100 | 87.5 | 0.3 | 12.2 |
| 3 | 120 | 25.5 | 25.5 | 23.3 | 0.2 | 1.9 |
| 4 | 130 | 55.3 | 55.3 | 50.5 | 0.5 | 4.3 |
| 5 | 140 | 88.5 | 88.5 | 75.5 | 1.1 | 11.9 |
| 6 | 150 | 100 | 100 | 22.4 | 1.6 | 73.1 |
| 7 | 160 | 100 | 100 | 6.8 | 0.9 | 92.3 |
Catalytic transfer hydrogenation of benzyl phenyl ether with different alcohols
Furthermore, other common alcohols were tested for the transfer hydrogenolysis of benzyl phenyl ether at 160 °C. Compared with iso-propanol, the transfer hydrogenolysis of benzyl phenyl ether in primary alcohols such as ethanol, n-propanol and n-butanol produced much lower conversions (Table S2,† entries 1–3 vs. 4), due to the difficulty of eliminating β-hydrides from primary alcohols on the surface of the catalyst.36 In addition, 2-butanol was inactive for the transfer hydrogenolysis of benzyl phenyl ether due to the larger steric hindrance in comparison with iso-propanol, which makes attack by the metal sites difficult (Table S2,† entries 4 vs. 5).Comparison of the activity of the three representative model compounds
The transfer hydrogenolysis of three model compounds – benzyl phenyl ether (α-O-4), 2-phenylethyl phenyl ether (β-O-4) and diphenyl ether (4-O-5) – were studied. The bond dissociation energy (BDE) of the aliphatic ether bonds of α-O-4 (218 kJ mol−1) is lower than the aliphatic ether bonds of β-O-4 (289 kJ mol−1) and the aryl ether bond (314 kJ mol−1) of 4-O-5.18 As listed in Table S3,† the reaction rate at substrate conversions below 20% decreased greatly in the order: benzyl phenyl ether (7.2 mmol gcata.−1 h−1), 2-phenylethyl phenyl ether (2.2 mmol gcata.−1 h−1), and diphenyl ether (1.6 mmol gcata.−1 h−1). This correlates well with the bond dissociation energies of these bonds (Table S3,† entries 1–3).18 Kinetic studies of the three model compounds were further studied (Fig. S3†), and their apparent activation energies were calculated to be 103, 130 and 147 kJ mol−1 for the cleavage of the benzyl phenyl ether (α-O-4), 2-phenylethyl phenyl ether (β-O-4) and diphenyl ether (4-O-5), respectively. The apparent activation energies for the cleavage of α-O-4, β-O-4, and 4-O-5 bonds increased almost linearly with their C–O BDEs (Fig. 4), indicating that the cleavage barrier of the aryl C–O bonds was heavily influenced by the C–O BDE. As shown in Table S3,† the lowest BDE of α-O-4 (218 kJ mol−1) corresponds well with the highest reaction rate (7.2 mmol gcata.−1 h−1) and the lowest apparent activation energy (Ea = 103 kJ mol−1). By comparison, much higher apparent activation energy was observed for the 2-phenylethyl phenyl ether (β-O-4, Ea = 130 kJ mol−1) and diphenyl ether (4-O-5, Ea = 147 kJ mol−1).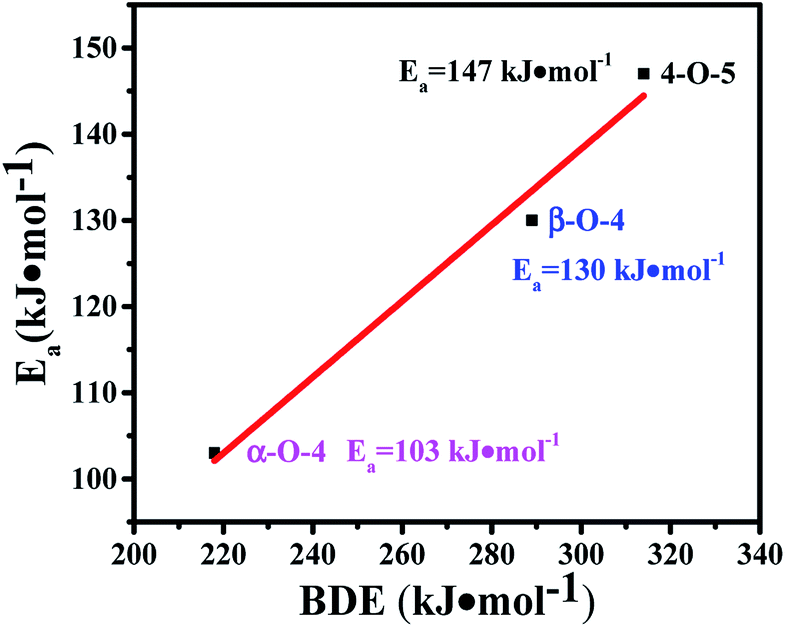 | ||
| Fig. 4 The relationship between apparent Ea and BDE for the three model compounds. | ||
Kinetics and reaction pathways of the transfer hydrogenolysis of the three model compounds
