
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances in the conversion of furfural into bio-chemicals through chemo- and bio-catalysis
Xu Zhangabc,
Siquan Xud,
Qinfang Libc,
Guilin Zhou *a and
Haian Xia*bc
*a and
Haian Xia*bc
aEngineering Research Center for Waste Oil Recovery Technology and Equipment, Ministry of Education, Chongqing Technology and Business University, Chongqing 400067, China. E-mail: dicpglzhou@ctbu.edu.cn; Fax: +86-25-85428873; Tel: +86-25-85427635
bJiangsu Provincial Key Lab for the Chemistry and Utilization of Agro-Forest Biomass, College of Chemical Engineering, Nanjing Forestry University, Nanjing 210037, China. E-mail: haxia@dicp.ac.cn
cJiangsu Co-Innovation Center of Efficient Processing and Utilization of Forest Resources, Nanjing Forestry University, Nanjing 210037, China
dSchool of Forestry, Anhui Agricultural University, Hefei, 230036, China
First published on 9th August 2021
Abstract
Furfural is a promising renewable platform molecule derived from hemi-cellulose, which can be further converted to fossil fuel alternatives and valuable chemicals due to its highly functionalized molecular structure. This mini-review summarizes the recent progress in the chemo-catalytic and/or bio-catalytic conversion of furfural into high-value-added chemicals, including furfurylamine, C6 carboxylic acid, i.e., furandicarboxylic acid, furfural alcohol, aromatics, levulinic acid, maleic acid, succinic acid, furoic acid, and cyclopentanone, particularly the advances in the catalytic valorization of furfural into useful chemicals in the last few years. The possible reaction mechanisms for the conversion of furfural into bio-chemicals are summarized and discussed. The future prospective and challenges in the utilization of furfural through chemo- and bio-catalysis are also put forward for the further design and optimization of catalytic processes for the conversion of furfural.
1. Introduction
With the depletion of fuel reserves and increasing demands for fuel, great efforts have been devoted to developing novel sustainable and renewable energies and resources.1–5 Biomass is the only renewable organic carbon source in nature, which is environmentally friendly, has abundant reserves and inexpensive, providing unique advantages for the production of fuels and industrially important chemicals.5–7 Furfural is a versatile platform molecule, and thus has received considerable attention in recent years. The presence of an aldehyde group in furfural makes it possible to further synthesize higher value chemicals and polymer monomers, including furandicarboxylic acid, furfurylamine, furfural alcohol, cyclopentanone, and levulinic acid and other molecules through oxidation, hydrogenation, and hydrolysis (Fig. 1).6 Hence, the high-value utilization of biomass-derived furfural is important for sustainable development in the future.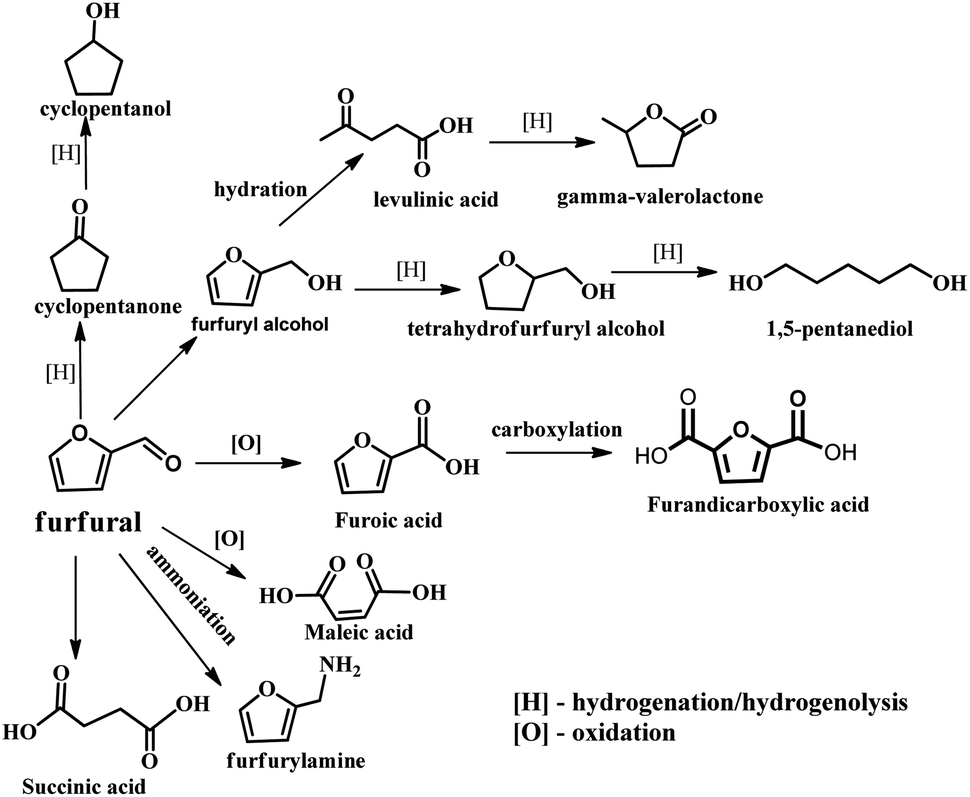 | ||
| Fig. 1 Furfural as a platform compound for diverse reactions. | ||
Furfural is produced from agricultural resources containing xylose or xylan, such as wood wastes and corn cob.8 The main producers of furfural are China (∼70% total production capacity), The Dominican Republic (Central Romana Corporation, 32 kton per year), and South Africa (20 kton per year). These three countries account for approximately 90% of the global furfural capacity (280 kton per year).9,10 In the past, most studies have been carried out to reveal the reaction mechanism for the conversion carbohydrates into furfural at the molecular level. As shown in Fig. 2, initially, the formation of furfural from hemicellulose-containing biomass includes the acid-catalyzed depolymerization of the hemicellulose component to form xylose. Subsequently, the isomerization of xylose to xylulose occurs followed by a dehydration reaction to yield furfural.11 To date, various reaction systems such as biphasic reaction systems and ionic liquids have been developed to boost the yield of furfural.
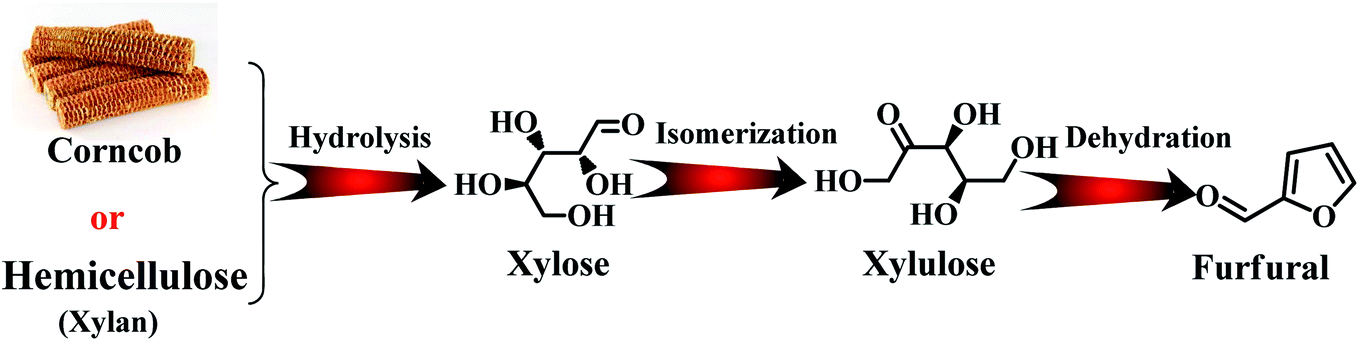 | ||
| Fig. 2 Conversion pathway of hemicellulose component into furfural. | ||
Some excellent reviews about the conversion of hemicellulose to furfural and the valorization of furfural into bio-fuels have been published.5,6,12–14 A systematic review of the conversion of xylan into furfural is beyond the scope of this manuscript. However, a comprehensive summary of latest advances in the valorization of furfural to value-added chemicals, especially the progress in the last five years, has not been reported to date. This mini-review summarizes the recent advances in the conversion of furfural into valuable chemicals through chemo- and bio-catalysis. This mini-review mainly focuses on the catalytic strategies for the valorization of furfural into useful chemicals including selective oxidation reactions, hydrogenation, hydrogenolysis, reductive amination, and aromatization. The valuable products, include maleic anhydrides, maleic acid, succinic acid, furoic acid, furfurylamine, furandicarboxylic acid, furfural alcohol, aromatics, levulinic acid, and cyclopentanone, especially for advancements in the catalytic conversion of furfural to useful chemicals in the last few years. The possible reaction mechanisms for the conversion of furfural into bio-chemicals are summarized and discussed. In addition, future prospects are also provided to highlight the challenges and opportunities for the valorization of furfural.
2. Catalytic conversion of furfural into value-added chemicals
2.1 Aerobic oxidation of furfural into carboxylic acid
In the past years, numerous heterogeneous catalysts have been applied to catalyze the oxidation of furfural to MA and FA. This reaction was studied by Yin et al. with phosphomolybdic acid as the catalyst in aqueous/organic biphasic reaction systems.19 In the biphasic reaction system, the aerobic oxidation of furfural took place in the aqueous phase, while the organic phase constituted a reservoir to gradually release the substrate to the aqueous phase through phase equilibrium. A 34.5% yield of MA with a selectivity of 68.6% was obtained with the furfural conversion of 50.4%.19 In addition, a possible reaction mechanism was also proposed, as illustrated in Fig. 3.19 Firstly, a furfural radical, compound 1, is formed through the first hydrogen abstraction by either oxygen or phosphomolybdic acid, and subsequently transfers an electron to phosphomolybdic acid to produce the furfural cation (compound 2). Secondly, the furfural cation is attacked by H2O to generate compound 3, which can undergo 1,4-rearrangement to form compound 4. Eventually, compound 4 is hydrolyzed to produce compound 5, which may be further oxidized to MA.19 However, the catalysts were prone to deactivation owing to the leaching of their active components. In the case of the Mo–V–O catalyst, a total yield of MA and FA of up to 65% was afforded in acetic acid solvent under 20 bar of O2, but the leaching of the V species into the solution resulted in a decrease in the yield of MA and FA in the catalyst recycling tests.21 Significant leaching of Ti was also observed for titanium silicate-1 (TS-1), which was used to catalyze the transformation of furfural to MA with a maximum yield of 78%.22 Recently, an iron-porphyrin catalyst was employed for the aerobic oxidation of furfural into MA in the aqueous phase. Under the optimized conditions, an MA yield of up to 56.1% with a selectivity of 73.8% was obtained with the furfural conversion of 76%.23 The FeT(p-Br)PPCl/SBA-15 catalyst exhibited good stability, which could be recycled at least five times without a remarkable loss in activity. Obviously, a great disadvantage of transition metal oxides such as V, Mo, and Ti oxides is that they are unstable in liquid reaction systems because of the leaching of the active metal species during the oxidation of furfural into MA. Thus, it is of great significance to design and develop some novel robust solid catalysts to boost the aerobic oxidation of furfural into MA via different strategies such as the use of “confined” catalysts embedded in zeolite and porous materials and strong metal–support interaction.
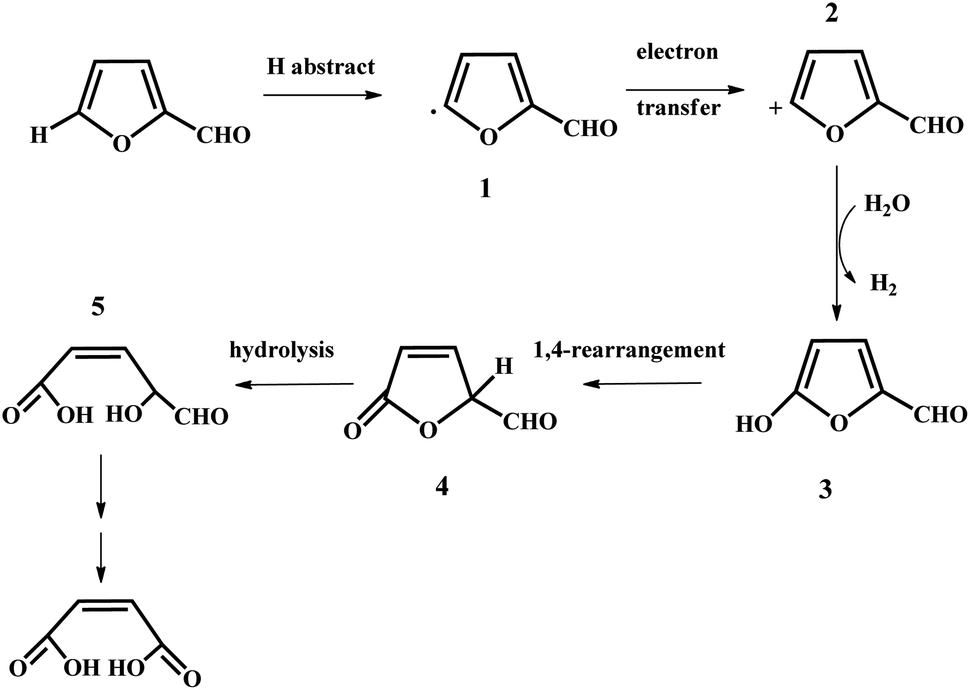 | ||
| Fig. 3 Proposed mechanism for the oxidation of furfural catalyzed by phosphomolybdic acid. Reprinted from ref. 19. Copyright 2011, the American Chemical Society. | ||
Considering the high cost and poor recyclability of transition metal oxide catalysts, some metal-free catalytic reaction systems have been employed to catalyze the oxidation of furfural to MA. Li et al. reported a facile metal catalyst-free system to transform furfural into MA with H2O2 as the oxidant and formic acid as the solvent, which afforded an unprecedented MA yield of 95%.15 Furthermore, this protocol is also efficient for the generation of MA from other platform molecules, such as HMF and HMF derivatives. Almost quantitative yield (99%) of MA was achieved from furan, and good to excellent yields of MA (77–91%) were also obtained as the furan ring was substituted with aldehyde groups such as HMF and DFF.15 When the furan ring was substituted with carboxylic acid and methyl groups, a low MA yield was obtained, implying that carboxylic acid and methyl groups have an unfavorable effect on the MA yield.15 Similarly, an innovative technique was disclosed by Vigier et al. for the valorization of furfural to generate MA and fumaric acid (FA) with an overall yield greater than 90% using betaine hydrochloride (BHC) with H2O2 as the oxidant.24 BHC could be recycled without significant loss in its activity after four successive cycles, and it could be used to directly convert xylose to MA and FA, thus offering a more cost-effective pathway for the production of MA and FA directly from sugars.24 Recently, a facile method was revealed for the oxidation of furfural to both MA and FA in a green deep eutectic solvent (DES) containing oxalic acid and choline chloride (ChCl), and a total yield of both MA and FA of up to 95.7% was obtained with the complete conversion of furfural under mild conditions.16 Besides the above-mentioned catalysts, some other catalysts and their catalytic properties are summarized in Table 1.
