
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
P-stereocontrolled synthesis of oligo(nucleoside N3′→O5′ phosphoramidothioate)s – opportunities and limitations†‡
Ewa Radzikowska *a,
Renata Kaczmareka,
Dariusz Korczyńskia,
Agnieszka Krakowiaka,
Barbara Mikołajczyka,
Janina Baraniaka,
Piotr Gugaa,
Kraig A. Wheelerb,
Tomasz Pawlaka and
Barbara Nawrota
*a,
Renata Kaczmareka,
Dariusz Korczyńskia,
Agnieszka Krakowiaka,
Barbara Mikołajczyka,
Janina Baraniaka,
Piotr Gugaa,
Kraig A. Wheelerb,
Tomasz Pawlaka and
Barbara Nawrota
aCentre of Molecular and Macromolecular Studies, Polish Academy of Sciences, Sienkiewicza 112, 90-363 Łódź, Poland. E-mail: eradziko@cbmm.lodz.pl
bWhitworth University, Department of Chemistry, 300 W. Hawthorne Rd., Spokane, WA 99251, USA
First published on 23rd September 2020
Abstract
3′-N-(2-Thio-1,3,2-oxathiaphospholane) derivatives of 5′-O-DMT-3′-amino-2′,3′-dideoxy-ribonucleosides (NOTP-N), that bear a 4,4-unsubstituted, 4,4-dimethyl, or 4,4-pentamethylene substituted oxathiaphospholane ring, were synthesized. Within these three series, NOTP-N differed by canonical nucleobases (i.e., AdeBz, CytBz, GuaiBu, or Thy). The monomers were chromatographically separated into P-diastereomers, which were further used to prepare NNPSN′ dinucleotides (3), as well as short P-stereodefined oligo(deoxyribonucleoside N3′→O5′ phosphoramidothioate)s (NPS-) and chimeric NPS/PO- and NPS/PS-oligomers. The condensation reaction for NOTP-N monomers was found to be 5–6 times slower than the analogous OTP derivatives. When the 5′-end nucleoside of a growing oligomer adopts a C3′-endo conformation, a conformational ‘clash’ with the incoming NOTP-N monomer takes place, which is a main factor decreasing the repetitive yield of chain elongation. Although both isomers of NNPSN′ were digested by the HINT1 phosphoramidase enzyme, the isomers hydrolyzed at a faster rate were tentatively assigned the RP absolute configuration. This assignment is supported by X-ray analysis of the protected dinucleotide DMTdGiBuNPSMeTOAc, which is P-stereoequivalent to the hydrolyzed faster P-diastereomer of dGNPST.
Introduction
Therapeutic properties of synthetic DNA oligonucleotides (PO-oligos) targeted against complementary DNA or mRNA molecules have been tested for more than 40 years.1 In principle, the probes should be characterized by high target affinity, efficient cellular uptake, and enhanced resistance towards nucleases. To make PO-oligos more stable towards nucleases, phosphorothioate analogs (PS-oligos, a dinucleotide NPSN′ (1) is shown in Chart 1) were introduced many years ago2 and remain of interest to the scientific community today.2e A single PO→PS substitution performed with a non-bridging oxygen atom produces a new stereogenic center, thus PS-oligos synthesized by the standard, marginally stereoselective, phosphoramidite or H-phosphonate methods are mixtures of hundreds or thousands of P-diastereomers.3 To date, P-stereodefined PS-oligos have been mainly prepared by (developed in this laboratory) the oxathiaphospholane approach4 (Otp, see Scheme 1) based on the use of P-diastereomerically pure 5′-O-DMT-nucleoside-3′-O-(2-thio-1,3,2-oxathiaphospholane)s or their 4,4-disubstituted analogs (OTP-N; R = H or R = Me or R,R = −(CH2)5–, Scheme 1). Other methods are available as well.5 With the exception of homopurine RP-PS-oligos, which with the RNA complement(s) form thermally stable parallel duplexes and triplexes with overall A conformation,6 both RP- and SP-PS-oligos possess unfavorable hybridization properties as compared to those observed for the unmodified oligonucleotides.7 However, their physico-chemical7–9 and biological10–12 properties often depend on the absolute configuration of the P-atoms.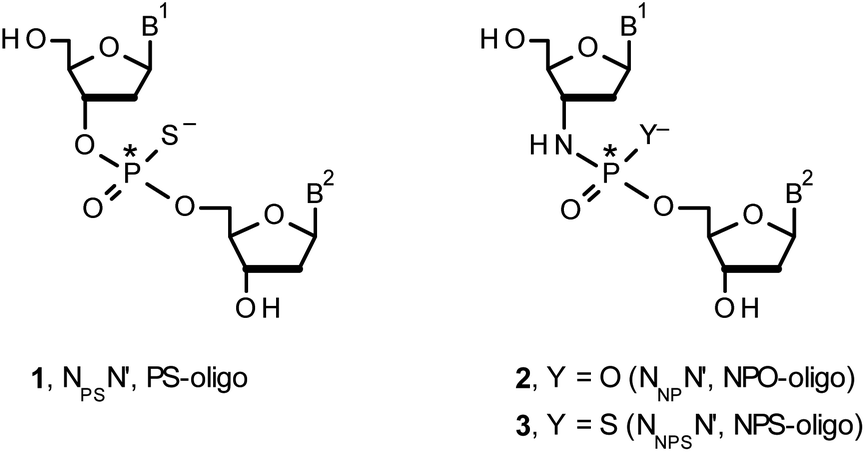 | ||
| Chart 1 B1,B2 = Ade, Cyt, Gua or Thy. The asterisks indicate the P-stereogenic centers. | ||
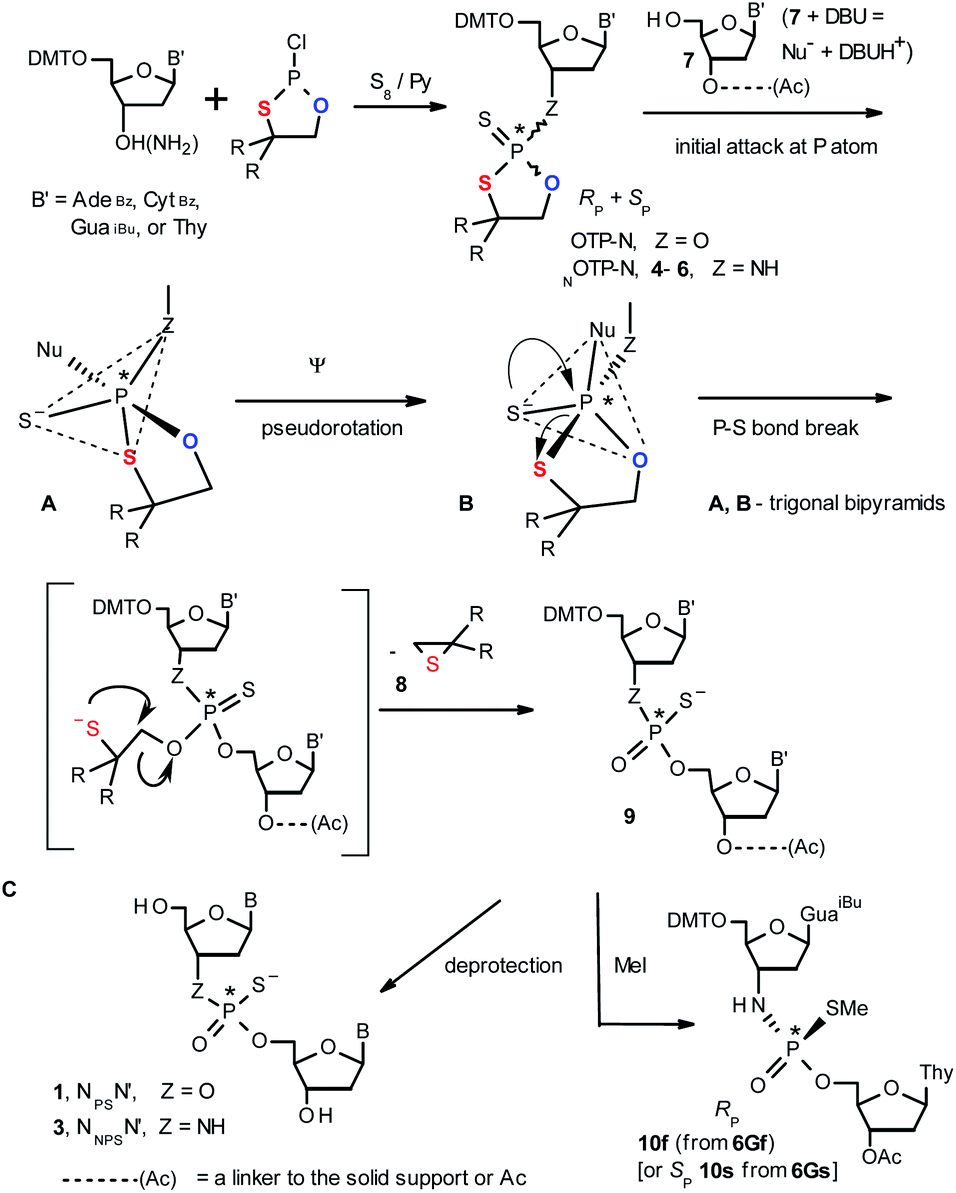 | ||
| Scheme 1 A mechanism of the DBU-promoted condensation step in Otp synthesis of NPSN′ or NNPSN′ with the use of a mixture of P-epimers of OTP-N (Z = O) or NOTP-N monomers (Z = NH), respectively. 4, R = H; 5, R = Me; 6, R,R = −(CH2)5–. B′ = AdeBz, CytBz, GuaiBu, or Thy. The codes 6Gf and 6Gs indicate fast- and slow-eluting P-diastereomers of Pm-NOTP-dG (vide infra). | ||
Another class of DNA analogs of considerable nucleolytic stability consists of P-achiral oligo(deoxyribonucleoside phosphoramidate)s13 (NPO-oligos, a dinucleotide NNPN′ (2) is shown in Chart 1), in which the 3′-oxygen atom is replaced by a nitrogen atom.14 CD measurements15 as well as high-resolution X-ray crystallographic data16 of duplexes formed by NPO-oligos and DNA or RNA strands revealed A-like conformations, which contribute to their high thermal stability.15 NPO-oligos were found to be allosteric inhibitors of telomerase, which is a ribonucleoprotein responsible for maintaining telomeres in nearly all eukaryotic cells.17
Oligo(deoxyribonucleoside phosphoramidothioate)s (NPS-oligos, a dinucleotide NNPSN′ (3) is shown in Chart 1 and in Scheme 1) were synthesized to combine the useful properties of PS- and NPO-oligos.18 NPS-oligos are P-chiral and the syntheses based on the widely used, yet of low stereoselectivity, phosphoramidite or H-phosphonate methodology generate stereorandom mixtures of P-epimers. Important biochemical findings about NPS-oligos§ prompted us to check if P-stereodefined NPS-oligos may be obtained in a variant form of the Otp methodology using the monomers 4–6 (NOTP-N, Z = NH: 4, R = H; 5, R = Me; 6, R,R = −(CH2)5–; B′ = AdeBz, CytBz, GuaiBu, or Thy, Scheme 1). Importantly, it was documented,19 that if Z = O the Otp condensation does not follow a stereoinvertive SN2P mechanism, but this is a stereoretentive process, where the initial attack of a nucleophile 7 takes place from the side opposite to the most electronegative atom attached to the phosphorus center (the oxygen atom in OTP-N, marked in blue; Scheme 1) to form a trigonal bipyramid A. Then, the permutational isomerization (a pseudorotation process) furnishes the bipyramid B, where the leaving thioalkyl group occupies the axial position, necessary for the cleavage of the P–S bond. The condensation process concludes with the elimination of episulfide 8 from the triester intermediate C. It was assumed, that NOTP-N would react with the 5′-OH group of 7 in an analogous manner to yield phosphoramidothioate diester 9 (Z = NH), with a final deprotection step producing NPS-oligos 3.