The time course of the products distribution was studied for the transfer hydrogenolysis of benzyl phenyl ether over the Ni/Al2O3-600 catalyst at 150 °C (Fig. 5a). Benzyl phenyl ether was fully consumed after 2 h at 150 °C. Interestingly, the slope of the conversion curve between the 4 reaction points (0–15 min, 15–30 min; 30–45 min; 45–60 min) became larger, indicating that a higher transfer hydrogenolysis rate was obtained. Generally, a higher reaction rate was observed at the earlier reaction stage because of the higher concentration of benzyl phenyl ether at the earlier reaction stage. The possible reason could be that NiO in the Ni/Al2O3-600 catalyst was gradually reduced to metallic nickel by iso-propanol within 60 min, which is the active site for transfer hydrogenation.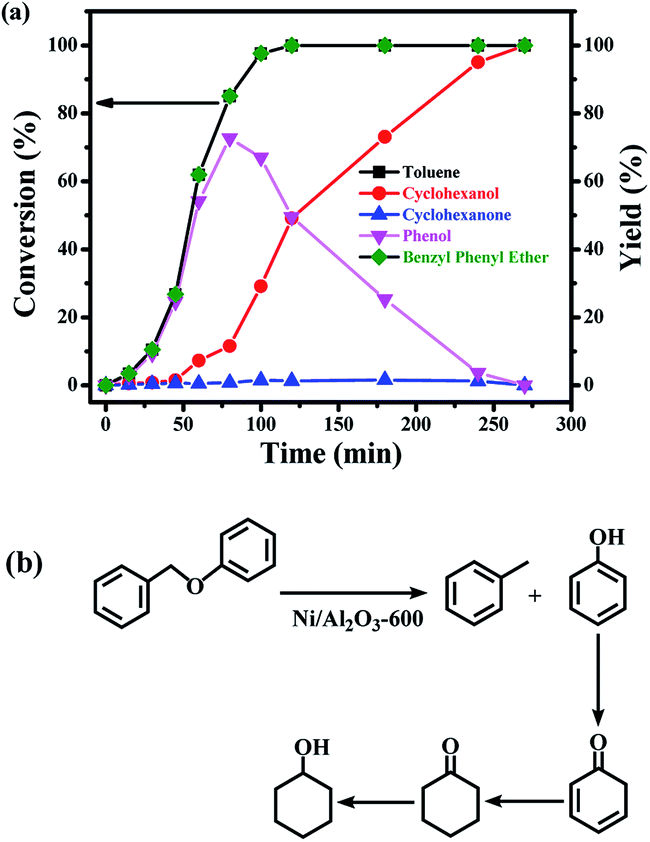 | ||
| Fig. 5 (a) The time course of the product distributions for the conversion of benzyl phenyl ether (α-O-4) over the Ni/Al2O3-600 catalyst. Reaction conditions: benzyl phenyl ether (α-O-4) (185 mg, 1 mmol), Ni/Al2O3-600 (20 mg), solvent (10 mL), 150 °C, stirring at 1000 rpm. (b) Reaction pathway for the cleavage of benzyl phenyl ether. | ||
Phenol and toluene are the major products early in the reaction. The further hydrogenation of the aromatic ring in toluene into methylcyclohexane was not observed, but phenol was further reduced to cyclohexanol with cyclohexanone as the intermediate. It has been reported that the hydrogenation of phenol into cyclohexanol does not occur with direct hydrogenation of the aromatic ring,38 cyclohexanone is the intermediate. According to the reported results,38 the –OH group possibly promotes the facile formation of cyclohexanone via keto–enol tautomerism as depicted in Fig. 5b. Therefore, the hydroxyl group in phenol is crucial for its hydrogenation, while toluene is stable under the reaction conditions.
The selectivity of cyclohexanol reached 100% after 4.5 h at 150 °C. The deep hydrogenolysis of cyclohexanol into cyclohexane was not observed, which is promoted by acid–metal bifunctional catalysts involving the acid-catalyzed dehydration of cyclohexanol into cyclohexene and the metal-catalyzed hydrogenation of cyclohexene into cyclohexane.37 Benzene and benzyl alcohol were not detected during the reaction process, which revealed that the C–O–C bond of benzyl phenyl ether was selectively cleaved at the position of the aliphatic carbon oxygen bond (Caliphatic–O) on the Ni surface, due to the much lower C–O bond dissociation energy barrier of the Caliphatic–O bond (Table S3,† entry 1), producing phenol and toluene (Fig. 5b). Given the above results, the plausible reaction route for the transfer hydrogenolysis of benzyl phenyl ether is depicted in Fig. 5b. Hydrogenolysis was the dominant and initial reaction pathway for the cleavage of benzyl phenyl ether (α-O-4) on the surface of Ni nanoparticles, affording toluene and phenol as the primary products (Fig. 5b). The as-formed phenol underwent the sequential hydrogenations to produce cyclohexanol.
