
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Emerging investigator series: a holistic approach to multicomponent EXAFS: Sr and Cs complexation in clayey soils†
Pieter
Bots
 *a,
M. Josick
Comarmond
b,
Timothy E.
Payne
b,
Katharina
Gückel‡
c,
Rebecca J.
Lunn
a,
Luca
Rizzo
a,
Alexandra E. P.
Schellenger
a and
Joanna C.
Renshaw
a
*a,
M. Josick
Comarmond
b,
Timothy E.
Payne
b,
Katharina
Gückel‡
c,
Rebecca J.
Lunn
a,
Luca
Rizzo
a,
Alexandra E. P.
Schellenger
a and
Joanna C.
Renshaw
a
aDepartment of Civil and Environmental Engineering, University of Strathclyde, Glasgow, G1 1XJ, UK. E-mail: pieter.bots@strath.ac.uk
bAustralian Nuclear Science and Technology Organisation, Lucas Heights, NSW 2234, Australia
cHelmholtz-Zentrum Dresden-Rossendorf, Institute of Resource Ecology, Bautzner Landstraße 400, 01328 Dresden, Germany
First published on 21st June 2021
Abstract
Strontium and caesium are fission products of concern at many nuclear legacy sites and Cs is additionally a significant consideration at sites in the aftermath of nuclear accidents and incidents. Such sites require long-term management to minimize the risk of such contaminants to the environment and the public. Understanding the geochemical speciation of Sr and Cs in situ in the soils and groundwater is essential to develop engineered management strategies. Here we developed and utilized a comprehensive approach to fitting the EXAFS of Sr and Cs adsorption to single mineral phases and a composite clayey soil. First, a shell-by-shell fitting strategy enabled us to determine that Sr surface complexes involve the formation of bidentate edge sharing complexes with anatase and illite-smectite, and form at the silicon vacancy sites at the kaolinite basal surfaces. Cs surface complexes form at the silicon vacancy sites at the illite-smectite and kaolinite basal surfaces. Second, using a subsequent holistic approach we determined the predominance of these complexes within a composite clayey soil. Sr was dominated by complexation with illite-smectite (72–76%) and to a lesser extent with kaolinite (25–30%) with negligible complexation with anatase, while Cs complexed roughly equally to both illite-smectite and kaolinite. The presented approach to fitting EXAFS spectra will strengthen predictive modelling on the behaviour of elements of interest. For example, the details on Sr and Cs speciation will enable predictive modelling to characterise their long-term behaviour and the design and validation of evidence-based engineering options for long-term management of nuclear legacy sites.
Environmental significanceLegacy sites contaminated with radioactive elements exist worldwide and their long-term management and remediation are subjects of public concern. In order to develop appropriate evidence-based engineering strategies, it is also essential to understand and predict the geochemical state(s) of contaminants at such sites. In this study we developed and utilized a comprehensive strategy examining (X-ray absorption) spectroscopy data on the geochemistry of strontium and caesium, two radioactive fission products of concern at many radioactive legacy sites. The information on the geochemistry of strontium and caesium can be utilized to develop numerical models on the long-term behaviour of strontium and caesium and to inform environmental engineering strategies to mitigate their risks and impacts. |
Introduction
Following more than 60 years of nuclear power generation, and a longer history of nuclear research, many legacy waste sites with stored radioactive wastes exist throughout the world. Such legacy sites include Sellafield (Cumbria, UK),1 Savannah River (NC, USA),2 and the Little Forest Legacy Site (NSW, Australia).3,4 The location for such sites was often chosen based on the local geology and soil composition. For example, the Little Forest Legacy Site was chosen based on the clayey nature of the soils and rocks present (∼50% kaolinite and illite-smectite), limiting water movement and migration of radioactive contaminants.3,4 Despite the clay-rich environments, radioactive contaminants (including fission products such as strontium and caesium, and actinides) have been detected in sediments, groundwater, surface runoff and vegetation surrounding such legacy sites.1,2,4–7 Furthermore, nuclear incidents such as the Fukushima Daiichi nuclear disaster have resulted in fission products, including caesium, entering the environment.8–13Transport of fission products like caesium (Cs) and strontium (Sr) through the environment is heavily impacted by the soil mineralogy (including clay minerals and metal oxides in the soils at the Little Forest Legacy Site3,4,7), their surface speciation and complexation with organic matter.10,11,14–19 Previous studies have identified multiple surface complexes for Sr and Cs complexed with mineral phases present within clayey soils using X-ray absorption spectroscopy and molecular dynamics techniques. Sr has been shown to complex with kaolinite through the formation of hydrated (outer-sphere) surface complexes at elevated concentration and inner-sphere complexes at lower concentrations.20,21 A wider range of surface complexes have been identified for Sr complexed with 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 clay minerals. This includes outer-sphere15,22–24 and inner-sphere surface complexes,22,25 and incorporation into the interlayer of illite15 and smectite.24,26 Sr has been identified to complex with titanium dioxide (common as an accessory mineral in clayey soils3,4) through forming inner-sphere surface complexes with anatase and rutile.27,28 Cs complexes strongly with clay minerals such as illite and smectite;29,30 however, identifying Cs complexation mechanisms with clay minerals is more challenging compared to Sr due to the limitations with respect to Cs X-ray absorption spectroscopy.19,31 Various studies have proposed the formation of outer-sphere surface complexes with clay minerals24,32–34 at relatively high concentrations. Recent molecular dynamics studies have identified Cs complexation with illite and kaolinite through inner-sphere surface complexes on the Si vacancies on the basal surfaces.35,36 This has now been supported through X-ray absorption spectroscopy on a clayey soil.19 Conversely, at trace concentrations Cs has been shown to incorporate into the interlayers of 2:1 clay minerals,24,25,31,37,38 and complexes to the frayed edge sites,29,30,39 dependent on the crystallinity of such clay minerals.39
:1 clay minerals. This includes outer-sphere15,22–24 and inner-sphere surface complexes,22,25 and incorporation into the interlayer of illite15 and smectite.24,26 Sr has been identified to complex with titanium dioxide (common as an accessory mineral in clayey soils3,4) through forming inner-sphere surface complexes with anatase and rutile.27,28 Cs complexes strongly with clay minerals such as illite and smectite;29,30 however, identifying Cs complexation mechanisms with clay minerals is more challenging compared to Sr due to the limitations with respect to Cs X-ray absorption spectroscopy.19,31 Various studies have proposed the formation of outer-sphere surface complexes with clay minerals24,32–34 at relatively high concentrations. Recent molecular dynamics studies have identified Cs complexation with illite and kaolinite through inner-sphere surface complexes on the Si vacancies on the basal surfaces.35,36 This has now been supported through X-ray absorption spectroscopy on a clayey soil.19 Conversely, at trace concentrations Cs has been shown to incorporate into the interlayers of 2:1 clay minerals,24,25,31,37,38 and complexes to the frayed edge sites,29,30,39 dependent on the crystallinity of such clay minerals.39
The above described complexation with mineral phases can inhibit the uptake of metals and contaminants by fungi and vegetation, impacting on their toxicity,40–43 and/or retain metals within their environment.43 Any engineering strategy designed to limit the environmental impacts of such fission products will impact on their surface speciation and consequentially on their mobility and toxicity within the environment.19 Thus, in order to develop evidence-based strategies to ensure long-term safety of contaminated sites (such as the Little Forest Legacy Site), we also need to develop a thorough understanding of the geochemical behaviour and surface speciation of Sr and Cs.19,44,45 Experimental and analytical information to fully understand the geochemical speciation of Sr and Cs within clayey soils is lacking. For example, no spectroscopic evidence exists for Sr complexation with anatase and Cs complexation with either illite or kaolinite. Furthermore, to comprehensively determine the surface complexes in multicomponent systems such as clayey soils (with multiple mineral phases competing for surface complexation) is challenging because spectroscopic techniques only provide an average coordination environment15,23 and interpreting such spectroscopic information is non-trivial.
Here, we utilised a comprehensive/dual approach to fitting the extended X-ray absorption fine structure (EXAFS) spectra on Sr and Cs surface complexation. Through a shell-by-shell approach to fitting the EXAFS we first determined the surface speciation of Sr and Cs with illite-smectite, kaolinite and anatase. Secondly, through a holistic fitting strategy of the Sr and Cs EXAFS spectra, we provide detailed information on the speciation of Sr and Cs in a clayey soil beyond an average coordination environment.
Experimental
All chemicals (NaCl, NaOH, HCl, SrCl2·6H2O and CsCl) used during the adsorption experiments were analytical grade reagents obtained from Sigma Aldrich, except for the Sr-85 and Cs-137 radiotracer stock solutions which were obtained from Eckert & Ziegler. A selection of clay, shaley clay, and shale samples were obtained from two cores (CH1a and CH30)7 from the Little Forest Legacy site. Details on these cores are described by Hankin46 and Payne et al.7 Adsorption experiments were performed on the <1 mm (CH1a) or the <2 mm (CH30) fraction of these samples. The mineralogy of these samples was assessed by XRD (X'Pert Pro diffractometer using Cu Kα1,2 radiation with a Ni filter) and subsequent quantitative Rietveld refinements using the PANanalytical analysis package High Score Pro for the CH30 sample; PANanlytical X'Pert Pro multi-purpose diffractometer using Fe filtered Co Kα radiation with quantitative analysis using Siroquant from Sietronics Ltd for the CH1a samples47 (Fig. S1†). The choice of minerals used during adsorption experiments on single mineral phases and a composite clayey soil was based on the mineralogical assessments of the clay and shale samples from the LFLS cores (goethite was chosen to represent iron oxides, anatase to represent titanium dioxides, kaolinite to represent kaolinite and dickite48–50 and illite-smectite mixed layer to represent illite, smectite and illite-smectite; Fig. S1†). These minerals were obtained from Sigma Aldrich except for the illite-smectite mixed layer (70/30 ordered), which was obtained from the Clay Mineral Society (special source clay: ISCz-1). The purity and composition of these minerals were assessed by XRD (Bruker D8 Advance with Davinci X-ray Diffractometer) and SEM microscopy (Hitachi SU-6600 Field Emission – Scanning Electron Microscopy). These preliminary analyses showed that anatase contained traces (1%) of rutile, kaolinite contained approximately 5% quartz and illite-smectite contained approximately 20% quartz, while quartz and goethite did not contain measurable trace amounts of contamination. Furthermore, no traces of Sr and Cs contamination were detected that could interfere with the subsequent adsorption experiments. These minerals were used during the adsorption experiments as received, either as single mineral adsorption experiments or mixed to simulate clayey soil with a composition comparable to the clayey soil at the Little Forest Legacy Site (45% quartz, 25% illite-smectite, 25% kaolinite, 3% goethite and 2% anatase).Sr and Cs adsorption to soil minerals and a clayey soil
Adsorption experiments were performed at pH 4–8 and a solid loading of 10 g l−1 in 10 mM NaCl. Two sets of adsorption experiments were performed on the single minerals and the composite clayey soil; at elevated concentrations of 25 ppm Sr and/or Cs (0.29 mM Sr and/or 0.19 mM Cs) and at trace/environmental concentrations of 20 Bq ml−1 Sr-85 and Cs-137 (2.7 × 10−10 mM Sr-85 and 4.6 × 10−8 mM Cs-137). A third set of adsorption experiments were performed at trace/environmental concentrations of 20 Bq ml−1 Sr-85 and Cs-137 on the samples from the LFLS cores. PHREEQC calculations showed that atmosphere CO2 had negligible impact on Sr and Cs speciation and did not induce supersaturation with respect to strontianite. Hence, no atmospheric controls were included in the experimental setup. 0.25 g of the single mineral, composite clayey soil or LFLS core sample was added to 50 ml polypropylene centrifuge tubes. Subsequently, 22 ml of 10 mM NaCl was added to the centrifuge tubes and the pH was adjusted to the desired pH values (4–8) using 10 mM NaOH or HCl. These suspensions were allowed to equilibrate on an orbital shaker (180 rpm) for at least 20 h. After this, the pH was checked and readjusted to the desired values. The equilibration and pH adjustment steps were repeated until the pH was relatively stable (e.g. within 0.1 pH units), after which the volume was corrected to 25 ml by adding small aliquots of 10 mM NaCl. These equilibrated suspensions were spiked with slightly acidified radiotracer stock solutions (5 kBq ml−1 Sr-85 and 5 kBq ml−1 Cs-137) for the experiments at trace concentrations of 20 Bq ml−1 Sr-85 and 20 Bq ml−1 Cs-137, or with slightly acidified 10000 ppm Sr and/or Cs stock solution for the experiments at 25 ppm Sr and/or Cs. Next, small aliquots of 10 mM NaOH were added to the adsorption experiments to counter acidification due to the addition of the stock solutions. These adsorption experiments were allowed to equilibrate for 48 h on an orbital shaker (180 rpm), after which the pH was measured, the suspensions were centrifuged at 8000 rpm for 20 minutes (to remove solids with diameters greater than 0.1 μm) and 10 ml aliquots were obtained using a syringe and a 0.1 μm HDPE syringe filter. These aqueous aliquots were stored at 4 °C prior to ion chromatography analyses for Sr and Cs (Metrohm CatAn Professional IC with a C6 cation exchange column and an eluent containing 4.5 mM HNO3 and 1.5 mM dipicolinic acid), or acidified with 10 μl concentrated HCl prior to analyses for Sr-85 and Cs-137 using γ-spectroscopy (ORTEC p-type high purity germanium detector (HPGe) coupled to ORTEC DSPec Pro, and ORTEC n-type reduced background HPGe detector coupled to an ORTEC DSPec Pro for the LFLS core samples), and the activity concentrations were decay corrected to the start of the adsorption experiments.
