
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
BODIPY as electron withdrawing group for the activation of double bonds in asymmetric cycloaddition reactions†
Andrea
Guerrero-Corella
 a,
Juan
Asenjo-Pascual
a,
Tushar Janardan
Pawar
e,
Sergio
Díaz-Tendero
bd,
Ana
Martín-Sómer
b,
Clarisa Villegas
Gómez
e,
José L.
Belmonte-Vázquez
e,
Diana E.
Ramírez-Ornelas
e,
Eduardo
Peña-Cabrera
e,
Alberto
Fraile
ac,
David Cruz
Cruz
*e and
José
Alemán
*ac
a,
Juan
Asenjo-Pascual
a,
Tushar Janardan
Pawar
e,
Sergio
Díaz-Tendero
bd,
Ana
Martín-Sómer
b,
Clarisa Villegas
Gómez
e,
José L.
Belmonte-Vázquez
e,
Diana E.
Ramírez-Ornelas
e,
Eduardo
Peña-Cabrera
e,
Alberto
Fraile
ac,
David Cruz
Cruz
*e and
José
Alemán
*ac
aOrganic Chemistry Department, Módulo 1, Universidad Autónoma de Madrid, Madrid-28049, Spain. E-mail: jose.aleman@uam.es; Web: http://www.uam.es/jose.aleman
bChemistry Department, Universidad Autónoma de Madrid, Madrid-28049, Spain
cInstitute for Advanced Research in Chemical Sciences (IAdChem), Universidad Autónoma de Madrid, Madrid-28049, Spain
dCondensed Matter Physics Center, IFIMAC, Universidad Autónoma de Madrid, 28049 Madrid, Spain
eChemistry Department, División de Ciencias Naturales y Exactas, Universidad de Guanajuato, Noria Alta S/N, 36050 Guanajuato, Gto, Mexico
First published on 20th March 2019
Abstract
In this work we have found that a BODIPY can be used as an electron withdrawing group for the activation of double bonds in asymmetric catalysis. The synthesis of cyclohexyl derivatives containing a BODIPY unit can easily be achieved via trienamine catalysis. This allows a new different asymmetric synthesis of BODIPY derivatives and opens the door to future transformation of this useful fluorophore. In addition, the Quantum Chemistry calculations and mechanistic studies provide insights into the role of BODIPY as an EWG.
Introduction
BODIPY dyes (BOron DIPYrromethene) are a remarkable family of fluorophores that have been studied in recent years due to their excellent robustness, and chemical- and photo-stability.1 The structure of the BODIPY derivatives is formed by two pyrrole units linked by a carbon and complexed with a di-substituted boron atom, mainly a BF2 motif, which forms the core scaffold (see top, Scheme 1). They show impressive spectroscopic properties such as narrow absorption and emission bands in the visible wavelength range, high fluorescence quantum yields and large molar absorption coefficients among others.1b,2 As a result of these interesting characteristics, this class of fluorophores has attracted a lot of attention due to their numerous applications, for instance, as labelling reagents, in the bioimaging of living cells,3 as radiotracers for positron emission tomography,4 photocatalysts5 or photodynamic therapy (PDT).6 In addition, the introduction of stereogenic centres in these type of structures is of great importance as it is possible to modulate the BODIPY photophysics. Therefore, chiroptical applications based on circular dichroism (CD) and circularly polarized luminescence (CPL) can be used in devices for optical storage and enantioselective CPL sensors, among others.7a,b | ||