Compared with the hydrogenolysis of benzyl phenyl ether (α-O-4), the hydrogenolysis conversion of diphenyl ethers was much more challenging.23 Fortunately, the transfer hydrogenolysis of diphenyl ether could be performed under mild reaction conditions (120–150 °C, Fig. S3b†). The time course of the products distribution for the hydrogenolysis of diphenyl ether over the Ni/Al2O3-600 catalyst at 150 °C is recorded in Fig. 6a. Six products were detected for the hydrogenolysis of benzyl phenyl ether. As shown in Fig. 6a, benzene (33.6% selectivity), phenol (28.7% selectivity) and cyclohexyl phenyl ether (32.2% selectivity) were detected as the three major products within 1 h, suggesting the transfer hydrogenolysis of diphenyl ether underwent a different reaction pathway in comparison with that of benzyl phenyl ether (α-O-4). Cyclohexyl phenyl ether should be generated from the hydrogenation of one aromatic ring in benzyl phenyl ether (Fig. 6b, route B). After 7 h, the conversion of diphenyl ether reached 100%, and the yield of benzene gradually increased to 76.3% after 7 h, and finally remained stable from 7 h to 12 h. These results suggest that benzene is generated from the direct cleavage of the diphenyl ether (4-O-5) as depicted in route A, while phenol was simultaneously produced and was further hydrogenated into cyclohexanol via cyclohexanone as the intermediate as discussed above in the reaction pathway for the hydrogenolysis of benzyl phenyl ether (α-O-4) in Fig. 6b. The stable benzene yield from 7 h to 12 h also revealed that the cyclohexyl phenyl ether did not undergo the pathway to produce benzene and cyclohexanol, and the deep hydrogenation of benzene into cyclohexane did not occur under our reaction conditions. The selective cleavage of the aliphatic C–O bond in cyclohexyl phenyl ether generated the cyclohexane and phenol, and phenol was finally hydrogenated into cyclohexane (Fig. 6b). The depicted reaction pathways in Fig. 6b clearly explain our results. At the end of the reaction time point, the yield of cyclohexanol was 100%, and the yields of benzene and cyclohexane were 74.9% and 25.1%, respectively. Cyclohexanol could be produced from route A and route B in Fig. 6b, therefore the 100% yield is achieved. However, benzene could only be produced from route A, and cyclohexane only from route B. Thus, the total yield of benzene and cyclohexane is 100%.
 | ||
| Fig. 6 (a) The time course of the product distributions for the conversion of diphenyl ether (4-O-5) over the Ni/Al2O3-600 catalyst. Reaction conditions: diphenyl ether (4-O-5) (170 mg, 1 mmol), Ni/Al2O3-600 (20 mg), solvent (10 mL), 150 °C, stirring at 1000 rpm. (b) Reaction pathway for the cleavage of diphenyl ether. | ||
Finally, the time course of the product distribution of the transfer hydrogenation of 2-phenylethyl phenyl ether (β-O-4) was also studied (Fig. 7a). Phenol and ethylbenzene were the main products within 1 h, generated from the cleavage of the aliphatic C–O bond in 2-phenylethyl phenyl ether (β-O-4) (route A, Fig. 7b). Phenol was again gradually hydrogenated into cyclohexanol via cyclohexanone as the intermediate, and the yield of ethylbenzene increased during the reaction process. These results indicate that ethylbenzene is stable. In fact, the further hydrogenation of ethylbenzene did not occur under the same reaction conditions. Similar to the hydrogenolysis of the diphenyl ether (4-O-5), the transfer hydrogenation of the aromatic ring in 2-phenylethyl phenyl ether without the cleavage of the ether (β-O-4) was also observed, generating cyclohexyl phenethyl ether (route B, Fig. 7b) and 2-cyclohexylethyl phenyl ether (route C, Fig. 7b), but the degree of the hydrogenation of the aromatic ring in 2-phenylethyl phenyl ether (β-O-4) was very low. Ethylcyclohexane as one of the products was also observed, and its yield increased during the reaction process. Ethylcyclohexane could be generated from the hydrogenolysis of 2-cyclohexylethyl phenyl ether, and phenol was simultaneously produced followed by the further hydrogenation into cyclohexanol (route B, Fig. 7b). The cleavage of cyclohexyl phenethyl ether produced ethylbenzene and cyclohexanol (route C). The proposed reaction pathway in Fig. 7b is consistent with our experiments. Cyclohexanol can be produced in three routes in Fig. 7b, thus it could be attained at 100% yield. The total yield of ethylbenzene and ethylcyclohexane should be 100% according to Fig. 7b, which is consistent with our experimental results.