X-ray absorption spectroscopy (XAS)
To determine the coordination environment of the Sr and Cs surface complexes, X-ray absorption spectra were collected for selected solid samples from the adsorption experiments (including relevant standards: Sr2+, SrCO3 and Cs+). These samples were obtained by centrifuging the suspensions at 8000 rpm for 20 minutes and decanting the supernatant. The remaining slurry was stored in a 2 ml cryo-tube with the slurry (held in place by compressed cotton wool as a filler and to absorb excess water from the slurry) prior to analyses at beamline B18 at Diamond Light Source.51 The Cs K-edge (35985 eV) was measured using a Si(311) monochromator and the Sr K-edge (16105 eV) was measured using a Si(111) monochromator. The energy of the X-rays was calibrated using an yttrium foil (for the Sr K-edge) and an antimony foil (for the Cs K-edge). The samples were analysed for their spectra in fluorescence mode using a 36-element Ge detector, and the standards were analysed in transmission mode. The spectra were processed and analysed using the Demeter software package.52 Athena was used for data reduction, background subtraction and linear combination fitting (LCF). Artemis was used to fit the Fourier transform (in R-space) of the EXAFS spectra. For detailed characterisation of Sr and Cs species in the analysed samples, the respective spectra were fitted using two complementary fitting strategies. (1) The spectra of the samples with the same mineral phase or the clayey soil (at pH 5–8) were fitted simultaneously. Here we used a shell-by-shell approach to determine the coordination environment of the surface complexes, the respective coordination numbers of the different backscatters, and the impact of pH on the coordination environments (this strategy and respective variables are described in Tables S1 and S3†). (2) The results of the shell-by-shell approach were subsequently used in a more holistic fitting approach. In this approach all spectra from the samples at the same pH value were fitted simultaneously (examples of this strategy and respective variables are described in Tables S2 and S3†) to identify (and quantify) the individual surface complexes (Sr or Cs adsorbed to illite-smectite, kaolinite and/or anatase) represented in the spectra from the respective clayey soil samples. This enabled the quantification of the contribution of individual (clay) minerals to the complexation (and mobility) of Sr and Cs within clayey soils. During both approaches, FEFF6 (ref. 53) was used to calculate the theoretical (initial) Sr and Cs scattering paths by developing complexation models using the crystallographic information files for anatase,54 kaolinite55 and illite,56 based on previous analytical and molecular dynamics results.15,20,21,27,31,35,36
Results and discussion
Sr and Cs adsorption to a composite clayey soil and its mineral components
The results from the adsorption experiments to the single mineral phases and the composite clayey soil are summarized in Tables S4 to S7† and visualized in Fig. 1. Briefly, preliminary adsorption experiments showed that Sr and Cs were not removed from solution during the adsorption experiments using either quartz or goethite (Tables S5 to S7†). This shows that under the investigated conditions Sr and Cs did not complex to quartz and goethite; thus these mineral phases are not included in the remainder of the results and discussion. Furthermore, the results from the adsorption experiments show that Sr and Cs both complexed strongly with the tested clay minerals while Sr additionally complexed strongly with anatase. The values for Sr and Cs adsorption with the mineral phases and the composite clayey soil samples correspond well with % adsorbed and distribution coefficients (KD values) determined in recent research on Sr and Cs complexation with clay minerals, metal oxides and clayey soils.1,15,16,18,19,23,27,29,30,39,57–63 The similarities with previous results on Sr complexation with clay minerals, metal oxides and clayey soils indicate that any stable Sr present in the mineralogical samples did not significantly affect Sr surface complexation at 20 Bq ml−1 Sr-85 to the single mineral phases or the composite clayey soil.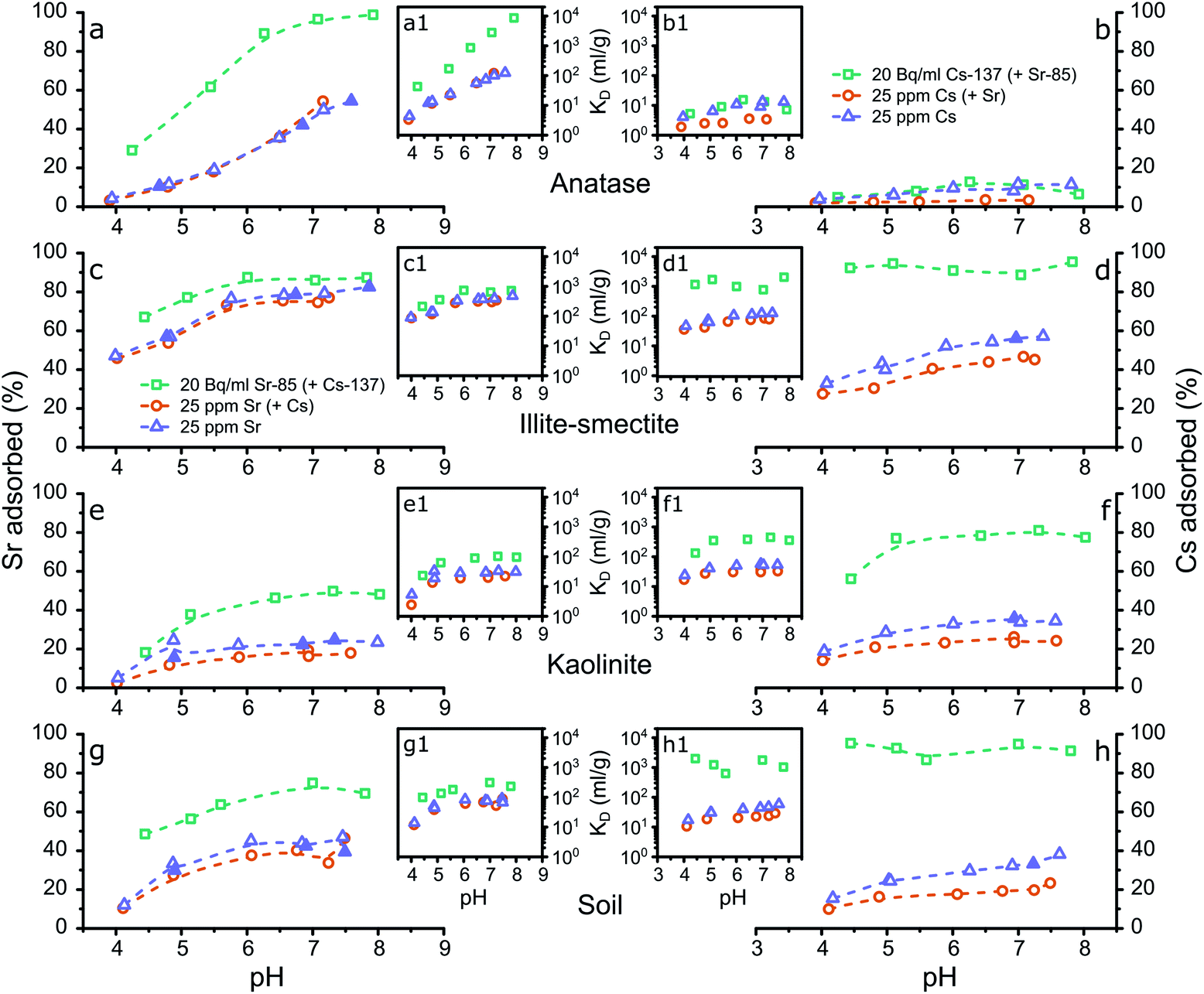 | ||
| Fig. 1 Graphical representation of the results from the aqueous analyses from the adsorption experiments with the % of strontium (a, c, e and g) or caesium (b, d, f and h) adsorbed to anatase (a and b), illite-smectite (c and d), kaolinite (e and f) and the composite clayey soil (g and h) plotted against the pH, including the respective distribution coefficients (a1–h1). The legends refer to the initial Sr or Cs concentrations (Tables S4 to S7†) and whether both Sr and Cs were present (Tables S4 and S5†) during the experimental set-up; the filled symbols represent the experiments that produced samples for XAS analyses (Table 1); the dashed lines are intended as a visual guide only. | ||
Conversely, adsorption of Sr to anatase is significant (Fig. 1a) and shows a strong dependence on the pH and on the Sr concentration in the experiments. In the adsorption experiments at elevated concentrations the KD values increase from <101 ml g−1 at pH 4, to approximately 102 ml g−1 at pH 8 (Tables S5 and S6†), and in the adsorption experiments at trace concentrations the KD values increase from <102 ml g−1 at pH 4 to approximately 104 ml g−1 at pH 8 (Fig. 1a1). These values are higher compared to the values by Ridley et al.,27 likely due to the difference in concentration used by Ridley et al. of 1–2.5 mM Sr, compared to a maximum of 25 ppm Sr (0.29 mM) in the presented experiments (Fig. 1a1, Tables S5 and S6†). This indicates that Sr complexes relatively weakly to anatase surfaces, but that the high surface area of anatase nanoparticles resulted in high KD values in the presented results.27 Finally, Cs present as a counter ion in solution did not significantly affect Sr adsorption to anatase.
Sr complexation with illite-smectite was significant, and show similar trends with varying pH values, at both trace and elevated Sr concentrations. At trace concentrations, up to ∼90% of Sr was removed from solution (Fig. 1c) with KD values of up to ∼700 ml g−1 (Fig. 1c1 and Table S4†), while at elevated concentration up to 80% of Sr was removed from solution (Fig. 1c) with KD values of up to ∼470 ml g−1 (Fig. 1c1, Tables S5 and S6†). These observations suggest that at trace and elevated concentrations the removal from solution occurs through similar mechanisms, such as the inner-sphere surface complexes identified by Fuller et al.15
Interestingly, at elevated Cs concentrations when Sr is present (as co-contaminant) a decrease of about 10% in the removal of Cs from solution by illite-smectite can be observed (Fig. 1d, Tables S5 and S7†). This suggests that Cs competes for surface complexation with Sr at elevated concentrations. This decrease in removal from solution was not observed for Sr in the presence of Cs as co-contaminant (Fig. 1c, Tables S5 and S6†), suggesting that Sr complexes more strongly with illite-smectite compared to Cs (at elevated concentrations).
