
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA molecular design strategy toward enzyme-activated probes with near-infrared I and II fluorescence for targeted cancer imaging†
Rongchen
Wang‡
a,
Jian
Chen‡
a,
Jie
Gao‡
b,
Ji-An
Chen‡
b,
Ge
Xu
a,
Tianli
Zhu
a,
Xianfeng
Gu
 b,
Zhiqian
Guo
a,
Wei-Hong
Zhu
a and
Chunchang
Zhao
*a
b,
Zhiqian
Guo
a,
Wei-Hong
Zhu
a and
Chunchang
Zhao
*a
aKey Laboratory for Advanced Materials and Feringa Nobel Prize Scientist Joint Research Center, Institute of Fine Chemicals, School of Chemistry and Molecular Engineering, East China University of Science and Technology, Shanghai, 200237, P. R. China. E-mail: zhaocchang@ecust.edu.cn
bDepartment of Medicinal Chemistry, School of Pharmacy, Fudan University, Shanghai, 201203, P. R. China
First published on 14th June 2019
Abstract
The advance of cancer imaging requires innovations to establish novel fluorescent scaffolds that are excitable and emit in the near-infrared region with favorable Stokes shifts. Nevertheless, the lack of probes with these optimized optical properties presents a major bottleneck in targeted cancer imaging. By coupling of boron dipyrromethene platforms to enzymic substrates via a self-immolative benzyl thioether linker, we here report a strategy toward enzyme-activated fluorescent probes to satisfy these requirements. This strategy is applicable to generate various BODIPY-based probes across the NIR spectrum via introducing diverse electron-withdrawing substituents at the 3-position of the BODIPY core through a vinylene unit. As expected, such designed probes show advantages of two-channel ratiometric fluorescence and light-up NIR (I and II) emission with large Stokes shifts upon enzyme activation, enabling targeted cancer cell imaging and accurate tumor location by real-time monitoring of enzyme activities. This strategy is promising in engineering activatable molecular probes suitable for precision medicine.
Introduction
Scientists are in enthusiastic pursuit of novel assay techniques for targeted cancer imaging that could provide valuable insight into cancer diagnosis, treatment, and management.1 Among the numerous analytical techniques, fluorescence imaging, due to its operational simplicity, high spatiotemporal resolution, and noninvasive capability, provides a powerful and popular tool for identification and enumeration of living cancers.2 In this context, the past decades have witnessed the rapid development of versatile molecular fluorescent probes for detection of cancer-related biomarkers.3 Particularly, activatable molecular probes that generate fluorescence signals only in response to enzyme biomarkers of cancers have been reported for successful cancer imaging.4 However, most of the conventional fluorescent probes with emission in the visible light region encounter several challenges regarding strong autofluorescence and the scattering of short-wavelength light in living tissues that significantly reduce the sensitivity of fluorescence imaging in vivo.5An attractive approach for the detection of cancer biomarkers in vitro and in vivo is optical imaging in the near-infrared (NIR) range, offering a number of advantages, including less autofluorescence background, lower tissue absorption, better tissue penetration, and higher in vivo spatial resolution compared to visible imaging.6 Nevertheless, construction of small-molecule NIR fluorescent probes remains a significant challenge. Currently, several NIR probes to detect enzyme activities have been built based on cyanine and rhodamine scaffolds.7 However, these known NIR probes suffer from undesirable small Stokes shifts, which generally lead to severe cross-talk between the excitation and emission spectra that significantly compromises sensitive detection and accurate analysis.8 Thus, the advance of cancer-biomarker imaging requires innovations to establish novel fluorescent scaffolds that are excitable and emit in the NIR region with large Stokes shifts.
Here, we report a molecular design strategy that establishes a universal platform to access diverse enzyme-activated NIR fluorescent probes with desired optical properties. These activatable probes were engineered by coupling of BODIPY platforms to enzymic substrates via a self-immolative benzyl thioether linker (Fig. 1). The use of the benzyl thioether linker as a self-immolative spacer instead of the conventional benzyl ether can be ascribed to three reasons: (1) the ease of synthetic accessibility via the aromatic nucleophilic substitution (SNAr) reaction by fully employing the nucleophilic feature of benzyl mercaptan derivatives; (2) as an excellent leaving group, the released aromatic thiol could facilitate the fragmentation of the self-immolative spacer after enzyme activation; and (3) increasing the electron-withdrawing power of the substituents attached to the BODIPY core generally leads to optically bathochromic shifts for thiol-substituted BODIPY dye,9 enabling creation of various dyes across the NIR spectrum by simply introducing diverse electron-withdrawing substituents at the 3-position of the thiol-substituted BODIPY core through a vinylene unit (Fig. 1). Specifically, thiol-substituted BODIPY has a characteristic thiol group for regulating the optical properties (Fig. 1). Thus, the fluorescence wavelength could be significantly changed through the thiol protection/deprotection approach. In this regard, by grafting a trigger onto the thiol moiety via a self-immolative linker, thiol-substituted BODIPYs can be modified to afford a panel of NIR fluorescent probes for various enzymes (Fig. 1). As expected, such designed probes showed the typical properties of BODIPY with absorption and emission in the visible region due to the diminished electron-donating ability of the sulfur atom. In the presence of enzymes, self-immolation was initiated to liberate thiol-substituted BODIPY with enhanced electron-donating ability of the sulfur atom, eventually leading to dyes that are excitable and emit in the NIR region with large Stokes shifts.