Early studies in this field showed that both P-epimers of (Rc)-2-(1-(α-naphthyl)ethyl)amino)-2-thio-1,3,2-oxathiaphospholane (Chart 2, structure D) in the presence of DBU reacted with primary alcohols to give products with stereochemical retention – i.e., the incoming alkoxyl group was found in the product at the position originally occupied by the endocyclic sulfur atom (marked in red in Scheme 1).20
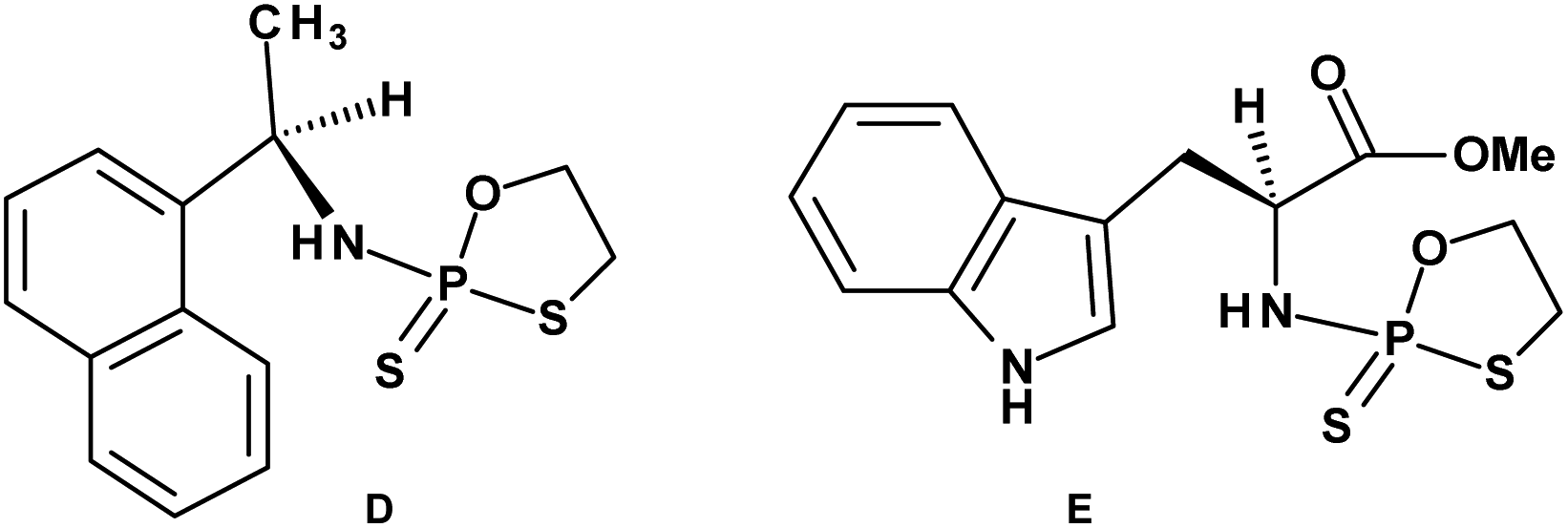 | ||
| Chart 2 (D) (Rc)-2-(1-(α-Naphthyl)ethyl)amino)-2-thio-1,3,2-oxathiaphospholane, (E) a 2-thio-1,3,2-oxathiaphospholane derivative of L-tryptophan methyl ester; NOTP-Trp. | ||
Several years ago such high stereoselectivity was confirmed in the reaction of the NOTP derivative of L-tryptophan methyl ester (NOTP-Trp, Chart 2, structure E) with O2′,O3′,N6-tribenzoyl-adenosine21 leading to the tribenzoylated derivative of AMPS-Trp-OMe. Similar stereoselective results were achieved from the reactions of P-diastereomers of the NOTP derivative of 3′-amino-3′-deoxy-thymidine (5, B′ = Thy) with 3′-O-acetyl-thymidine, which (after deprotection) furnished the TNPST dimers.22
Once obtained, homopurine RP-NPS-oligos with the anticipated intrinsic C3′-endo conformation (vide supra) could be used to effectively stabilize the previously described parallel duplexes and triplexes.6
Here we show that using P-diastereomerically pure NOTP-N monomers results in P-stereodefined chimeric PO/NPS- or PS/NPS-oligos with the NPS-nucleotides introduced in “alternate” positions. Additional experiments revealed several structural factors responsible for the observed low repetitive yields in the solid phase synthesis of uniformly modified NPS-oligos.
Results and discussion
Preparation of P-diastereomerically pure NOTP-N monomers 4–6
Our earlier experiments with OTP-N monomers (Z = O, Scheme 1) showed that in terms of chromatographic separability of P-diastereomers, those 4,4-unsubstituted (R = H) were most troublesome, followed by the more convenient compounds bearing R = Me and the most useful 4,4-pentamethylene substituted congeners.4 The same was observed for their LNA (Locked Nucleic Acids) analogs.¶In the present work three sets of NOTP-N monomers (4,4-unsubstituted, Un-, 4; 4,4-dimethyl substituted, Dm-, 5; and 4,4-pentamethylene substituted, Pm-NOTP-N, 6; Scheme 2) were obtained from the 5′-O-DMT derivatives of 3′-amino-2′,3′-dideoxy-ribonucleosides. However, chromatographic separation of P-diastereomers was achieved only for 4A, 5T, 5C, and 6G (the suffixes A, G, C or T indicate the Ade, Gua, Cyt or Thy nucleobases, respectively). The chromatographic details related to the separation of the fast- and slow-eluting P-diastereomers are given in Table 1, and the relative mobility is reflected in their codes by an f or s suffix, respectively (e.g. 4Af or 4As, Scheme 2). The relevant HR MS, 31P NMR, 1H NMR, and 13C NMR spectra are shown in Data Sets S1–S4 and Fig. S1–S4 (ESI).‡ Attempts to separate other NOTP-Ns, e.g. 5A or 6A, were unsuccessful with the data for unresolved 4–6 given in Table S1 (ESI).‡ [Note: The pro-RP and pro-SP descriptors, shown in Scheme 2, indicate the absolute configuration of P-atom in the internucleotide phosphoramidothioate moiety formed upon condensation of a given P-diastereomer of NOTP-N with the 5′-OH group of a nucleoside/nucleotide. The assignment was based on the differences in rates of hydrolysis of P-diastereomeric NNPSN′ dinucleotides 3 (Scheme 1) with HINT1 phosphoramidase (vide infra)]. It should be noted, that stereochemically equivalent pro-SP OTP-N and NOTP-N monomers (Scheme 2, F and G, respectively) have opposite absolute configurations of the phosphorus stereogenic centers. Obviously, the same relationship is valid for the pro-RP pairs.
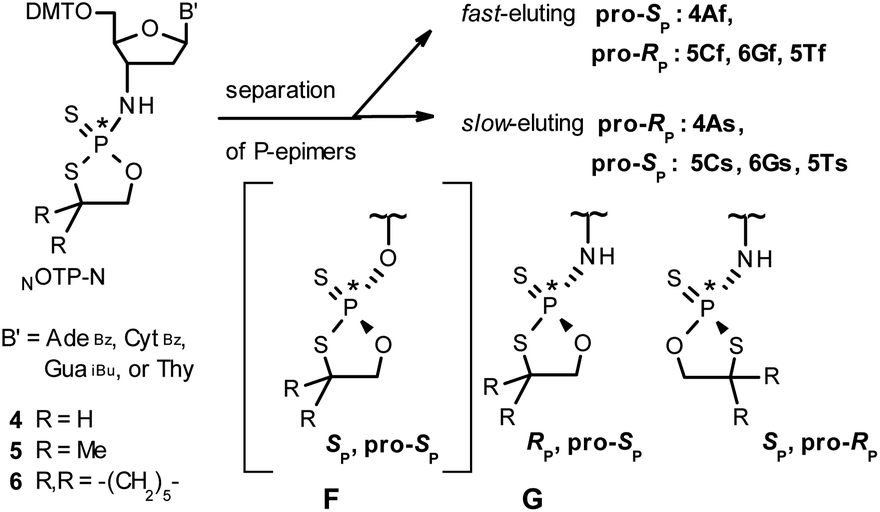 | ||
| Scheme 2 Effects of silica gel separation of P-diastereomers of 2-thio-1,3,2-oxathiaphospholane monomers 4–6. The descriptors pro-RP and pro-SP indicate the absolute configuration of P-atom in the subsequently formed internucleotide phosphoramidothioate linkages. The structure F is given only for comparison with G. | ||
| B′, R,R | Yielda (%) | MW calc. (Da) | TOF MS ES (m/z)b | Code | Rfc | 31P NMRd (δ, ppm) |
|---|---|---|---|---|---|---|
a Total yield of the isolated mixture of isomers; yields of the separated fast- and slow-eluting isomers, respectively; ‘flash’ chromatography on silica gel 200–300 mesh was performed unless otherwise stated.b [M + H+] ions, measured for the unresolved mixture of P-epimers.c HP TLC plates.d In CDCl3.e Silica gel 60H, 0→1% MeOH/CHCl3, v/v.f 0→2% MeOH/CHCl3, v/v.g CHCl3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH 50:1, v/v.h AcOEt:hexane 1:1, v/v.i Only a minute amount of 4Af was isolated and in further experiments a 1:2 mixture of 4Af and 4As was used.j CHCl3:MeOH 20:1, v/v, double development.k CHCl3:MeOH 20:1, v/v.l AcOEt:hexane 1:1, v/v. :MeOH 50:1, v/v.h AcOEt:hexane 1:1, v/v.i Only a minute amount of 4Af was isolated and in further experiments a 1:2 mixture of 4Af and 4As was used.j CHCl3:MeOH 20:1, v/v, double development.k CHCl3:MeOH 20:1, v/v.l AcOEt:hexane 1:1, v/v. |
||||||
| AdeBz, H,H | 83 | 794 | 795.2191; 817.2008 [M + Na+] 100% | 4Afi | 0.50 | 95.92 |
| 24/12e | 4Asi | 0.48j | 95.25 | |||
| CytBz, Me,Me | 79 | 798 | 799.2385 | 5Cf | 0.70 | 97.74 |
| 19/26f | 5Cs | 0.65k | 97.25 | |||
| GuaiBu, –(CH2)5– | 84 | 844 | 845.2920 | 6Gf | 0.58 | 96.92 |
| 52/26g | 6Gs | 0.47k | 96.94 | |||
| Thy, Me,Me | 88 | 709 | 732.1951, [M + Na+] | 5Tf | 0.65 | 96.69 |
| 22/22h | 5Ts | 0.58l | 96.58 | |||
Tentative assignment of the absolute configuration at P-atoms in separated diastereomers of NOTP-Ns 4–6
During the course of earlier works on stereocontrolled synthesis of PS-oligos, the experiments of enzymatic hydrolysis of dinucleoside phosphorothioates NPSN′ (1) with RP-specific snake venom phosphodiesterase (svPDE) and SP-specific Nuclease P1 (ref. 23) provided sufficient data to establish an empirical rule that the fast-eluting Un-OTP-N (Z = O, B’ = AdeBz, CytBz, GuaiBu,DPC, or Thy) are precursors of SP-NPSN′, contrary to fast-eluting Dm- and Pm-OTP-N congeners, which yield RP-NPSN′.4a,19 This relationship was later confirmed for the Pm-OTP derivatives of LNA nucleosides.24,25 Unfortunately, although NPO-oligos were hydrolyzed by svPDE to a measurable extent (after 4.5 h incubation ∼50% of NPO-T10 remained intact;13a after 24 h incubation phosphoramidate TNPT was hydrolyzed in 22% yield, data not shown), phosphoramidothioates 3 when treated with svPDE or Nuclease P1 remained intact.A few years ago, chromatographically separated 5Tf and 5Ts (R = Me, B′ = Thy) and 3′-O-acetylated thymidine were used in the synthesis of P-diastereomeric DMTTNPSTOAc (5′-O-DMT and 3′-O-Ac protected precursors of 3, Chart 3).22 Both anionic DMTTNPSTOAc diesters were then stereoretentively S-methylated to yield the corresponding P-diastereomeric triesters DMTTNPSMeTOAc (3Mf and 3Ms, Chart 3). At that point, 2D NMR ROESY experiments suggested that, contrary to the aforementioned empirical rule, the relative orientation of the sulfur, phosphoryl oxygen and thymidine O5′ atoms around the phosphorus atom in the S-methylated triester 3Mf (Chart 3, obtained from the fast-eluting 5Tf) is equivalent to that in phosphorothioate SP-1, which is hydrolyzed by Nuclease P1. Consequently, 3Ms obtained from the slow-eluting 5Ts is P-stereoequivalent to RP-1, which is hydrolyzed by svPDE. Although the O3′→N3′ replacement does change the priority of substituents (O3′ – priority 2, N3′ – priority 4), by virtue of the formalism of the Cahn-Ingold-Prelog rules, not only the relative orientations but also the absolute configurations of P atoms in SP-1, and 3Mf are the same.