Sr removal from solution follows similar trends at the trace concentration and the elevated concentration experiments. At trace concentration, from pH 4 to 8, ∼20 to 50% was removed from solution (KD: 20–100 ml g; Table S4†) and at elevated concentration from pH 4 to 8, ∼2 to 25% was removed from solution (KD: 2–30 ml g; Tables S5 and S6†). This suggests that similar mechanisms at trace and elevated concentrations induced the removal from solution, such as the formation of hydrated surface complexes on the kaolinite surfaces.20,21 In comparison to Sr complexation with illite-smectite, the KD values are lower for Sr complexation with kaolinite (Fig. 1c1 and e1, and Tables S4 to S6†) and the difference in removal of Sr from solution between the trace and elevated concentration experiments (Fig. 1c and e, and Tables S4 to S6†) is smaller for Sr complexation with illite-smectite. This indicates that Sr complexation with illite-smectite is stronger compared to kaolinite, and that at elevated concentration complexation to illite-smectite is less limited by available surface sites compared to kaolinite.
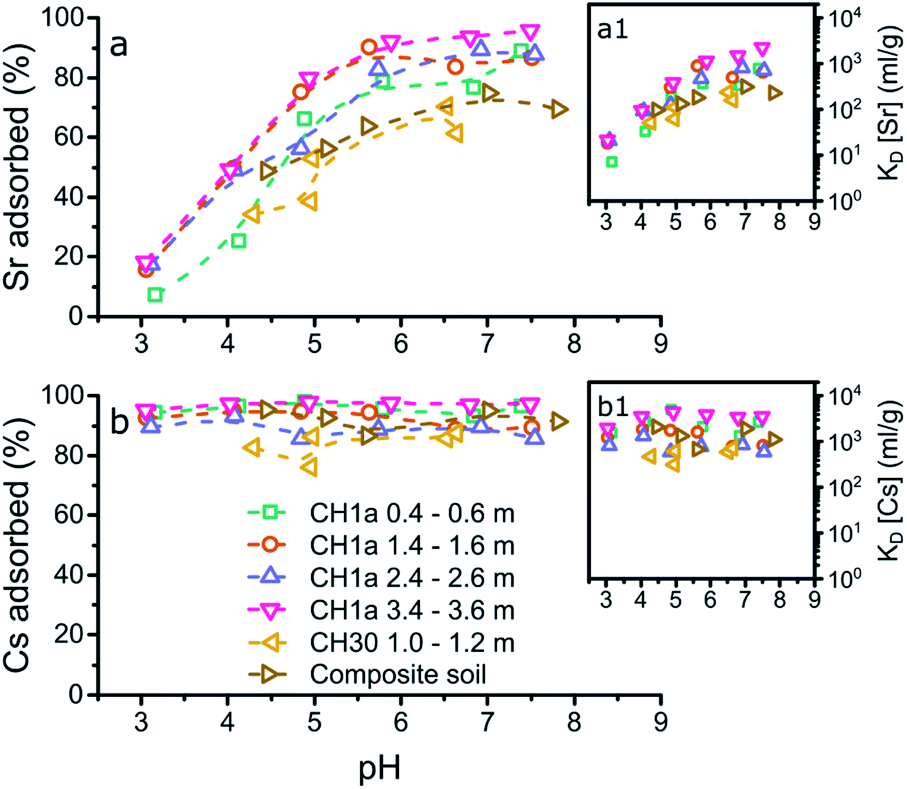 | ||
| Fig. 2 Graphical representation of the results from the aqueous analyses from the adsorption experiments to the composite clayey soil and the LFLS core samples (initial concentrations of 20 Bq ml−1 Sr-85 and Cs-137) with the % of Sr (a) or Cs (b) adsorbed plotted against pH, and the respective KD values (a1 and b1). The legend represents the location46 and depth of the LFLS core samples used in the adsorption experiments. | ||
In contrast to the adsorption of Cs to the LFLS core samples, Sr adsorption to most of the LFLS core samples was higher compared to its adsorption to the composite clayey soils (Fig. 2a) with the exception of the LFLS core sample ‘CH30 1.0–1.2 m’ where adsorption was similar to that for the composite clayey soils (Fig. 2a). The clay mineral composition of ‘CH30 1.0–1.2 m’ was most similar to the composite clayey soil sample (with ∼20% kaolinite and dickite48,50 and ∼24% illite-smectite) while the CH1a samples all had relatively higher kaolinite (28–31%) and illite-smectite, illite and smectite contents (34–46%) (Fig. S1†). This suggests that the somewhat higher adsorption in the CH1a core samples is likely due to the higher proportion of these clay minerals. This confirms our previous interpretation that the fraction of clay minerals govern the adsorption of Sr to the composite clayey soils and the LFLS core samples.
Finally, the similarities (and differences) in the adsorption of Sr and Cs to the composite clayey soil sample and the LFLS core samples (Fig. 2) highlight the applicability of using a composite clayey soil sample to mimic Sr and Cs adsorption at legacy nuclear sites such as the LFLS. Furthermore, this comparison validates the use of illite-smectite to represent illite-smectite, illite and smectite phases, and kaolinite to represent kaolinite and dickite (due to their crystallographic similarities48,50) in natural clay, shaley clay, and shale samples.
X-ray absorption spectroscopy of Sr and Cs complexation with a composite clayey soil and its mineral components
To understand the surface complexation mechanisms, and enable quantification of the contribution of the individual (clay) mineral phases to the speciation of Sr and Cs within clayey soils, we analysed a selection of samples for their X-ray absorption spectra. These samples are described in Table 1, and in Fig. 1 with filled symbols.| Sample | pHfinal | Final aqueous conc. (ppm) | Solid conc. (μg g−1) | K D (ml g−1) |
|---|---|---|---|---|
| Sr anatase | 4.66 | 22.3 | 271 | 12 |
| Sr anatase | 6.85 | 14.4 | 1047 | 73 |
| Sr anatase | 7.59 | 11.5 | 1401 | 121 |
| Sr illite-smectite | 4.77 | 10.6 | 1430 | 134 |
| Sr illite-smectite | 6.74 | 5.3 | 1962 | 371 |
| Sr illite-smectite | 7.86 | 4.5 | 2123 | 468 |
| Sr kaolinite | 4.89 | 20.8 | 386 | 19 |
| Sr kaolinite | 6.85 | 19.3 | 554 | 29 |
| Sr kaolinite | 7.33 | 19.2 | 645 | 33 |
| Sr soil | 4.90 | 17.2 | 746 | 43 |
| Sr soil | 6.90 | 14.2 | 1056 | 75 |
| Sr soil | 7.49 | 15.1 | 1012 | 67 |
| Cs illite-smectite | 6.67 | 10.9 | 1415 | 130 |
| Cs kaolinite | 6.86 | 16.1 | 901 | 56 |
| Cs soil | 7.01 | 16.7 | 847 | 50 |
Fig. 3 summarizes the X-ray absorption near edge structure (XANES) spectra for Sr adsorbed to the mineral phases and composite clayey soil samples at pH 5 (a), 7 (b) and 8 (c) and for Cs at pH 7 (d), including aqueous standards (Sr2+ and Cs+) and strontianite (SrCO3). The XANES for strontianite is distinct from the other XANES, and includes a broad minimum in the XANES between 16130 and 16140 eV (dotted line, Fig. 3a) and a broader white line at 16120 eV (dash-dotted line; Fig. 3a–c). This demonstrates that no strontianite formed during adsorption experiments and the XANES spectra of the samples represent adsorption of Sr to the solid phases. Furthermore, there are only minor differences in the XANES spectra for the aqueous Sr standard and the spectra for the samples with Sr adsorbed to anatase and illite-smectite. This suggests that Sr speciation in these samples is similar to Sr2+ and likely dominated by coordination with oxygen (water).
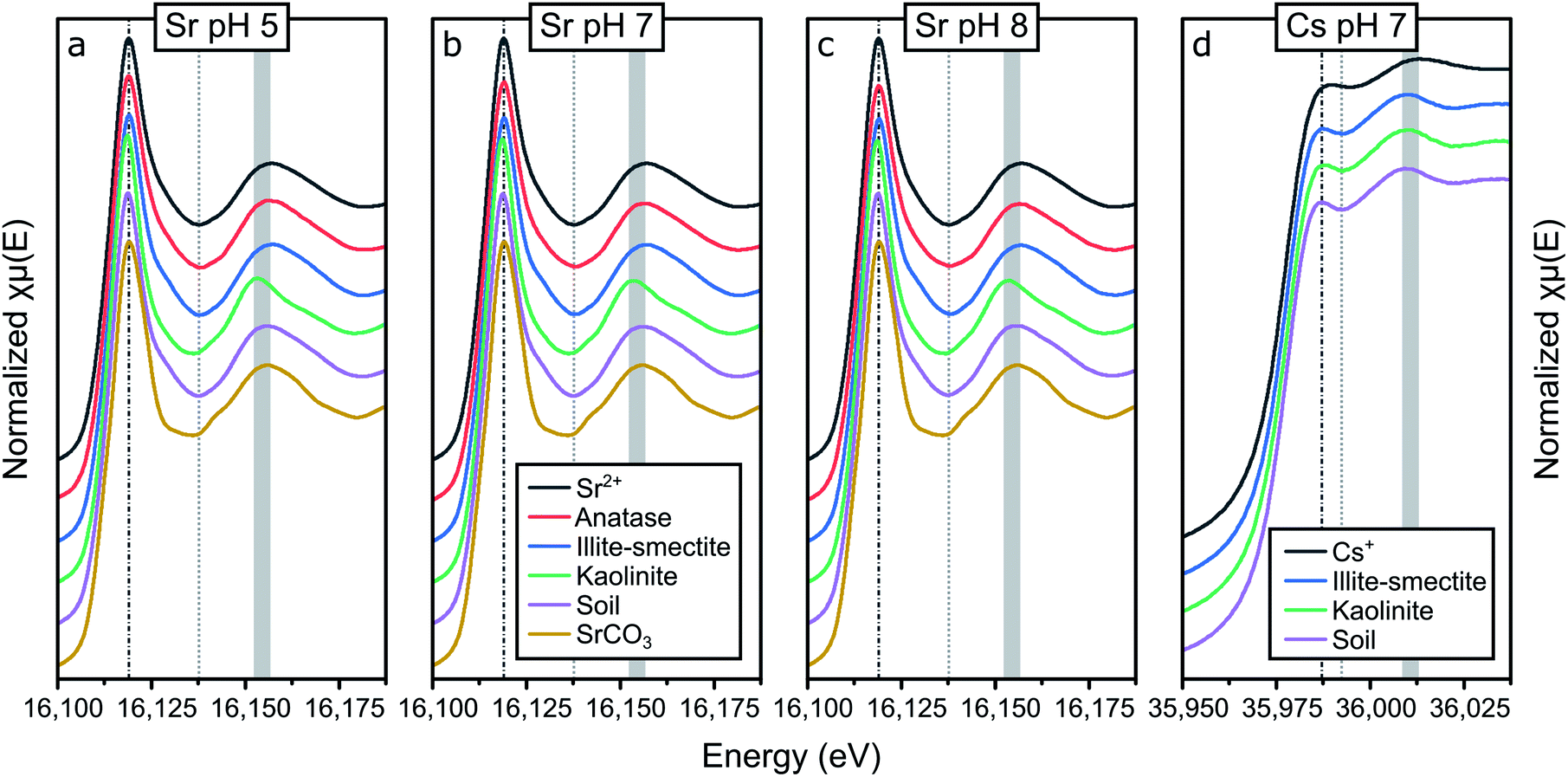 | ||
| Fig. 3 Summary of the K-edge XANES spectra for Sr adsorbed to anatase, illite-smectite, kaolinite and a composite clayey soil at pH 5 (a), pH 7 (b) and pH 8 (c) and Cs adsorbed to illite-smectite, kaolinite and a composite clayey soil (d); also plotted are in-house standards for comparison: aqueous Sr2+ and SrCO3 (strontianite; a–c) and aqueous Cs+ (d) as (in-house) standards, the vertical dash-dotted line highlights the position of the white line, the dotted line highlights the first absorption minimum and the broad grey band highlights the (variation in the energy position of the) first multiple scattering feature in the XANES to emphasize differences in the XANES spectra. | ||
It should be noted that traces of strontium within the structure of kaolinite have been shown to potentially significantly impact the resulting X-ray absorption spectra.20 Hence we also analysed an aliquot of kaolinite without Sr adsorbed to its surface and compared this to the XANES for the samples with Sr adsorbed to kaolinite (Fig. S2†). This comparison shows that the Sr absorption edge is ∼2 orders of magnitude lower in the pure kaolinite sample compared to the samples with Sr adsorbed to kaolinite (Fig. S2a†). Additionally, the multiple scattering features in the XANES of the blank kaolinite are identical to those in the XANES of blank kaolinite as measured by Parkman et al.20 and different from the samples with Sr adsorbed to kaolinite, specifically with respect to the first maximum at 16155 eV post white line (16120 eV; Fig. S2b†). These observations highlight that any strontium within the structure of kaolinite did not significantly affect the presented X-ray absorption spectra for Sr adsorbed to kaolinite. We are thus confident that the XANES and EXAFS collected on the samples with Sr adsorbed to kaolinite represent Sr surface complexes only. Relatively large differences can be observed between the spectra for Sr adsorbed to kaolinite compared to Sr2+ and Sr adsorbed to anatase and illite-smectite. This difference includes a more intense white line (dash-dotted line, Fig. 3a–c), and a shift in energy and change in shape of the multiple scattering features in the XANES (dashed and broad grey lines, Fig. 3a–c). This suggests that in these samples the Sr coordination environment is distinct from Sr2+ and Sr adsorbed to anatase and illite-smectite, and consequently, the mechanism of Sr adsorption to kaolinite is different compared to anatase and illite-smectite.
