
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmplified detection of nucleic acids and proteins using an isothermal proximity CRISPR Cas12a assay†
Yongya
Li
ab,
Hayam
Mansour
bc,
Colton J. F.
Watson
d,
Yanan
Tang
*a,
Adam J.
MacNeil
d and
Feng
Li
 *ab
*ab
aKey Laboratory of Green Chemistry & Technology of Ministry of Education, College of Chemistry, Analytical & Testing Center, Sichuan University, 29 Wangjing Road, Chengdu, Sichuan 610064, China. E-mail: yanantang@scu.edu.cn
bDepartment of Chemistry, Centre for Biotechnology, Brock University, St. Catharines, Ontario L2S 3A1, Canada. E-mail: fli@brocku.ca
cDepartment of Cell Biology, National Research Center, 12622, Egypt
dDepartment of Health Sciences, Brock University, St. Catharines, Ontario L2S 3A1, Canada
First published on 4th January 2021
Abstract
Herein, we describe an isothermal proximity CRISPR Cas12a assay that harnesses the target-induced indiscrimitive single-stranded DNase activity of Cas12a for the quantitative profiling of gene expression at the mRNA level and detection of proteins with high sensitivity and specificity. The target recognition is achieved through proximity binding rather than recognition by CRISPR RNA (crRNA), which allows for flexible assay design. A binding-induced primer extension reaction is used to generate a predesigned CRISPR-targetable sequence as a barcode for further signal amplification. Through this dual amplification protocol, we were able to detect as low as 1 fM target nucleic acid and 100 fM target protein isothermally. The practical applicability of this assay was successfully demonstrated for the temporal profiling of interleukin-6 gene expression during allergen-mediated mast cell activation.
Introduction
Isothermal amplification is playing increasingly important roles in nucleic acid and protein detection and quantification within diverse clinical and biological settings.1–3 Comparing to the classic polymerase chain reaction (PCR), isothermal nucleic acid amplification can be more rapid and efficient but with no need for bulky thermocycling equipment. Therefore, isothermal assays are ideal PCR-alternatives for applications such as rapid screening of disease biomarkers and point-of-care testing (POCT).1–3Recent advances in microbial clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated (CRISPR-Cas) enzymes offer exciting opportunities for developing novel isothermal nucleic acid amplification assays that are both sensitive and specific.4–18 In particular, the combination of recombinase polymerase amplification (RPA) with the unique target-binding induced indiscriminative single-stranded DNase (ssDNase) activity of Cas12 and Cas13 has led to the development of a series of isothermal assays, such as specific high-sensitivity enzymatic reporter unlocking (SHERLOCK)4–6 and DNA endonuclease-targeted CRISPR trans reporter (DETECTR).7 We have also introduced an isothermal plasmonic CRISPR assay by integrating Cas12a with loop-mediated isothermal amplification (LAMP) and plasmonic nanoparticles.8 So far, the detection of each specific genetic marker requires the existence of a protospacer-adjacent motif (PAM) domain and the design of a specific CRISPR RNA (crRNA). Herein, we introduce a new isothermal amplification assay principle, where target recognition is achieved through proximity hybridization17–21 rather than crRNA binding. As such, Cas12a is decoupled from the target sequence and can be used as a universal amplifier for both DNA and RNA sequences with no requirement of PAM domain. Moreover, the proximity binding principle also makes it possible to expand Cas12a-based assays to non-nucleic-acid targets, such as proteins. The practical usefulness of this novel isothermal proximity CRISPR Cas12a assay (iPCCA) was demonstrated by quantitatively profiling the temporal changes of interleukin-6 (IL-6) gene expression during allergen-mediated mast cell activation, as well as the quantification of IL-6 proteins.