| Entry | Catalyst | Oxidant | T (°C)/t (h) | Furfural con. (%) | MA yield (%) | TON (mmol g−1) | Ref. |
|---|---|---|---|---|---|---|---|
| 1 | FeT(p-Br)PPCl/SBA-15 | O2 | 90/6 | 76.0 | 56.1 | 45.6 | 23 |
| 2 | FeT(p-Cl)PPCl | O2 | 90/6 | 49.6 | 33.0 | — | 25 |
| 3 | VO–NH2–GO | O2 | 90/4 | 99.8 | 95.3 | 20 | 26 |
| 4 | CaCuP2O7 | O2 | 115/18 | 62 | 31.8 | 3.1 | 27 |
| 5 | TS-1 | H2O2 | 80/4 | 100 | 62.0 | 84.8 | 28 |
| 6 | Betaine hydrochloride (BHC) | H2O2 | 100/4 | — | 61 | — | 24 |
| 7 | — | H2O2 | 50/24 | 100 | 95.7 | — | 16 |
| 8 | N-doped carbon | H2O2 | 80/5 | 100 | 61.0 | 20 | 29 |
| 9 | SnCl4 | H2O2 | 120/10 | 20.8 | 20.8 | — | 30 |
| 10 | P-doped carbon | H2O2 | 60/10 | 100 | 76.3 | 12.7 | 31 |
| 11 | KBr/g-C3N4 | H2O2 | 100/3 | 100 | 70.4 | 20 | 32 |
| 12 | KBr–KOH | H2O2 | 90/3 | 100 | 72.4 | — | 33 |
| 13 | ZrP2O7–(VO)2P2O7/KIT-6 | H2O2 | 70/1 | 91.2 | 29.2 | 150 | 34 |
| 14 | V2O5/SnO2 | H2O2 | 70/20 | 100 | 60 | 12 | 35 |
| 15 | Cu–Pd@HPC | H2O2 | 50/4 | 100 | 97.8 | — | 36 |
As mentioned above, in contrast to heterogeneous catalysts, homogeneous catalysts exhibit higher stability and selectivity, but the reaction systems require the use of the expensive H2O2 as the oxidant. Obviously, it is more desirable and economical to use O2 as the oxidizer in the oxidation of furfural to MA. However, it remains a great challenge to use O2 as the oxidant in the aerobic oxidation of furfural over non-noble metal oxide catalysts under mild reaction conditions. Thus, it is promising to develop novel photo- and electro-catalytic, even photoelectrocatalytic techniques for the oxidation of furfural to MA in the future.
It has been reported that MAnh may be generated with 5-HMF as the starting material.43 A high yield of MA of up to 79% was obtained over a cheap V2O5 catalyst.43 However, although the yield of MAnh from pure 5-HMF is good, the high cost of 5-HMF hinders the production of MAnh on an industrial scale.18,44 Alternatively, furfural possess a similar structure as 5-HMF and may act as another feedstock for the production of MAnh. Compared with HMF with a limited production volume, furfural is regarded as a more desirable starting material for the production of MAnh.
In the past few years, several catalytic reaction systems including homogeneous and heterogeneous catalysis have been applied to catalyze the oxidation of furfural to MA. Yin et al. explored a new homogeneous oxidation strategy in which H5PV2Mo10O40 and Cu(CF3SO3)2 were used to catalyze the aerobic oxidation of furfural to MAnh and furanone.38 Under the optimal conditions, an MAnh yield of 54% was obtained with 7.5% of 5-acetoxyl-2(5H)-furanone. Furthermore, a plausible mechanism for the oxidation of furfural was proposed, in which three different reaction routes were put forward. They suggested that the dominant oxidation pathway is initiated by hydrogen abstraction from the 5-position of furfural, and then MAnh and 5-acetoxyl-2(5H)-furanone are produced simultaneously. As is well known, homogeneous catalysis has some drawbacks in terms of product separation and environmentally friendliness, especially for catalyst recycling.
In comparison with homogeneous catalysis, heterogeneous catalytic systems have more advantages in the separation of products and catalyst recycling. In 2012, Ojeda et al. reported a heterogeneous catalytic system, giving an MAnh yield as high as 73% through the gas oxidation of furfural at 593 K over VOx/Al2O3.45 It was shown that furfural undergoes tandem oxidation to generate furan, 2-furanone, and eventually MAnh. It was revealed that Al2O3-supported polyvanadates are more active than monovandates (VO4) and V2O5 oxides in the production of MAnh and furan.45 Zhang and co-workers reported that plate-like vanadium phosphorous oxides (VPO) exhibited good activity in the oxidation of furfural to MAnh in the gas phase.41 An MAnh yield of up to 97% was achieved at 10 vol% of furfural in the feed with air as the oxidant at 340 °C. The catalyst also showed good stability with no remarkable loss in activity and selectivity toward MAnh during 25 h reaction. Based on the product distribution analysis, 2-furoic acid was proposed as the main side-product and was also a key intermediate for the synthesis of MAnh over the VPO catalyst. The possible reaction pathway for the gas oxidation of furfural to MAnh was proposed, as depicted in Fig. 4.41 The authors assumed that the oxidation of furfural to 2-furoic acid is the rate-limiting step for the oxidation of furfural over the VPO catalyst because furan was detected in a very trace amount or not produced.41
 | ||
| Fig. 4 Possible reaction pathway for the oxidation of furfural to MAnh on VPO catalyst. Reprinted from ref. 41. Copyright 2018 Wiley. | ||
Granados et al. studied the deactivation process and mechanism for the gas phase oxidation of furfural to MAnh over V2O5/γ-Al2O3 catalysts.46 The results showed that the catalyst was readily deactivated because of the formation of maleates and resins on its surface, especially when the fresh catalyst initially contacted the reaction feedstock at a low temperature. Moreover, increasing the temperature from low to high did not lead to the removal of the deposits, but boosted their deposition. Thus, the yield of MAnh never exceeded 30% because of the deactivation of the catalyst. In contrast, when the fresh catalyst initially contacted the feedstock at high temperature, the deposition rate of maleates and resins remarkably slowed down. Therefore, the yield of MAnh significantly increased and the deactivation rate was reduced substantially.46 In addition, VOx immobilized on Schiff base-modified graphene oxide was utilized to oxidize furfural to MAnh with 95.3% yield under the optimized reaction conditions.26 The oxidation reaction results of furfural to MAnh are summarized in Table 2.
| Entry | Catalyst | Reaction type | T (°C)/t (h) | Oxidant (MPa) | Conversion of furfural (%) | MAnh yield (%) | Ref. |
|---|---|---|---|---|---|---|---|
| 1 | H5PV2Mo10O40·xH2O | Liquid/CH3CN | 110/3 | O2 (20 bar) | 98.7 | 54.0 | 47 |
| 2 | H6PV3Mo9O40·xH2O | Liquid/CH3CN | 110/3 | O2 (20 bar) | 98.7 | 54.0 | 47 |
| 3 | V2O5/Al2O3 | Gas-phase | 310 | 10% O2/He | 100 | 86 | 40 |
| 4 | VPOHT | Gas-phase | 340 | 10% O2/He | 99.2 | 97.9 | 41 |
| 5 | Mo4VO14 | liquid/AcOH | 120/16 | O2 (20 bar) | 100 | 65 | 48 |
| 6 | VOx/Al2O3 | Gas-phase | 320 | O2 | 100 | 73 | 37 |
| 7 | VOx/Al2O3 | Gas-phase | 300 | O2 | — | 55 | 42 |
| 8 | VO–NH2–GO | liquid/AcOH | 90/4 | O2 (20 bar) | 99.8 | 95.3 | 26 |
In addition to the gas-phase oxidation of furfural to MA, solid–liquid reaction systems exhibit good performances in the production of MA. For example, a binary Mo–V metal oxide was used to catalyze the aerobic oxidation of furfural to MA, and an MA yield of 65% was afforded on the Mo4VO14 catalyst under the optimal reaction conditions.21 However, the recyclability test of the Mo4VO14 catalyst showed that a certain amount of V and Mo components leaked into the solution, thereby leading to a decrease in the activity and selectivity toward MAnh. Unlike the gas-phase oxidation of furfural, the solid–liquid reaction easily results in catalyst deactivation, particularly for transition metal oxide catalysts used in the reaction. Thus, it is crucial to develop novel, cheap, robust solid catalysts for the oxidation of furfural to MA in the future.
Considering the wide application of SA, the development of green, high-effective chemical synthesis techniques is an important topic. In the past, SA was generated via hydration and/or hydrogenation of petroleum-derived maleic anhydride or maleic acid.53 Recently, bio-based MAnh or MA was obtained via the oxidation of HMF, furfural, and levulinic acid (LA). However, complex processes make it an uneconomic pathway if the bio-based MA hydrogenation reaction is used in the production of SA. Thus, the direct production of SA from cheap and available bio-platform molecules via economical and environmentally friendly pathways is of great significance.
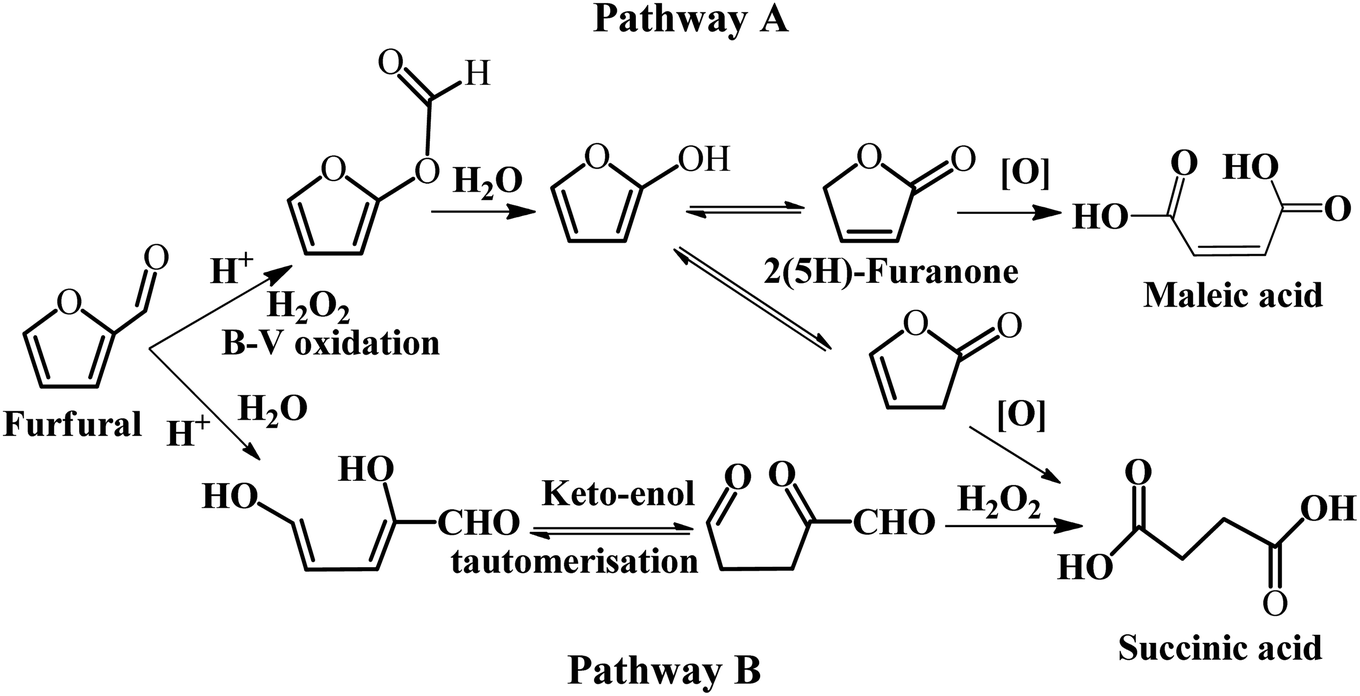
Some homogeneous catalysts, such as formic acid, have been employed in the selective oxidation of furfural. A summary of the metal-free catalysts for the selective oxidation of furfural to SA is presented in Table 3. Wang et al. studied the selective oxidation of furfural to 2(5H)-furnanone, SA, and MA with formic acid as the oxidant in an aqueous/organic biphasic system.54 In this reaction system, the total yield of SA and MA was 15–20%, while the main product was 2(5H)-furanone with excellent yield when formic acid was used as the catalyst. A possible explanation is that formic acid reacts with H2O2 to form performic acid with strong oxidizing property. The plausible reaction pathway was proposed based on the reaction kinetics studies, as illustrated in Fig. 5. In pathway A, furfural firstly undergoes the Baeyer–Villiger rearrangement reaction to produce 2-formyloxyfuran, and then it is hydrolyzed to form 2-hydroxyfuran and its tautomers, 2(5H)-furannone and 2(3H)-furannone, which may be further converted to MA and SA, respectively. In pathway B, furfural is subject to open cyclization to form a dienol, which can undergo ketonization to diketo aldehyde. Subsequently, the diketo aldehyde is oxidized to SA by H2O2. It should be noted that pathway A and B co-exist in the oxidation of furfural in the biphasic system.