The XANES for the samples on Sr adsorption to soil appear to be intermediate to the XANES of Sr adsorbed to anatase or illite-smectite and kaolinite, suggesting that Sr likely adsorbs to multiple mineral phases within the composite clayey soil. This is confirmed by linear combination fitting (LCF)52 of the XANES spectra where the best fit (with the lowest R-factor and reduced χ2; Table S9†) included multiple minerals phases. Furthermore, when strontianite (SrCO3) was included in the LCF, there was an increase in both the R-factor and the reduced χ2 (Table S9†). This statistically confirms our earlier observation from visual inspection of the XANES spectra (Fig. 3a–c) that no strontianite formed during the adsorption experiments. Interestingly, the R-factor and the reduced χ2 did not show much variation between the LCF that included anatase, illite-smectite, or both (due to the similarities in the XANES spectra; Table S9† and Fig. 3), thus further investigation of the EXAFS was required to fully characterise the Sr surfaces species in the composite clayey soil samples. Finally, no visible variation in the XANES spectra at different pH values can be observed; this indicates limited variation in the Sr surface complexes at pH 5–8.
XAS spectra for the Cs adsorption samples were only collected for the samples at pH 7 (due to time limitations on Cs XAS analyses),19 and are visualized in Fig. 3d. For Cs, the XANES of the aqueous standard is significantly different compared to the XANES of all three adsorption samples with respect to the shape and energy position of the white line (dash-dotted line, Fig. 3d) and the multiple scattering features (dotted and broad grey lines, Fig. 3d). This indicates that the Cs local coordination environment is comparable in all three adsorption samples and distinct from aqueous Cs. Due to the similarities between the Cs XANES spectra of the adsorption samples, LCF did not provide meaningful insights into the speciation of Cs in the composite clayey soil sample (the XANES of the soil sample could be fitted as identical to the XANES from the kaolinite sample and to the XANES from the illite-smectite sample). Thus further investigation of the EXAFS was required to fully characterise the Cs surfaces species in the composite clayey soil samples.
Surface speciation of Sr and Cs
Initially, we attempted to fit the EXAFS spectra for Sr adsorbed to anatase according to the tridentate Sr surface complex on terraces of the (101) anatase surfaces and a Sr–O coordination number of 6 and a total Sr–Ti coordination number of 5 as identified from density functional theory modelling by Ridley et al.27 This did not appear to result in a valid fit. Rather, all EXAFS spectra for strontium adsorbed to anatase could be fit with a single Sr–O scattering path with a radial distance of 2.595–2.597 Å, and a coordination number of 8.5–9.3 (Table S10†). This coordination number closely matches that of hydrated Sr2+,19–22,65,66 suggesting that Sr complexed with anatase is more strongly hydrated compared to the species identified by Ridley et al.27 Additionally, a single Sr–Ti scattering path could be fit with a radial distance of 3.58 Å, and a coordination number of 0.5–0.9 (Table S10†). This coordination environment corresponds with our conceptual model of Sr complexed with octahedral Ti in anatase through a bidentate edge sharing surface complex (as visualised in Fig. 4) similar to the species previously identified for Zn complexes with anatase.67 The difference between this Sr bidentate edge sharing surface complex (Fig. 4) and the (tridentate) surface complex identified by Ridley et al.27 is likely caused by the different approaches used to derive the local coordination environment for Sr complexed with anatase (spectroscopic analyses on experimental samples vs. density function theory modelling, respectively). Interestingly, the shell-by-shell fitting strategy resulted in a progressively smaller Ti coordination number from 0.9 to 0.5 when the pH decreased from 8 to 5, visualised by a concurrent decrease in the amplitude of the feature in the FT at 3 Å (dash-dotted line Fig. S3e†). This observation could suggest that with a decrease in pH a larger fraction of Sr does not exhibit a Sr–Ti scattering path in their respective EXAFS. We tentatively ascribe this to two surface complexes (with different ratios at different pH values) with an outer-sphere surface complex more dominant at lower pH values causing the decrease in the Sr–Ti coordination number. This increase in the dominance of the possible outer-sphere surface complex is possibly linked or due to the lower adsorption and KD values at lower pH values (Fig. 1 and Table 1).
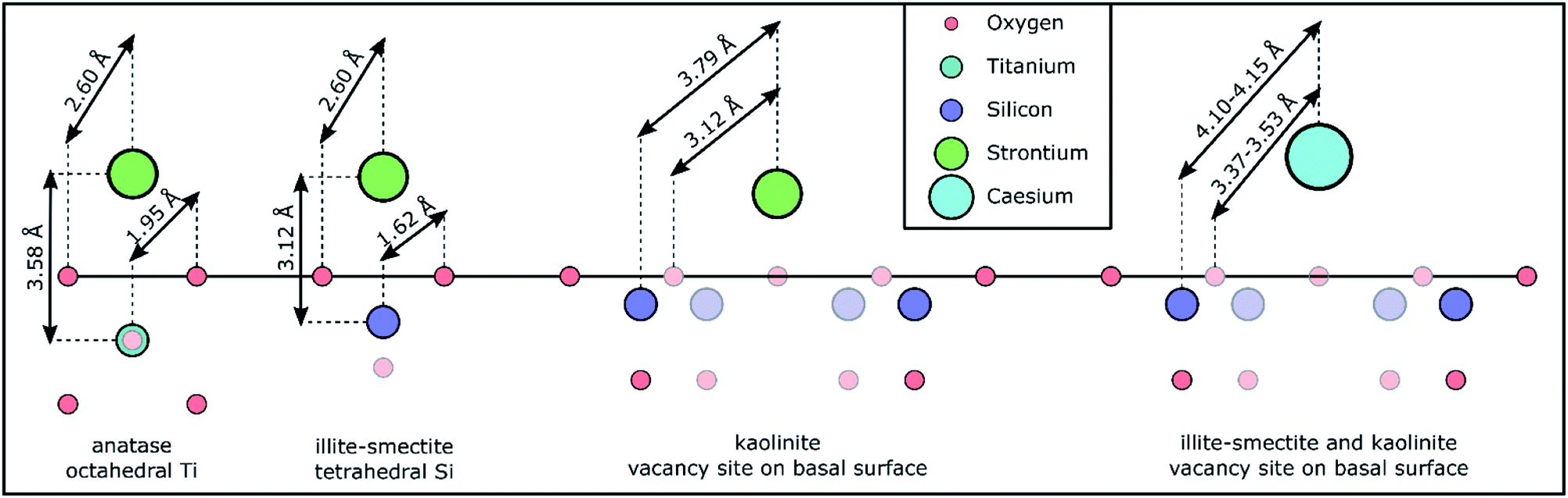 | ||
| Fig. 4 Schematic representation of the Sr and Cs coordination environments/chemical speciation based on the (conceptual) models used for the shell-by-shell fitting strategy and the respective results (Tables S10 and S14†) to the EXAFS spectra and the crystallographic distances for Ti–O in anatase and Si–O in illite from the respective crystallographic information files.54,56 The faded symbols represent atoms either in a closer or more distant plane compared to Sr or Cs; the hydration sphere around Sr and Cs is omitted to improve the clarity of the schematic. The schematic is roughly to scale. | ||
The shell-by-shell fitting strategy on the spectra for Sr adsorption to the mixed layered illite-smectite phase resulted in a best fit with a Sr–O scattering path at 2.602–2.604 Å, and a coordination number of 8.9–9.3. Additionally, a Sr–Si scattering path could be fit at 3.12 Å, and a coordination number of 0.9–1.0. A Sr–Si scattering path of 3.12 Å is short compared to previously identified Sr–Si scattering paths for Sr complexed with illite (at 3.33–3.39 Å).15 This could be due to the differences in the experimental conditions (i.e. elevated pH, Sr surface coverage).15 Nevertheless, the inclusion of the Sr–Si scattering path at 3.12 Å did statistically improve the fits to the EXAFS spectra (Table S11†). This relatively short Sr–Si scattering path could potentially be due to interferences of multiple surface complexes. However, if multiple surface complexes were present, modelling suggests that these surface complexes vary in dominance between pH 5 and 8,62,68 and would result in varying coordination environments (e.g. coordination number or radial distance) as a function of pH (as observed for the increase in the dominance of the potential outer-sphere surface complex on anatase with a decrease in the pH). Thus, the similarity of the Sr–Si coordination number at all pH values suggests that Sr complexation to illite-smectite was dominated by a single surface complex. It is however possible that a second surface complex does impact on the Sr surface complexation to illite-smectite as a minor surface complex of the total surface complexation.69 Such minor complexes are unlikely to significantly affect the fit to the EXAFS spectra; this could however be confirmed by utilizing similar constraints in a shell-by-shell fitting strategy (Table S1†) on adsorption samples with varying Sr concentration rather than pH. Additionally, a Sr–Si scattering path of 3.12 Å corresponds with our (conceptual) model of Sr complexed to illite-smectite as a bidentate edge sharing surface complex with tetrahedral Si (as visualized in Fig. 4). We are thus confident that the Sr EXAFS represent one dominant surface complex. In comparison to the anatase bidentate edge sharing surface complex, the shorter Sr–Si scattering path than 3.58 Å (for Sr–Ti, Table S10†) in a similar adsorption mechanism is to be expected from a difference in the crystal radii between octahedral Ti (0.75 Å) and tetrahedral Si (0.4 Å).70
During the shell-by-shell fitting strategy to the EXAFS spectra for Sr adsorbed to kaolinite a bidentate edge sharing surface complex, as identified for Sr complexed with illite-smectite and anatase, did not provide an adequate fit (as indicated by the interpretation of the XANES spectra). Rather the best fit included three scattering paths. This includes two Sr–O scattering paths with a radial distance of 2.60 Å and 3.12 Å and a coordination number of 8.8–9.0 and 2.9–3.6, respectively. Further, a Sr–Si scattering path was required at 3.79 Å, and a coordination number of 5.5–6.6 (Tables S10 and S11†). First, due to the relatively large Sr coordination to O in this coordination environment, we initially considered the possibility of a ternary surface complex with carbonate, akin to ternary uranyl complexes.71 However, at pH 5–8, aqueous Sr carbonate and bicarbonate complexes only account for 10−4 to 10−1 % of the total aqueous Sr (as identified using PHREEQC72); it is unlikely that ternary Sr–CO3 surface complexes would dominate. Furthermore, the coordination environment, with respect to the coordination numbers of the Sr scattering paths, is stable. Conversely, the dominance of any ternary complex involving carbonate would vary either in dominance or the KD value would vary significantly; neither is observed in the presented EXAFS or the results from the adsorption experiments (Fig. 1). Finally, attempting to fit the EXAFS using a (conceptual) model for a ternary Sr–CO3 surface complex did not result in a chemically reasonable fit with respect to the coordination number and/or radial distances expected from our conceptual model. We are thus confident of the presence of both Sr–O scattering paths. The existence of these two Sr–O scattering paths and an average Si coordination number of 5.9, confirms the interpretation of the XANES spectra, that the Sr coordination environment is more closely associated to the mineral surface and less hydrated compared to Sr adsorbed to anatase and illite-smectite. The radial distances of 3.12 Å for the second Sr–O scattering path and the radial distance of 3.79 Å and respective average coordination number of 5.9 for the Sr–Si scattering paths correspond to our conceptual model of Sr complexed with