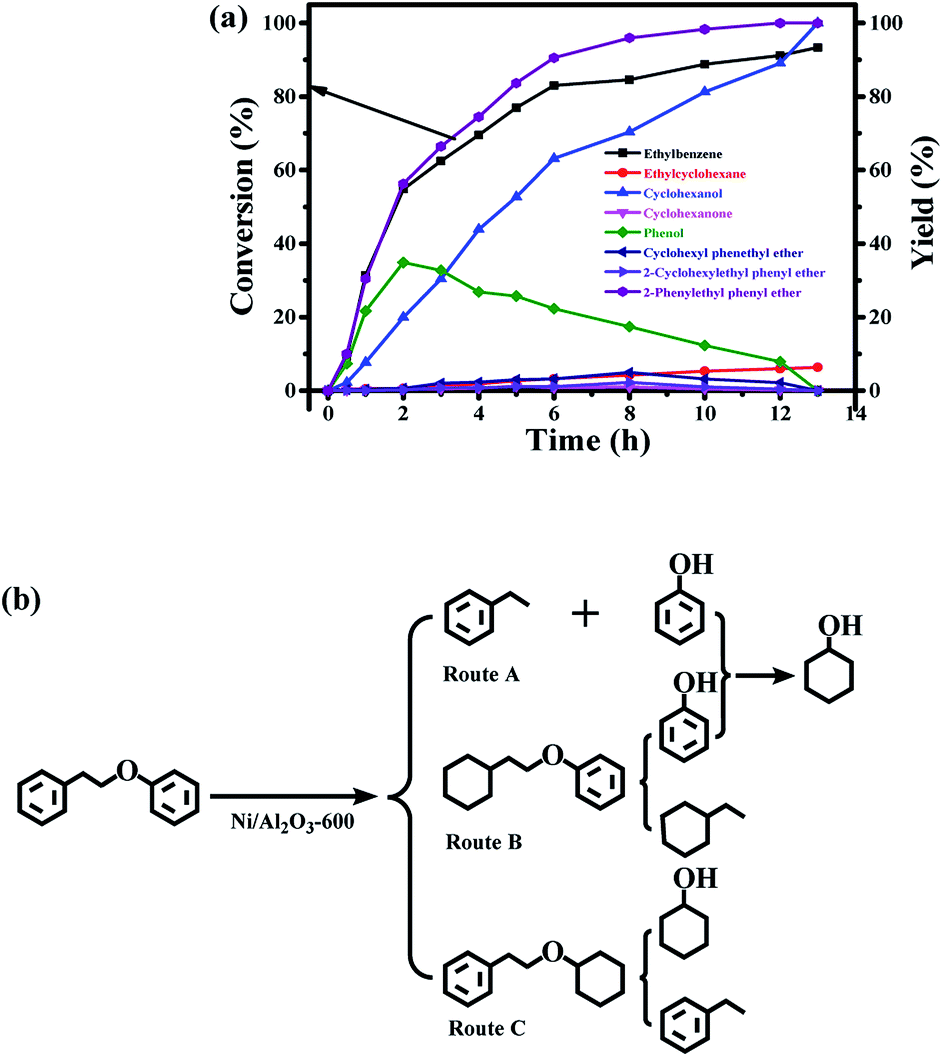 | ||
| Fig. 7 (a) The time course of the product distributions for the conversion of 2-phenylethyl phenyl ether (β-O-4) over the Ni/Al2O3-600 catalyst. Reaction conditions: 2-phenylethyl phenyl ether (β-O-4) (198 mg, 1 mmol), Ni/Al2O3-600 (20 mg), solvent (10 mL), 170 °C, stirring at 1000 rpm, (b) reaction pathway for the cleavage of 2-phenylethyl phenyl ether. | ||
In summary, the Caliphatic–O–Caromatic linkages (α-O-4, benzyl phenyl ether and β-O-4, 2-phenylethyl phenyl ether) are mainly cleaved on the Caliphatic–O side due to the weaker bond energy of Caliphatic–O bonds. The Caromatic–O–Caromatic linkage in the diphenyl ether underwent transfer hydrogenolysis as well. In addition, the cleavage of the Caliphatic–O in benzyl phenyl ether (α-O-4) was much easier due to its lower activation energy than those of the Caliphatic–O bonds in 2-phenylethyl phenyl ether and Caromatic–O–Caromatic linkage in diphenyl ether. At the same time, the hydrogenation of the aromatic rings was not observed in benzyl phenyl ether. However, the hydrogenation of aromatic rings in 2-phenylethyl phenyl ether and diphenyl ether occurred at higher reaction temperatures. The partial hydrogenation products of 2-phenylethyl phenyl ether and diphenyl ether also underwent the cleavage of Caliphatic–O to generate the hydrogenolysis products. Phenol was further hydrogenated into cyclohexanol, while the aromatic rings in benzene, toluene, and ethylbenzene inert under our reaction conditions.
Catalytic transfer hydrogenolysis cleavage of lignin
Given the excellent results in model compounds and the kinetic study, dioxasolv beech lignin was further used to prove the transfer hydrogenolysis activity of Ni/Al2O3-600 by iso-propanol. The depolymerisation results of total oil yield and identified monomers yield are shown in Table S4.† 69.2% of aromatic liquid oil with 13.4% of identified monomers (i.e., product 1–9, Table S4†) was obtained over Ni/Al2O3-600, demonstrating that the lignin could also be effectively depolymerized without external hydrogen.2D HSQC NMR was employed to better estimate the structure change of lignin before and after reaction. The linkage region of lignin and the corresponding lignin oil are illustrated in Fig. 8a and c. It was found that all the A (β-O-4), B (β-5/α-O-4), C (β–β) and lignin-bound Hibbert’s ketone (LBHK) in lignin oil experienced notable decrease in area after reaction compared with that in the lignin sample (Fig. 8cvs.Fig. 8a), indicating the versatility of our catalytic system. In particular, a significant decrease in area of A and LBHK linkages was observed. In detail, 45 per C9 units was attributed to A in lignin before reaction (Fig. 8a), while the datum was 12 in lignin oil after reaction (Fig. 8c), suggesting that major β-O-4 bonds in lignin were cleaved over Ni/Al2O3-600. A similar trend was obtained in LBHK linkage, the number of which fell to 3 in lignin oil from 13 in lignin. The above phenomenon implies that all the linkages could be attacked by the Ni/Al2O3-600 catalyst, while A and LBHK linkages to be destructed due to the relatively lower BDE, as described above compared with B and C linkages. No clear difference of S:G ratio was observed in Fig. 8b and d. The above results further confirm the high transfer hydrogenolysis of our catalytic system in lignin depolymerisation.