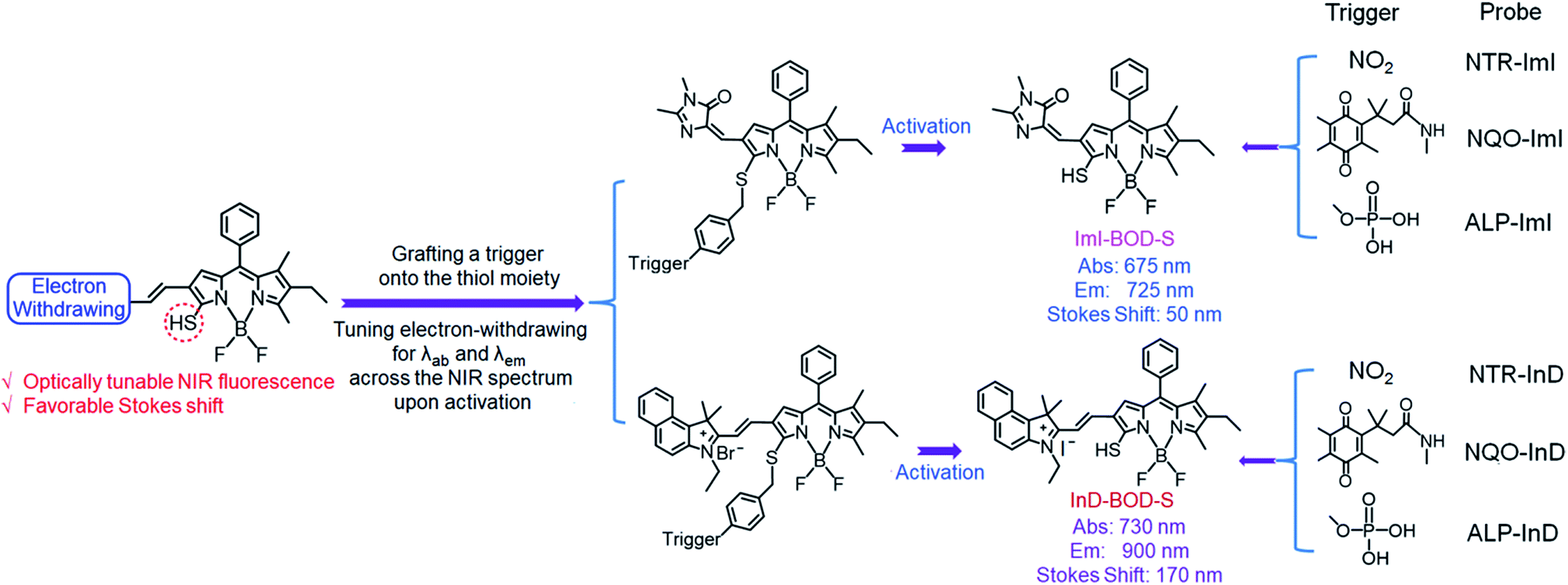 | ||
| Fig. 1 Schematic representation of the design strategy for diverse enzyme-activated NIR fluorescent probes and their chemical structures. | ||
Results and discussion
The self-immolative chemistry based strategy has mainly focused on the liberation of phenolic or amine-containing payloads,10 while relatively few reports have focused on analyte-mediated release of desired compounds containing thiol groups.11 To explore the potential of using the self-immolative pathway for fragmentation of the benzyl thioether linker to release thiol substituted BODIPYs, two nitroreductase (NTR) and NAD(P)H:quinone oxidoreductase isozyme 1 (NQO1) activated probes were first designed by coupling of BODIPY platforms to enzymic substrates (nitro group and trimethyl-locked quinone propionic amide) via a benzyl thioether linker. The detailed synthetic procedures are depicted in Schemes 1 and S1–S3.† High-resolution mass spectrometry (HRMS) experiments showed the successful conversion of probes NTR-ImI and NQO-ImI to ImI-BOD-S (Fig. S1†). These results suggested that NTR/NQO1 can trigger the formation of the p-aminobenzyl thioether linker that is capable of undergoing a 1,6-elimination reaction to release the desired fluorophore (Fig. 2). Similarly, the fragmentation of the p-hydroxybenzyl thioether linker could also lead to release of thiol substituted BODIPYs, as evidenced by the probe ALP-ImI wherein alkaline phosphatase (ALP) induced selective dephosphorylation and spontaneous 1,6-elimination.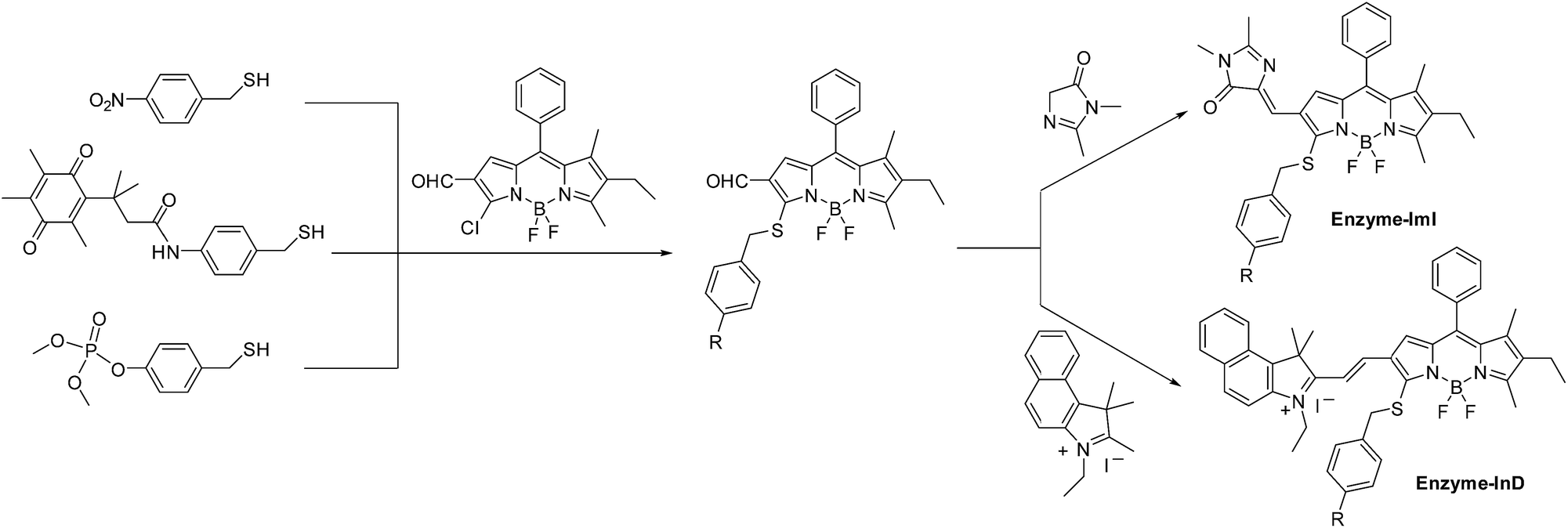 | ||
| Scheme 1 The synthetic procedures for enzyme-activated probes. | ||
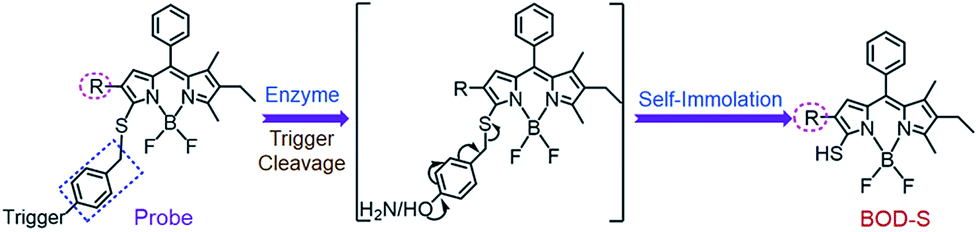 | ||
| Fig. 2 Self-immolative pathway for fragmentation of the benzyl thioether linker to release thiol substituted BODIPYs. | ||