Results and discussion
The assay principle is illustrated in Fig. 1. Two hybridization probes P1 and P2 are designed to have a short 6 nt complementary domain (red domain), so that they do not hybridize to each other in the absence of the target. The hybridization of the two probes to the same target brings them into proximity and thus induces the formation of a three-way junction. In the presence of a polymerase, a primer extension reaction occurs and produces a double-stranded DNA (dsDNA) barcode that can be recognized by a pre-designed crRNA. The ssDNase activity of Cas12a is then activated. Amplified fluorescence signal is achieved by adding a short ssDNA substrate labelled with a fluorophore at the 5′ end and a quencher at the 3′ end, respectively. As the generation of the DNA barcode is quantitatively determined by the amount of the original target, quantitative results can be obtained in the form of a critical time τ to reach a threshold fluorescence value (Fig. 2).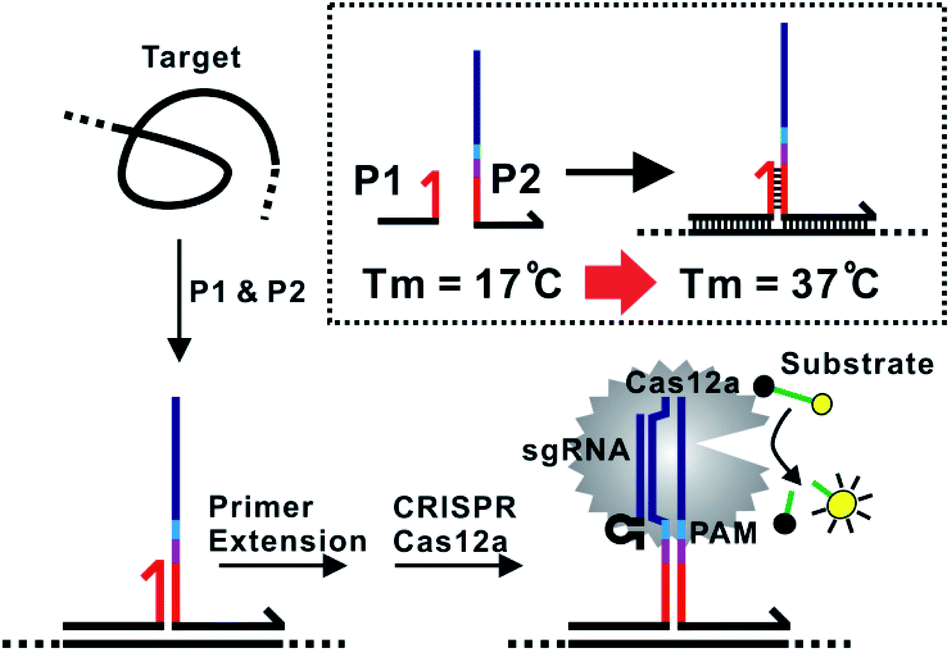 | ||
| Fig. 1 Schematic illustration of the proximity CRISPR Cas12a assay for the amplified detection of nucleic acids. | ||
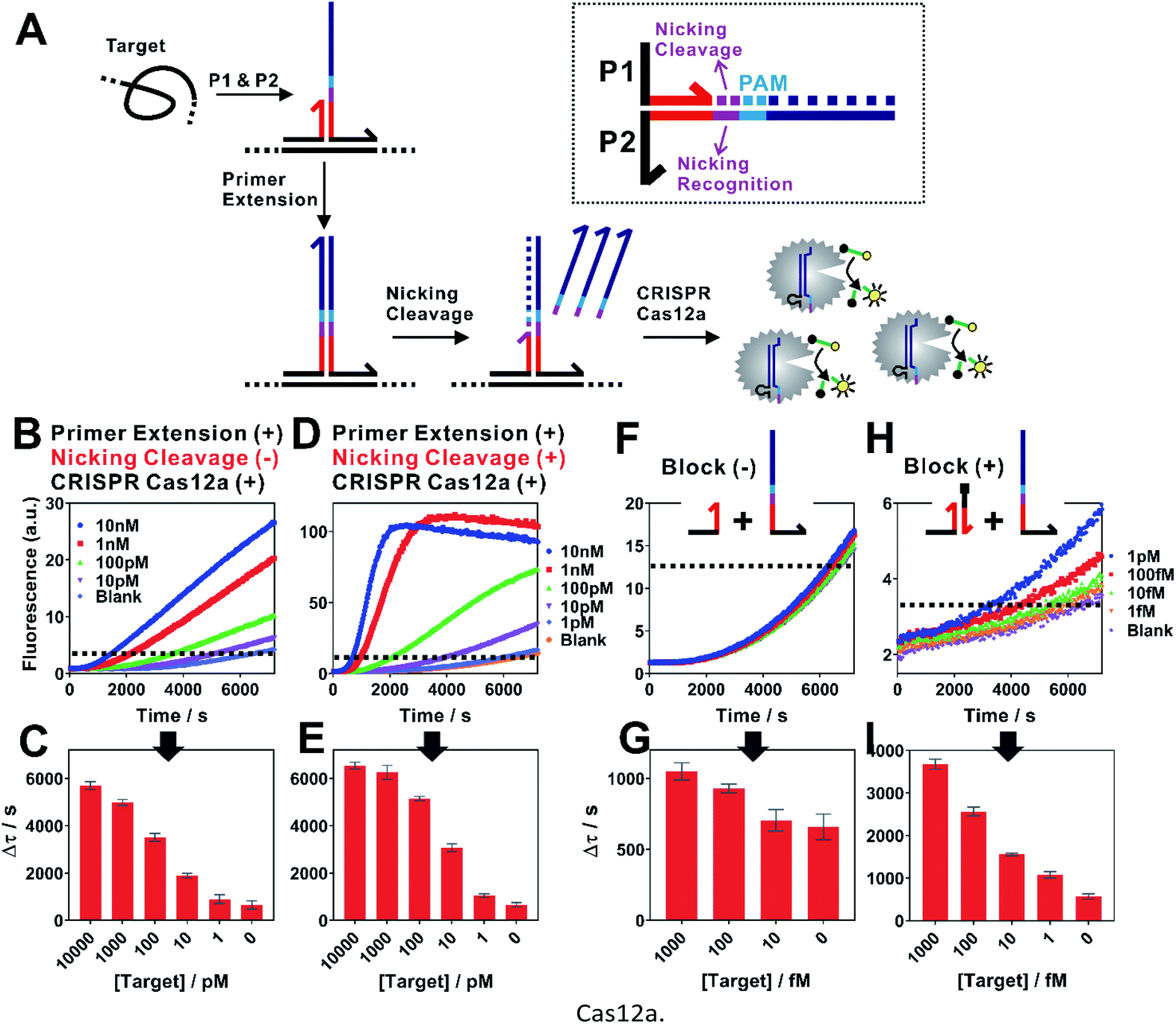 | ||
| Fig. 2 Isothermal proximity CRISPR Cas12a assay for the detection of nucleic acids. (A) Schematic illustration of the integration of proximity CRISPR Cas12a assay with nicking cleavage for further signal amplification. (B and C) Detection of a synthetic nucleic acid target using proximity primer extension followed by CRISPR Cas12a amplification. Once measuring the fluorescence increase in real-time (B), we set a threshold (dashed line) to determine the critical time τ, which is the minimal time to reach the threshold. A calibration curve was then established by plotting Δτ (Δτ = 7200 s − τ) as a function of target concentrations (C). (D and E) Detection of nucleic acid target by integrating the proximity CRISPR Cas12a assay with nicking