
Layer-by-layer coated nanoliposomes for oral delivery of insulin†
Yiming
Zhang
a,
Gordon Minru
Xiong
a,
Yusuf
Ali
b,
Bernhard O.
Boehm
b,
Ying Ying
Huang
 *a and
Subbu
Venkatraman
*a
*a and
Subbu
Venkatraman
*a
aSchool of Materials Science and Engineering, Nanyang Technological University, Blk N4.1, Nanyang Avenue, Singapore 639798. E-mail: yingyinghuang@ntu.edu.sg; assubbu@ntu.edu.sg; Fax: +65 6790 9081; Tel: +65 6316 8976
bLee Kong Chian School of Medicine, Clinical Sciences Building, 11 Mandalay Road, Singapore 308232
First published on 11th November 2020
Abstract
Crossing the intestinal epithelial cell barrier safely and reaching the blood with therapeutic levels of bioactive insulin have been the ultimate goal of oral insulin delivery. The optimum way to overcome the barrier lies in the design of an efficient high drug loading carrier, that can protect insulin from the harsh Gastrointestinal (GI) environment and enhance its uptake and transport by epithelial cells. In the present study, we developed a multi-layered insulin loading strategy on an anionic nanoliposome surface based on electrostatic interaction with chitosan. The layer-by-layer (LbL) coated nanoliposomes achieved high insulin loading (10.7% by weight) and offered superior protection with limited release in simulated gastric fluid (SGF) (about 6% in 1 h), simulated intestinal fluid (SIF) (2% in two weeks), and phosphate buffered saline (PBS) (5% in two weeks). Intracellular imaging revealed that the LbL coated liposomes were internalized and intracellularly trafficked towards the basolateral side of the Caco-2 monolayer. Transported insulin demonstrated retention of bioactivity while crossing the epithelial barrier in the glucose uptake study in 3T3 L1-MBX adipocytes. In rat studies, oral administration of the formulation resulted in rapid absorption with a peak in plasma insulin levels 0.5 h post oral gavaging. This technology thus serves as a promising platform for potential oral insulin applications.
1. Introduction
An oral route of insulin delivery offers the advantage of non-invasiveness and direct delivery to the liver because it mimics the physiological pathway taken by pancreatic insulin, reducing the risk of hypoglycaemia and hyperinsulinemia, and thus has been the most desired insulin therapy in people with diabetes.1 However, delivery of protein drugs via the oral route is challenging because of the chemical and physical barriers in the GI tract. The acidic pH of the stomach and the hydrolytic enzymes in the GI tract degrade any large molecular weight proteins approaching the intestinal surface, lowering the bioavailability of orally administered insulin. Furthermore, the gaps between adjacent epithelial cells are sealed by tight junctions which limit the permeation to only small hydrophobic molecules (<700 Da) and even smaller sized hydrophilic molecules (<200 Da).2 Large molecular weight protein drugs have no chance of crossing the epithelial cell barrier naturally without altering the epithelial membrane and its tight junctions even if they have survived the harsh conditions of the stomach.Current efforts for oral delivery of insulin have revolved around the use of permeation enhancers and insulin analogues.3 For example, Novo Nordisk's oral insulin 338 (I338) is a modified insulin analogue formulated in a tablet together with the permeation enhancer sodium caprate.4 Similarly, Oramed's ORMD-0801 technology involves an enteric coated capsule loaded with permeation enhancer ethylenediaminetetraacetic acid (EDTA) and bare insulin.5 The permeation enhancer plays a part in modulating the tight junction by transiently opening up the intercellular junctional space, allowing the paracellular diffusion of insulin. However, long term reliance on permeation enhancers for treating chronic diseases such as diabetes brings an unavoidable compromise on safety, which has limited the progress of these technologies in the clinical trials. The liposome based hepatic-directed vesicle (HDV) produced by Diasome Pharmaceuticals Inc. is the only nanotechnology to reach clinical phase III trials in the field of oral insulin.6,7 The technology utilizes a liver targeting moiety to alter the surface of the nanoliposome (<150 nm). By targeting the hepatocytes, a much lower dose of insulin would be required to control glycemia. Despite the HDV technology being able to stimulate the liver's involvement in hepatic glucose uptake and prevent hypoglycaemia events, a major drawback of current (injected) insulin regimens, the drug loading remained limited with only 1% drug encapsulation by the nanoliposome.7 There is a need to design a nanocarrier that can be absorbed with minimal alteration of the epithelial membrane, and at the same time capable of loading a large amount of insulin for achieving the desired therapeutic effect.
To overcome the barriers of delivering therapeutic doses via the oral route, protein drugs need to be protected by carriers during their transit through the GI tract and the carriers need to be small enough to initiate cellular uptake and transport across the intestinal epithelial cells. The characteristics of a nanocarrier that are needed for delivering proteins in therapeutic amounts include: (1) high loading capacity for the protein,8,9 (2) ability to protect the protein from the GI environment,10 and (3) ability to enhance cellular uptake and transport by the intestinal epithelial cells.2 Carriers such as liposomes have a long history of application in drug delivery, owing to their biocompatibility as well as their capacity to accommodate both hydrophilic and hydrophobic drugs. Early findings in 1976 demonstrated the possibility of oral insulin by encapsulating it in a liposome, however, low solubility of the protein adversely affected its entrapment and as a result most of the drug was electrostatically associated on the surface.11 To date, there is no liposome-based oral insulin formulation that meets the therapeutic levels required for daily administration. To improve the insulin loading and thereby the bioavailability, we propose the use of surface coating of nanoliposomes, taking advantage of the high surface area to volume ratio of nanoparticles (NP). Surface modification of liposomes can in principle, alter the properties of the liposomes in terms of their stability, release in GI environment, mucosal adherence, drug loading, and all of which contribute to improved bioavailability.12 Keeping these in mind, we report on a LbL-coated nanoliposome that is able to deliver insulin across intestinal epithelial cells.
The layer-by-layer approach can enhance drug loading using multiple alternating layers of protein and counter-ionic polyelectrolyte, and sustained release is achievable based on the rate of defoliation of the top layers.13 By coating the protein on a spherical nanoparticle surface which has a surface area of 4πr2, drug loading will increase exponentially as the particle size (r) increases. This allows enormous room for customizing the drug loading based on the desired therapeutic level, and it can simply be achieved by altering the number of drug layers on the surface. Such an approach brings new insight into loading of challenging macromolecular hydrophilic drugs as the loading will no longer be restricted by the limited core capacity but will be extending into the vast aqueous environment outside the nanoparticle. In our previous study, we have demonstrated that siRNA loaded LbL nanoparticles initiated cellular uptake and the internalized particle was capable of endosomal escape, resulting in 60% SPARC-gene knock down in FibroGRO cells.14 Furthermore, Deng et al. reported that a single layer of siRNA on the nanoparticle surface was able to load 3500 siRNA molecules and co-delivery of the siRNA with the doxorubicin loaded liposome enhanced the serum half-life of up to 28 hours and efficacy by 4 fold in vitro.15 In our system, we propose the use of a layer-by-layer drug delivery system that allows for higher drug loading, especially of macromolecular protein drugs, and has the potential for protecting bioactives including siRNA in the GI environment.13,14
Chitosan has been widely used in oral drug delivery because of its cationic nature which can increase the mucoadhesiveness and residence time of the nanocarrier for enhanced endocytic uptake in the GI tract.16 The polymer itself also acts as a permeation enhancer, which is able to interact with tight junctions for enhanced permeation of orally administered drugs.17,18 The use of chitosan in the LbL system increases tissue residence time and promotes cellular uptake, and slow defoliation of the polymer layers can also promote paracellular diffusion of the insulin released from the underlying layers of the LbL, due to the ability of chitosan to transiently open tight junctions.19 We hypothesize that LbL loading not only helps to preserve the protein structure by eliminating the exposure to extreme solvent conditions, but also enables high protein loading while retaining the nano dimensions of the carrier. This will be of great importance in developing an oral delivery form for insulin, as we show in this paper.