 | ||
Chart 3 Orientation of the sulfur, phosphoryl oxygen and thymidine O5′ atoms around the phosphorus atoms: Panel A, in phosphorothioate SP-1, ref. 23; Panel B, 2D NMR ROESY based assignment (here challenged by X-ray analysis of 10f, Scheme 1) in the S-methylated SP-3Mf, ref. 22 (the phosphoramidothioate diester precursor was obtained from fast-eluting 5Tf). The Arabic numerals 1–4 indicate the priority of substituents around the phosphorus atoms according to the Cahn–Ingold–Prelog rules. Note: to establish the priority of substituents, the formal double bond between the phosphoryl oxygen atom and the phosphorus atom (P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) should be considered a single one. O) should be considered a single one. | ||
This violation of the empirical rule “fast-eluting Dm- and Pm-OTP-N → RP-NPSN” could not be explained by the C3′-endo conformation of the sugar moiety in 3′-amino-2′,3′-dideoxy-nucleosides, because the C3′-endo locked LNA-derived oxathiaphospholane monomers adhere to that rule.24 In the present work, this unexpected NMR-based assignment was challenged by a successful crystallographic experiment, which showed the rule-obeying RP absolute configuration (Fig. 1) for a non-ionic DMTdGiBuNPSMeTOAc amidodiester (10f, Scheme 1, ESI‡) obtained from the fast-eluting 6Gf (a Pm-derivative) after S-methylation of the negatively charged amidoester intermediate DMTdGiBuNPSTOAc (9). Importantly, compound 10f crystallized spontaneously, without the typically applied slow evaporation. The complete crystal structure is shown in Fig. S7 (ESI)‡ with crystallographic details summarized in Table S2 (ESI).‡
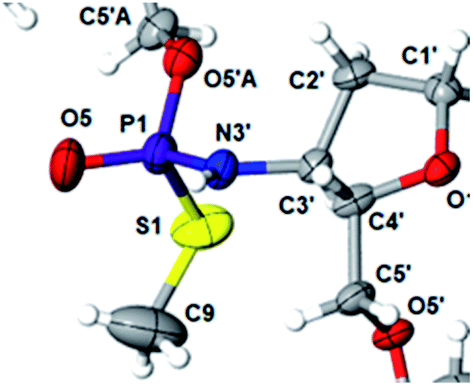 | ||
| Fig. 1 Crystal structure of DMTdGiBuNPSMeTOAc 10f showing partial structure and spatial orientation of substituents around the phosphorus stereogenic center (marked P1). Compound 10 was obtained from the monomer 6Gf (fast-eluting 5′-O-DMT-3′-amino-2′,3′-dideoxy-N6-iBu-guanosine-3′-N-(2-thio-4,4-pentamethylene-1,3,2-oxathiaphospholane). The oxygen atom marked O5′A belongs to the thymidine residue. | ||
The opposite absolute configurations of P-atoms determined by the dinucleotide derivatives 3Mf and 10f, obtained from the fast-eluting Dm-5Tf (the NMR data) and from the fast-eluting Pm-6Gf (the X-ray data), respectively, seemed to be a contradiction. Although it is possible that the Dm-NOTP monomers, unlike Dm-OTP monomers, behave differently from the Pm-analogs, the contradiction could not be left unaddressed. Because attempts at crystallization of other NNPSN′ dimers (protected or unprotected) or their NOTP-N precursors were unsuccessful, we turned our attention to HINT1 phosphoramidase. This enzyme belongs to a family of hydrolases and transferases characterized by the presence of the histidine triad H–X–H–X–H–X–X–X (HIT) motif at their catalytic center, where H is a histidine residue and X is a hydrophobic amino acid.26 The HINT1 catalyzed in vitro hydrolysis of the P–N bond releases (d)NMP or (d)NMPS from their phosphoramidate or phosphoramidothioate conjugates with amino acids, e.g., from a conjugate of L-tryptophan amide and AMPS (AMPS-Trp-NH2||, Scheme 3).21,27
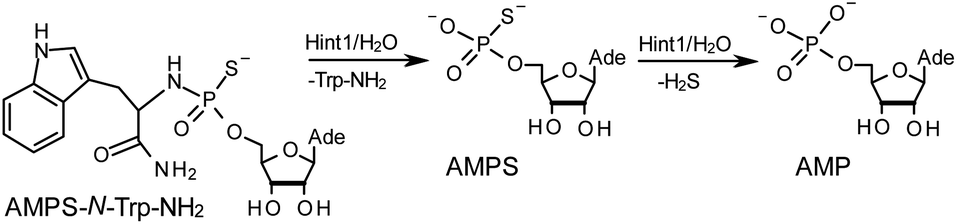 | ||
| Scheme 3 The HINT1 catalyzed hydrolysis of adenosine-5′-O-((L-tryptophan amide-Nα-yl)-phosphoramidothioate) (AMPS-Trp-NH2), followed by desulfuration of the AMPS intermediate leading to the formation of adenosine-5′-O-phosphate. | ||
It is known that the hydrolysis of the P–N bond in the RP isomer of AMPS-Trp-NH2 is approximately 4 times faster than the same reaction using the SP counterpart.21 The released (d)NMPS is further desulfured to form (d)NMP and hydrogen sulfide, and this secondary nucleotide product must be taken into account during HPLC-based quantification of the products of hydrolysis. The rates of desulfuration of (d)NMPS decrease in the following order: AMPS, GMPS ≥ CMPS > UMPS > dAMPS, dGMPS ≫ dCMPS, TMPS.28 One can assume that this decreasing order of affinity would also be reflected in the rates of hydrolysis of the RP and SP series of (d)NMPS-conjugates. To determine the absolute configuration at P-atoms in the oligomers obtained from the separated fast- and slow-eluting P-diastereomers of 4–6, model NNPSN′ dimers 11–17 (see Table 2) were prepared using the solid phase synthesis method. Due to the small difference of Rf values, only a minute amount of 4Af was isolated and in further experiments a 1:2 mixture of 4Af and 4As was used. [Notes: (a) To prevent the DBU-promoted intramolecular cleavage of the standard LCAA (long chain alkylamino) linker, a modified solid-phase support (-$) was used, in which the 3′-OH group of nucleoside was attached to the CPG (controlled pore glass) beads via a sarcosine linker [–COCH2CH2CON(CH3)CH2CO–LCAA–CPG].29 (b) To assess relative reactivity of NOTP-N and OTP-N monomers, HOdG-$ was elongated with an equimolar mixture of NOTP-T and OTP-T, and the resultant mixture consisted of TNPSdG and TPSdG at ca. 1:5 ratio.]
We recognized that the d(NNPSN′) dimers, as well as TNPOT, were hydrolyzed by recombinant human HINT1 phosphoramidase. In an initial rate assay, the substrates were treated at 37 °C until 10% degradation was detected by RP HPLC. The observed hydrolysis rates are given in Fig. 2 and Table 3. [Note: because the phosphoramidate conjugates of TMPS are the least effective substrates for HINT1 (vide supra) the rates of hydrolysis of 16 and 17 were very low; therefore, for better visualization the corresponding four bars in Fig. 2 were increased by a factor of 10.] Assuming that the hydrolysis of NNPSN′ mechanistically follows that observed for AMPS-Trp-NH2, the RP-NNPSN′ dimers should be hydrolyzed faster than the SP counterparts. Based on this assumption, the HPLC analysis allowed assignment of the RP absolute configuration to d(ANPSA) obtained from slow-eluting 4As, and to d(CNPSA), TNPSdA, TNPSdG, TNPST, dGNPST, and d(GNPSG) obtained from fast-eluting 5Cf, 5Tf, and 6Gf. This conclusion is contradictory to the earlier assignment from NMR data, but is consistent with the result of X-ray analysis shown in Fig. 1. Since the NMR based assessment of absolute configuration is less reliable than the crystallographic analysis, we claim that the resolved NOTP-N monomers follow the earlier observations made for OTP-N, so the fast-eluting 4Af is a precursor of SP-NNPSN′, and fast-eluting Dm- and Pm-NOTP-N yield RP-NNPSN′.
