
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe recent development of donepezil structure-based hybrids as potential multifunctional anti-Alzheimer's agents: highlights from 2010 to 2020
Rzgar Tawfeeq Kareem
a,
Fahimeh Abedinifar
 b,
Evan Abdolkareem Mahmood
c,
Abdol Ghaffar Ebadi
d,
Fatemeh Rajabi
*e and
Esmail Vessally
e
b,
Evan Abdolkareem Mahmood
c,
Abdol Ghaffar Ebadi
d,
Fatemeh Rajabi
*e and
Esmail Vessally
e
aDepartment of Chemistry, College of Science, University of Bu Ali Sina, Hamadan, Iran
bSchool of Chemistry, College of Science, University of Tehran, Tehran, Iran
cCollege of Health Sciences, University of Human Development, Sulaimaniyah, Kurdistan region of Iraq
dDepartment of Agriculture, Jouybar Branch, Islamic Azad University, Jouybar, Iran
eDepartment of Chemistry, Payame Noor University, P.O. Box 19395-3697, Tehran, Iran. E-mail: f_rajabi@pnu.ac.ir
First published on 16th September 2021
Abstract
Dementia is a term used to define different brain disorders that affect memory, thinking, behavior, and emotion. Alzheimer's disease (AD) is the second cause of dementia that is generated by the death of cholinergic neurons (especially acetylcholine (ACh)), which have a vital role in cognition. Acetylcholinesterase inhibitors (AChEI) affect acetylcholine levels in the brain and are broadly used to treat Alzheimer's. Donepezil, rivastigmine, and galantamine, which are FDA-approved drugs for AD, are cholinesterase inhibitors. In addition, scientists are attempting to develop hybrid molecules and multi-target-directed ligands (MTDLs) that can simultaneously modulate multiple biological targets. This review highlights recent examples of MTDLs and fragment-based strategy in the rational design of new potential AD medications from 2010 onwards.
 Rzgar Tawfeeq Kareem | Rzgar Tawfeeq Kareem was born in Sulaimaniyah, Iraq, in 1988. He received his B.S. degree in pure chemistry from Garmian University, Iraq, and his M.S. degree in analytical chemistry from the University of Siirt, Turkey. Now, he is completing his PhD degree at the University of Bu Ali Sina. |
 Fahimeh Abedinifar | Fahimeh Abedinifar was born in 1983 in Qazvin, Iran. She received her M.Sc. in organic chemistry from IKIU University in 2009 under the guidance of Prof. M. Alavi. She carried out her PhD degree under the supervision of Dr M. Farnia at Tehran university in 2018. She collaborated as a postdoctoral researcher with Dr Larijani's research team at the Institute of Endocrinology and Metabolism Clinical Sciences, Tehran University of Medical Sciences, Iran, from 2019 to 2020. Her research interests include the synthesis of biologically active heterocycles and application of new catalysts in multicomponent reactions. |
 Evan Abdolkareem Mahmood | Evan Abdolkareem Mahmood was born in Erbil, Kurdistan Region, Iraq, in 1990. She is a Biochemistry lecturer. She lives in Iraq/Kurdistan Region. About her educational background, she got a Bachelor degree of Chemistry from University of Sulaimaniyah, College of Science. She completed her Master Degree in Biochemistry at Yüzüncü Yil University at Van city, Turkey. She is an Assistant Lecturer at University of Human Development, College of Health Science. Also, she worked as a Head of Pharmacy department at National Institute in Sulaimaniyah/Iraq. Her research interests include health, environment, chemistry, clinical biochemistry science. |
 Abdol Ghaffar Ebadi | Dr Abdol Ghaffar Ebadi finished his doctoral degree in Environmental Biotechnology (Algology) from Tajik Academy of Sciences. Now he is a researcher in TAS in Tajikistan and faculty member at the Islamic Azad University of Jouybar in Mazandaran. Dr Ebadi published more than 400 scientific papers in qualified international journals and attended more than 50 international conferences. He has cooperation with many research project teams around world such as China, Malaysia, and Thailand. His interests are Environmental Biotechnology, Biochemistry, Gene pathways in the phytoremediation processes. |
 Fatemeh Rajabi | Dr Fatemeh Rajabi was born in Qazvin, Iran. She received her doctoral degree in organic chemistry at the Sharif University of Technology in 2005. In 2004, she received a British Council Scholarship and joined Prof. James Clark research group at the University of York (UK) in the Green Chemistry Centre of Excellence where the group works at the frontiers of modern chemical research in the areas of clean synthesis, catalysis, novel materials and application of renewable resources. She has become a faculty member at the Payame Noor University since 2006. In 2010 and 2017, she received DAAD Research Stays for University Academics and Scientists and joined Prof. Werner Thiel research group at the Technische Universität Kaiserslautern, Germany, where they work on the synthesis and characterization of homogeneous and heterogeneous (photo)catalysts and the elucidation of reaction mechanisms and structure/activity relationships. In 2012–2013, 2014, and 2018 she received a prestigious Alexander Von Humboldt Fellowship and was re-invited to join Prof. Werner Thiel research group and establish long collaboration. Her main research interests is the synthesis of hybrid organic-inorganic mesoporous silica-based organometallic complexes applied to green and sustainable catalysis. |
 Esmail Vessally | Esmail Vessally was born in Sharabiyan, Sarab, Iran, in 1973. He received his B.S. degree in pure chemistry from university of Tabriz, Tabriz, Iran, and his M.S. degree in organic chemistry from Tehran university, Tehran, Iran, in 1999 under the supervision of Prof. H. Pirelahi. He completed his PhD degree in 2005 under the supervision of Prof. M. Z. Kassaee. Now he is working at Payame Noor University as Professor in organic chemistry. His research interests include theoretical organic chemistry, new methodologies in organic synthesis and spectral studies of organic compounds. |
1. Introduction
Alzheimer's disease (AD) is a progressive, chronic, and incurable neurological disorder that affects millions of people worldwide. According to the World Alzheimer report, there are currently more than 30 million people with Alzheimer's disease, with estimates that this number will increase up to 115 million in 2050.1 The symptoms of this disease consist of memory loss, challenges in planning or solving problems, changes in mood and personality, and decreased judgment or decision-making abilities.2 Cholinergic neuron death, diminishing neurotransmitter acetylcholine (ACh) levels in the brain,3 metal dyshomeostasis, oxidative stress,4 deficiency in steroid hormones, higher inflammatory mediators, deposition of β-amyloid (Aβ),5 and tau-protein6 have been mentioned as the main causes of AD.Since this disease has a multifactorial nature, there is no effective treatment. Multi-target-directed ligands (MTDLs) are single molecules that can simultaneously modulate multiple targets in neurodegenerative disorders. Multi-target-directed ligands (MTDLs) generally are designed by connecting the pharmacophores of target ligands and are produced by chemical syntheses. Degeneration in the basal forebrain leads to a deficiency in central cholinergic transmission, which is believed to be a significant neurochemical and pathological characteristic of AD.7 The cholinergic hypothesis is one of the oldest hypotheses that takes the role of ACh role into account in animal and human behavior. Even though the cholinergic hypothesis is the most developed, the other hypothesis, Aβ peptide aggregation, describes how this aggregation could affect the AChE enzyme and is considered the most important. Hydrolysis of acetylcholine in the brain occurs by two types of cholinesterases (ChE): acetylcholinesterase enzyme (AChE) and butyrylcholinesterase (BuChE). Acetylcholinesterase enzyme (AChE) possesses two main binding sites for drug interaction, the catalytic site and the peripheral anionic site (PAS) (Fig. 1).8
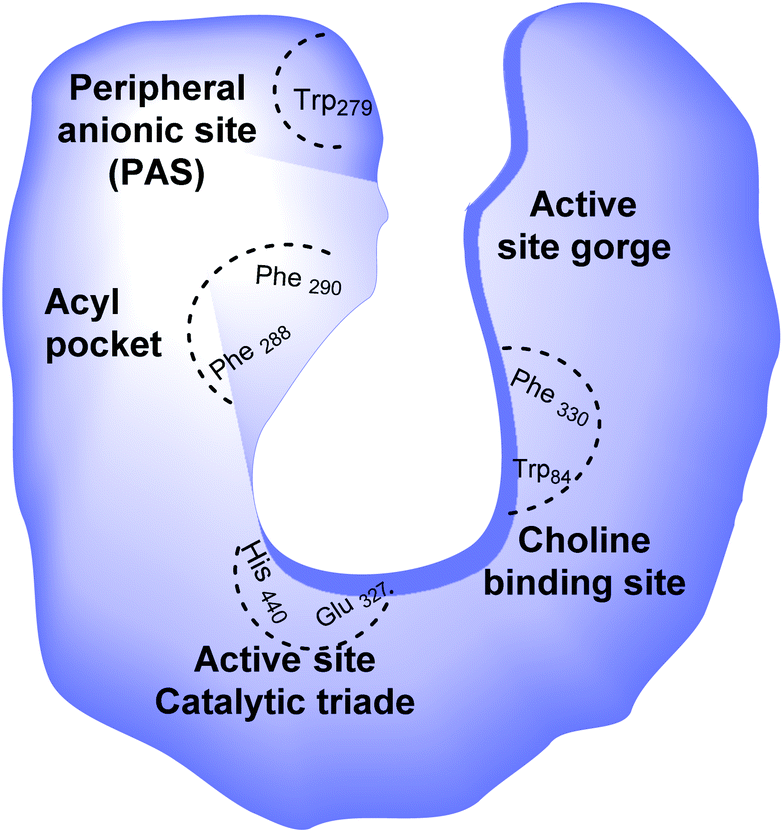 | ||
| Fig. 1 Schematic representation of AChE binding sites; His: histidine, Glu: glutamate, Phe: phenylalanine, Trp: tryptophan. | ||
In the amyloid aggregation process, the AChE anionic binding site is believed to be of paramount importance. Hence, this site is targeted as an alternative site by AChE inhibitors, which function as multi-target ligands that cause a decrease in the formation of senile plaque. Therefore, cholinomimetic therapy with anticholinergic drugs is the main approach in treating AD. The acetylcholinesterase enzyme inhibitor (AChEI) increases endogenous AChE levels and enhances cholinergic neurotransmission in the brain. Among the investigated acetylcholinesterase inhibitors, donepezil (2-((1-benzylpiperidin-4-yl)methyl)-5,6-dimethoxy-2,3-dihydro-1H-inden-1-one) is of most highly effective and well-known FDA-approved drug (Fig. 2).9 Donepezil consists of dimethoxy indanone, which is connected to N-benzylpiperidine via a methylene linker.
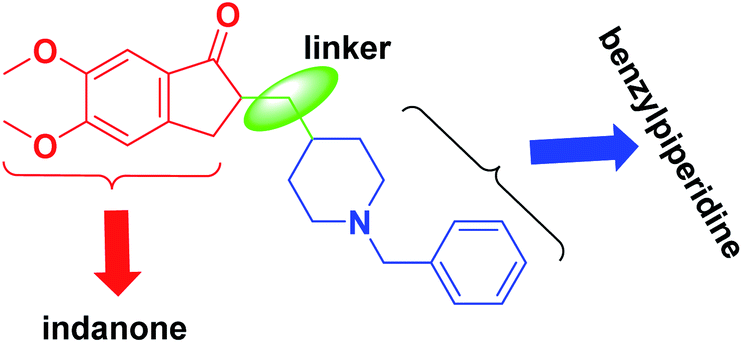 | ||
| Fig. 2 Structure of donepezil. | ||
A molecular docking investigation of donepezil showed that the indanone moiety fuses with the PAS of AChE, and the benzylic part links with the catalytic anionic site (CAS).10 The N-benzylpiperidine group is situated onto the narrowest active-site cavity section. It interacts with four amino acid residues, Tyr-70, Asp-72, Tyr-121, and Tyr-334, in the anionic site.11 A large number of reviews have been published on Alzheimer's topics.12,13 Still, this review summarizes recent findings in AD research by emphasizing simple synthetic compounds with modified donepezil structures that act as MTDLs. We focused on the current literature and produced a collection of all hybrids of N-saturated heterocyclic compounds based on the structure of donepezil that were published from 2010 until now. We categorized this review into main sections of donepezil derivatives with different linkers for ease of reading, replacing indanone with heterocycles, and inserting piperazine/pyrrolidine instead of piperidine to produce donepezil-like structures. This current review will provide insights into the design and development of novel therapeutics for treating AD.