cleavage. (F and G) The detection of target at concentrations from 1 fM to 1 pM using the proximity CRISPR Cas12a assay integrated with nicking cleavage. To further push the detection limit to lower target concentrations, a blocking DNA was introduced to suppress the background (H and I). Each error bar represents one standard deviation from triplicate analyses. | ||
When examining the performance of iPCCA using a synthetic DNA target, we were able to detect as low as 1 pM target (Fig. 2B and C). The obtained limit of detection (LOD) is comparable with that using direct crRNA recognition and Cas12a cleavage (Fig. S1†), suggesting that the binding-induced primer extension can effectively translate the detection of a target sequence to the production of the DNA barcode for the subsequent Cas12a-mediated amplification.
To improve the assay sensitivity, we further coded the DNA barcode with a nicking cleavage domain (Fig. 2A). Specifically, P2 was designed to contain a nicking recognition domain (purple) adjacent to PAM. Upon primer extension, a sequence that contains both the barcode for Cas12a activation and a nicking cleavage domain was generated. In the presence of a nicking endonuclease, a ssDNA barcode was released through nicking cleavage and a new round of primer extension was triggered. As such, each target could trigger the production of multiple ssDNA barcodes and thus further amplifies the detection signal upon integration with the ssDNase activity of Cas12a.
As shown in Fig. 2D, the dual amplification strategy significantly enhanced fluorescence signals comparing to a single-step Cas12a amplification. Upon optimization (Fig. S2–S4†), we were able to push the LOD to 100 fM (Fig. 2E and G), which corresponds to a 10-times improvement. Meanwhile, the dual amplification also led to higher fluorescence background, making it difficult to further distinguish targets of concentrations lower than 100 fM (Fig. 2F).