The objective of this study was to improve the loading and transport of insulin across intestinal epithelial cells by coating insulin layer-by-layer onto the liposome surface with the help of cationic chitosan. Human epithelial colorectal adenocarcinoma cells (Caco-2) were used as the in vitro model to study the uptake and transport of LbL-liposomes because of their ability to establish apical-to-basal polarity.20 Chitosan was used for the alternating cationic layers to hold the insulin (negative charge at the pH employed), with the outermost layer being cationic (chitosan). We hypothesized that the cationic outermost layer of the nanoparticle would facilitate trans-cellular transport across the Caco-2 cells, while the “permeation-enhancing” effect of chitosan (free or attached to NPs) would facilitate the para-cellular transport of free insulin.21 The uptake of insulin loaded LbL-liposomes was analysed by confocal microscopy and the amount of transported insulin was quantified by human insulin ELISA. Bioactivity of the transported insulin was investigated by glucose uptake assay in matured 3T3 L1-MBX adipocytes. Finally, in vivo absorption of insulin with the assistance of LbL coated liposomes was demonstrated following oral gavage in Wistar rats.
2. Results
2.1. Fabrication and characterization of insulin loaded LbL-liposomes
The limited loading of macromolecular drugs by nanoliposomes has been hampering its translation for the delivery of therapeutic proteins such as insulin. Diasome pharmaceuticals’ HDV (Hepatocyte-Directing Vesicle) liposome which is currently in clinical trials, encapsulates only 1% of insulin with the majority staying in the free form outside the carrier.7 To improve loading, a layer-by-layer approach was used to modify the nanoliposome surface with multilayers of oppositely charged insulin and chitosan layers, using an anionic liposome core (HSPC/DPPG) (Fig. 1). The liposome aqueous core was loaded with insulin during thin film rehydration before surface modification. Since delaying the release of insulin in the GI environment and promoting cellular uptake were desired for the multi-layered nanoparticle, chitosan was coated as the odd layers to serve as the limiting factor for controlling the diffusion of insulin from both the aqueous core and the multilayers. Chitosan of 3 different molecular weights namely chitosan 15 kDa, chitosan 190–50 kDa, and chitosan 310–190 kDa were used to compare the differences in drug loading capacity. Chitosan with the highest loading capacity was selected for subsequent in vitro and in vivo studies. | ||
| Fig. 1 Schematic drawing of LbL coated liposome and the proposed molecular interaction between insulin and 3 different molecular weights of chitosan. | ||
The DLS measurement shows charge reversal when the anionic liposome surface was modified with chitosan of all three different molecular weights and the pattern repeats as the coating is continued for the subsequent insulin and chitosan layers (Fig. 2). This indicates that the coated liposomes were stabilized by the repelling force generated by the vesicle surface charge and the coating technique is effective regardless of the number of repeating units of the cationic polymer.22,23 Comparing the hydrodynamic radius between the LbL-liposomes coated with the three molar masses of chitosan, increasing hydrodynamic radius was observed as the number of layers increased, and the increment was larger for LbL-liposomes coated with higher molecular weight chitosan. The trend line for each graph in Fig. 2 shows that the average thickness per layer of coating increased from 2.2 nm to 7.4 nm as the polymer molar mass increased from 15 kDa to 310–190 kDa. This is likely due to “excess” cationic charge on the longer chains, which may result in uneven binding to the negatively charged insulin, resulting in coiling and extension into space of the chitosan layer.24 Interestingly, the hydrodynamic radius of both the insulin layers and chitosan layers showed generally an increasing trend as the number of layers increased; if examined separately, however, the size increment after insulin layer addition is generally smaller than that of the chitosan layers. This could be due to the structural difference between chitosan and insulin. Chitosan is a long chain of unbranched polymer consisting of repeating units of amino groups with pKa 6.5, making the polymer positively charged in weakly acidic conditions.25 On the other hand, insulin is a protein that has its unique 3-dimensional shape with an overall negative charge when dissolved in a buffer of pH higher than its isoelectric point. The deposition of chitosan layers on the liposome surface results in partial attachment of the long polymer chains with a portion of it extending out into the solution.26 This creates binding space for the incorporation of insulin, making the protein layer more likely to be condensed because of the molecular interactions such as intra-molecular hydrogen bonding and hydrophobic interaction, and resulting in a smaller size increment for the insulin coating compared to that of chitosan.
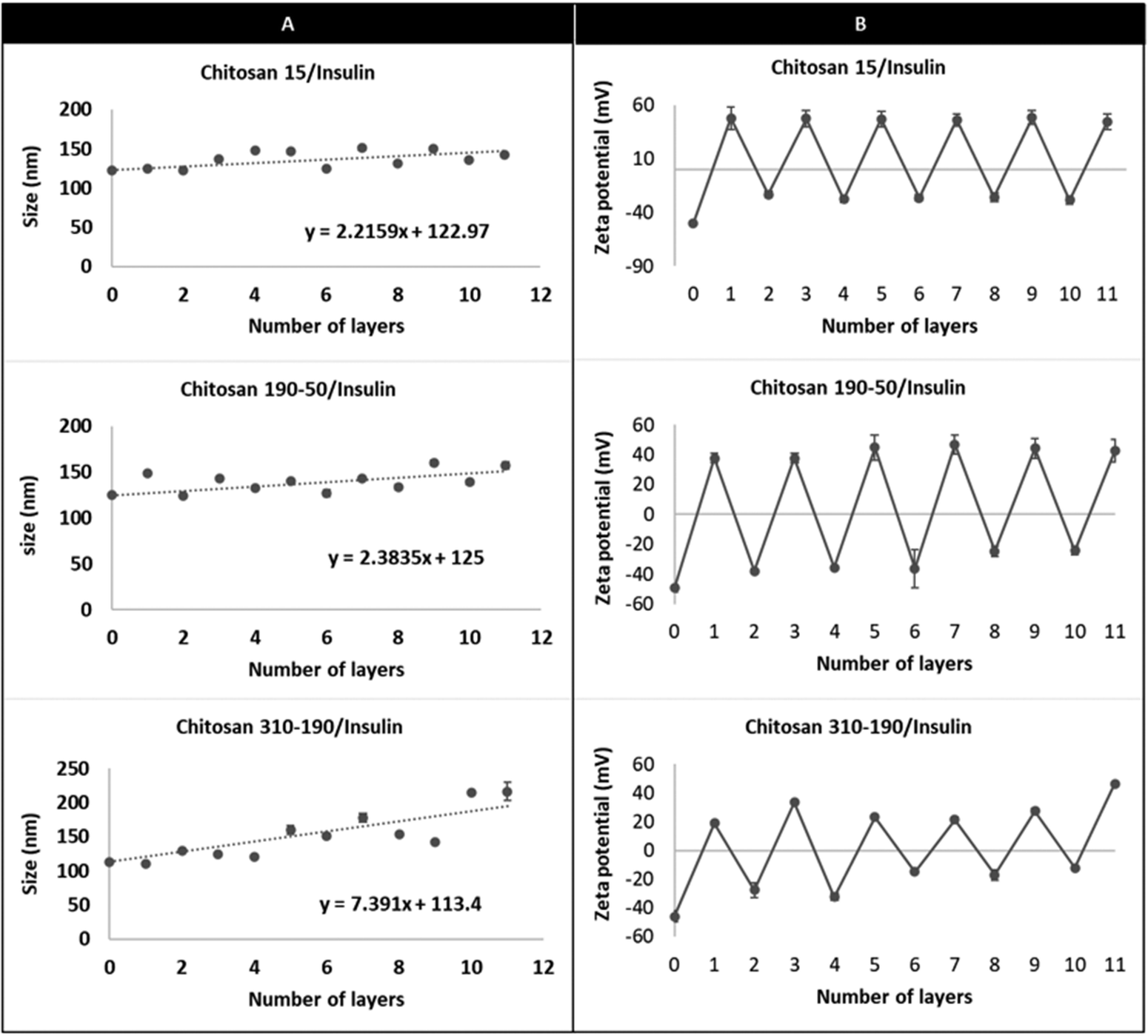 | ||
| Fig. 2 DLS characterization of size and zeta potential of the LbL coated liposome fabricated using alternating layers of chitosan and insulin. Effect of increasing coating layers of insulin and different molecular weights of chitosan (chitosan 15, chitosan 190–50, and chitosan 310–190) on (A) hydrodynamic size and (B) zeta potential. | ||
2.2. Loading of insulin in LbL coated liposome
Two groups of the LbL coated liposome were studied for insulin loading using a fluorescence-based method. The first group addresses the effect of increasing the number of coating layers on the loading of LbL-liposomes, whereas the second group addresses the effect of different molecular weights of chitosan on the loading of LbL-liposomes while keeping the number of layers constant. In the first group, loading of insulin for the LbL coated HSPC/DPPG liposomes (L3, L5 and L11) was significantly higher (*p < 0.05) compared to the uncoated HSPC/DPPG liposomes (L0) in Fig. 3a. L11 (11-layers) with 5 layers of insulin coating on the liposome surface, significantly improved the loading by 13× compared to the conventional thin film rehydration method. While, comparing the loading of insulin between the LbL-liposomes prepared from different molecular weights of chitosan, the loading of LbL-liposomes prepared from chitosan 310–190 reached 10.7% by weight which was significantly higher (*p < 0.05) compared to LbL-liposomes prepared from chitosan 190–50 and chitosan 15 (Fig. 3b). This is simply due to increases in charge density that enables condensation of more insulin molecules per layer. | ||
| Fig. 3 Effect of increasing coating layers and polymer length on insulin loading. (a) Comparison between loading after 3, 5, and 11 layers of chitosan 310–190 per insulin coating and insulin loaded HSPC/DPPG liposome core (L0). (b) Comparison between loading of 11 layers of LbL fabricated with different molecular weight of chitosan/insulin. The data are represented by mean ± standard deviation (n = 3, *P < 0.05). | ||