| Scheme 1 Background and present work in the [4 + 2] cycloaddition reaction via trienamine with alkenyl BODIPY derivatives (BP = BODIPY). | ||
Different modes of functionalization of BODIPY dyes have been described in the literature. They present eight different positions that can be modulated, causing changes and modifications of the spectral and photochemical properties.1,7c Initial studies into the reactivity and derivatization of these important building blocks have been carried out by Werz,8a,b Ziessel,8c Shinokubo,8d Burgess,8e Liras,8f Bröring,8g de la Moya,8h,i and us.8j
However, in spite of these efforts, very little is known about the catalytic asymmetric synthesis of BODIPY derivatives. Two main reactivities can be found: aromatic type reactivities (see top Scheme 1 coloured green, pink and blue), which are related to the direct regioselective halogenations that can be performed at different positions,9 aromatic substitutions,10 as well as cross-coupling reactions;11 and reactivity at the methyl of the methylene bridge, the most acidic position (see top Scheme 1 coloured red), although the number of these examples is scarce.1a,11 This latter position can be deprotonated and can react with diethyl ketomalonate,8a or aldehydes.12 Moreover, de la Moya group have shown that boron functionalization can be easily achieved as well, introducing different alcohol or amine derivatives.8h,i
One of the most used strategies to polarize double bonds in asymmetric catalysis is the employment of Electron Withdrawing Groups (EWGs, middle Scheme 1), which decrease the energy of the LUMO, thus favouring the interaction with the HOMO of the nucleophile. This strategy has been widely used for Michael-type nucleophilic additions or stepwise [4 + 2] cycloadditions. For this latter reaction, trienamine catalysis13 has shown to be one of the most prominent strategies,14 using double bonds activated with nitro,14c,d azlactones14a or cyanoacetate groups14b as dienophiles (middle Scheme 1). These authors have described this [4 + 2] reaction as an asynchronous cycloaddition,15via a Michael addition followed by an intramolecular iminium ion reaction. In all these examples, very strong EWGs, e.g. nitro group, or two nitriles, at the double bond were used in order to achieve the desired reactivity. Therefore, based on electron-withdrawing character of the BODIPY core,16 we wondered if it would be possible to use this interesting fluorescent moiety as an EWG of a double bond located at the 8-position to perform an asymmetric [4 + 2] cycloaddition (bottom Scheme 1). In this work, we describe the catalytic asymmetric synthesis of chiral BODIPY cyclohexane derivatives, using trienamine aminocatalysis via a Diels–Alder reaction (Scheme 1c). In addition, the optical properties of the adducts and DFT calculations, which explain the mechanism and the role of the BODIPY as an EWG have been performed.