| Entry | Catalyst | Reaction type | T (°C)/t (h) | Oxidant | Conversion of furfural (%) | SA yield (%) | Ref. |
|---|---|---|---|---|---|---|---|
| a CD – β-cyclodextrin.b GO – acidic metal-free graphene oxide. | |||||||
| 1 | SO3H-CDa | 60/1.5 | H2O2 | 100 | 81.2 | 53 | |
| 2 | Amberlyst-15 | Toluene | — | H2O2 | — | 52.3 | 49 |
| 3 | Sulfated zirconia | — | 75 | H2O2 | 100 | 86.0 | 52 |
| 4 | GOb | H2O | 70/24 | H2O2 | 99.8 | 97.9 | 50 |
| 5 | SO3H-carbon | H2O | 80/1 | H2O2 | 100 | 93.0 | 51 |
 | ||
| Fig. 5 Possible reaction pathways for the oxidation of furfural. B–V refers to the Baeyer–Villiger reaction. Reprinted from ref. 54. Copyright 2016, Elsevier. | ||
Recently, solid acids have been used to catalyze the oxidation of furfural to SA with H2O2 as the oxidant, such as Amberlyst-15 cation exchange resin,49,55 sulfonated graphene oxide (GO),50 –SO3H-functionalized polymer,51,53 and sulfated zirconia.56 An innovative pathway has been reported for the conversion of hemicellulose prehydrolysate to SA using Amberlyst-15 as the catalyst and H2O2 as the oxidant in a biphasic system.49 In the biphasic system, a key intermediate, furfural, which was produced from the dehydration of xylose, was oxidized to SA with a maximum yield of 52.3%. This work exhibits the potential for the valorization of inexpensive hemicellulose to the value-added SA. Lv and co-workers synthesized a highly active graphene oxide (GO) catalyst decorated with –SO3H groups for the oxidative conversion of furfural to SA with H2O2 as the oxidant.50 A maximum SA yield of 97.9% was achieved under the optimized reaction conditions. They revealed that GO with –SO3H groups exhibited superior activity and SA selectivity for the oxidation of furfural than other common acid catalysts, such as HCl, H2SO4, and ZSM-5. Moreover, the GO catalyst has good reusability with no remarkable loss in furfural conversion and SA yield after six consecutive recycles. The decrease in the furfural oxidation activity was attributed to the leaching of the oxygenated-containing groups and –SO3H groups, as evidenced by the XPS results. However, the active sites of the catalysts are the –SO3 groups and the oxidizing agent is the expensive H2O2.50 In addition, Murzin and co-workers reported the oxidation of furfural over sulfated zirconia in the presence of H2O2, and many products including MA, SA, furoic acid, and 2(5H)-furanone were generated, but the SA selectivity was low. Thus, more efforts should be devoted to developing more robust and effective catalytic systems besides the use of –SO3H group-grafted polymers or their functionalized catalysts with the use of the inexpensive O2 or air as the oxidant instead of the expensive H2O2 for the oxidation of furfural to SA in the future.
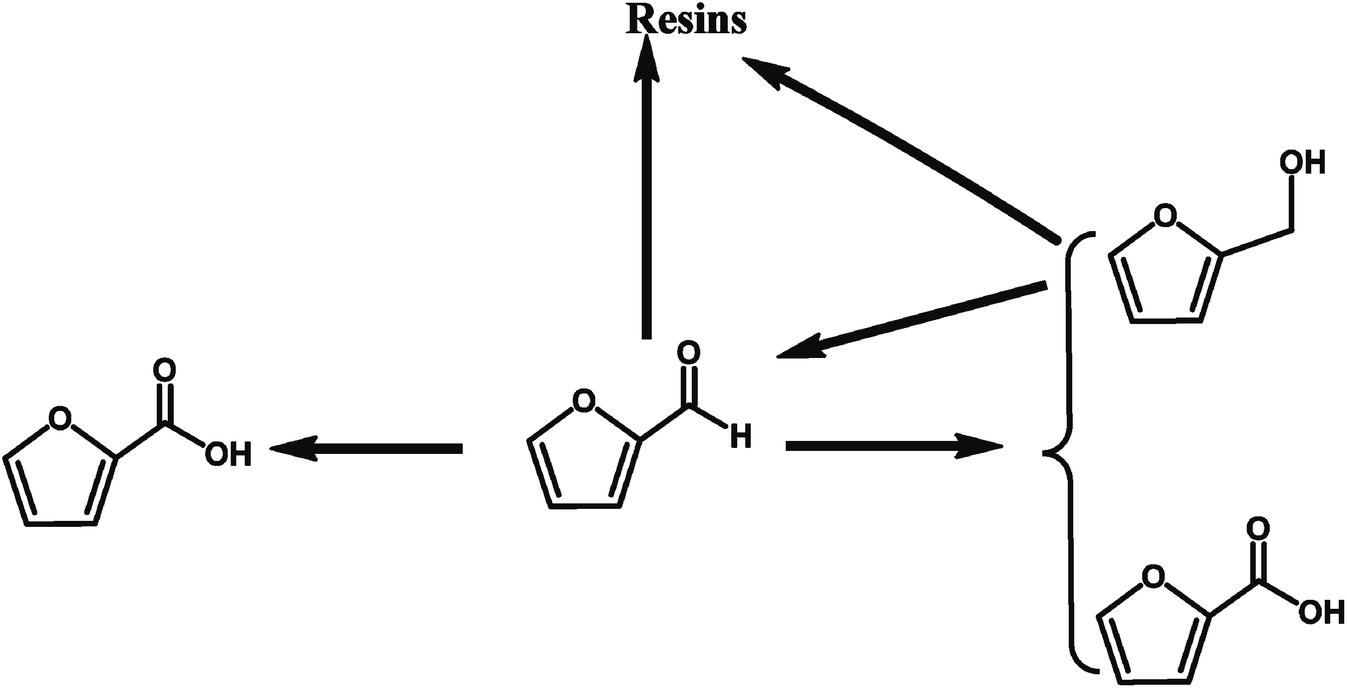
2.1.4.1 Selective oxidation of furfural to FCA through chemical conversion. Various heterogeneous catalysts such as Ag2O/CuO,58 AuPd/Mg(OH)2,57 and Au/ZTC59 catalysts, have been utilized to catalyze the oxidation of furfural into FCA. A summary of the solid catalysts for the selective oxidation of furfural to FCA is provided in Table 4. Giordano et al. reported the excellent performances of Au/ZTC, a carbon support synthesized using a carbon replica of BETA zeolite, in the aerobic oxidation of furfural to FCA in the absence of base.59 An 89% furfural conversion with full selectivity toward FCA was achieved in the absence of base, which provides a green approach for the production of FCA from furfural. Hutchings et al. reported that a 1% AuPd/Mg(OH)2 catalyst catalyzed the selective oxidation of furfural to FCA with a yield of 84.5% in the presence of NaOH.57 They revealed that the reaction proceeds via many different pathways, and the addition of NaOH promotes the Cannizzaro reaction between the furfural compounds, leading to stoichiometric quantities of furfural alcohol and FCA, as illustrated in Fig. 6. The catalyst enhances both the direct conversion of aldehyde to the acid and the tandem oxidation of furfural alcohol to furfural and then to FCA.57 However, this reaction requires the addition of NaOH as the base catalyst to promote the selective oxidation of furfural to FCA, making it difficult to separate the products downstream and increasing the production costs. Compared to the current industrial process through the Cannizzaro reaction catalyzed by an inorganic base, the use of solid catalysts for the selective oxidation of furfural to FCA has more advantages such as easy separation of the products and higher FCA yields.
| Entry | Catalyst | Reaction type | T (°C)/t (h) | Oxidant | Conversion of furfural (%) | SA yield (%) | Ref. |
|---|---|---|---|---|---|---|---|
| a ZTC – zeolite-templated carbons.b ALDHs – aldehyde dehydrogenases; buffer – KH2PO4/K2HPO4, pH = 8.5.c Buffer – KH2PO4/K2HPO4, pH = 7. | |||||||
| 1 | Au/ZTCa | 120/6 | O2 | 89 | 89 | 63 | |
| 2 | N-heterocyclic carbene | DMSO | 40/6 | O2 | >99 | >99 | 64 |
| 3 | AuPd/Mg(OH)2 | H2O | 30 | O2 | 99.5 | 84.5 | 57 |
| 4 | ALDHsb | Buffer | 40 | — | >99 | >99 | 62 |
| 5 | E. coli | Bufferc | 30 | — | 100 | 99 | 61 |
 | ||
| Fig. 6 Proposed reaction routes for the selective oxidation of furfural to FCA over AuPd/Mg(OH)2 in the presence of NaOH. Reprinted from ref. 57. Copyright 2017, The Royal Society of Chemistry. | ||
2.1.4.2 Selective oxidation of furfural to FCA through bio-catalysis. Besides the chemical synthesis of FCA, Peng et al. reported an efficient one-pot chemo-enzymatic approach for the conversion of rice straw to form FCA through a tandem catalyst with Sn-sepiolite and whole-cell harboring horse liver alcohol dehydrogenase (HLADH) under mild reaction conditions.60 The Sn-sepiolite could effectively convert rice straw to furfural in 42.2% yield at 170 °C for 20 min. Subsequently, the resulting furfural could be completely oxidized to FCA with E. coli TS whole-cells within 96 h at room temperature. In addition, the inorganic catalyst and immobilized TS whole-cell biocatalyst were recycled and reused. Li and co-workers also reported a biocatalytic route for the selective oxidation of furfural to FCA using recombinant E. coli cells harboring ASPDH from C. testosteroni SC1588.61 An FCA yield as high as 99% was achieved under the optimized reaction conditions. Moreover, the recombinant strain showed good tolerance to FF up to 100 mM.61 Mutti et al. presented a study on the oxidation of furfural to FCA using three recombinant aldehyde dehydrogenases (ALDHs).62 A very high FCA yield of >99% could be obtained in phosphate buffer at pH 8.5 and 40 °C.62 However, for the biocatalytic oxidation of furfural toward FCA, the long-term stability should be further improved through reaction engineering strategies. In addition, the enhanced substrate tolerance is highly favorable for industrial application in future.