2.3. Release and stability of the LbL coated liposome in simulated bodily fluid
During the GI transit, insulin experiences a variation of pH from a highly acidic pH 1.2 in the stomach, to pH 6.8 in the small intestine, to finally a neutral pH 7.4 if the drug overcomes all the barriers and reaches the blood. LbL coated liposomes are targeted at protecting insulin during its transit through the GI tract and facilitating its absorption in the small intestine. In order to study the effectiveness of these particles in protecting insulin and the stability of these particles during the GI transit, in vitro release studies were assessed in simulated gastric fluid (SGF pH 1.2), simulated intestinal fluid (SIF pH 6.8), and Phosphate-buffered saline (PBS pH 7.4).First, let us consider a 11-layer LbL-coated nanoparticle with 5 layers of insulin and 6 layers of chitosan. If the 5 layers are distributed equally, each layer is 20% of the total amount of insulin. At pH 1.2 (SGF), about 6% insulin is released in one hour (Fig. 4a), implying that the chitosan has swollen enough to allow for some release of insulin. At the higher pH of 6.8 (SIF), this release is much slower, with less than 0.2% of insulin being released in 8 hours, consistent with lowered swelling of the chitosan (lower degree of ionization) at pH 6.8 compared to pH 1.2 (100% ionization). Turning to the effect of layering (Fig. 4b), we find that the percentage of insulin released is roughly the same, regardless of it being 3-layers (1 insulin layer) or 5-layers (2 insulin layers). The 3-layered system, however, releases somewhat higher percentage over 7 days, as would be expected. Nevertheless for 11-layered systems, the release is very slow at pH 6.8 implying that the insulin stays mostly encapsulated while in the small intestine. The release is similar at pH 7.4, as would be expected of a low degree of ionization of the chitosan. Looking at the zeta potential data (Fig. 4d), it appears that the defoliation of the outermost chitosan layer is completed in 7 days. Chitosan is a long chain of unbranched polymer consisting of repeating units of amino groups with pKa 6.5, making the polymer positively charged at pH 1.2. The human recombinant insulin used in this study has an isoelectric point (pI) of 7, which becomes positively charged when the surrounding pH is lower than its pI. At pH 1.2, defoliation or drug release is faster because both chitosan and insulin are positively charged, creating repulsion force which accelerates the penetration of water and charged ions into the underlying layers. At SIF pH 6.8 and pH 7.4 which is above the pKa of chitosan's amino groups, both chitosan and insulin should be neutrally charged. Defoliation or drug release is slowed down due to the hydrophobic interaction between chitosan and insulin which prevents water penetration.
 | ||
| Fig. 4 Release and stability of the LbL coated liposome in SGF 1.2, SIF 6.8, and PBS 7.4 (a) release of 11-layers LbL coated liposome in SGF pH 1.2 and SIF pH 6.8, (b) comparison of release between 3 and 5 layer LbL coated liposome and uncoated liposome (L0) in PBS pH 7.4. (c) Hydrodynamic size and (d) zeta potential of 3, 5 layer LbL coated liposome and uncoated liposome (L0) in PBS pH 7.4. | ||
2.4. Caco-2 uptake and transport of LbL coated liposome
In order to determine the biological relevance of the fabricated insulin-chitosan liposomes, the LbL coated liposome formulation was tested on human epithelial colorectal adenocarcinoma cells in culture (Caco-2 cells) to investigate the ability to be taken up and transported across the intestinal epithelial cells in vitro. Caco-2 cells were seeded on a glass coverslip and maintained for 21 days with medium replacement every 2 days before being treated with the LbL coated liposomes. The lipid bilayer of the liposome inner core (11-layered system) was fluorescently tagged with coumarin-6 for monitoring intracellular trafficking (Fig. 5). Confocal microscopy revealed that these nano-sized LbL coated liposomes were heavily up-taken by Caco-2 cells and the nanoparticles translocated to the basal side of the monolayer after 4 hours of treatment as shown in the 3D rendering in Fig. 5b. The Z-stack analysis allowed visualization of the monolayer on different planes (from the apical to the basolateral surface) and thus showed the intracellular position of the endocytosed nanoparticles at different depths. Images at different Z-positions, which represent the distance away from the bottom of the monolayer, reflected that the endocytosed nanoparticles concentrated near the bottom of the monolayer. As the Z-position decreased from 12.03 μm to 1.9 μm, the green fluorescence from the nanoparticles gradually increased (Fig. 5a), indicating that the nanoparticles have been intracellularly trafficked from the apical side to the basal side of the caco-2 cells. The outermost layer of the LbL coated liposome (11-layered) is cationic, which enables cellular uptake due to favourable interaction with the negatively charged cell surface.19 | ||
| Fig. 5 Cellular uptake and transport of LbL coated liposome by Caco-2 cells. (a) Confocal microscopy imaging of Caco-2 monolayers at different z-positions. Green represent LbL coated liposome fluorescently tagged with coumarin-6 in the liposome lipid bilayer. Red represents immunostaining for Claudin-1. Blue represent DAPI staining. (b). 3D rendering of the stack of confocal images with the orientation representing apical to basal. Inset represents schematic drawing illustrating the position of the fluorescence tag in the lipid bilayer of the liposome core. (c) Cumulative transport of insulin across Caco-2 cells over 4 hours. Amount of insulin measured by ELISA. (d) TEER measurement during the transport study. (e) Alamar blue assay of Caco-2 cells immediately performed after the transport study. | ||
Transport of insulin across the Caco-2 cells was studied by ELISA (Fig. 5c). The amount of insulin being transported across the Caco-2 cells with the help of the LbL carrier was more than 3× higher than bare insulin. TEER measurement during the 4 hours of transport indicated that no significant changes to the tight junctions occurred during the uptake and transport of the LbL coated liposomes as the TEER values remained at a value above 500 Ω cm2 during and 24 hours after treatment (Fig. 5d). Since the particles are stable in SIF conditions with minimal insulin release, and the tight junction was intact as indicated by the increasing TEER values, this enhanced transport of insulin is likely to be the result of carrier facilitated endocytic uptake of the LbL coated liposome, followed by exocytosis of intracellularly released insulin while crossing the Caco-2 monolayer. Alamar Blue assay was performed immediately after the treatment to investigate cytotoxicity of the LbL nanoparticles. The results showed that the percentage cell viability was higher when cells were treated with insulin alone or LbL nanoparticles compared to cells treated with HBSS control (Fig. 5e). This increase in percentage viability could be due to the growth promoting effect of insulin released from the LbL nanoparticles, further indicating that the LbL formulation was safe for use in vitro.