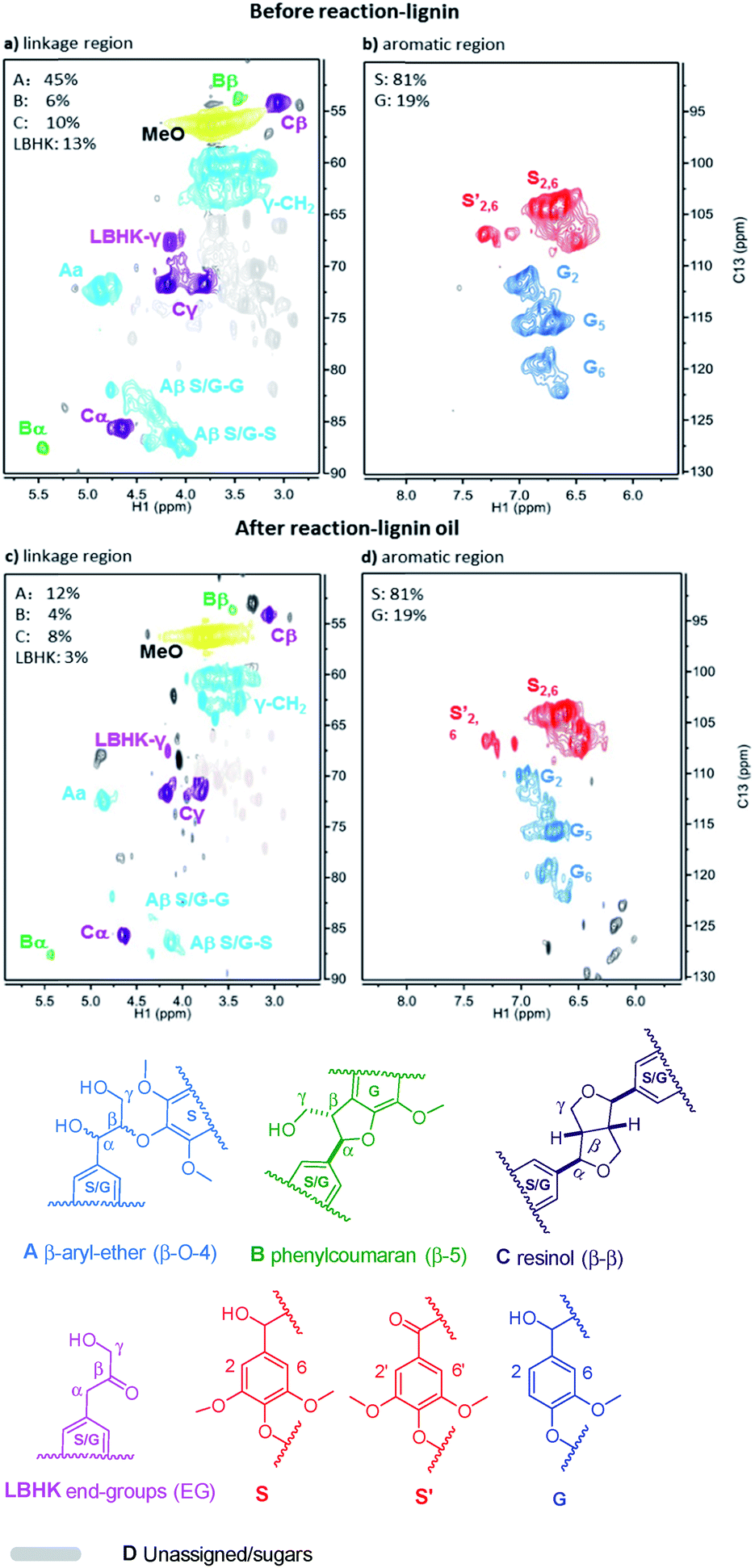 | ||
| Fig. 8 2D HSQC NMR spectra of beech lignin in DMSO-d6 before reaction (a and b); and lignin oil after reaction (c and d). Note: the percentage of linkages A, B, C and LBHK was calculated based on per 100 C9 unit, see Fig. S4† for the detailed integration process. | ||
Mechanisms of the cleavage of the C–O bond for the three model compounds
Although it is difficult to get an accurate mechanism for the transfer hydrogenolysis of C–O bonds in lignin model compounds, we would like to provide further insights into this transformation. As we know, the hydrogen in the hydroxyl group (–OH) of iso-propanol is active (H+), and can move freely. It is reported that Ni/Al2O3 catalyst contains acid and basic sites (Niδ+–Oδ− or Alδ+–Oδ−),39 and the H+ in the O–H group of iso-propanol can be combined with the basic sites (Oδ−, Scheme 2). In fact, Oδ− containing material such as α-cyclodextrin (α-CD) or molecular sieves have been reported to serve as basic sites (combing with H+) to promote the heterolytic cleavage of H2 molecules.40 According to the above experiments and analysis, a plausible mechanism is proposed for the transfer hydrogenolysis of an ether bond. The dissociated H+ would combine with the oxygen atom in the ether bond (C–O), and then the phenol molecule breaks away. Three carbon cations (phenylium, phenylmethylium, phenylethlium) are hence generated. Our results in Table 1 confirm that the active sites are on the metallic nickel, and the metallic nickel activates the C–H bond connected by the hydroxyl group to generate the active species (Ni–H−). Then the hybrid (H−) would combine with the carbon cations (phenylium, phenylmethylium, phenylethlium) to generate benzene, toluene and ethylbenzene, respectively. Similarly, the hydrogenation of phenol by the H+ and H− species generated from iso-propanol over the Ni/Al2O3-600 catalyst produced cyclohexanol via cyclohexanone as the intermediate.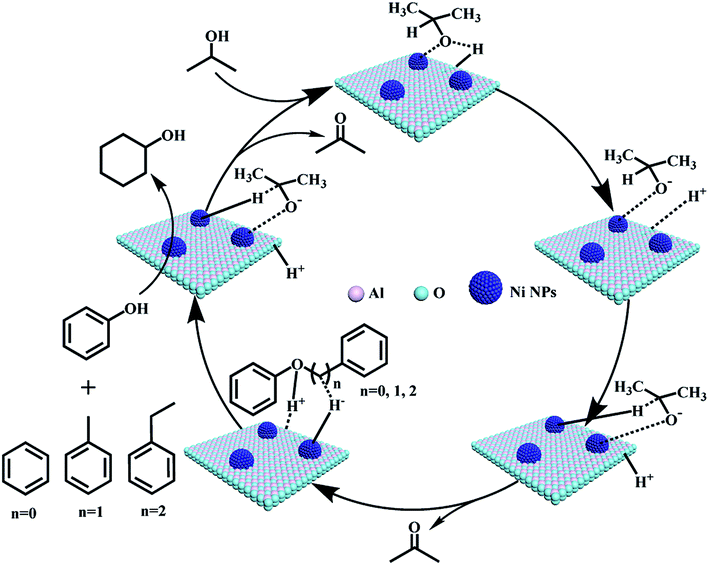 | ||
| Scheme 2 Proposed mechanisms for cleavage of C–O bonds in the α-O-4, β-O-4, and 4-O-5 model compounds. | ||
Catalyst stability
As a heterogeneous catalyst, ability to recycle is important.42,43 The recycling of the Ni/Al2O3-600 catalyst was studied during the transfer hydrogenation of benzyl phenyl ether at 150 °C. One important advantage is that the catalyst is easily separated from the reaction mixture owing to the magnetic properties of Ni particles, which simplifies the tedious recycling procedures via centrifugation and filtration for non-magnetic catalysts. As shown in Fig. 9, the Ni/Al2O3-600 catalyst is stable and shows no obvious decrease in activity and selectivity after five catalytic cycles. ICP measurements show that there is no leaching of Ni into the reaction solution. These results suggest that the Ni/Al2O3-600 catalyst is highly stable during the reaction process without loss of its activity.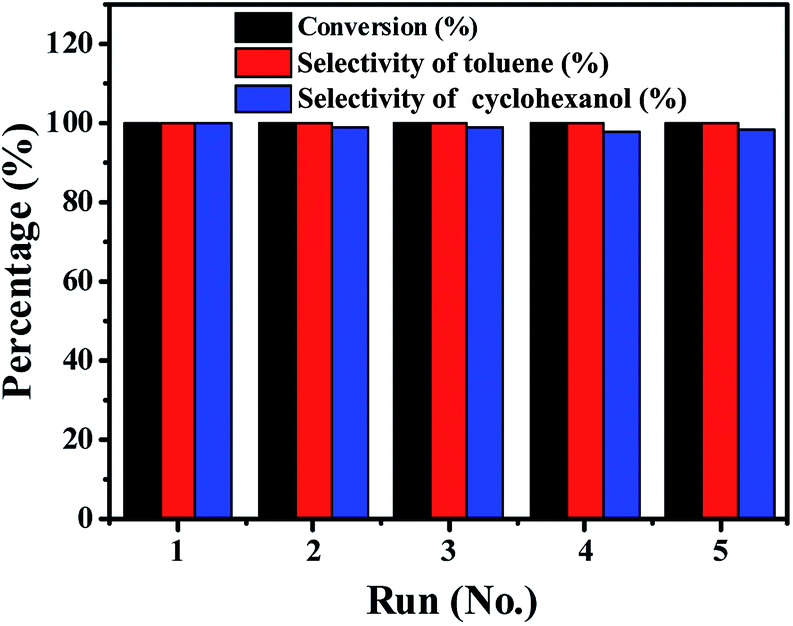 | ||