Results and discussion
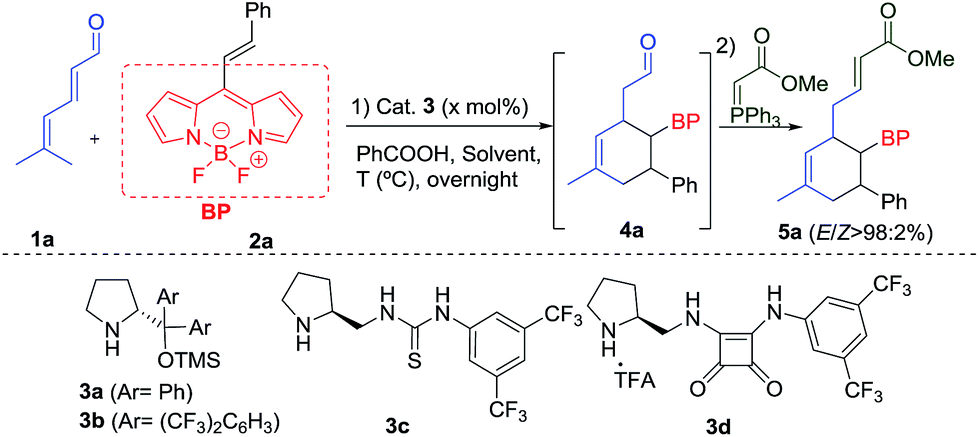
We started the present study with the reaction between the dienal 1a and the BODIPY 2a in the presence of the Jørgensen–Hayashi catalyst 3a in chloroform at room temperature. We found that the reaction gave the desired product 5a with a very low conversion (entry 1, Table 1). In order to improve this preliminary result, we tested the addition of benzoic acid as an additive (entry 2), increasing the conversion to 32%. Latterly, when the temperature was increased to 45 °C, full conversion was achieved (entry 3). Following this, different aminocatalysts 3b–d were tried (entries 4–6). Interestingly, the bulkiest catalyst 3b or the hydrogen bond type catalysts 3c and 3d did not provide any conversion to the product 5a. Different solvents under 3a catalysis were then examined (entries 7–10). Chlorinated solvents and THF gave only modest results, but very apolar solvents such as toluene and p-xylene provided full conversions and very high enantioselectivities (94 and 96% ee). The catalytic loading was decreased to 10 mol%, achieving the product 5a with 96% ee, full conversion, and 82% isolated yield (entry 11). However, when the catalyst loading was 5 mol%, only 10% conversion was found (entry 12). Once the best conditions had been determined, we carried out the scope of the reaction using different aldehydes 1 and BODIPYs 2 (Table 2).|
|
|||||
|---|---|---|---|---|---|
| Entry | Cat. [mol%] | Solvent | Temp (°C) | Convb (%) | eec (%) |
| a 0.05 mmol of 2a, 0.13 mmol of 1a in 0.5 mL of the indicated solvent and the same amount of PhCO2H as catalyst loading. b Conversion and Z/E ratio determined by 1H NMR analysis of the crude mixture. c Determined by SFC. d Without PhCO2H. e Complex mixture. f Not determined. g Isolated yield after FC in brackets. | |||||
| 1d | 3a (20) | CHCl3 | rt | 9 | — |
| 2 | 3a (20) | CHCl3 | rt | 32 | 92 |
| 3 | 3a (20) | CHCl3 | 45 | 100 | 84 |
| 4 | 3b (20) | CHCl3 | 45 | n.r. | — |
| 5 | 3c (20) | CHCl3 | 45 | n.r. | — |
| 6 | 3d (20) | CHCl3 | 45 | n.r. | — |
| 7 | 3a (20) | CH2Cl2 | 45 | 15 | n.d.f |
| 8 | 3a (20) | THF | 45 | c.m.e | — |
| 9 | 3a (20) | Toluene | 45 | 100 | 94 |
| 10 | 3a (20) | p-Xylene | 45 | 100 | 96 |
| 11 | 3a (10) | p-Xylene | 45 | 100 (82) | 96 |
| 12 | 3a (5) | p-Xylene | 45 | 10 | n.d.f |
| a Conditions: 0.1 mmol of 2, 0.25 mmol of 1, 10 mol% of 3a and 10 mol% of PhCO2H in 1.0 mL of p-xylene. Enantiomeric excess determined by SFC. |