We reason that an important source of high fluorescence background is the transient binding between P1 and P2, which was then stabilized by primer extension. To address this issue, we rationally designed a blocking DNA that competitively consumes P1 and thus prevents the transient binding between P1 and P2 (Fig. 2H). Specifically, the blocking DNA was designed to contain the identical short complementary domain as P2 but with an additional 6 nt polydT domain at the 5′ end. As the blocking DNA was supplied in much higher concentration than that of P2, unreacted P1 favoured the binding with the blocking probe, which was then locked through primer extension. In the presence of a target, the target-specific binding brought P1 and P2 into proximity and thus outcompeted the blocking DNA, which allowed the target-specific amplification. Therefore, this blocking strategy was found to effectively reduce the background, allowing the detection of as low as 1 fM target DNA (Fig. 1H and I).
We next challenged practical applicability of iPCCA to real biological samples. One advantage of the proximity recognition mechanism is that the target is not limited to DNA. RNA can also be recognized directly through proximity hybridization without the need for reverse transcription. Therefore, we demonstrated the use of iPCCA for profiling the temporal changes of IL-6 gene expression at the mRNA level during allergen-mediated mast cell activation (Fig. 3). IL-6 has a wide variety of activities on immune cell function and on the replication and differentiation of many cell types.22 It is also a mediator of inflammation and a potential therapeutic target to treat inflammatory disease.23 Therefore, rapid and quantified profiling of IL-6 gene expression during allergic reactions holds great potential for understanding and diagnosing diseases such as mastocytosis, anaphylaxis, and asthma.22–24
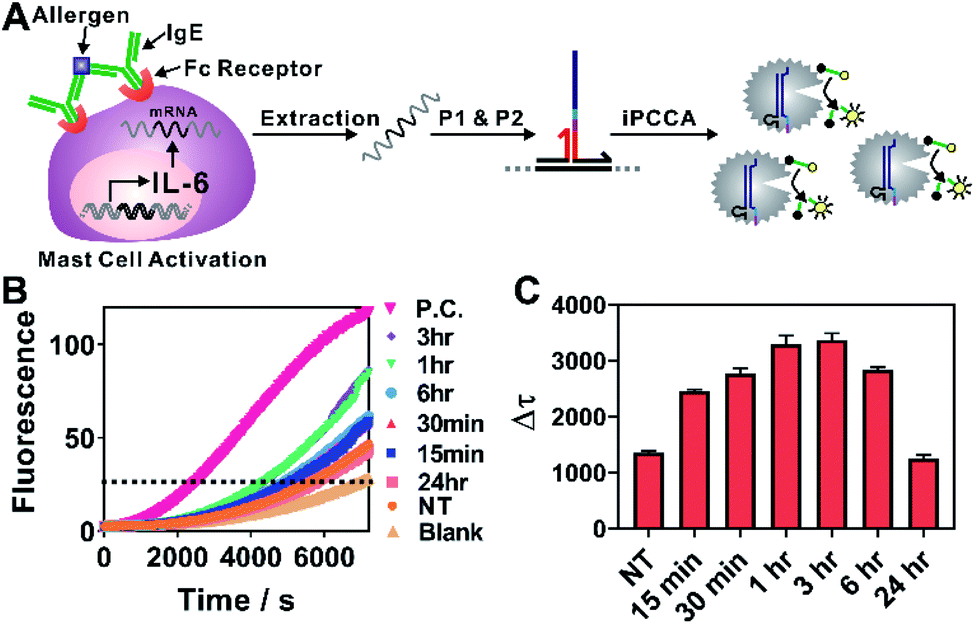 | ||
| Fig. 3 (A) Schematic illustration of quantitative profiling of IL-6 expression during allergen-mediated mast cell activation using iPCCA. (B) Real-time monitoring of the detection of IL-6 mRNA at varying stimulation time points. A threshold was set to determine the critical time τ. Δτ was then determined using Δτ = 7200 s − τ. (C) Δτ was normalized against total RNA for samples collected at each stimulation time point and then plotted to determine the temporal changes of IL-6 gene expression during allergen-mediated mast cell activation. Each error bar represents one standard deviation from triplicate analyses. | ||
As shown in Fig. 3A, the allergen-mediated mast cell activation was achieved by sensitizing bone marrow-derived mast cells (BMMCs) from wild-type C57BL/6 mice with trinitrophenyl (TNP)-specific IgE and then stimulating the cells using TNP-BSA (allergen) and stem cell factor (SCF) (100 ng mL−1) for varying time points (0 min, 15 min, 30 min, 1 h, 3 h, 6 h, and 24 h). Total RNA was then isolated and quantified directly using iPCCA. We observed repeatedly that the expression level of IL-6 gene increased upon mast cell activation, peaking at 1–3 h (Fig. 3B, C and S5†). These observations are highly consistent with our previous observations using reverse transcription and PCR,25,26 suggesting that iPCCA holds the potential to replace PCR as an isothermal alternative for quantifying genetic markers.