| Fig. 9 Catalytic cycling tests of Ni/Al2O3-600 catalyst during the hydrogenation of benzyl phenyl ether. Reaction conditions: benzyl phenyl ether (α-O-4) (185 mg, 1 mmol), Ni/Al2O3-600 (20 mg), solvent (10 mL), 150 °C, stirring at 1000 rpm, 5 h. | ||
Conclusions
In this study, we developed an effective method for the transfer cleavage of the C–O bond in model compounds as well as beech lignin using iso-propanol as the hydrogen donor over Ni/Al2O3-T catalysts. The catalytic activity is greatly influenced by the reduction temperature of the NiAl-layered double hydroxide precursor, which greatly affects the nickel nanoparticle size as well as the metallic nickel composition. The Ni/Al2O3-600 catalyst shows the highest catalytic activity for the transfer hydrogenolysis reactions, as it demonstrates an appropriated nanoparticle size and a relatively high ratio of metallic nickel. Kinetic studies reveal that the transfer hydrogenolysis activity of the three model compounds decreases in the following order: benzyl phenyl ether (α-O-4), 2-phenylethyl phenyl ether (β-O-4) and diphenyl ether (4-O-5), which linearly correspond to their binding energy and the activation energies. A plausible mechanism is proposed for the transfer hydrogenolysis of ether bonds over the Ni/Al2O3-600 catalyst by the use of iso-propanol as the hydrogen donor, in which the proton (H+) and hybrid (H−) were the active sites for the cleavage of the C–O bonds. Reaction pathway studies revealed that aromatic alkanes and cyclohexanol are the major products for the transfer cleavage of the lignin model compounds, and a whole reaction pathway is depicted for each model compound. The present work provides a deep understanding of the excellent activity of Ni/Al2O3 in cleaving both lignin models and lignin without external hydrogen. This study may also inspire research on the use of non-noble metal catalysts for the production of other value-added chemicals from biomass-derivatives by transfer hydrogenation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported by the Special Fund for Basic Scientific Research of Central Colleges, South-Central University for Nationalities (CZR18001 and YCZW15100), the Natural Science Foundation of Hubei Province (No. 2017CFB432) and the National Natural Science Foundation of China (No. 21872175, 21690080, 21690083, 21878288).Notes and references