|---|
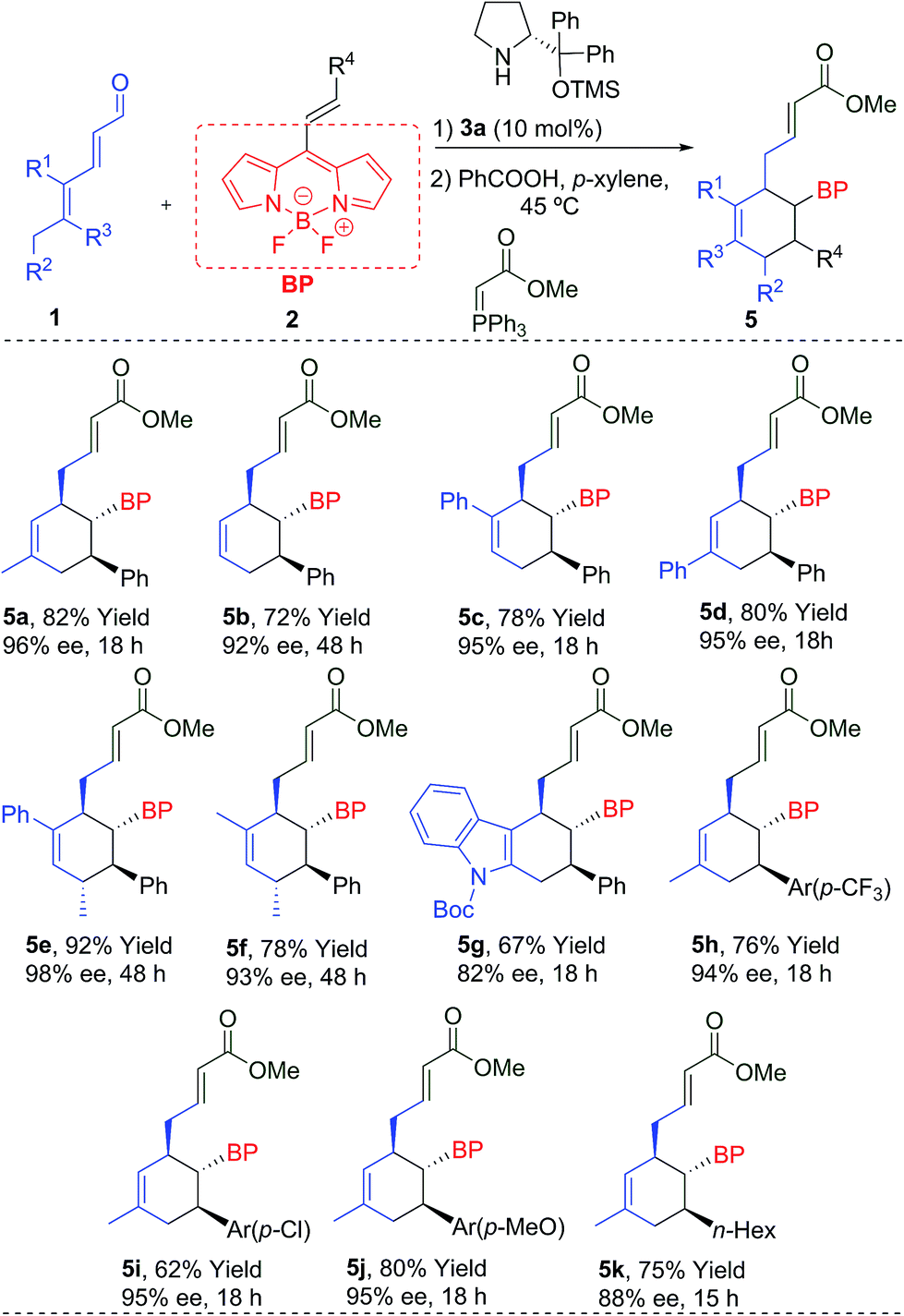
|
The reaction worked even when all substituents were hydrogen (R1 = R2 = R3 = H), giving 5b with a 92% ee and a 72% yield. The cyclohexanes 5c and 5d, with different substitutions at R1 or R3 (Ph), were also obtained with excellent yields and enantioselectivities after 18 h. An additional stereogenic centre can be also obtained using 1e (R1 = Ph, R2 = R3 = Me) and 1f (R1 = R2 = R3 = Me), that allows access to the products 5e and 5f with complete stereocontrol at the four stereogenic centres. An interesting indole derivative 5g was obtained with a very good ee and a good yield. The use of different EDGs (p-MeO) and EWGs (p-CF3 or p-Cl) at the aromatic ring of the double bond, gave the final products 5h–j with good results. Aliphatic derivatives (5k) were also tolerated. We also measured the absorption and emission spectra of these new BODIPYs (5a–k), which are comparable with other previously related derivatives,1b,7 described in the literature (top-right, Fig. 1b).
 | ||
| Fig. 1 (a) Energy (in eV) of the frontier molecular orbitals calculated for trienamine A, and the BODIPY 2a for the endo and exo approaches. (b) Absorption and emission (dash-line) spectra of BODIPYs 5a–k (see ESI for details†). (c) Gibbs free energy profile of the endo-[4 + 2] cycloaddition of the trienamine formed from 1b and catalyst 3a to the double bond 2a. The reactive part is highlighted in orange and the shadow wraps the catalyst. Energies in kcal mol−1. Geometry optimization was carried out at the M06-2X/6-31G(d,p) level of theory and single point energies including solvent at the SMD(p-xylene)/M06-2X/6-31+G(d,p) level of theory. | ||
The absolute configuration was determined by derivatization of the intermediate 4a, yielding the olefin 8 with concomitant bromination of the cyclohexene double bond (Scheme 2). Therefore, we assigned the configuration of compounds 5 as 1′S, 2′S, 3′R using X-ray analysis.17
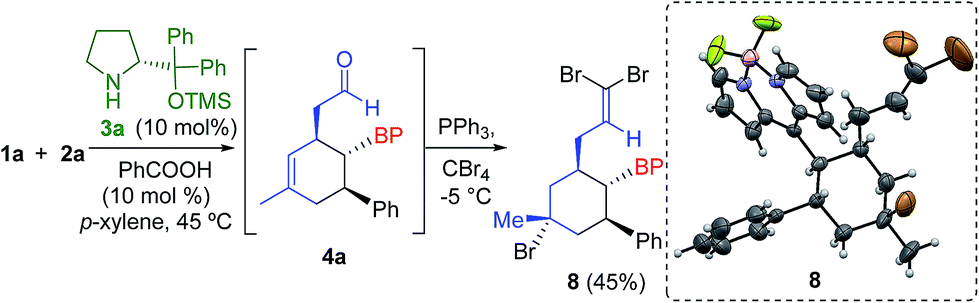 | ||
| Scheme 2 Derivatization of compound 4a and X-ray analysis of compound 8. | ||
In order to shed light onto the reactivity, we performed a Frontier Molecular Orbital (FMO) analysis, using the density functional theory (DFT),18 frequently employed to explain the reactivity in pericyclic reactions19 as in the one presented here (for more details see ESI†). The orbitals of the reagents in the ground state are used to predict the way the reaction proceeds (both orientation and reaction rate). The addition of trienamine A (1b) to the double bond of BODIPY 2a is governed mainly by the overlap between the Highest Occupied Molecular Orbital of the nucleophile (HOMOtrienamine) and the Lowest Unoccupied Molecular Orbital of the electrophile (LUMOdienophile), leading