2.5. Glucose uptake by 3T3 L1-MBX
3T3 L1-MBX fibroblasts were differentiated into adipocytes for the study of glucose uptake of the transported insulin. 3T3 L1-MBX fibroblasts were maintained in DMEM containing 10% fetal bovine serum and 1% antibiotic-antimicotic and used for differentiation within 10 passages. Cells were maintained in maintenance medium (DMEM containing 3% fetal bovine serum and 1% antibiotic-antimicotic) for 4 days prior to treatment with maintenance medium containing insulin (1 μg mL−1), isobutylxanthine (0.5 mM), dexamethansone (1 μM), and rosiglitazone (2 μM) to the maintenance medium, followed by treatment with maintenance medium containing insulin (1 μg mL−1). Medium was replaced back with maintenance medium after 2 days and maintained for another 8–11 days with medium replacement every 2 days until the glucose uptake experiment was performed. Morphologically, the elongated fibroblast gradually altered into spherical fat storing cells with big intracellular vesicles and reduced size of the cytoplasm (Fig. 6a–d). Oil red staining indicated that the cells had been fully differentiated into adipocytes (Fig. 6e–f). Upon maturation, the fully differentiated 3T3 L1-MBX cells were treated with the basal compartment fluid collected from the transport study. An insulin titration curve was first established by measuring the glucose response of matured adipocytes when treated with known concentrations of insulin. From Fig. 6g, a typical sigmoidal curve was obtained indicating that the cells were responsive to glucose when treated with the insulin used for fabricating LbL coated liposome. In Fig. 6h, the luminescence detected for cells treated with the transport fluid was about 3.5× higher than the cells treated with PBS negative control. The glucose response was correlated to 1–100 nM of insulin based on the insulin titration curve, suggesting that a substantial amount of insulin after crossing the intestinal epithelial cells was bioactive in triggering the glucose uptake in adipocytes. The results from the glucose uptake study suggests that LbL coated liposomes were effective at protecting the insulin during its intracellular trafficking by Caco-2 cells, and the transported insulin has retained its bioactivity while crossing the intestinal epithelial Caco-2 monolayer. | ||
| Fig. 6 Differentiation of 3T3 L1-MBX fibroblast into adipocytes for the bioactivity study of transported insulin. (a)–(d) Morphology of the 3T3 L1-MBX fibroblast after 0, 12, 14, 25 days of differentiation. (e) and (f) Oil red staining of matured 3T3 L1-MBX adipocytes. (g) and (h) Insulin response curve and glucose uptake of transported insulin performed on matured 3T3 L1-MBX adipocytes. The data are represented by mean ± standard deviation (n = 3, *P < 0.05). | ||
2.6. Lyophilization of LbL coated liposome
Lyophilisation was carried out to increase the shelf-life of the formulation for extended storage stability. Reconstituted lyophilized formulation was used throughout the animal study. The LbL coated liposome also demonstrated excellent stability during lyophilisation when an appropriate cryo-protectant was added (Fig. 7). Comparing between two common cryo-protectants sucrose and trehalose, increasing the concentration ensures better stability during lyophilisation and size recovery upon reconstitution in water. Trehalose served as a better cryo-protectant compared to sucrose when applied at 5% during lyophilisation since the size recovered closest to the original size with only 27.9 nm increment upon reconstitution. Trehalose has been used as a cryoprotectant in stabilizing nanoparticles during freeze drying due to its advantages such higher glass transition temperature Tg, lower hygroscopicity and the ability to form flexible hydrogen bonds with nanoparticles which allows easy removal of these sugars from the nanoparticle surface after lyophilisation.27,28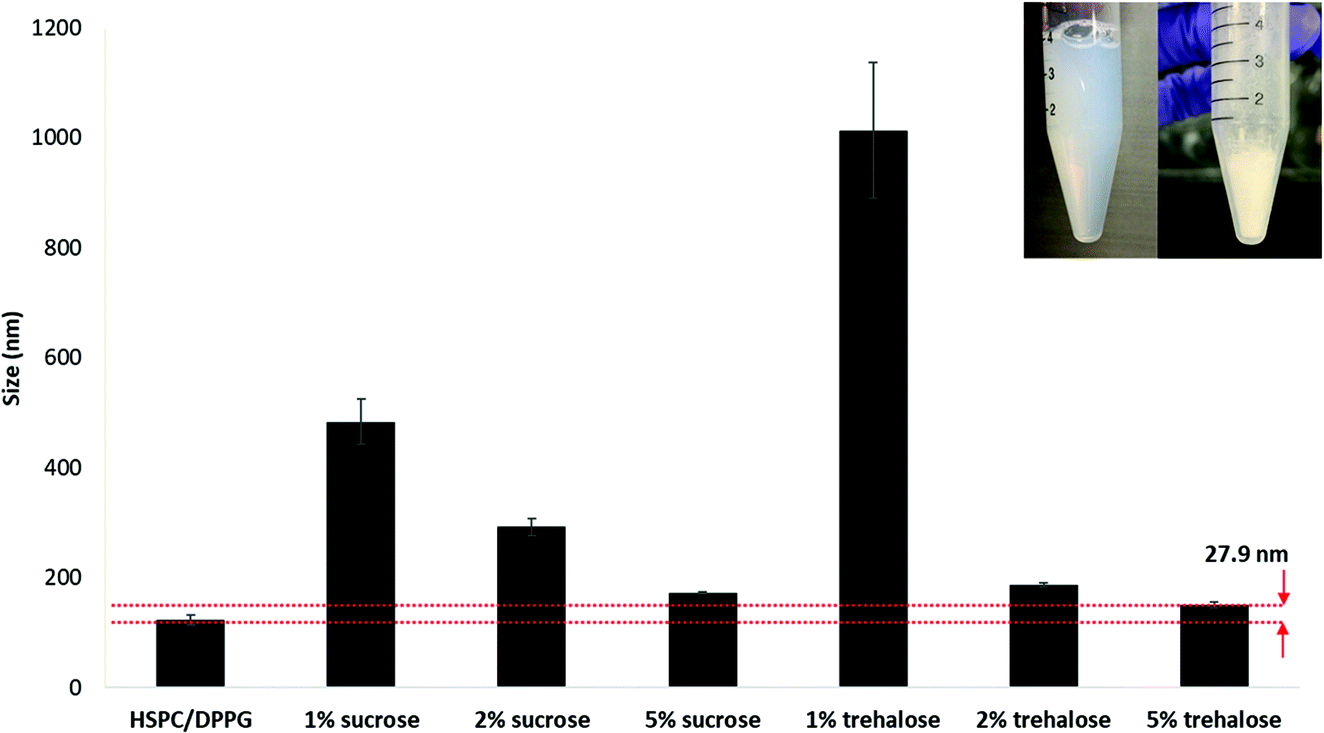 | ||
| Fig. 7 Lyophilization of LbL coated liposome with varying concentrations of cryoprotectants, sucrose and trehalose. | ||
2.7. Pharmacokinetics of plasma insulin level in vivo
To study the pharmacokinetics of insulin absorption and elimination, a total of 12 Wistar rats with 4 animals per treatment group was used. The LbL coated liposome formulations were fed to over-night fasted rats via oral gavage in solution and capsule form. Human insulin ELISA (Mercodia, Sweden) with no cross-reactivity to endogenous rat insulin (0.7%) was used to measure the blood distribution of human insulin loaded inside the LbL coated liposome. Lyophilized LbL-liposome formulation was applied at maximum dosage in both solution and capsule form to select the group with a positive outcome, based on which a potential method of delivery will be selected for dosage optimization and efficacy test. Oral administration of 320 IU kg−1 insulin loaded chitosan 310–190 LbL nanoparticles (11-layered system) in solution resulted in a rapid increase in plasma insulin concentration which peaked at 0.5 hours with maximum absorption of close to 3 mIU L−1 and a subsequent decrease to the baseline level within the next 3.5 hours due to elimination (Fig. 8). On the other hand, no significant insulin was detected in the plasma when the same formulation was administered in an enteric coated capsule at 43 IU kg−1. One possible reason could be that the formulation loaded inside the capsule was not released during the short passage time in the GI of the rat, causing the formulation to be excreted before being absorbed. It is also possible that the dose given in the capsule was not high enough to deliver measurable levels of insulin in the plasma. Furthermore, the volume and concentration of the solution group might also influence the relationship between applied dosage and plasma insulin level. Considering that in an animal model, absorption of intact orally administered protein drugs is virtually impossible as macromolecules will be digested into their simplest units before absorption, any amount of insulin detected in the blood means the LbL nanoparticles has overcame significant amount of hurdles in delivering insulin across intestinal epithelial cells. | ||
| Fig. 8 In vivo pharmacokinetics study of insulin loaded in LbL coated liposome orally administered to Wistar rats. | ||
3. Discussion
In the present study, we present a facile layer-by-layer method for loading large amounts of insulin on the surface of a nanoliposome. The LbL nanoparticles outperform the conventional liposome in terms of (1) drug loading, (2) protection against GI environment, and (3) penetration of intestinal epithelium with retention of bioactivity. The release of insulin from the inner layers in this LbL system is dictated by the speed of defoliation of the outer layer, and this defoliation (or swelling) is slower at higher pH, where the chitosan is less ionized. Conversely, the release is expected to be higher in the SGF, with a pH of 1.2, where the chitosan is highly ionized and therefore swells to a larger extent. Thus, it is expected that about 6% of the insulin in the nanoparticle may be lost in the stomach, but 94% should be still encapsulated as it passes into the small intestine. This loss may be decreased by using a capsule with enteric coating that has to be precisely controlled for releasing the content at the right section of the small intestine.Most of the existing nanomedicines suffer from low drug loading with almost all formulations, having the drug content being less than 10% of the NP weight.29 This has been a stumbling block in translation,8 because even if sufficient absorption of NPs occurs in the small intestine, drug bioavailability is still inadequate for therapeutic effect. For example, Diasome pharmaceuticals’ HDV liposome can only hold 1 IU of insulin per 1 mg of HDV, resulting in a formulation consisting of 99% free insulin and only 1% is associated with the carrier.7 Trying to load insulin into the core of the nanoliposome has not met with any success because of (1) the large size of the protein, (2) difficulty of driving insulin into the core, and (3) limited aqueous core capacity of the liposome. Due to the abovementioned reasons, protein tends to sit on the surface of the liposome instead of moving inside the core during thin film rehydration. Other methods such as active loading have been extensively explored, but due to the size, charge, and hydrophilic nature of the protein, little success has been achieved as diffusion across the lipid bilayer after carrier formation was almost impossible.