As iPCCA relies on proximity binding for target recognition, it holds the potential for detecting non-nucleic-acid target by modifying P1 and P2 with affinity ligands.19–21 To test this hypothesis, we engineered P1 and P2 with polyclonal antibodies that bind to IL-6 protein through well-established streptavidin-biotin conjugation chemistry.27 The two probes, termed as P1′ and P2′, can bind to the same IL-6 protein but at different epitopes. Therefore, the affinity interactions among IL-6 and the two antibodies will bring P1′ and P2′ into proximity and thus trigger subsequent iPCCA for amplification and detection (Fig. 4A). As shown in Fig. 4B and C, we were able to detect as low as 100 fM IL-6 proteins in an isothermal and wash-free manner.
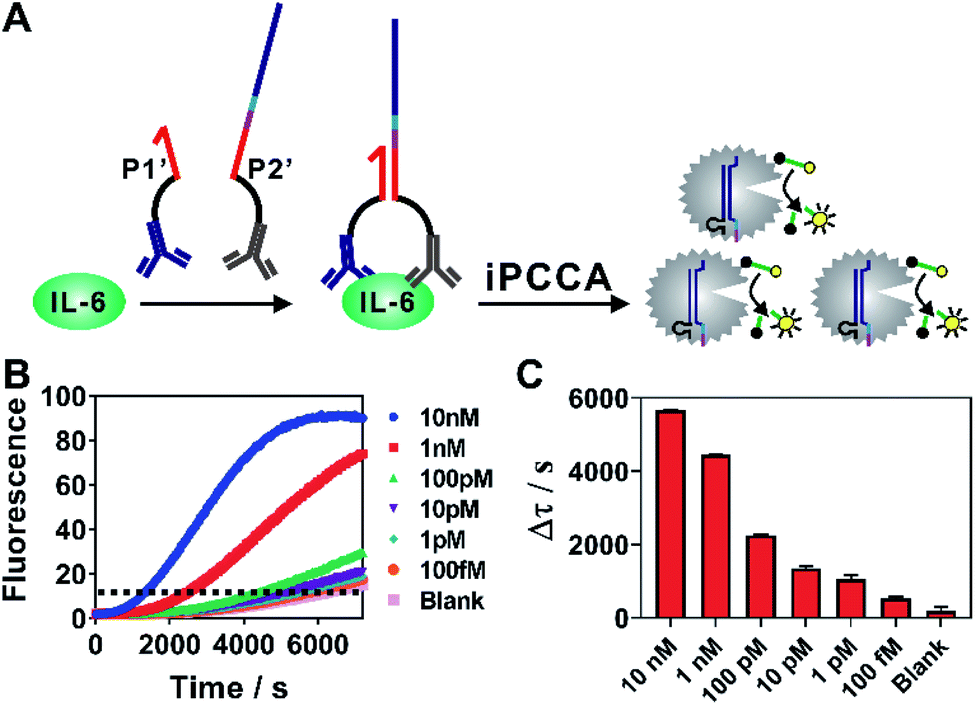 | ||
| Fig. 4 (A) Schematic illustration of iPCCA for amplified detection of IL-6 protein. (B) Real-time monitoring of the detection of IL-6 protein at varying concentrations. A threshold was set to determine the critical time τ. Δτ was then determined using Δτ = 7200 s − τ. (C) Protein quantification using Δτ as a function of IL-6 protein concentrations. Each error bar represents one standard deviation from triplicate analyses. | ||
Conclusions
In conclusion, we have introduced a novel isothermal proximity CRISPR Cas12a assay (iPCCA) capable of detecting genetic markers, levels of gene expression, as well as proteins. Because the target recognition is achieved through proximity binding of hybridization probes to the target sequence, iPCCA enables Cas12a as a universal amplifier for both DNA and RNA targets without the need for a PAM domain. The proximity binding principle also further expands the use of iPCCA to non-nucleic-acid target, such as proteins. Owing the sensitivity, flexibility and simplicity, our iPCCA approach enriches the current toolbox of CRISPR diagnostics (CRISPR Dx) by offering new mechanisms for target recognition and signal amplification and thus holds the potential for wide uses in molecular diagnostics.Materials and methods
Materials
EnGen Lba Cas12a (Cpf1), 10X NEBuffer™ 2.1 buffer, Klenow fragment (3′ → 5′ exo-), 10X NEBuffer™ 2, deoxynucleotide (dNTP) solution mix, nicking endonuclease (Nb.BbvCI) were purchased from New England Biolabs Ltd. (Whitby, ON, Canada). Streptavidin from Streptomyces avidinii and biotin were purchased from Sigma (Oakville, ON, Canada). IL-6 protein and polyclonal anti-IL-6 antibodies were purchased from Thermo fisher Scientific (Mississauga, ON, Canada). Human serum, magnesium chloride hexahydrate (MgCl2·6H2O), and 100× Tris–EDTA (TE, pH 7.4) buffer were purchased from Sigma-Aldrich (Mississauga, ON, Canada). NANOpure H2O (>18.0 MΩ), purified using an Ultrapure Mili-Q water system, was used for all experiments. All DNA samples and the guide RNAs were Integrated DNA Technologies (Coralville, IA) and purified using high-performance liquid chromatography.Nucleic acid detection using iPCCA