to the correct orientation. In addition, the HOMO–LUMO energy gap is related to the reaction rate (k), which is enhanced when the gap decreases.20 Therefore, if we compare the reaction of two different electrophiles with the same nucleophile, the lowest HOMO–LUMO gap will explain the highest catalytic efficiency. The energy difference between the frontier orbitals in the reaction of A with 2a, the energy gap (ΔE = LUMO−HOMO) was higher, and too large, for the exo- than for the endo-approach (3.40 and 3.19 eV respectively, top-left, Fig. 1a).21 Therefore, we only considered the reaction energy profile for the endo-approach (bottom, Fig. 1c).
We found that the reaction takes place in a stepwise fashion (bottom, Fig. 1c) as reported in previous examples in the literature.15 Once the pre-association complex (PAC) is formed,22 the first C–C bond between the terminal carbon of the trienamine A and the β-carbon of 2a is formed with a barrier of 9.5 kcal mol−1 (TS_1), which is the stereoselective limiting step barrier. Then, after a series of rotations with negligible energy cost (from the intermediate Int-1 to Int-1′) it forms the second C–C bond with a barrier of 7.9 kcal mol−1 (TS_2) to yield the final adduct which is easily cleaved, via hydrolysis, releasing the catalyst 3a and the desired product 4a.
Finally, to study the relative reactivity of BODIPY 2a, we compared its reaction with other known dienophiles in trienamine chemistry such as the nitrostyrene.14d The reaction of dienal 1a with nitroalkene 9 yielded 10 with a 26% conversion after 24 h under the same reaction conditions (top, Scheme 3). We then carried out a competitive reaction between 2a and 9, and found that only the BODIPY derivative reacted, without any traces of product 10, thus highlighting the higher reactivity of 2a. The origin of this notable difference in the reactivity was analyzed with the frontier orbitals of trienamine A, and dienophiles 2a and 9 (bottom, Scheme 3). The HOMO orbital of trienamine A is delocalized over the two central double bonds, between the nitrogen and the terminal nucleophilic carbon atom that will attack the β position of the BODIPY double bond. The LUMO orbital of 2a is delocalized over the BODIPY with an important contribution at the β position of the double bond and without any contribution at the α position. This explains the regioselectivity, as the β carbon of 2a is the first to react. However, in the case of nitroalkene 9, the LUMO orbital is fully delocalized through the molecule with contributions from α and β carbon atoms. In addition, the HOMO–LUMO gap is much lower for 2a (3.19 eV) than for 9 (3.58 eV). This means that the BODIPY is a better EWG than the NO2 for this reaction and explains the higher reactivity of the BODIPY derivatives 2 when compared with nitrostyrene 9.