We then assessed the spectroscopic properties of probes NQO-ImI, NTR-ImI and ALP-ImI in the absence and presence of NQO1, NTR and ALP in aqueous solutions. For example, free NQO-ImI showed the typical features of BODIPY with absorption and emission at 557 nm and 635 nm (ϕ = 0.017, Table S1†). Upon treatment with NQO1 (25 μg mL−1) and the cofactor NADH (500 μM), a new absorption band around 675 nm emerged gradually (Fig. 3a), while the original absorption band of NQO-ImI at 557 nm was attenuated significantly, showing a remarkable red-shift of 118 nm. Interestingly, the fluorescence titration experiments demonstrated that NQO-ImI allowed the assay of NQO1 in both two-channel ratiometric and turn-on NIR fluorescence modes. Reaction of NQO-ImI and NQO1 elicited a time-dependent quenching of the fluorescence at 635 nm when the excitation wavelength was 557 nm (Fig. 3b). Fortunately, upon excitation at 650 nm or 675 nm, a new NIR emission at 725 nm (ϕ = 0.074, Table S1†) with the Stokes shift of 50 nm was activated concomitantly (Fig. 3c), exhibiting a nearly 105-fold fluorescence intensity enhancement. In this context, the fluorescence intensity ratio at 635 and 725 nm (I725/I635) showed a 316-fold enhancement upon enzyme-catalyzed complete conversion (Fig. S2†). Of note, negligible fluorescence enhancement was found upon the pretreatment of NQO1 with an inhibitor dicoumarol (Fig. S2†), indicative of the critical role of NQO1 in activation of NQO-ImI. Consistent with other reported NQO1 probes,4h NQO-ImI unambiguously exhibited high specificity for NQO1 activity (Fig. S3†). In buffer solution at pH 7.4, the kinetic values of NQO-ImI against NQO1 showed a Michaelis–Menten constant (Km) of 18.42 μM and maximum rate (Vmax) of 64.10 μM min−1 (Fig. S4†). Similar optical responsiveness was observed for probes NTR-ImI and ALP-ImI with the exception of activation by NTR and ALP, respectively (Fig. S5–S10†). It should be noted that the maximal excitation and emission changed slightly upon varying the testing conditions. In addition, these designed probes exhibited good optical responsiveness to enzymes within a physiological pH range (pH 8 to approximately 6; Fig. S11†).