In our technology, coating only 5 layers of insulin on the liposome surface using Chitosan 310–190 kDa results in a 10.7% loading by weight which is considered relatively high for a nanocarrier. Increasing the number of coating layers results in increasing amount of loaded insulin per particle; this implies that even if insulin release rates remain the same, greater cumulative amounts of insulin are in fact released with the higher loading. Furthermore, since insulin is loaded on the surface of a spherical liposome, loading of insulin will be size and surface area dependent. The surface area of a spherical nanoparticle is 4πr2, which is directly related to insulin loading and will have an exponential relationship with the particle radius. Loading will increase as the size of the particle increases, which will provide a larger surface area to accommodate more insulin as the number of layers increase. In addition, most current nanocarriers only load insulin inside the core, which has a limited capacity and drug loading becomes extremely challenging when the carrier size is down to the nanometer. In contrast, the biggest advantage of the present technology is that a new approach supporting loading of protein on the nanocarrier surface was developed, the successful application of which allows enormous room for improving loading by extending the protein layer into the vast outer aqueous space, significantly changing the way a protein drug can be loaded. The timeframe of release of the LbL coated nanoliposome is superior in SGF, SIF, and PBS due to the direct complexation of insulin which stabilizes the protein within the layers via electrostatic interactions. The stability of the LbL coated liposome was excellent in PBS 7.4 with only 50–60 nm increase in size over a period of 4 weeks at 37 °C (Fig. 4c and d). This was not achieved in the well-established chitosan TPP nanoparticles as the particles were unstable at pH 7 and above with almost 60% release at pH 7.4 within a few hours. As a result, the compromised particle also loses its ability to be taken up by cells.30,31
The LbL coated nanoliposome has a cationic outermost layer that facilitates its association, uptake and transport by the intestinal epithelial cells, specifically Caco-2 cells. In our study, chitosan was selected as a coating layer to improve insulin loading because of its ability to form ionic complexes with negatively-charged drugs, and its biodegradability as well as reported biocompatibility. In a couple of studies, free chitosan has been reported to act as a permeation enhancer, enabling increased transport via the paracellular pathway: enhanced enteric absorption of insulin and a hypoglycemic effect was observed after oral administration in mice32 and rats.33 It was postulated that chitosan enhances paracellular transport of insulin by mediating with the tight junction protein claudin-4, thereby opening up the junctional space for the passage of insulin.34 However, our studies do not see an enhancement of paracellular transport by chitosan-coated nanoliposomes. TEER measurement during the in vitro transport study (Fig. 5d) showed that LbL coated liposomes assist the transport of insulin mainly via transcellular transport primarily due to two reasons. Firstly, no free chitosan was present in our formulation to interact with tight junction proteins because after the final step of coating, all the free polymers were removed by ultracentrifugation. Secondly, the chances of the top chitosan layer defoliating and then interacting with tight junctions to enhance localized paracellular transport of released insulin were very low as seen from the slow release, and slow defoliation, as gauged by the zeta potential (Fig. 4d). In addition, it is not likely that the LbL coated liposome with a size of about 200 nm, is able to pass through the paracellular space, because the tight junctional space when fully opened by any enhancers is only 20 nm.35 TEER values first decreased 1 hour after treatment and subsequently increased from 2 to 4 hours, following the same pattern as HBSS control (buffer without any nanoparticles), suggesting that the tight junction was not involved to cause any paracellular transport of insulin. The transported insulin detected in the basal compartment was due to transcellular transport of the endocytosed insulin loaded LbL coated liposome. This observation was aligned with confocal analysis which further confirmed that the endocytosed particles entered the cytoplasm and travelled towards the basal side of the cell (Fig. 5a–b).
The amount of insulin transported across the Caco-2 monolayer was 3-fold higher when loaded in the LbL coated liposome compared to bare insulin solution. Bioactivity of the transported insulin can be directly measured by its ability to trigger glucose uptake in adipocytes. In type I diabetes, the body's own immune cells destroy the insulin producing β cells, as a result glucose cannot enter the adipose or muscle cells for adenosine triphosphate (ATP) production. Bioactive insulin would be able to bind to the insulin receptor on the adipocyte surface to initiate the entry of 2-deoxyglucose (2DG) and accumulation of deoxyglucose-6-phosphate (2DG6P) inside the cell, which will be converted to a luminescent signal for detection.36 In our study, the transported insulin retained its bioactivity while crossing the Caco-2 monolayer as demonstrated by the glucose uptake by matured 3T3 L1-MBX adipocytes. This is encouraging because one of the most challenging problems for oral drug delivery is to prevent drug degradation and to retain bioactivity during GI transit and intestinal absorption.
A preliminary feasibility study in an animal model demonstrated that the lyophilized formulation assisted the absorption of insulin in rats when oral gavaged in solution form. Lyophilisation of the LbL coated liposome with 5% trehalose preserves the particle and ensures longer shelf life. Rapid absorption of the formulation results in a peak in plasma insulin 0.5 hour post oral gavaging at 3 mIU L−1 (Fig. 8). In contrast, absorption of insulin-loaded Eudragit® RS nanoparticles prepared using an emulsion technique, was prolonged with a broad peak of absorption between 6–8 hours.37 The study reported the absorption was due to mucoadhesion of the nanoparticles which gave out localized delivery of insulin on the small intestinal wall. Although similar levels of plasma insulin were detected (∼5 mIU L−1) with Eudragit® RS nanoparticles of lower dose (50 IU kg−1), it is worth noting that the plasma insulin peak was broad and was too close to that of the basal level to give a conclusive evidence of significant absorption. On the other hand, the present technology resulted in rapid absorption (0.5 h) with a sharp peak in plasma insulin at about 3 μIU mL−1 after oral administration of LbL-liposome with only 5 layers of insulin, and the duration in the blood stream was short (up till 4 hours) due to rapid elimination, demonstrating a proof of concept. This plasma insulin can be further increased just by repeating the number of alternating insulin and chitosan layers on the liposome surface. Theoretically, a total of 182 alternating layers (91 layers of insulin) could be coated on the liposomes surface while keeping the size of the particle <500 nm for optimal cellular uptake.38 This will bring significant clinical importance for the purpose of blood glucose reduction.