- A. M. Robinson, J. E. Hensley and J. W. Medlin, ACS Catal., 2016, 6, 5026–5043 CrossRef CAS
.
- A. E. Settle, L. Berstis, N. A. Rorrer, Y. Roman-Leshkov, G. T. Beckham, R. M. Richards and D. R. Vardon, Green Chem., 2017, 19, 3468–3492 RSC
- W. Boerjan, J. Ralph and M. Baucher, Annu. Rev. Plant Biol., 2003, 54, 519–546 CrossRef CAS PubMed
- Z. H. Sun, B. Fridrich, A. de Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678 CrossRef CAS PubMed
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. A. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 8164–8215 CrossRef CAS PubMed
- Q. N. Xia, Z. J. Chen, Y. Shao, X. Q. Gong, H. F. Wang, X. H. Liu, S. F. Parker, X. Han, S. H. Yang and Y. Q. Wang, Nat. Commun., 2016, 7, 11162 CrossRef PubMed
- Q. Song, F. Wang, J. Y. Cai, Y. H. Wang, J. J. Zhang, W. Q. Yu and J. Xu, Energy Environ. Sci., 2013, 6, 994–1007 RSC
- R. Ma, W. Y. Hao, X. L. Ma, Y. Tian and Y. D. Li, Angew. Chem., Int. Ed., 2014, 53, 7310–7315 CrossRef CAS PubMed
-
Y. R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, Boca Raton, FL, 2007 Search PubMed
- J. M. Nichols, L. M. Bishop, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2010, 132, 12554–12555 CrossRef CAS PubMed
- S. Son and F. D. Toste, Angew. Chem., Int. Ed., 2010, 49, 3791–3794 CrossRef CAS PubMed
- A. G. Sergeev and J. F. Hartwig, Science, 2011, 332, 439–443 CrossRef CAS PubMed
- Y. L. Ren, M. J. Yan, J. J. Wang, Z. C. Zhang and K. S. Yao, Angew. Chem., Int. Ed., 2013, 52, 12674–12678 CrossRef CAS PubMed
- J. G. Zhang, J. Teo, X. Chen, H. Asakura, T. Tanaka, K. Teramura and N. Yan, ACS Catal., 2014, 4, 1574–1583 CrossRef CAS
- J. G. Zhang, H. Asakura, J. van Rijn, J. Yang, P. Duchesne, B. Zhang, X. Chen, P. Zhang, M. Saeys and N. Yan, Green Chem., 2014, 16, 2432–2437 RSC
- M. Wang, H. Shi, D. M. Camaioni and J. A. Lercher, Angew. Chem., Int. Ed., 2017, 56, 2110–2114 CrossRef CAS PubMed
- M. Zaheer, J. Hermannsdorfer, W. P. Kretschmer, G. Motz and R. Kempe, ChemCatChem, 2014, 6, 91–95 CrossRef CAS
- J. He, C. Zhao and J. A. Lercher, J. Am. Chem. Soc., 2012, 134, 20768–20775 CrossRef CAS PubMed
- V. Molinari, C. Giordano, M. Antonietti and D. Esposito, J. Am. Chem. Soc., 2014, 136, 1758–1761 CrossRef CAS PubMed
- A. G. Sergeev, J. D. Webb and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 20226–20229 CrossRef CAS PubMed
- V. M. Roberts, V. Stein, T. Reiner, X. Li, A. Lemonidou, X. Li and J. A. Lercher, Chem.–Eur. J., 2011, 17, 5939–5948 CrossRef CAS PubMed