For a typical test, a 50 μL reaction mixture contained 5 μL of 100 nM P1, 5 μL of 100 nM P2, 5 μL of 200 nM blocking DNA, 10 μL of varying concentrations of genetic target, 3.3 mmol of dNTPs, 5 units of Klenow fragment and 0.5 unit of nicking endonuclease in 1X NEBuffer™ 2. The solution was incubated at 37 °C for 20 min. A 50 μL enzyme solution which contains 30 nM of Cas12a, 30 nM of gRNA and 60 nM of signal reporter in 1X NEBuffer™ 2.1 was added. Fluorescence was measured immediately after transferring the reaction mixture to a 96-well microplate and kept measuring every 30 s for 2 hours at 37 °C using a SpectraMax i3 multi-mode microplate reader (Molecular Devices) with excitation/emission at 485/515 nm.Mast cell culture and activation
Primary mast cell cultures were established by isolating bone marrow from the tibia and femur of wild-type C57BL/6 mice (Charles River) and inducing differentiation with IL-3 (from Wehi-3b cells, American Type Culture Collection – ATCC) and PGE2 (Sigma) conditioned culture media. The BMMCs were cultured twice weekly by collection, centrifugation, and resuspension of cells in fresh media, with cell viability confirmed by NucBlue stain (Life Technologies, R37605) and cell growth measured on a Countess II FL (Life Technologies Inc., AMQAF1000). Cells were maintained at a density of 0.5 × 106 cells per mL and incubated at 37 °C with 5% carbon dioxide (CO2). For IgE-mediated activation, BMMCs were sensitized overnight with TNP-specific IgE harvested from TIB-141 cells (ATCC) and the following day unbound IgE was washed away with RPMI 1640. Prior to stimulation, the cells were resuspended in RPMI 1640 supplemented with 10% FBS and 1% pen/strep. Cells were then separated for various time points (15 min, 30 min, 1 h, 3 h, 6 h, 24 h) and stimulated with 100 ng mL−1 TNP-BSA (Biosearch Technologies, Novato, CA) under SCF potentiation at 100 ng mL−1 (Peprotech, Dollard des Ormeaux, QC).RNA isolation and IL-6 gene expression analysis using iPCCA
2–3 × 106 BMMCs were sensitized, washed and stimulated as described above. RNA was isolated using the RNeasy Plus kit (Qiagen) according to the manufacturer's suggested protocol. Briefly, at each time point, cells were centrifuged at 1500 rpm for 5 min at 4 °C and cell pellets were lysed in 350 μL of RLT Plus lysis buffer with 10 μL mL−1 2-mercaptoethanol. RNA was eluted in RNase-free water and quantified using NanoDrop. IL-6 mRNA was then measured directly from total RNA by iPCCA using the abovementioned protocol. The final Δτ for each stimulation time point was normalized against the total RNA using the following equation: Δτnormalized = Δτmeasured × [total RNA]/[total RNA]max, where [total RNA] represents the total RNA for each specific sample and [total RNA]max represents the maximum amount of total RNA in the sample series.IL-6 detection using iPCCA