kaolinite at the vacancy sites on the basal surface of kaolinite (Fig. 4), where the Sr–O scattering path at 3.12 Å represents surface oxygen backscatters (from tetrahedral Si) and the Sr–O scattering path at 2.60 Å represents oxygen in the first hydration sphere of Sr, and the relatively large Sr coordination to O is due to the small interatomic distances between the O from the Si tetrahedrons surrounding the Si vacancies on the kaolinite basal surface (Fig. 4). The identification of Sr complexation at the vacancy sites on the kaolinite basal surfaces is different compared to previous studies where instead hydrated surface complexes were identified more similar to the species we identified for Sr complexation with illite-smectite and anatase.20,21 The difference in the identified Sr surface complex on kaolinite could be due to variations in reaction time (24–48 h), in Sr concentration/surface coverage,20,21 in the surface chemistry of different kaolinite sources (English China Clays plc.20 Clay Mineral Society21 and Sigma Aldrich), or the presence of trace mineral phases such as titanium dioxide, which can significantly impact the surface speciation of metals as shown for uranyl adsorption to kaolinite (Clay Mineral Society, source clay: KGa-1 and KGa-1B).73 However, we should note that the previously identified Sr surface complexes were based on fits that only included a single (first) Sr–O scattering path to the Fourier filtered EXAFS,20,21 rather than a fit to the EXAFS over a larger k- and R-range (Fig. S3 and Table S11†). It is thus also possible, or likely, that improved EXAFS data quality has enabled us to refine the Sr surface complex beyond what was possible over two decades ago.20,21
The shell-by-shell fitting strategy to the spectra on Sr adsorption to the composite clayey soil resulted in best fits with two scattering paths. These fits include a Sr–O scattering path at 2.608–2.609 Å with a coordination number of 10.1–10.6, and a Sr–Si scattering path at 3.79 Å with a coordination number of 1.1–1.2 (Table S10†). Such a coordination environment could correspond to a monodentate edge sharing surface complex at a relatively large angle between Sr–O and the tetrahedral Si–O (as the radial distances of Sr–Si (3.79 Å) is smaller compared to the sum of the Sr–O and Si–O distances, Fig. 4) and corresponds to previously identified coordination environments for Sr adsorbed to clayey materials and soils.15,19,22 However, the inferred monodentate edge sharing surface complex corresponds to none of the Sr surface complexes identified for Sr adsorption with the single mineral phases (Fig. 4). Additionally, while the radial distance of the Sr–Si scattering path corresponds to the radial distance of the Sr–Si scattering path for Sr adsorbed to kaolinite (Table S10†) the Si coordination number is lower (1.1–1.2 instead of 5.5–6.6) and only a single Sr–O scattering path could be fit compared to two Sr–O scattering paths for Sr adsorbed to kaolinite (Table S10†). Furthermore, the coordination environment as identified by the shell-by-shell fitting strategy correspond neither to the coordination environments for Sr adsorbed to anatase nor illite-smectite (Table S10†). This indicates that for more complex samples with multiple possible coordination environments the employed shell-by-shell fitting strategy did not provide sufficiently detailed information to determine the mechanisms of Sr interaction with the mineral phases within such samples. Hence, to provide detailed information on Sr speciation within these composite clayey soil samples, we first attempted a linear combination fitting (LCF) strategy on the EXAFS in k-space.74 The results of this LCF strategy are summarized in Table S9.† The LCF using Sr complexed to anatase, illite-smectite and kaolinite as standards resulted in statistically better fit (lower R-factor and reduced χ2) compared to using Sr complexed to kaolinite and illite-smectite only. This suggests anatase could contribute up to 20% to the surface complexation of Sr in the composite clayey soil samples. However, similar to the LCF on the XANES spectra, the R-factor and reduced χ2 of the LCF on the EXAFS in k-space show little variation between several combinations of standards, even when including strontianite or aqueous Sr2+ in the LCF. This suggests that the results from the LCF to the EXAFS could be significantly impacted by noise in the spectra. Hence, we developed and employed a second, holistic fitting strategy to the Sr EXAFS spectra.
The holistic fitting approach (Table S2†) was based on the conceptual models developed from the shell-by-shell fitting strategy on Sr adsorption to the single mineral phases (Table S10†) shown in Fig. 4. During this second approach, all Sr EXAFS spectra from the adsorption samples at one pH value were fitted simultaneously. During these fits, the coordination numbers for scattering paths representative of the Sr complexes with anatase, illite-smectite and kaolinite were fixed to the values determined during the shell-by-shell fitting strategy (Table S10†). Furthermore, the EXAFS from the composite clayey soil samples were fitted as a combination of the fits to the Sr adsorbed to anatase, illite-smectite and kaolinite to identify the fraction of Sr adsorbed to these mineral phases (fAnatase, fIllite, fKaolinite) within the analysed composite clayey soil samples, enabling the EXAFS of these composite clayey soil samples to be fitted with just 4 variables describing 3 surface complexes (ΔE0, fAnatase, fIllite, and fKaolinite; Table S2†). The results from the holistic fitting strategy are visualised in Fig. 5 and described in Tables S13† and 2.
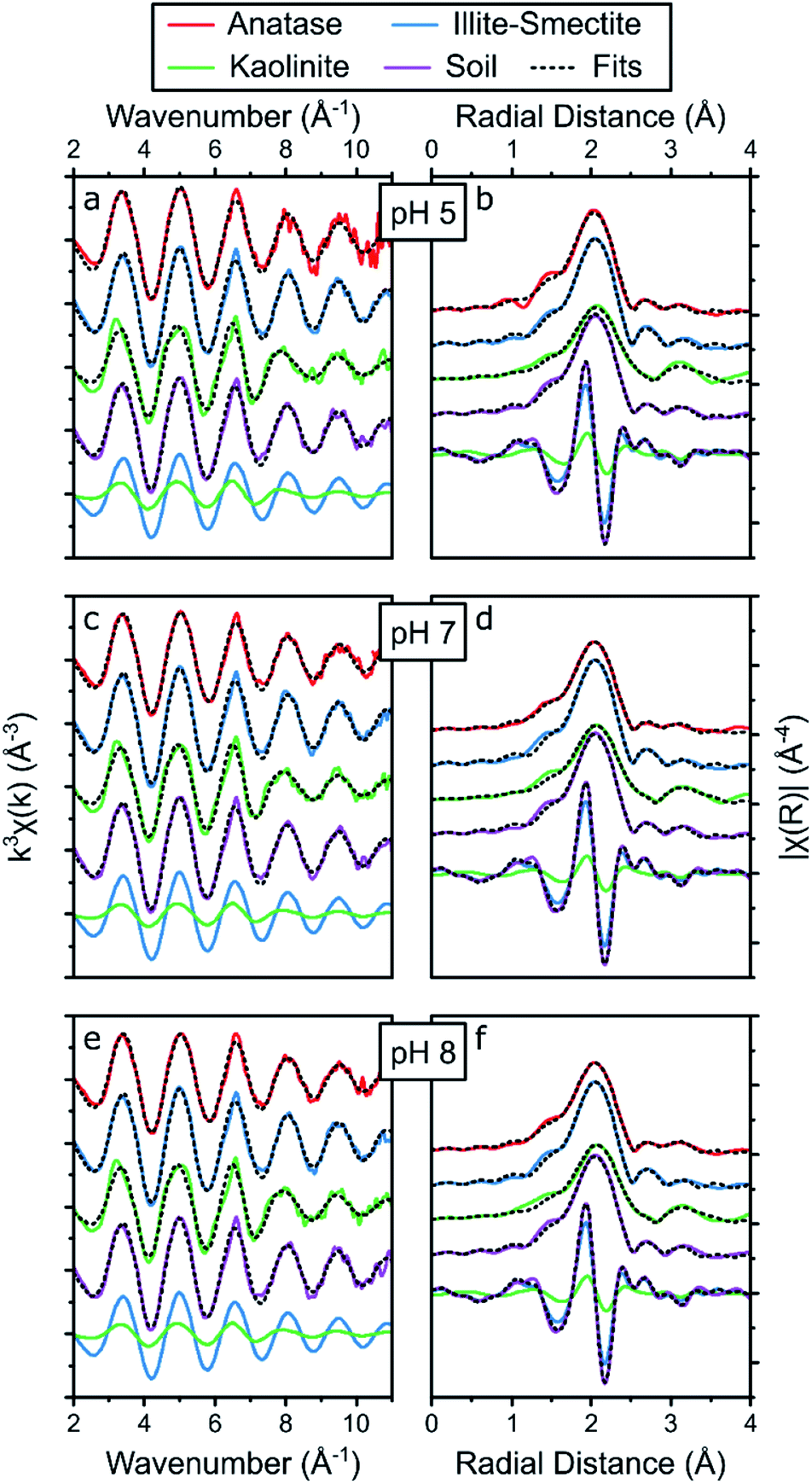 | ||
| Fig. 5 Summary of the Sr K-edge EXAFS (a, c and e) and the respective Fourier transform (b, d and f) of the adsorption samples at pH 5 (a, b), 7 (c, d) and 8 (e, f), including the results from the holistic fitting strategy (black dotted lines, Table 2) and the respective contribution of the EXAFS and FT of Sr adsorbed to illite-smectite and kaolinite to the fits to the EXAFS of Sr adsorbed to the soil samples at the bottom of each panel. | ||
| Anatase | Illite-smectite | Kaolinite | Soil | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| pH 5 | pH 7 | pH 8 | pH 5 | pH 7 | pH 8 | pH 5 | pH 7 | pH 8 | pH 5 | pH 7 | pH 8 | |
| a S 0 2 represents the amplitude correction factor (fixed to 1), C.N. the coordination number (fixed, the errors on the C.N. are estimated to be ∼25%), ΔE0 the energy shift, R the radial distance and σ2 the Debye–Waller factor, the numbers in parentheses are the uncertainties as calculated by Artemis,52 the C.N. were fixed based on the shell by shell fits (Table S10). b The fitting parameters to the soil samples (besides the energy shift, ΔE0) represent the fraction of Sr in the sample complexed with anatase (fAnatase), illite-smectite (fIllite) and kaolinite (fKaolinite; Table S2) rather than describing each scattering paths separately; including Sr complexed to anatase (fAnatase) as a fitting parameter did not statistically improve the fit to the soil sample (Table S12), hence the fAnatase was fixed to 0. | ||||||||||||
| S 0 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| ΔE0 (eV)a | −3.8(10) | −3.0(4) | −2.8(4) | −1.8(3) | −1.9(3) | −1.8(3) | −7.2(8) | −6.3(7) | −5.8(8) | −3.1(4) | −2.7(3) | −2.7(4) |
| Sr–O | Sr–O | Sr–O(1) | f Anatase | |||||||||
| C.N.a | 9.3 | 8.7 | 8.5 | 8.9 | 9.3 | 9.1 | 8.8 | 8.9 | 9.0 | 0 | 0 | 0 |
| R (Å)a | 2.59(1) | 2.596(4) | 2.596(8) | 2.601(3) | 2.603(3) | 2.603(3) | 2.600(8) | 2.600(7) | 2.606(8) | |||
| σ (Å2)a | 0.0100(7) | 0.0099(3) | 0.0099(3) | 0.0080(2) | 0.0087(2) | 0.0087(2) | 0.0111(4) | 0.0110(4) | 0.0110(4) | |||
| Sr–Ti | Sr–Si | Sr–O(2) | f Illite | |||||||||
| C.N.a | 0.5 | 0.7 | 0.9 | 1.0 | 0.9 | 0.9 | 3.6 | 3.1 | 2.9 | 0.72(9) | 0.76(9) | 0.76(11) |
| R (Å)a | 3.52(15) | 3.57(4) | 3.59(3) | 3.11(2) | 3.11(2) | 3.11(2) | 3.11(1) | 3.11(1) | 3.12(2) | |||
| σ (Å2)a | 0.02(2) | 0.017(6) | 0.017(5) | 0.013(3) | 0.014(3) | 0.014(3) | 0.010(2) | 0.012(2) | 0.012(3) | |||
| Sr–Si | f Kaolinite | |||||||||||
| C.N.a | 6.6 | 5.7 | 5.5 | 0.30(11) | 0.25(11) | 0.25(13) | ||||||
| R (Å)a | 3.79(2) | 3.80(2) | 3.80(2) | |||||||||
| σ (Å2)a | 0.020(2) | 0.020(2) | 0.020(2) | |||||||||
First, when refining fAnatase during the holistic fitting strategy the fraction of Sr adsorbed to of anatase was refined to 0–2% at pH 7 and 8, and to 49% percent at pH 5 (Table S13†). This contradicts the results from the experiments on Sr adsorption to anatase, where adsorption of Sr decreases significantly when the pH lowers from 8 to 5 (Fig. 1, Tables S5 and S7†). Secondly, at pH 5, the radial distance of the Sr–Ti scattering path (3.42 Å, Table S13†) was significantly smaller compared to the result from the shell-by-shell fitting strategy (3.58 Å, Table S10†). Finally, the f-test for EXAFS75 shows that including Sr adsorbed to anatase did not significantly improve the fit (Table S12†). These three observations suggest, or even highlight, that anatase did not significantly impact on the surface complexation of Sr in composite clayey soils, even though the LCF to the XANES and EXAFS spectra suggest a potential contribution from Sr surface complexes to anatase of up to 26% in these samples (Table S9†). This discrepancy is likely because the XANES spectra for Sr adsorbed to anatase and illite-smectite were nearly identical (Fig. 3), while the local coordination environment within the conceptual models for Sr complexation with anatase and illite-smectite are different (Fig. 4). This, furthermore, appears to confirm that the results from the LCF to the EXAFS are significantly impacted by noise in the spectra. Hence, during the final holistic fitting strategy we fixed fAnatase to 0, thus enabling the EXAFS of these composite clayey soil samples to be fitted with just 3 variables describing 2 surface complexes (ΔE0, fIllite, and fKaolinite; Table S2;† results summarized in Fig. 5 and Table 2).