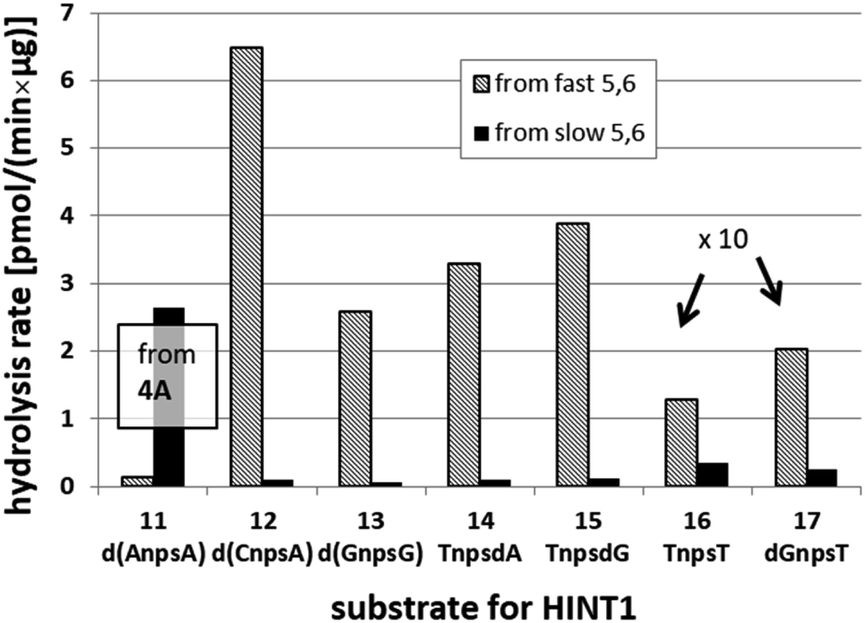 | ||
| Fig. 2 Rates of hydrolysis of NPS-dinucleotides 11–17 (data from Table 2). The descriptions fast and slow refer to the relative chromatographic mobility of NOTP-N precursors. For better visualization the heights of bars depicting the rates of hydrolysis of 16 and 17 have been scaled up by a factor of 10. | ||
| NOTP-N | d(NNPSN′) | NOTP-N substrate | |
|---|---|---|---|
| Fast | Slow | ||
| a Because a 1:2 mixture of 4Af and 4As was used in synthesis of 11, the corresponding 1:2 mixture of P-diastereomers was formed and P-diastereomerically pure 11 were obtained by RP HPLC separation of the fully deprotected compounds. |
|||
| 4A | 11 d(ANPSA)a | 0.135 ± 0.007 | 2.65 ± 0.30 |
| 5C | 12 d(CNPSA) | 6.48 ± 0.36 | 0.107 ± 0.025 |
| 6G | 13 d(GNPSG) | 2.585 ± 0.019 | 0.063 ± 0.007 |
| 5T | 14 TNPSdA | 3.296 ± 0.631 | 0.099 ± 0.010 |
| 5T | 15 TNPSdG | 3.883 ± 0.651 | 0.125 ± 0.023 |
| 5T | 16 TNPST | 0.129 ± 0.160 | 0.034 ± 0.007 |
| 6G | 17 dGNPST | 0.202 ± 0.044 | 0.026 ± 0.004 |
| NA | TNPOT | 7.171 ± 1.225 | |
Factors affecting the yield of condensation using NOTP-N
The previous experiments performed in our laboratory showed that, although the yield of the first coupling step of 6T (a mixture of P-diastereomers) with the 5′-OH group of a deoxyribonucleoside attached to a solid support was acceptable (ca. 90%), the yields associated with the subsequent coupling steps decreased at least by half.30 No improvement was observed when longer reaction times (600 s) or double delivery of the monomer (the second delivery after washing and drying of the support) were applied.:4, v/v) the condensation yield dropped even more (25–30%, assessed by the DMT+ assay, Fig. 3, Exp. #1), whereas the condensation with OTP-N (R = H) in the same solvent mixture was >90% effective (data not shown).
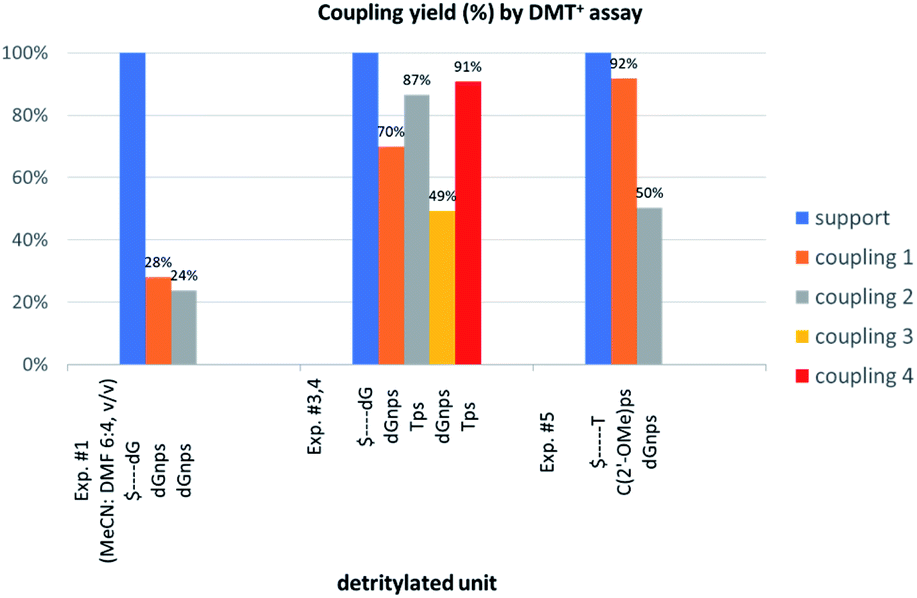 | ||
| Fig. 3 Coupling yields in the consecutive condensation steps, assessed by DMT+ assay at 504 nm. Absorptions of the DMT+ cation released from the nucleosides attached to the support (blue bars) are taken as 100%. | ||
Because chemically NPS-oligos and PS-oligos differ by the presence of a 3′-NH amide group (a potent donor of hydrogen bonding) instead of 3′-O ester atom (a weak acceptor), one might assume that the former may be involved in aggregation, which potentially may be disrupted by the coordination of the 3′-NH with a strong non-nucleophilic amine. Because DBU is a sterically hindered amine and perhaps unable to provide this coordination, we tried to use a mixture of DBU and other less sterically demanding amines. The coupling reactions were performed at the 1 μmol scale (in all experiments thymidine was attached to the support) using 20-fold molar excess of each of four NOTP-N monomer 4, 50-fold excess of DBU and, starting from the second coupling, 50-fold excess of the additional amine. Unexpectedly, we noted a significantly increased repetitive yield of condensation only for 4T (67–88%, assessed by the DMT+ cation assay, Table 4). For unknown reasons, the best and the worst results were obtained using NEt3 and structurally similar NBu3, respectively. It should be noted that although in the past triethylamine was found to promote the condensation of much more reactive OTP-N monomers, the rate of coupling was low and several hours were necessary to complete the process.19
| Amine | pKa | Repetitive yield (%) | Overall yield (%) |
|---|---|---|---|
| NBu3 | 10.89 | 67 | 33 |
| NEt3 | 10.76 | 88 | 68 |
| DIPEA | 10.50 | 76 | 44 |
| DMAP | 9.20 | 78 | 48 |
| Collidine | 7.48 | 74 | 36 |
| NOTP-N | Nucleobase | |||
|---|---|---|---|---|
| AdeBz | CytBz | GuaiBu | Thy | |
| 4 | 78 | 86 | 84 | 88 |
| 5 | 21 | 71 | 63 | 81 |
| 6 | 8 | 60 | 40 | 66 |
Additionally, increasing the steric bulk of C4 substituents on the oxathiaphospholane ring results in a decrease in coupling efficiency. This phenomenon was most profound for 4A, 5A, and 6A, and using 6A the trimer d(ANPSANPSA) was obtained in less than 10%. Fortunately, much more reactive 4A can be resolved onto P-epimers. On the contrary, 4G and 5G are much more reactive than 6G, but only the epimers of the latter monomer (6Gf and 6Gs) are separable. Interestingly, there are significant differences in the coupling yields for 6Gf and 6Gs (Table 2, 75% vs. 46%, 84% vs. 69%) and this observation prompted us to perform some calculations (using a Gaussian 16 software (ref. 31)) to assess their geometries. As expected, it was found that both compounds predominantly exist in a C3′-endo conformation. Then we focused our attention on the accessibility of the phosphorus centers for the initial attack of the nucleophile leading to the formation of the bipyramid A (Scheme 1). The views along the P–O bonds (the P and O atoms are green and red, respectively) are presented in Fig. 4. Comparing the areas inside the yellow circles one can notice that the steric hindrance is much lower in SP NOTP-dG, so the more reactive 6Gf can be tentatively assigned the SP absolute configuration. Consequently, 6Gf should be a precursor for RP-NNPSN′ dinucleotides. This assignment is consistent with the crystallographic data collected for 10f and with the results of Hint1 promoted hydrolysis of 13 and 17, but cannot be considered decisive.
 | ||
| Fig. 4 Visualization of the lowest energy conformers of 6G (RP Pm-NOTP-dG and SP Pm-NOTP-dG). The data were obtained using the Gaussian 16 package. Heteroatom labeling: P-green, O-red, S-yellow, N-navy blue. | ||
Working earlier with LNA-derived OTP monomers (OTP-NLNA, which adopt the profound C3′-endo conformation), we observed (Exp. #2) that in the synthesis of per-(PS-LNA) oligonucleotides even double condensation of OTP-NLNA to HONLNAPS…-$ was ineffective and a DMT+ cation absorption virtually decayed after the 5th cycle (K. Jastrzębska, P. Guga, unpublished data); whereas, such a condensation with the 5′–OH–DNA-$ proceeded with >94% efficacy.24 As mentioned earlier, molecular modeling showed that poorly reacting 6Gf and 6Gs predominantly exist in a C3′-endo conformation. To verify whether the most reactive NOTP-T monomers 5Tf and 5Ts adopt that conformation (characteristic of 3′-amino-2′,3′-dideoxyribonucleosides), we performed NMR analysis. Unexpectedly, the recorded 2D 1H–1H COSY and 1H–13C EDITED-HSQC spectra showed 3JH1′,H2′ = 6.5 Hz, which according to literature data32 indicate a pseudorotation phase P = 90° or 195°. The former value is characteristic of a rare O4′-endo conformer, whereas the latter indicates a C2′-endo structure. To distinguish these options, a 3JH3′,H4′ value would be useful, but its measurement was significantly more complicated and neither analysis of the multiplicity of H3′ and H4′ signals nor attempts at simulation of the full spin system were successful. The molecular modeling experiments performed for RP-5T (5Ts, slightly less effective than 5Tf as shown in Table 2) showed that starting with C2′-endo, C3′-endo, or O4′-endo conformations the geometry optimization always ended at the C2′-endo conformation (Fig. 5), characteristic of the OTP-N monomers.