In another study, the serum insulin level peaked at about 50 mIU L−1 5 hours after the oral administration of the powdered form of TPP chitosan nanoparticles loaded in enteric capsules.31 From their stability study, these pH dependent nanoparticles aggregated at pH 7 and above rapidly releasing more than 60% of insulin within 4 hours at pH 7.4. The pH in the intestinal tract varied from pH 6.6 ± 0.5 in the proximal small intestine to pH 7.5 ± 0.4 in the terminal ileum.39 By the end of 3 hours when the capsule releases the formulation, it was expected to aggregate and lose its ability to enhance cellular uptake and transport, the only possible reason for the absorption was paracellular transport with the aid of disintegrated chitosan polymer from the destabilized formulation. Premature release of insulin in the GI tract may expose the protein in the enzyme-rich brush border environment of the small intestine, which indicates that the carrier was incapable of protecting its payload during intestinal penetration. Indeed, the TPP chitosan nanoparticle was not optimized at transcellular transport of nanoparticles across the intestinal epithelium, but functions as a permeation enhancer which was released due to particle instability in the intestinal pH to permit paracellular diffusion of insulin. A major drawback of using permeation enhancer is the potential damage to the intestinal lining.6 Major pharmaceutical companies such as Novo Nordisk's I388 and Oramed's ORMD-0801 have their technology focused on permeation enhancers namely sodium caprate and ethylenediaminetetraacetic acid (EDTA), respectively.4,5 However, being the potential treatment of a chronic metabolic disease whereby repeated once-daily administration is inevitable, prolonged exposure of the intestine to high local concentrations of sodium caprate or EDTA imposes unavoidable safety concerns. In addition, bioavailability of orally administered I338 remained low, and it was hypothesized that the unabsorbed insulin might result in increased risk of proliferative effects (cancer-causing) in localized areas of the gastrointestinal tract due to direct exposure to high levels of I338. Due to these safety related issues, long-term safety trials remain necessary to evaluate the possible outcomes of persistent exposure of the intestine to high local concentrations of permeation enhancers.
In comparison, the present technology delivers insulin via a transcellular pathway which is safer and more desirable because absorption take place without disrupting the tight junctions, which is important in maintaining the barrier function. The LbL coated liposome was able to protect insulin during intestinal penetration and the loading can be further improved by increasing the number of insulin layers. Considering the protein nature of insulin and its usual destiny after oral administration, the current in vivo finding is encouraging for further translation. In summary, these results (in vitro and in vivo) indicate the potential of the LbL technology using chitosan and insulin as an oral delivery system for treating diabetes mellitus.
4. Conclusion
A layer by layer technique of surface modifying liposomes with alternating layers of chitosan and insulin was developed, leading to a liposome-based nanocarrier with high insulin loading (>10% by weight). The LbL coated liposome demonstrated excellent stability for a 4 weeks study in PBS pH 7.4 at 37 °C. The outermost chitosan layer of the LbL coated liposome facilitated cellular uptake and transport by Caco-2 cells and the transported insulin demonstrated retention of bioactivity through glucose uptake assay performed on 3T3 L1-MBX adipocytes. These LbL coated liposomes were able to protect insulin during its GI transit and ensure its insulin payload reached the systemic blood circulation, as verified in a pharmacokinetic study in a rat model, thus indicating the potential application of these nanoparticles in the field of oral protein delivery.5. Experimental section
5.1. Materials
Hydrogenated soybean phosphatidylcholine (HSPC), and 1,2-dipalmitoyl-sn-glycero-3-phosphoglycerol, sodium salt (DPPG) were purchased from Coatsome. Chitosan of molecular weight 15 kDa (chitosan 15), 190–50 kDa (chitosan 190–50), and 310–190 kDa (chitosan 310–190) was obtained from Sigma-Aldrich. Human recombinant insulin, Triton X-100, coumarin-6, carbonate–bicarbonate buffer, HBSS, HEPES, NaHCO3, and 12-well Transwell inserts were purchased from Sigma-Aldrich. ELISA kits were purchased from Mercodia. Caco-2 and 3T3 L1-MBX cells were purchased from ATCC. Alexa Fluor (AF) 647 was purchased from Thermo Fisher. Glucose uptake-Glo™ assay was purchased from Promega.5.2. Liposome fabrication and insulin loading
100 nm liposomes were synthesized following a thin film rehydration method. Briefly, a known amount of HSPC lipids were weighed and dissolved in chloroform and methanol solvents in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio in a round bottom flask. Fluorescent lipid coumarin-6 was added at a 0.5 mol% for the transport study of empty liposomes. Anionic lipid DPPG was added into the organic solvent mixture at 10 mol% for the preparation of a negatively charged surface. Solvents were maintained at constant temperature at 60 °C and allowed to evaporate under a stepwise reduction in pressure and finally stabilized at 20 mbar for an hour in a rotary evaporator (BUCHI Rotavapor® R-100). Insulin solution (10 mg mL−1) containing sodium phosphate dibasic was adjusted to pH 4 using 1 M HCL. A gradient dilution intended to improve the loading was achieved by rehydrating the thin film following a gradient dilution. Generally, insulin solution of 500 μL of 10 mg mL−1, 5 mg mL−1, 2.5 mg mL−1, and 1 mL of 1.25 mg mL−1 was first added to the thin film in a step wise manner with a 5 min interval in between each addition. Subsequently, 2.5 mL of carbonate-bicarnonate buffer (CBB) (pH 9.6) was added to dilute the liposome solution so that the lipid concentration of the final solution remains at 20 mM.
:1 ratio in a round bottom flask. Fluorescent lipid coumarin-6 was added at a 0.5 mol% for the transport study of empty liposomes. Anionic lipid DPPG was added into the organic solvent mixture at 10 mol% for the preparation of a negatively charged surface. Solvents were maintained at constant temperature at 60 °C and allowed to evaporate under a stepwise reduction in pressure and finally stabilized at 20 mbar for an hour in a rotary evaporator (BUCHI Rotavapor® R-100). Insulin solution (10 mg mL−1) containing sodium phosphate dibasic was adjusted to pH 4 using 1 M HCL. A gradient dilution intended to improve the loading was achieved by rehydrating the thin film following a gradient dilution. Generally, insulin solution of 500 μL of 10 mg mL−1, 5 mg mL−1, 2.5 mg mL−1, and 1 mL of 1.25 mg mL−1 was first added to the thin film in a step wise manner with a 5 min interval in between each addition. Subsequently, 2.5 mL of carbonate-bicarnonate buffer (CBB) (pH 9.6) was added to dilute the liposome solution so that the lipid concentration of the final solution remains at 20 mM.
5.3. Quantification of encapsulation efficiency and loading efficiency
Fabricated 100 nm liposomes were ultracentrifuged (Thermo Fisher, SORVALL WX ultra) at 50000 rpm, 4 °C for 30 min. The concentration of un-encapsulated insulin in the supernatant, CrhINS,SN (mg mL−1), was determined using microBCA assay against a standard curve obtained by serial dilution of insulin of known concentration. The ratio of insulin concentration in the supernatant CrhINS,SN (mg mL−1) to the total insulin concentration CrhINS,TOT (mg mL−1) can be used to calculate the percentage encapsulation efficiency (EE %) using eqn (1): | (1) |
Following the ultracentrifugation step, the pellets were separated from the supernatant and re-suspended in DI water. Complete resuspension was achieved by vortex mixing. Thereafter, the liposomal suspension was transferred to a pre-weighed microtube and frozen overnight in a −80 °C freezer before being placed in a manifold freeze-dryer for 24–48 h. The mass of the insulin-loaded liposomes was determined from the difference between the mass of microtubes containing powdered liposomes and that of empty microtubes. Percentage loading efficiency (LE %) was calculated using eqn (2):
 | (2) |
To calculate the loading of insulin in the layers on the surface of the liposome, insulin was fluorescently tagged with AF647 (Thermo Fisher Scientific) following the manufacturer's protocol before being coated as the layers on the liposome surface. Standards were prepared by serially diluting known concentrations of the AF647 tagged insulin. The AF647 fluorescence intensity of the final coated particle was compared to that of the particle before coating using a fluorescent microplate reader (Tecan Infinite 200). Coumain-6 serves as an internal standard for normalization of the liposome to ensure equal amounts were used for comparison. Excitation and emission were set at 650 nm and 680 nm for AF647, while they were 480 nm and 530 nm for coumain-6. The mass of insulin loaded as layers on the surface of the liposome was calculated based on the difference in fluorescence before and after coating using fluorescence intensity of serial dilutions of known concentrations of insulin. The mass of the total insulin loaded LbL coated liposome was measured by resuspending the ultracentrifuged LbL coated liposome in DI and freeze dried in a pre-weighed microtube. The loading of insulin using the LbL coated liposome was calculated based on eqn (3).
 | (3) |
5.4. LbL coating of liposomes
Anionic HSPC liposomes containing 10 mol% DPPG lipids were coated with alternating layers of chitosan and insulin based on electrostatic interaction. Odd layers were positively charged chitosan of three molecular weights namely, chitosan 15, chitosan 190–50, and chitosan 310–190. Even layers were negatively charged insulin prepared in CBB buffer. Briefly, odd layers were coated by mixing 0.1% (w/v) chitosan solution in acetic acid (0.1% v/v) with 1 mM liposomes, followed by ultracentrifugation at 50000 rpm, 4 °C for 60 min to pellet down the coated particles. Subsequently, the pellets were resuspended in acidic water (pH 1–2) before injecting into 1 mg ml−1 insulin solution in CBB buffer for the even layers. The process was repeated until 11 layers of coating were achieved for all three molecular weights of chitosan.
5.5. Dynamic light scattering characterization
The liposome suspension was diluted 100 times with deionized (DI) water for analysing its size and zeta potential using a Zetasizer Nano1 (Malvern Instruments, Malvern, UK). Disposable polystyrene cuvettes were used for measuring size, while a folded capillary cell was used for measuring the charge of liposomes.5.6. Release study
The release study was performed on LbL coated liposomes in SGF pH 1.2, SIF pH 6.8, and PBS pH 7.4 at 37 °C under constant stirring. LbL coated liposomes were placed in a dialysis bag with pore size 300 kDa. Release samples were collected and quantified by ELISA.5.7. Cellular uptake study
Caco-2 cellular uptake was studied by flow cytometry and confocal microscopy. Caco-2 cells were seeded in 6-well plate with glass cover slip, maintained for at least 21 days before treating with coumarin-6 tagged LbL liposomes. After 4 hours of treatment, cells grown on the coverslip in the 6-well plate were washed twice with PBS, fixed with ice cold methanol and perforated with 0.1% Triton-X, before staining immunostaining with anti-claudin-1 antibodies. After staining, coverslips were inverted and mounted using VectaShield mounting media onto a microscopy slide for imaging by ZEISS confocal laser scanning microscope LSM 710.5.8. Transport study
To establish an in vitro cell model for the transport study, Caco-2 cells were grown on 12-well Transwell inserts with 0.4 μm pore size with TEER values monitored every second day for the development of a monolayer for 21 days. Caco-2 cells were cultured in T75 flasks with Dulbecco's Modified Eagle's Medium (DMEM) high glucose medium containing 20% FBS, 1% Pen strep, and 1% essential amino acids and harvested when the cells were 70–80% confluent. Cells were seeded at 300000 cells per well on a 12 mm permeable membrane support. Medium was changed every other day post seeding and transepithelial electrical resistance (TEER) values were recorded using epithelial voltohmmeters (EVOM) equipped with “chopstick” electrodes (World Precision Instruments, Sarasota, FL). The transport experiment was performed 21 days post-seeding when the TEER values reached above 300 Ω cm2. Hanks’ balanced salt solution was prepared by dissolving 9.7 g of HBSS, 4.7 g of HEPES, and 0.35 g of NaHCO3 in 1 litre of DI water. 1 M NaOH was used to adjust the pH of the solution to pH 7.4 and the buffer solution was sterile filtered using a 0.22 μm pore size membrane. The transport experiment was carried out following an established protocol.40 Briefly, 500 μL of 1 mM LbL coated liposomes were added in the apical compartment and 1.5 mL of the buffer were added into the basal compartment of the 12 well insert. Sampling was performed at 0, 1st, 2nd, 3rd, and 4th hour with complete buffer replacement from the basal compartment. Alamar Blue (Biorad) assay was performed immediately after the transport study on Caco-2 cells cultured on a Transwell membrane. Briefly, Caco-2 cells after treatment were incubated with 50 μl of Alamar blue dye diluted with PBS at 37 °C for 6 hours. The results were obtained using a microplate reader using an excitation wavelength of 560 nm and emission wavelength of 590 nm.
5.9. Glucose uptake study
3T3 L1-MBX fibroblasts were differentiated into adipocytes following the recommended protocol from the Glucose Uptake-Glo™ kit. 3T3 L1-MBX fibroblasts were maintained in DMEM containing 10% fetal bovine serum and 1% antibiotic-antimicotic and used for differentiation within 10 passages. On day 1, cells were seeded at 20000 cells per 100 μl in a 96-well plate and maintained in maintenance medium (DMEM containing 3% fetal bovine serum and 1% antibiotic-antimicotic) for 4 days. On day 5, medium was replaced with 100 μl of differentiation medium-I which was prepared by adding differentiation drugs including insulin (1 μg mL−1), isobutylxanthine (0.5 mM), dexamethansone (1 μM), and rosiglitazone (2 μM) to the maintenance medium. Medium was replaced every 2 days. On day 12, medium was replaced with 100 μl of differentiation medium-II which was prepared by adding insulin (1 μg mL−1) to the maintenance medium. On day 14, medium was replaced back with maintenance medium and maintained for another 8–11 days with medium replacement every 2 days until the glucose uptake experiment was performed. Insulin response was evaluated for glucose uptake assay upon maturation of the 3T3 L1-MBX adipocyte, and luminescence of cells treated with transported insulin and PBS control was measured using a microplate reader (Tecan Infinite 200). Cells were treated with known concentrations of insulin for generating the standard insulin response curve for comparison.
5.10. Quantification of transported insulin
The amount of insulin in the collected buffer from the transport study was quantified for transported insulin by ELISA (Mercodia), which was performed according to the manufacturer's protocol using a known amount of human recombinant insulin (Sigma Aldrich) as the standard.5.11. Pharmacokinetics study in vivo
The pharmacokinetics of orally administered LbL coated liposome formulation was evaluated in adult nondiabetic male Wistar rats that had undergone 12 hours fasting. For the solution group, lyophilized formulation was reconstituted in DI water and administered at 320 IU kg−1 before oral gavage dosing. Lyophilized formulation and bare insulin powder were loaded in enteric coated size 9 capsules (Torpac) and dosed at 43 IU kg−1. Blood was collected by inserting a tail vein catheter into the lateral tail vein using a BD Insyte 22-gauge needle. After inserting the catheter, the samples were withdrawn from the catheter. To maintain the patency at each sample 100 μL of heparinized saline (10 U mL−1) was flushed. At each time point when the catheter cap was opened and flushed with heparinized saline, the initial 2 drops of blood was discarded and 200 to 300 μL of blood was collected. After sample collection the cap was closed by flushing with 200 μL of heparinized saline. The same procedure was repeated for all time points. Blood was collected in micro tubes and kept on ice and centrifuged after collecting all samples. The micro tubes were centrifuged at 5000 rcf for 10 min at 40 °C to separate the plasma. The plasma was collected into two aliquots of approximately equal volume. The plasma was transferred immediately to −80 °C. Permission to operate animal experiments was obtained with Institutional Animal Care and Use Committee (IACUC) Service Protocol number #181313, entitled for evaluation of novel compounds to assess toxicity and efficacy of pharmacokinetic/pharmacodynamic parameters in mouse models. This study was performed in strict accordance with the NIH guidelines for the care and use of laboratory animals (NIH Publication No. 85-23 Rev. 1985) and was approved by the Institutional Animal Care and Use Committee (Singapore).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NITHM interdisciplinary diabetes and metabolic diseases grant and HealthTech NTU ID & MDP Gap Funding. The authors would like to acknowledge interdisciplinary graduate school of Nanyang Technological University for scholarship support and thank Dr Soak Kuan Lai from the School of Biological Sciences for her assistance in confocal microscopy.References
- E. Arbit and M. Kidron, J. Diabetes Sci. Technol., 2009, 3(3), 562–567, DOI:10.1177/193229680900300322.
- M. C. Chen, K. Sonaje, K. J. Chen and H. W. Sung, Biomaterials, 2011, 32(36), 9826–9838, DOI:10.1016/j.biomaterials.2011.08.087.