2.2 Catalytic transformation of furfural to C6 carboxylic acid
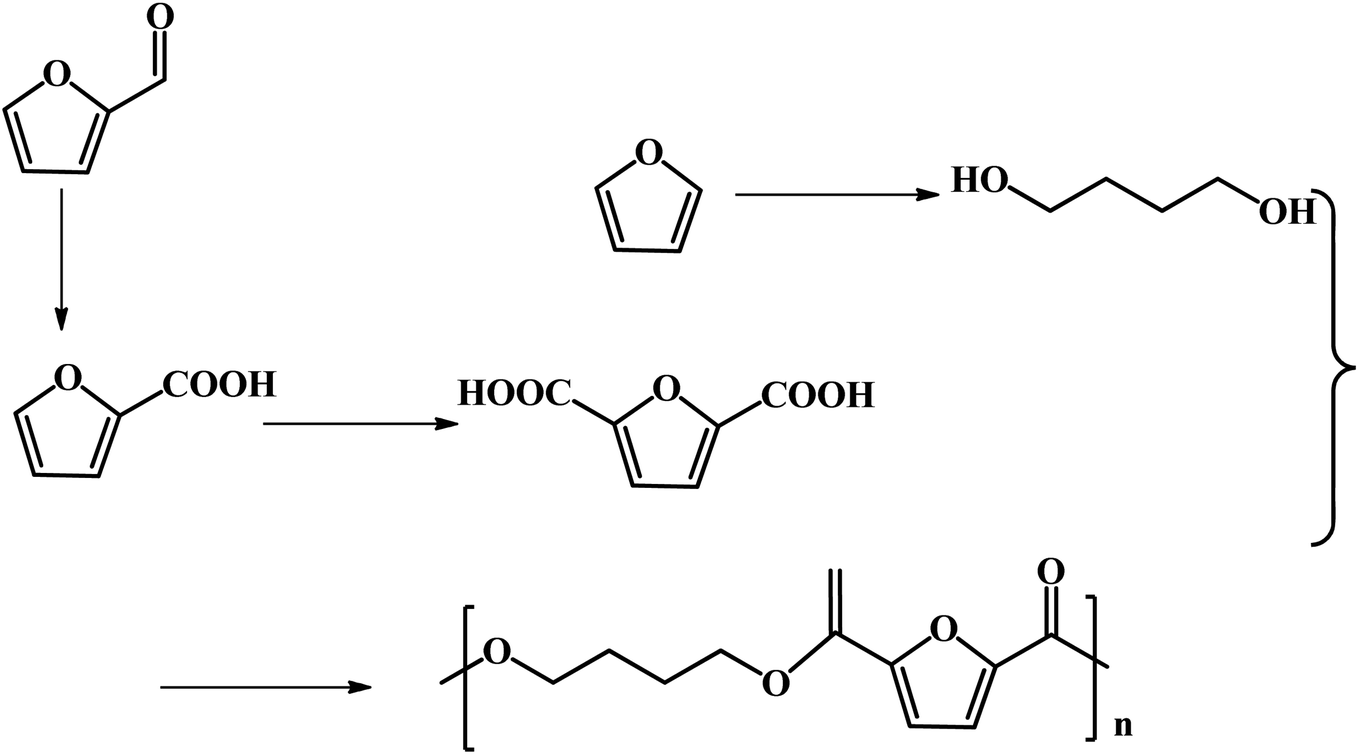
FDCA-based polyesters represent one of the important types of furan-based polymers. At present, the production of FDCA is mainly through the catalytic oxidation of HMF.65 However, as mentioned above, HMF has high production costs, which impedes the large-scale commercial production of downstream products such as FDCA and DFF. Furfural, a commercial bulk biomass-based chemical, is a more desirable starting material in the production of FDCA than HMF. Efforts have been undertaken to explore some novel pathways for the generation of FDCA from furfural to address the bottleneck of HMF production.Fu and co-workers reported the multistep catalytic conversion of furfural to poly(butylene 2,5-furandicarboxylate) (PBF) via the disproportionation of furoate to furan and 2,5-furandicarboxylate with total carbon utilization, as shown in Fig. 7.66 The reaction pathway includes four steps as follows: firstly, the oxidation of furfural to furoate is catalyzed. Secondly, the resulting furoate undergoes disproportionation to give rise to furan and 2,5-furandicarboxylate. Subsequently, furan is subjected to hydrogenation, and then hydrolysis to form 1,4-butanediol (1,4-BDO). Finally, polymerization of 1,4-BDO with 2,5-FDCA is performed to form PBF. The critical step of this pathway is the selective disproportionation of furoate into furan and 2,5-furandicarboxylate, which provides a key linkage between commercialized furfural and the furan-based polymer monomer FDCA.66
 | ||
| Fig. 7 Multistep conversion of furfural to the 2,5-furandicarboxylic acid-based polyester PBF. Reprinted from ref. 66. Copyright 2013 Wiley. | ||
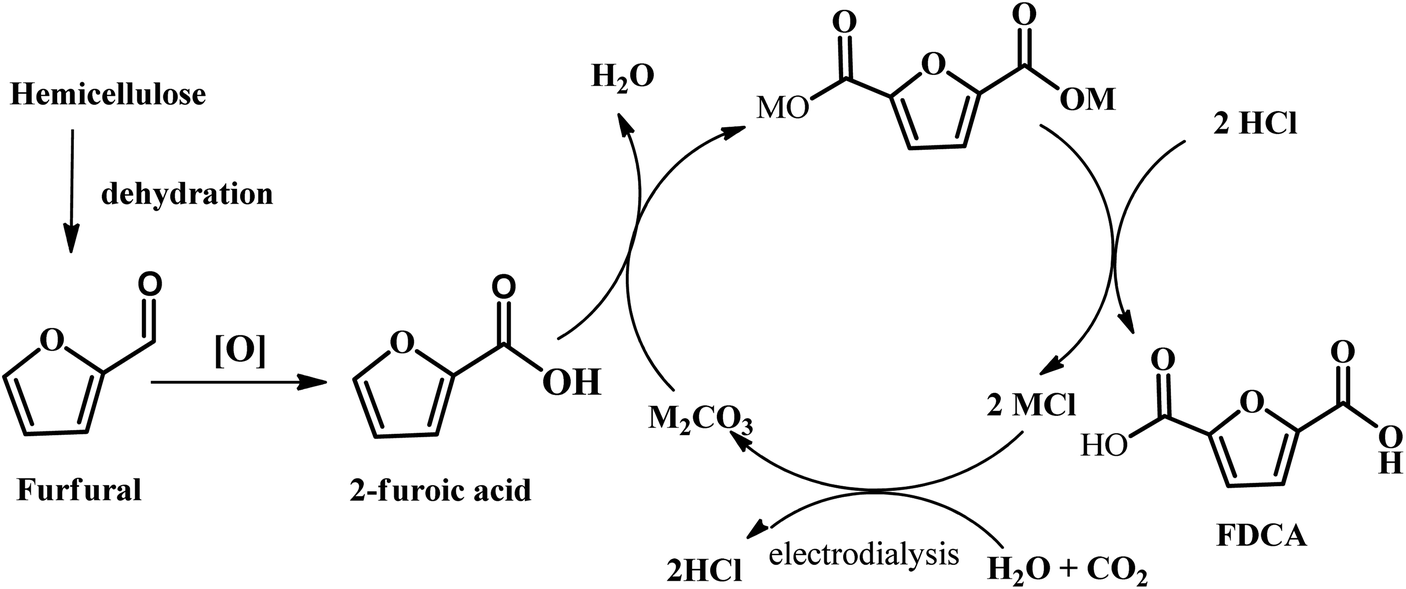
Another emerging pathway has been reported by Kanan et al., where a C–H carboxylation reaction was firstly carried out to generate 2-furonic acid and CO2 in high yield followed by protonation to convert 2-furoic acid to FDCA (see Fig. 8).67 This reaction was performed by heating a salt mixture consisting of caesium furoate (the deprotonated form of 2-furoic acid) and Cs2CO3 under a CO2 atmosphere. A maximum FDCA yield of 89% was achieved on a 1–10 mmol scale after 5 h of reaction, with only 5% degradation product. After carboxylation, protonation with strong acid (e.g., HCl) in water precipitated the FDCA product and an aqueous salt solution (e.g., MCl). The stoichiometric base and acid consumed in this synthesis could be regenerated via bipolar membrane electrodialysis of the salt solution. This CO32−-promoted carboxylation pathway to FDCA has a number of great potential advantages over conventional hexose-based routes. Firstly, the oxidation of furfural into 2-furoic acid is easier than oxidizing HMF to FDCA. Next, no solvents or transition metal catalysts are required in this strategy, which is an environmentally friendly route to synthesize FDCA. Considering that 2-furoic acid can be readily generated from lignocellulose, CO32−-promoted C–H carboxylation opens a new avenue to convert inedible biomass and CO2 into a valuable chemical.
 | ||
| Fig. 8 Schematic illustration of the overall diagram for the production of FDCA from biomass through furfural using CO32−-promoted C–H carboxylation. Recovery of the alkali cations is achieved by protonation of the FDCA salt and bipolar membrane electrodialysis of the resulting alkali chloride salt. Reprinted from ref. 67. Copyright 2016, The Royal Society of Chemistry. | ||
Recently, Liu and co-workers reported an integrated process for the full conversion of lignocellulosic materials and CO2 to form FDCA using a single-atom Co on N-doped carbon catalyst (Co SAS/N@C).68 During the carboxylation reaction of furfural, Cs2CO3 was employed as a base additive, which boosted the carboxylation reaction of the 5-position of furoic acid. An FDCA yield of 71.7% with a selectivity of 82.9% was obtained from the oxidation-derived product of furfural, which is comparable to the previous work (76% FDCA yield with 82.6% selectivity). Moreover, the Co SAS catalyst was found to afford an FA yield as high as 96.9% in the aerobic oxidation of furfural to FA, outperforming the state-of-the-art non-noble metal catalysts.68
In addition to the disproportionation and carboxylation routes of furoate, Yin et al. explored a new approach to synthesize FDCA from 2-furoic acid through consecutive bromination, esterification, carbonylation, and hydrolysis with 65% total yield in four steps and above 80% isolated yield in each step (Fig. 9).69 The first step, i.e., bromination, was performed by reacting furoic acid with bromine to obtain 5-bromo-furoic acid with a yield of 86%. In the next step, ethyl 5-bromo-furan-2-carboxylate (3) was feasibly generated by refluxing 5-bromo-furoic acid (2) in ethanol with the use of concentrated H2SO4, which afforded 87% yield of isolated product. In the third step, carbonylation of 5-bromo-furan-2-carboxylate was conducted using a homogeneous Pd catalyst, and a high yield of 91% was obtained. In the final step, the hydrolysis of diethyl furan-2,5-dicarboxylate (4) was conducted in the presence of H2SO4 at 160 °C for 8 h. After the solution was cooled to room temperature, a white solid precipitate was obtained after filtration, resulting in 96% isolated yield. It is noteworthy that this strategy has some disadvantages, such as the use of a toxic bromine in the bromination step and use of H2SO4 as the catalyst, which are environmentally unfriendly agents. Overall, this approach offers an alternative way to produce FDCA for the sustainable utilization of furfural and CO.
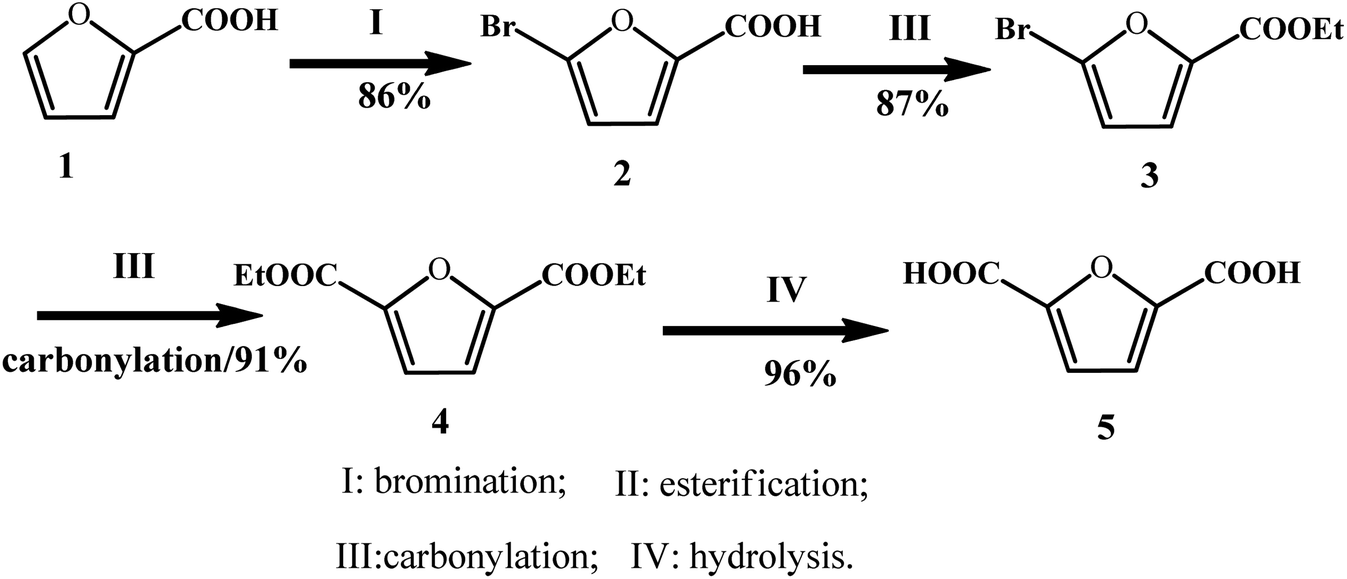 | ||
| Fig. 9 Synthetic strategies for FDCA from furoic acid. Reprinted from ref. 69. Copyright 2017, the American Chemical Society. | ||
2.3 Catalytic conversion of furfural to furfurylamine
It was reported that Au, Pt and Ir colloids supported on sulfonic acid-functionalized silica were employed for the reductive amination of furfural using aniline.78 This was proposed that the acidic SO3H sites facilitate the formation of imine and subsequent hydrogenation to amine by metal particles. However, the yield was relatively low due to the PVP molecules covering the metal active sites, thus leading to low activity.78 Chatterjee et al. provided a more efficient approach for the amination of furfural, which afforded 92% furfurylamine selectivity over the commercial 5% Rh/Al2O3 catalyst, with the use of 28% ammonia and molecular hydrogen as the reactants.71 They further revealed that the hydrogenation of imines to furfurylamine is the main reaction pathway, while the transformation of the Schiff base-type intermediate to furfural amine and secondary amines is the minor pathway (see Fig. 10). However, the activity of the catalyst declined after five successive recycles, which could be due to the presence of some adsorbed species (e.g., amine) blocking the active sites.71 This study offers an environmentally friendly pathway for the synthesis of amines, which can also be further extended to the reductive amination of other aldehydes. Recently, a similar work was reported by Vigiera et al., where a facile and efficient protocol for the synthesis of furfural-derived amines was developed with a high yield in the presence of methyl isobutyl ketone, NH3, H2, and Ru/C catalyst.79 The catalytic system can effectively hydrogenate C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds and the reductive amination of carbonyl groups under mild reaction conditions.
C bonds and the reductive amination of carbonyl groups under mild reaction conditions.