The final holistic fitting strategy to the Sr EXAFS spectra from the (anatase) illite-smectite and kaolinite adsorption samples resulted in radial distances and Debye–Waller factors within error of the values obtained from the shell-by-shell fitting strategy (Tables 2 and S10†). This emphasizes that the holistic fitting strategy of the Sr EXAFS from the composite clayey soil adsorption samples could accurately describe the coordination environments for Sr complexation with illite-smectite and kaolinite, developed through the shell-by-shell fitting strategy of the Sr EXAFS from the single mineral adsorption samples. This is furthermore confirmed by the f-test75 on whether the addition of the separate scattering paths improved the goodness of fit parameters (Table S12†), where all but one scattering path (Sr–Ti at pH 5) were statistically significant. The low statistical significance of the addition of the Sr–Ti scattering path at pH 5 is likely due to either the relatively noisy data (of the EXAFS for Sr adsorbed to anatase at pH 5) or the lower coordination number for this scattering path at pH 5. Consequently, these similarities and observations reaffirm that Sr complexation in the composite clayey soil samples is dominated by adsorption to kaolinite and illite-smectite, and highlight that the holistic fitting strategy could successfully and confidently be used to fit the EXAFS of the composite clayey soil samples as a combination of the fits to the EXAFS of the samples with Sr adsorbed to single mineral phases. This enabled us to use the holistic fitting strategy to identify and quantify the fraction of Sr adsorbed to these mineral phases (fIllite, fKaolinite; Table 2, Fig. 5 and Table S12†).
The results from the final holistic fitting strategy (Table 2) show that the EXAFS of the soil sample could be best fitted with 25–30% Sr adsorbed to kaolinite and 72–76% to illite-smectite with no significant or systematic variations as a function of the pH (Table 2). This shows that even though the shell-by-shell fitting strategy resulted in a best fit with a Sr–Si scattering path at 3.79 Å, suggesting that Sr speciation was dominated by Sr adsorbed to kaolinite, this accounts for only 25–30% of the total Sr surface complexes, with the remaining adsorbed to illite-smectite. This corresponds well with the observations from the adsorption experiments, where Sr adsorption is 3–4 times higher for illite-smectite compared to kaolinite at all pH values (Fig. 1, Tables S5 and S7†) and the composite clayey soil contains equal amounts of kaolinite and illite-smectite (Fig. S1†). Additionally, the holistic fitting strategy highlights that (because of the low anatase content in the composite clayey soil samples, Fig. S1†) Sr complexation with anatase did not significantly contribute to the Sr complexation in composite clayey soil. Finally, this holistic fitting strategy enabled us to (more) accurately and confidently fit several coordination environments to the Sr EXAFS from the composite clayey soil sample (beyond possible with only LCF to the Sr EXAFS and/or XANES) without the need to fit an excessive amount of scattering paths. This strategy successfully provided detailed information on the speciation of Sr in a relatively complex matrix.
The shell-by-shell fits to the EXAFS of Cs adsorbed to illite-smectite included two Cs–O scattering paths at 3.18 and 3.55 Å with coordination numbers of 8 and 4 (respectively), and a Cs–Si scattering path at 4.11 Å with a coordination number of 5 (Table S14†). Such a coordination environment is different compared to retention of Cs within the interlayer of illite31 and to outer sphere surface complexation with smectite.32 Rather, this coordination environment corresponds to Cs complexed at the Si vacancy sites in the basal surface of illite-smectite (visualised in Fig. 4) as determined by Kerisit et al.35 (Table S14†) and identified for Cs complexation in a clayey soil.19
The shell-by-shell fit to the EXAFS of Cs adsorbed to kaolinite was very similar to illite-smectite and included two Cs–O scattering paths at 3.15 and 3.37 Å with coordination numbers of 7 and 5 (respectively) and a single Cs–Si scattering path at 4.14 Å with a coordination number of 6. This shows that Cs also complexes with kaolinite at the Si vacancy sites on the basal surfaces (Fig. 4) and corresponds with the coordination environment determined by Vasconcelos et al.36 (Table S14†). It is interesting to note that the largest difference between the fits to the EXAFS of Cs adsorbed to illite-smectite and to kaolinite is the radial distance of the second Cs–O scattering path (3.55 vs. 3.37 Å). We suggest that this is due to differences in the orientation and/or disorder of the tetrahedral Si in the basal surfaces of illite-smectite and kaolinite.55,56
Based on the similarities of the fits to the EXAFS of Cs adsorbed to illite-smectite and kaolinite, it is unsurprising that the shell-by-shell fit to the EXAFS of Cs adsorbed to the composite clayey soil sample is similar to both these fits with values roughly average for Cs adsorbed to illite-smectite and to kaolinite (Table S14†). This included two Cs–O scattering paths at 3.14 and 3.43 Å with coordination numbers of 8 and 4 (respectively) and a Cs–Si scattering path at 4.12 Å with a coordination number of 5.5. The values for these scattering paths are within error of our previous interpretation of Cs complexation with clayey soil;19 the minor differences in the radial distances and the Debye–Waller factors are due to different constraints used during the respective fitting strategies. The similarities reaffirm that surface complexation at the basal surface of the clay minerals dominated Cs complexation in the composite clayey soil sample.19,35,36 As the values for fit to the EXAFS of Cs adsorbed to the composite clayey soil sample are roughly average for Cs adsorbed to illite-smectite and to kaolinite, this does not provide much detail on the relative roles of Cs complexation with illite-smectite compared to kaolinite within the EXAFS of the composite clayey soil sample. Thus, we performed a similar holistic fitting strategy for the EXAFS of the Cs adsorbed to the composite clayey soil sample to the strategy previously used for the EXAFS of Sr adsorbed to the composite clayey soil samples (as described in Table S3†).
The results from the holistic approach to fitting the Cs EXAFS are described in Table 3 and visualized in Fig. 6 (and the goodness of fit parameters are described in Table S15†). First, the radial distances and Debye–Waller factors for Cs complexed to kaolinite and illite-smectite are within error of the results from the shell-by-shell fitting strategy (Tables 3 and S14†). Furthermore, the goodness of fit parameters show that to fit the EXAFS of Cs adsorbed to the clayey soil sample, both Cs complexed with illite-smectite and kaolinite were required in the fit, and that Cs complexed at the Si vacancy sites of the basal surfaces of illite-smectite and kaolinite contribute roughly equally to the overall coordination environment of Cs in the composite clayey soil samples (Table 3). For comparison, the adsorption experiments show that Cs complexed to illite-smectite, kaolinite, and the composite clayey soil samples exhibit KD values in the same order of magnitude (130, 56 and 50 mg l−1, respectively, Fig. 1d, f and h, Tables 1, S5 and S7†) and no adsorption of Cs occurred to the other phases within the composite clayey soil samples (Fig. 1, Tables S5, and S7†). As discussed above, this suggests that both clay minerals contribute to the complexation of Cs in the composite clayey soil sample, which corresponds to the results from the holistic fitting strategy and provides confidence in these results (Table 3). This is distinct from the observations from Payne et al.29 who identified that Cs mobility in natural clayey soils was governed by complexation at the illite frayed edge sites (at trace Cs concentrations), and by Cornell30 who highlights KD values for complexation with illite significantly higher than kaolinite. We suggest that this difference was caused by the elevated concentrations required for EXAFS analyses39 and due to the presence or absence of competing cations. For example, we identified that Sr competes for complexation with Cs on illite-smectite and kaolinite (Fig. 1d, f and h), and complexes to the same Si vacancy sites as Cs on kaolinite (Fig. 4). Furthermore, we suggest that variations in the structure and crystallinity39 of illite-smectite and kaolinite (and their surfaces) from different materials sources used in the presented and previous research29,30 may also contribute to the differences in the distribution of Cs as calculated for the composite clayey soil sample (Table 3) and from previous research.29,30 We have to note, however, that the error on these values is relatively large (40%, Table 3). This is likely due to the similarities between the local coordination environments of Cs complexed at the Si vacancy sites on the basal surface of illite-smectite and to kaolinite, resulting in small differences in the respective EXAFS (the only noticeable difference is a dampening in the features in the EXAFS for Cs complexed with illite-smectite, k = 6 and 7.5 Å−1; dashed vertical lines Fig. 6a).