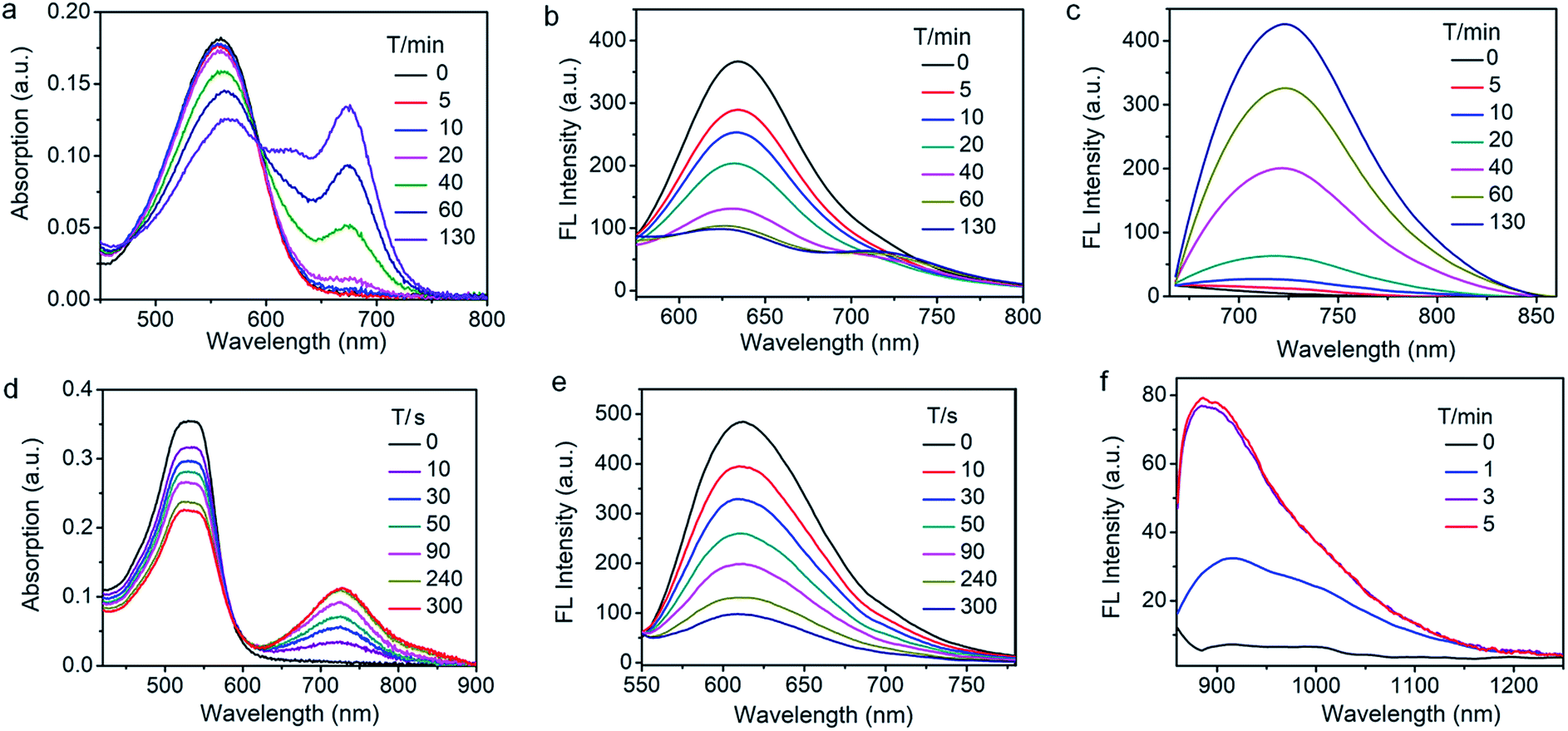 | ||
Fig. 3 Time-dependent spectral changes of NQO-ImI and NTR-InD in the presence of enzymes. (a) Absorption, (b) fluorescence quenching (λex = 557 nm) and (c) NIR-I fluorescence turn-on (λex = 650 nm) of NQO-ImI upon addition of NQO1 (25 μg mL−1) in buffer (DMSO/PBS, v/v, 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8, pH 7.4) at 37 °C. (d) Absorption, (e) fluorescence quenching (λex = 535 nm) and (f) NIR-II fluorescence turn-on (λex = 730 nm) of NTR-InD upon addition of NTR (20 μg mL−1) in buffer (DMSO/Tris–HCl, v/v, 2:8, pH 7.4) at 37 °C. :8, pH 7.4) at 37 °C. (d) Absorption, (e) fluorescence quenching (λex = 535 nm) and (f) NIR-II fluorescence turn-on (λex = 730 nm) of NTR-InD upon addition of NTR (20 μg mL−1) in buffer (DMSO/Tris–HCl, v/v, 2:8, pH 7.4) at 37 °C. | ||
To further red-shift the absorption and emission and enlarge the Stokes shift, a stronger electron-withdrawing unit 3-ethyl-1,1,2-trimethyl-1H-benz[e]indolium instead of 1,2-dimethyl-1H-imidazol-5(4H)-one was appended to the BODIPY core through a vinylene unit, and probes NTR-InD, NQO-InD and ALP-InD were thus obtained. As predicted, enzymes initiated self-immolation in these probes and eventually led to formation of InD-BOD-S that was excitable (730 nm) and emitted (900 nm) in the NIR region with a large Stokes shift of 170 nm (Fig. 3 and S12–S16†). Upon reaction with enzyme NTR (20 μg mL−1), generation of NIR absorption at 730 nm was noted with a concomitant decrease of the band at 535 nm (Fig. 3d). Such treatment with enzymes made the emission at 612 nm (excitation at 535 nm) completely quenched within 5 min (Fig. 3e). Gratifyingly, a robust increase in the fluorescence intensity at 900 nm was observed when the excitation wavelength was 730 nm, producing a 12-fold increase (Fig. 3f). Importantly, these probes could exhibit enzyme-activated emission with a fluorescence tail in the region (1000–1300 nm) of NIR-II, which is preferable for in vivo cancer imaging as NIR-II imaging enables high-resolution bioimaging with deep-tissue penetration.12 In addition, these probes and the NIR products ImI-BOD-S and InD-BOD-S showed good stability under testing conditions (Fig. S17 and S18†), indicative of the suitability for in vivo imaging. Collectively, all these optical advantages of such designed probes, including light-up NIR-II emissions with a large stokes shift of 170 nm, make them a superior platform for in vivo cancer imaging.