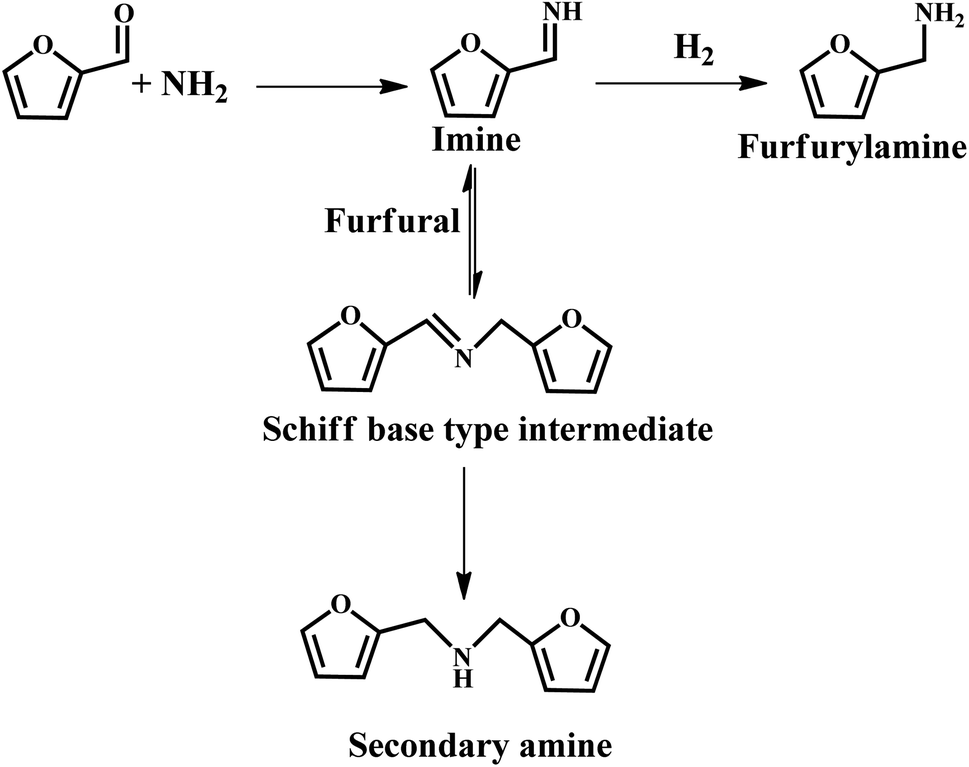 | ||
| Fig. 10 Possible reaction path for the formation of furfurylamine under the studied reaction conditions. Reprinted from ref. 71. Copyright 2016, The Royal Society of Chemistry. | ||
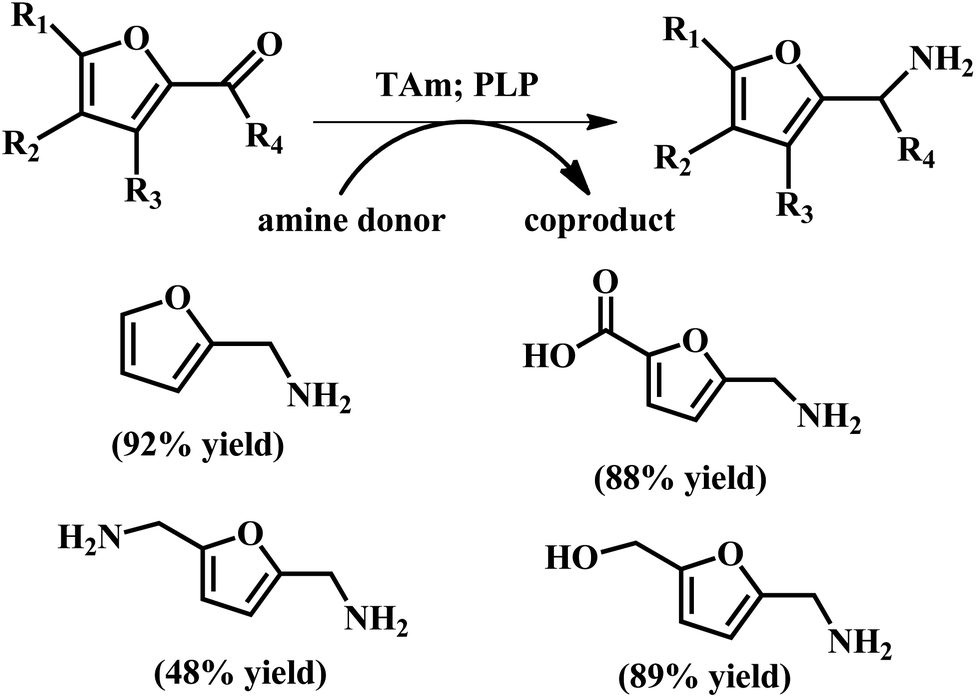 | ||
| Fig. 11 Conversion of aldehydes and ketones to produce amines using TAms with co-factor pyridoxal-5-phosphate (PLP). The reaction conditions: Chromobacterium violaceum DSM30191 (CV-TAm); 200 μL total reaction volume; isopropylamine (IPA) (100 mM), PLP (1 mM), potassium phosphate buffer (100 mM, pH 8 for IPA), amine acceptor (10 mM when IPA was used) and crude cell lysate (20 μL) at 35 °C with IPA for 24 h. Reprinted from ref. 72. Copyright 2017, The Royal Society of Chemistry. | ||
2.4 Hydrogenation of furfural to furfuryl alcohol (FOL)
Furfuryl alcohol is the most important hydrogenated product derived from furfural and accounts for nearly 62% of the annual downstream products of furfural.11 Currently, its industrial production in the liquid or gas phase is mainly through the selective hydrogenation of furfural.81 Although the cost-effective gas reaction process is more suitable for industrial applications, this process only gives a low yield of FOL, which cannot be properly addressed.11 Therefore, the production of FOL in the liquid phase has become the trend of current research.For liquid phase hydrogenation, a variety of catalysts can achieve an FOL yield greater than 90%. The commonly used catalysts are Cu–Cr catalysts.82 Although this type of catalyst can give high FOL yields, the catalyst tends to deactivation because of the strong adsorption of byproducts on its active metal sites, and the Cr element is toxic. Furthermore, the hydrogenation process of furfural with Cu–Cr catalysts is environmentally unfriendly. Thus, in recent years, environmentally benign catalysts for the hydrogenation of furfural have been extensively explored using green reaction systems, and some excellent works are summarized in Table 5.
| Entry | Catalyst | Solvent | T (°C)/t (h) | H2 (MPa) | Furfural con. (%) | FOL yield (%) | TOF (h−1) | Ref. |
|---|---|---|---|---|---|---|---|---|
| a PES refers to polyethersulfone.b SAs refer to single atom catalysts. | ||||||||
| 1 | Cu–SiO2–I | 2-Propanol | 110 | 10 | 66.3 | — | 0.009 | 83 |
| 2 | 3% Pt/SiC–C | H2O | 25/3 | 1.0 | 80.2 | 26.5 | — | |
| 3 | Pd/H-UiO-6 | H2O | 60/3 | 0.5 | >99 | >99 | 66.7 | 84 |
| 4 | Pt/(Fe,Co)-BTC | 2-Propanol | 70/2 | 1.0 | 98.0 | 97.0 | 1044 | 85 |
| 5 | PtNi nanoframes | 2-Propanol | 100/1 | 1 | 100 | 99 | 2.56 | 86 |
| 6 | Pd–Cu/MgO | H2O | 110/1.3 | 0.6 | 100 | 99 | — | 87 |
| 7 | Ru/UiO-66 | H2O | 20/4 | 0.5 | 95 | 94 | — | 88 |
| 8 | Fe(NiFe)O4–SiO2 | Heptane | 90/4 | 2 | 94 | 93 | — | 89 |
| 9 | Ru-PESa | H2O | 70/7 | 0.7 | 26.1 | >99 | 48![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
90 |
| 10 | Cu/C | 2-Propanol | 170/3 | — | 75.0 | 72.5 | 14.4 | 91 |
| 11 | Pt–Sn/SiO2 | 2-Propanol | 100/8 | — | 100 | 96 | — | 92 |
| 12 | Ni–SAs/NSb | 2-Propanol | 130/3 | — | 95.6 | 92.5 | 623 | 93 |
| 13 | PtSn@HMSNs | 2-Propanol | 100/5 | 1.0 | 100 | 94.5 | — | 94 |
Li et al. used a Pd/H-UiO-6 catalyst to catalyze the hydrogenation of furfural to FOL and a high yield was achieved under mild reaction conditions.84 The Pd–Cu/MgO and Ru/UiO-66 catalysts afforded excellent FOL yields of 99% and 94%, respectively.87,88 Halilu et al. synthesized low-cost, recyclable Fe(NiFe)O4–SiO2 for the hydrogenation of furfural, which resulted in 93% FOL yield in heptane solvent.89 In addition, some durable, efficient catalysts such as Cu–Fe oxides,95 Ni–Fe alloys,96,97 and PtSn alloy94 have also been employed in the hydrogenation of furfural to produce FOL in recent years. It is highly desirable to design and develop cheap, robust, efficient catalysts for the hydrogenation of furfural, but noble metal catalysts should not be the first choice because they are expensive and increasingly exhausted.
Besides the use of H2 as a hydrogen source, catalytic transfer hydrogenation (CTH) is a potential alternative protocol for the hydrogenation of furfural to FOL.93,98–101 Zr-based catalysts,102–105 CuNi@C,101 single-atom Ni catalyst,93 metal–organic framework (MOF) materials,101,103,106,107 noble metal catalysts such as Pd, Pt, and Ru,108–111 and transition metal catalysts such as Fe oxides112 have been used to catalyze the reaction. MOF-808 has been shown to be the most active catalyst in the CTH of FUR to FOL among the Zr-MOFs due to the facile modification of the metal node mediating the acid–base properties by the introduction of coordinatively unsaturated sites, hydroxyl and methoxy groups in the framework of the Zr-MOF, which favor the adsorption of furfural and 2-propanol. For the in-depth understanding of the CTH mechanism, a computational study based on DFT was conducted, and two feasible geometries of Zr nodes were proposed, i.e., singly and doubly modified Zr nodes of MOF-808.103 It was disclosed that the hydride transfer from alkoxy carbon to carbonyl carbon is the rate-limiting step based on DFT studies.103 Considering that the hydrogenation of furfural is a relatively easy reaction, some cheap and robust supported catalysts are more desirable for the reaction compared to expensive noble metal catalysts.
2.5 Furfural to cyclopentanone (CPO)
Cyclopentanone (CPO) is a versatile compound derived from furfural, which is widely used in the synthesis of fuels, pharmaceuticals, rubber chemicals, fungicides, and flavor and fragrance chemicals.113–116 It is well-accepted that the most important reaction step in the conversion of furfural to cyclopentanone is the hydrogenative ring-rearrangement of furfural, but the reaction mechanism is still under debate, and various reaction routes and reaction intermediates have been proposed, which may be dependent on the catalyst type, reaction conditions, etc.116–121 In general, furfural is firstly hydrogenated to produce furfuryl alcohol, followed by a ring-opening reaction to give 2-pentene-1,4-dione (2-PED) accompanied by the formation of a tetrahydrofurfuryl alcohol (THFA) byproduct. Subsequently, 2-PED undergoes an aldol reaction to give 4-hydroxy-2-cyclopentenone (HCPEO), and then further hydrogenated to form 3-hydroxycyclopentanone (HCPO). Eventually, HCPO is converted to CPO by hydrogenation.115 The possible reaction pathway is displayed in Fig. 12.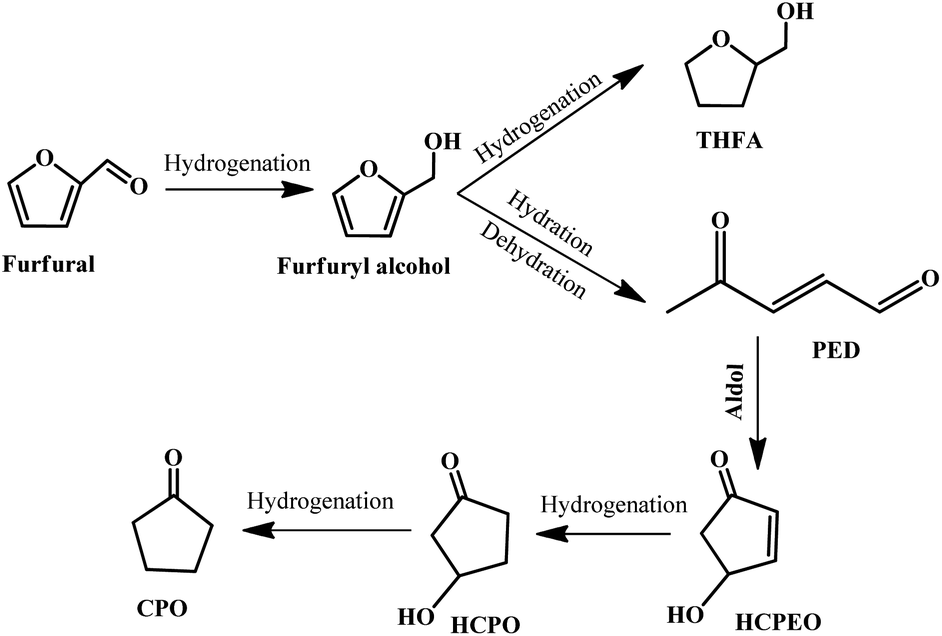 | ||
| Fig. 12 Reaction pathway for the hydrogenative ring-rearrangement of furfural into cyclopentanone. Reprinted from ref. 115. Copyright 2019, Elsevier. | ||
Various monometallic catalysts such as supported Cu,121–123 Pt,124 Ru,125,126 and bimetallic catalysts, e.g., supported Co–Ni,127 Cu–Co,128 and Pd–Co,129 have been shown to have high catalytic activity for the transformation of furfural to CPO, and their catalytic performances are listed in Table 6. Among the catalysts used in this reaction, Cu-based catalysts are the most thoroughly studied because of their low cost and high catalytic activity. Wang and co-workers demonstrated that a CuNi0.5@C bimetallic catalyst fabricated via the pyrolysis of Cu-based metal–organic frameworks provided a high CPO yield of 96.9% with the almost complete conversion of furfural.118 2-Cyclopentenone is prospected as the reaction intermediate involved in the reaction,118 which is consistent with the assumption by some researchers that 4-hydroxy-2-cyclopentenone is the reaction intermediate.115,130
| Entry | Catalyst | Solvent | T (°C)/t (h) | H2 pressure (MPa) | Conversion of furfural (%) | CPO yield (%) | Ref. |
|---|---|---|---|---|---|---|---|
| 1 | CuZnAl-LDH | H2O | 140/2 | 2 | 100 | 86.5 | 131 |
| 2 | Cu/ZrO2 | H2O | 150/4 | 1.5 | 100 | 91.3 | 121 |
| 3 | Cu/Fe3O4 | H2O | 170/4 | 3 | 100 | 91 | 123 |
| 4 | CuNi@C | H2O | 130/5 | 5 | 99.3 | 96.9 | 132 |
| 5 | Cu–Co@C-500 | H2O | 150 | 0.5 | 90.2 | 128 | |
| 6 | PtCo/C | Toluene/H2O | 180 | 1 | 100 | 75 | 133 |
| 7 | Pt/N-doped carbon | H2O | 150/4 | 3 | >99 | >76 | 124 |
| 8 | Pd–Co@UIO-66 | H2O | 120/2 | 3 | >99 | 96 | 116 |
| 9 | Pd/FeZn-DMC | H2O | 150 | 4 | 100 | 96.6 | 114 |
| 10 | Co–Ni/TiO2 | H2O | 150 | 4 | 100 | 53.3 | 127 |
| 11 | Ru/CNTs | H2O | 160/3 | 1 | 99 | 91 | 126 |
In addition to Cu-based catalysts, other noble metal catalysts also possess appreciable activity for the generation of CPO from furfural.114,116,120,124–126,134–136 Anatase TiO2-supported Au nanoparticles were found to effectively inhibit the side reaction and achieve nearly 100% CPO yield in the conversion of furfural to CPO.134 Deng et al. showed that a double-metal cyanide (DMC)-supported bi-functional Pd catalyst had high efficiency for the hydrogenative ring-rearrangement of furfural to cyclopentanone.114 It has been revealed that Lewis acid sites play an important role, e.g., Pd/FeZn-DMC with moderate Lewis acidity gave a cyclopentanone yield of 96.6%, whereas Pd/FeNi-DMC and Pd/FeCo-DMC with weak Lewis acidity afforded a furfuryl alcohol yield of 91.2%.