- T. A. S. Aguirre, D. Teijeiro-Osorio, M. Rosa, I. S. Coulter, M. J. Alonso and D. J. Brayden, Adv. Drug Delivery Rev., 2016, 106, 223–241, DOI:10.1016/j.addr.2016.02.004.
- I. B. Halberg, K. Lyby, K. Wassermann, T. Heise, E. Zijlstra and L. Plum-Mörschel, Lancet Diabetes Endocrinol., 2019, 7(3), 179–188, DOI:10.1016/S2213-8587(18)30372-3.
- R. Eldor, E. Arbit, A. Corcos and M. Kidron, PLoS One, 2013, 8(4), e59524, DOI:10.1371/journal.pone.0059524.
- H. Iyer, A. Khedkar and M. Verma, Diabetes, Obes. Metab., 2010, 12(3), 179–185, DOI:10.1111/j.1463-1326.2009.01150.x.
- W. B. Geho, H. C. Geho, J. R. Lau and T. J. Gana, J. Diabetes Sci. Technol., 2009, 3(6), 1451–1459, DOI:10.1177/193229680900300627.
- S. Shen, Y. Wu, Y. Liu and D. Wu, Int. J. Nanomed., 2017, 12, 4085–4109, DOI:10.2147/ijn.s132780.
- Application 6 - Design of Nanoparticles for Oral Delivery of Peptide Drugs, in Nanoparticle Technology Handbook, ed. M. Hosokawa, K. Nogi, M. Naito and T. Yokoyama, Elsevier, Amsterdam, 2008, pp. 442–450 Search PubMed.
- M. S. Alqahtani, A. Alqahtani, A. Al-Thabit, M. Roni and R. Syed, J. Mater. Chem. B, 2019, 7(28), 4461–4473, 10.1039/C9TB00594C.
- H. M. Patel and B. E. Ryman, FEBS Lett., 1976, 62(1), 60–63, DOI:10.1016/0014–5793(76)80016–6.
- T. X. Nguyen, L. Huang, M. Gauthier, G. Yang and Q. Wang, Nanomedicine, 2016, 11(9), 1169–1185, DOI:10.2217/nnm.16.9.
- Y. F. Tan, Y. S. Lee, L.-F. Seet, K. W. Ng, T. T. Wong and S. Venkatraman, Expert Opin. Drug Delivery, 2018, 15(10), 937–949, DOI:10.1080/17425247.2018.1518426.
- Y. F. Tan, R. C. Mundargi, M. H. Chen, J. Lessig, B. Neu, S. S. Venkatraman and T. T. Wong, Small, 2014, 10(9), 1790–1798, DOI:10.1002/smll.201303201.
- Z. J. Deng, S. W. Morton, E. Ben-Akiva, E. C. Dreaden, K. E. Shopsowitz and P. T. Hammond, ACS Nano, 2013, 7(11), 9571–9584, DOI:10.1021/nn4047925.
- X.-J. Du, J.-L. Wang, S. Iqbal, H.-J. Li, Z.-T. Cao, Y.-C. Wang, J.-Z. Du and J. Wang, Biomater. Sci., 2018, 6(3), 642–650, 10.1039/C7BM01096F.
- A. M. Sadeghi, F. A. Dorkoosh, M. R. Avadi, M. Weinhold, A. Bayat, F. Delie, R. Gurny, B. Larijani, M. Rafiee-Tehrani and H. E. Junginger, Eur. J. Pharm. Biopharm., 2008, 70(1), 270–278, DOI:10.1016/j.ejpb.2008.03.004.
- Y. J. Zhang, C. H. Ma, W. L. Lu, X. Zhang, X. L. Wang, J. N. Sun and Q. Zhang, Acta Pharmacol. Sin., 2005, 26(11), 1402–1408, DOI:10.1111/j.1745–7254.2005.00174.x.
- M. Amidi, E. Mastrobattista, W. Jiskoot and W. E. Hennink, Adv. Drug Delivery Rev., 2010, 62(1), 59–82, DOI:10.1016/j.addr.2009.11.009.
- C. Schreider, G. Peignon, S. Thenet, J. Chambaz and M. Pinçon-Raymond, J. Cell Sci., 2002, 115(3), 543–552 CAS.
- A. Bernkop-Schnürch and S. Dünnhaupt, Eur. J. Pharm. Biopharm., 2012, 81(3), 463–469, DOI:10.1016/j.ejpb.2012.04.007.
- J. Hierrezuelo, A. Sadeghpour, I. Szilagyi, A. Vaccaro and M. Borkovec, Langmuir, 2010, 26(19), 15109–15111, DOI:10.1021/la102912u.
- I. Szilagyi, G. Trefalt, A. Tiraferri, P. Maroni and M. Borkovec, Soft Matter, 2014, 10(15), 2479–2502, 10.1039/C3SM52132J.
- H. W. Walker and S. B. Grant, J. Colloid Interface Sci., 1996, 179(2), 552–560, DOI:10.1006/jcis.1996.0249.
- G. C. Ritthidej, Chapter 3 - Nasal Delivery of Peptides and Proteins with Chitosan and Related Mucoadhesive Polymers, in Peptide and Protein Delivery, ed. C. van der Waalse, Academic Press, Boston, 2011, pp. 47–68 Search PubMed.
- O. Mertins and R. Dimova, Langmuir, 2011, 27(9), 5506–5515, DOI:10.1021/la200553t.
- P. Fonte, S. Soares, A. Costa, J. C. Andrade, V. Seabra, S. Reis and B. Sarmento, Biomatter, 2012, 2(4), 329–339, DOI:10.4161/biom.23246.
- L. M. Crowe, D. S. Reid and J. H. Crowe, Biophys. J., 1996, 71(4), 2087–2093, DOI:10.1016/s0006-3495(96)79407-9.
- S. Shen, Y. Wu, Y. Liu and D. Wu, Int. J. Nanomed., 2017, 12, 4085–4109, DOI:10.2147/IJN.S132780.
- Y.-H. Lin, F.-L. Mi, C.-T. Chen, W.-C. Chang, S.-F. Peng, H.-F. Liang and H.-W. Sung, Biomacromolecules, 2007, 8(1), 146–152, DOI:10.1021/bm0607776.
- Z. He, J. L. Santos, H. Tian, H. Huang, Y. Hu, L. Liu, K. W. Leong, Y. Chen and H.-Q. Mao, Biomaterials, 2017, 130, 28–41, DOI:10.1016/j.biomaterials.2017.03.028.
- Z.-H. Wu, Q.-N. Ping, Y. Wei and J.-M. Lai, Acta Pharmacol. Sin., 2004, 25(7), 966–972 CAS.
- H. Takeuchi, H. Yamamoto, T. Niwa, T. Hino and Y. Kawashima, Pharm. Res., 1996, 13(6), 896–901, DOI:10.1023/a:1016009313548.
- T. H. Yeh, L. W. Hsu, M. T. Tseng, P. L. Lee, K. Sonjae, Y. C. Ho and H. W. Sung, Biomaterials, 2011, 32(26), 6164–6173, DOI:10.1016/j.biomaterials.2011.03.056.
- K. Sonaje, Y.-H. Lin, J.-H. Juang, S.-P. Wey, C.-T. Chen and H.-W. Sung, Biomaterials, 2009, 30(12), 2329–2339, DOI:10.1016/j.biomaterials.2008.12.066.
- M. P. Valley, N. Karassina, N. Aoyama, C. Carlson, J. J. Cali and J. Vidugiriene, Anal. Biochem., 2016, 505, 43–50, DOI:10.1016/j.ab.2016.04.010.
- C. Damgé, P. Maincent and N. Ubrich, J. Controlled Release, 2007, 117(2), 163–170, DOI:10.1016/j.jconrel.2006.10.023.
- M.-C. Chen, K. Sonaje, K.-J. Chen and H.-W. Sung, Biomaterials, 2011, 32(36), 9826–9838, DOI:10.1016/j.biomaterials.2011.08.087.
- D. F. Evans, G. Pye, R. Bramley, A. G. Clark, T. J. Dyson and J. D. Hardcastle, Gut, 1988, 29(8), 1035–1041, DOI:10.1136/gut.29.8.1035.
- I. Hubatsch, E. G. Ragnarsson and P. Artursson, Nat. Protoc., 2007, 2(9), 2111–2119, DOI:10.1038/nprot.2007.303.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr06104b |
| This journal is © The Royal Society of Chemistry 2021 |