After demonstrating the low cytotoxicity of our designed probes toward living cells (Fig. S19†), we then testified their capability for selectively identifying cancer cells by real-time tracking of enzyme activities. For evaluation of NTR activity, A549 cells were exposed to the probe NTR-ImI because such cells are known to overexpress NTR under hypoxic conditions.13 The treatment of A549 cells with NTR-ImI under normoxic conditions (20% O2) led to bright fluorescence signals in the green channel and relatively moderate fluorescence signals in the red channel (Fig. 4). The fluorescence ratio of the red channel to green channel was found to be 0.8. In contrast, this fluorescence ratio (Ired/Igreen) showed a time-dependent enhancement when A549 cells were incubated with NTR-ImI under hypoxia (1% O2), for example, Ired/Igreen was markedly increased to 1.6 when the incubation time was 1 h, suggesting significant activation of NTR-ImI by NTR to produce ImI-BOD-S in hypoxic cancer cells. These imaging experiments demonstrated that NTR-ImI is capable of monitoring NTR activity in dual-color imaging modality.
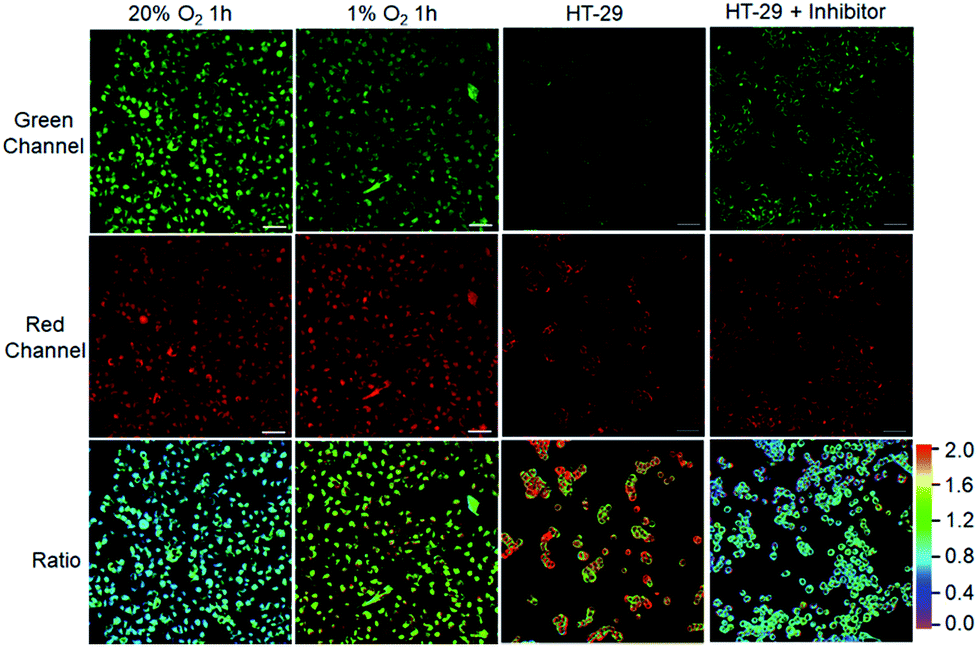 | ||
| Fig. 4 Visualization of cancer cells by confocal microscopy imaging. A549 cells cultured with NTR-ImI (10 μM) under various oxygen concentration conditions. HT-29 cells or HT-29 cells pretreated with dicoumarol (0.5 mM) for 2 h were incubated with NQO-ImI (20 μM) for 2 h. Green channel at 600–670 nm with λex = 514 nm excitation, red channel at 700–755 nm using λex = 633 nm; ratio image was Fred/Fgreen. Scale bar = 50 μm. | ||
To explore the feasibility of visualization of cancer cells based on NQO1 activity, imaging experiments with HT-29 cells were performed. As shown in Fig. 4, HT-29 cells (high NQO1 activity)13 exhibited relatively faint fluorescence in the green channel and strong fluorescence in the red channel, resulting in a significant red-to-green signal ratio of 1.8. In contrast, addition of dicoumarol, an enzyme inhibitor, led to bright fluorescence in green and moderate emission in red channels. Such treatment with dicoumarol induced the red-to-green signal ratio to be significantly lowered, indicative of the fluorescence imaging signal indeed initiated by NQO1 activity. All these results implied that the designed probes can serve as a promising tool for targeted cancer cell imaging by real-time monitoring of enzyme activities in living cells.
The ability of the probes for in vivo targeted cancer visualization was subsequently evaluated. Fig. 5 illustrates the NIR imaging results of A549 subcutaneous xenograft nude mice treated with NTR-ImI and NTR-InD via intratumoral injection, respectively. Immediately after in situ probe administration, remarkable NIR fluorescence was collected specifically in the tumor region (Fig. 5a and b), while barely detectable fluorescence lighted up in the normal injection site, indicative of the rapid and specific activation of probes by tumor-derived enzymes. The fact that dicoumarol, an inhibitor of NTR, effectively attenuated the fluorescence signals in the tumor also demonstrated the vital role of NTR in tumors for selective activation of probes for targeted cancer imaging. The fluorescence signals in the tumor site gradually increased overtime and eventually reached a plateau after 60 min post-injection. Interestingly, the fluorescence intensity of the tumor over the normal site (T/N) in NTR-InD treated mice was much higher than that of NTR-ImI treated ones at each post-injection time point (Fig. 5c), presumably due to greatly reduced autofluorescence and photon scattering in the NIR-II region.12 It was clear that the T/N of NIR-II from NTR-InD injected mice was 10.6 (60 min), while that of NIR-I in NTR-ImI treated ones was only 1.5 (60 min). Furthermore, the advantage of NIR-II imaging with intrinsically deeper penetration over NIR-I imaging was also tested in a simulated deep-tissue setting (Fig. 5d and S20†). The results showed that the bright NIR-II signal from NTR-InD could be used to sensitively visualize A549 tumors even at a depth of 7 mm. In contrast, the tumor NIR-I fluorescence signal from NTR-ImI was undetectable above 3 mm. These results suggest that NIR-II imaging is reliable to provide precise location of tumors within deep tissues, which is difficult to achieve by visible and traditional NIR-I fluorescence imaging. It should be noted that cancers can also be rapidly visualized and differentiated by real-time and in vivo tracking of NQO1 activity with probes NQO-ImI and NQO-InD as a proof-of-concept (Fig. S21†). Taken together, these cancer biomarker-activated molecular probes could provide a powerful and popular tool for accurate identification of living cancers.