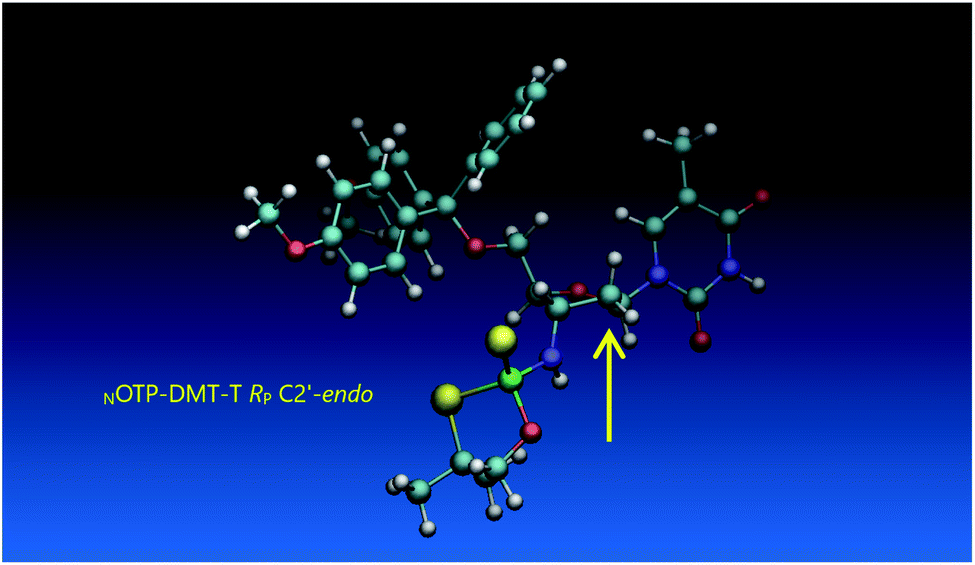 | ||
| Fig. 5 Visualization of the lowest energy conformer of RP-5T (the C2′-endo atom is marked with an arrow). The data were obtained using the Gaussian 16 package. Heteroatom labeling: P-green, O-red, S-yellow, N-navy blue. | ||
Analogous calculations were performed for (SP)-5′-O-DMT-N6-benzoyl-2′-deoxyadenosine-3′-O-(2-thio-4,4-pentamethylene-1,3,2-oxathiaphospholane) (SP Pm-OTP-dA, slightly less reactive than the RP counterpart), and (RP)-5′-O-DMT-N6-benzoyl-3′-amino-2′,3′-dideoxy-adenosine-3′-N-(2-thio-4,4-pentamethylene-1,3,2-oxathiaphospholane) (RP Pm-NOTP-dA, RP-6A, the least effective monomer as shown in Table 5). [Note: looking along the P–O3′ bond in SP Pm-OTP-dA and along the P–N3′ bond in RP-6A, despite of the opposite absolute configurations of phosphorus atoms (SP vs. RP), all other substituents attached to the P-atoms are positioned with similar spatial orientations, so that both compounds are P-stereochemically equivalent.] Independent of the implemented solvent (acetonitrile or chloroform), the results were very similar and showed (see the yellow circles) that the approach of the nucleophile along the P–O bond in SP Pm-OTP-dA is virtually unrestricted (Fig. 6, an upper panel). Importantly, a similar marginally restricted approach is predicted for efficiently reacting 5Ts (Fig. 6, a bottom panel). On contrary, in a poorly reacting C3′-endo (RP)-3′-amino-2′,3′dideoxy-adenosine analog (a middle panel) such the approach is substantially blocked by the DMT propeller.
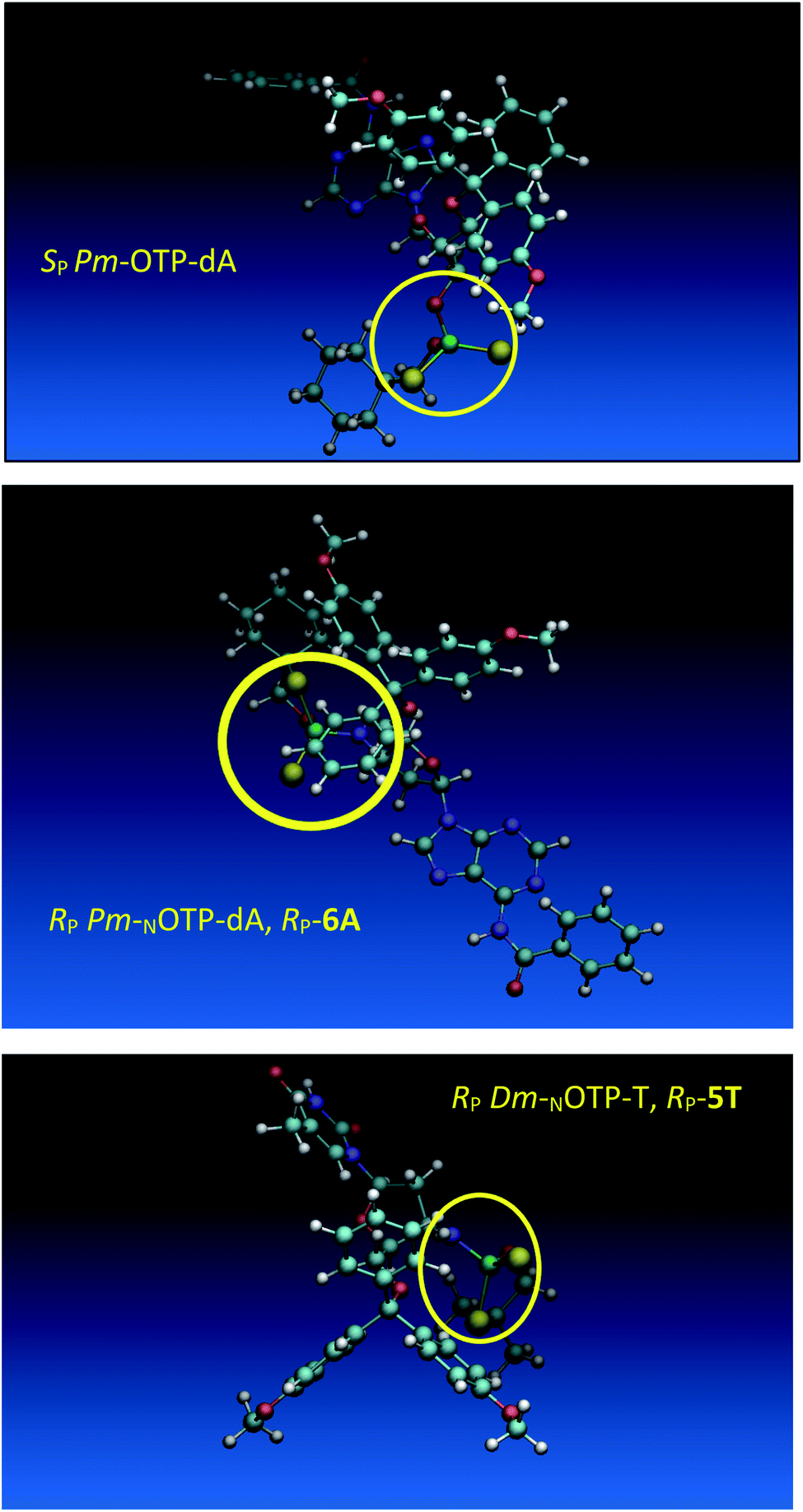 | ||
| Fig. 6 Predicted by molecular modeling the lowest energy conformations of: SP Pm-OTP-dA, an upper panel; RP-6A, a middle panel; RP-5T, a bottom panel. All three compounds are P-stereochemically equivalent (vide supra). | ||
Indications of ‘conformational clash’
In next three condensation experiments we used monomers of low coupling potency, i.e., a 6Gf + 6Gs mixture. If HOTPSd(GNPSG)-$ is condensed with an NOTP-G monomer (Exp. #3), the product is formed in 49% yield (Fig. 3), while only 40% yield was noted for elongation of HOd(GNPSG)-$ (Table 5). If HOd(GNPSG)-$ or HOd(GNPSTPSGNPSG)-$ are elongated using an OTP-T monomer (C2′-endo) the efficacy is close to 90% (Exp. #4). If HOC(2’-OMe)PST-$ (the 2′-OMe unit exists in the C3′-endo conformation) reacts with an NOTP-dG monomer (Exp. #5), ca. 50% efficacy is observed.Important information comes from a paper by Hodgson and co-workers, which describes 1H NMR conformational analysis of TNPST dinucleotide, which is an anionic S-alkyl phosphoramidothiolate compound bearing a 5′-deoxy-5′-thio-thymidine residue (ST) at the 3′-end.33 They found that the 3′-amino-2′,3′-dideoxy-ribose ring adopts the C3′-endo conformation, while the ring of the ST unit retains the C2′-endo conformation. Thus, the C3′-endo conformation is not transmitted downstream to the ST DNA unit and the preserved neighboring conformations C3′-endo/C2′-endo lead to the lowest overall energy of the TNPST system. This indicates that the C3′-endo/C2′-endo conformation of both nucleosides is energetically favored over the C3′-endo/C3′-endo conformation, which is observed in RNA/DNA hybrids. This observation may explain the greater reaction efficiency of NOTP-N with a growing oligomer bearing the 5′-end DNA unit (the first coupling step and the result of Exp. #3). At this point, one can mention DMTdGiBuNPSMeTOAc (10f), which also adopted the C3′-endo/C2′-endo conformation (Fig. S8, ESI‡), and spontaneous crystallization of which indicates remarkably low energy of this system.
Thus one can conclude that if the 5′-end segment of a growing oligomer (HONNPS…-$, HONLNAPS…-$ in Exp. #2, or HOC(2’-OMe)PS…-$ in Exp. #5) and an incoming monomer (NOTP-N or OTP-NLNA) adopt the C3′-endo conformation there is a more crowded space around the phosphorus atom. This conformational ‘clash’ may result in a decrease of reaction rate that promotes additional side-reactions and may heighten the damaging effect of the aforementioned intrinsic lower reactivity of the thiophosphoramide NOTP-N monomers compared to OTP-N. This decrease of coupling efficiency is much less severe if the incoming monomer exists in the C2′-endo conformation (Exp. #4 and relatively high reactivity of 5T).