2.6 Hydrogenolysis of furfural to pentanediol
Pentanediol, which is considered to be a promising bio-fuel, monomer of polyesters, and fine chemical intermediate, can be generated via the one-pot reaction of furfural or the hydrogenolysis of THFA.137–139 However, compared to THFA hydrogenolysis, the direct transformation of furfural to pentanediol is more challenging because it is very difficult to achieve high selectivity toward pentanediol and it tends to yield many byproducts.138,140Much effort has been focused on exploring some novel catalysts for the hydrogenolysis of furfural to pentanediol. For instance, a Pd–Ir-ReOx/SiO2 bi-functional catalyst was used for the one-pot hydrogenolysis of furfural to 1,5-pentanediol with THFA as the intermediate and gave a maximum 1,5-pentanediol yield of 71.4%.141 Hydrotalcite-supported Pt nanoparticles achieved a 1,5-pentanediol yield of up to 73% from furfural without any additives.142 Besides 1,5-pentanediol and 1,2-pentanediol, 1,4-pentanediol (1,4-PeD) could also be produced by the direct hydrogenolysis of furfural catalyzed by hydroxyapatite-supported Pt–Mo bimetallic catalysts.143
2.7 Aromatization of furfural to aromatics
Aromatics are an important type of chemicals, especially for the production of polymers, solvents, pharmaceutical, fragrances, etc. Aromatics can be produced from fossil resources, either via the catalytic reforming of naphtha or steam cracking of hydrocarbons.5,6,144 This is an emerging and alternative way to produce aromatics using biomass as starting materials. Bio-based aromatics can be typically produced via the catalytic fast pyrolysis of lignocellulose, depolymerization or cracking of lignin, and conversion of carbohydrates via the furanic platform.144–146 In many cases, these processes mainly generate benzene, toluene, and xylene (BTX) through sequential Diels–Alder (DA)/aromatization reactions with ethylene over acid catalysts. Bio-based BTX can be further converted without any modification of current downstream processes and applications.To date, very few studies have been reported on the production of functionalized and industrially relevant aromatics from biomass, such as benzonitrile, benzaldehyde, and benzylamine derivatives. Among the biomass-derived aromatics, meta-xylylenediamine (MXD) is an important chemical, which is widely used in the production of polymers including polyamide and polyurethane. To date, MXD is produced at 100 kT per year from fossil feedstocks. Recently, the renewable MXD was synthesized from biomass-derived furfural and acrylonitrile.147 However, the D–A reaction of furfural with acrylonitrile did not occur because the electron-withdrawing formyl group in furfural is thermodynamically unfavorable with a significantly positive free energy according to DFT results. Therefore, a novel strategy was employed via the acetalization derivatization of furfural with ethylene glycol to give 2-(furan-2-yl)-1,3-dioxolane, which afforded a thermodynamically favorable reaction when it reacted with acrylonitrile (as shown in Fig. 13).147 Subsequently, aromatization of D–A with acrylonitrile gave 86% yield of aromatic cycloadducts catalyzed by CH3ONa in DMSO. After aromatization, the meta-aromatic isomer underwent deprotection in the presence of HCl to produce ethylene glycol and meta-cyanobenzaldehyde in quantitative yield. Finally, reductive amination of the aldehyde with ammonia using RANEY® Co yielded 70% MXD. Moreover, all the steps (protection, DA/aromatization, deprotection and hydrogenation) are highly selective and chemical intermediates can be directly used in the next step without purification. Therefore, this novel strategy provides a sustainable, alternative way to produce MXD with high yield from biomass-derived furfural and acrylonitrile.
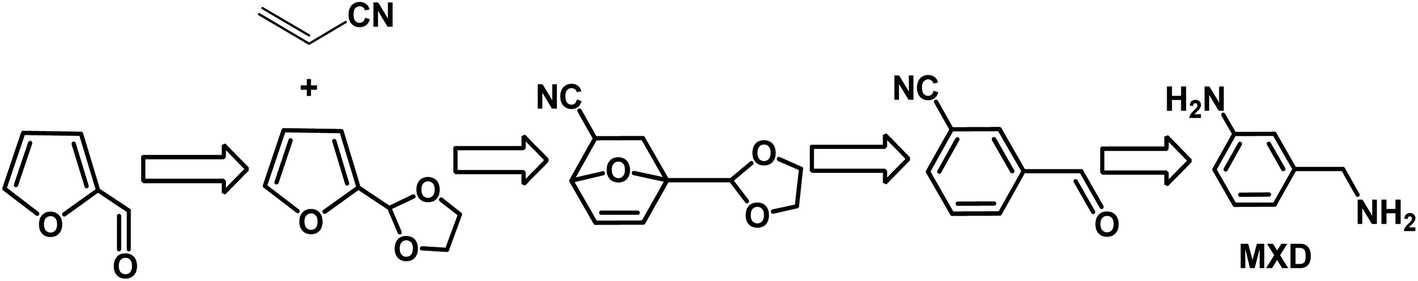 | ||
| Fig. 13 Pathways to meta-xylylenediamine (MXD) from furfural and acrylonitrile. Reprinted from ref. 147. Copyright 2018, Wiley. | ||
2.8 Furfural to other useful chemicals
Tetrahydrofurfuryl alcohol (THFA) is a liquid compound with a high boiling point, which is transparent and completely miscible with water. It is a green solvent for agricultural applications, electronic cleaners and printing inks.148 THFA can be produced directly from furfural. The typical approach for the production of THFA from furfural is a two-step hydrogenation reaction through the furfuryl alcohol intermediate over Cu–Cr and noble metal catalysts separately.149 However, this cumbersome process increases the production cost of THFA. Recently, the synthesis of THFA from furfural via a “one-pot method” addressed this problem. For example, in the study conducted by Rode et al., 95% THFA yield was achieved in the presence of an Fe catalyst by direct hydrogenation of furfural using a “one-pot process”.150Levulinic acid can also be produced from furfural, where the conversion of furfural to LA firstly involves the selective hydrogenation of furfural to form FA, followed by hydrolysis to yield levulinic acid.11 Maldonado et al. used different experimental methods to study the hydrolysis of FA to LA with Amberlyst-15 in the aqueous phase.151 Different reaction pathways were systematically investigated in this process, in which 4,5,5-trihydroxypentan-2-one was considered to be a key intermediate.151 Sulfuric acid was employed to hydrolyze hemicellulose to LA in a bi-phasic reactor consisting of alkylphenol solvents and water.152 The bi-phasic reaction system effectively suppressed the undesirable polymerization reactions and obtained a yield of 72%. Recently, an LA yield greater than 70% was achieved using H-ZSM-5 as a catalyst in an H2O/tetrahydrofuran (THF) biphasic reaction system.153
γ-Butyrolactone (GBL), an important intermediate, is widely used as a fine chemical, medicine, etc. At present, GBL is produced in the petrochemical industry such as the hydrogenation of maleic anhydride. Wang et al. reported a two-step approach for the synthesis of GBL from furfural.154 Firstly, furfural oxidation was conducted to form 2-furanone with HCOOH as the catalyst and H2O2 as the oxidant. Subsequently, the purified 2-furanone was further converted to GBL via hydrogenation reaction. The oxidation products include maleic acid and succinic acid besides 2-furanone, and formic acid was found to give a high yield of 2-furanone of up to 61.5%. It was revealed that the TOF values of supported group VIII metals for the hydrogenation of 2-furanone exhibit a volcano-type correlation with the d-band center relative to the Fermi level, and Pd/SiO2 shows the best performance. This work provides a novel approach for transforming biomass-derived furfural to a useful chemical. It is of great importance to develop heterogeneous catalysts to afford a higher yield of 2-furanone in the first step, and thus decrease its downstream separation cost to realize the large-scale production of GBL in the future.
3. Conclusions and outlook
This review presented a comprehensive overview on the latest advancements in the catalytic valorization of furfural into value-added chemicals through chemo- and bio-catalysis approaches, focusing on the development of catalysts and the reaction mechanisms of furfural conversion. The wide utilization of biomass-derived furfural opens a new avenue to reduce the dependence on oil and fossil resources and provides a sustainable development for mankind. Despite the great progress achieved in the efficient utilization of furfural to produce useful chemicals including furandicarboxylic acid, aromatics, and cyclopentanone, and the fact that some bottlenecks have been addressed and some emerging strategies have been exploited, there are still critical challenges that hinder the utilization of furfural to produce value-added chemicals on a large scale.(1) The bio-catalytic conversion of furfural provides a green protocol to valuable chemicals. Compared to chemo-catalysis, bio-catalysis has some advantages including high product selectivity and environmentally friendly nature, but it has some limitations, such as low productivity and low feedstock concentration, leading to extensive energy consumption for product separation and purification in the downstream processes. Thus, it is urgent to develop some novel processes by integrating bio-catalysis and chemo-catalysis to make full use of their advantages and overcome their drawbacks in the transformation of biomass into useful chemicals.
(2) Significant challenges remain in designing and developing industrially feasible reaction pathways through heterogeneous catalysts and/or bio-catalysts for the conversion of furfural into useful chemicals including diacids, diols, aromatics, and cyclopentanone. To date, most research on the utilization of furfural is still in its infancy, such as the production of furandicarboxylic acid, aromatics, succinic acid, and cyclopentanone. Thus, great efforts need to be further undertaken to meet the commercial-scale application requirements. Recently, artificial intelligence (AI)-driven synthesis has been shown to be a very strong tool in organic synthesis, materials design, etc. This novel technique will remarkably facilitate the screening of excellent catalysts and the development of efficient and economic pathways for the valorization of furfural to value-added chemicals.
(3) In general, the catalytic conversion of furfural to bio-chemicals requires cascade reactions, especially for hydrogenation and hydrodeoxygenation, which are usually conducted under harsh reaction conditions at high temperatures and pressures. These harsh reaction conditions result in the degradation of furfural to produce other by-products or humins, which will increase the production cost of the desired products. In addition, the hydrothermal reaction conditions easily lead to leaching of the active components and even deactivation of solid catalysts. Thus, it is of great significance to develop efficient and robust heterogeneous catalysts for the conversion of furfural. Recently, the photo-, electro-, and photoelectron-catalytic conversion of biomass into bio-fuels and bio-chemicals have attracted increasing attentions due to their mild reaction conditions, high selectivity, and environmentally friendly nature. The development of photoelectrochemical processes can minimize the energy input, while affording high conversion efficiency and selectivity. It can be expected that some innovative reaction pathways and novel valuable bio-chemicals will be yielded through these new techniques.
(4) It is necessary to have an in-depth understanding of the reaction mechanism for further improving the design and catalytic properties of solid catalysts. Besides the identification of the key reaction intermediates, the surface/interface reaction kinetics also need thorough studies. However, it is a significant challenge to in situ dynamically identify and follow the evolution of reaction intermediates, especially under the harsh hydrothermal reaction conditions. Thus, in situ and even operando characterization techniques and theoretical calculation may act as useful tools for this goal, which can propel our understanding of the structure–activity relationship of catalysts and promote the development of high-efficient catalysts for the valorization of furfural.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by open foundation of the Engineering Research Center for Waste Oil Recovery Technology and Equipment, Ministry of Education (KFJJ2019045) and the Key Research & Development Program of Jiangsu Province (BE2020039).References
- R. Luque, L. Herrero-Davila, J. M. Campelo, J. H. Clark, J. M. Hidalgo, D. Luna, J. M. Marinas and A. A. Romero, Energy Environ. Sci., 2008, 1, 542–564 RSC
.