- V. M. Roberts, S. Fendt, T. Reiner, X. Li, A. A. Lemonidou, X. Li and J. A. Lercher, Appl. Catal., B, 2010, 95, 71–77 CrossRef CAS
- C. Zhao and J. A. Lercher, Angew. Chem., Int. Ed., 2012, 51, 5935–5940 CrossRef CAS PubMed
- F. Gao, J. D. Webb and J. F. Hartwig, Angew. Chem., Int. Ed., 2016, 55, 1474–1478 CrossRef CAS PubMed
- M. J. Gilkey and B. J. Xu, ACS Catal., 2016, 6, 1420–1436 CrossRef CAS
- F. Alonso, P. Riente and M. Yus, Acc. Chem. Res., 2011, 44, 379–391 CrossRef CAS PubMed
- J. G. Zhang, Green Energy & Environ., 2018, 3, 328–334 Search PubMed
- K. Barta, T. D. Matson, M. L. Fettig, S. L. Scott, A. V. Iretskii and P. C. Ford, Green Chem., 2010, 12, 1640–1647 RSC
- X. Wang and R. Rinaldi, Energy Environ. Sci., 2012, 5, 8244–8260 RSC
- L. He, Y. Q. Huang, A. Q. Wang, X. D. Wang, X. W. Chen, J. J. Delgado and T. Zhang, Angew. Chem., Int. Ed., 2012, 51, 6191–6194 CrossRef CAS PubMed
- K. Vijayakrishna, K. T. P. Charan, K. Manojkumar, S. Venkatesh, N. Pothanagandhi, A. Sivaramakrishna, P. Mayuri, A. S. Kumar and B. Sreedhar, ChemCatChem, 2016, 8, 1139–1145 CrossRef CAS
- Y. F. Zhao, B. Zhao, J. J. Liu, G. B. Chen, R. Gao, S. Y. Yao, M. Z. Li, Q. H. Zhang, L. Gu, J. L. Xie, X. D. Wen, L. Z. Wu, C. H. Tung, D. Ma and T. R. Zhang, Angew. Chem., Int. Ed., 2016, 55, 4215–4219 CrossRef CAS PubMed
- Z. X. Xu, N. Wang, W. Chu, J. Deng and S. Z. Luo, Catal. Sci. Technol., 2015, 5, 1588–1597 RSC
- X. Kong, R. Zheng, Y. Zhu, G. Ding, Y. Zhu and Y. W. Li, Green Chem., 2015, 17, 2504–2514 RSC
- T. T. Huang, Q. Y. Peng, W. J. Shi, J. D. Xu and Y. Fan, Appl. Catal., B, 2018, 230, 154–164 CrossRef CAS
- Z. Yang, Y. B. Huang, Q. X. Guo and Y. Fu, Chem. Commun., 2013, 49, 5328–5330 RSC
- S. Roy, G. Mpourmpakis, D. Y. Hong, D. G. Vlachos, A. Bhan and R. J. Gorte, ACS Catal., 2012, 2, 1846–1853 CrossRef CAS
- K. L. Luska, P. Migowski, S. El Sayed and W. Leitner, Angew. Chem., Int. Ed., 2015, 54, 15750–15755 CrossRef CAS PubMed
- H. Chen, S. He, M. Xu, M. Wei, D. G. Eyans and X. Duan, ACS Catal., 2017, 7, 2735–2743 CrossRef CAS
- T. Mahdi and D. W. Stephan, Angew. Chem., Int. Ed., 2015, 54, 8511–8514 CrossRef CAS PubMed
- H. W. Guo, D. M. Miles-Barrett, A. R. Neal, T. Zhang, C. Z. Li and N. J. Westwood, Chem. Sci., 2018, 9, 702–711 RSC
- Y. S. Ren, Z. L. Yuan, K. L. Lv, J. Sun, Z. H. Zhang and Q. Chi, Green Chem., 2018, 20, 4946–4956 RSC
- Z. L. Yuan, B. Liu, P. Zhou, Z. H. Zhang and Q. Chi, Catal. Sci. Technol., 2018, 8, 4430–4439 RSC
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc00691e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |