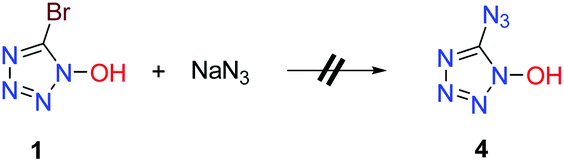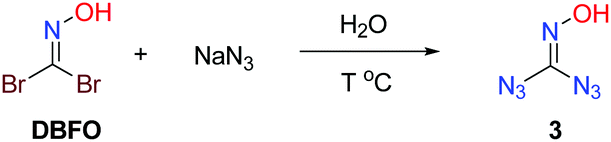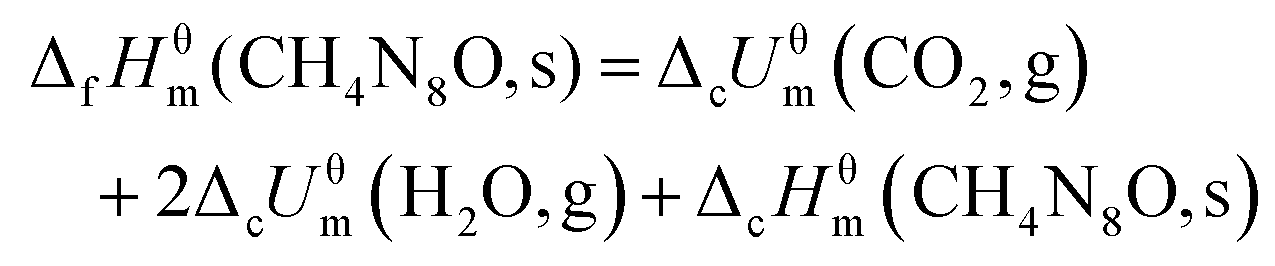DOI:
10.1039/D1QO00123J
(Research Article)
Org. Chem. Front., 2021,
8, 2420-2428
Engaging DBFO as a C1N1 “two-atom synthon” in [3 + 2] cycloaddition reaction: synthesis of the energetic material 5-azidotetrazolate 1N-oxide†
Received
23rd January 2021
, Accepted 7th March 2021
First published on 23rd March 2021
Abstract
We report a formal [3 + 2] annulation of dibromoformaloxime (DBFO) and sodium azide for the synthesis of tetrazole-based energetic materials. Several energetic salts were prepared and fully characterized by X-ray diffraction, Raman spectroscopy, multinuclear NMR spectroscopy, elemental analysis, differential scanning calorimetry (DSC) and impact and friction sensitivity testing. The heat of formation of nitrogen-rich salts 8 and 9 was calculated by experiments, and detonation parameters were estimated using the EXPLO5 software. This is the first example of using DBFO as a unique C1N1 “two-atom synthon” in the synthesis of tetrazoles. Furthermore, this reaction not only unlocks a different strategy for 5-azidotetrazole synthesis, but also exploits a new reactivity of DBFO.
Introduction
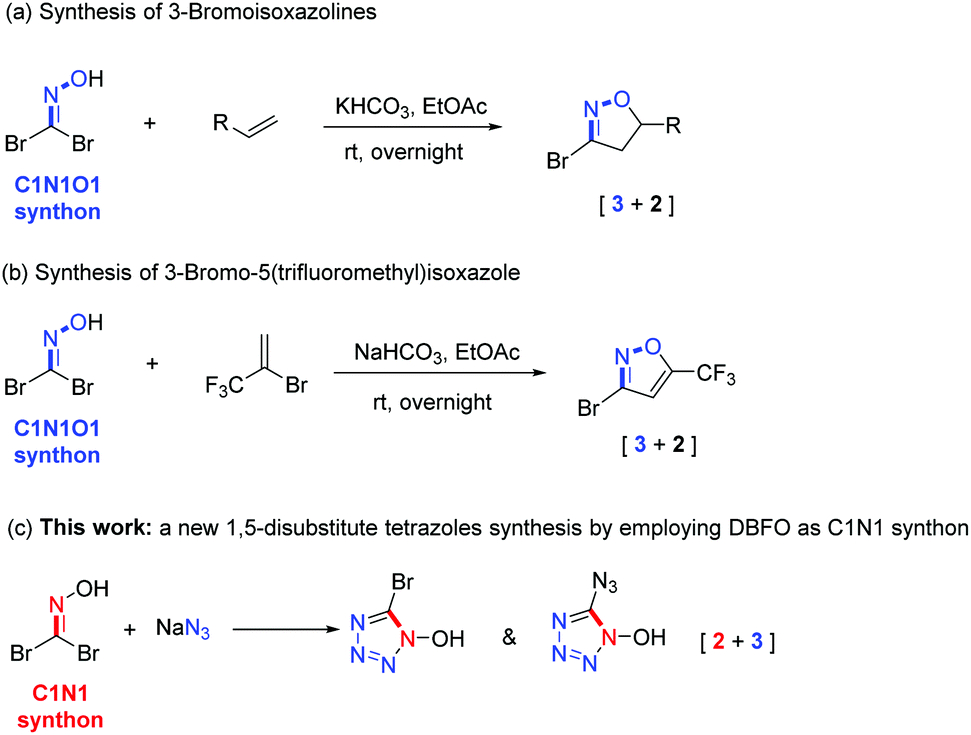
Dibromoformaldoxime (DBFO)1 is a highly versatile and important synthon which has been widely used in organic chemistry due to the high reactivity of its nitrile oxide intermediate species generated in situ by HBr elimination.2,3 Arguably, the most important application of DBFO is the synthesis of various N-heterocyclic compounds,4 particularly multi-substituted isoxazole or isoxazoline.5 For example, in 2017, Battilocchio et al.6 reported a multistep continuous process for the preparation of DBFO as a precursor to generate 3-bromoisoxazolines (Scheme 1a). Very recently, the Chalyk group7 developed a one-pot metal-free [3 + 2] cycloaddition strategy for the preparation of 5-trifluoromethylisoxazoles in a regioselective manner by employing DBFO as an halogenoxime (Scheme 1b). Although these transformations of DBFO offer additional chances for enriching the diversity of the heterocyclic motif construction, they remain limited to [3 + 2] cycloaddition using DBFO as a C1N1O1 “three-atom synthon” under basic conditions or metal catalysis. To the best of our knowledge, exploration of a new role of DBFO as a C1N1 “two-atom synthon” to form a tetrazole heterocyclic ring has not been reported and is a highly interesting new task.
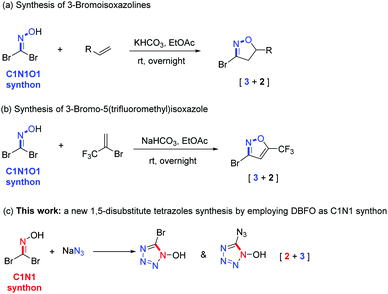 |
| | Scheme 1 Five-member N-heterocycle synthesis using geminal halides DBFO. | |
The synthesis of nitrogen-rich heterocyclic substances has recently been the topic of interest in the study of energetic materials.8 It has long been established that tetrazole is the preferred framework of choice for the design of new energetic nitrogen-rich compounds due to its outstanding benefit of merging a high nitrogen content with good thermal stabilities.9,10 Inspired by previous works,11 heterocycle-based α-azido-oximes can convert into the corresponding tetrazoles via an intramolecular cyclization under the catalysis of HCl(g). Given the reactive C–Br bonds in DBFO that can serve as adjustable handles for further functionalization, we envisioned that DBFO might participate in a substitution reaction with sodium azide to form an azido substituted oxime intermediate, followed by annulation to build the 1,5-disubstituted tetrazole framework (Scheme 1c).
Results and discussion
Synthesis of 5-bromotetrazol-1-ol (1) and its ammonium salt (2)
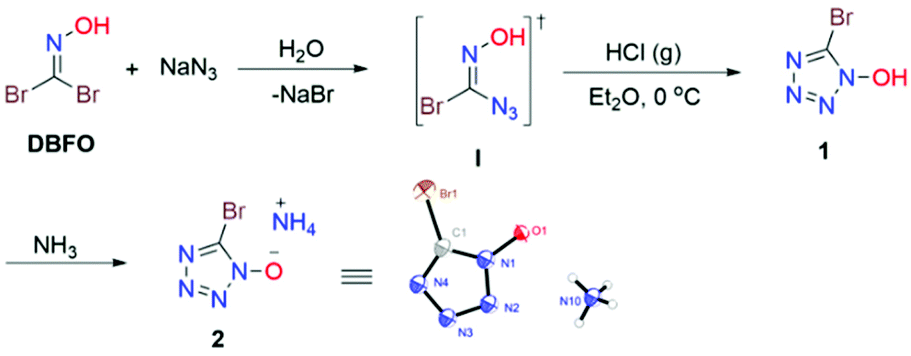
Our studies began with the reaction of DBFO and sodium azide in water at 0 °C. When DBFO (10 mmol) was treated with an equivalent amount of sodium azide, we could detect the monoazido substituted α-bromo oxime intermediate I formed by thin-layer chromatography (TLC) and we separated it as an off-white solid. The resulting azide I readily underwent isomerization to 1 (5-bromo-tetrazol-1-ol) upon treatment with gaseous hydrogen chloride in ether. The structure of 1 was determined indirectly from the crystal structure of its ammonium salt (2) obtained by treating 1 with aqueous ammonia in ethanol (Scheme 2).
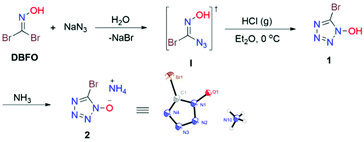 |
| | Scheme 2 Synthetic route to the target compounds. | |
Synthesis of 5-azidotetrazol-1-ol (4)
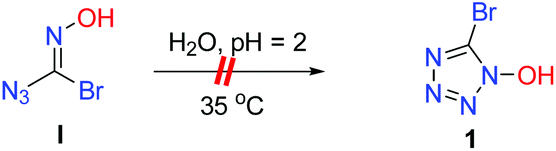
5-Azidotetrazole compounds are notorious for their tendencies toward instability (extremely sensitive toward external stimuli (friction, shock, etc.)), which poses great technical challenges in terms of their synthesis and isolation.12,13 Despite more than 80 years of research, the successful development of new synthesis routes towards 5-azidotetrazole compounds continues to prove uniquely challenging in synthetic chemistry. To further demonstrate the synthetic utility of 1, we tried to combine 1 with sodium azide to synthesize 5-azidotetrazole molecules. Unfortunately, the bromo atom at the C5 position in the tetrazole ring cannot be replaced by an azido ion (N3−) (Scheme 3).
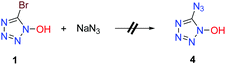 |
| | Scheme 3 Late modification of 1. | |
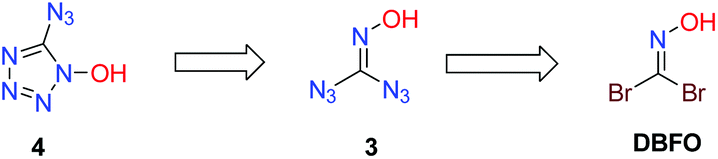
Retrosynthetically, a symmetric disubstituted oxime strategy was envisioned (Scheme 4). The conversion of 1,1-diazidoformaldoxime 3 to the target molecule 4 can be realized through an intramolecular annulation. The synthesis of 3 can be readily achieved by treatment of DBFO with an excess of sodium azide.
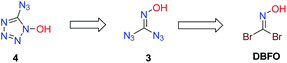 |
| | Scheme 4 Retrosynthetic analysis of 5-azidotetrazole-1-ol. | |
Accordingly, our optimization commenced with the screening of temperatures to obtain the desired product. Water was used as a solvent to activate the reaction. After a brief screening of temperature, we found conditions to work best with water as the solvent at 35 °C (Table 1, entries 1–5).
Table 1 Representative optimization conditionsa
|
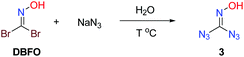
|
| Entry |
Solvent |
T (°C) |
Time (h) |
Yieldb (%) |
|
Reaction conditions: DBFO (5 mmol), NaN3 (10.5 mmol, 2.1 equiv.), and H2O 10 mL, unless otherwise noted.
Isolated yields.
|
| 1 |
H2O |
0 |
>48 |
<10 |
| 2 |
H2O |
15 |
>48 |
36 |
| 3 |
H2O |
25 |
18 |
67 |
| 4 |
H2O |
35 |
4 |
84 |
| 5 |
H2O |
45 |
3.5 |
79 |
Structure of the disubstituted product: 1,1-diazidoformaldoxime (3) or 5-azidotrazol-1-ol (4)?
Multinuclear NMR spectroscopy (13C) of the product was performed to determine its structure. The product appeared as two 13C peaks near δ (ppm) = 144, 156; thus we speculated that there might be an equilibrium between 1,1-diazidoformaldoxime (3) and 5-azidotrazol-1-ol (4) in the solution (Fig. 1).
 |
| | Fig. 1 Equilibrium between 3 and 4. | |
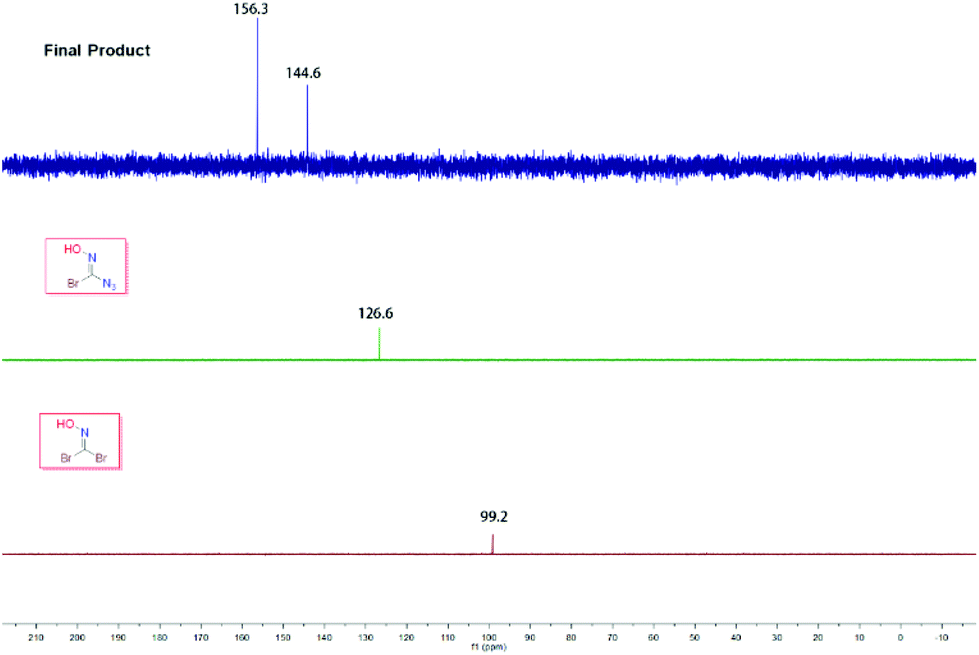
In order to explore the abnormal signals of the final product which were not caused by the starting material 1,1-dibromoformaldoxime or the intermediate species I, we carefully studied the 13C NMR spectra of these compounds. From a comparison, it is preliminarily concluded that this phenomenon was not caused by the impurity of the sample (Fig. 2).
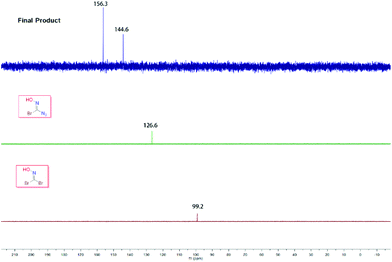 |
| | Fig. 2
13C NMR overlay chart of DBFO, intermediate I and the final product. | |
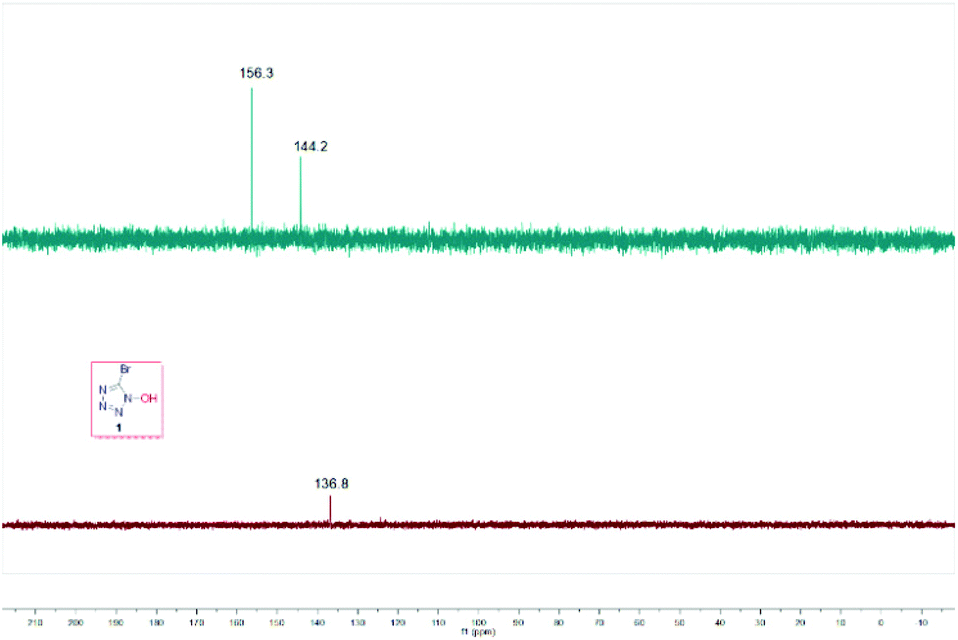
On the other hand, we observed that the pH value of the reaction mixture decreased to 2 as the reaction proceeded. To further validate whether the intermediate I can undergo an intramolecular cyclization to form the corresponding product 1, the control experiment of I was carried out under the standard reaction conditions (Fig. 3). The results showed that intramolecular cyclization of intermediate I failed to proceed under standard reaction conditions, and the 13C NMR comparison diagram of the two products revealed that they are three different kinds of carbon (Fig. 4).
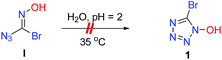 |
| | Fig. 3 Control experiment: intermediate I under acid catalysis. | |
 |
| | Fig. 4
13C NMR chart of 1 and the final product. | |
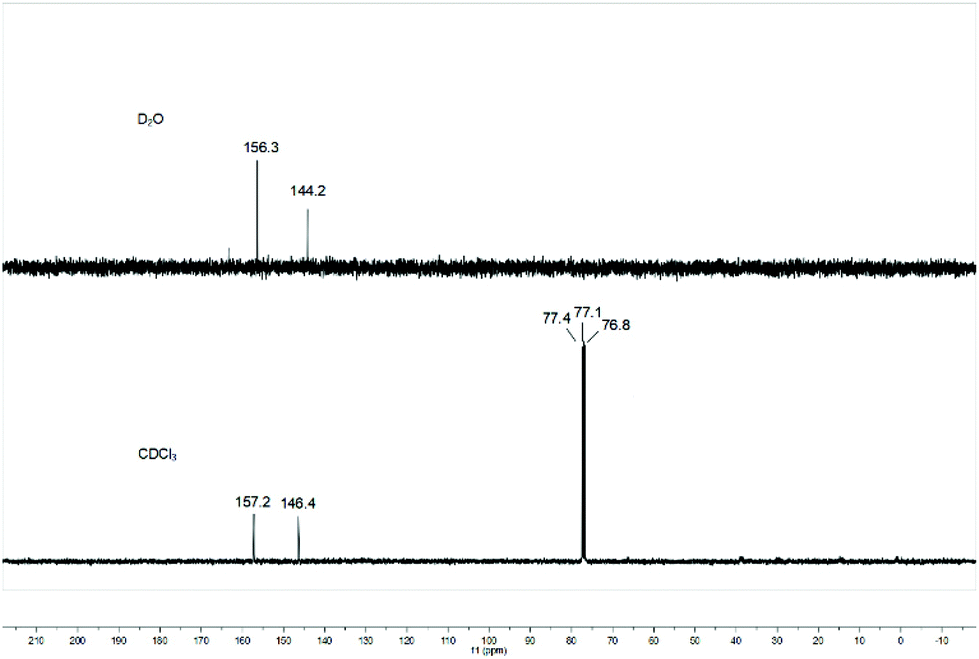
The relationship between the deuterated solvent and the equilibrium of 1,1-diazidoformaloxime (3) and 5-azidotrazol-1-ol (4) was also investigated. When the aprotic solvent deuterated chloroform was used as solvent, the 13C NMR spectrum of the product still showed two kinds of carbon peaks, which indicated that there is no correlation between the deuterated solvent and equilibrium of 3 and 4 (Fig. 5).
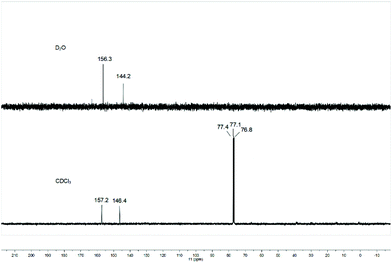 |
| | Fig. 5
13C NMR of the product in CDCl3 or D2O. | |
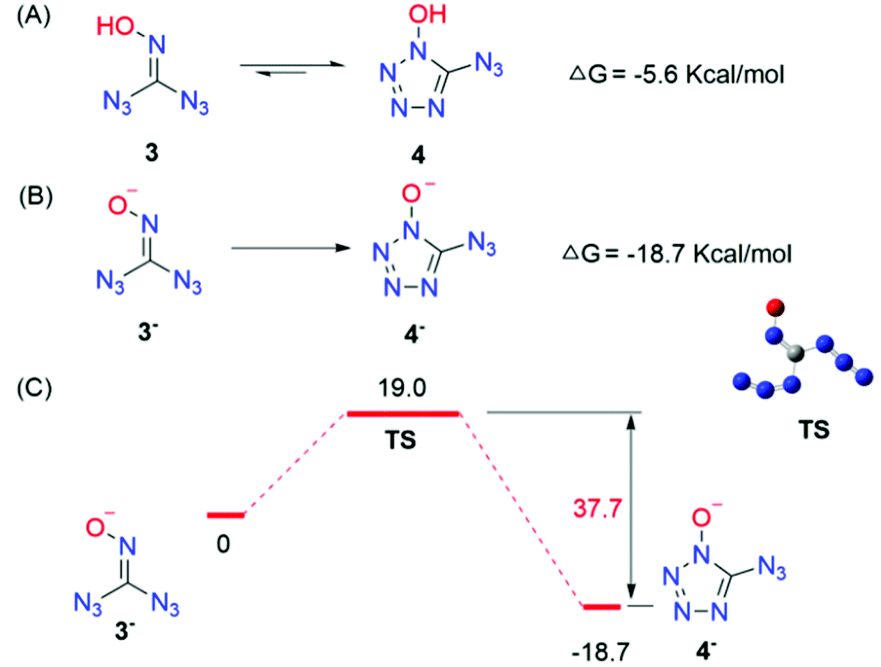
In addition, DFT calculations were performed to gain further insight into the thermodynamic and kinetic features of the mechanistic pathway between 3 and 4. Scheme 5 details the thermodynamics of ring-closing of 3. The energetics of N–N bond formation (ΔG = −5.6 kcal mol−1) are exergonic, resulting in a thermodynamically favourable ring-closing/N–N bond formation sequence; the lowest energy transition states for the N–N bond formation were also computed. The transition state was located with an activation barrier of 15.2 kcal mol−1, which means that 3 can easily transfer to 4 at room temperature and the reverse holds true as well. Moreover, the rate constant k revealed that low temperature would retard the equilibrium while higher temperature would promote the transformation from 3 to 4 (see the ESI†). In particular, the 13C NMR spectra of alkali metal salts of 4 showed only one kind of carbon while ammonium and guanidinium displayed two kinds of carbon, which revealed that a free proton might play an important role in the equilibrium between 3 and 4. DFT calculations showed that the deprotonated product 3− can transfer to the corresponding substance 4− irreversibly, which may explain the presence of only one kind of carbon in alkali metal salts (Scheme 5B and C). All in all, these pieces of evidence clearly indicated that there must be an equilibrium between 3 and 4.
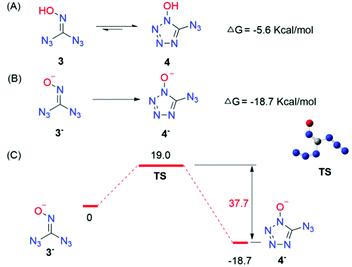 |
| | Scheme 5 (A) Calculated thermodynamics for equilibrium between 3 and 4. (B) Calculated thermodynamics for 3− to 4−. (C) Energy profile for the lowest-energy transition state. | |
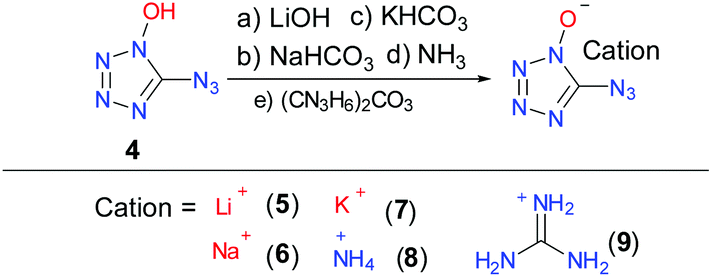
Synthesis of 5-azidotetrazolate 1N-oxide
Interestingly, when the diazido-substituted product of DBFO was treated with a weak base, such as NaHCO3, the crystal structure of the deprotonated product demonstrated that there was only one structure, while the salts of 3 cannot be obtained. Thus, the salts of 4 were synthesized and characterized to study their energetic properties. The lithium (5), sodium (6), potassium (7) and guanidinium (9) salts of 4 were prepared by dissolving it in H2O and reacting with the corresponding base. In the case of the ammonium salt (8), 4 was dissolved in ethyl ether and ammonia solution (ca. 7 N in methanol) was added slowly and the final product was collected by filtration (Scheme 6). Saturated solutions were slowly evaporated to give suitable crystals in methanol or methanol/water mixtures to obtain crystals of 5, 6, 7, 8 and 9, making X-ray structure analysis.
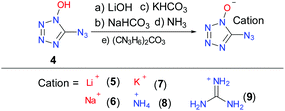 |
| | Scheme 6 Synthesis of 1N-hydroxyl-5-azidotetrazole as well as its alkali metal and selected nitrogen-rich salts. | |
Crystallography
5-Azidotetrazolate 1N-oxide anions in all compounds described in this work have nearly identical geometries. The N–N and C–N bond lengths within the tetrazole ring are all between typical single and double bond lengths.14 The bond length between O1 and N4(1) (from 1.308 Å to 1.342 Å) is shorter than that of an N–O single bond (1.45 Å) and longer than that of an N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond (1.17 Å), indicating the N–O bond has a significant N-oxide character.
O double bond (1.17 Å), indicating the N–O bond has a significant N-oxide character.
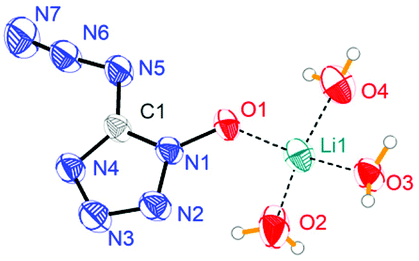
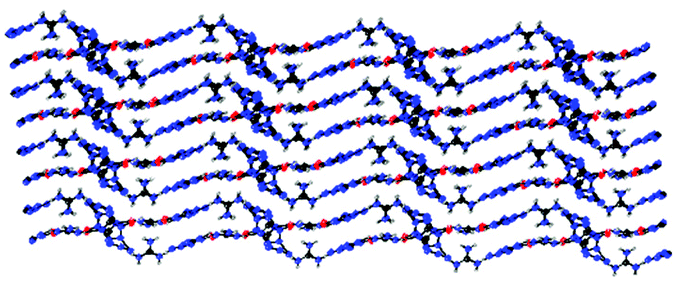
Lithium 5-azidotetrazolate 1N-oxide (5·3H2O) crystallizes in the monoclinic space group P2(1)/n with four molecules in the unit cell and its density is 1.4841 g cm−3 (Fig. 6). In the crystal structure of 5·3H2O, six different hydrogen bonds were found, with five of them interacting with oxygen and nitrogen atoms of four different 5-azidotetrazolate N-oxide rings.
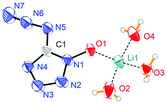 |
| | Fig. 6 Molecular moiety of lithium 5-azidotetrazolate 1N-oxide 5·3H2O. Thermal ellipsoids represent the 50% probability level. | |
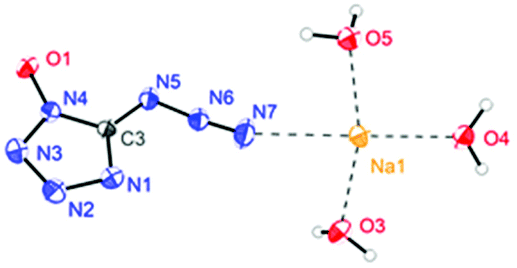
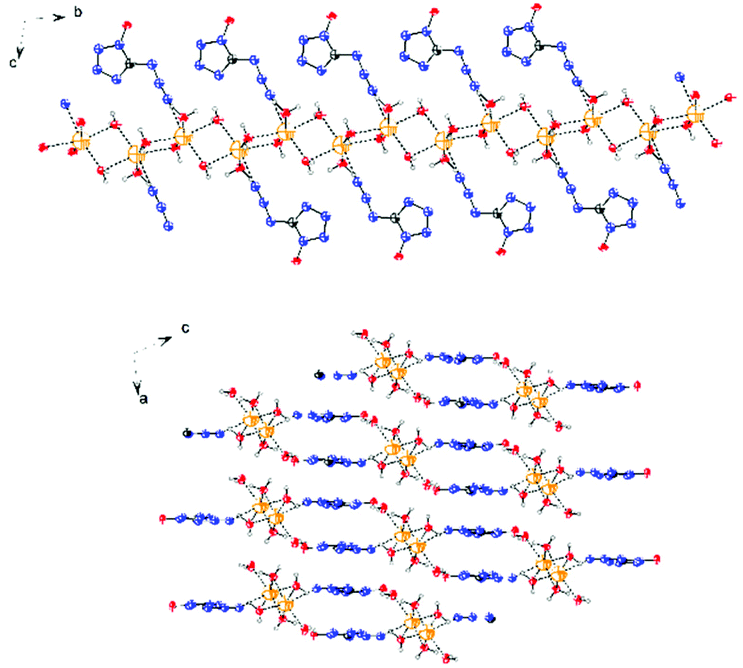
The molecular structure of 6·3H2O, which falls in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with a density of 1.6696 g cm−3 (296 K) and two molecules in the unit cell, is shown in Fig. 7. The nitrogen atom N7 of the azido group participates as a weak hydrogen bond acceptor and shows coordination distances of N7–Na1 = 2.7388(18) Å. Due to the presence of water, the packing of 6·3H2O is dominated by a strong hydrogen bond network, and the water molecules bridge the extended molecular moiety to layers along the a–b plane by the hydrogen bonds (Fig. 8).
with a density of 1.6696 g cm−3 (296 K) and two molecules in the unit cell, is shown in Fig. 7. The nitrogen atom N7 of the azido group participates as a weak hydrogen bond acceptor and shows coordination distances of N7–Na1 = 2.7388(18) Å. Due to the presence of water, the packing of 6·3H2O is dominated by a strong hydrogen bond network, and the water molecules bridge the extended molecular moiety to layers along the a–b plane by the hydrogen bonds (Fig. 8).
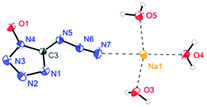 |
| | Fig. 7 Coordination of sodium cations in the packing of 6·3H2O. Thermal ellipsoids represent the 50% probability level. | |
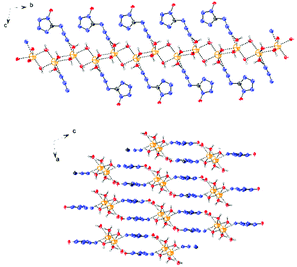 |
| | Fig. 8 View of the layers along the a and b axes in the structure of 6·3H2O. | |
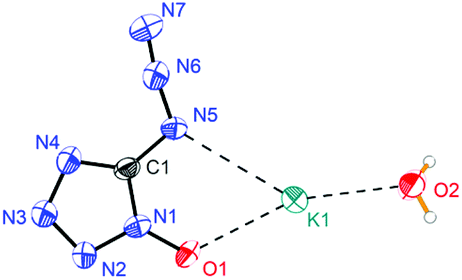
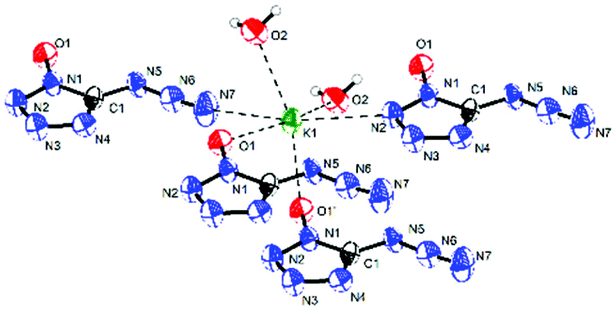
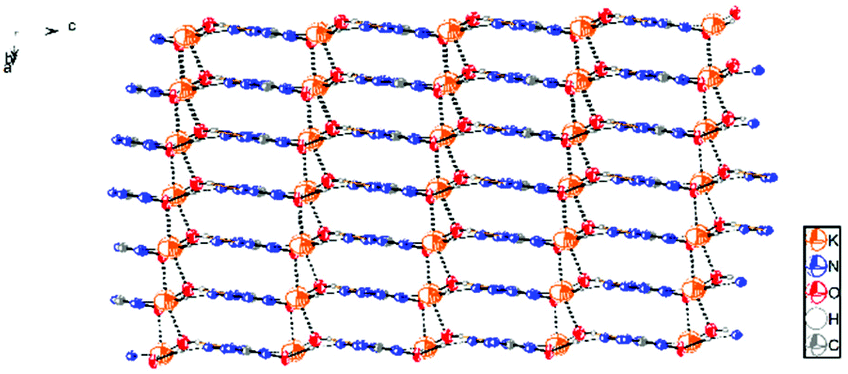
Potassium 5-azidotetrazolate 1N-oxide monohydrate (7·H2O) crystallizes in the triclinic space group P1(1) with one molecule in the unit cell (Fig. 9). The presence of water decreases the density to 1.7312 g cm−3. The potassium cations are coordinated octahedrally by the nitrogen atoms N2, N2, O1 and O1′ of four different CN7O anions and two water molecules. The coordination distances range from 2.75 to 3.07 Å (Fig. 10). The packing structure of 7 is influenced by the strong hydrogen bonds with the aid of water molecules, which shows π–π stacking (Fig. 11).
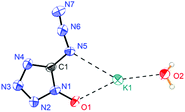 |
| | Fig. 9 Asymmetric unit of 7·H2O. | |
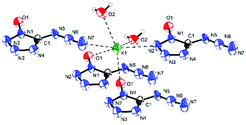 |
| | Fig. 10 Coordination of the potassium atoms in the structure of 7·H2O. | |
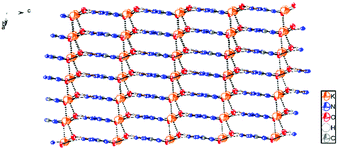 |
| | Fig. 11 Packing diagram of 7·H2O. Hydrogen bonds are shown as black dots. | |
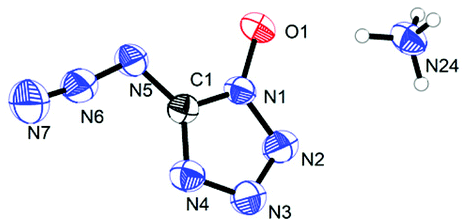
Ammonium 5-azidotetrazolate 1N-oxide (8) crystallizes in the monoclinic space group P21/c with twelve molecules in the unit cell and a density of 1.6130 g cm−3. The molecular moiety is shown in Fig. 12.
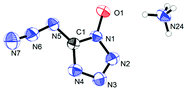 |
| | Fig. 12 Molecular unit of ammonia 5-azidotetrazolate 1N-oxide 8. Thermal ellipsoids represent the 50% probability level. | |
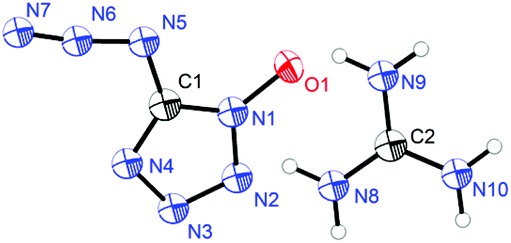
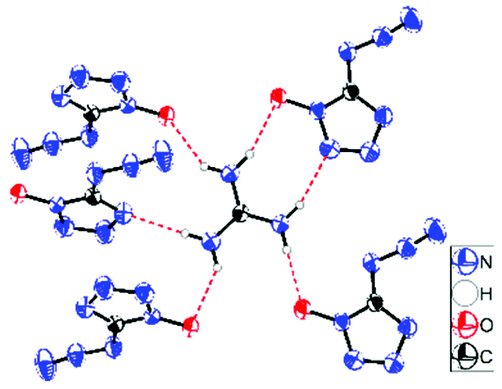
Guanidinium 5-azidotetrazolate 1N-oxide (9) falls in the common monoclinic space group C2/c, with a density of 1.6100 g cm−3 (Fig. 13). A view of the intermolecular hydrogen bonds of the guanidinium cations is illustrated in Fig. 14. None of the nitrogen atoms of the azide group participates in the formation of the layers. The guanidinium cations with C–N bond lengths between 1.295 and 1.301 Å are observed. These distances are shorter than the C–N single bonds (1.47 Å) but slightly longer than the CN double bonds (1.22 Å), which indicated the delocalization of the positive charge. Moreover, the packing of 9 is strongly influenced by the six different hydrogen bonds (Table S15†). The crystal packing of this molecule shows a wave-like stacking mode, which partly accounts for its low sensitivities towards impact and friction (Fig. 15).
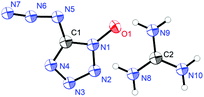 |
| | Fig. 13 Molecular unit of guanidinium 5-azidotetrazolate 1N-oxide 9. | |
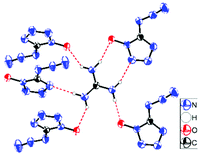 |
| | Fig. 14 Hydrogen bonds in the packing of 9. | |
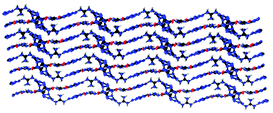 |
| | Fig. 15 Packing diagram of 9. Hydrogen bonds between layers are omitted. | |
Thermal behaviour
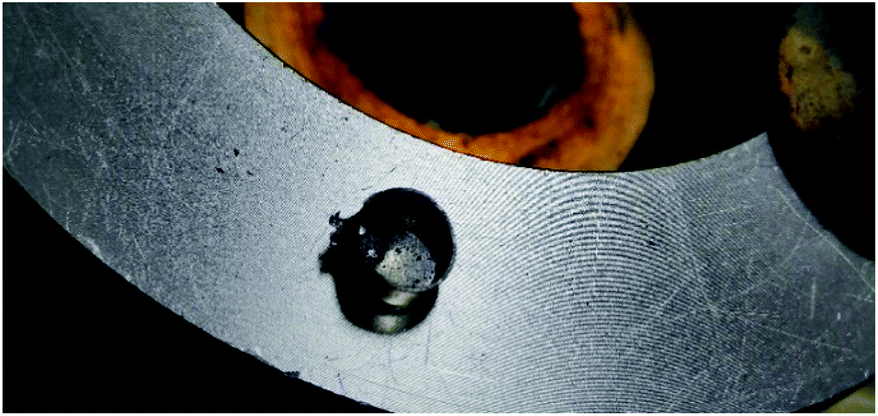
The thermal behaviours of 2, 5, 6, 7, 8 and 9 were investigated using a differential scanning calorimeter (DSC) at a heating rate of 5 °C min−1 with approximately 0.2–1.0 mg of material. Ammonium salt (2) is the most stable; it decomposes at 180.64 °C with no prior melting. The anhydrous lithium salt 5 has two broad decomposition peaks at 91.16 and 210.16 °C, respectively. The sodium salt 6·3H2O loses hydration water at 83.65 °C, and undergoes brief decomposition at 110.73 °C, followed by a broad decomposition peak at 185.43 °C. The potassium salt 7·H2O decomposes violently at 117.83 °C with no prior melting or hydration water loss, where it detonates with enough force to decimate the aluminium pan (Fig. 16). Within the nitrogen-rich derivatives 8 and 9, the guanidinium salt (9) is more stable. It decomposes at 123.11 °C with no prior melting, 9 °C higher than the ammonia salt 8.
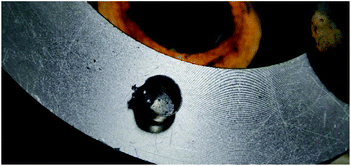 |
| | Fig. 16 Damage sustained to the DSC sample pan after 0.45 mg of 7·H2O detonated. | |
Sensitivities
For initial safety testing, the impact and friction sensitivity of the prepared nitrogen-rich salts were determined using standard BAM methods.15 The sensitivities of all compounds are highly dependent on the hydrogen bond network.16 Anhydrous salts 5 and 7–9 are very sensitive compounds, but the ammonium salt 2 (>40 J impact, 288 N friction) and sodium salt 5·3H2O (>40 J impact, 144 N friction) are not. The lithium salt (6) containing no water has the highest insensitivity (<1 J impact, <5 N friction), while the trihydrate compound 6·3H2O shows an impact >40 J and a friction of 168 N. The potassium monohydrate salt 7·H2O has an impact sensitivity of 1.0 J and a friction sensitivity of 10 N. The guanidinium salt 9 has a relatively low sensitivity towards impact (5 J) and friction (168 N) and is less sensitive than the ammonia cation 8 (1 J, <5 N). As far as we know, the energetic salt 9 probably is the safest known nitrogen-rich 5-azidotetrazole compound towards impact and friction. The “enhanced” low sensitivities of 9 may be attributed to the regular crystal packing structure of the system.
Heats of formation
The constant-volume combustion energy of ammonia 5-azidotetrazole 1N-oxide (8) and guanidinium 5-azidotetrazole 1N-oxide (9) is determined by oxygen bomb calorimetry (OZM BAC500). Approximately 500 mg of 8 or 9 were pressed to form a tablet to ensure better combustion. The recorded data are the average of five single measurements. The calorimeter is calibrated by the combustion of certified benzoic acid under an oxygen atmosphere at a pressure of 30.5 bar.
The average experimental value for the constant volume combustion energies (ΔcUθm) of 8 is −10580.33 J g−1 (−1507.23 kJ mol−1). The combustion reaction equation (1) and energy of combustion equation (2) are listed as follows.
The combustion reaction equation,
| | | CH4N8O(s) + 1.5O2(g) → CO2(g) + 2H2O + 4N2(g) | (1) |
The energy of combustion equation,
| | | ΔcHθm(CH4N8O,s) = ΔcUθm + ΔnRT | (2) |
where Δ
n is the change in the number of gas products during the reaction process,
R = 8.314 J mol
−1 K
−1, and
T = 298.15 K. According to
eqn (1) and (2), the calculated combustion enthalpy of ammonium 5-azidotetrazole 1
N-oxide (
8) is −1515.95 kJ mol
−1. Based on the calculated combustion enthalpy of
8 and the known enthalpies of formation of the combustion products (CO
2(g) = −393.51 kJ mol
−1), (H
2O(l) = −285.83 kJ mol
−1), the standard formation enthalpy of
8 is back-calculated from its combustion
equation (3). On the basis of Hess's law in thermochemical
equation (3), the standard enthalpy of formation (Δ
fHθm) of
8 was calculated to be 550.78 kJ mol
−1.
| | 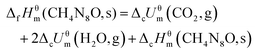 | (3) |
Likewise, the standard enthalpy of formation (ΔfHθm) of 9 was calculated to be 278.61 kJ mol−1 with constant volume combustion energies ΔcUθm (9, s) = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 384.84 J g−1.
384.84 J g−1.
The calculated heats of formation of compounds 8 and 9 are summarized in Table 2. The calculation of the detonation parameters (detonation velocity and pressure) of nitrogen-rich compounds 8 and 9 was performed with the program package EXPLO5 (version 6.01) and the following values were obtained:17detonation pressure of 29.8 GPa and 21.7 GPa, and detonation velocity of 8946 m s−1 and 7984 m s−1, respectively
Table 2 Properties of compounds 8 and9
| |
8
|
9
|
|
Sumof nitrogen and oxygen content.
X-ray densities at 298 K.
Calculated detonation velocity.
Calculated detonation pressure.
Heat of formation.
Temperature of decomposition (onset).
Impact sensitivity.
Friction sensitivity.
|
| N + Oa [%] |
88.86 |
83.85 |
|
ρ
[g cm−3] |
1.613 |
1.610 |
|
V
detc [m s−1] |
8946 |
7984 |
|
P
[GPa] |
29.8 |
21.7 |
| ΔfHθe [kJ mol−1] |
550.78 |
278.61 |
|
T
decf [°C] |
114.24 |
123.11 |
| ISg [J] |
1 |
5 |
| FSh [N] |
<5 |
168 |
Conclusions
We have developed an intramolecular [3 + 2] cycloaddition of 1,1-dibromoformaldoxime for the synthesis of 5-substitute tetrazole 1N-oxide 1 and 4. The alkali metal and nitrogen-rich salts of 4 were obtained. The structures of compounds 2, and 5–9 were confirmed by single crystal X-ray diffraction. Compounds 7–9 are extremely sensitive to physical stimuli according to their sensitivity testing. Except for the sodium salts 5·3H2O and 6·3H2O, guanidinium 5-azidotetrazolate 1N-oxide (9) was found to be the most insensitive nitrogen-rich compound containing a 5-azidotetrazole moiety toward mechanical sensitivity (FS: 168 N, IS: 5 J). Compounds 8 and 9 possess high positive heats of formation at 550.78 kJ mol−1 and 278.61 kJ mol−1, and detonation velocity values of 8946 m s−1 and 7984 m s−1, respectively. This is the first example of using DBFO as a C1N1 “two-atom synthon” to form 1,5-disubstituted tetrazoles by an intramolecular [3 + 2] strategy. Furthermore, this new reaction not only exploits a new reactivity of DBFO but also enriches the diversity of the tetrazole synthesis.
Experimental
Caution: The compounds in this work are highly sensitive energetic substances that could explode under certain conditions (e.g., impact, friction or electric discharge). Appropriate safety precautions, such as the use of safety shields in a fume hood and personal protection equipment (safety glasses, face shields, earplugs, as well as gloves), should always be taken when handling these materials.
Materials and methods
All reagents were purchased from Sigma-Aldrich or Energy Chemicals and were used without further purification; the starting material DBFO was synthesized using the method reported in the literature.18 The 1H and 13C NMR spectra were recorded on a Bruker AVL300 spectrometer; chemical shifts were determined with respect to external SiMe4 (1H: 400.0 MHz, 13C: 100.0 MHz). IR spectra were recorded on a PerkinElmer Spectrum BX-FTIR spectrometer. Elemental analysis was performed on an Elementar Vario El III analyzer. Differential scanning calorimetry (DSC) analyses were performed on a METTLER Toledo DSC 3; decomposition temperatures of products were measured under the same conditions at a 5 °C min−1 heating rate. Impact and friction sensitivity measurements were performed using a standard BAM Fall hammer and a BAM friction tester.
X-ray crystallography
The data for 2 and 5–9 were collected with a Bruker three-circle platform diffractometer equipped with a SMART APEX II CCD detector. A Kryo-Flex low-temperature device was used to keep the crystals at a constant 298(2) K during data collection. The data collection and initial unit cell refinement were performed using APEX2 (v2010.3-0).19 Data reduction was performed using SAINT (v7.68A) and XPREP (v2008/2).20 The structure was solved and refined with the aid of the programs in the SHELXTL-plus (v2008/4) system of programs.21 The full-matrix least-squares refinement on F2 included atomic coordinates and anisotropic thermal parameters for all non-H atoms. The structure was solved by direct methods with SHELXS-97 and expanded by the Fourier technique.22 The nonhydrogen atoms were refined anisotropically. The hydrogen atoms were located and refined.
Computational methods
All density functional theory (DFT) calculations were performed using Gaussian 09. Geometry optimizations and frequency calculations were performed at the B3LYP/6-311G(d,p) level of theory. Normal vibrational mode analysis confirmed that the optimized structures are minima or transition structures. Transition structures are verified by intrinsic reaction coordinate (IRC) calculations. The reaction rate constants k between compounds 3 and 4 under different temperature conditions (173.15–373.15 K) were calculated using canonical variational transition state theory with the Wigner tunneling correction (CVT/Wigner).23
Synthesis
5-Bromotetrazol-1-ol (1) and its ammonium salt (2).
To a solution of DBFO (2.03 g, 10 mmol) in H2O (15.0 mL) was added sodium azide (650 mg, 10.0 mmol, 1.0 eq.) portionwise at 0 °C; the reaction mixture was then stirred for 0.5 h. The solution was extracted with diethyl ether (50.0 mL). The combined organic layer was dried over anhydrous Na2SO4, filtered, and concentrated in vacuo to give an off-white solid (1.52 g, 92%). To this crude product in ether (20 mL) was bubbled gaseous HCl over 1.5 h. The reaction mixture was sealed and stirred for 20 h at 20 °C. The resulting mixture was poured into cold water (30 mL) and then extracted with ethyl acetate (20 × 4 mL). The combined organic layer was dried over anhydrous Na2SO4, filtered, and concentrated under vacuum to 1/3 volume. Ammonia (7 N in methanol) was added to the final solution at 0 °C and the ammonium salt was collected by filtration to obtain a white solid (1.59 g, 95%). 1H NMR (400 MHz, CH3OH-d4): δ 5.07 (s, 4H). 13C NMR (100 MHz, CH3OH-d4) δ 135.8. IR(ν): 609.7, 629.8, 669.0, 748.7, 985.4, 1194.2, 1206.4, 1225.9, 1437.2, 1460.4, 1690.0, 1890.1, 2866.1, 3026.5. DSC (5 °C min−1): 180.64 °C (onset). Elemental analysis calcd (%) for CH4BrN5O (181.98 g mol−1): C 6.60, H 2.22, N 38.38; found: C 6.49, H 2.32, N 38.30. BAM impact: >40 J; and BAM friction: 288 N.
5-Azidotetrazol-1-ol (4).
To a solution of DBFO (1.0, 5.0 mmol, 1.0 eq.) in H2O (10.0 mL) was added sodium azide (0.682 g, 10.5 mmol, 2.1 eq.) portionwise at room temperature. The reaction mixture was then stirred for 4 h at 35 °C and 20% sulfuric acid (2.0 mL) was added upon cooling. The solution was extracted with diethyl ether (120.0 mL). The combined organic layer was dried over anhydrous Na2SO4, filtered, and concentrated in vacuo to give a colourless viscous liquid (0.53 g, yield: 84%). 13C NMR (100 MHz, CDCl3) δ 156.3, 144.2.
Lithium 5-azidotetrazolate 1N-oxide (5).
Compound 4 (1.27 g, 10 mmol) was dissolved in methanol (5 mL) and LiOH solid (10 mmol) was added. The solution was allowed to concentrate under vacuum and the product was dried under vacuum, thereby yielding anhydrous 2 (0.97 g, 72%). 13C NMR (100 MHz, DMSO-d6): δ 138.8. Raman: 3378 (w), 2170 (m), 1539 (m), 1500 (vs), 1331 (m), 1280 (w), 1206 (w), 1121 (w), 831 (vw), 773 (vw), 689 (vw). DSC (5 °C min−1): 91.16 °C (onset), 210.16 (onset). Elemental analysis calcd (%) for CLiN7O (133.00 g mol−1): C 9.03, N 73.72; found: C 8.98, N 73.59. For anhydrous product, BAM impact: <1 J and BAM friction: <5 N; for crystal (trihydrate compound) of 2·3H2O, BAM impact: >40 J and BAM friction: 168 N.
Sodium 5-azidotetrazolate 1N-oxide (6·3H2O).
Compound 4 (1.27 g, 10 mmol) was dissolved in cold water (5 mL) and NaHCO3 solid (10 mmol) was added portion-wise. The solution was allowed to evaporate, and the resulting white powder was recrystallized from MeOH to yield 6·3H2O (1.71 g, 84%). 13C NMR (100 MHz, D2O): δ 142.7. Raman: 2166 (m), 1549 (m), 1507 (vs), 1439 (s), 1379 (s), 1290 (w), 1251 (w), 1228 (w), 1156 (w), 1012 (w), 854 (w), 682 (m), 580 (vw), 468 (vw), 270 (vw), 152 (w). Elemental analysis calcd (%) for CH6N7NaO4 (203.09 g mol−1): C 5.91, H 2.98, N 48.28; found: C 5.88, H 2.89, N 48.20. DSC (5 °C min−1): 83.65 °C (onset, –H2O), 110.73 °C (onset). BAM impact: >40 J; and BAM friction: 144 N.
Potassium 5-azidotetrazolate 1N-oxide (7·H2O).
Compound 4 (254 mg, 2 mmol) was dissolved in cold water (2 mL) and KHCO3 powder (2 mmol) was added portion-wise. The solution was allowed to slowly evaporate, thereby yielding 7·H2O (245 mg, 67%). 13C NMR (100 MHz, D2O): δ 141.3. Raman: 2149 (m), 1540 (s), 1500 (vs), 1475 (m), 1332 (s), 1279 (m) 1251 (w), 1207 (m), 1151 (vw), 1031 (vw), 776 (vw), 677 (vw), 437 (vw), 148 (w). Elemental analysis calcd (%) for CH2KN7O2 (183.17 g mol−1): C 6.56, N 53.53; found: C 6.63, N 53.47. DSC (5 °C min−1): 117.83 °C (onset). BAM impact: 1 J; and BAM friction: <5 N.
Ammonium 5-azidotetrazolate 1N-oxide (8).
Compound 4 (0.64 g, 5 mmol) was dissolved in Et2O (50 mL) and the mixture was then cooled to 0 °C. NH3 solution (7 N in MeOH, 1 mL) was added dropwise to the solution. After stirring for 5 min, the reaction mixture was filtered and the filter cake was washed with a small amount of Et2O, and dried in air to afford 8 as a white solid (0.63 g, 87%). 1H NMR (400 MHz, CH3OH-d4) δ 5.48 (s, 4H). 13C NMR (100 MHz, CH3OH-d4): δ 159.0, 141.5. Raman: 3008 (s), 1539 (s), 1482 (m), 1474 (m), 1350 (s) 1306 (m) 1269 (m), 1097 (m), 582 (vw), 204 (m), 126 (s). Elemental analysis calcd (%) for CH4N8O (144.10 g mol−1): C 8.34, H 2.80, N 77.76; found: C 8.39, H 2.80, N 77.69. DSC (5 °C min−1): 114.24 °C (decomp). BAM impact: 1 J; and BAM friction: <5 N.
Guanidinium 5-azidotetrazolate 1N-oxide (9).
Compound 4 (1.27 g, 10 mmol) was dissolved in cold water (5 mL) and guanidine carbonate (1.21 g, 10 mmol) was added portionwise. The solution was allowed to evaporate, and the resulting white powder was recrystallized from MeOH to yield 9 (1.56 g, 84%). 1H NMR (400 MHz, DMSO-d6) δ 7.14 (s, 6H); 13C NMR (100 MHz, DMSO-d6): δ 158.5, 138.9. Raman: 2162 (s), 1539 (w), 1496 (vs), 1440 (s), 1379 (s) 1261 (vw), 1133 (w), 1011 (m), 694 (w), 569 (vw), 442 (vw), 233 (m), 126 (w). Elemental analysis calcd (%) for C2H6N10O (186.14 g mol−1): C 12.91, H 3.25, N 75.25; found: C 12.82, H 3.24, N 75.33. DSC (5 °C min−1): 123.11 °C (onset). BAM impact: 5 J; and BAM friction: 168 N.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are thankful to NSAF (U1830134) and NSFC (21905023 and 21911530096) for their generous financial support.
Notes and references
-
(a) R. N. Hanson and F. A. Mohamed, An expedient synthesis of (±)-2-amino-3-(3-hydroxy-5-methylisoxazol-4-yl)propionic acid hydrobromide via a 3-bromoisoxazole intermediate, J. Heterocycl. Chem., 1997, 34, 345–348 CrossRef CAS;
(b) G. W. Kabalka and M. S. Reddy, Sodium percarbonate, e-EROS Encycl. Reagents Org. Synth., 2008, 1–5 CAS;
(c) Z. Siddiqi, W. C. Wertjes and D. Sarlah, Chemical Equivalent of Arene Monooxygenases: Dearomative Synthesis of Arene Oxides and Oxepines, J. Am. Chem. Soc., 2020, 142, 10125–10131 CrossRef CAS PubMed.
- application in total synthesis, see:
(a) Y. Kitabayashi, T. Fukuyama and S. Yokoshima, Synthesis of the [7–5–5] tricyclic core of Daphniphyllum alkaloids, Org. Biomol. Chem., 2018, 16, 3556–3559 RSC;
(b) Y. Yoshii, H. Tokuyama and D. Y. K. Chen, Total Synthesis of Actinophyllic Acid, Angew. Chem., Int. Ed., 2017, 56, 12277–12281 CrossRef CAS PubMed;
(c) L. Leng, X. Zhou, Q. Liao, F. Wang, H. Song, D. Zhang, X.-Y. Liu and Y. Qin, Asymmetric Total Syntheses of Kopsia Indole Alkaloids, Angew. Chem., Int. Ed., 2017, 56, 3703–3707 CrossRef CAS PubMed;
(d) H.-X. Huang, S.-J. Jin, J. Gong, D. Zhang, H. Song and Y. Qin, Studies of a Diazo Cyclopropanation Strategy for the Total Synthesis of (-)-Lundurine A, Chem. – Eur. J., 2015, 21, 13284–13290 CrossRef CAS PubMed;
(e) G.-L. Cheng, X.-Y. Wang, R. Zhu, C.-W. Shao, J.-M. Xu and Y.-F. Hu, Total Synthesis of (-)-Cocaine and (-)-Ferruginine, J. Org. Chem., 2011, 76, 2694–2700 CrossRef CAS PubMed.
-
(a) A. Alizadeh, B. Farajpour, K. Amir Ashjaee Asalemi and S. Taghipour, Diastereoselective Synthesis of Coumarin-Based Fused Heterocycles via Intramolecular Diels-Alder and 1,3-Dipolar Cycloaddition Reactions, ChemistrySelect, 2020, 5, 9834–9838 CrossRef CAS;
(b) H. Yazdani and A. Bazgir, Lewis Acid Catalyzed Regio- and Diastereoselective Synthesis of Spiroisoxazolines via One-Pot Sequential Knoevenagel Condensation/1,3-Dipolar Cycloaddition Reaction, Synthesis, 2019, 1669–1679 CAS;
(c) B. A. Chalyk, A. S. Sosedko, D. M. Volochnyuk, A. A. Tolmachev, K. S. Gavrilenko, O. S. Liashuk and O. O. Grygorenko, Regioselective synthesis of isoxazole and 1,2,4-oxadiazole-derived phosphonates via [3 + 2] cycloaddition, Org. Biomol. Chem., 2018, 16, 9152–9164 RSC;
(d) D. M. Kuznetsov and A. G. Kutateladze, Step-Economical Photoassisted Diversity-Oriented Synthesis: Sustaining Cascade Photoreactions in Oxalyl Anilides to Access Complex Polyheterocyclic Molecular Architectures, J. Am. Chem. Soc., 2017, 139, 16584–16590 CrossRef CAS PubMed.
- For the furoxan framework, see:
(a) R. Matsubara, A. Ando, H. Hasebe, H. Kim, T. Tsuneda and M. Hayashi, Synthesis and synthetic application of chloro- and bromofuroxans, J. Org. Chem., 2020, 85, 5959–5972 CrossRef CAS PubMed For the isoxazole motif, see:
(b) H. Yazdani, E. Kaul, A. Bazgir, D. Maysinger and A. Kakkar, Telodendrimer-based macromolecular drug design using 1,3-dipolar cycloaddition for applications in biology, Molecules, 2020, 25, 857 CrossRef CAS PubMed;
(c) M. Girardin, S. J. Dolman, S. Lauzon, S. G. Ouellet, G. Hughes, P. Fernandez, G. Zhou and P. D. O'Shea, Development of a Practical Synthesis of Stearoyl-CoA Desaturase (SCD1) Inhibitor MK-8245, Org. Process Res. Dev., 2011, 15, 1073–1080 CrossRef CAS For oxadiazoles, see:
(d) G. R. Humphrey and S. H. B. Wright, A novel synthesis of 3-bromo-1,2,4-oxadiazoles, J. Heterocycl. Chem., 1989, 26, 23–24 CrossRef CAS.
-
(a) W. Chen, B. Wang, N. Liu, D. Huang, X. Wang and Y. Hu, Tandem Synthesis of 3-Halo-5-Substituted Isoxazoles from 1-Copper(I) Alkynes and Dihaloformaldoximes, Org. Lett., 2014, 16, 6140–6143 CrossRef CAS PubMed;
(b) F. Tamborini, L. Mastronardi, B. Lo Presti, C. De Micheli, P. Conti and A. Pinto, Synthesis of L-tricholomic acid analogs and pharmacological charaacterization at ionotropic glutamate receptors, ChemistrySelect, 2017, 2, 10295–10299 CrossRef;
(c) S. Bruno, A. Pinto, G. Paredi, L. Tamborini, C. De Micheli, V. La Pietra, L. Marinelli, E. Novellino, P. Conti and A. Mozzarelli, Promiscuity and Selectivity in Covalent Enzyme Inhibition: A Systematic Study of Electrophilic Fragments, J. Med. Chem., 2014, 57, 7465–7471 CrossRef CAS PubMed.
- C. Battilocchio, F. Bosica, S. M. Rowe, B. L. Abreu, E. Godineau, M. Lehmann and S. V. Ley, Continuous Preparation and Use of Dibromoformaldoxime as a Reactive Intermediate for the Synthesis of 3-Bromoisoxazolines, Org. Process Res. Dev., 2017, 21, 1588–1594 CrossRef CAS.
- B. A. Chalyk, K. V. Hrebeniuk, Y. V. Fil, K. S. Gavrilenko, A. B. Rozhenko, B. V. Vashchenko, O. V. Borysov, A. V. Biitseva, P. S. Lebed, I. Bakanovych, Y. S. Moroz and O. O. Grygorenko, Synthesis of 5-(Fluoroalkyl)isoxazole Building Blocks by Regioselective Reactions of Functionalized Halogenoximes, J. Org. Chem., 2019, 84, 15877–15899 CrossRef CAS PubMed.
-
(a) O. T. O'Sullivan and M. J. Zdilla, Properties and Promise of Catenated Nitrogen Systems As High-Energy-Density Materials, Chem. Rev., 2020, 120, 5682–5744 CrossRef PubMed;
(b) L. L. Fershtat and N. N. Makhova, 1,2,5−Oxadiazole–Based High–Energy–Density Materials: Synthesis and Performance, ChemPlusChem, 2019, 85, 13–42 CrossRef;
(c) J. T. Wu, J. Xu, W. Li and H. B. Li, Coplanar Fused Heterocycle–Based Energetic Materials, Propellants, Explos., Pyrotech., 2020, 45, 536–545 CrossRef CAS;
(d) Y. Qu and S. P. Babailov, Azo-linked high-nitrogen energetic materials, J. Mater. Chem. A, 2018, 6, 1915–1940 RSC.
-
(a) S. Chen, Y. Liu, Y. Feng, X. Yang and Q. Zhang, 5,6-Fused bicyclic tetrazolo-pyridazine energetic materials, Chem. Commun., 2020, 56, 1493–1496 RSC;
(b) H. Huo, J. Zhang, J. Dong, L. Zhai, T. Guo, Z. Wang, F. Bi and B. Wang, A promising insensitive energetic material based on a fluorodinitromethyl explosophore group and 1,2,3,4-tetrahydro-1,3,5-triazine: synthesis, crystal structure and performance, RSC Adv., 2020, 10, 11816–11822 RSC;
(c) C. Bian, X. Dong, X. Zhang, Z. Zhou, M. Zhang and C. Li, The unique synthesis and energetic properties of a novel fused heterocycle: 7-nitro-4-oxo-4,8-dihydro-[1,2,4]triazolo[5,1-d][1,2,3,5]tetrazine 2-oxide and its energetic salts, J. Mater. Chem. A, 2015, 3, 3594–3601 RSC.
-
(a) Y. Tang, H. Yang, B. Wu, X. Ju, C. Lu and G. Cheng, Synthesis and characterization of a stable, catenated N11 energetic salt, Angew. Chem., Int. Ed., 2013, 52, 4875–4877 CrossRef CAS PubMed;
(b) X. X. Zhao, S. H. Li, Y. Wang, Y. C. Li, F. Q. Zhao and S. P. Pang, Design and synthesis of energetic materials towards high density and positive oxygen balance by N-dinitromethyl functionalization of nitroazoles, J. Mater. Chem. A, 2016, 4, 5495–5504 RSC.
-
(a) C. Yan, K. Wang, T. Liu, H. Yang, G. Cheng and Q. Zhang, Exploiting the energetic potential of 1,2,4-oxadiazole derivatives: combining the benefits of a 1,2,4-oxadiazole framework with various energetic functionalities, Dalton Trans., 2017, 46, 14210–14218 RSC;
(b) W. H. Lee and K. Kwon, Safe Synthesis of TKX–50 Using an Insensitive Intermediate, Propellants, Explos., Pyrotech., 2019, 44, 1478–1481 CrossRef CAS.
-
(a)
W. Friedrich and K. Flick, DE719135, 1942;
(b)
W. Friedrich, GB51906919400315, 1940;
(c)
W. Friedrich and K. Flick, US217978319391114, 1939;
(d) T. M. Klapötke, D. G. Piercey and J. Stierstorfer, Chemistry, 2011, 17, 13068–13077 CrossRef PubMed.
- K. Banert, S. Richter, D. Schaarschmidt and H. Lang, Well known or new? Synthesis and structure assignment of binary C2N14 compounds reinvestigated, Angew. Chem., Int. Ed., 2013, 52, 3499–3502 CrossRef CAS PubMed.
-
(a) X. Li, Q. Sun, Q. Lin and M. Lu, [N-N=N-N]-linked fused triazoles with π-π stacking and hydrogen bonds: Towards thermally stable, Insensitive, and highly energetic materials, Chem. Eng. J., 2021, 406, 126817 CrossRef CAS;
(b) D. R. Wozniak, B. Salfer, M. Zeller, E. F. C. Byrd and D. G. Piercey, Tailoring Energetic Sensitivity and Classification through Regioisomerism, Org. Lett., 2020, 22, 9114–9117 CrossRef CAS PubMed.
-
(a)
http://www.bam.de
;
(b) A 20 mg sample was subjected to a drop-hammer test with a 5 or 10 kg dropping weight. The impact sensitivity was characterized according to the UN recommendations (insensitive, >40 J; less sensitive, 35 J; sensitive, 4 J; very sensitive, 3 J).
-
(a) R. Bu, F. Jiao, G. Liu, J. Zhao and C. Zhang, Categorizing and Understanding Energetic Crystals, Cryst. Growth Des., 2021, 21, 3–15 CrossRef CAS.
-
M. Sućeska, EXPLO5, Version 6.01, Brodarski Institute, Zagreb, Croatia, 2013 Search PubMed.
- P. A. Wade, J. F. Bereznak, B. A. Palfey, P. J. Carroll, W. P. Dailey and S. Sivasubramanian, Diastereofacial selectivity studies on 3-alkenyl-4,5-diphenyl-4,5-dihydroisoxazoles, J. Org. Chem., 1990, 55, 3045–3051 CrossRef CAS.
- D. E. Chavez and M. A. Hiskey, Synthesis of the bi-heterocyclic parent ring system 1,2,4-triazolo[4,3-b][1,2,4,5]tetrazine and some 3,6-disubstituted derivatives, J. Heterocycl. Chem., 1998, 35, 1329–1332 CrossRef CAS.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Phys. Chem., 1993, 98, 5648–5652 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213 CrossRef CAS.
-
D. R. Lide, CRC Handbook of Chemistry and Physics, 88th Edition (Internet Version 2008), CRC Press/Taylor and Francis, Boca Raton, FL, 2007–2008 Search PubMed.
- D. G. Truhlar and B. C. Garrett, Variational transition state theory, Annu. Rev. Phys. Chem., 1984, 35, 159–189 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C-NMR and DSC spectra and crystallographic data. CCDC 2044151–2044155 and 2057903. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1qo00123j |
|
| This journal is © the Partner Organisations 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a,
Qi
Zhang
*a,
Qi
Zhang