Attempts at solid phase synthesis of P-stereodefined NPS-oligo and chimeric NPS/PS and NPS/PO oligomers
The low repetitive yields encountered in the syntheses of NNPSNNPSN with a random configuration of phosphorus atoms (Table 5) were confirmed (by the DMT+ assay) by use of resolved P-epimers (see the column “Yield” in Table 6 for several examples). This circumstance rendered the syntheses of longer P-stereodefined NPS-oligo difficult. Also, to avoid extensive hydrolysis of phosphoramidothioate linkages upon treatment with 50% aqueous acetic acid34 at the detritylation step, performed after the otherwise beneficial DMT-ON RP HPLC purification, the last step of the solid support synthesis included the use of an anhydrous 3% solution of DCA in methylene chloride for detritylation. In the case of short oligomers 18–20 (up to heptamers), careful RP HPLC purification furnished the products of satisfactory purity, as assessed by PAGE and MALDI-TOF MS analyses (data not shown). The earlier mentioned lower efficiencies of condensation for the purine monomers, as well as those for the slow-eluting compounds, were confirmed in the syntheses summarized in Table 6. Only homo-thymidine oligomers 19 and 20 were isolated in appreciable amounts, while the samples of 18f and 18s allowed only for MALDI-TOF MS analysis.| Sequence | Code | NOTP-N substrate | Yielda (%) | Amountb (OD) |
|---|---|---|---|---|
| a The yield calculated from the absorbance measured (at 504 nm) for the last dimethoxytrityl cation released compared to the initial value.b After double HPLC purification on a C18 reverse phase column.c An unresolved mixture of P-epimers. | ||||
| d(GNPSGNPSG) | 18f | 6Gf | 17 | 0.5 |
| 18s | 6Gs | 8 | 0.5 | |
| TNPS(TNPS)4T | 19f | 5Tf | 37 | 4.2 |
| 19s | 5Ts | 26 | 3.0 | |
| TNPS(TNPS)5T | 20m | 5Tc | 16 | 9.8 |
| TNPS(TNPS)8T | 21m | 5Tc | 21 | 5.4 |
| 21f | 5Tf | 17 | 7.5 | |
This modified approach was unsuccessful for the decamers 21 as neither RP nor IE HPLC purification provided products of acceptable purity. Therefore, the DMT-tagged oligomers 21 were isolated by means of RP HPLC using DMT-ON parameters. The final detritylation step was performed using 10% aqueous solution of dichloroacetic acid for 10 minutes, and the final RP HPLC purification was performed. The HPLC profile and the PAGE electropherogram recorded for 21f are shown in Fig. 7, while the relevant 31P NMR and MALDI-TOF MS spectra are shown in Fig. S8 (ESI).‡ Unfortunately, because of very low repetitive condensation yields, all attempts to synthesize 21 using 5Ts were unsuccessful. This may indicate that 21m obtained from unresolved 5T predominantly contained the NPS linkages having the P atoms of RP absolute configuration, but we were unable to confirm this assumption experimentally. Regretfully, at the time being, the field of antisense applications is inaccessible for uniformly modified P-stereodefined NPS-oligomers, although search for more effective conditions of condensation is going on. However, having resolved the problems with determination of the absolute configurations in the prepared monomers, one can use them in synthesis of precisely tailored probes (e.g. for enzymatic studies) bearing P-stereodefined NPS-units in a few preselected positions. As mentioned earlier, elongation of oligomers having at the 5′-end an NNPS unit using an OTP-N monomer is more effective and a PS/NPS chimeric TPSdGNPSTPSdGNPSdG oligomer (see observation #4 and Fig. 3, a middle plot) was obtained in 27% overall yield (assessed by the DMT+ cation assay). This is an acceptable result when considering the known low efficacy of condensation (see Table 2) of Pm-NOTP-dG.
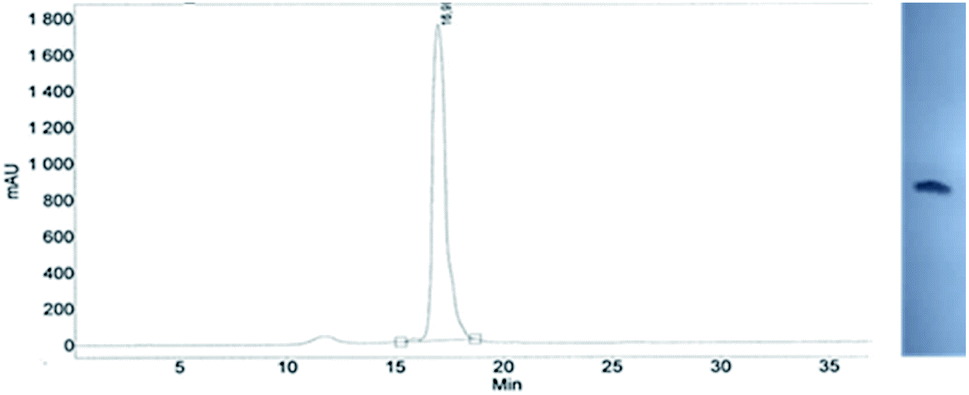 | ||
| Fig. 7 Analysis of 21f obtained from 5Tf. (Left) An RP HPLC profile (DMT-OFF); (right): an electropherogram (20% PAGE). | ||
Finally, we decided to explore the possibility of making chimeric NPS/PO oligomers. In an attempt to synthesize the 9-mer of the sequence 5′-DMTTPO(TPO)4(TNPS)3T-3′, the NPS-oligo fragment TNPSTNPSTNPST-$ was assembled using the unresolved 4T, with NEt3 added in second and third coupling (Scheme S1, ESI‡). This TNPSTNPSTNPST-$ core was intended to be elongated with the phosphate units using the phosphoramidite method of DNA synthesis. The standard protocol of the latter method could not be used, because past work in our group on the synthesis of PS/PO chimeras indicated that the anionic internucleotide phosphorothioate linkage is quantitatively converted to the phosphate derivative. This process occurs upon contact with an iodine–base–water solution (which is routinely applied to oxidize the just formed phosphite triester), as well as with many other reagents such as camphorsulfonyloxaziridine,35 alkyl hydroperoxides36 or peracids.37 To a certain extent, this problem was resolved by S-alkylation of the PS-oligo core with 2-nitrobenzyl bromide to form the corresponding triester, which can be eventually deprotected with a thiophenolate anion. However, this process has only been optimized for d(NPSN) dinucleotides.38 Our later studies showed that the destructive PS→PO exchange in PS-oligo is avoided if the PIII→PV conversion is performed using t-Bu-OOSiMe3 (ref. 39) (0.33 M in CH3CN, 30 min, room temperature).40 In the present work, the 31P NMR and MALDI-TOF MS measurements revealed that under these conditions the phosphoramidothioate linkage in DMTTNPSTOAc also remained unaffected (data not shown). The 9-nt DMTTPO(TPO)4(TNPS)3T-3′, was obtained in 42% yield as assessed from the DMT+ cation assay. Its identity was confirmed by MALDI-TOF MS analysis (Fig. S9, ESI‡). Using 3H-1,2-benzodithiol-3-one 1,1-dioxide (a Beaucage reagent), ((dimethylamino-methylidene)amino)-3H-1,2,4-dithiazoline-3-thione (DDTT) or phenylacetyl disulfide to sulfurize the P atom in the phosphite triester linkage (formed after the phosphoramidite coupling step) the corresponding PS/NPS chimeras can be obtained.
Conclusions
In an attempt to synthesize P-stereodefined oligo(deoxyribonucleoside N3′→O5′ phosphoramidothioate)s (NPS-) and chimeric NPS/PO- and NPS/PS-oligomers, 3′-N-(2-thio-1,3,2-oxathiaphospholane) derivatives of 5′-O-DMT-3′-amino-2′,3′-dideoxy-ribonucleosides (NOTP-N), that bear a 4,4-unsubstituted, 4,4-dimethyl, or 4,4-pentamethylene substituted oxathiaphospholane rings, were synthesized. The four monomers 4A, 5C, 5T, and 6G were chromatographically separated into P-diastereomers, which were used in synthesis of dinucleotides NNPSN′. One of the intermediate compounds, i.e., DMTdGiBuNPSTOAc, was converted into the S-Me derivative, which spontaneously crystallized. Analysis of the relevant diffraction data and the results of stereoselective HINT1 catalyzed hydrolysis of several P-diastereomerically pure NNPSN′ provided an evidence that the earlier NMR based assignment of absolute configuration of the P-atom in DMTTNPSMeTOAc was incorrect. Mechanistic studies revealed that the relatively low repetitive yield observed during the synthesis of uniformly modified NPS-oligos is caused by a conformational ‘clash’ between adopting a C3′-endo conformation the 5′-end nucleoside (3′-amino-2′,3′-dideoxy-, but also 2′-OMe-, or LNA-nucleoside) of a growing oligomer and the incoming NOTP-N monomer. This effect is less severe if the 5-end nucleoside (e.g. a DNA unit) adopts a C2′-endo conformation. Also, an NPS-core can be effectively elongated using the phosphoramidite approach giving rise to chimeric NPS/PO-oligomers.Experimental section
Analytical equipment
1H NMR and 31P NMR spectra were recorded using Bruker AV-200 (200 MHz for 1H) or DRX-500 (500.13 MHz for 1H, 125.75 MHz for 13C) instruments, with TMS or 85% H3PO4 used as external standards. High-resolution mass spectra (HRMS) were recorded using a Synapt G2 Si mass spectrometer (Waters) equipped with an ESI source and a quadrupole-time-of-flight mass analyzer. The measurements were performed in negative or positive ion modes, with the capillary and sampling cone voltage set to 2.7 kV and 20 V, respectively. The source temperature was 110 °C. To ensure satisfactory accuracy, data were collected in a centroid mode and the readings were corrected during acquisition using leucine enkephalin as an external reference (Lock-SprayTM), which generated the reference ions at m/z 554.2615 Da ([M − H]−) in the negative ESI mode and at m/z 556.2771 Da ([M + H]+) in a positive ESI mode. The data sets were processed using the MassLynx 4.1 software (Waters). The FAB-MS spectra (13 keV, Cs+) were recorded on a Finnigan MAT 95 spectrometer, in positive and negative ion modes. MALDI-TOF MS analyses of oligonucleotides were performed using a Voyager-Elite instrument (PerSeptive Biosystems Inc., Framingham, MA) operating in the reflector mode with the detection of negative ions. All UV absorption measurements were carried out in a 1 cm path-length cell, using a double beam spectrophotometer (CINTRA 10e, GBC, Dandenong, Australia), equipped with a silicon photo-diode detector.Deprotected dinucleoside (N3′→O5′) phosphoramidothioates were isolated using a binary Varian HPLC system, consisting of two PrepStar 218 pumps and a ProStar 325 UV/VIS detector set at 260 nm. A reverse phase HPLC column (PRP-1, C18, 7 μm, 305 × 7 mm, Hamilton, Reno, NV) was eluted with a gradient of CH3CN (1% min−1) in 0.1 M TEAB (pH 7.3) at a 2.5 mL min−1 flow rate.
Analytical RP HPLC runs were performed using a Kinetex® 5 μm column 100 Å (4.6 × 250 mm, Phenomenex) at a 1 mL min−1 flow rate, buffer A, 0.05 M TEAB pH 7.5; buffer B, 40% CH3CN in 0.05 M TEAB; a gradient 0 to 40% B over 30 min.
X-ray data were collected on a Bruker APEX III D8 Venture dual microsource system using phi and omega scans with graphite monochromatic Cu Mo Kα (λ = 1.54178 Å) radiation.