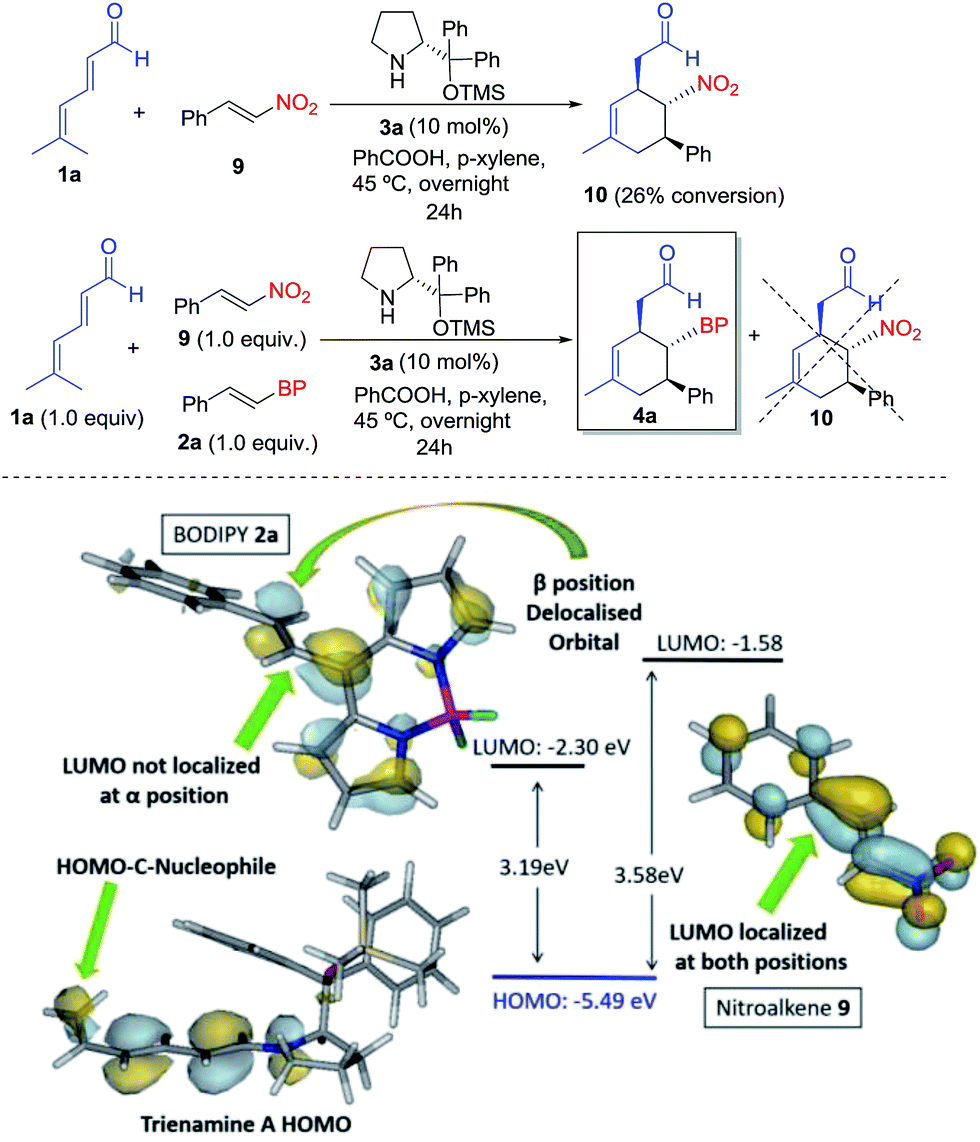 | ||
| Scheme 3 Top: reaction and reactivity comparison of BODIPY 2a with nitroalkene 9. Bottom: orbital analysis of 2a, 9 and trienamine A. | ||
Conclusions
In conclusion, we have shown that the BODIPY can be used as an electron withdrawing group for the activation of double bonds in asymmetric catalysis. Indeed, the BODIPY acts as a stronger EWG than the nitro group. In this work, we have applied this characteristic for the synthesis of asymmetric cyclohexyl derivatives via trienamine catalysis, that contain a BODIPY unit in their structure, allowing a new functionalization of these fluorophores. In addition, we have been able to explain the observed reactivity with Quantum Chemistry calculations, confirming the role of the BODIPY as an EWG in the double bond. The new reactivity here presented can be used in the future for further asymmetric transformations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Spanish Government (CTQ2015-64561-R, CTQ2016-76061-P), CONACYT (project supported by the Fondo Sectorial de Investigación para la Educación) and PRODEP (Mexico) are acknowledged. We acknowledge allocation of computing time at the CCC-UAM. A. G. C. thanks MINECO (FPI) and T. J. P. CONACYT for PhD fellowships, respectively. A. M. S. thanks CAM for a postdoctoral contract (2016-T2/IND-1660). The authors wish to thank ''Comunidad de Madrid'' for its support to the FotoArt-CM Project (S2018/NMT-4367) through the Program of R&D activities between research groups in Technologies 2013, co-financed by European Structural Funds.Notes and references
-
(a) N. Boens, B. Verbelen and W. Dehaen, Eur. J. Org. Chem., 2015, 6577 CrossRef CAS
; (b) G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184 CrossRef CAS PubMed
-
(a) N. Boens, V. Leen and W. Dehaen, Chem. Soc. Rev., 2012, 41, 1130 RSC
-
(a) S. Kolemen and E. U. Akkaya, Coord. Chem. Rev., 2018, 354, 121 CrossRef CAS
-
(a) K. Chansaenpak, B. Vabre and F. P. Gabbaï, Chem. Soc. Rev., 2016, 45, 954 RSC
-
(a) A. Turksoy, D. Yildiz and E. U. Akkaya, Coord. Chem. Rev., 2019, 379, 47 CrossRef CAS
-
(a) X. Guo, X. Li, X.-C. Liu, P. Li, Z. Yao, J. Li, W. Zhang, J.-P. Zhang, D. Xue and R. Cao, Chem. Commun., 2018, 54, 845 RSC
-
(a) E. M. Sánchez-Carnerero, A. R. Agarrabeitia, F. Moreno, B. L. Maroto, G. Muller, M. J. Ortiz and S. de la Moya, Chem.–Eur. J., 2015, 21, 13488 CrossRef PubMed
-
(a) L. J. Patalag, J. A. Ulrichs, P. G. Jones and D. B. Werz, Org. Lett., 2017, 19, 2090 CrossRef CAS PubMed