To prepare probes for IL-6 protein, we mixed 25 μL of 2.5 μM biotinylated DNA probes with equal volume of 2.5 μM streptavidin and then incubated the solution at 37 °C for 30 min. We then added 50 μM of 1.25 μM biotinylated IL-6 polyclonal antibodies. The solution was incubated for another 30 min, followed by a dilution to 250 nM with a solution containing 20 mM Tris buffer, 0.01% BSA, and 1 mM biotin. For a typical test, a 50 μL reaction mixture contained 5 μL of 100 nM P1′, 5 μL of 100 nM P2′, 5 μL of 200 nM blocking DNA, 10 μL of varying concentrations of IL-6 protein, 3.3 mmol of dNTPs, 5 units of Klenow fragment and 0.5 unit of nicking endonuclease in 1X NEBuffer™ 2. The solution was incubated at 37 °C for 20 min. A 50 μL enzyme solution which contains 30 nM of Cas12a, 30 nM of gRNA and 60 nM of signal reporter in 1X NEBuffer™ 2.1 was added. Fluorescence was measured immediately after transferring the reaction mixture to a 96-well microplate and kept measuring every 30 s for 2 hours at 37 °C using a SpectraMax i3 multi-mode microplate reader (Molecular Devices) with excitation/emission at 485/515 nm.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The protocols in this study were approved by the Bioscience Research Ethics Board of the Brock University, in accordance with the guidelines of the Canadian Council on Animal Care. We thank the Fundamental Research Funds for the Central Universities (No. YJ201975), the National Natural Science Foundation of China (No. 22074099; No. 22006104), the Natural Sciences and Engineering Research Council of Canada (RGPIN05240), and the Ontario Ministry of Research, Innovation and Science for the financial support.Notes and references
- H. Zhang, F. Li, B. Dever, X.-F. Li and X. C. Le, Chem. Rev., 2013, 113, 2812–2841 CrossRef CAS.
- Y. Zhao, F. Chen, Q. Li, L. Wang and C. Fan, Chem. Rev., 2015, 115, 12491–12545 CrossRef CAS.
- P. Craw and W. Balachandran, Lab Chip, 2012, 12, 2469–2486 RSC.
- J. S. Gootenberg, O. O. Abdauueh, J. W. Lee, P. Essletzbichler, A. J. Dy, J. Joung, V. Verdine, N. Donghia, N. M. Daringer, C. A. Frejie, C. Myhrvold, R. P. Bhattacharyya, J. Livny, A. Regev, E. V. Koonin, D. T. Hing, P. C. Sabeti, J. J. Collions and F. Zhang, Science, 2017, 356, 438–442 CrossRef CAS.
- J. S. Gootenberg, O. O. Abudayyeh, M. J. Kellner, J. Joung, J. J. Collins and F. Zhang, Science, 2018, 360, 439–444 CrossRef CAS.
- C. Myhrvold, C. A. Freije, J. S. Gootenberg, O. O. Abudayyeh, H. C. Metsky, A. F. Durbin, M. J. Kellner, A. L. Tan, L. M. Paul, L. A. Parham, K. F. Garcia, K. G. Barnes, B. CHak, A. Mondini, M. L. Nogueira, S. Isern, S. F. Michael, I. Lorenzana, N. L. Yozwiak, B. L. MacInnis, I. Bosch, L. Gehrke, F. Zhang and P. C. Sabeti, Science, 2018, 360, 444–448 CrossRef CAS.
- J. S. Chen, E. Ma, L. B. Harrington, M. D. Costa, X. Tian, J. M. Palefsky and J. A. Doudna, Science, 2018, 360, 436–439 CrossRef CAS.
- R. Zhou, Y. Li, T. Dong, Y. Tang and F. Li, Chem. Commun., 2020, 56, 3536–3538 RSC.
- S. Y. Li, Q. X. Cheng, J. M. Wang, X. Y. Li, Z. L. Zhang, S. Gao, R. B. Cao, G. P. Zhao and J. Wang, Cell Discovery, 2018, 4, 20 CrossRef.