Materials
Silica gel chromatography media were supplied by MERCK. TLC silica gel 60 plates were used for routine analyses, while HPTLC silica gel 60 plates were used for the assessment of the chromatographic separability of P-diastereomers of the oxathiaphospholane derivatives; all plates contained a UV F254 indicator. Silica gel 60, 200–300 mesh, was used for routine open column chromatographic purification. Protected 3′-amino-2′,3′-dideoxy-ribonucleosides were purchased from Carbosynth Limited (Compton, United Kingdom) or Pharma Waldhof (Germany). 2-Chloro-1,3,2-oxathiaphospholane and its 4,4-substituted analogs were prepared according to the published methods.4b Acetonitrile (HPLC grade) used in the syntheses of oligonucleotides was dried over 3 Å molecular sieves until the residual moisture content dropped below 10 ppm (by Karl-Fischer coulometry).Phosphitylation/sulfurization of 5′-O-DMT-nucleobase-protected 3′-amino-2′,3′-dideoxy-ribonucleosides with 2-chloro-1,3,2-oxathiaphospholane or its 4,4-substituted analogs – a general procedure
To a suspension of 5′-O-DMT-nucleobase-protected 3′-amino-2′,3′-dideoxy-nucleosides (1 mmol, the 3′-amino analogs of dABz, dGiBu, T, or dCBz) and elemental sulfur (2 mmol) in dry pyridine (4 mL), appropriate 2-chloro-1,3,2-oxathiaphospholane compound (R = H or Me, or R,R = −(CH2)5–, 1.2 mmol)4b was added dropwise using a gas-tight Hamilton syringe. The mixture was stirred at room temperature until the nucleoside disappeared (ca. 3 h, monitored by TLC). The solvent was evaporated and the residue was dissolved in acetonitrile (5 mL). Excess sulfur was filtered off and the filtrate was condensed in vacuo. The product (a mixture of P-diastereoisomers) was isolated by “flash” silica gel chromatography (230–400 mesh) using a column eluted with a linear 0→3% gradient of methanol in chloroform containing 0.1% pyridine. The mixtures of P-diastereomers of 4–6 were isolated in good yield (49–88%).Separation of the P-diastereomers of NOTP-N 4–6
A solution of the appropriate monomer 4–6 in 1.0 mL of the eluent specified in Table 1 was applied to a silica gel column (200 g, 75 × 2 cm). The column was eluted with 300 mL of the eluent and fractions at 10–12 mL were collected and analyzed on HP TLC plates. Yields and 31P NMR chemical shifts of the isolated P-epimers are given in Table 1.“In solution” synthesis of DMTdGiBuNPSMeTOAc (10)
To a solution of 5′-O-DMT-N2-isobutyryl-3′-amino-2′,3′-dideoxy-guanosine-3′-N-(2-thio-4,4-pentamethylene-1,3,2-oxathiaphospholane) (6Gf, 0.844 g, 1 mmol) in anhydrous acetonitrile (5 mL), a solution of 3′-O-acetyl-thymidine (0.284 g, 1 mmol) and DBU (0.182 g, 1.2 mmol) in anhydrous acetonitrile (3 mL) was added. After 3 h, the reaction mixture was concentrated under reduced pressure. The residue was applied on a silica gel column, which was eluted with a linear 0→30% gradient of methanol in chloroform with 1% of pyridine. The product was isolated in 83% yield (31P NMR (CDCl3): δ 58.53 ppm; FAB MS: m/z 999.4 (M−1)−). To its solution in a mixture of anhydrous acetonitrile (4 mL) and pyridine (1 mL), N,N-diisopropylethylamine (646 mg, 5 mmol) was added, followed by methyl iodide (710 mg, 5 mmol). The mixture was kept at room temperature overnight and concentrated under reduced pressure. The residue was applied on a silica gel column, which was eluted with a linear 0→10% gradient of methanol in chloroform containing 0.2% of pyridine. The triester was obtained in 63% yield (31P NMR (CDCl3): δ 36.06 ppm; ESI MS: m/z 1013.32, (M−1)−). It crystallized at room temperature from a 50:1 (v/v) CHCl3/MeOH mixture containing 4% of pyridine.
Crystallographic analysis of compound 10
Data sets were corrected for Lorentz and polarization effects as well as absorption. The criterion for observed reflections is I > 2σ(I). Lattice parameters were determined from least-squares analysis and reflection data. Empirical absorption corrections were applied using SADABS.41 structures were solved by direct methods and refined by full-matrix least-squares analysis on F2 using X-SEED42 equipped with SHELXT.43 All non-hydrogen atoms were refined anisotropically by full-matrix least-squares on F2 using the SHELXL44 program. H atoms attached to oxygens were located in difference Fourier maps and refined isotropically with independent O–H distances. The remaining H atoms were included in idealized geometric positions with Uiso = 1.2Ueq of the atom to which they were attached (Uiso = 1.5Ueq for methyl groups). Molecular configurations were compared to estimated Flack parameters.45 The acetate group and one of the methanol solvate molecules exhibit two-part disorder with occupancies refined to 64:36 and 53:47, respectively. One of the methoxy groups exhibits three-part disorder with occupancies refined to 40:37:23. These disordered groups were refined with a mixture of restraints to approximate idealized geometries.
HINT1 catalyzed cleavage of the NNPSN′ 11–17
The human Hint1 (HINT1) protein was expressed from the plasmid pSGA02 (ref. 46) in an E. coli strain BL 21* and was purified using AMP-agarose (Sigma-Aldrich) affinity chromatography according to the published procedure.27 The homogenous enzyme preparations were dialyzed against 20 mM Tris and 150 mM NaCl buffer (pH 7.5), and the resultant solutions were concentrated to a protein concentration 10 mg mL−1 and stored at −80 °C.For the hydrolysis of 11–17, to the 50 μM solutions of the substrates prepared in 20 mM HEPES-Na, 0.5 mM MgCl2 buffer (pH 7.2) HINT1 (1–13 μg, with the intention to provide rate of hydrolysis in pmol min−1 μg−1 protein) was added and the reaction mixtures (of total volume 20 μl) were incubated for 30–120 min at 37 °C. Then, the reaction mixtures were quenched by cooling on ice and analyzed by RP HPLC on a Kinetex column (5 μm C18, 100 Å, 250 × 4.6 mm; Phenomenex) with mobile phases A: 0.05 M TEAB pH 7.5; and B: 40% CH3CN in 0.05 M TEAB delivered in a gradient from 0% to 17% B over 15 min, at a flow rate of 1 mL min−1. Quantification was performed by integration of peaks of the substrate and products (including desulfured phosphate species) taking into account the number of chromophores in them. As the reference compounds appropriate nucleoside 5′-O-phosphorothioates and 5′-O-phosphates (dAMPS/dAMP, dGMPS/dGMP, TMPS/TMP) and 3′-amino-2,3′-dideoxy-nucleosides were used. Each experiment was performed in at least triplicate.
Introduction of N3′→O5′ phosphoramidothioate units during solid-phase synthesis of oligonucleotides
Syntheses were carried out manually using CPG-support, to which a 5′-O-DMT-nucleoside unit (1 μmol loaded) was attached with the succinyl-sarcosinyl type linker. A single cycle of chain elongation consisted of the following steps: (1) detritylation [3% dichloroacetic acid in methylene chloride (5 mL); 120 s]; (2) wash [acetonitrile (5 mL), methylene chloride (5 mL)] + drying; (3) coupling [a solution of NOTP-N (20 μmol) and DBU (50 μmol) (+50 μmol of NEt3 for NOTP-T) in 0.15 mL of acetonitrile, freshly premixed; 900 s]; (4) wash [acetonitrile (5 mL), methylene chloride (5 mL)] + drying; (6) capping, [acetic anhydride/DMAP/2,6-lutidine/tetrahydrofuran (0.15 mL), 120 s]; (7) wash [acetonitrile (5 mL), methylene chloride (5 mL)] + drying.The coupling efficiency was controlled by measurement of the DMT+ absorption at 504 nm. The cleavage from the support and nucleobase deprotection were performed with 30% NH4OHaq at 55 °C for 8 h. Because post-synthetic detritylation with aqueous acetic acid would destroy the oligomer, after the oligonucleotide chain assembly was completed the “on-instrument” detritylation step was executed. Alternatively, after RP HPLC purification of DMT-tagged oligomers, the DMT group was removed with 10% aqueous solution of dichloroacetic acid for 10 minutes. All oligomers were purified by RP HPLC (a DMT-OFF procedure) and MALDI-TOF MS gave satisfactory results.
Molecular modeling
All calculations have been performed using a Gaussian 16 (G16) package.31 In the calculations aimed at comparison of (SP)-5′-O-DMT-N6-benzoyl-2′-deoxyadenosine-3′-O-(2-thio-4,4-pentamethylene-1,3,2-oxathiaphospholane) and (RP)-5′-O-DMT-N6-benzoyl-3′-amino-2′,3′dideoxy-adenosine-3′-N-(2-thio-4,4-pentamethylene-1,3,2-oxathiaphospholane) (presented in Fig. 6), convergence to 10−8 in the energy and to 10−6 in the density matrix was used. An ultrafine grid with 75 radial shells and 302 angular points was employed. The geometry was optimized without any symmetry restrictions. The geometries of the compounds were generated de novo using multiple starting conformations and minimized using the polarizable continuum model (PCM) of the solvent. In two experiments acetonitrile and chloroform were set as the solvent with the standard parameter of dielectric constant ε of 35.7 and 4.7, respectively.47 Optimizations were performed using the hybrid Hartree–Fock/Density Functional Theory (HF/DFT) method PBE0 (named also as PBE1PBE)48 and the 6–311++G** basis set.49 Each stationary point was characterized by calculating the harmonic vibration frequencies in order to verify that they have no imaginary frequency. Only the lowest energy structures were taken for the further evaluation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr Wojciech J. Stec and Dr Sergei Gryaznov for encouraging and helpful discussions which prompted us to launch the project. This work was financially supported by statutory funds of Centre of Molecular and Macromolecular Studies, Polish Academy of Sciences, Łódź, Poland, and by National Centre of Science, Poland, grant UMO-2015/19/B/ST5/03116 to P. G. The support of X-ray instrumentation by the NSF-MRI grant CHE1827313 (to K. A. W.) is gratefully acknowledged. The computational resources were partially provided by the Polish Infrastructure for Supporting Computational Science in the European Research Space (PL-GRID).Notes and references
- P. C. Zamecnik and M. L. Stephenson, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 280 CrossRef CAS; J. Kurreck, Eur. J. Biochem., 2003, 270, 1628 CrossRef; R. Kole, A. R. Krainer and S. Altman, Nat. Rev. Drug Discovery, 2012,, 11, 12 Search PubMed.
- (a) F. Eckstein, Antisense Nucleic Acid Drug Dev., 2000, 10, 117 CrossRef CAS; (b) P. Guga and M. Koziołkiewicz, Chem. Biodiversity, 2011, 8, 1642 CrossRef CAS; (c) F. Eckstein, Nucleic Acid Ther., 2014, 24, 374 CrossRef CAS; (d) D. E. Volk and G. L. R. Lokesh, Biomedicines, 2017, 5, 41 CrossRef; (e) X. Shen and D. R. Corey, Nucleic Acids Res., 2018, 46, 1584 CrossRef CAS.
- A. Wilk and W. J. Stec, Nucleic Acids Res., 1995, 23, 530 CrossRef CAS.
- (a) W. J. Stec, B. Karwowski, M. Boczkowska, P. Guga, M. Koziołkiewicz, M. Sochacki, M. Wieczorek and J. Błaszczyk, J. Am. Chem. Soc., 1998, 120, 7156 CrossRef CAS; (b) P. Guga and W. J. Stec, in Current Protocols in Nucleic Acid Chemistry, ed. S. L. Beaucage, D. E. Bergstrom, G. D. Glick and R. A. Jones, John Wiley and Sons, Hoboken, N.J., 2003, p. 4.17.1 Search PubMed.