-
(a) C. S. Gutsche, B. F. Hohlfeld, K. J. Flanagan, M. O. Senge, N. Kulak and A. Wiehe, Eur. J. Org. Chem., 2017, 3187 CrossRef CAS
-
(a) X. Zhou, C. Yu, Z. Feng, Y. Yu, J. Wang, E. Hao, Y. Wei, X. Mu and L. Jiao, Org. Lett., 2015, 17, 4632 CrossRef CAS PubMed
- For cross-coupling at C-8, see e.g.:
(a) V. Leen, P. Yuan, L. Wang, N. Boens and W. Dehaen, Org. Lett., 2012, 14, 6150 CrossRef CAS PubMed
-
(a) E. Palao, A. R. Agarrabeitia, J. Bañuelos-Prieto, T. Arbeloa López, I. López-Arbeloa, D. Armesto and M. J. Ortiz, Org. Lett., 2013, 15, 4454 CrossRef CAS PubMed
-
(a) H. B. Hepburn, L. Dell'Amico and P. Melchiorre, Chem. Rev., 2016, 16, 1787 CAS
-
(a) K. S. Halskov, T. K. Johansen, R. L. Davis, M. Steurer, F. Jensen and K. A. Jørgensen, J. Am. Chem. Soc., 2012, 134, 12943 CrossRef CAS PubMed
- A. Dieckmann, M. Breugst and K. N. Houk, J. Am. Chem. Soc., 2013, 135, 3237 CrossRef CAS PubMed
- Y. Liu, X. Lv, M. Hou, Y. Shi and W. Guo, Anal. Chem., 2015, 87, 11475 CrossRef CAS PubMed
- CCDC 1880124 contains crystallographic data of compound 8.†.
- Geometry optimization were performed with the M06-2X functional and 6-31G(d,p) basis set. Harmonic frequencies and thermodynamic corrections were computed at the same level. More accurate values of the final energy were computed over the geometries previously obtained with the same functional and a larger basis set 6-31+G(d,p), also including solvent effects (p-xylene) with the SMD solvation model. All simulations were carried out with the Gaussian09 program. ESI† for more details.
-
I. Fleming, Frontier Orbitals and Organic Chemical Reactions, Wiley, London, 1978 Search PubMed
- J. D. Bradley and G. C. Gerrans, J. Chem. Educ., 1978, 55, 437 CrossRef
- The orbital energies are taken from separated reagents (infinite distance). However, we have further verified that in the pre-association complex (with the two reagents interacting before the first C–C bond formation), the energy gap barely changes (for endo is 3.19 versus 3.20 eV and for exo 3.40 vs. 3.39 eV).
- For a study about the importance of preassociated complexes in an aminocatalytic case, see:
(a) E. M. Arpa, M. Frías, C. Alvarado, J. Alemán and S. J. Díaz-Tendero, J. Mol. Catal. A: Chem., 2016, 423, 308 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1880124. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc00959k |
| This journal is © The Royal Society of Chemistry 2019 |