| Illite-smectite | Kaolinite | Soil | |
|---|---|---|---|
| pH 7 | pH 7 | pH 7 | |
| a S 0 2 represents the amplitude correction factor (fixed to 1), C.N. the coordination number, ΔE0 the energy shift, R the radial distance and σ2 the Debye–Waller factor, the numbers in parentheses are the uncertainties as calculated by Artemis,52 the C.N. for O and Cs were fixed based on molecular dynamics for Cs complexation at the Si vacancy sites of the basal plane of illite (kerisit) or kaolinite (vasconcelos). b The fitting parameters to the soil sample (besides the energy shift, ΔE0) represent the fraction of Cs in the sample complexed with illite-smectite (fIllite) and kaolinite (fKaolinite; Table S3) rather than single scattering paths. | |||
| S 0 2 | 1 | 1 | 1 |
| ΔE0 (eV)a | −1.4(29) | 4.7(31) | 1.4(33) |
| Cs–O(1) | f Illite | ||
| C.N.a | 8 | 7 | 0.5(4) |
| R (Å)a | 3.16(3) | 3.15(4) | |
| σ (Å2)a | 0.022(3) | 0.014(3) | |
| Cs–O(1) | f Kaolinite | ||
| C.N.a | 4 | 5 | 0.5(4) |
| R (Å)a | 3.53(5) | 3.38(4) | |
| σ (Å2)a | 0.022(3) | 0.014(3) | |
| Cs–Si | |||
| C.N.a | 5 | 6 | |
| R (Å)a | 4.10(4) | 4.15(4) | |
| σ (Å2)a | 0.016(2) | 0.013(2) | |
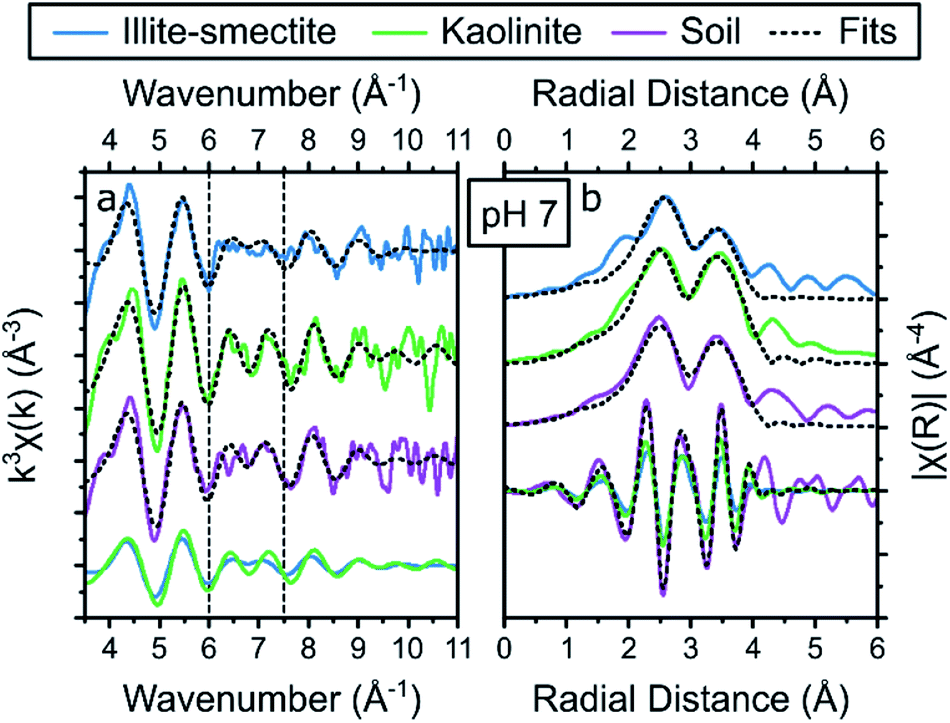 | ||
| Fig. 6 Summary of the Cs K-edge EXAFS (a) and the respective Fourier transform (b) of the adsorption samples at pH 7, including the results from the holistic fitting strategy (black dotted lines, Table 3) and the respective contribution of the EXAFS and FT of Cs adsorbed to illite-smectite and kaolinite to the fits to the EXAFS of Cs adsorbed to the soil samples at the bottom of each panel. | ||
Concluding remarks
Through adsorption experiments, Sr and Cs EXAFS analyses and subsequent detailed shell-by-shell fitting of the EXAFS, we have provided detailed experimental/analytical information on the surface speciation of Sr and Cs with clay minerals and anatase (TiO2). We identified that Sr complexes with anatase and illite-smectite through bidentate edge-sharing complexes with the octahedral titanium (in anatase, Fig. 4) and tetrahedral silicon (in illite-smectite, Fig. 4). We furthermore identified that Sr complexes with kaolinite and Cs with illite-smectite and kaolinite through inner-sphere complexes at the silicon vacancies on the basal surfaces of these clay minerals (Fig. 4). In combination with spectra of for example Cs in the interlayer of illite,31 the presented spectra could subsequently be utilized (as standards) for spectra of clay minerals with Cs (and/or Sr) complexed at multiple sites, e.g. to determine the coordination environment of Cs (and/or Sr) at the frayed edge sites29,30,39 of clay minerals. Furthermore, by utilizing a holistic approach to fitting Sr and Cs EXAFS in composite clayey soil samples we highlight that Sr and Cs dominantly complex with the clay minerals in clayey soils (kaolinite and illite-smectite). These complexes thus govern the mobility of Sr and Cs at contaminated sites with such clayey soils (e.g. the Little Forest Legacy Site in Australia).3,4,19The presented comprehensive/dual strategy to interpreting the EXAFS spectra, using a combination of fitting approaches, provides an additional strategy to fitting EXAFS of multicomponent samples (such as soils). Through utilizing conceptual models on the local coordination environment of potential standards, based on an initial shell-by-shell strategy, this holistic fitting strategy enables the determination of the validity of the standards used to fit the EXAFS of multicomponent samples (as highlighted above for the use of Sr adsorbed to anatase as standard). This subsequently enables the extraction of more accurate information from EXAFS spectra (e.g. quantifying multiple species present in multicomponent samples) compared to a shell-by-shell fitting strategy alone,1,15 or linear combination fits (LCF) of (noisy) EXAFS spectra.74 Additionally, we suggest that if this holistic fitting approach does not result in a satisfactory or chemically feasible fit to the EXAFS of a multicomponent sample, it is likely that a conceptual model of a local coordination environment is missing. Such information is not readily available when performing LCF to the EXAFS spectra. This strategy complements statistical methodologies to provide qualitative information on the adsorption governing mineral phases (e.g. at trace concentrations where XAS analyses are unfeasible)29,39 and existing strategies to interpret XANES and EXAFS spectra from multicomponent systems, such as LCF (Table S9†) of the XANES,52ab initio modelling of EXAFS spectra combined with LCF thereof,76 and iterative target transformation Fourier analyses.77,78
The combination of such statistical29,39 and (the presented) spectroscopy strategies will strengthen predictive modelling of the long-term behaviour of elements of interest, including moving toward “smart” KD values.79,80 Such predictive modelling can assist with extrapolating results from elevated concentrations (required for EXAFS analyses) to more environmentally relevant/trace concentrations. We suggest that the presented information on the Sr and Cs speciation within a composite clayey soil (including the complete results from the adsorption experiments at trace and elevated concentrations on these solids, Fig. 1 and Tables S4 to S8†) will enable and support surface complexation and reactive transport modelling of Sr and Cs. For example, incorporating the presented information in surface complexation modelling could be used to determine the relative importance of Cs and/or Sr retention in the illite-smectite interlayers24–26,31,37,38 and their frayed edges sites29,30,39 at trace Cs and Sr concentrations (e.g. in the composite clayey soils and the LFLS core samples Fig. 1 and 2). Such (evidence-based) modelling efforts will enable predicting the long-term behaviour of Sr and Cs at a wide range of concentrations, and enable predicting the impacts of any environmental engineering strategies at contaminated legacy sites.80,81 Such predictive modelling is essential in the design of evidence-based engineering strategies for the remediation and long-term management of contaminated (legacy) sites.19
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to acknowledge Diamond Light Source for awarding us with XAS beamtime at the Core XAS beamline (B18) under grant number: SP17114 and Dr Gianantonio Cibin for his assistance during XAS beamtime. We would also like to acknowledge the EPSRC ICASE award: EP/L505663/1 with financial support from the Nuclear Decommissioning Authority to support L. Rizzo and the EPSRC funded DISTINCTIVE consortium: EP/L014041/1. Finally, we thank Sangeeth Thiruvoth for assistance with tracer stock preparations, Lida Mokhber Shahin and Jennifer Harrison for their assistance with the radiochemical analyses, Jennifer Harrison and Pichamon Sarakan for their assistance with the adsorption experiments to ‘CH30 1.0–1.2 m’, Henri Wong for the ICP-AES analyses, Dr Dioni Cendón for the mineralogical assessment of the CH30 sample, and Dr Vinzenz Brendler for the preliminary discussions on the adsorption of radionuclides to clay minerals and requirements for potential surface complexation modelling.Notes and references
- S. H. Wallace, S. Shaw, K. Morris, J. S. Small, A. J. Fuller and I. T. Burke, Appl. Geochem., 2012, 27, 1482–1491 CrossRef CAS.
- N. L. Hakem, J. A. Apps, G. J. Moridis and I. Al Mahamid, Radiochim. Acta, 2004, 92, 419–432 CrossRef CAS.
- T. E. Payne, Backgro (Australian Nuclear Science and Technology Organisation) und Report on the Little Forest Burial Ground Legacy Waste Site; ANSTO/E-780, Australian Nuclear Science and Technology Organisation, 2012 Search PubMed.
- T. E. Payne, J. J. Harrison, C. E. Hughes, M. P. Johansen, S. Thiruvoth, K. L. Wilsher, D. I. Cendón, S. I. Hankin, B. Rowling and A. Zawadzki, Environ. Sci. Technol., 2013, 47, 13284–13293 CrossRef CAS PubMed.
- D. I. Cendón, C. E. Hughes, J. J. Harrison, S. I. Hankin, M. P. Johansen, T. E. Payne, H. Wong, B. Rowling, M. Vine, K. Wilsher, A. Guinea and S. Thiruvoth, Aust. J. Earth Sci., 2015, 62, 123–141 CrossRef.
- N. L. Hakem, I. Al Mahamid, J. A. Apps and G. J. Moridis, J. Radioanal. Nucl. Chem., 2000, 246, 275–278 CrossRef CAS.
- T. E. Payne, J. J. Harrison, D. I. Cendon, M. J. Comarmond, S. Hankin, C. E. Hughes, M. P. Johansen, A. Kinsela, L. M. Shahin, A. Silitonga, S. Thiruvoth and K. L. Wilsher, J. Environ. Radioact., 2020, 211, 106081 CrossRef CAS PubMed.
- H. Mukai, T. Hatta, H. Kitazawa, H. Yamada, T. Yaita and T. Kogure, Environ. Sci. Technol., 2014, 48, 13053–13059 CrossRef CAS.
- V. Sanial, K. O. Buesseler, M. A. Charette and S. Nagao, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 11092–11096 CrossRef CAS PubMed.
- P. K. Thai, Y. Suka, M. Sakai, K. Nanko, J. H. Yen and H. Watanabe, Environ. Sci.: Processes Impacts, 2015, 17, 1157–1163 RSC.
- T. Wakahara, Y. Onda, H. Kato, A. Sakaguchi and K. Yoshimura, Environ. Sci.: Processes Impacts, 2014, 16, 2580–2591 RSC.
- S. Otosaka and Y. Kato, Environ. Sci.: Processes Impacts, 2014, 16, 978–990 RSC.
- B. Grambow and M. Mostafavi, Environ. Sci.: Processes Impacts, 2014, 16, 2472–2476 RSC.
- H. Ashworth, L. Abrahamsen-Mills, N. Bryan, L. Foster, J. R. Lloyd, S. Kellet and S. Heath, Environ. Sci.: Processes Impacts, 2018, 20, 956–964 RSC.
- A. J. Fuller, S. Shaw, C. L. Peacock, D. Trivedi and I. T. Burke, Langmuir, 2016, 32, 2937–2946 CrossRef CAS PubMed.
- A. J. Fuller, S. Shaw, C. L. Peacock, D. Trivedi, J. S. Small, L. G. Abrahamsen and I. T. Burke, Appl. Geochem., 2014, 40, 32–42 CrossRef CAS.
- T. Missana, M. Garcia-Gutierrez and U. Alonso, Phys. Chem. Earth, 2008, 33, S156–S162 CrossRef.
- T. Missana, M. García-Gutiérrez, A. Benedicto, C. Ayora and K. De-Pourcq, Appl. Geochem., 2014, 49, 95–102 CrossRef CAS.
- P. Bots, J. C. Renshaw, T. E. Payne, M. J. Comarmond, A. E. P. Schellenger, M. Pedrotti, E. Calì and R. J. Lunn, Environ. Sci.: Nano, 2020, 7, 1481–1495 RSC.
- R. H. Parkman, J. M. Charnock, F. R. Livens and D. J. Vaughan, Geochim. Cosmochim. Acta, 1998, 62, 1481–1492 CrossRef CAS.