- A. Wilk, A. Grajkowski, L. R. Phillips and S. L. Beaucage, J. Am. Chem. Soc., 2000, 122, 2149 CrossRef CAS; N. Oka, M. Yamamoto, T. Sato and T. Wada, J. Am. Chem. Soc., 2008, 130, 16031 CrossRef; K. W. Knouse, J. N. deGruyter, M. A. Schmidt, B. Zheng, J. C. Vantourout, C. Kingston, S. E. Mercer, I. M. Mcdonald, R. E. Olson, Y. Zhu, C. Hang, J. Zhu, C. Yuan, Q. Wang, P. Park, M. D. Eastgate and P. S. Baran, Science, 2018, 361, 1234 CrossRef; N. Iwamoto, D. C. D. Butler, N. Svrzikapa, S. Mohapatra, I. Zlatev, D. W. Y. Sah, Meena, S. M. Standley, G. Lu, L. H. Apponi, M. Frank-Kamenetsky, J. J. Zhang, C. Vargeese and G. L. Verdine, Nat. Biotechnol., 2017, 35, 845 CrossRef.
- P. Guga, M. Boczkowska, M. Janicka, A. Maciaszek, S. Kuberski and W. J. Stec, Biophys. J., 2007, 92, 2507 CrossRef CAS; P. Guga, M. Janicka, A. Maciaszek, B. Rębowska and G. Nowak, Biophys. J., 2007, 93, 3567 CrossRef; A. Maciaszek, A. Krakowiak, M. Janicka, A. Tomaszewska-Antczak, M. Sobczak, B. Mikołajczyk and P. Guga, Org. Biomol. Chem., 2015, 13, 2375 RSC.
- M. Boczkowska, P. Guga and W. J. Stec, Biochemistry, 2002, 41, 12483 CrossRef CAS.
- M. Boczkowska, P. Guga, B. Karwowski and A. Maciaszek, Biochemistry, 2000, 39, 11057 CrossRef CAS.
- P. A. Frey and R. D. Sammons, Science, 1985, 228, 541 CrossRef CAS.
- L. Benimetskaya, J. L. Tonkinson, M. Koziołkiewicz, B. Karwowski, P. Guga, R. Zelser, W. J. Stec and C. A. Stein, Nucleic Acids Res., 1995, 23, 4239 CrossRef CAS.
- A. Krieg, P. Guga and W. J. Stec, Oligonucleotides, 2003, 13, 491 CrossRef CAS.
- T. Inagawa, H. Nakashima, B. Karwowski, P. Guga, W. J. Stec, H. Takeuchi and H. Takaku, FEBS Lett., 2002, 528, 48 CrossRef CAS.
- (a) S. Gryaznov, T. Skorski, C. Cucco, M. Nieborowska-Skorska, C. Y. Chiu, D. Lloyd, J. K. Chen, M. Koziolkiewicz and B. Calabretta, Nucleic Acids Res., 1996, 24, 1508 CrossRef CAS; (b) S. M. Gryaznov, Biochim. Biophys. Acta, 1999, 1489, 131 CrossRef CAS; (c) T. Skorski, D. Perrotti, M. Nieborowska-Skorska, S. M. Gryaznov and B. Calabretta, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 3966 CrossRef CAS.
- S. M. Gryaznov and J. K. Chen, J. Am. Chem. Soc., 1994, 116, 3143 CrossRef CAS; J. P. Schrum, A. Ricardo, M. Krishnamurthy, J. C. Blain and J. W. Szostak, J. Am. Chem. Soc., 2009, 131, 14560 CrossRef; E. C. Izgu, S. S. Ohab and J. W. Szostak, Chem. Commun., 2016, 52, 3684 RSC.
- S. M. Gryaznov, D. H. Lloyd, J.-K. Chen, R. G. Schultz, A. DeDionisio, L. Ratmeyer and W. D. Wilson, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 5798 CrossRef CAS.
- V. Tereshko, S. M. Gryaznov and M. Egli, J. Am. Chem. Soc., 1998, 120, 269 CrossRef CAS.
- R. Pruzan, K. Pongracz, K. Gietzen, G. Wallweber and S. M. Gryaznov, Nucleic Acids Res., 2002, 30, 559 CrossRef CAS.
- L. Kers, J. Stawiński and A. Kraszewski, Tetrahedron, 1999, 55, 11579 CrossRef CAS; K. Pongracz and S. M. Gryaznov, Tetrahedron Lett., 1999, 40, 7661 CrossRef; A. Asai, Y. Oshima, Y. Yamamoto, T. Uochi, H. Kusaka, S. Akinaga, Y. Yamashita, K. Pongracz, R. Pruzan, E. Wunder, M. Piatyszek, S. Li, A. C. Chin, C. B. Harley and S. Gryaznov, Cancer Res., 2003, 63, 3931 Search PubMed.
- W. J. Stec, A. Grajkowski, A. Kobylańska, B. Karwowski, M. Koziołkiewicz, K. Misiura, A. Okruszek, A. Wilk, P. Guga and M. Boczkowska, J. Am. Chem. Soc., 1995, 117, 12019 CrossRef CAS.
- B. Uznański, A. Grajkowski, B. Krzyżanowska, A. Kaźmierkowska, W. J. Stec, M. W. Wieczorek and J. Błaszczyk, J. Am. Chem. Soc., 1992, 114, 10197 CrossRef.
- A. Krakowiak, R. Kaczmarek, J. Baraniak, M. Wieczorek and W. J. Stec, Chem. Commun., 2007, 2163 RSC.
- R. Kaczmarek, S. Kaźmierski, T. Pawlak, E. Radzikowska and J. Baraniak, Tetrahedron, 2016, 72, 803 CrossRef CAS.
- F. Eckstein, P. M. J. Burgers and D. H. Hunneman, J. Biol. Chem., 1979, 254, 7476 Search PubMed; F. Eckstein, P. M. J. Burgers, B. K. Sathyanarayana and W. Saenger, Eur. J. Biochem., 1979, 100, 585 CrossRef; S. J. Benkovic and F. R. Bryant, Biochemistry, 1979, 18, 2825 CrossRef; F. Eckstein, B. V. L. Potter and B. Connolly, Biochemistry, 1983, 22, 1369 CrossRef.
- B. Karwowski, A. Okruszek, J. Wengel and W. J. Stec, Bioorg. Med. Chem. Lett., 2001, 11, 1001 CrossRef CAS.
- K. Jastrzębska, A. Maciaszek, R. Dolot, G. Bujacz and P. Guga, Org. Biomol. Chem., 2015, 13, 10032 RSC.
- J. Martin, M. V. St-Pierre and J. F. Dufour, Biochim. Biophys. Acta, 2011, 1807, 626 CrossRef CAS.
- P. Bieganowski, P. N. Garrison, S. C. Hodawadekar, G. Faye, L. D. Barnes and C. Brenner, J. Biol. Chem., 2002, 277, 10852 CrossRef CAS.
- M. Ozga, R. Dolot, M. Janicka, R. Kaczmarek and A. Krakowiak, J. Biol. Chem., 2010, 285, 40809 CrossRef CAS.
- T. Brown, C. E. Pritchard, G. Turner and S. A. Salisbury, J. Chem. Soc. Chem. Commun., 1989, 891 RSC.
- J. Baraniak, D. Korczyński and W. J. Stec, J. Org. Chem., 1999, 64, 4533 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox. Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- J. P. Marino, H. Schwalbe and Ch. Griesinger, Acc. Chem. Res., 1999, 32, 614 CrossRef CAS.
- L. P. Conway, S. Mikkola, A. C. O'Donoghue and D. R. W. Hodgson, Org. Biomol. Chem., 2016, 14, 7361 RSC.
- M. Ora, M. Murtola, S. Ahoa and M. Oivanen, Org. Biomol. Chem., 2004, 2, 593 RSC.
- T. Wada, A. Mochizuki, Y. Sato and M. Sekine, Tetrahedron Lett., 1998, 39, 7123 CrossRef CAS.
- Y. Hayakawa, M. Uchiyama and R. Noyori, Tetrahedron Lett., 1986, 27, 4191 CrossRef CAS.
- K. Ogilvie and M. J. Nemer, Tetrahedron Lett., 1981, 22, 2531 CrossRef CAS.
- B. Nawrot, B. Rębowska, K. Cieślińska and W. J. Stec, Tetrahedron Lett., 2005, 46, 6641 CrossRef CAS.
- G. M. Salamończyk, M. Kuźnikowski and E. Poniatowska, Tetrahedron Lett., 2002, 43, 1747 CrossRef.
- E. Radzikowska and J. Baraniak, Org. Biomol. Chem., 2015, 13, 269 RSC.
- G. M. Sheldrick, SADABS (2016/2)—Program for Area Detector Absorption Corrections, University of Göttingen, Göttingen, Germany, 2016 Search PubMed.
- L. J. Barbour, J. Supramol. Chem., 2001, 1, 189 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2015, 71, 3 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
- H. D. Flack, Acta Crystallogr., 1983, 39, 876 CrossRef.
- S. Ghosh and J. M. Lowenstein, Gene, 1996, 176, 249 CrossRef CAS.
- S. Miertus, E. Scrocco and J. Tomasi, J. Chem. Phys., 1981, 55, 117 Search PubMed; J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef CAS.
- Z. Gunnur Dikmen, T. Ozgurtas, S. M. Gryzanov and B.-S. Herbert, Biochim. Biophys. Acta, 2009, 1792, 240 CrossRef CAS; B.-S. Herbert, G. C. Gellert, A. Hochreiter, K. Pongracz, W. E. Wright, D. Zielinska, A. C. Chin, C. B. Harley, J. W. Shay and S. M. Gryaznov, Oncogene, 2005, 24, 5262 CrossRef.
- E. H. Blackburn, Mol. Cancer Res., 2005, 3, 477 CrossRef CAS; J. W. Shay and W. E. Wright, Nat. Rev., 2006, 5, 577 Search PubMed; J. M. Y. Wong and K. Collins, Lancet, 2003, 362, 983 CrossRef.
Footnotes |
| † Dedicated to Prof. Dr Wojciech J. Stec on the occasion of his 80th birthday. |
| ‡ Electronic supplementary information (ESI) available: HR MS, 31P NMR, 1H NMR, and 13C NMR spectra of separated P-epimers of 4–6; MS and 31P NMR data for unresolved monomers 4–6; 31P NMR, 1H NMR, 13C NMR spectra, details of crystallographic analysis, and crystal structure of DMTdGiBuNPSMeTOAc obtained from 6Gf; selected data for NPS- and chimeric NPS/PO oligomers. CCDC 1939477. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra04987e |
| § Noteworthy, an NPS-oligomer bearing a lipid tag (GRN163L50) was found to be a potent inhibitor of human telomerase,51 which is an enzyme highly active in ∼85% of known human tumor cells, whereas in normal cells its activity is marginal. |
| ¶ Unpublished results. The rank was determined during work on ref. 25. |
| || AMPS-Trp-NH2 was concomitantly obtained from AMPS-Trp-OMe during ammonolysis of the protecting benzoyl groups in the adenosine moiety. |
| ** It must be pointed out that all the supports used were loaded with a given nucleoside to a similar extent (29–33 μmol g−1), and before the first condensation the support was extensively capped (Ac2O/DMAP/2,6-lutidine/tetrahydrofuran) to exclude unspecific coupling. |
| This journal is © The Royal Society of Chemistry 2020 |