- K. Pardee, A. A. Green, M. K. Takahashi, D. Braff, G. Lambert, J. W. Lee, T. Ferrante, D. Ma, N. Donghia, M. Fan, N. M. Daringer, I. Bosch, D. M. Dudley, D. H. O'Connor, L. Gehrke and J. J. Collins, Cell, 2016, 165, 1255–1266 CrossRef CAS.
- C. Yuan, T. Tian, J. Sun, M. Hu, X. Wang, E. Xiong, M. Cheng, Y. Bao, W. Lin, J. Jiang, C. Yang, Q. Chen, H. Zhang, H. Wang, X. Wang, X. Deng, X. Liao, Y. Liu, Z. Wang, G. Zhang and X. Zhou, Anal. Chem., 2020, 92, 4029–4037 CrossRef CAS.
- W. Zhou, L. Hu, L. Ying, Z. Zhao, P. K. Chu and X. F. Yu, Nat. Commun., 2018, 9, 5012 CrossRef.
- X. Y. Qiu, L. Y. Zhu, C. S. Zhu, J. X. Ma, T. Hou, X. M. Wu, S. S. Xie, L. Min, D. A. Tan, D. Y. Zhang and L. Zhu, ACS Synth. Biol., 2018, 7, 807–813 CrossRef CAS.
- M. Hu, C. Yuan, T. Tian, X. Wang, J. Sun, E. Xiong and X. Zhou, J. Am. Chem. Soc., 2020, 142, 7506–7513 CrossRef CAS.
- L. B. Harrington, D. Burstein, J. S. Chen, D. Paez-Espino, E. Ma, I. P. Witte, J. C. Cofsky, N. C. Kyrpides, J. F. Banfield and J. A. Doudna, Science, 2018, 362, 839–842 CrossRef CAS.
- Y. Li, X. Teng, K. Zhang, R. Deng and J. Li, Anal. Chem., 2019, 91, 3989–3996 CrossRef CAS.
- H. Gao, K. Zhang, X. Teng and J. Li, Trends Anal. Chem., 2019, 121, 115700 CrossRef CAS.
- K. Zhang, R. Deng, X. Teng, Y. LI, Y. Sun, X. Ren and J. Li, J. Am. Chem. Soc., 2018, 140, 11293–11301 CrossRef CAS.
- S. Fredriksson, M. Gullberg, J. Jarvius, C. Olsson, K. Pietras, S. M. Gustafsdottir, A. Ostman and U. Landegren, Nat. Biotechnol., 2002, 20, 473–477 CrossRef CAS.
- M. Lundberg, A. Eriksson, B. Tran, E. Assarsson and S. Fredriksson, Nucleic Acids Res., 2011, 39, e102 CrossRef CAS.
- F. Li, H. Zhang, Z. Wang, X. Li, X. F. Li and X. C. Le, J. Am. Chem. Soc., 2013, 135, 2443–2446 CrossRef CAS.
- S. Akira, T. Hirano, T. Taga and T. Kishimoto, FASEB J., 1990, 4, 2860–2867 CrossRef CAS.
- M. Hashizume, S.-L. Tan, J. Takano, K. Ohsawa, I. Hasada, A. Hanasaki, I. Ito, M. Mihara and K. Nishida, Int. Rev. Immunol., 2015, 34, 265–279 CrossRef CAS.
- S. F. Stone, C. Cotterell, G. K. Isbister, A. Holdgate and S. G. A. Brown, J. Allergy Clin. Immunol., 2009, 124, 786–792 CrossRef CAS.
- C. J. F. Watson, A. R. R. Maguire, M. M. Rouillard, R. W. E. Crozier, M. Yousef, K. M. Bruton, V. A. Fajardo and A. J. MacNeil, J. Leukocyte Biol., 2020, 107, 649–661 CrossRef CAS.
- T. Dong, H. Mansour, H. Hu, G. A. Wang, C. J. F. Watson, M. Yousef, G. Matamoros, A. L. Sanchez, A. J. MacNeil, P. Wu and F. Li, Anal. Chem., 2020, 92, 6456–6461 CrossRef CAS.
- Y. Tang, Y. Lin, X. Yang, Z. Wang, X. C. Le and F. Li, Anal. Chem., 2015, 87, 8063–8066 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc06113a |
| This journal is © The Royal Society of Chemistry 2021 |