- N. Sahai, S. A. Carroll, S. Roberts and P. A. O'Day, J. Colloid Interface Sci., 2000, 222, 198–212 CrossRef CAS PubMed.
- S. H. Wallace, S. Shaw, K. Morris, J. S. Small and I. T. Burke, Environ. Sci. Technol., 2013, 47, 3694–3700 CrossRef CAS PubMed.
- T. Cole, G. Bidoglio, M. Soupioni, M. O'Gorman and N. Gibson, Geochim. Cosmochim. Acta, 2000, 64, 385–396 CrossRef CAS.
- A. Yamaguchi, M. Tanaka, Y. Kurihara and Y. Takahashi, J. Radioanal. Nucl. Chem., 2018, 317, 545–551 CrossRef CAS.
- K. Tamura, T. Kogure, Y. Watanabe, C. Nagai and H. Yamada, Environ. Sci. Technol., 2014, 48, 5808–5815 CrossRef CAS.
- T. H. Wang, T. E. Payne, J. J. Harrison and S. P. Teng, J. Radioanal. Nucl. Chem., 2015, 304, 95–105 CrossRef CAS.
- M. K. Ridley, M. L. Machesky and J. D. Kubicki, Langmuir, 2015, 31, 703–713 CrossRef CAS PubMed.
- W. R. Bower, K. Morris, J. F. W. Mosselmans, O. R. Thompson, A. W. Banford, K. Law and R. A. D. Pattrick, J. Hazard. Mater., 2016, 317, 97–107 CrossRef CAS PubMed.
- T. E. Payne, W. K. Bertram, T. Itakura and M. D. Raven, Radiochim. Acta, 2002, 90, 705–711 CrossRef CAS.
- R. M. Cornell, J. Radioanal. Nucl. Chem., 1993, 171, 483–500 CrossRef CAS.
- A. J. Fuller, S. Shaw, M. B. Ward, S. J. Haigh, J. F. W. Mosselmans, C. L. Peacock, S. Stackhouse, A. J. Dent, D. Trivedi and I. T. Burke, Appl. Clay Sci., 2015, 108, 128–134 CrossRef CAS.
- W.-T. Liu, S.-C. Tsai, T.-L. Tsai, C.-P. Lee and C.-H. Lee, Environ. Sci.: Processes Impacts, 2019, 21, 930–937 RSC.
- S. M. Park, J. Lee, E. K. Jeon, S. Kang, M. S. Alam, D. C. W. Tsang, D. S. Alessi and K. Baek, Geoderma, 2019, 340, 49–54 CrossRef CAS.
- B. C. Bostick, M. A. Vairavamurthy, K. G. Karthikeyan and J. Chorover, Environ. Sci. Technol., 2002, 36, 2670–2676 CrossRef CAS PubMed.
- S. Kerisit, M. Okumura, K. M. Rosso and M. Machida, Clays Clay Miner., 2016, 64, 389–400 CrossRef CAS.
- I. F. Vasconcelos, B. A. Bunker and R. T. Cygan, J. Phys. Chem. C, 2007, 111, 6753–6762 CrossRef CAS.
- T. Okumura, K. Tamura, E. Fujii, H. Yamada and T. Kogure, Microscopy, 2014, 63, 65–72 CrossRef CAS PubMed.
- T. Kogure, K. Morimoto, K. Tamura, H. Sato and A. Yamagishi, Chem. Lett., 2012, 41, 380–382 CrossRef CAS.
- J. Hwang, S. Choung, W. Shin, W. S. Han and C.-M. Chon, Water, 2021, 13, 409 CrossRef.
- T. Ohnuki, F. Sakamoto, N. Kozai, K. Nanba, H. Neda, Y. Sasaki, T. Niizato, N. Watanabe and T. Kozaki, Environ. Sci.: Processes Impacts, 2019, 21, 1164–1173 RSC.
- L. H. Dietterich, C. Gonneau and B. B. Casper, Ecol. Appl., 2017, 27, 1862–1875 CrossRef PubMed.
- K. Tanaka, T. Kanasashi, C. Takenaka and Y. Takahashi, Sci. Total Environ., 2021, 755, 142598 CrossRef CAS.
- E. W. Katengeza, Y. Sanada, K. Yoshimura, K. Ochi and T. Iimoto, Environ. Sci.: Processes Impacts, 2020, 22, 1566–1576 RSC.
- S. Choi, P. A. O'Day, N. A. Rivera, K. T. Mueller, M. A. Vairavamurthy, S. Seraphin and J. Chorover, Environ. Sci. Technol., 2006, 40, 2608–2614 CrossRef CAS PubMed.
- J. Chorover, S. Choi, P. Rotenberg, R. J. Serne, N. Rivera, C. Strepka, A. Thompson, K. T. Mueller and P. A. O'Day, Geochim. Cosmochim. Acta, 2008, 72, 2024–2047 CrossRef CAS.
- S. Hankin, Little Forest Burial Ground-geology, geophysics and well installation 2009–2010, Report ANSTO/E-781, Australian Nuclear Science and Technology Organisation, 2012 Search PubMed.
- M. D. Raven and P. G. Self, XRD report—Bulk and clay fraction analysis of soil samples for ANSTO, CSIRO Land and Water XRD Report D2381, CSIRO, 2011 Search PubMed.
- G. W. Brindley, C. C. Kao, J. L. Harrison, M. Lipsicas and R. Raythatha, Clays Clay Miner., 1986, 34, 239–249 CrossRef CAS.
- A. W. Fordham, Clays Clay Miner., 1973, 21, 175–184 CrossRef CAS.
- A. Plancon, R. F. Giese Jr, R. Snyder, V. A. Drits and A. S. Bookin, Clays Clay Miner., 1989, 37, 203–210 CrossRef CAS.
- A. J. Dent, G. Cibin, S. Ramos, S. A. Parry, D. Gianolio, A. D. Smith, S. M. Scott, L. Varandas, S. Patel, M. R. Pearson, L. Hudson, N. A. Krumpa, A. S. Marsch and P. E. Robbins, J. Phys.: Conf. Ser., 2013, 430, 012023 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- B. Ravel and J. J. Rehr, J. Phys. IV, 1997, 7, C2-229–C2-230 Search PubMed.
- I. Djerdj and A. M. Tonejc, J. Alloys Compd., 2006, 413, 159–174 CrossRef CAS.
- R. A. Young and A. W. Hewat, Clays Clay Miner., 1988, 36, 225–232 CrossRef CAS.
- V. Drits, J. Srodon and D. D. Eberl, Clays Clay Miner., 1997, 45, 461–475 CrossRef CAS.
- K. Brix, C. Hein, A. Haben and R. Kautenburger, Appl. Clay Sci., 2019, 182, 105275 CrossRef CAS.
- M. M. Gouda, Y. H. Dawood, A. A. Zaki, H. A. Salam Ibrahim, M. R. El-Naggar and A. Gad, J. Geochem. Explor., 2019, 206, 106366 CrossRef CAS.
- S. Kasar, S. Mishra, Y. Omori, S. K. Sahoo, N. Kavasi, H. Arae, A. Sorimachi and T. Aono, J. Soils Sediments, 2020, 20, 392–403 CrossRef CAS.
- A. S. Semenkova, T. R. Polyakova, D. K. Korob, I. F. Seregina, I. V. Mikheev, V. V. Krupskaya, A. Y. Romanchuk and S. N. Kalmykov, Radiochemistry, 2019, 61, 619–624 CrossRef CAS.
- T. Missana, A. Benedicto, M. García-Gutiérrez and U. Alonso, Geochim. Cosmochim. Acta, 2014, 128, 266–277 CrossRef CAS.
- B. Siroux, A. Wissocq, C. Beaucaire, C. Latrille, C. Petcut, J. Calvaire, M. Tabarant, M. F. Benedetti and P. E. Reiller, Appl. Geochem., 2018, 99, 65–74 CrossRef CAS.
- A. D. Ebner, J. A. Ritter and J. D. Navratil, Ind. Eng. Chem. Res., 2001, 40, 1615–1623 CrossRef CAS.
- C. B. Durrant, J. D. Begg, A. B. Kersting and M. Zavarin, Sci. Total Environ., 2018, 610–611, 511–520 CrossRef CAS PubMed.
- J. L. Littlewood, S. Shaw, C. L. Peacock, P. Bots, D. Trivedi and I. T. Burke, Cryst. Growth Des., 2017, 17, 1214–1223 CrossRef CAS.
- P. A. O'Day, M. Newville, P. S. Neuhoff, N. Sahai and S. A. Carroll, J. Colloid Interface Sci., 2000, 222, 184–197 CrossRef PubMed.
- Y. Yang, H. Chen and C. Ye, Adsorption, 2013, 19, 1019–1025 CrossRef CAS.
- A. Wissocq, C. Beaucaire and C. Latrille, Appl. Geochem., 2018, 93, 167–177 CrossRef CAS.
- M. H. Bradbury and B. Baeyens, Experimental and modelling investigations on Na-illite: acid-base behaviour and the sorption of strontium, nickel, europium and uranyl, 2005 Search PubMed.
- R. D. Shannon and C. T. Prewitt, J. Inorg. Nucl. Chem., 1970, 32, 1427–1441 CrossRef CAS.
- D. R. Brookshaw, R. A. D. Pattrick, P. Bots, G. T. W. Law, J. R. Lloyd, J. F. W. Mosselmans, D. J. Vaughan, K. Dardenne and K. Morris, Environ. Sci. Technol., 2015, 79, 13139–13148 CrossRef PubMed.
- D. L. Parkhurst and C. A. J. Appelo, User's guide to PHREEQC (version 2)—a computer program for speciation, batch-reaction, one-dimensional transport, and inverse geochemical calculations, U.S. Geological Survey, Denver, Colorado, 1999 Search PubMed.
- T. E. Payne, J. A. Davis, G. R. Lumpkin, R. Chisari and T. D. Waite, Appl. Clay Sci., 2004, 26, 151–162 CrossRef CAS.
- A. Voegelin, S. Pfister, A. C. Scheinost, M. A. Marcus and R. Kretzschmar, Environ. Sci. Technol., 2005, 39, 6616–6623 CrossRef CAS PubMed.
- L. Downward, C. H. Booth, W. W. Lukens and F. Bridges, AIP Conf. Proc., 2007, 129–131 CrossRef CAS.
- M. E. McBriarty, S. Kerisit, E. J. Bylaska, S. Shaw, K. Morris and E. S. Ilton, Environ. Sci. Technol., 2018, 52, 6282–6290 CrossRef CAS PubMed.
- A. Roßberg, T. Reich and G. Bernhard, Anal. Bioanal. Chem., 2003, 376, 631–638 CrossRef.
- A. Rossberg, K.-U. Ulrich, S. Weiss, S. Tsushima, T. Hiemstra and A. C. Scheinost, Environ. Sci. Technol., 2009, 43, 1400–1406 CrossRef CAS PubMed.
- V. Brendler, T. Arnold, S. Nordlinder, H. Zänker and G. Bernhard, Speciation and Sorption for Risk Assessment: Modelling and Database Applications, in Geochemical Processes, 2002, pp. 79–94 Search PubMed.
- M. Stockmann, J. Schikora, D. A. Becker, J. Flügge, U. Noseck and V. Brendler, Chemosphere, 2017, 187, 277–285 CrossRef CAS PubMed.
- C. Joseph, J. Mibus, P. Trepte, C. Müller, V. Brendler, D. M. Park, Y. Jiao, A. B. Kersting and M. Zavarin, Sci. Total Environ., 2017, 575, 207–218 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Summary of the results from the mineralogical assessment of the LFLS core samples, the full results of the Sr and Cs adsorption experiments and detailed information on the EXAFS fitting strategies and the respective fits. See DOI: 10.1039/d1em00121c |
| ‡ Current address: Helmholtz Zentrum München GmbH, German Research Center for Environmental Health, Department Infrastructure, Ingolstädter Landstraβe 1, 85764 Neuherberg, Germany. |
| This journal is © The Royal Society of Chemistry 2021 |