- L. Xiao, Q. Zhang, P. Chen, L. Chen, F. Ding, J. Tang, Y.-J. Li, C.-T. Au and S.-F. Yin, Appl. Catal., B, 2019, 248, 380–387 CrossRef CAS
- Z. Xiang, J. Liang, H. M. Morgan Jr, Y. Liu, H. Mao and Q. Bu, Bioresour. Technol., 2018, 247, 804–811 CrossRef CAS PubMed
- Q. Bu, K. Chen, W. Xie, Y. Liu, M. Cao, X. kong, Q. Chu and H. Mao, Bioresour. Technol., 2019, 291, 121860 CrossRef CAS
- S. Chen, R. Wojcieszak, F. Dumeignil, E. Marceau and S. Royer, Chem. Rev., 2018, 118, 11023–11117 CrossRef CAS
- L. T. Mika, E. Csefalvay and A. Nemeth, Chem. Rev., 2018, 118, 505–613 CrossRef CAS PubMed
- M. Hong, J. Min, S. Wu, J. Li, J. Wang, L. Wei, Z. Ling, K. Li and S. Wang, Appl. Organomet. Chem., 2020, 34, e5411 CrossRef CAS
- D. E. Resasco, S. Sitthisa, J. Faria, T. Prasomsri and M. P. Ruiz, in Solid Waste as a Renewable Resource: Methodologies, Apple Academic Press, 2015, pp. 103–144 Search PubMed
- R. Mariscal, P. Maireles-Torres, M. Ojeda, I. Sadaba and M. Lopez Granados, Energy Environ. Sci., 2016, 9, 1144–1189 RSC
- A. D. K. Deshan, L. Atanda, L. Moghaddam, D. W. Rackemann, J. Beltramini and W. O. S. Doherty, Front. Chem., 2020, 8, 859 Search PubMed
- X. Li, P. Jia and T. Wang, ACS Catal., 2016, 6, 7621–7640 CrossRef CAS
- S. Peleteiro, S. Rivas, J. L. Alonso, V. Santos and J. C. Parajó, Bioresour. Technol., 2016, 202, 181–191 CrossRef CAS PubMed
- C. Xu, E. Paone, D. Rodriguez-Padron, R. Luque and F. Mauriello, Chem. Soc. Rev., 2020, 49, 4273 RSC
- R. Gerardy, D. P. Debecker, J. Estager, P. Luis and J.-C. M. Monbaliu, Chem. Rev., 2020, 120, 7219–7347 CrossRef CAS PubMed
- X. Li, B. Ho, D. S. Lim and Y. Zhang, Green Chem., 2017, 19, 914–918 RSC
- Y. Ni, Z. Bi, H. Su and L. Yan, Green Chem., 2019, 21, 1075–1079 RSC
- H. Xia, H. Hu, S. Xu, K. Xiao and S. Zuo, Biomass Bioenergy, 2018, 108, 426–432 CrossRef CAS
- C. Cheng, X. Zhang, Z. Fu and Z. Yang, J. Phys.: Condens. Matter, 2018, 30, 475201 CrossRef
- H. Guo and G. Yin, J. Phys. Chem. C, 2011, 115, 17516–17522 CrossRef CAS
- N. Alonso-Fagúndez, M. L. Granados, R. Mariscal and M. Ojeda, ChemSusChem, 2012, 5, 1984–1990 CrossRef
- X. Li, B. Ho and Y. Zhang, Green Chem., 2016, 18, 2976–2980 RSC
- N. Alonso-Fagúndez, I. Agirrezabal-Telleria, P. Arias, J. Fierro, R. Mariscal and M. L. Granados, RSC Adv., 2014, 4, 54960–54972 RSC
- Y. Xie, Y. Huang, C. Wu, W. Yuan, Y. Xia, X. Liu and H. Wang, Mol. Catal., 2018, 452, 20–27 CrossRef CAS
- N. Araji, D. D. Madjinza, G. Chatel, A. Moores, F. Jerome and K. D. O. Vigier, Green Chem., 2017, 19, 98–101 RSC
- Y. Huang, C. Wu, W. Yuan, Y. Xia, X. Liu, H. Yang and H. Wang, J. Chin. Chem. Soc., 2017, 64, 786–794 CrossRef CAS
- G. Lv, C. Chen, B. Lu, J. Li, Y. Yang, C. Chen, T. Deng, Y. Zhu and X. Hou, RSC Adv., 2016, 6, 101277–101282 RSC
- T. Sotak, M. Hronec, M. Gal, E. Dobrocka and J. Skriniarova, Catal. Lett., 2017, 147, 2714–2723 CrossRef CAS
- Y. Lou, S. Marinkovic, B. Estrine, W. Qiang and G. Enderlin, ACS Omega, 2020, 5, 2561–2568 CrossRef CAS
- C. V. Nguyen, J. R. Boo, C.-H. Liu, T. Ahamad, S. M. Alshehri, B. M. Matsagar and K. C. W. Wu, Catal. Sci. Technol., 2020, 10, 1498–1506 RSC
- Q. Yu, R. Bai, F. Wang, Q. Zhang, Y. Sun, Y. Zhang, L. Qin, Z. Wang and Z. Yuan, J. Chem. Technol. Biotechnol., 2020, 95, 751–757 CrossRef CAS
- H. Zhang, S. Wang, H. Zhang, J. H. Clark and F. Cao, Green Chem., 2021, 23, 1370–1381 RSC
- T. Yang, W. Li, Q. Liu, M. Su, T. Zhang and J. Ma, Bioresour, 2019, 14, 5025–5044 CAS
- T. Yang, W. Li and A. T. Ogunbiyi, Mol. Catal., 2021, 504, 111488 CrossRef CAS
- M. Rezaei, A. N. Chermahini, H. A. Dabbagh, M. Saraji and A. Shahvar, J. Environ. Chem. Eng., 2019, 7, 102855 CrossRef
- P. M. Malibo, P. R. Makgwane and P. G. Baker, ChemistrySelect, 2020, 5, 6255–6267 CrossRef CAS
- X. Zhao, X. Kong, F. Wang, R. Fang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 10842–10849 CrossRef CAS PubMed
- N. Alonso-Fagundez, M. Lopez Granados, R. Mariscal and M. Ojeda, ChemSusChem, 2012, 5, 1984–1990 CrossRef CAS
- J. Lan, Z. Chen, J. Lin and G. Yin, Green Chem., 2014, 16, 4604 RSC
- X. Li and Y. Zhang, Green Chem., 2016, 18, 643–647 RSC
- N. Alonso-Fagundez, M. Ojeda, R. Mariscal, J. L. G. Fierro and M. Lopez Granados, J. Catal., 2017, 348, 265–275 CrossRef CAS
- X. Li, J. Ko and Y. Zhang, ChemSusChem, 2018, 11, 612–618 CrossRef CAS
- P. Santander, L. Bravo, G. Pecchi and A. Karelovic, Appl. Catal., A, 2020, 595, 117513 CrossRef CAS
- W. Jia, Z. Si, Y. Feng, X. Zhang, X. Zhao, Y. Sun, X. Tang, X. Zeng and L. Lin, ACS Sustainable Chem. Eng., 2020, 8, 7901–7908 CrossRef CAS
- H. Xia, S. Xu, X. Yan and S. Zuo, Fuel Process. Technol., 2016, 152, 140–146 CrossRef CAS
- N. Alonso-Fagundez, M. L. Granados, R. Mariscal and M. Ojeda, ChemSusChem, 2012, 5, 1984–1990 CrossRef CAS PubMed
- N. Alonso-Fagundez, M. Ojeda, R. Mariscal, J. L. G. Fierro and M. L. Granados, J. Catal., 2017, 348, 265–275 CrossRef CAS
- J. Lan, Z. Chen, J. Lin and G. Yin, Green Chem., 2014, 16, 4351–4358 RSC
- X. Li, B. Ho and Y. Zhang, Green Chem., 2016, 18, 2976–2980 RSC
- S. S. Dalli, T. J. Tilaye and S. K. Rakshit, Ind. Eng. Chem. Res., 2017, 56, 10582–10590 CrossRef CAS
- W. Zhu, F. Tao, S. Chen, M. Li, Y. Yang and G. Lv, ACS Sustainable Chem. Eng., 2019, 7, 296–305 CrossRef CAS
- U. Thubsuang, S. Chotirut, K. Nuithitikul, A. Payaka, N. Manmuanpom, T. Chaisuwan and S. Wongkasemjit, J. Colloid Interface Sci., 2020, 565, 96–109 CrossRef CAS PubMed
- D. Y. Murzin, F. Saleem, J. Warna and T. Salmi, Chem. Eng. J., 2020, 382, 122811 CrossRef
- P. Maneechakr and S. Karnjanakom, Energy Convers. Manag., 2017, 154, 299–310 CrossRef CAS
- X. Li, X. Lan and T. Wang, Catal. Today, 2016, 276, 97–104 CrossRef CAS
- H. Choudhary, S. Nishimura and K. Ebitani, Appl. Catal., A, 2013, 458, 55–62 CrossRef CAS
- D. Y. Murzin, E. Bertrand, P. Tolvanen, S. Devyatkov, J. Rahkila, K. Eranen, J. Warna and T. Salmi, Ind. Eng. Chem. Res., 2020, 59, 13516–13527 CrossRef CAS
- M. Douthwaite, X. Huang, S. Iqbal, P. J. Miedziak, G. L. Brett, S. A. Kondrat, J. K. Edwards, M. Sankar, D. W. Knight, D. Bethell and G. J. Hutchings, Catal. Sci. Technol., 2017, 7, 5284–5293 RSC
- Q. Tian, D. Shi and Y. Sha, Molecules, 2008, 13, 948–957 CrossRef CAS PubMed
- G. Papanikolaou, P. Lanzafame, S. Perathoner, G. Centi, D. Cozza, G. Giorgianni, M. Migliori and G. Giordano, Catal. Commun., 2021, 49, 106234 CrossRef
- B. Peng, C.-L. Ma, P.-Q. Zhang, C.-Q. Wu, Z.-W. Wang, A.-T. Li, Y.-C. He and B. Yang, Green Chem., 2019, 21, 5914–5923 RSC
- S.-S. Shi, X.-Y. Zhang, M.-H. Zong, C.-F. Wang and N. Li, Mol. Catal., 2019, 469, 68–74 CrossRef CAS
- T. Knaus, V. Tseliou, L. D. Humphreys, N. S. Scrutton and F. G. Mutti, Green Chem., 2018, 20, 3931–3943 RSC
- G. Bharath, K. Rambabu, A. Hai, F. Banat, S. Rajendran, D. D. Dionysiou and P. L. Show, Fuel, 2021, 290, 119826 CrossRef CAS
- N. K. Gupta, A. Fukuoka and K. Nakajima, ACS Sustainable Chem. Eng., 2018, 6, 3434–3442 CrossRef CAS
- A. Lolli, S. Albonetti, L. Utili, R. Amadori, F. Ospitali, C. Lucarelli and F. Cavani, Appl. Catal., A, 2015, 504, 408–419 CrossRef CAS
- T. Pan, J. Deng, Q. Xu, Y. Zuo, Q.-X. Guo and Y. Fu, ChemSusChem, 2013, 6, 47–50 CrossRef CAS
- A. Banerjee, G. R. Dick, T. Yoshino and M. W. Kanan, Nature, 2016, 531, 215–219 CrossRef CAS
- H. Zhou, H. Xu, X. Wang and Y. Liu, Green Chem., 2019, 21, 2923–2927 RSC
- S. Zhang, J. Lan, Z. Chen, G. Yin and G. Li, ACS Sustainable Chem. Eng., 2017, 5, 9360–9369 CrossRef CAS
- K. A. P. Payne, S. A. Marshall, K. Fisher, M. J. Cliff, D. M. Cannas, C. Yan, D. J. Heyes, D. A. Parker, I. Larrosa and D. Leys, ACS Catal., 2019, 9, 2854–2865 CrossRef CAS PubMed
- M. Chatterjee, T. Ishizaka and H. Kawanami, Green Chem., 2016, 18, 487–496 RSC
- A. Dunbabin, F. Subrizi, J. M. Ward, T. D. Sheppard and H. C. Hailes, Green Chem., 2017, 19, 397–404 RSC
- K. Zhou, B. Chen, X. Zhou, S. Kang, Y. Xu and J. Wei, ChemCatChem, 2019, 11, 5562–5569 CrossRef CAS
- J. He, L. Chen, S. Liu, K. Song, S. Yang and A. Riisager, Green Chem., 2020, 22, 6714–6747 RSC
- C. Dong, H. Wang, H. Du, J. Peng, Y. Cai, S. Guo, J. Zhang, C. Samart and M. Ding, Mol. Catal., 2020, 482, 110755 CrossRef CAS
- X. Chen, Y. Liu and J. Wang, Ind. Eng. Chem. Res., 2020, 59, 17008–17025 CrossRef CAS
- X. Chen, S. Song, H. Li, G. Gozaydin and N. Yan, Acc. Chem. Res., 2021, 54, 1711–1722 CrossRef CAS
- J. J. Martínez, E. Nope, H. Rojas, M. H. Brijaldo, F. Passos and G. Romanelli, J. Mol. Catal. A: Chem., 2014, 392, 235–240 CrossRef