 | ||
| Fig. 5 Targeted cancer visualization in the A549 tumor-bearing mouse model. (a) Time-dependent NIR-I imaging of mice injected with NTR-ImI (30 nmol) or NTR-ImI + dicoumarol (0.3 mmol). (b) Time-dependent NIR-II imaging of mice injected with NTR-InD (30 nmol) or NTR-InD + dicoumarol (0.3 mmol). (c) The fluorescence intensity of the tumor over the normal injection site in (a) and (b) via region of interest analysis. (d) Validation of the superiority of NIR-II over NIR-I imaging using NTR-InD and NTR-ImI in a simulated deep-tissue setting. | ||
Live subject statement
Animal experiments were carried out in compliance with Chinese legislation on the Use and Care of Research Animals, and guidelines established by Fudan University Animal Studies Committee for the Care and Use of laboratory animals. All experimental procedures were approved by this committee.Conclusions
In summary, we have established a universal platform to access diverse enzyme-activated NIR fluorescent probes for targeted cancer imaging. These activatable probes were constructed by coupling of BODIPY platforms to enzymic substrates via a self-immolative benzyl thioether linker. As proof-of-concept examples, enzymes (NTR, NQO1, and ALP) initiated the formation of the p-amino/hydroxybenzyl thioether linker and spontaneous 1,6-elimination reactions to release thiol-substituted BODIPYs that can be facilely derivatized via tuning the electron-withdrawing power of the substituents appended at the 3-position of the BODIPY core. In this way, the design strategy allows access to novel fluorescent scaffolds that are excitable and emit in the NIR (I and II) region with favorable Stokes shifts. Interestingly, such designed probes were capable of targeted cancer cell imaging and differentiation by real-time monitoring of enzyme activities. More importantly, in vivo targeted cancer imaging implied that these biomarker-activated molecular probes could provide a powerful tool for accurate identification. In light of the promising features, further study toward the optimization of the molecular structure to endow probes with good water solubility by appending hydrophilic units or utilizing the supramolecular assembly approach is currently underway. To our knowledge, enzyme-activated probes with the aforementioned favorable responsiveness are highly limited. It is anticipated that our design approach can be generalizable for customization of a wide range of activatable probes with optimized optical properties, thus facilitating targeted cancer diagnostics.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We gratefully acknowledge the financial support by the National Natural Science Foundation of China (21874043 and 21672062), the Shanghai Municipal Science and Technology Major Project (Grant No. 2018SHZDZX03) and the Programme of Introducing Talents of Discipline to Universities (B16017).Notes and references
- (a) A. Razgulin, N. Ma and J. Rao, Chem. Soc. Rev., 2011, 40, 4186 RSC; (b) R. R. Zhang, A. B. Schroeder, J. J. Grudzinski, E. L. Rosenthal, J. M. Warram, A. N. Pinchuk, K. W. Eliceiri, J. S. Kuo and J. P. Weichert, Nat. Rev. Clin. Oncol., 2017, 14, 347 CrossRef CAS PubMed; (c) M. Fernández, F. Javaid and V. Chudasama, Chem. Sci., 2018, 9, 790 RSC.
- (a) X. H. Li, X. H. Gao, W. Shi and H. M. Ma, Chem. Rev., 2014, 114, 590 CrossRef CAS PubMed; (b) Y. M. Yang, Q. Zhao, W. Feng and F. Y. Li, Chem. Rev., 2013, 113, 192 CrossRef CAS PubMed; (c) H. Zhu, J. L. Fan, J. J. Du and X. J. Peng, Acc. Chem. Res., 2016, 49, 2115 CrossRef CAS PubMed; (d) L.-Y. Niu, Y.-Z. Chen, H.-R. Zheng, L.-Z. Wu, C.-H. Tung and Q.-Z. Yang, Chem. Soc. Rev., 2015, 44, 6143 RSC; (e) H. S. Jung, X. Chen, J. S. Kim and J. Yoon, Chem. Soc. Rev., 2013, 42, 6019 RSC; (f) D. Wu, A. C. Sedgwick, T. Gunnlaugsson, E. U. Akkaya, J. Yoon and T. D. James, Chem. Soc. Rev., 2017, 46, 7105 RSC.