2. Donepezil-like derivatives with different linkers
One strategy in designing new acetylcholinesterase inhibitors (AChEI) is to create different linkers in the donepezil structure. Costanzo et al. synthesized indanonylidenyl precursors of donepezil through condensing the indanone nucleus and derivatives of N-benzylpiperidine-4-carboxaldehyde in the presence of a basic resin, Amberlyst A-26. The biological assessment indicated that compounds 1 and 2 demonstrated higher dual activity and decreased values of IC50 toward the AChE enzyme and BACE-1 enzyme as compared to donepezil (Fig. 3).14 Greunen et al. synthesized two libraries of possible AChE inhibitors according to the AChE molecular skeleton by replacing the piperidine ring with N-saturated ring systems in different sizes and introducing various linker piperidine rings to the indanone in donepezil. Replacement of a methyl linker with an ester moiety resulted in a complete loss of activity in all but one case, where 5,6-dimethoxy-1-oxo-2,3-dihydro-1H-inden-2-yl-1-benzylpiperidine-4-carboxylate 3 yielded 0.03 μM IC50 towards AChE in vitro (Fig. 3).15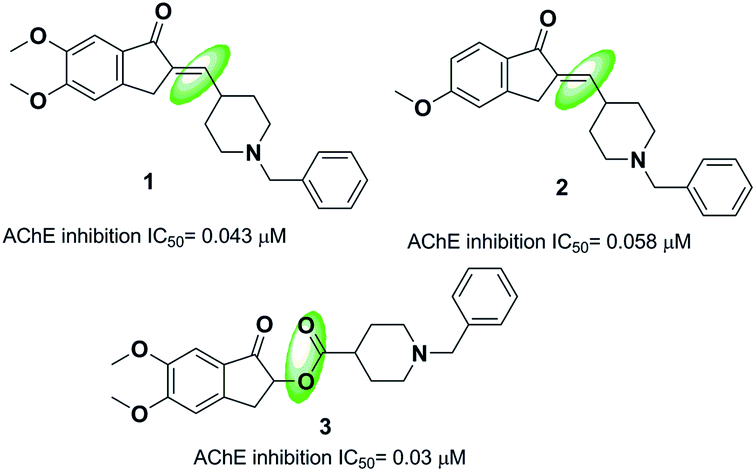 | ||
| Fig. 3 Donepezil analogs with different linkers. | ||
3. Donepezil-like derivatives with the replacement of the indanone part
As mentioned in the introduction, donepezil consists of an indanone moiety. Replacement of this part with various compounds has led to new hybrids of donepezil with various inhibitory activities.3.1. Replacement of indanone with aromatics
Yan et al. produced a new series of derivatives by fusing donepezil and curcumin pharmacophores under aldol condensation.16 They investigated the AChE inhibitor activity, Aβ-aggregation, metal chelation, and antioxidant activities. Among the synthetic targets, compound 4 showed the highest selectivity ratio of 66.3 for BuChE/AChE, which was considerably better than the reference tacrine and galantamine with a selectivity ratio of 0.15 and 25.3 and slightly worse than donepezil with a selectivity ratio of 85.4. Compound 4 also demonstrated potent inhibition of self-induced Aβ aggregation (Fig. 4). Košak et al. developed a class of N-benzylpiperidine carbamates as viable anti-Alzheimer agents. Compound 5a showed selective hBuChE and hMAO-B inhibitor activity (with an IC50 of 0.178 μM), but 5b exhibited selective hAChE inhibition (Fig. 4).17 In 2017, Monjas prepared L- and D-glutamic acid derivatives with the N-benzylpiperidine fragment in the γ-position in the presence of Mucor miehei and Candida antarctica lipases in high yield. The multifunctional profile of synthesized compound 6 exhibited stronger inhibition of human ChE activity (Fig. 4).18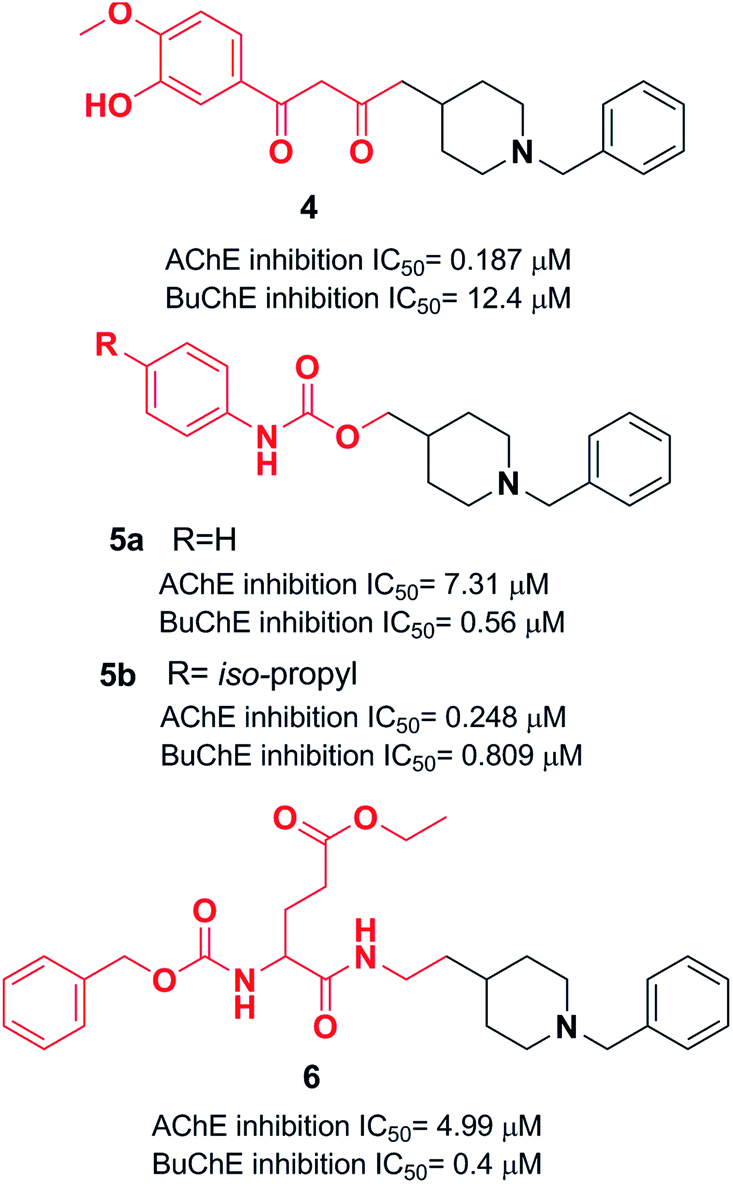 | ||
| Fig. 4 Donepezil analogs with aromatic molecules. | ||
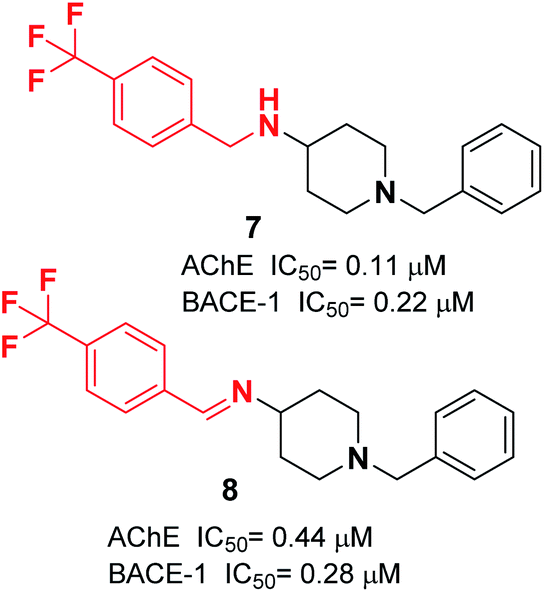
At ambient temperature, the treatment of 1-benzylpiperidin-4-amine with disparate aromatic aldehydes under glacial acetic acid in absolute ethanol generated imine hybrids. The reduction of imines in NaBH4 produced benzylamine derivatives in good yield (Fig. 5). These two series of amine derivatives were assessed as multi-functional AChE and BACE-1 inhibitors having moderate to excellent activities. Compared with donepezil, which exhibited 42.1-fold AChE selectivity (with an IC50 of 0.44 μM for AChE, and IC50 of 0.28 μM for BACE-1), the selective inhibition of compound 7 was 28.2-fold for AChE, and it displayed self-induced as well as AChE-induced Aβ aggregation inhibition in the thioflavin T-assay. Docking studies revealed that 7 and 8 elicited similar interactions at the PAS, except for additional π–π stacking and π–cation interactions with the Tyr341 residue. The benzyl portion of the compounds exhibited interactions with Trp86 (π–π stacking or π–cation) and Glu202 (electrostatic) residues at anionic subsites (Fig. 5).19
 | ||
| Fig. 5 Structure of donepezil-based benzylamine derivatives. | ||
3.2. Replacement of indanone with ferulic acid derivatives
Ferulic acid, which is found in plant cell walls, is a hydroxycinnamic acid with pharmacological activities.20 In a study carried out by Dias et al., the synthesis, design, and pharmacological assessment of novel molecular hybrids of feruloyl–donepezil were reported. The N-benzylpiperidine pharmacophore of donepezil is responsible for its sufficient recognition through AChE and the subunit feruloyl, which exists in curcumin and ferulic acid. Based on the in vitro results, all the compounds mentioned demonstrated potent AChE inhibitory activity, moderate antioxidant properties, neuroprotection of neuronal cells against the oxidative process, and significant metal chelation of Fe2+ and Cu2+. Compound 9 had the greatest AChE inhibitor potency (Fig. 6).21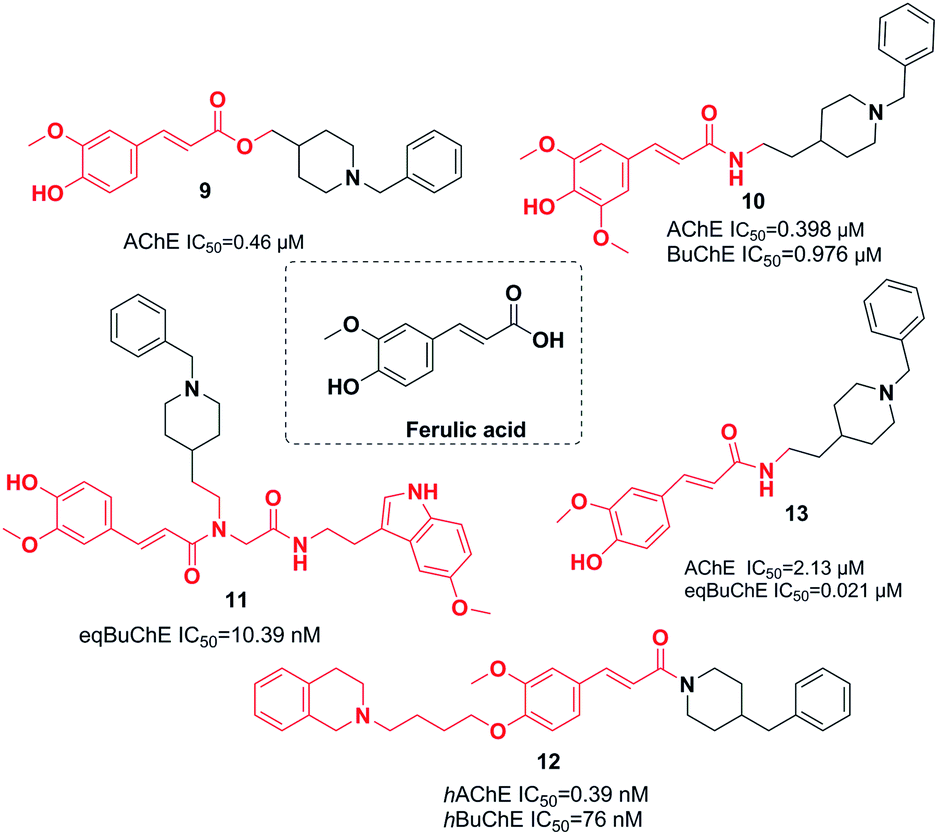 | ||
| Fig. 6 Structure of most potent donepezil–ferulic acid hybrids. | ||
In 2016, the design and synthesis of other donepezil–ferulic acid hybrids were carried out. The synthetic molecules were assessed as MTDLs against AD. An in vitro assay demonstrated that there was moderate AChE and BuChE inhibition by compound 10. This compound exhibited considerable antioxidant activity (1.78 Trolox equivalents through the ABTS method, with an IC50 of 24.9 μM through the DPPH method) (Fig. 6).22 Benchekroun et al. prepared donepezil–ferulic acid hybrids (DFAHs) through one-pot Ugi-4CR in low to moderate yield. They found that hybrid (E)-N-(2-(1-benzylpiperidin-4-yl)ethyl)-3-(4-hydroxy-3-methoxyphenyl)-N-(2-((2-(5-methoxy-1H-indol-3-yl)ethyl)amino)-2-oxoethyl)acrylamide 11 was a selective eqBuChE, and the oxygen radical-absorbing capacity of this compound was 8.71 μmol (Fig. 6).23
Sang et al. synthesized new derivatives of ferulic acid-O-alkylamine and then evaluated them against AD. Based on in vitro studies, all exhibited considerable inhibitory activity toward BuChE, acceptable self-induced aggregation (Aβ), and functioned as viable antioxidants. Specifically, compound 12 was a satisfactory AChE inhibitor with the greatest BuChE inhibition potency. It exhibited an inhibitory impact upon self-induced aggregation of Aβ1-42 (50.8%) and self-induced aggregation of Aβ1-42 (38.7%), moderate antioxidant activity with 0.55 eq. of Trolox, acceptable protection against H2O2-induced PC12 cell injury, and low toxicity (Fig. 6).22 Estrada et al. linked cinnamic-related structures to N-benzylpiperidine. Compound 13 in series demonstrated moderate inhibition of hAChE with an IC50 of 0.39 and inhibition of hBuChE with an IC50 of 76 nM. The existence of the p-hydroxy group was necessary for inhibition of the cinnamic fragment (Fig. 6).24
3.3. Donepezil-like derivatives with replacement of indanone with heterocycles
 | ||
| Fig. 7 Donepezil–flavonoid hybrids. | ||
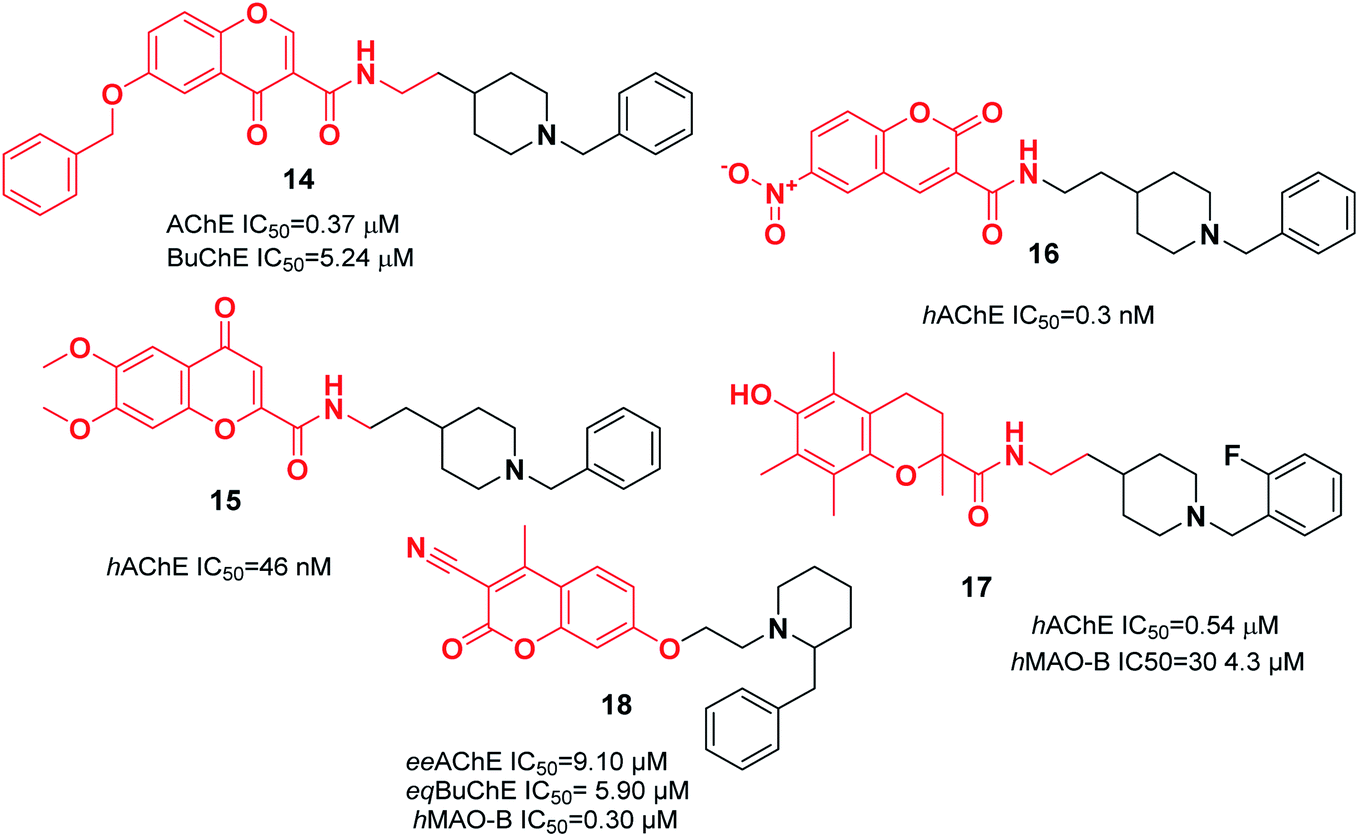
In a research study performed by Asadipour et al., derivatives of coumarin-3-carboxamide were synthesized that were connected to N-benzylpiperidine. Based on the results, almost all the compounds exerted potent activity against AChE in the nM concentration range. Among them, compound 16, with a N-methyl carboxamide linker and 6-nitro substituent, demonstrated the most potent activity and the highest selectivity of 26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 with 46-fold more potency than the standard drug donepezil against AChE (Fig. 7).27 To synthesize multifunctional anti-AD agents, donepezil and Trolox pharmacophores were fused into one molecule by Cai et al. Among the targets, N-(2-(1-(2-fluorobenzyl)piperidin-4-yl)ethyl)-6-hydroxy-2,5,7,8-tetramethylchromane-2-carboxamide 17 exhibited balanced functions with an acceptable inhibitory effect upon hAChE and hMAO-B, considerable antioxidant activity with an IC50 of 41.33 μM, outstanding copper chelation, Aβ1–42 aggregation inhibition impact, and less hepatotoxicity toward cell lines HePG2, PC12, and BV-2 (Fig. 7).28 The design and synthesis of 7-substituted coumarin derivatives were carried out by Joubert et al. These compounds retained the N-benzylpiperidine function of donepezil (ChE inhibitor) and the coumarin structure (MAO-B inhibitor) connected at the 7-position by an alkyl ether linkage. According to the biological assay results, compound 18 had the highest multifunctional agent potency, and exhibited a satisfactory eeAChE inhibition and potent selective hMAO-B inhibition (SI > 33). According to the molecular modeling, compound 18 simultaneously binds to the PAS, mid-gorge, and CAS of AChE and BuChE (Fig. 7).29