- S. Jiang, W. Ramdani, E. Muller, C. Ma, M. Pera-Titus, F. Jerome and K. De Oliveira Vigiera, ChemSusChem, 2020, 13, 1699–1704 CrossRef CAS
- Q. Wu, M. Jing, Y. Wei, Z. Zhao, X. Zhang, J. Xiong, J. Liu, W. Song and J. Li, Appl. Catal., B, 2019, 244, 628–640 CrossRef CAS
- R. Rao, A. Dandekar, R. Baker and M. Vannice, J. Catal., 1997, 171, 406–419 CrossRef CAS
- D. Liu, D. Zemlyanov, T. Wu, R. J. Lobo-Lapidus, J. A. Dumesic, J. T. Miller and C. L. Marshall, J. Catal., 2013, 299, 336–345 CrossRef CAS
- M. M. Villaverde, N. M. Bertero, T. F. Garetto and A. J. Marchi, Catal. Today, 2013, 213, 87–92 CrossRef CAS
- R. Fang, L. Chen, Z. Shen and Y. Li, Catal. Today, 2021, 368, 217–223 CrossRef CAS
- W. Sun, L. Luo, J. Li, X. Tian, D. Yan and Y. Zhu, Catal. Lett., 2021 DOI:10.1007/s10562-021-03656-y
- J. Wu, C. Liu, Y. Zhu, X. Song, C. Wen, X. Zhang, C. Wang and L. Ma, J. Energy Chem., 2021, 60, 16–24 CrossRef
- K. Fulajtárova, T. Soták, M. Hronec, I. Vávra, E. Dobročka and M. Omastová, Appl. Catal., A, 2015, 502, 78–85 CrossRef
- Q. Yuan, D. Zhang, L. van Haandel, F. Ye, T. Xue, E. J. Hensen and Y. Guan, J. Mol. Catal. A: Chem., 2015, 406, 58–64 CrossRef CAS
- A. Halilu, T. H. Ali, A. Y. Atta, P. Sudarsanam, S. K. Bhargava and S. B. Abd Hamid, Energy Fuels, 2016, 30, 2216–2226 CrossRef CAS
- G. Bagnato, A. Figoli, C. Ursino, F. Galiano and A. Sanna, J. Mater. Chem. A, 2018, 6, 4955–4965 RSC
- C. Wang, Y. Liu, Z. Cui, X. Yu, X. Zhang, Y. Li, Q. Zhang, L. Chen and L. Ma, ACS Sustainable Chem. Eng., 2020, 8, 12944–12955 CrossRef CAS
- A. B. Merlo, V. Vetere, J. F. Ruggera and M. L. Casella, Catal. Commun., 2009, 10, 1665–1669 CrossRef CAS
- Y. Fan, C. Zhuang, S. Li, Y. Wang, X. Zou, X. Liu, W. Huang and G. Zhu, J. Mater. Chem. A, 2021, 9, 1110–1118 RSC
- T. Xiao, P. Yan, K. Li, C. Yang, H. Yu, J. Wang, H. Yin and S. Zhou, Ind. Eng. Chem. Res., 2021, 60, 6078–6088 CrossRef CAS
- K. Yan and A. Chen, Fuel, 2014, 115, 101–108 CrossRef CAS
- T. Wang, A. Hu, H. Wang and Y. Xia, J. Chin. Chem. Soc., 2019, 66, 1610–1618 CrossRef CAS
- Y. Shao, J. Wang, K. Sun, G. Gao, C. Li, L. Zhang, S. Zhang, L. Xu, G. Hu and X. Hu, Renewable Energy, 2021, 170, 1114–1128 CrossRef CAS
- F. Li, S. Jiang, J. Huang, Y. Wang, S. Lu and C. Li, New J. Chem., 2020, 44, 478–486 RSC
- T. Wang, J. Du, Y. Sun, X. Tang, Z.-J. Wei, X. Zeng, S.-J. Liu and L. Lin, Chin. Chem. Lett., 2021, 32, 1186–1190 CrossRef CAS
- P. Hou, M. Ma, P. Zhang, J. Cao, H. Liu, X. Xu, H. Yue, G. Tian and S. Feng, New J. Chem., 2021, 45, 2715–2722 RSC
- F. Li, S. Jiang, Y. Wang, J. Huang and C. Li, Russ. J. Phys. Chem. A, 2021, 95, 68–79 CrossRef
- T. Wang, A. Hu, G. Xu, C. Liu, H. Wang and Y. Xia, Catal. Lett., 2019, 149, 1845–1855 CrossRef CAS
- A. H. Valekar, M. Lee, J. W. Yoon, J. Kwak, D.-Y. Hong, K.-R. Oh, G.-Y. Cha, Y.-U. Kwon, J. Jung, J.-S. Chang and Y. K. Hwang, ACS Catal., 2020, 10, 3720–3732 CrossRef CAS
- J. Hao, L. Han, Y. Sha, X. Yu, H. Liu, X. Ma, Y. Yang, H. Zhou and Q. Liu, Fuel, 2019, 239, 1304–1314 CrossRef CAS
- Y. Wang, D. Zhao, R. Liang, K. S. Triantafyllidis, W. Yang and C. Len, Mol. Catal., 2021, 499, 111199 CrossRef CAS
- J. Sittiwong, S. Boonmark, W. Nunthakitgoson, T. Maihom, C. Wattanakit and J. Limtrakul, Inorg. Chem., 2021, 60, 4860–4868 CrossRef CAS PubMed
- M. Qiu, T. Guo, R. Xi, D. Li and X. Qi, Appl. Catal., A, 2020, 602, 117719 CrossRef CAS
- S. Nishimura, T. Shimura and K. Ebitani, J. Taiwan Inst. Chem. Eng., 2017, 79, 97–102 CrossRef CAS
- F. Li, W. Zhu, S. Jiang, Y. Wang, H. Song and C. Li, Int. J. Hydrogen Energy, 2020, 45, 1981–1990 CrossRef CAS
- P. Panagiotopoulou, N. Martin and D. G. Vlachos, J. Mol. Catal. A: Chem., 2014, 392, 223–228 CrossRef CAS
- A. S. Nagpure, P. Gogoi, N. Lucas and S. V. Chilukuri, Sustainable Energy Fuels, 2020, 4, 3654–3667 RSC
- S. Jiang, J. Huang, Y. Wang, S. Lu, P. Li, C. Li and F. Li, J. Chem. Technol. Biotechnol., 2021, 96, 639–649 CrossRef CAS
- M. Hronec, K. Fulajtárova, T. Liptaj, N. Prónayová and T. Soták, Fuel Process. Technol., 2015, 138, 564–569 CrossRef CAS
- X. Li, Q. Deng, S. Zhou, J. Zou, J. Wang, R. Wang, Z. Zeng and S. Deng, J. Catal., 2019, 378, 201–208 CrossRef CAS
- X. Li, Q. Deng, L. Zhang, J. Wang, R. Wang, Z. Zeng and S. Deng, Appl. Catal., A, 2019, 575, 152–158 CrossRef CAS
- Y. Wang, C. Liu and X. Zhang, Catal. Lett., 2020, 150, 2158–2166 CrossRef CAS
- Y. Yang, Z. Du, Y. Huang, F. Lu, F. Wang, J. Gao and J. Xu, Green Chem., 2013, 15, 1932–1940 RSC
- Y. Wang, S. Sang, W. Zhu, L. Gao and G. Xiao, Chem. Eng. J., 2016, 299, 104–111 CrossRef CAS
- K. Fulajtarova, M. Hronec, T. Liptaj, N. a. Pronayova and T. Sotak, J. Taiwan Inst. Chem. Eng., 2016, 66, 137–142 CrossRef CAS
- N. S. Date, S. E. Kondawar, R. C. Chikate and C. V. Rode, ACS Omega, 2018, 3, 9860–9871 CrossRef CAS
- Y. Zhang, G. Fan, L. Yang and F. Li, Appl. Catal., A, 2018, 561, 117–126 CrossRef CAS
- M. Zhou, J. Li, K. Wang, H. Xia, J. Xu and J. Jiang, Fuel, 2017, 202, 1–11 CrossRef CAS
- P. Pan, W.-Y. Xu, T.-J. Pu, X.-D. Wang, X.-J. Pei, F. Tang and Y.-S. Feng, ChemistrySelect, 2019, 4, 5845–5852 CrossRef CAS
- X. Liu, B. Zhang, B. Fei, X. Chen, J. Zhang and X. Mu, Faraday Discuss., 2017, 202, 79–98 RSC
- R. Fang, H. Liu, R. Luque and Y. Li, Green Chem., 2015, 17, 4183–4188 RSC
- Y. Liu, Z. Chen, X. Wang, Y. Liang, X. Yang and Z. Wang, ACS Sustainable Chem. Eng., 2017, 5, 744–751 CrossRef CAS
- Y. Li, X. Guo, D. Liu, X. Mu, X. Chen and Y. Shi, Catalysts, 2018, 8, 193 CrossRef
- W. Gong, C. Chen, H. Zhang, G. Wang and H. Zhao, ACS Sustainable Chem. Eng., 2018, 6, 14919–14925 CrossRef CAS
- Y. Wang, C. Liu and X. Zhang, Catal. Lett., 2020, 150, 2158–2166 CrossRef CAS
- X.-L. Li, J. Deng, J. Shi, T. Pan, C.-G. Yu, H.-J. Xu and Y. Fu, Green Chem., 2015, 17, 1038–1046 RSC
- B. Ren, C. Zhao, L. Yang, G. Fan and F. Li, Appl. Surf. Sci., 2020, 504, 144364 CrossRef
- Y. Wang, S. Sang, W. Zhu, L. Gao and G. Xiao, Chem. Eng. J., 2016, 299, 104–111 CrossRef CAS
- M. Dohade and P. L. Dhepe, Catal. Sci. Technol., 2018, 8, 5259–5269 RSC
- G.-S. Zhang, M.-M. Zhu, Q. Zhang, Y.-M. Liu, H.-Y. He and Y. Cao, Green Chem., 2016, 18, 2155–2164 RSC
- M. Hronec, K. Fulajtarova, I. Vavra, T. Sotak, E. Dobrocka and M. Micusik, Appl. Catal., B, 2016, 181, 210–219 CrossRef CAS
- Q. Deng, X. Wen and P. Zhang, Catal. Commun., 2019, 126, 5–9 CrossRef CAS
- D. Sun, S. Sato, W. Ueda, A. Primo, H. Garcia and A. Corma, ChemInform, 2016, 47, 2579–2597 Search PubMed
- S. Liu, Y. Amada, M. Tamura, Y. Nakagawa and K. Tomishige, Catal. Sci. Technol., 2014, 4, 2535–2549 RSC
- K. Zhang, X.-L. Li, S.-Y. Chen, H.-J. Xu, J. Deng and Y. Fu, ChemSusChem, 2018, 11, 726–734 CrossRef CAS PubMed
- K. Huang, Z. J. Brentzel, K. J. Barnett, J. A. Dumesic, G. W. Huber and C. T. Maravelias, ACS Sustainable Chem. Eng., 2017, 5, 4699–4706 CrossRef CAS
- W. Xu, H. Wang, X. Liu, J. Ren, Y. Wang and G. Lu, Chem. Commun., 2011, 47, 3924–3926 RSC
- T. Mizugaki, T. Yamakawa, Y. Nagatsu, Z. Maeno, T. Mitsudome, K. Jitsukawa and K. Kaneda, ACS Sustainable Chem. Eng., 2014, 2, 2243–2247 CrossRef CAS
- T. Mizugaki, Y. Nagatsu, K. Togo, Z. Maeno, T. Mitsudome, K. Jitsukawa and K. Kaneda, Green Chem., 2015, 17, 5136–5139 RSC
- Z. Zhang, J. Song and B. Han, Chem. Rev., 2016, 117, 6834–6880 CrossRef
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS
- H. Guo, Z. Qi, Y. Liu, H. Xia, L. Li, Q. Huang, A. Wang and C. Li, Catal. Sci. Technol., 2019, 9, 2144–2151 RSC
- I. Scodeller, S. Mansouri, D. Morvan, E. Muller, K. d. O. Vigier, R. Wischert and F. Jerome, Angew. Chem., Int. Ed., 2018, 57, 10510–10514 CrossRef CAS PubMed
- K. Yan, G. Wu, T. Lafleur and C. Jarvis, Renewable Sustainable Energy Rev., 2014, 38, 663–676 CrossRef CAS
- J. G. Stevens, R. A. Bourne, M. V. Twigg and M. Poliakoff, Angew. Chem., 2010, 122, 9040–9043 CrossRef
- N. S. Biradar, A. M. Hengne, S. N. Birajdar, P. S. Niphadkar, P. N. Joshi and C. V. Rode, ACS Sustainable Chem. Eng., 2013, 2, 272–281 CrossRef
- G. M. G. Maldonado, R. S. Assary, J. Dumesic and L. A. Curtiss, Energy Environ. Sci., 2012, 5, 6981–6989 RSC
- E. I. Gürbüz, S. G. Wettstein and J. A. Dumesic, ChemSusChem, 2012, 5, 383–387 CrossRef PubMed
- M. A. Mellmer, J. M. R. Gallo, D. Martin Alonso and J. A. Dumesic, ACS Catal., 2015, 5, 3354–3359 CrossRef CAS
- X. Li, X. Lan and T. Wang, Green Chem., 2016, 18, 638–642 RSC
| This journal is © The Royal Society of Chemistry 2021 |