- (a) F. Wang, G. Xu, X. Gu, Z. Wang, Z. Wang, B. Shi, C. Lu, X. Gong and C. Zhao, Biomaterials, 2018, 159, 82 CrossRef CAS PubMed; (b) R. T. K. Kwok, C. W. T. Leung, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2015, 44, 4228 RSC; (c) D. Ye, A. J. Shuhendler, L. Cui, L. Tong, S. S. Tee, G. Tikhomirov, D. W. Felsher and J. Rao, Nat. Chem., 2014, 6, 519 CrossRef CAS PubMed; (d) H. Zhang, J. Fan, J. Wang, S. Zhang, B. Dou and X. Peng, J. Am. Chem. Soc., 2013, 135, 11663 CrossRef CAS PubMed; (e) Z. Luo, R. An and D. Ye, ChemBioChem, 2018, 20, 474 CrossRef PubMed; (f) Y. Song, T. Tian, Y. Shi, W. Liu, Y. Zou, T. Khajvand, S. Wang, Z. Zhu and C. Yang, Chem. Sci., 2017, 8, 1736 RSC; (g) H.-B. Cheng, Y.-M. Zhang, Y. Liu and J. Yoon, Chem, 2019, 5, 553 CrossRef CAS; (h) J. Zhang, X. Chai, X.-P. He, H.-J. Kim, J. Yoon and H. Tian, Chem. Soc. Rev., 2019, 48, 683 RSC.
- (a) H.-W. Liu, L. Chen, C. Xu, Z. Li, H. Zhang, X.-B. Zhang and W. Tan, Chem. Soc. Rev., 2018, 47, 7140 RSC; (b) X. Wu, W. Shi, X. Li and H. Ma, Angew. Chem., Int. Ed., 2017, 56, 15319 CrossRef CAS PubMed; (c) H. Shi, R. T. K. Kwok, J. Liu, B. Xing, B. Z. Tang and B. Liu, J. Am. Chem. Soc., 2012, 134, 17972 CrossRef CAS PubMed; (d) F. Wang, Y. Zhu, L. Zhou, L. Pan, Z. Cui, Q. Fei, S. Luo, D. Pan, Q. Huang, R. Wang, C. Zhao, H. Tian and C. Fan, Angew. Chem., Int. Ed., 2015, 54, 7349 CrossRef CAS PubMed; (e) Y. Liu, J. Tan, Y. Zhang, J. Zhuang, M. Ge, B. Sh, J. Li, G. Xu, S. Xu, C. Fan and C. Zhao, Biomaterials, 2018, 173, 1 CrossRef CAS PubMed; (f) Y. Urano, M. Sakabe, N. Kosaka, M. Ogawa, M. Mitsunaga, D. Asanuma, M. Kamiya, M. R. Young, T. Nagano and P. L. Choyke, Sci. Transl. Med., 2011, 3, 110ra119 Search PubMed; (g) Y. Li, Y. Sun, J. Li, Q. Su, W. Yuan, Y. Dai, C. Han, Q. Wang, W. Feng and F. Li, J. Am. Chem. Soc., 2015, 137, 6407 CrossRef CAS PubMed; (h) S. U. Hettiarachchi, B. Prasai and R. L. McCarley, J. Am. Chem. Soc., 2014, 136, 7575 CrossRef CAS PubMed; (i) S. Luo, Y. Liu, F. Wang, Q. Fei, B. Shi, J. An, C. Zhao and C.-H. Tung, Analyst, 2016, 141, 2879 RSC; (j) H.-W. Liu, K. Li, X.-X. Hu, L. Zhu, Q. Rong, Y. Liu, X.-B. Zhang, J. Hasserodt, F.-L. Qu and W. Tan, Angew. Chem., Int. Ed., 2017, 56, 11788 CrossRef CAS PubMed; (k) A. Chevalier, Y. Zhang, O. M. Khdour, J. B. Kaye and S. M. Hecht, J. Am. Chem. Soc., 2016, 138, 12009 CrossRef CAS PubMed; (l) K. Gu, W. Qiu, Z. Guo, C. Yan, S. Zhu, D. Yao, P. Shi, H. Tian and W.-H. Zhu, Chem. Sci., 2019, 10, 398 RSC; (m) B. Shi, X. Gu, Q. Fei and C. Zhao, Chem. Sci., 2017, 8, 2150 RSC; (n) X. Li, C.-Y. Kim, S. Lee, D. Lee, H.-M. Chung, G. Kim, S.-H. Heo, C. Kim, K.-S. Hong and J. Yoon, J. Am. Chem. Soc., 2017, 139, 10880 CrossRef CAS PubMed.
- J. V. Frangioni, Curr. Opin. Chem. Biol., 2003, 5, 626 CrossRef.
- (a) Kenry, Y. Duan and B. Liu, Adv. Mater., 2018, 1802394 CrossRef PubMed; (b) L. Yuan, W. Lin, K. Zheng, L. He and W. Huang, Chem. Soc. Rev., 2013, 42, 622 RSC.
- (a) Q. Miao, D. C. Yeo, C. Wiraja, J. Zhang, X. Ning, C. Xu and K. Pu, Angew. Chem., Int. Ed., 2018, 57, 1256 CrossRef CAS PubMed; (b) K. Gu, Y. Xu, H. Li, Z. Guo, S. Zhu, S. Zhu, P. Shi, T. D. James, H. Tian and W.-H. Zhu, J. Am. Chem. Soc., 2016, 138, 5334 CrossRef CAS PubMed; (c) M. Sakabe, D. Asanuma, M. Kamiya, R. J. Iwatate, K. Hanaoka, T. Terai, T. Nagano and Y. Urano, J. Am. Chem. Soc., 2013, 135, 409 CrossRef CAS PubMed; (d) Q. A. Best, B. Prasai, A. Rouillere, A. E. Johnson and R. L. McCarley, Chem. Commun., 2017, 53, 783 RSC; (e) J. Ning, T. Liu, P. Dong, W. Wang, G. Ge, B. Wang, Z. Yu, L. Shi, X. Tian, X. Huo, L. Feng, C. Wang, C. Sun, J. Cui, T. D. James and X. Ma, J. Am. Chem. Soc., 2019, 141, 1126 CrossRef CAS PubMed; (f) H. Li, Y. Li, Q. Yao, J. Fan, W. Sun, S. Long, K. Shao, J. Du, J. Wang and X. Peng, Chem. Sci., 2019, 10, 1619 RSC.