300 with 46-fold more potency than the standard drug donepezil against AChE (Fig. 7).27 To synthesize multifunctional anti-AD agents, donepezil and Trolox pharmacophores were fused into one molecule by Cai et al. Among the targets, N-(2-(1-(2-fluorobenzyl)piperidin-4-yl)ethyl)-6-hydroxy-2,5,7,8-tetramethylchromane-2-carboxamide 17 exhibited balanced functions with an acceptable inhibitory effect upon hAChE and hMAO-B, considerable antioxidant activity with an IC50 of 41.33 μM, outstanding copper chelation, Aβ1–42 aggregation inhibition impact, and less hepatotoxicity toward cell lines HePG2, PC12, and BV-2 (Fig. 7).28 The design and synthesis of 7-substituted coumarin derivatives were carried out by Joubert et al. These compounds retained the N-benzylpiperidine function of donepezil (ChE inhibitor) and the coumarin structure (MAO-B inhibitor) connected at the 7-position by an alkyl ether linkage. According to the biological assay results, compound 18 had the highest multifunctional agent potency, and exhibited a satisfactory eeAChE inhibition and potent selective hMAO-B inhibition (SI > 33). According to the molecular modeling, compound 18 simultaneously binds to the PAS, mid-gorge, and CAS of AChE and BuChE (Fig. 7).29
 | ||
| Fig. 8 Structure of phthalimide–dithiocarbamate hybrids. | ||
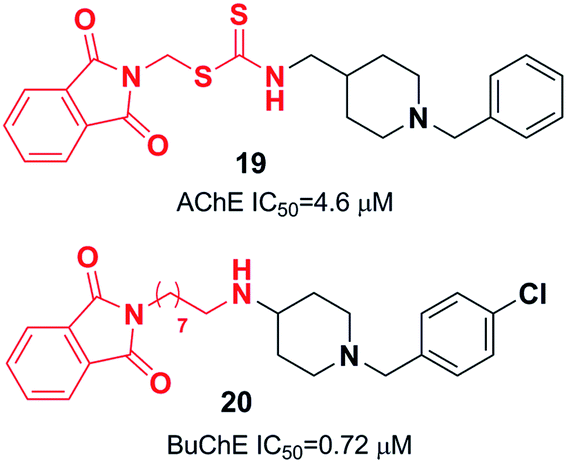
Substituting the indanone fragment of donepezil with the benzamide or 2-picolinamide moiety introduced a novel MTDL that exhibited potent inhibitory activities towards MAO-A, MAO-B, and AChE. Compound 21, N-(2-(1-benzylpiperidin-4-yl)ethyl)-5-chloropicolinamide, was the most potent agent with moderate cholinesterase inhibition activity in comparison with galanthamine as a reference (Fig. 9). It showed satisfactory inhibitory activity toward MAO-B with an IC50 of 3.14 μM and IC50 of 13.4 μM for MAO-A. Also, 21 can be considered a metal chelator, and it has the ability to cross the BBB with a low level of toxicity to PC12 cells.32
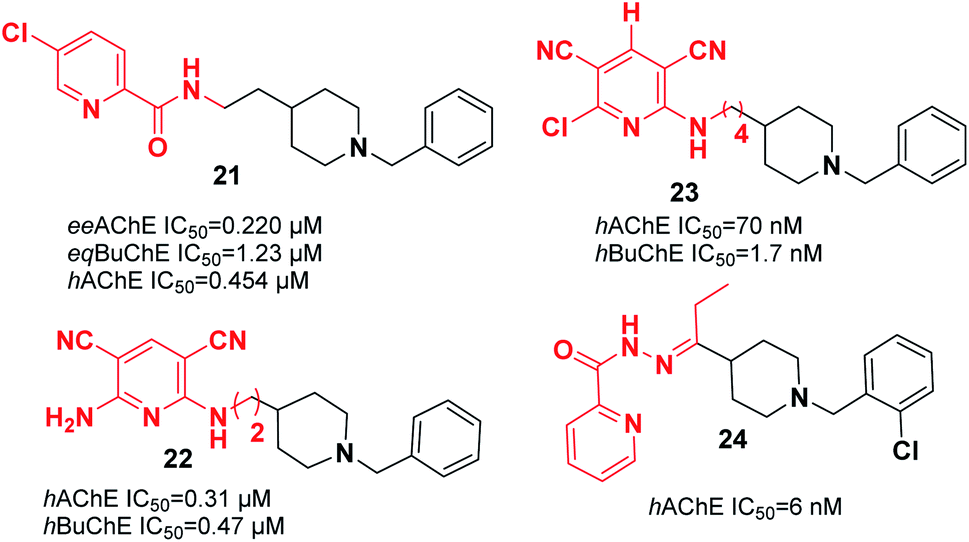 | ||
| Fig. 9 Pyridine–donepezil hybrids with ChEI activity. | ||
Samadi et al. attached the N-benzylpiperidine moiety of donepezil to 2-aminopyridine derivatives and synthesized a novel series of considerably potent donepezil–aminopyridine hybrids. The most potent hAChE inhibitor was 2-amino-6-((3-(1-benzylpiperidin-4-yl)propyl)amino)pyridine-3,5-dicarbonitrile pyridonepezil 22, and its activity was 2.6-fold less compared to donepezil for hBuChE (IC50 (hAChE) = 9.4 nM; IC50 (hBuChE) = 6.6 μM) (Fig. 9).33 In 2013, Samadi et al. developed 6-chloro-pyridonepezils and evaluated them against hAChE and hBuChE.34 According to the biological evaluation, the novel compounds are inhibitors of cholinesterase in the submicromolar range. Particularly, 6-chloro-pyridonepezil 23 was the most potent target, being 625-fold more selective for hAChE than hBuChE. It can be considered a selective dual AChEI for further pharmacological developments in AD treatment (Fig. 9). Novel molecules were produced through the interaction of the N-benzylpiperidine moiety of donepezil with pyridine hydrazide, which exhibited greater AChE inhibition and showed higher selectivity over BuChE. For the most active compound 24, the IC50 was 6 nM with the capability to chelate cupric ion (Fig. 9).35
It was reported that a new hybrid of donepezil-8-hydroxyquinoline was a multifunctional molecule that can be used to treat AD. Among the molecules, racemic α-aminonitrile-4-(1-benzylpiperidin-4-yl)-2-(((8-hydroxyquinolin-5-yl)methyl)(prop-2-yn-1-yl)amino)butane nitrile 25 was the most promising compound and exhibited mixed-type AChE inhibitory effects and metal-chelating characteristics (Fig. 10).36
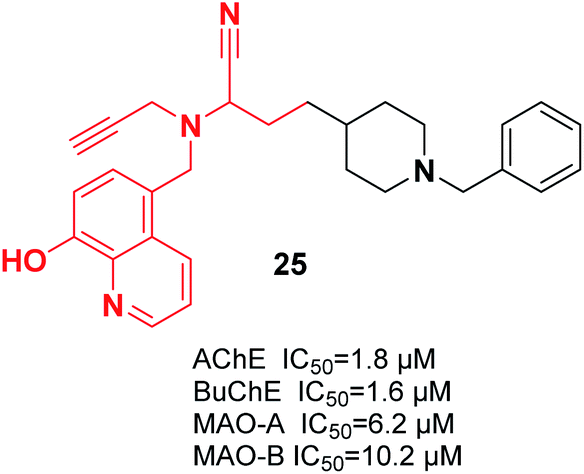 | ||
| Fig. 10 Structure of a donepezil-8-hydroxyquinoline derivative as a mixed-type AChE inhibitor. | ||
Donepezil-quinolone carboxamide analogs were synthesized by Pudlo et al. Derivative 26a with the methoxy group and 26b with the hydroxy group were potent and highly selective AChE inhibitors (Fig. 11).37
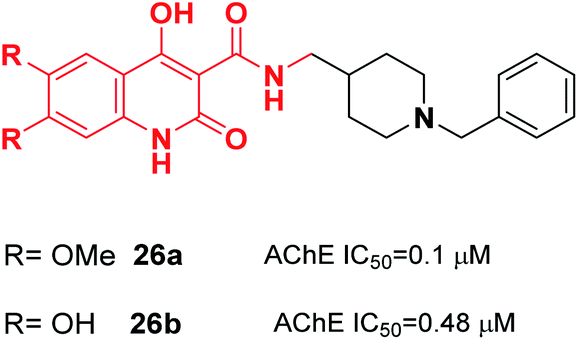 | ||
| Fig. 11 Structure of a quinolone–benzylpiperidine derivative as a new acetylcholinesterase inhibitor. | ||
Wang et al. produced novel N-benzylpiperidine-indole hybrids with a carboxamide linker. Compound 27 with a two-carbon spacer exhibited satisfactory AChE and BuChE inhibition, with an average inhibition (56.3%) of Aβ aggregation at 20 μM, and satisfactory antioxidant activity (3.28 Trolox equivalent by ORAC assay). Moreover, 27 chelated metal ions, decreased the death of PC12 cells caused by oxidative stress, and was able to cross the BBB. The molecular docking of 27 represents a mixed-type inhibitor that simultaneously binds with the CAS and PAS of AChE (Fig. 12).38 Based on the benzylpiperidine portion of donepezil and the indole derivative of propargylamine, various multi-target molecules were synthesized by Bautrisa-Aguilera et al. Among the synthetic compounds, N-((5-(3-(1-benzylpiperidin-4-yl)propoxy)-1-methyl-1H-indol-2-yl)methyl)prop-2-yn-1-amine 28 was the most potent inhibitor (Fig. 12).39
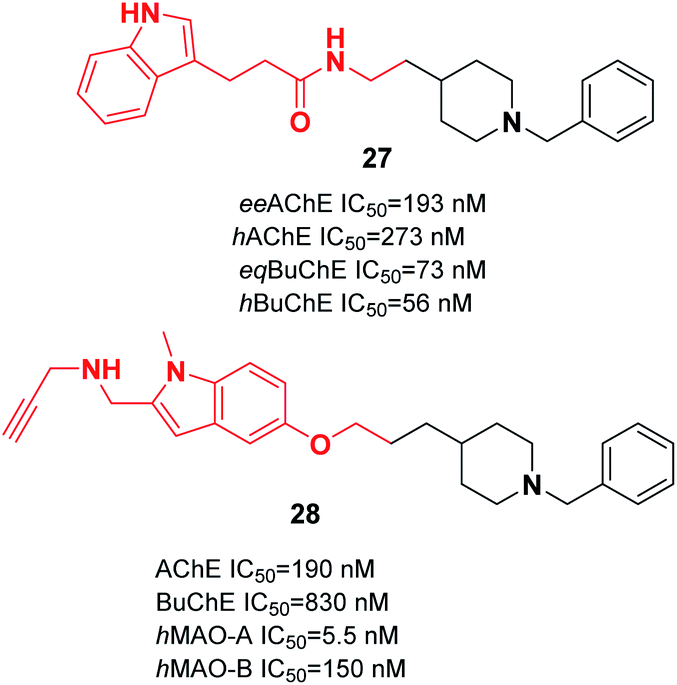 | ||
| Fig. 12 Multitarget molecules based on donepezil–indole hybrids. | ||
Piemontese et al. conjugated the benzylpiperidine part of donepezil with the benzimidazole bioactive molecule. The biological assessment showed that new targets have potential anti-Aβ aggregation capacity, antioxidant activity, and metal-chelating properties. The synthetic lead 29 showed satisfactory AChE inhibition activity and moderate inhibition of Aβ1–42 self-mediated aggregation, as well as metal-chelating ability. Benzimidazole hybrid 29 exhibited greater inhibition of Cu-induced aggregation of Aβ1–42 as well as enhanced antioxidant capacity (Fig. 13)40 based on the molecular docking assessment with compound 29 superimposed in the CAS as compared with donepezil. Recently, Chaves et al. synthesized 2-hydroxyphenyl benzimidazole derivative 30. They illustrated that the fluorine atoms and hydroxy groups on the benzene ring resulted in higher cholinergic AChE activity and increased β-amyloid (Aβ) aggregation (self-induced 61%, Cu-induced 71%) (Fig. 13).41
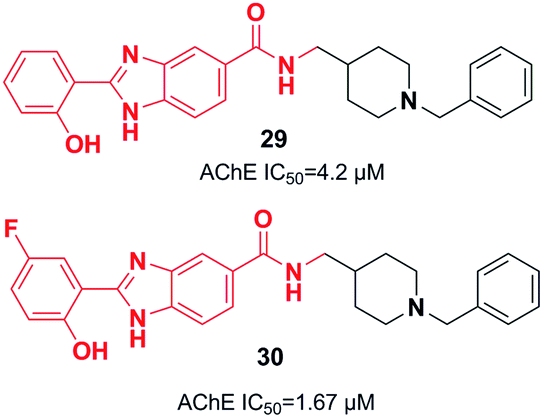 | ||
| Fig. 13 Hydroxy benzimidazole–donepezil hybrids that are multi-target-directed ligands. | ||
Mo et al. reported the synthesis of benzylpiperidine-linked 1,3-dimethylbenzimidazolinones. The synthesized compounds 31a and 31b containing F or Br on the meta-position of the benzyl ring showed submicromolar IC50 values for AChE and BuChE. Additionally, these compounds demonstrated a neuroprotective impact upon H2O2-induced oxidative damage to PC12 cells as well as antioxidant activity based on the DPPH assay (Fig. 14).42
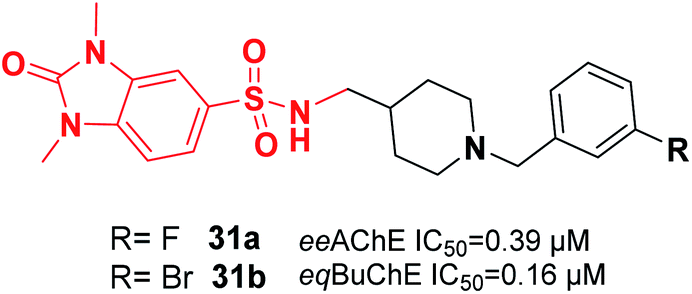 | ||
| Fig. 14 Structure of a dimethylbenzimidazolinone–benzylpiperidine hybrid. | ||
Kilic and colleagues focused on pyridazine derivatives. First, they synthesized 6-chloro-N-(2-substituted ethyl)pyridazine-3-carboxamide intermediates from 6-chloropyridazine-3-carboxylic acid and ethylamine derivatives. Next, they employed the Suzuki cross-coupling reaction for the carboxamide intermediate with a suitable phenylboronic acid derivative to obtain the corresponding N-(2-substituted ethyl)-6-phenylpyridazine-3-carboxamide derivatives. The biological assay revealed that pyridazine-3-carboxamide derivative 32 exerted dual cholinesterase inhibitory effects, with an IC50 of 0.16 μM for AChE and IC50 of 9.80 μM for BuChE (Fig. 15).43 The molecular docking presents the interactions of a pyridazine ring with the active-site gorge of AchE and the Trp279 residue in the peripheral anionic site and an anisole ring with a water molecule.
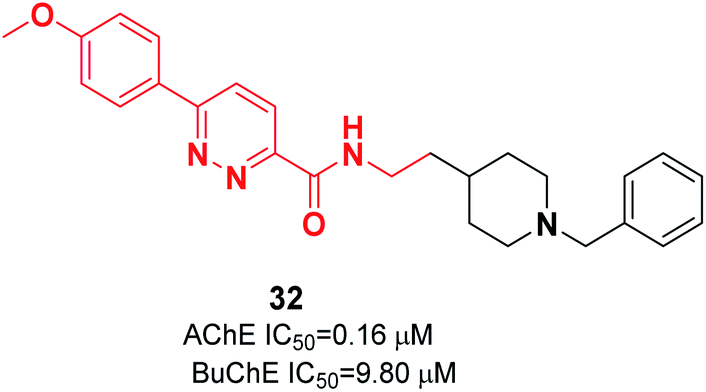 | ||
| Fig. 15 Structure of a phenylpyridazine-3-carboxamide-benzylpiperidine hybrid. | ||
Vila et al. reported synthesizing donepezil analogs using a phthalazin-1(2H)-one scaffold as potent human ChEIs. The biological assessment showed that the ChE inhibitory potency and selectivity of AChE/BuChE were affected by the structural modifications to a great extent. Also, the length of the chain between the N-benzylpiperidine fragment and the phthalazinone moiety impacted the inhibition of AChE. Compound 33 behaved as a dual cholinesterase inhibitor towards the AChE and BuChE enzymes (Fig. 16).44
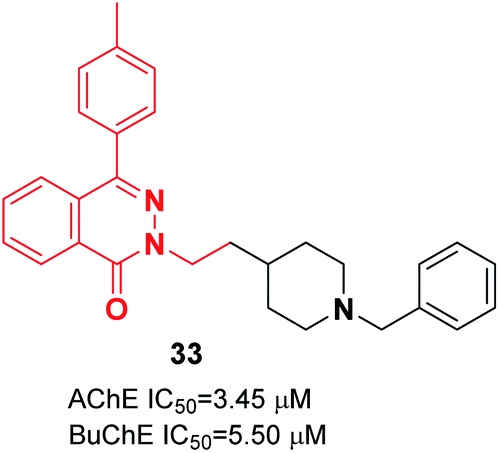 | ||
| Fig. 16 Structure of a phthalazin-1(2H)-one-donepezil hybrid. | ||
In 2019, Greunen et al. attached the indanone part of donepezil to different commercial heterocycle amines under mild conditions in the presence of catalytic 4-(dimethylamino)pyridine (4-DMAP) to obtain carboxamide derivatives in low to good yield. Among the selected targets, 1-benzyl-N-(1-methyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazol-4-yl)piperidine-4-carboxamide 34 afforded in vitro AChE inhibition with an IC50 value of 5.94 μM (Fig. 17).45
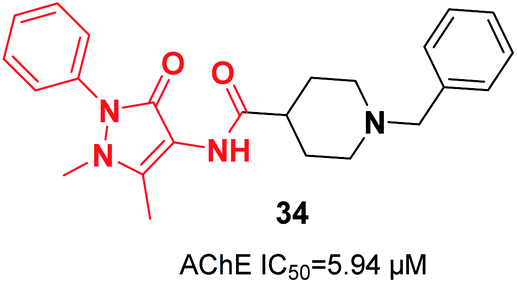 | ||
| Fig. 17 Structure of a pyrazole-benzylpiperidine derivative. | ||
De Andrade et al. replaced the 5,6-dimethoxy-1-indanone moiety of the donepezil structure with triazoles. This change resulted in a reduction in hAChE inhibition and an increase in hBuChE inhibitor activity. Compound 35 was the most potent (0.17 nM) selective hBuChE inhibitor (>58000-fold) and showed no cytotoxicity (Fig. 18).46
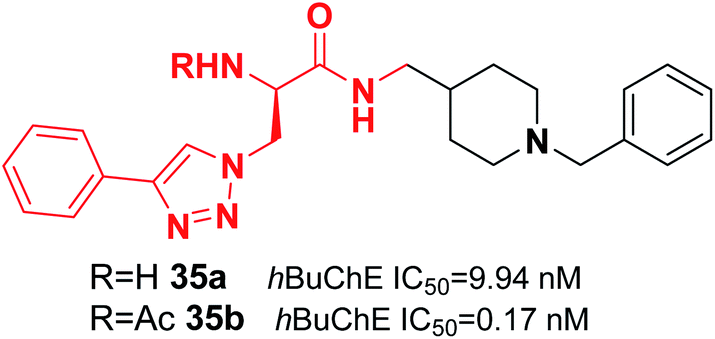 | ||
| Fig. 18 Structure of an azido 1,2,3-triazoles-benzylpiperidine analog as a selective BuChE inhibitor. | ||
In 2019, El-Sayed constructed pyrrolizine-benzylpiperidine hybrids. In vitro assay indicated that in this series, N-(1-benzylpiperidin-4-yl)-4-oxo-4,5,6,7-tetrahydro-3H-pyrimido[5,4-a]pyrrolizine-9-carboxamide 36 exhibited dual inhibitory impact upon hAChE and hBuChE in the submicromolar range. Also, compound 36 demonstrated cytotoxicity lower or similar to that of donepezil against a normal human hepatic (THLE2) cell line and human neuroblastoma (SH-SY5Y). Based on in vivo studies, the compound decreased the cognitive dysfunction of scopolamine-induced AD mice (Fig. 19).47
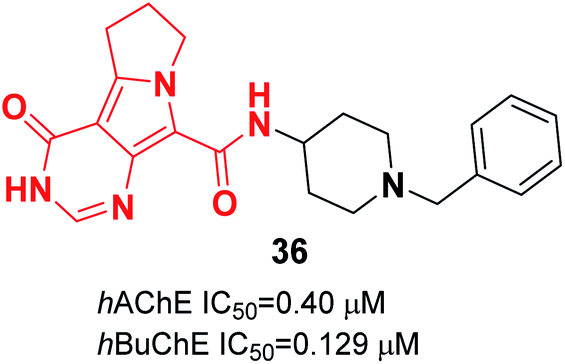 | ||
| Fig. 19 Structure of a pyrrolizine–benzylpiperidine hybrid. | ||
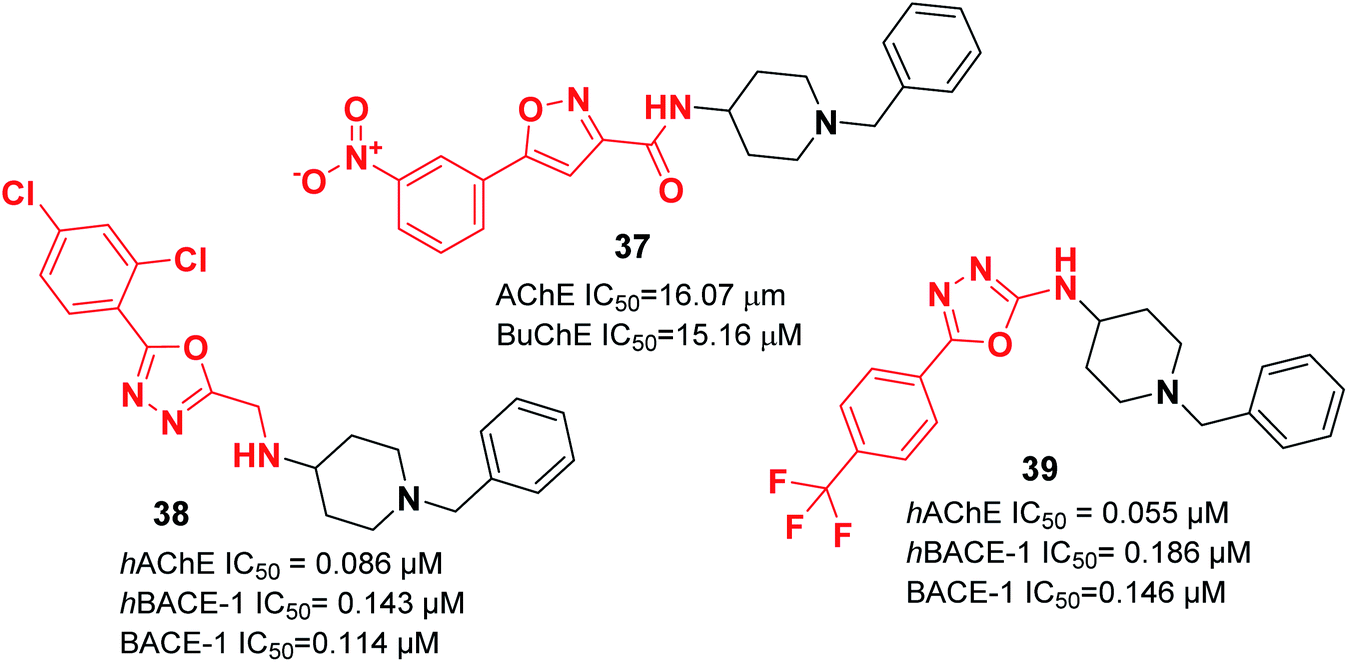 | ||
| Fig. 20 Structure of isoxazole/oxadiazole–benzylpiperidine hybrids. | ||
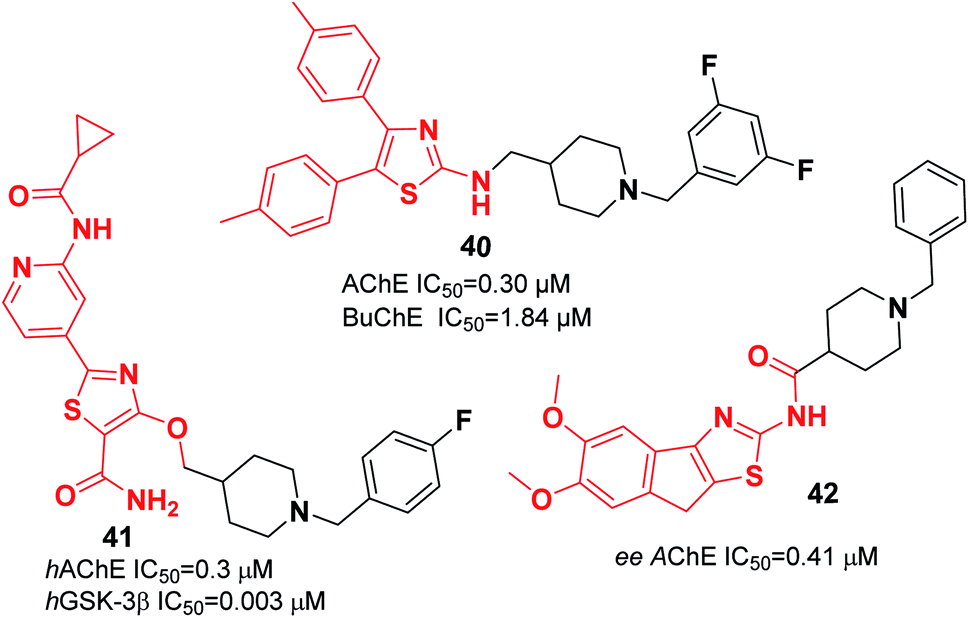 | ||
| Fig. 21 Structure of thiazole-benzylpiperidine scaffolds. | ||
Recently, the design and synthesis of new types of pyridinethiazole with benzylpiperidine hybrids were carried out as GSK-3β/AChE dual-target inhibitors. Among these scaffolds, 41 was the most encouraging choice, with an IC50 of 0.3 μM for hAChE and IC50 of 0.003 μM for hGSK-3β (Fig. 21).51 The Greunen group synthesized 1-benzyl-N-(5,6-dimethoxy-8H-indeno[1,2-d]thiazol-2-yl) piperidine-4-carboxamide 42, which exhibited AChE inhibitory activity (IC50 = 0.41 μM) (Fig. 21).45
3.4. Replacement of indanone with a lipophilic structure
Estrada et al. combined the lipoic acid structure with N-benzylpiperidine to obtain novel MTDLs. Lipoic acid-based hybrids were synthesized through coupling reaction between the corresponding amine derivative and (R,S)-LA or enantiopure (R)-LA or (S)-LA in a microwave oven at 120 °C, using 1,1′-carbonyldiimidazole (CDI) as an activating agent. The most potent ligand 43 demonstrated significant inhibitory activity against ChEs (Fig. 22).52 | ||
| Fig. 22 A lipoic acid-based benzylpiperidine hybrid. | ||
3.5. Miscellaneous MTDLs
An organoselenium compound with biological properties is ebselen (2-phenyl-1,2-benzisoselenazol-3(2H)-one).53 Luo et al. connected ebselen to the pharmacophores of the AChEIs for producing novel hybrid molecules. Compounds 44 and 45 exhibited the highest potency against AChE (Fig. 23).54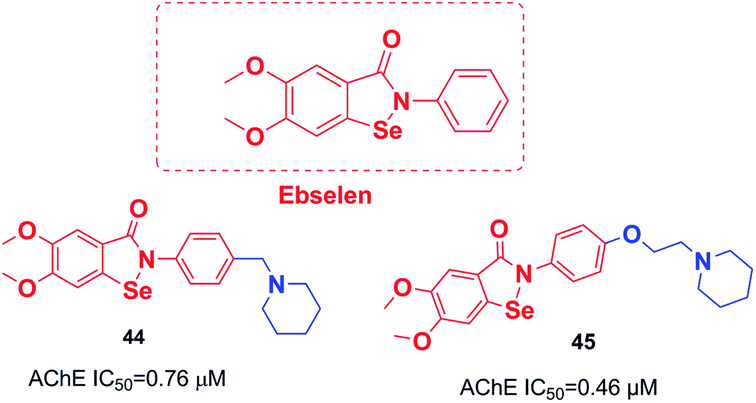 | ||
| Fig. 23 Ebselen-piperidine hybrids with AChE inhibitor activity. | ||
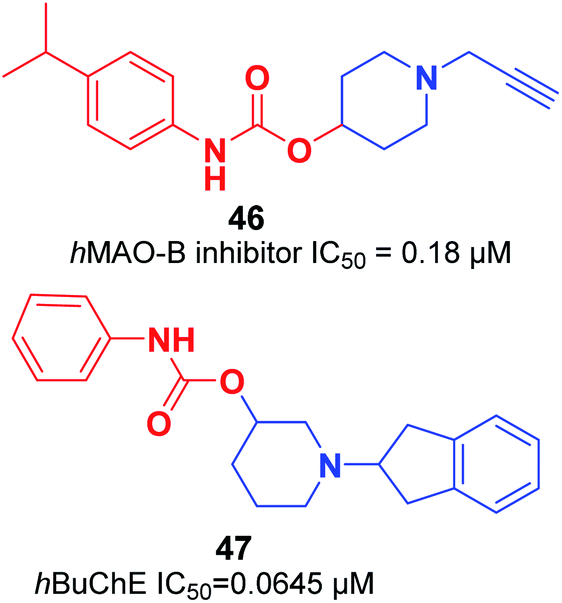
Košak et al. developed a class of N-alkylpiperidine carbamates as viable agents against AD, with the most promising compounds being 46 and 47. N-Propargylpiperidine 46 revealed hMAO-B inhibitor activity (IC50 = 0.18 μM), and compound 47 acted as a selective hBuChE inhibitor with an IC50 of 0.0645 μM (Fig. 24).17
 | ||
| Fig. 24 N-alkylpiperidine carbamates as new inhibitors. | ||
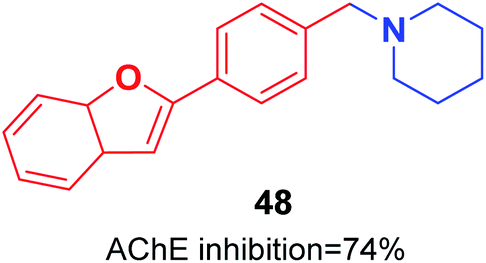
Pouramiri et al. synthesized a library of 2-aryl benzofuran derivatives through four-step reactions from 2-hydroxybenzyl alcohol in good yield. In comparison with donepezil, the synthesized compound 48 with the piperidine moiety exhibited the most optimal AChE inhibition activity of 74% at 23 μM (Fig. 25).55
 | ||
| Fig. 25 2-Arylbenzofuran derivative anticholinesterase agent. | ||
Cai et al. investigated the role of the 4-N-phenylaminoquinoline core in AChE and BuChE inhibition. They synthesized a series of novel derivatives of 4-methyl-piperidine-4-N-phenylaminoquinoline from the commercially available vanillic acid via ten steps. The biological evaluation indicated that compound 49 with a para methoxy group was more highly potent against AChE than galanthamine (Fig. 26).56
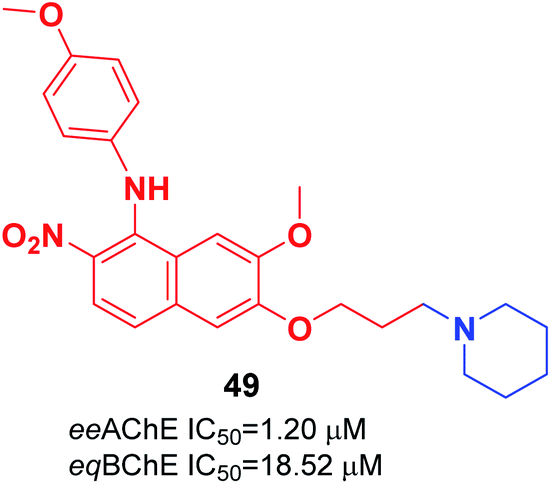 | ||
| Fig. 26 Structure of 4-N-phenylaminoquinoline–piperidine hybrid. | ||
In 2018, the synthesis of a novel series of aryl acyl hydrazones was carried out by Viegas et al. according to the pharmacophoric N-benzyl-piperidine subunit of donepezil, the substituted hydroxy-piperidine fragment, and an acyl hydrazone linker. Among N-benzyl-piperidine acyl hydrazone derivatives, compound 50 showed satisfactory inhibition of electric eel (Electrophorus electricus) AChE, COX-1 (64%), and COX-2 (53%) (Fig. 27).57 Özer et al. carried out the synthesis of N′-2-(4-benzylpiperidin-yl)acyl hydrazone derivatives. Based on the enzyme inhibition assay results, compound 51 with the 4-ethoxybenzylidene group was the most active, with an IC50 of 53.1 μM for AChE enzyme and IC50 of 67.3 μM BuChE enzyme (Fig. 27).58
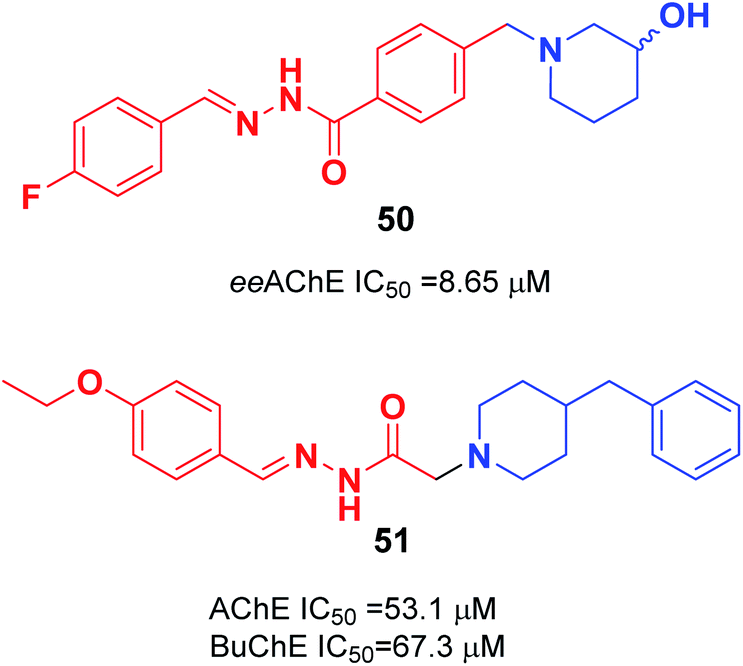 | ||
| Fig. 27 Structure of aryl acyl hydrazones based on N-benzyl-piperidine. | ||
The design and the synthesis of donepezil-hydrazine nicotinates were performed by Zurek et al. in 2013. All the products showed a higher affinity for AChE than BuChE. Compound 52 was the most effective inhibitor of AChE (IC50 = 1.087 × 10−5 μM). The molecular modeling was performed using Cache software and showed that compound 52 binds to AChE with a total score of −181.939 (Fig. 28).59
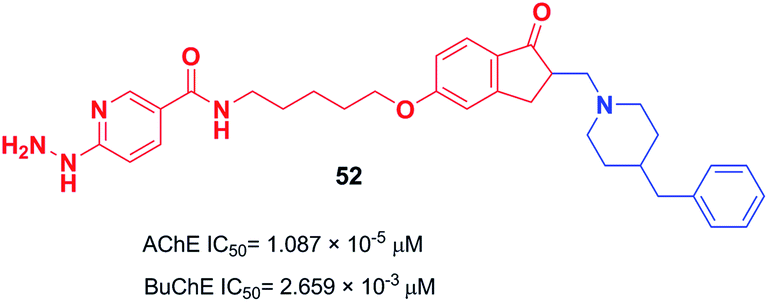 | ||
| Fig. 28 Structure of donepezil–hydrazine nicotinate hybrid 52. | ||
Gupta et al. replaced the 1-indanone moiety with N-benzylated-pyrrolidin-2-one and found that 3-(4-(4-fluorobenzoyl)-piperidin-1-yl)-1-(4-methoxybenzyl)-pyrrolidin-2-one 53 exhibited an excellent anti-Alzheimer's profile (Fig. 29).60
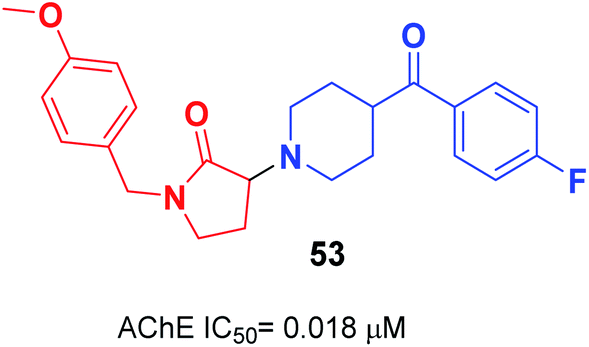 | ||
| Fig. 29 Structure of N-benzylated-pyrrolidin-2-one-piperidine hybrid 53. | ||
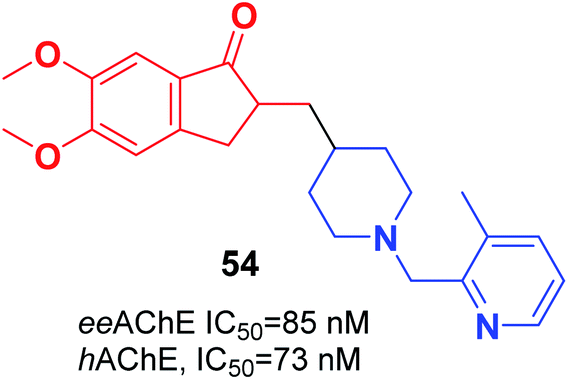
Wang et al. focused on modifying the benzyl group of donepezil using several disparate fragments, including quinolone, pyridine, and hydroxyquinoline, for better interaction with dual binding sites of AChE. Based on the screening results, all the compounds demonstrated a potent AChE inhibition with values of IC50 in the nanomolar range, satisfactory antioxidant activities, Aβ interaction, and BBB penetration. (5,6-dimethoxy-2-((1-((3-methylpyridin-2-yl)methyl)piperidin-4-yl)methyl)-2,3-dihydro-1H-inden-1-one) 54 displayed outstanding inhibition of AChE, satisfactory metal chelation, and inhibitory impact upon self-induced (18.5%), hAChE-induced (72.4%), and Cu2+-induced (46.3%) aggregation of Aβ1–42 at 20 μM (Fig. 30).61
 | ||
| Fig. 30 Structure of a 3-hydroxypyridinaldoxime-indanone derivative. | ||
4. Derivatives with replacement of the piperidine part of donepezil
4.1. Indanone–piperazine hybrids
The N-substituted piperazine moiety mimics the N-benzylpiperidine fragment from donepezil. In 2016, Saglik and coworkers synthesized novel donepezil analogs by replacing the piperidine part with 4-substituted piperazines. Among the compounds, 4-dimethylamino piperazine derivative 55 displayed prominent inhibition of AChE and nontoxicity towards the NH/3T3 cell line (Fig. 31).62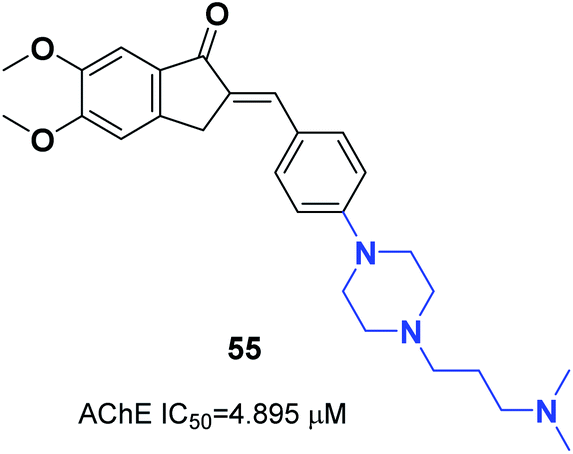 | ||
| Fig. 31 Structure of indenone–piperazine hybrid 55. | ||
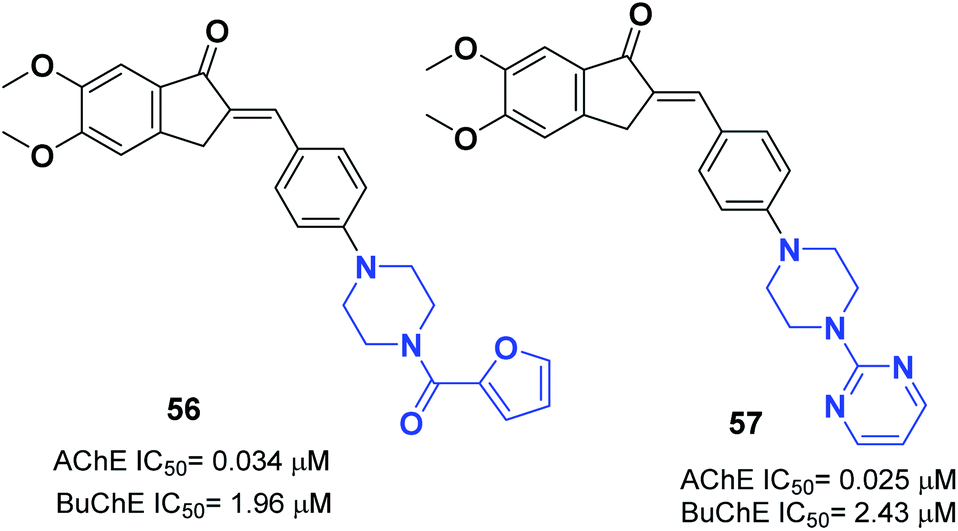
In 2017, Mishra attached the indanone part of donepezil to the 4-substituted piperazin-1-yl-benzylidene moiety from commercially available substituted piperazine and 4-fluoro benzaldehyde at 100 °C followed by the Knoevenagel condensation reaction to link the indanone moiety with various substituted piperazines through a methylene linker. Compounds 56 and 57 displayed excellent AChE inhibitory ability with an IC50 = 0.034 μM and 0.025 μM and Aβ fibril aggregation inhibition activity of 80.4 and 81.6%, respectively (Fig. 32).63
 | ||
| Fig. 32 The structure of piperazin–benzylidene hybrids. | ||
4.2. O-Heterocycle–piperazine hybrids
Gulcan designed and synthesized 6H-benzo[c]chromen-6-one and 7,8,9,10-tetrahydro-benzo[c]chromen-6-one derivatives (Fig. 33).64 Replacement of indanone with chromene and piperidine with piperazine led to an increase in the inhibitory activity of compound 58. In addition, a three-carbon atom spacer between the benzo[c]chromen-6-one moiety and piperazine is ideal for cholinesterase inhibition within this group of compounds.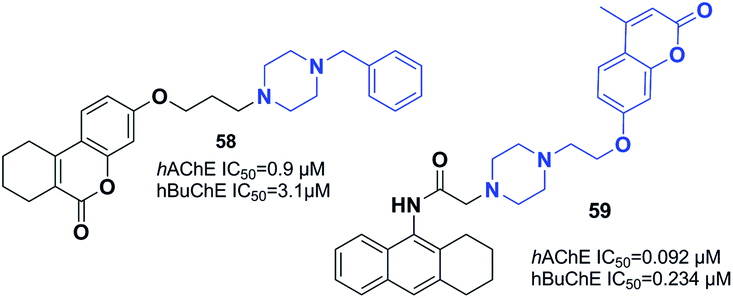 | ||
| Fig. 33 Structure of chromene/coumarin–piperazine hybrids. | ||
Tacrine is another FDA-approved ChE inhibitor that has become an extensively used scaffold in recent years. In 2013, Xie et al. synthesized a novel MTDL by connecting tacrine, coumarins, and piperazine. Among the synthetic compounds, 59 with the methyl on the 4-position of chromene showed the greatest AChE inhibition, and an acceptable BuChE inhibition and Aβ aggregation inhibition (67.8%, 20 μM). Moreover, 59 was a satisfactory metal chelator. Based on molecular modeling and kinetic studies, 59 was a mixed type of inhibitor that binds to the CAS, PAS, and mid-gorge site of AChE at the same time (Fig. 33).65
4.3. N-Heterocycle–piperazine hybrids
Mohammadi et al. designed phthalimid-piperazine scaffolds. They employed the Gabriel synthetic reaction of phthalic anhydride with N-aminoethylpiperazine to obtain 2-(2-(piperazin-1-yl)ethyl)isoindoline-1,3-dione with 61% yield. In addition, equimolar quantities of isoindoline derivatives were reacted with the appropriate benzyl chloride to obtain the corresponding targets. Biological assessment of anticholinesterase activity determined that products with electron-withdrawing groups showed greater potency, and compound 60 with a chloro substituent on the ortho position of the phenyl ring exhibited the greatest AChE inhibition with an IC50 of 0.91 μM (Fig. 34).66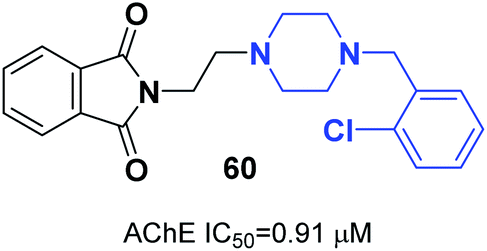 | ||
| Fig. 34 Structure of phthalimide–piperazine hybrid 60. | ||
Replacement of the indanone part of donepezil with indoline-2,3-dione generated a novel scaffold with greater activity than that of donepezil. Compound 61 (1-(2-(4-(2-fluorobenzyl)piperazin-1-yl)acetyl)indoline-2,3-dione) demonstrated greater inhibitory activity compared to donepezil because of the 2-fluorobenzyl replacement and the acetamido spacer. This molecule remarkably binds to the H-bond receptor with Phe288, leading to more optimal pharmacological activity (Fig. 35).67
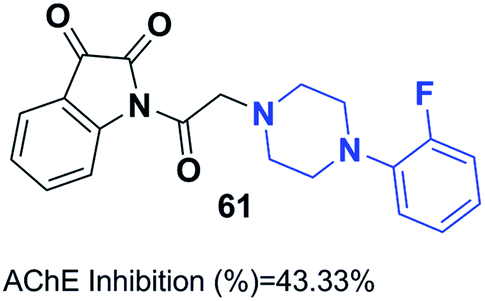 | ||
| Fig. 35 Structure of (1-(2-(4-(2-fluorobenzyl)piperazin-1-yl)acetyl)indoline-2,3-dione) 61. | ||
Prati et al. proposed and designed a new series of 8-hydroxyquinoline-donepezil-like hybrids with multi-targeting activities towards key AD targets. The new hybrid 7-((4-(2-methoxybenzyl) piperazin-1-yl)methyl)-8-hydroxyquinoline 62 inhibited Aβ self-aggregation, exhibited the highest potency for chelating copper(II) and zinc(II), and exerted the highest in vitro antioxidant activity. For compound 62, significant BBB penetration, negligible cytotoxicity in T67 cells, and acceptable toxicity in primary human umbilical vein endothelial cells (HUVECs) accompanied its multi-target profile (Fig. 36).68 The interaction of 8-hydroxyquinoline and donepezil hybrid molecules with cholinesterase and monoamine oxidase enzymes was evaluated by Yang et al.69 Compound 63 had considerable inhibitory impacts against self-induced aggregation of Aβ1–42 (IC50 = 5.64 μM) and potential antioxidant properties (2.63 Trolox equivalents) (Fig. 36). Furthermore, 63 was capable of chelating biometals, inhibiting aggregation of Cu2+/Zn2+, and penetrating the BBB in vitro.
 | ||
| Fig. 36 Structure of 8-hydroxyquinoline-donepezil-like molecules. | ||
Korabecny developed novel ChEIs based on 7-methoxytacrine-donepezil-like structures. The synthesized compounds showed cholinesterase inhibitory activity, and the values of IC50 were in the micromolar to sub-micromolar scale range toward enzymes of human and animal origin. N-(2-{4-[(4-Bromophenyl)methyl]piperazin-1-yl}ethyl)-7-methoxy-1,2,3,4-tetrahydroacridin-9-amine trihydrochloride 64 was the most promising compound (Fig. 37).70 In 2016, Sepsova et al. evaluated the interaction of 7-methoxy-N-(2-(4-(3-methyl benzyl)piperazin-1-yl)ethyl)-1,2,3,4-tetrahydroacridin-9-amine with AChE and cholinergic (muscarinic and nicotinic) receptors. The results showed that compound 65 acts as an AChE inhibitor (Fig. 37).71 Więckowska et al. designed (phenyl sulfonyl-1H-indol-4-yl piperazin-1-yl)hexyl)-1,2,3,4-tetrahydroacridin-9-amine, and biological evaluation indicated that compound 66 had a HT6 antagonist (Kb = 27 nM) (Fig. 37).72
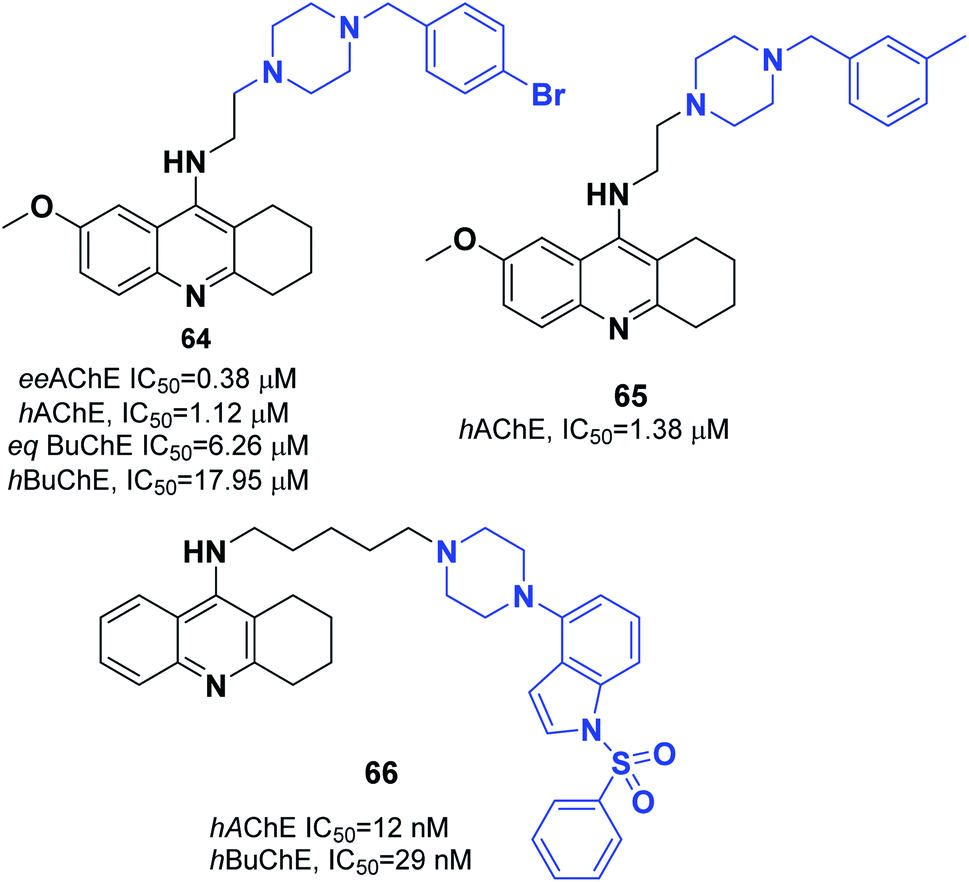 | ||
| Fig. 37 Structure of acridine–piperazine hybrids. | ||
Molecular hydrides of the 5,6-biphenyl-3-oxo-1,2,4-triazine nucleus and substituted piperazines were synthesized and assessed for inhibition of ChE. Compound 67 (5,6-diphenyl-3-oxo-2-(3-(4-benzylpiperazin-1-yl)propyl)-1,2,4-triazine) with three carbon atoms connected to the benzylpiperazine terminal group demonstrated inhibition potency toward AChE with an IC50 of 0.2 μM. Based on docking models, the benzyl group engaging at the lowest part of the enzyme gorge plays a crucial role in interaction with CAS residues of AChE (Fig. 38).73
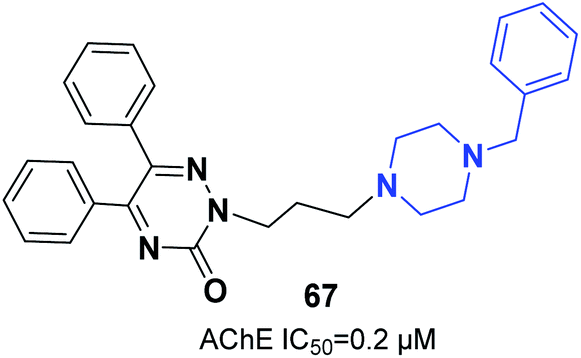 | ||
| Fig. 38 Structure of 3-oxo-1,2,4-triazine-piperazine hybrid 67. | ||
4.4. Isoxazole/oxazole–piperazine hybrids
Saeedi and colleagues designed new isoxazoles linked to the moiety of phenylpiperazine and evaluated the AChE inhibitory effect of targets through Ellman's method as compared to donepezil and rivastigmine references. Among the synthetic compounds, (5-(2-chlorophenyl)isoxazol-3-yl)(4-phenylpiperazin-1-yl)methanone 68 was the most potent candidate (Fig. 39).74 Tripathi et al. explored the synthesis of 2-pyridylpiperazine hybrids and 5-phenyl-1,3,4-oxadiazoles with considerable inhibitory possibilities for AD. Compound 69 with a 2,4-difluoro replacement at the end phenyl ring was considered the most viable lead, with cholinesterase inhibition and β-secretase-1 activity. Based on an analysis of enzyme kinetics, 69 demonstrated a mixed-type inhibition toward hAChE, with a Ki of 0.030 μM. Compound 69 showed considerable deposition of propidium iodide from the PAS of hAChE, penetration in PAMPA, neuroprotective capability against the SH-SY5Y neuroblastoma cell line, and outstanding BBB (Fig. 39).75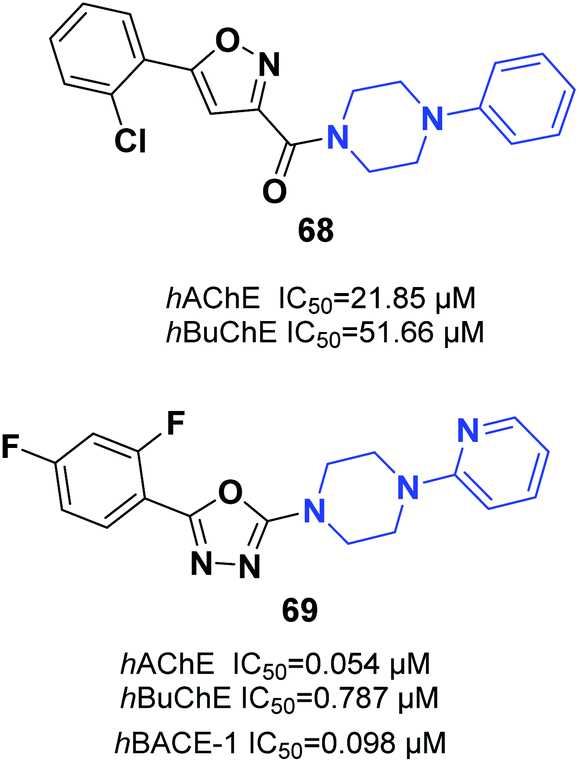 | ||
| Fig. 39 Isoxazole/oxazole–piperazine hybrids. | ||
4.5. Thiophene–piperazine hybrids
The synthesis of new thiophene derivatives and evaluation of their biological inhibition were carried out by Ismail et al. 2-(2-(4-(4-Methoxyphenyl)piperazin-1-yl)acetamido)-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxamide 70 demonstrated inhibition of 60% in comparison with only 40% inhibition for donepezil (Fig. 40).76 The amide linker in this scaffold led to extra binding to the receptor through three different H-bonds with Phe288 and resulted in increased pharmacological activity.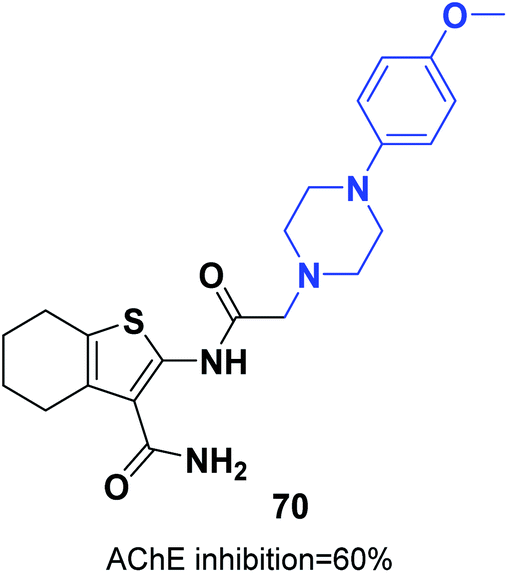 | ||
| Fig. 40 Structure of thiophene-piperazine 70. | ||
4.6. Modified scaffolds based on pyrrolidine
One of the latest studies on MTDLs was carried out by Choubey et al. They designed derivatives of N-benzyl pyrrolidine and biologically assessed their ability to treat AD. Among the synthesized leads, 71a and 71b demonstrated proportionate enzymatic inhibition against cholinesterases. They also showed significant PAS-AChE binding ability, outstanding BBB permeation, neuroprotective activity against Aβ-induced stress, and possible disassembly of Aβ aggregates (Fig. 41).77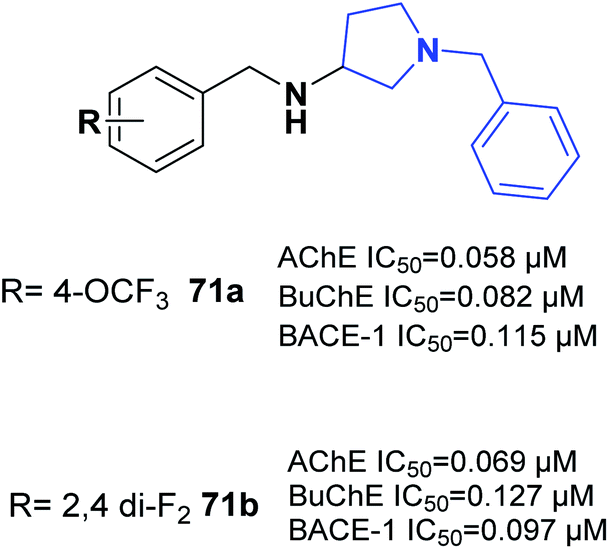 | ||
| Fig. 41 Structure of benzylamine-pyrrolidine derivative 71. | ||
Wichur et al. synthesized novel multifunctional ligands to investigate their inhibitory effects on BuChE, β-secretase, and amyloid-β (Aβ), and their antioxidant, aggregation of tau protein, and metal-chelating characteristics. All the targets exhibited dual anti-aggregating properties towards Aβ and tau protein (72a: 45% for Aβ, 53% for tau; 72b: 49% for Aβ, 54% for tau). Compound 72a with an IC50 of 2.39 μM and 72ab with an IC50 of 1.94 μM exhibited the highest inhibitory potency against BuChE (Fig. 42).78
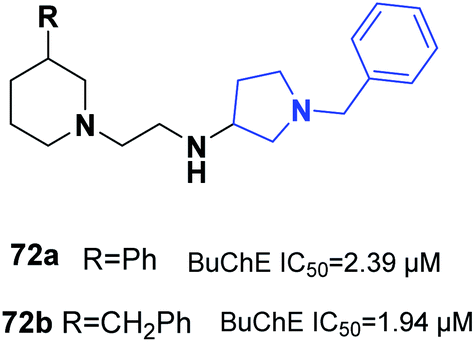 | ||
| Fig. 42 Structure of piperazine-benzyl pyrrolidine derivatives. | ||
5. Conclusion
The enhancement of ACh would be considered an effective method to compensate for the deficiency of acetylcholine, and most treatment strategies for AD are focused on enhancing cholinergic neurotransmission. However, there are severe side effects to FDA-approved drugs for AD, including vomiting, nausea, diarrhea, bradycardia, abnormal dreams, and fatigue. Because of AD's complex pathophysiology, there is an urgent need to discover and develop efficient new therapeutic agents with minimal side effects to effectively combat AD.In this review, we discussed the structural modification of donepezil as described in articles published from 2012–2020. This review is categorized into sections according to the changes made to the donepezil linker, replacement of the indanone moiety with O-containing heterocycles or N-heterocyclic moieties, and replacement of the piperidine moiety with piperazine or pyrrolidine structures. In addition, we tried to outline different synthetic small molecules based on the structure of donepezil and describe detailed structure–activity relationships.
Studies showed that attaching chromones with piperidine will produce ChE inhibitors and selective hMAO inhibition. Also, the type of spacer modulates additional activities, including metal-chelating characteristics. Furthermore, using hydroxyquinoline instead of the indanone part of donepezil leads to multi-target properties. Hybrid molecules from tetrahydroacridin-9-amines and piperazine will exhibit the best dual ChE activities and are vital for the effective treatment of AD. This review will assist researchers and medicinal chemists in designing novel, target-based, and advanced donepezil analogs with advantageous biological properties for effective AD therapy (Table 1).
| Compound number | AChE | BuChE | Inhibition of Aβ | BACE-1 | Metal chelation | MAO | Non-toxicity | Reference |
|---|---|---|---|---|---|---|---|---|
| 1 | * | * | 14 | |||||
| 2 | * | * | 14 | |||||
| 3 | * | 15 | ||||||
| 4 | * | * | * | 16 | ||||
| 5 | * | * | * | 17 | ||||
| 6 | * | * | 18 | |||||
| 7 | * | * | * | 19 | ||||
| 8 | * | * | * | 19 | ||||
| 9 | * | * | * | * | 21 | |||
| 10 | * | * | 22 | |||||
| 11 | * | 23 | ||||||
| 12 | * | * | * | 22 | ||||
| 13 | * | * | 24 | |||||
| 14 | * | * | * | * | 25 | |||
| 15 | * | * | 26 | |||||
| 16 | * | 27 | ||||||
| 17 | * | * | * | * | 28 | |||
| 18 | * | * | * | 29 | ||||
| 19 | * | 30 | ||||||
| 20 | * | * | 31 | |||||
| 21 | * | * | * | * | * | * | 32 | |
| 22 | * | * | 33 | |||||
| 23 | * | * | 34 | |||||
| 24 | * | * | * | 35 | ||||
| 25 | * | * | * | 36 | ||||
| 26 | * | 37 | ||||||
| 27 | * | * | * | * | * | 38 | ||
| 28 | * | * | * | * | 39 | |||
| 29 | * | * | * | 40 | ||||
| 30 | * | * | 41 | |||||
| 31 | * | * | 42 | |||||
| 32 | * | * | 43 | |||||
| 33 | * | * | 44 | |||||
| 34 | * | 45 | ||||||
| 35 | * | 46 | ||||||
| 36 | * | * | * | 47 | ||||
| 37 | * | * | 48 | |||||
| 38 | * | * | * | * | * | 49 | ||
| 39 | * | * | * | * | * | 49 | ||
| 40 | * | * | * | 50 | ||||
| 41 | * | 51 | ||||||
| 42 | * | 45 | ||||||
| 43 | * | * | 52 | |||||
| 44 | * | 54 | ||||||
| 45 | * | 54 | ||||||
| 46 | * | 17 | ||||||
| 47 | * | 17 | ||||||
| 48 | * | 55 | ||||||
| 49 | * | * | 56 | |||||
| 50 | * | 57 | ||||||
| 51 | * | * | 58 | |||||
| 52 | * | * | 59 | |||||
| 53 | * | 60 | ||||||
| 54 | * | * | 61 | |||||
| 55 | * | * | 62 | |||||
| 56 | * | * | * | 63 | ||||
| 57 | * | * | * | 63 | ||||
| 58 | * | * | 64 | |||||
| 59 | * | * | * | * | 65 | |||
| 60 | * | 66 | ||||||
| 61 | * | 67 | ||||||
| 62 | * | * | * | * | 68 | |||
| 63 | * | * | 69 | |||||
| 64 | * | * | 70 | |||||
| 65 | * | 71 | ||||||
| 66 | * | * | 72 | |||||
| 67 | * | 73 | ||||||
| 68 | * | * | 74 | |||||
| 69 | * | * | * | * | 75 | |||
| 70 | * | 76 | ||||||
| 71 | * | * | * | 77 | ||||
| 72 | * | * | * | 78 |
Abbreviations
| ABTS | (2,2′-Azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) |
| ACh | Acetylcholine |
| AChE | Acetylcholinesterase |
| AChEI | Acetylcholinesterase inhibitor |
| AD | Alzheimer's disease |
| BACE-1 | β-Secretase-1 |
| BuChE | Butyrylcholinesterase |
| CAS | Catalytic anionic site |
| ChE | Cholinesterase |
| DFH | Donepezil–flavonoid hybrid |
| eeAChE | Electrophorus electricus acetylcholinesterase |
| FRAP | Ferric reducing antioxidant power |
| Glu | Glutamate |
| GSK-3β | Glycogen synthase kinase-3β |
| His | Histidine |
| 5-HT | 5-Hydroxytryptamine (serotonin) |
| 5-LOX | 5-Lipoxygenase |
| MAO | Monoamine oxidase |
| MAOI | Monoamine oxidase inhibitor |
| MTDL | Multitarget-directed ligand |
| PAS | Peripheral anionic site |
| Phe | Phenylalanine |
| QSAR | Quantitative structure–activity |
| SI | Selectivity index |
| σ-1R | Sigma-1 receptor |
| Trp | Tryptophan |
Conflicts of interest
There are no conflicts to declare.References
- P. Scheltens, B. De Strooper, M. Kivipelto, H. Holstege, G. Chételat, C. E. Teunissen, J. Cummings and W. M. van der Flier, Lancet, 2021, 397, 1577–1590 CrossRef CAS
.
- E. Scarpini, P. Scheltens and H. Feldman, Lancet Neurol., 2003, 2, 539–547 CrossRef CAS PubMed
- V. N. Talesa, Mech. Ageing Dev., 2001, 122, 1961–1969 CrossRef CAS PubMed
- X. Huang, R. D. Moir, R. E. Tanzi, A. I. Bush and J. T. Rogers, Ann. N. Y. Acad. Sci., 2004, 1012, 153–163 CrossRef CAS PubMed
- P. Seeman and N. Seeman, Synapses, 2011, 1297, 1289–1297 CrossRef PubMed
- M. Coedert, M. C. Spillantini, D. Rutherford and R. A. Crowther, Neuron, 1989, 3, 519–526 CrossRef
- P. T. Francis, A. M. Palmer, M. Snape and G. K. Wilcock, J. Neurol., Neurosurg. Psychiatry, 1999, 66, 137–147 CrossRef CAS PubMed
- T. L. Rosenberry, X. Brazzolotto, I. R. MacDonald, M. Wandhammer, M. Trovaslet-Leroy, S. Darvesh and F. Nachon, Molecules, 2017, 22, 1–21 CrossRef PubMed
- M. Rodrigues Simoes, F. Dias Viegas, M. Moreira, M. Freitas Silva, M. Riquiel, P. da Rosa, M. Castelli, M. dos Santos, M. Soares and C. Viegas, Mini-Rev. Med. Chem., 2014, 14, 2–19 CrossRef PubMed
- J. Cheung, M. J. Rudolph, F. Burshteyn, M. S. Cassidy, E. N. Gary, J. Love, M. C. Franklin and J. J. Height, J. Med. Chem., 2012, 55, 10282–10286 CrossRef CAS PubMed
- H. Sugimoto, Y. Yamanishi, Y. Iimura and Y. Kawakami, Curr. Med. Chem., 2000, 7, 303–339 CrossRef CAS PubMed
- S. Uddin, A. Al Mamun, T. Kabir, G. Ashraf and M. N. Bin-Jumah, Mol. Neurobiol., 2021, 58, 281–303 CrossRef PubMed
- S. Maramai, M. Benchekroun, M. T. Gabr and S. Yahiaoui, BioMed Res. Int., 2020, 2020, 5120230 Search PubMed
- P. Costanzo, L. Cariati, D. Desiderio, R. Sgammato, A. Lamberti, R. Arcone, R. Salerno, M. Nardi, M. Masullo and M. Oliverio, ACS Med. Chem. Lett., 2016, 7, 470–475 CrossRef CAS PubMed
- D. G. Van Greunen, W. Cordier, M. Nell, C. Van Der Westhuyzen, V. Steenkamp, J. Panayides and D. L. Riley, Eur. J. Med. Chem., 2017, 127, 671–690 CrossRef CAS PubMed
- J. Yan, J. Hu, A. Liu, L. He, X. Li and H. Wei, Bioorg. Med. Chem., 2017, 25, 2946–2955 CrossRef CAS PubMed
- U. Košak, N. Strašek, D. Knez, M. Jukič, S. Žakelj, A. Zahirović, A. Pišlar, X. Brazzolotto, F. Nachon, J. Kos and S. Gobec, Eur. J. Med. Chem., 2020, 197, 112282 CrossRef PubMed
- L. Monjas, M. P. Arce, R. León, J. Egea, C. Pérez, M. Villarroya, M. G. López, C. Gil, S. Conde and M. Isabel, Eur. J. Med.
Chem., 2017, 130, 60–72 CrossRef CAS PubMed
- P. Sharma, A. Tripathi, P. N. Tripathi, S. K. Prajapati, A. Seth, M. K. Tripathi, P. Srivastava, V. Tiwari, S. Krishnamurthy and S. K. Shrivastava, Eur. J. Med. Chem., 2019, 167, 510–524 CrossRef CAS PubMed
- S. Ou and K. Kwok, J. Sci. Food Agric., 2004, 1269, 1261–1269 CrossRef
- K. S. T. Dias, C. T. de Paula, T. dos Santos, I. N. O. Souza, M. S. Boni, M. J. R. Guimarães, F. M. R. da Silva, N. G. Castro, G. A. Neves, C. C. Veloso, M. M. Coelho, I. S. F. de Melo, F. C. V. Giusti, A. Giusti-Paiva, M. L. da Silva, L. E. Dardenne, I. A. Guedes, L. Pruccoli, F. Morroni, A. Tarozzi and C. Viegas, Eur. J. Med. Chem., 2017, 130, 440–457 CrossRef CAS PubMed
- W. Xu, X. B. Wang, Z. M. Wang, J. J. Wu, F. Li, J. Wang and L. Y. Kong, MedChemComm, 2016, 7, 990–998 RSC
- M. Benchekroun, L. Ismaili, M. Pudlo, V. Luzet, T. Gharbi, B. Refouvelet and J. Marco-Contelles, Future Med. Chem., 2015, 7, 15–21 CrossRef CAS PubMed
- M. Estrada, C. Herrera-Arozamena, C. Pérez, D. Viña, A. Romero, J. A. Morales-García, A. Pérez-Castillo and M. I. Rodríguez-Franco, Eur. J. Med. Chem., 2016, 121, 376–386 CrossRef CAS PubMed
- X. B. Wang, F. C. Yin, M. Huang, N. Jiang, J. S. Lan and L. Y. Kong, RSC Med. Chem., 2020, 11, 225–233 RSC
- M. E. Valencia, C. Herrera-arozamena, L. De Andrés, J. A. Morales-garcía, A. Pérez-castillo, E. Ramos, A. Romero, D. Viña, M. Yáñez, E. Laurini, S. Pricl and M. I. Rodríguez-, Eur. J. Med. Chem., 2018, 156, 534–553 CrossRef PubMed
- A. Asadipour, M. Alipour, M. Jafari, M. Khoobi, S. Emami, H. Nadri, A. Sakhteman, A. Moradi, V. Sheibani, F. Homayouni Moghadam, A. Shafiee and A. Foroumadi, Eur. J. Med. Chem., 2013, 70, 623–630 CrossRef CAS PubMed
- P. Cai, S. Fang, X. Yang and J. Wu, ACS Chem. Neurosci., 2017, 8, 2496–2511 CrossRef CAS PubMed
- J. Joubert, G. B. Foka, B. P. Repsold, D. W. Oliver, E. Kapp and S. F. Malan, Eur. J. Med. Chem., 2017, 125, 853–864 CrossRef CAS PubMed
- M. Asadi, M. Ebrahimi, M. Mohammadi-Khanaposhtani, H. Azizian, S. Sepehri, H. Nadri, M. Biglar, M. Amanlou, B. Larijani, R. Mirzazadeh, N. Edraki and M. Mahdavi, Chem. Biodiversity, 2019, 16, e1900370 CrossRef CAS PubMed
- A. Więckowska, K. Więckowski, M. Bajda, B. Brus, K. Sałat, P. Czerwińska, S. Gobec, B. Filipek and B. Malawska, Bioorg. Med. Chem., 2015, 23, 2445–2457 CrossRef
- F. Li, Z.-M. Wang, J.-J. Wu, J. Wang, S.-S. Xie, J.-S. Lan, W. Xu, L.-Y. Kong and X.-B. Wang, J. Enzyme Inhib. Med. Chem., 2016, 31, 41–53 CrossRef CAS PubMed
- A. Samadi, M. Estrada, C. Pérez, M. I. Rodríguez-franco, I. Iriepa, I. Moraleda, M. Chioua and J. Marco-contelles, Eur. J. Med. Chem., 2012, 57, 296–301 CrossRef CAS PubMed
- A. Samadi, M. D. L. F. Revenga, C. Pérez, I. Iriepa, I. Moraleda, M. I. Rodríguez-Franco and J. Marco-Contelles, Eur. J. Med. Chem., 2013, 67, 64–74 CrossRef CAS PubMed
- Y. Zhou, W. Sun, J. Peng, H. Yan, L. Zhang, X. Liu and Z. Zuo, Bioorg. Chem., 2019, 93, 103322 CrossRef PubMed
- L. Wang, G. Esteban, M. Ojima, O. M. Bautista-Aguilera, T. Inokuchi, I. Moraleda, I. Iriepa, A. Samadi, M. B. H. Youdim, A. Romero, E. Soriano, R. Herrero, A. P. Fernández Fernández, Ricardo-Martínez-Murillo, J. Marco-Contelles and M. Unzeta, Eur. J. Med. Chem., 2014, 80, 543–561 CrossRef CAS PubMed
- M. Pudlo, V. Luzet, L. Ismaïli, I. Tomassoli, A. Iutzeler and B. Refouvelet, Bioorg. Med. Chem., 2014, 22, 2496–2507 CrossRef CAS PubMed
- J. Wang, Z. M. Wang, X. M. Li, F. Li, J. J. Wu, L. Y. Kong and X. B. Wang, Bioorg. Med. Chem., 2016, 24, 4324–4338 CrossRef CAS PubMed
- O. M. Bautista-aguilera, G. Esteban, I. Bolea, K. Nikolic, D. Agbaba, I. Moraleda, I. Iriepa, A. Samadi, E. Soriano, M. Unzeta and J. Marco-contelles, Eur. J. Med. Chem., 2014, 75, 82–95 CrossRef CAS PubMed
- L. Piemontese, D. Tomás, A. Hiremathad, V. Capriati, E. Candeias, S. M. Cardoso, S. Chaves and M. A. Santos, J. Enzyme Inhib. Med. Chem., 2018, 33, 1212–1224 CrossRef CAS PubMed
- S. Chaves, S. Resta, F. Rinaldo, M. Costa, R. Josselin, K. Gwizdala, L. Piemontese, V. Capriati, A. R. Pereira-santos, S. M. Cardoso and M. Am, Molecules, 2020, 25, 985 CrossRef CAS PubMed
- J. Mo, T. Chen, H. Yang, Y. Guo, Q. Li, Y. Qiao, H. Lin, F. Feng, W. Liu, Y. Chen, Z. Liu and H. Sun, J. Enzyme Inhib. Med. Chem., 2020, 35, 330–343 CrossRef CAS PubMed
- B. Kilic, H. O. Gulcan, F. Aksakal, T. Ercetin, N. Oruklu, E. Umit Bagriacik and D. S. Dogruer, Bioorg. Chem., 2018, 79, 235–249 CrossRef CAS PubMed
- N. Vila, P. Besada, D. Viña, M. Sturlese, S. Moro and C. Terán, RSC Adv., 2016, 6, 46170–46185 RSC
- D. G. van Greunen, C. Johan van der Westhuizen, W. Cordier, M. Nell, A. Stander, V. Steenkamp, J. L. Panayides and D. L. Riley, Eur. J. Med. Chem., 2019, 179, 680–693 CrossRef CAS PubMed
- P. de Andrade, S. P. Mantoani, P. S. Gonçalves Nunes, C. R. Magadán, C. Pérez, D. J. Xavier, E. T. S. Hojo, N. E. Campillo, A. Martínez and I. Carvalho, Bioorg. Med. Chem., 2019, 27, 931–943 CrossRef CAS PubMed
- N. A. E. El-Sayed, A. E. S. Farag, M. A. F. Ezzat, H. Akincioglu, İ. Gülçin and S. M. Abou-Seri, Bioorg. Chem., 2019, 93, 103312 CrossRef PubMed
- M. Saeedi, P. Felegari, A. Iraji, R. Hariri, A. Rastegari, S. S. Mirfazli, N. Edraki, O. Firuzi, M. Mahdavi and T. Akbarzadeh, Arch. Pharm., 2020, 1–12 CAS
- P. Sharma, A. Tripathi, P. N. Tripathi, S. Sen Singh, S. P. Singh and S. K. Shrivastava, ACS Chem. Neurosci., 2019, 10, 4361–4384 CrossRef CAS PubMed
- M. Shidore, J. Machhi, K. Shingala, P. Murumkar, M. K. Sharma, N. Agrawal, A. Tripathi, Z. Parikh, P. Pillai and M. R. Yadav, J. Med. Chem., 2016, 59, 5823–5846 CrossRef CAS PubMed
- X. Jiang, Y. Wang, C. Liu, C. Xing, Y. Wang, W. Lyu, S. Wang, Q. Li, T. Chen, Y. Chen, F. Feng, W. Liu and H. Sun, Bioorg. Med. Chem., 2021, 30, 115940 CrossRef CAS PubMed
- M. Estrada, C. Pérez, E. Soriano, E. Laurini, M. Romano, S. Pricl, J. A. Morales-García, A. Pérez-Castillo and M. I. Rodríguez-Franco, Future Med. Chem., 2016, 8, 1191–1207 CrossRef CAS PubMed
- T. Schewe, Gen. Pharmacol., 1995, 26, 1153–1169 CrossRef CAS PubMed
- Z. Luo, L. Liang, J. Sheng, Y. Pang, J. Li, L. Huang and X. Li, Bioorg. Med. Chem., 2014, 22, 1355–1361 CrossRef CAS PubMed
- B. Pouramiri, M. Mahdavi, S. Moghimi, L. Firoozpour, H. Nadri, A. Moradi, E. Tavakolinejad-Kermani, A. Asadipour and A. Foroumadi, Lett. Drug Des. Discovery, 2016, 13, 897–902 CrossRef CAS
- R. Cai, L. N. Wang, J. J. Fan, S. Q. Geng and Y. M. Liu, Bioorg. Chem., 2019, 93, 103328 CrossRef PubMed
- F. Pereira, D. Viegas, M. D. F. Silva, M. Divino, R. Castelli, M. M. Riquiel, R. P. Machado, S. Macedo, D. Oliveira, É. P. Morais, V. S. Gontijo, F. M. R. Silva, D. D. Alincourt, N. G. Castro, A. Gilda, A. Giusti-paiva, F. C. Vilela, L. Orlandi, M. P. Veloso, L. Felipe, L. Coelho, M. Ionta, W. De Oliveira, C. Junior, P. Maria and Q. Bellozi, Eur. J. Med. Chem., 2018, 147, 48–65 CrossRef PubMed
- E. Özturan Özer, O. Unsal Tan, K. Ozadali, T. Küçükkilinç, A. Balkan and G. Uçar, Bioorg. Med. Chem. Lett., 2013, 23, 440–443 CrossRef PubMed
- E. Żurek, P. Szymański and E. Mikiciuk-Olasik, Drug Res., 2013, 63, 137–144 CrossRef PubMed
- M. Gupta, M. Ojha, D. Yadav, S. Pant and R. Yadav, ACS Chem. Neurosci., 2020, 11, 2849–2860 CrossRef CAS PubMed
- Z. M. Wang, P. Cai, Q. H. Liu, D. Q. Xu, X. L. Yang, J. J. Wu, L. Y. Kong and X. B. Wang, Eur. J. Med. Chem., 2016, 123, 282–297 CrossRef CAS PubMed
- B. N. Sağlık, S. Ilgın and Y. Özkay, Eur. J. Med. Chem., 2016, 124, 1026–1040 CrossRef PubMed
- C. B. Mishra, S. Kumari, A. Manral, A. Prakash, V. Saini, A. M. Lynn and M. Tiwari, Eur. J. Med. Chem., 2017, 125, 736–750 CrossRef CAS PubMed
- H. O. Gulcan, S. Unlu, I. Esiringu, T. Ercetin, Y. Sahin, D. Oz and M. F. Sahin, Bioorg. Med. Chem., 2014, 22, 5141–5154 CrossRef CAS PubMed
- S. S. Xie, X. B. Wang, J. Y. Li, L. Yang and L. Y. Kong, Eur. J. Med. Chem., 2013, 64, 540–553 CrossRef CAS PubMed
- A. Mohammadi-farani, A. Ahmadi, H. Nadri and A. Aliabadi, Daru, J. Pharm. Sci., 2013, 21, 47–56 CrossRef CAS PubMed
- M. M. Ismail, M. M. Kamel, L. W. Mohamed and S. I. Faggal, Molecules, 2012, 17, 4811–4823 CrossRef CAS PubMed
- F. Prati, C. Bergamini, R. Fato, O. Soukup, J. Korabecny, V. Andrisano, M. Bartolini and M. L. Bolognesi, ChemMedChem, 2016, 11, 1284–1295 CrossRef CAS PubMed
- X. Yang, P. Cai, Q. Liu, J. Wu, Y. Yin, X. Wang and L. Kong, Bioorg. Med. Chem., 2018, 26, 3191–3201 CrossRef CAS PubMed
- J. Korabecny, R. Dolezal, P. Cabelova, A. Horova, E. Hruba, J. Ricny, L. Sedlacek, E. Nepovimova, K. Spilovska, M. Andrs, K. Musilek, V. Opletalova, V. Sepsova, D. Ripova and K. Kuca, Eur. J. Med. Chem., 2014, 82, 426–438 CrossRef CAS PubMed
- C. Sepsova Vendula, Z. Karasova Jana, T. Gunnar, J. Daniel and J. Korabecny, et al., Gen. Physiol. Biophys., 2014, 34, 189–200 CrossRef PubMed
- A. Więckowska, M. Kołaczkowski, A. Bucki, J. Godyń, M. Marcinkowska, K. Więckowski, P. Zaręba, A. Siwek, G. Kazek, M. Głuch-Lutwin, P. Mierzejewski, P. Bienkowski, H. Sienkiewicz-Jarosz, D. Knez, T. Wichur, S. Gobec and B. Malawska, Eur. J. Med. Chem., 2016, 124, 63–81 CrossRef PubMed
- P. N. Tripathi, P. Srivastava, P. Sharma, M. K. Tripathi, A. Seth, A. Tripathi, S. N. Rai, S. P. Singh and S. K. Shrivastava, Bioorg. Chem., 2019, 85, 82–96 CrossRef CAS PubMed
- M. Saeedi, D. Mohtadi-Haghighi, S. S. Mirfazli, M. Mahdavi, R. Hariri, H. Lotfian, N. Edraki, A. Iraji, O. Firuzi and T. Akbarzadeh, Chem. Biodiversity, 2019, 16, e1800433 CrossRef PubMed
- A. Tripathi, P. K. Choubey, P. Sharma, A. Seth, P. N. Tripathi, M. K. Tripathi, S. K. Prajapati, S. Krishnamurthy and S. K. Shrivastava, Eur. J. Med. Chem., 2019, 183, 111707 CrossRef PubMed
- M. M. Ismail, M. M. Kamel, L. W. Mohamed, S. I. Faggal and M. A. Galal, Molecules, 2012, 17, 7217–7231 CrossRef CAS PubMed
- P. K. Choubey, A. Tripathi, P. Sharma and S. K. Shrivastava, Bioorg. Med. Chem., 2020, 28, 115721 CrossRef CAS PubMed
- T. Wichur, A. Więckowska, K. Więckowski, J. Godyń, J. Jończyk, Á. D. R. Valdivieso, D. Panek, A. Pasieka, R. Sabaté, D. Knez, S. Gobec and B. Malawska, Eur. J. Med. Chem., 2020, 187, 111916 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2021 |