- (a) T.-B. Ren, W. Xu, W. Zhang, X.-X. Zhang, Z.-Y. Wang, Z. Xiang, L. Yuan and X.-B. Zhang, J. Am. Chem. Soc., 2018, 140, 7716 CrossRef CAS PubMed; (b) Q. Fei, X. Gu, Y. Liu, B. Shi, H. Liu, G. Xu, C. Li, P. Shi and C. Zhao, Org. Biomol. Chem., 2017, 15, 4072 RSC.
- (a) C. Zhao, X. Zhang, K. Li, S. Zhu, Z. Guo, L. Zhang, F. Wang, Q. Fei, S. Luo, P. Shi, H. Tian and W.-H. Zhu, J. Am. Chem. Soc., 2015, 137, 8490 CrossRef CAS PubMed; (b) L.-Y. Niu, Y.-S. Guan, Y.-Z. Chen, L.-Z. Wu, C.-H. Tung and Q.-Z. Yang, J. Am. Chem. Soc., 2012, 134, 18928 CrossRef CAS PubMed.
- (a) H. Chen, X. He, M. Su, W. Zhai, H. Zhang and C. Li, J. Am. Chem. Soc., 2017, 139, 10157 CrossRef CAS PubMed; (b) J. Yan, S. Lee, A. Zhang and J. Yoon, Chem. Soc. Rev., 2018, 47, 6900 RSC; (c) A. Alouane, R. Labruere, T. Le Saux, F. Schmidt and L. Jullien, Angew. Chem., Int. Ed., 2015, 54, 7492 CrossRef CAS PubMed; (d) P. Cheng, J. Zhang, J. Huang, Q. Miao, C. Xu and K. Pu, Chem. Sci., 2018, 9, 6340 RSC; (e) Z. Luo, Z. Huang, K. Li, Y. Sun, J. Lin, D. Ye and H.-Y. Chen, Anal. Chem., 2018, 90, 2875 CrossRef CAS PubMed; (f) Z. Luo, L. Feng, R. An, G. Duan, R. Yan, H. Shi, J. He, Z. Zhou, C. Ji, H.-Y. Chen and D. Ye, Chem.–Eur. J., 2017, 23, 14778 CrossRef CAS PubMed.
- (a) G. Zlokarnik, P. A. Negulescu, T. E. Knapp, L. Mere, N. Burres, L. Feng, M. Whitney, K. Roemer and R. Y. Tsien, Science, 1998, 279, 84 CrossRef CAS PubMed; (b) B. Xing, A. Khanamiryan and J. Rao, J. Am. Chem. Soc., 2005, 127, 4158 CrossRef CAS PubMed.
- (a) G. Xu, Q. Yan, X. Lv, Y. Zhu, K. Xin, B. Shi, R. Wang, J. Chen, W. Gao, P. Shi, C. Fan, C. Zhao and H. Tian, Angew. Chem., Int. Ed., 2018, 57, 3626 CrossRef CAS PubMed; (b) A. L. Antaris, H. Chen, K. Cheng, Y. Sun, G. Hong, C. Qu, S. Diao, Z. Deng, X. Hu, B. Zhang, X. Zhang, O. K. Yaghi, Z. R. Alamparambil, X. Hong, Z. Cheng and H. Dai, Nat. Mater., 2016, 15, 235 CrossRef CAS PubMed; (c) Y. Jiang and K. Pu, Adv. Biosyst., 2018, 2, 1700262 CrossRef; (d) W. Wang, Z. Ma, S. Zhu, H. Wan, J. Yue, H. Ma, R. Ma, Q. Yang, Z. Wang, Q. Li, Y. Qian, C. Yue, Y. Wang, L. Fan, Y. Zhong, Y. Zhou, H. Gao, J. Ruan, Z. Hu, Y. Liang and H. Dai, Adv. Mater., 2018, 30, 1800106 CrossRef PubMed; (e) B. Shi, Q. Yan, J. Tang, K. Xin, J. Zhang, Y. Zhu, G. Xu, R. Wang, J. Chen, W. Gao, T. Zhu, J. Shi, C. Fan, C. Zhao and H. Tian, Nano Lett., 2018, 18, 6411 CrossRef CAS PubMed; (f) Q. Miao and K. Pu, Adv. Mater., 2018, 1801778 CrossRef PubMed; (g) Y. Sun, C. Qu, H. Chen, M. He, C. Tang, K. Shou, S. Hong, M. Yang, Y. Jiang, B. Ding, Y. Xiao, L. Xing, X. Hong and Z. Cheng, Chem. Sci., 2016, 7, 6203 RSC; (h) F. Ding, Y. Zhan, X. Lu and Y. Sun, Chem. Sci., 2018, 9, 4370 RSC.
- (a) T. Guo, L. Cui, J. Shen, W. Zhu, Y. Xu and X. Qian, Chem. Commun., 2013, 49, 10820 RSC; (b) W. C. Silvers, B. Prasai, D. H. Burk, M. L. Brown and R. L. McCarley, J. Am. Chem. Soc., 2013, 135, 309 CrossRef CAS PubMed; (c) W. S. Shin, M.-G. Lee, P. Verwilst, J. H. Lee, S.-G. Chi and J. S. Kim, Chem. Sci., 2016, 7, 6050 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Procedures for synthesis, characterization data, and supplementary figures. See DOI: 10.1039/c9sc02093d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |