
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of a heterobimetallic actinide nitride and an analysis of its bonding†
Selena L.
Staun
a,
Guang
Wu
a,
Wayne W.
Lukens
 *b and
Trevor W.
Hayton
*a
*b and
Trevor W.
Hayton
*a
aDepartment of Chemistry and Biochemistry, University of California, Santa Barbara, Santa Barbara, California 93106, USA. E-mail: hayton@chem.ucsb.edu
bChemical Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, California 94720, USA. E-mail: wwlukens@lbl.gov
First published on 15th November 2021
Abstract
Reaction of [K(DME)][Th{N(R)(SiMe2CH2)}2(NR2)] (R = SiMe3) with 1 equiv. of [U(NR2)3(NH2)] (1) in THF, in the presence of 18-crown-6, results in formation of a bridged uranium–thorium nitride complex, [K(18-crown-6)(THF)2][(NR2)3UIV(μ-N)ThIV(NR2)3] (2), which can be isolated in 48% yield after work-up. Complex 2 is the first isolable molecular mixed-actinide nitride complex. Also formed in the reaction is the methylene-bridged mixed-actinide nitride, [K(18-crown-6)][K(18-crown-6)(Et2O)2][(NR2)2U(μ-N)(μ–κ2-C,N–CH2SiMe2NR)Th(NR2)2]2 (3), which can be isolated in 34% yield after work-up. Complex 3 is likely generated by deprotonation of a methyl group in 2 by [NR2]−, yielding the new μ-CH2 moiety and HNR2. Reaction of 2 with 0.5 equiv. of I2 results in formation of a UV/ThIV bridged nitride, [(NR2)3UV(μ-N)ThIV(NR2)3] (4), which can be isolated in 42% yield after work-up. The electronic structure of 4 was analyzed with EPR spectroscopy, SQUID magnetometry, and NIR-visible spectroscopy. This analysis demonstrated that the energies of 5f orbitals of 4 are largely determined by the strong ligand field exerted by the nitride ligand.
Introduction
The study of actinide–nitrogen multiple bonding has had a considerable impact on our understanding of covalency and f orbital participation in the actinides.1–7 Notable advances in this area include the discovery of the first terminal actinide nitride by Liddle,8–11 the synthesis of the uranium poly(imido) complexes by Bart,12–14 and the isolation of a trans bis(nitrido) uranium complex by Kraus.15 These studies have revealed the remarkable ability of high valent uranium to engage its 5f orbitals in bonding. Moreover, complexes containing An![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds are known to mediate a variety of important and challenging small molecule transformations.16–18 For example, Mazzanti's U(III) nitride [K3{U(OSi(OtBu)3)3}2(μ-N)] is able to mediate the capture and functionalization of N2.19,20 Likewise, the U(IV) nitride, [Cs{U(OSi(OtBu)3)3}2(μ-N)], can activate both H2 (ref. 21) and CO.22
N bonds are known to mediate a variety of important and challenging small molecule transformations.16–18 For example, Mazzanti's U(III) nitride [K3{U(OSi(OtBu)3)3}2(μ-N)] is able to mediate the capture and functionalization of N2.19,20 Likewise, the U(IV) nitride, [Cs{U(OSi(OtBu)3)3}2(μ-N)], can activate both H2 (ref. 21) and CO.22
Among these complexes, the terminal U(V) nitride, [U(TrenTIPS)(N)]− (TrenTIPS = N(CH2CH2NSiiPr3)3), is especially notable.8,10,11 Its high symmetry and 5f1 electronic configuration enabled a detailed electronic structure analysis via electron paramagnetic resonance (EPR) spectroscopy, magnetic susceptibility measurements, and UV-vis/NIR spectroscopy, anchored by ab initio calculations.8 These studies indicated that the electronic structure of the U(V) nitride is dominated by the U–Nnitride interaction, which strongly destabilizes the fσ and fπ orbitals. In fact, several groups, including ours, have used the favorable properties of the 5f1 configuration to perform similar bonding analyses, which have significantly advanced our understanding of An–L bonding in high-valent systems.23–25 However, widespread access to terminal actinide nitrides has proven elusive.18,26 The majority of reported U(V) nitrides feature bridged nitride ligands that are capped by U(IV) ions,27–30 which complicates the subsequent electronic structure analysis.31 These observations point to the need for new synthetic routes to these materials, which would enable synthetic control over oxidation state, symmetry, and capping group identity. In this regard, we recently reported the synthesis of the first thorium nitride complex, [K(18-crown-6)(THF)2][(R2N)3Th(μ-N)Th(NR2)3] (R = SiMe3) via reaction of the Th metallacycle, [Th{N(R)(SiMe2)CH2}(NR2)2], with NaNH2.32 This complex could also be generated by addition of the parent amide complex, [Th(NR2)3(NH2)], to the Th bis(metallacycle), [K(DME)][Th{N(R)(SiMe2CH2)}2(NR2)]. These reactions are unique because they use NH3 as the nitride source, instead of [N3]−, which is typically the nitrogen source used to form actinide nitrides.16,18,26 Thus, we hypothesized that a similar synthetic approach could be used to generate a uranium nitride with the desired oxidation state and symmetry properties.
Herein, we report the synthesis of the first isolable heterobimetallic actinide nitride complex, [K(18-crown-6)(THF)2][(NR2)3UIV(μ-N)ThIV(NR2)3] (R = SiMe3), formed by reaction of the known thorium bis(metallacycle), [K(DME)][Th{N(R)(SiMe2CH2)}2(NR2)],33 with the uranium parent amide complex, [U(NR2)3(NH2)]. In addition, we explored the redox chemistry of the U(IV)/Th(IV) nitride, which permitted access to the U(V)/Th(IV) nitride, [(NR2)3UV(μ-N)ThIV(NR2)3]. We also report a detailed spectroscopic analysis of [(NR2)3UV(μ-N)ThIV(NR2)3], which was enabled by the 5f1 configuration of its U(V) center and the closed shell configuration of the capping Th(IV) ion.
Results and discussion
Addition of 3 equiv. of NH3, as a 0.4 M solution in THF, to a cold (−25 °C) THF solution of the known uranium metallacycle, [U{N(R)(SiMe2CH2)}(NR2)2] (R = SiMe3),34 results in the formation of [U(NR2)3(NH2)] (1), which can be isolated in 69% yield as dark brown-orange blocks after work-up (eqn (1)).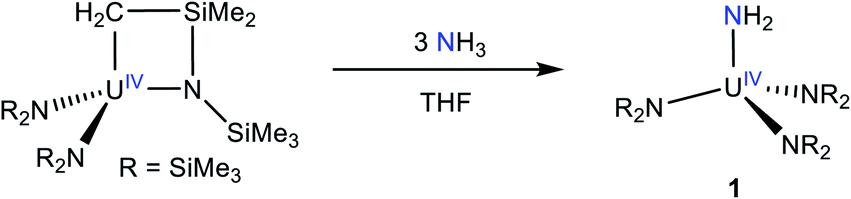 | (1) |
Complex 1 is highly soluble in pentane, benzene, Et2O, and THF. Furthermore, 1 is stable as a C6D6 solution for 24 h with minimal signs a decomposition. The 1H NMR spectrum of 1 in benzene-d6 features a sharp resonance at −2.95 ppm, which is assignable to the SiMe3 groups (Fig. S1†). The protons assignable to the –NH2 ligand could not be located in the 1H NMR spectrum. For comparison, the 1H chemical shifts for the parent amide ligands in [U(NH2)(TrenTIPS)] and [{1,2,4-C5H2tBu3}2U(NH2)2], appear at −5.03 ppm and −34 ppm, respectively.11,35 However, the IR spectrum of 1 features a prominent N–H stretching mode at 3336 cm−1 (Fig. S12†), providing support for the presence of the NH2 ligand. For comparison, this mode is observed at 3321, 3342, and 3364 cm−1 for the isostructural Th, Zr, and Hf analogues, respectively.32,36 Complex 1 was also characterized by X-ray crystallography (see ESI† for more details). It is isomorphous with its Th analogue.32
Previously, we had shown that reaction of the thorium parent amide complex, [Th(NR2)3(NH2)], with the known thorium bis(metallacycle), [K(DME)][Th{N(R)(SiMe2CH2)}2(NR2)], generated the thorium bridged nitride complex, [(NR2)3Th(μ-N)Th(NR2)3].32 Thus, we hypothesized that the reaction of 1 with [K(DME)][Th{N(R)(SiMe2CH2)}2(NR2)],33 would result in formation of a mixed-actinide nitride. Gratifyingly, reaction of 1, 18-crown-6, and [K(DME)][Th{N(R)(SiMe2CH2)}2(NR2)], in THF for 24 h at room temperature does indeed result in formation of the mixed-actinide bridged nitride, [K(18-crown-6)(THF)2][(NR2)3U(μ-N)Th(NR2)3] (2), which can be isolated as pale orange blocks in 48% yield after work-up (Scheme 1). Also formed in the reaction is [K(18-crown-6)][K(18-crown-6)(Et2O)2][(NR2)2U(μ-N)(μ–κ2-C,N–CH2SiMe2NR)Th(NR2)2]2 (3), which can be isolated from the supernatant as golden brown needles in 34% yield (Scheme 1). Significantly, complexes 2 and 3 are the first hetero-bimetallic nitrido complex reported for the actinides and rare examples of hetero-bimetallic actinide complexes of any type. Previously reported hetero-bimetallic actinide complexes include [(R2N)3Th(μ-CC)U(NR2)3],37 [UO2An(H2O)2{CH2(PO3)(PO3H)}2] (An = Th, Np, Pu),38,39 and 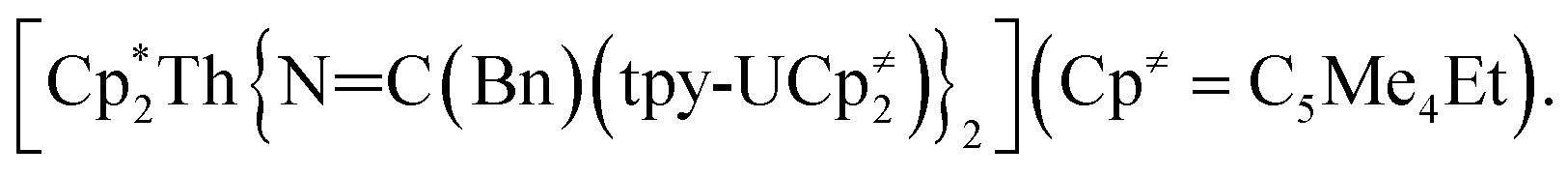 40 In addition, complex 2 is isostructural with the previously reported homo-bimetallic nitrides, [Na(18-crown-6)(Et2O)][(R2N)3Th(μ-N)Th(NR2)3] and [NBu4][(R2N)3U(μ-N)U(NR2)3].30,32
40 In addition, complex 2 is isostructural with the previously reported homo-bimetallic nitrides, [Na(18-crown-6)(Et2O)][(R2N)3Th(μ-N)Th(NR2)3] and [NBu4][(R2N)3U(μ-N)U(NR2)3].30,32
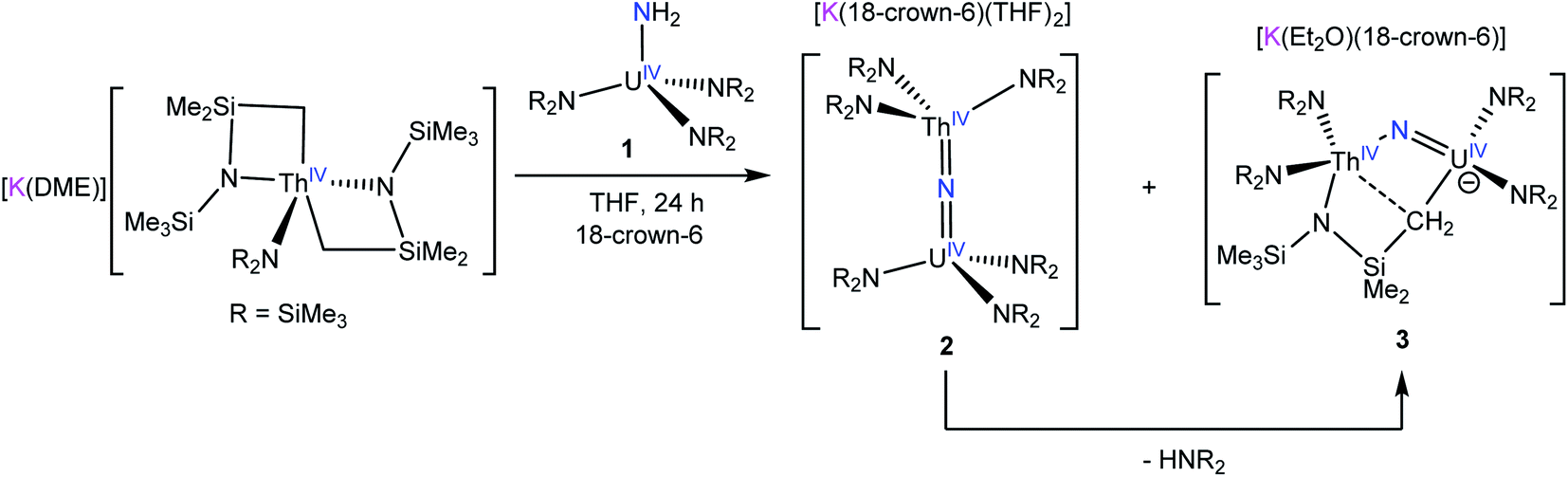 | ||
| Scheme 1 Synthesis of complexes 2 and 3. | ||
Complex 2 crystallizes in the monoclinic space group C2/m (Fig. 1) as a discrete cation/anion pair. Each actinide center features a pseudo-tetrahedral coordination geometry. In addition, the uranium and thorium atoms are mutually disordered over both metal sites in a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 ratio. The An1–Nnitride–An1* linkage is linear (180°), whereas the An1–Nnitride bond length is 2.1037(9) Å (Table 1). For comparison, [Na(18-crown-6)(Et2O)][(R2N)3Th(μ-N)Th(NR2)3] features comparable Th–Nnitride distances (Table 1).32 Similar An–N distances are also observed for [NBu4][(R2N)3U(μ-N)U(NR2)3] (e.g., U–Nnitride = 2.076(5) Å, 2.083(5) Å, and 2.08(2) Å).30 Finally, the An1–Nsilylamide bond lengths are 2.415(7) and 2.440(8) Å, which are similar to the An–Nsilylamide distances observed for [Na(18-crown-6)(Et2O)][(R2N)3Th(μ-N)Th(NR2)3].32 Unfortunately, the disorder extant in the structure of 2 does not allow us to definitively confirm the presence of both Th and U in the molecule; however, our spectroscopic analysis, coupled with the synthesis of 4 from 2, clearly support our hetero-bimetallic formulation (see below for more discussion).
:50 ratio. The An1–Nnitride–An1* linkage is linear (180°), whereas the An1–Nnitride bond length is 2.1037(9) Å (Table 1). For comparison, [Na(18-crown-6)(Et2O)][(R2N)3Th(μ-N)Th(NR2)3] features comparable Th–Nnitride distances (Table 1).32 Similar An–N distances are also observed for [NBu4][(R2N)3U(μ-N)U(NR2)3] (e.g., U–Nnitride = 2.076(5) Å, 2.083(5) Å, and 2.08(2) Å).30 Finally, the An1–Nsilylamide bond lengths are 2.415(7) and 2.440(8) Å, which are similar to the An–Nsilylamide distances observed for [Na(18-crown-6)(Et2O)][(R2N)3Th(μ-N)Th(NR2)3].32 Unfortunately, the disorder extant in the structure of 2 does not allow us to definitively confirm the presence of both Th and U in the molecule; however, our spectroscopic analysis, coupled with the synthesis of 4 from 2, clearly support our hetero-bimetallic formulation (see below for more discussion).
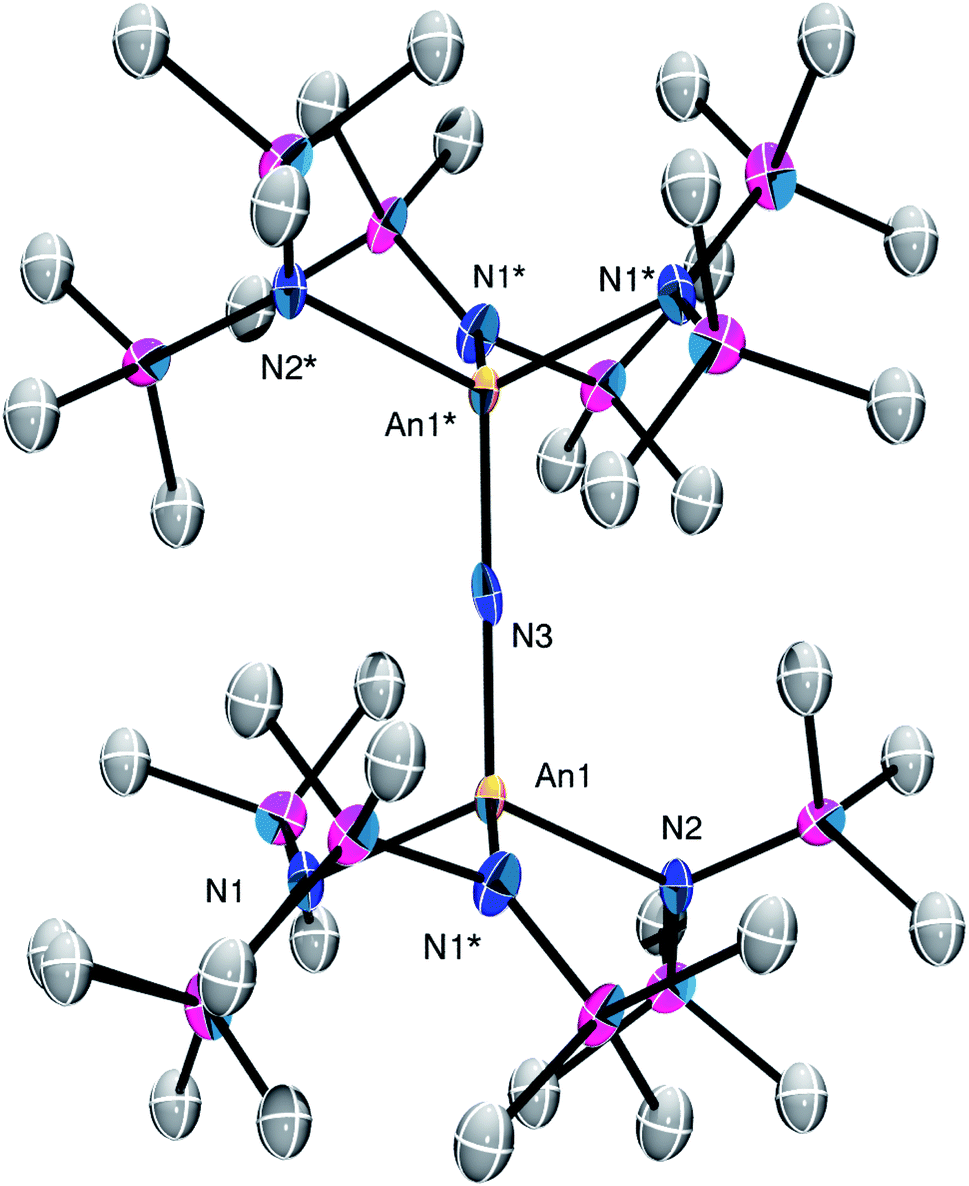 | ||
| Fig. 1 Solid-state molecular structure of 2 shown with 50% probability ellipsoids. [K(18-crown-6)(THF)2]+ counterion and hydrogen atoms are removed for clarity. | ||
| 2 | [(R2N)3Th(μ-N)Th(NR2)3]− | [(R2N)3U(μ-N)U(NR2)3]− | 4·2C5H12 | [(R2N)3U(μ-N)U(NR2)3] | 5Li | |
|---|---|---|---|---|---|---|
| An–Nnitride | 2.1037(9) | 2.14(2) | 2.076(5) | 2.10(1) | 2.080(5) | 1.815(6) |
| 2.11(2) | 2.083(5) | 2.17(1) | 2.150(5) | |||
| 2.08(2) | ||||||
| An–Namide | 2.415(7) | 2.41(1) | 2.366(5) | 2.30(1) | 2.274(4) | 2.341(6) |
| 2.440(8) | 2.41(1) | 2.347(4) | 2.31(1) | 2.271(4) | 2.364(5) | |
| 2.41(1) | 2.340(5) | 2.30(1) | 2.277(4) | 2.346(5) | ||
| 2.41(1) | 2.350(5) | 2.30(1) | 2.268(4) | |||
| 2.41(1) | 2.354(4) | 2.31(1) | 2.272(4) | |||
| 2.40(1) | 2.365(5) | 2.30(1) | 2.283(4) | |||
| 2.338(5) | ||||||
| 2.342(4) | ||||||
| 2.364(5) | ||||||
| An–Namine | 2.665(5) | |||||
| An–N–M | 180 | 179(1) | 178.7(2) | 177.9(6) | 179.4(3) | 172.1(5) |
| 180 | ||||||
| Nnitride–An–Namide | 110.3(1) | 109.9(7) | 113.9(2) | 115.2(4) | 115.0(2) | 113.0(3) |
| 112.7(2) | 109.9(7) | 111.0(2) | 113.9(4) | 112.7(2) | 106.3(2) | |
| 111.7(7) | 111.0(2) | 111.1(4) | 114.5(2) | 111.6(3) | ||
| 110.6(7) | 112.4(2) | 111.9(4) | 114.4(2) | |||
| 110.2(7) | 112.0(2) | 114.8(4) | 113.9(2) | |||
| 109.8(7) | 113.5(2) | 115.0(4) | 114.2(2) | |||
| 111.9(1) | ||||||
| 111.8(1) | ||||||
| 112.8(1) | ||||||
| Namide–An–Namide | 110.4(3) | 108.5(5) | 107.8(2) | 103.4(3) | 104.7(2) | 110.8(2) |
| 106.5(2) | 108.1(5) | 105.9(2) | 106.6(4) | 103.8(2) | 103.5(2) | |
| 108.4(5) | 107.0(2) | 105.9(4) | 105.0(2) | 111.7(2) | ||
| 108.9(5) | 106.3(2) | 105.6(4) | 105.5(2) | |||
| 107.7(5) | 106.6(2) | 105.1(4) | 103.4(2) | |||
| 109.6(5) | 105.5(2) | 103.5(4) | 104.2(2) | |||
| 106.1(2) | ||||||
| 107.0(2) | ||||||
| 106.9(2) |
Complex 2 is insoluble in pentane and benzene, but is soluble in Et2O and THF. It is stable as a THF-d8 solution at room temperature for at least 1 h, showing minimal signs of decomposition over this time. The 1H NMR spectrum of 2 in THF-d8 features a broad resonance at 9.93 ppm, which we have assigned to the Th-bound silylamide ligands. Also present in this spectrum is a sharp singlet at 0.38 ppm, which we attribute to the presence of a small amount of the homobimetallic thorium nitride, [K(solvent)][(R2N)3Th(μ-N)Th(NR2)3].32 On cooling this sample to −10 °C, the broad feature resolves into two resonances at 8.71 ppm and 13.81 ppm, which are assignable to the Th-bound endo and exo SiMe3 groups. Resonances assignable to the U-bound silylamide ligands are not observed in this spectrum, presumably due to paramagnetic broadening. On further cooling to −75 °C, the peaks assignable to 2 resolve into 9 broad resonances (12 resonances are expected), ranging from 23.39 to −98.95 ppm. We also observe signals in this spectrum that are assignable to the homobimetallic uranium nitride, [K(solvent)][(R2N)3U(μ-N)U(NR2)3], as evidenced by broad singlets at −62.27, −37.78, and −14.82 ppm.30 Complexes 2, [(R2N)3U(μ-N)U(NR2)3]−, and [(R2N)3Th(μ-N)Th(NR2)3]− are present in this sample in a ca. 20:3.3:1 ratio (Fig. S2 and S8†), according to the integrations of the methyl resonances in this spectrum. Finally, the UV-vis spectrum of 2 in THF exhibits two weak absorptions at 326 (ε = 260) and 370 nm (ε = 200), which accounts for its pale orange color (Fig. S16†). For comparison, [(R2N)3Th(μ-N)Th(NR2)3]− is colourless, whereas [(R2N)3U(μ-N)U(NR2)3]− is reportedly tan.30,32
Complex 3 crystalizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (Fig. 2). It exhibits a U–Nnitride distance of 2.002(4) Å, a Th–Nnitride distance of 2.160(5) Å, and a U–Nnitride–Th angle of 122.2(2)°. Similar nitride metrical parameters are observed in the isostructural U(IV)/U(IV) analogue, [Na(DME)2(TMEDA)][(NR2)2U(μ-N)(μ-CH2SiMe2NR)U(NR2)2], and are indicative of a localized Th–NU bonding motif.27 Complex 3 also features a μ-CH2 group, formed by deprotonation of a SiMe3 group, which is bound to both metal centers. The U1–C12 distance (2.525(6) Å) is consistent with that expected for a U(IV)–C single bond,41–43 but the Th1–C12 distance (2.962(5) Å) is more indicative of a weak electrostatic interaction. Also present in the lattice are [K(18-crown-6)]+ and [K(18-crown-6)(Et2O)2]+ cations. The [K(18-crown-6)]+ cation forms a bridging interaction between two [(NR2)2U(μ-N)(μ-CH2SiMe2NR)Th(NR2)2]− anions via the methyl groups of their N(SiMe3)2 ligands, whereas the [K(18-crown-6)(Et2O)2]+ cation is a discrete entity. Importantly, the isolation of 3 further supports heterobimetallic formulation of 2, given that 3 is most likely derived from 2 (see below for more discussion), and given that its crystal structure unambiguously supports the presence of both U and Th ions (as evidenced by its disparate U–Nsilylamide and Th–Nsilylamide distances).
(Fig. 2). It exhibits a U–Nnitride distance of 2.002(4) Å, a Th–Nnitride distance of 2.160(5) Å, and a U–Nnitride–Th angle of 122.2(2)°. Similar nitride metrical parameters are observed in the isostructural U(IV)/U(IV) analogue, [Na(DME)2(TMEDA)][(NR2)2U(μ-N)(μ-CH2SiMe2NR)U(NR2)2], and are indicative of a localized Th–NU bonding motif.27 Complex 3 also features a μ-CH2 group, formed by deprotonation of a SiMe3 group, which is bound to both metal centers. The U1–C12 distance (2.525(6) Å) is consistent with that expected for a U(IV)–C single bond,41–43 but the Th1–C12 distance (2.962(5) Å) is more indicative of a weak electrostatic interaction. Also present in the lattice are [K(18-crown-6)]+ and [K(18-crown-6)(Et2O)2]+ cations. The [K(18-crown-6)]+ cation forms a bridging interaction between two [(NR2)2U(μ-N)(μ-CH2SiMe2NR)Th(NR2)2]− anions via the methyl groups of their N(SiMe3)2 ligands, whereas the [K(18-crown-6)(Et2O)2]+ cation is a discrete entity. Importantly, the isolation of 3 further supports heterobimetallic formulation of 2, given that 3 is most likely derived from 2 (see below for more discussion), and given that its crystal structure unambiguously supports the presence of both U and Th ions (as evidenced by its disparate U–Nsilylamide and Th–Nsilylamide distances).
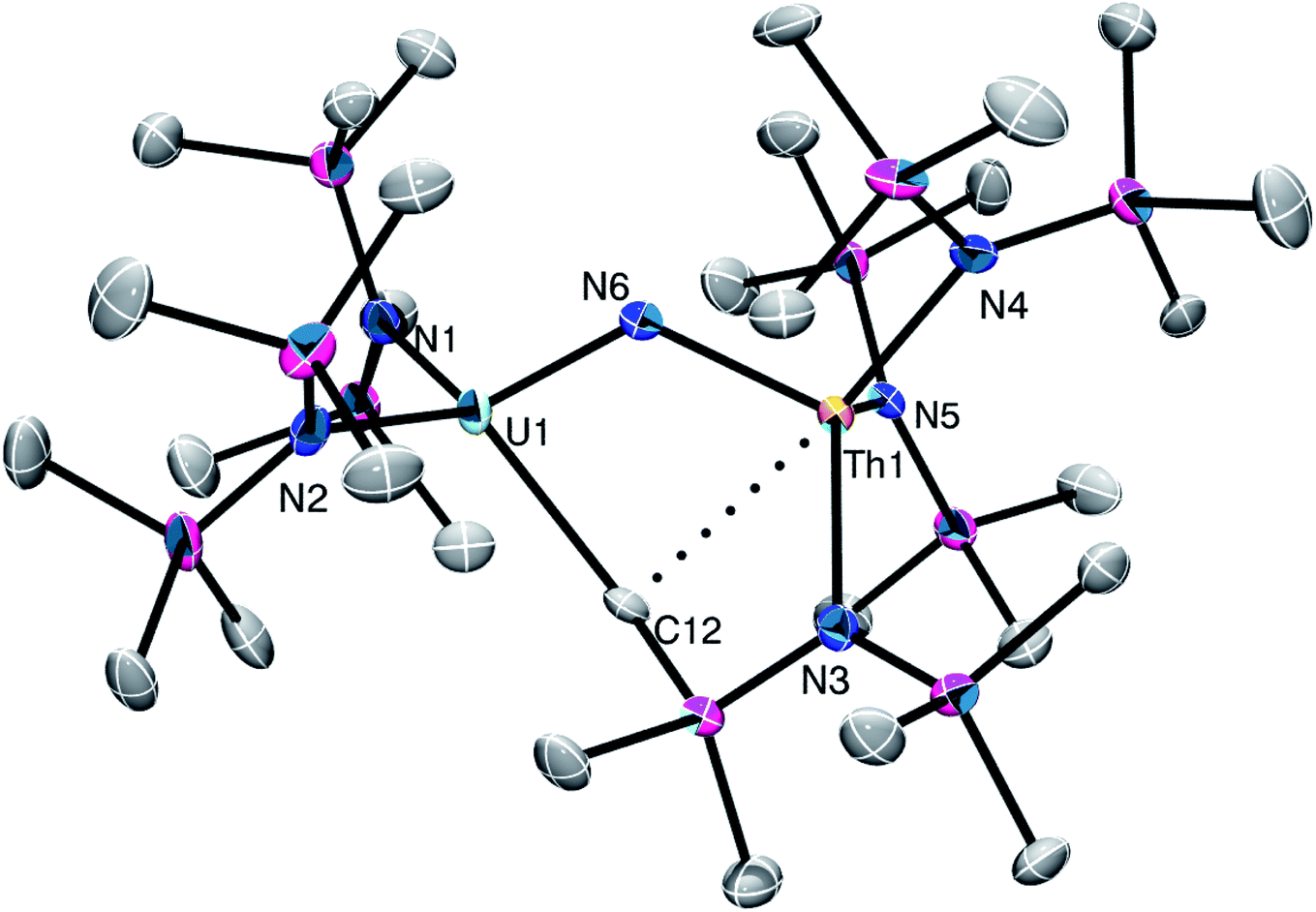 | ||
| Fig. 2 Solid-state molecular structure of 3 shown with 50% probability ellipsoids. [K(18-crown-6)]+ cation, [K(18-crown-6)(Et2O)2]+ cation, and hydrogen atoms removed for clarity. Selected bond lengths (Å) and angles (°): U1–N6 = 2.002(4), Th1–N6 = 2.160(5), U1–N1 = 2.366(5), U1–N2 = 2.349(5), Th1–N3 = 2.389(5), Th1–N4 = 2.395(5), Th1–N5 = 2.408(5), U1–C12 = 2.525(6), Th1–C12 = 2.962(5), U1–N6–Th1 = 122.2(2). | ||
The 1H NMR spectrum of 3 in THF-d8 features two broad resonances at −8.90 ppm and 10.51 ppm, which are assignable to the two expected N(SiMe3)2 environments. Also present in this spectrum are broad resonances at −42.43 and 5.95 ppm, which are assignable to the SiMe2 and SiMe3 environments of the μ-CH2SiMe2NR ligand, respectively. The CH2 environment was not observed, presumably due to paramagnetic broadening. In addition, we observe small amounts of the previously reported homobimetallic uranium bent nitride, [K(solvent)][(NR2)2U(μ-N)(μ-CH2SiMe2NR)U(NR2)2] in the sample,27 as well as small amounts of [K(solvent)][U{N(R)(SiMe2CH2)}2(NR2)] (Fig. S3 and S4†).41 These three species are present in this sample in a ca. 21:3.5:1 ratio.
Complex 2 is formally related to 3via deprotonation of a methyl group by [NR2]−, yielding the μ-CH2 moiety and HNR2. Deprotonation of an N(SiMe3)2 ligand to form the CH2SiMe2NR group is relatively common.27,44–47 Indeed, Mazzanti and co-workers recently demonstrated that [NBu4][(R2N)3U(μ-N)U(NR2)3] converts to [NBu4][(R2N)2U(μ-N)(μ-CH2SiMe2NR)U(NR2)2] on standing at room temperature,30 suggesting that complex 2 is also an intermediate in the formation of 3. The generation of the homobimetallic nitrides, [K(solvent)][(R2N)3Th(μ-N)Th(NR2)3], [K(solvent)][(R2N)3U(μ-N)U(NR2)3], and [K(solvent)][(NR2)2U(μ-N)(μ-CH2SiMe2NR)U(NR2)2], during the synthesis of 2 and 3 is more challenging to explain. To rationalize their formation, we suggest that methylene protonation by complex 1 is reversible, which permits transient formation of [Th(NR2)3(NH2)], along with formation of [K(solvent)][U{N(R)(SiMe2CH2)}2(NR2)]. The latter is observed in the reaction mixture in small quantities. These two species can then react with two starting materials, 1 and [K(DME)][Th{N(R)(SiMe2CH2)}2(NR2)], to generate the homobimetallic products. However, this is a minor reaction pathway, as the two major products from the reaction are the heterobimetallic complexes, 2 and 3, according to our analysis of the 1H NMR spectroscopic data. Consistent with our hypothesis, reaction of 1 with the uranium bis(metallacycle), [Na(DME)][U{N(R)(SiMe2CH2)}2(NR2)],42 in THF-d8, results in formation of the homobimetallic uranium bridged nitride, [K(solvent)][(R2N)3U(μ-N)U(NR2)3] (Fig. S10†). Curiously though, addition of the thorium parent amide, [Th(NR2)3(NH2)], to [Na(DME)][U{N(R)(SiMe2CH2)}2(NR2)] does not result in nitride formation, even upon heating to 50 °C (Fig. S6 and S7†).
We next explored the oxidation of 2, in an effort to generate a U(V)/Th(IV) bridged nitride complex. Thus, addition of 0.5 equiv. of I2 to a cold (−25 °C) THF solution of 2 resulted in formation of [(NR2)3UV(μ-N)ThIV(NR2)3] (4), which could be isolated as dark brown blocks in 42% yield after work-up (eqn (2)). The room temperature 1H NMR spectrum of 4 in THF-d8 features broad singlets at −12.47 ppm and 4.79 ppm, which are assignable to the two unique SiMe3 environments (Fig. S5†). The resonance at −12.47 ppm is substantially broader than that at 4.79 ppm, suggesting that it is assignable to the silylamide ligands attached to the paramagnetic U(V) center. In contrast, the 1H NMR spectrum recorded at −75 °C features nine broad singlets ranging from 47.78 to −39.77 ppm. The number of resonances can be rationalized by assuming restricted rotation of the An–N and N–Si bonds, which would result in 12 unique methyl environments, suggesting that three resonances are too broad to be observed. Similar behavior was observed for [(NR2)3UIV(μ-O)UIV(NR2)3].48 Signals for the homobimetallic uranium nitride, [(NR2)3UV(μ-N)UIV(NR2)3] are also evident in this spectrum (Fig. S9†). Complex 4 and [(NR2)3UV(μ-N)UIV(NR2)3] are present in a 12:1 ratio in this sample.
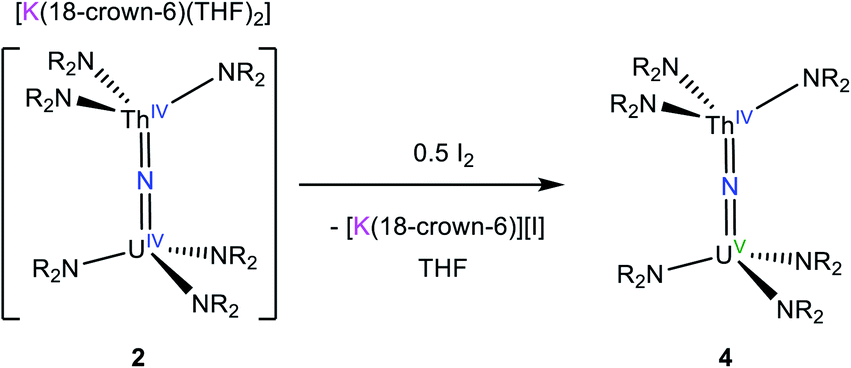 | (2) |
Complex 4 crystalizes in the monoclinic space group P21/n (Fig. 3) as the pentane solvate, 4·2C5H12. In the solid-state, each metal center features a pseudo-tetrahedral coordination geometry. Additionally, the uranium and thorium atoms were refined over both metal sites in a 50:50 ratio. The An–Nnitride bond lengths are 2.10(1) Å and 2.17(1) Å, whereas the average An–Nsilylamido distance is 2.30 Å. The An–Nnitride–An linkage remains linear (177.9(6)°). For comparison, the isostructural U(IV)/U(V) nitride, [(R2N)3U(μ-N)U(NR2)3],30 features comparable An–N bond distances (e.g., 2.080(5) Å and 2.150(5) Å); however, the bridged U(V) nitride, [U(TrenTIPS)(μ-N)Li(12-crown-4)] (5Li) (Chart 1),8 features a much shorter U–Nnitride distance (ca. 1.81 Å), but comparable U–Nsilylamide distances (ca. 2.35 Å). Despite the different nitride metrical parameters, 5Li makes a good point of comparison with 4 because its nitride ligand is capped with a diamagnetic Li+ cation and the U(V) center features identical C3v symmetry (see below for further comparisons).
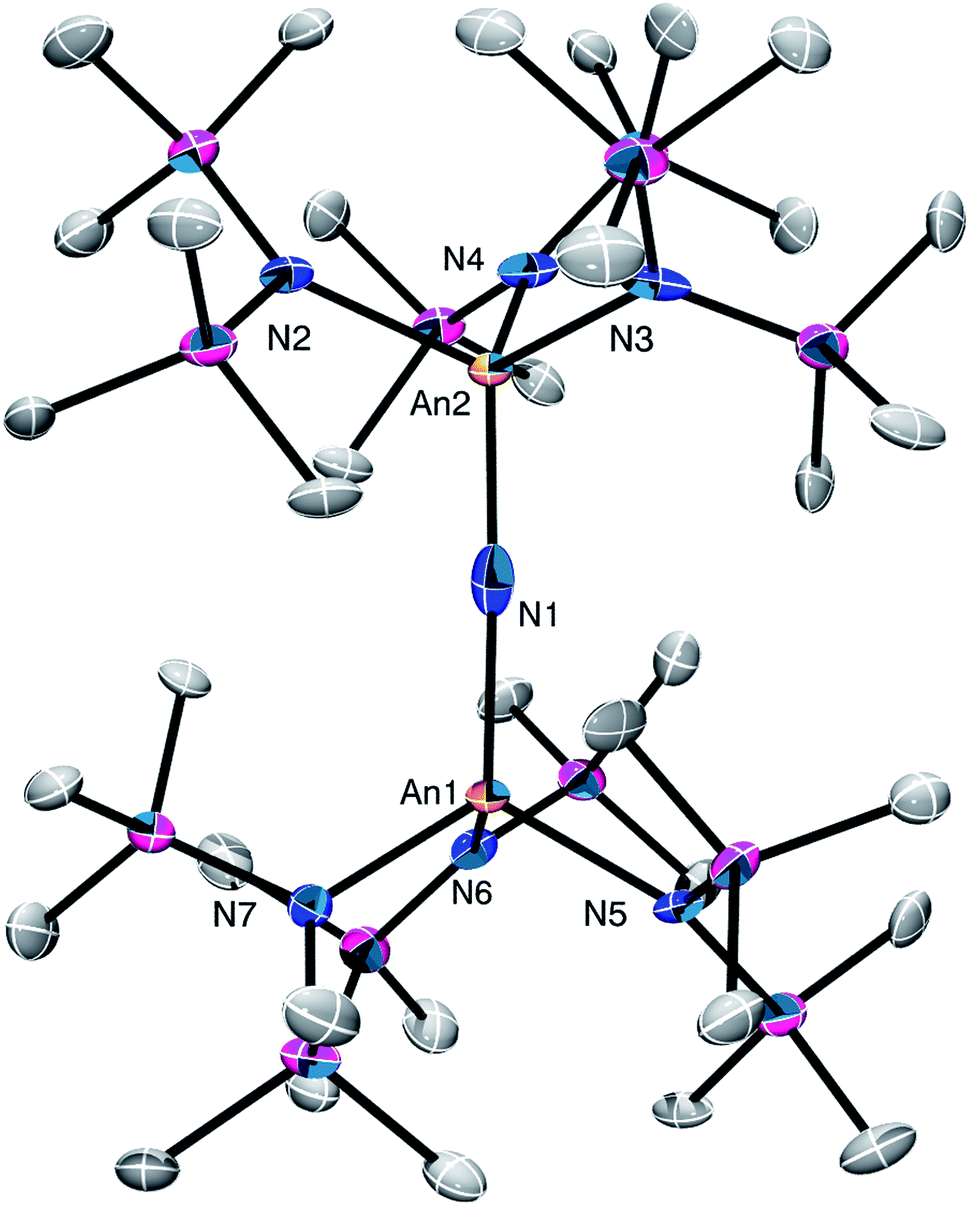 | ||
| Fig. 3 Solid-state molecular structure of 4·2C5H12 shown with 50% probability ellipsoids. Hydrogen atoms removed for clarity. | ||
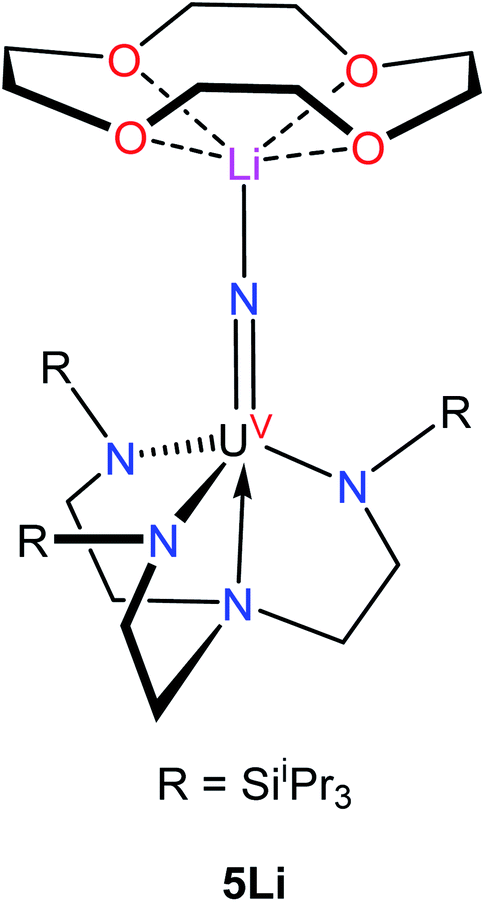 | ||
| Chart 1 | ||
Variable-temperature superconducting quantum interference device (SQUID) magnetometry was performed on a powdered sample of 4 to confirm the oxidation state assignment of the uranium center (Fig. 4, top). The sample was contaminated with a ferromagnetic impurity, which is common for air sensitive complexes due to the ubiquity of ferrites, especially from the surfaces of stainless steel laboratory equipment. The uncorrected magnetic susceptibility of 4 is shown in Fig. S19,† whereas the corrected data are shown in Fig. 4. As is evident from Fig. 4, the data at different fields are in good agreement once corrected for the presence of a ferromagnetic impurity. There are three regions of the susceptibility curve worth noting. Below 50 K, the value of χT decreases sharply with decreasing temperature due to saturation of the magnetization of 4 at high field and low temperature (the magnetization is no longer in the region of the Brillouin function that is linear with respect to the magnetic field). From 50 K to ∼160 K, χT increases linearly with increasing temperature, which indicates that a single state is thermally populated. Above ∼160 K, χT is no longer linear with respect to temperature, which indicates that at least one excited state becomes thermally populated. Complex 4 exhibits a χT value of 0.5 cm3 K mol−1 at 300 K. For comparison, the χT value of 5Li at 300 K is 0.35 cm3 K mol−1. In addition, the slope of χT for 5Li is linear above 160 K (unlike that of 4), and the slope is smaller than that of 4. All of these factors indicate that the lowest lying excited states of 4 are lower in energy than those of 5Li. This difference is presumably a consequence of the slightly different coordination geometries imposed by the –N(SiMe3)2 and TrenTIPS ligands. For further comparison, the χT value of [(R2N)3UV(μ-N)UIV(NR2)3] at 300 K is much higher (ca. 1.9 cm3 K mol−1), consistent with its 5f1/5f2 formulation.30
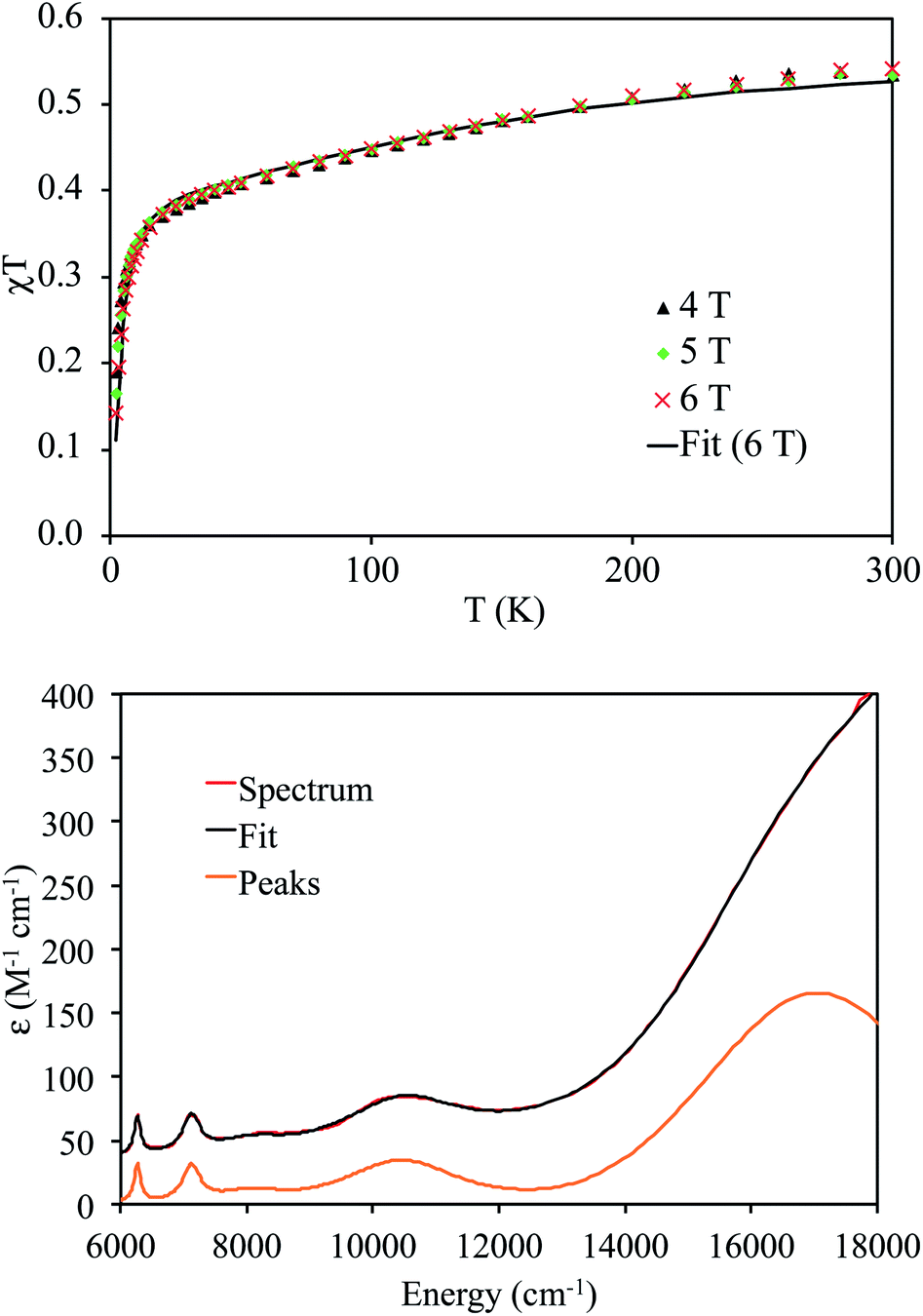 | ||
| Fig. 4 Top: magnetic moment of 4versus temperature with ferromagnetic impurity correction. Bottom: NIR-visible spectrum of 4 (THF solution, 10.44 mM) and the peaks from the fit without the tail due to an absorption at higher energy. | ||
The NIR-visible spectrum of 4 is shown in Fig. 4, along with a fit using pseudo-Voigt peaks (energies are in Table 2). It features three readily apparent peaks, at 6264, 7122, and 10455 cm−1. A fourth peak at ca. 17000 cm−1 is seen as a poorly resolved shoulder on the tail of a more intense peak at higher energy. The latter peak is presumably due to ligand-to-metal charge transfer. A small peak around 8000 cm−1 is also present; however, this peak is too weak to be assigned to 4. The spectrum is similar to that recorded for 5Li, although there are some qualitative differences. In particular, the highest energy peak in 5Li is higher in energy than that of 4 (Table 2), while the corresponding energies of the other peaks are slightly higher in energy for 4vs.5Li. These differences are a consequence of the stronger U–Nnitride bond in 5Li, as will be discussed in more detail below. We also recorded an EPR spectrum of a powdered sample of 4 at 4 K. This spectrum displays a single peak at 3.58, which is assigned to g‖ (Fig. S21†). The other g value, which would be assignable to g⊥, likely resides below 0.7 and is outside the range of the spectrometer. Complex 5Li features a similar g‖ value (3.74), which indicates that the electronic ground state is similar.8
| Parameter | Data | Calculation | 5Li |
|---|---|---|---|
| a g‖ was not used in fitting. | |||
| χ (10 K) (SI) | 4.15 × 10−5 | 4.25 × 10−5 | |
| χ (50 K) (SI) | 1.03 × 10−5 | 1.04 × 10−5 | |
| χ (100 K) (SI) | 5.64 × 10−6 | 5.66 × 10−6 | |
| χ (200 K) (SI) | 3.20 × 10−6 | 3.14 × 10−6 | |
| χ (300 K) (SI) | 2.27 × 10−6 | 2.19 × 10−6 | |
| Peak 1 (cm−1) | 17057 |
17000 |
18000 |
| Peak 2 (cm−1) | 10455 |
10029 |
8900 |
| Peak 3 (cm−1) | 7122 | 7365 | 6900 |
| Peak 4 (cm−1) | 6264 | 6287 | 6060 |
| |g‖|a | 3.58 | 3.52 | 3.74 |
The magnetic susceptibility measurements and excited state energies in Table 2 were fit with a crystal field model in the program CONDON 3.0,50 using the crystal field splitting and spin–orbit coupling framework developed previously by Liddle and co-workers for their series of closely-related U(V) nitrides.8 The results are given in Table 3 and the calculated magnetic susceptibility is shown in Fig. 4. The value of g‖ was not included in the model and was calculated from the ground state wavefunction as previously done for cerium double nitrate by Judd.51 The calculated value of g‖ is in good agreement with the observed value. The calculated values of the magnetic susceptibility and the energies of the f–f transitions are also in good agreement with the measured values.
The energies and compositions of the 5f states of 4 are given in Table 4, along with energies and compositions of the 5f orbitals. The energies of the analogous 5f states in 5Li are also given in Table 4.8 Not surprisingly, given their similar molecular structures, the electronic structures of 4 and 5Li are similar and are largely determined by the strong U–Nnitride interaction. In addition, we find that 4 has a very similar ground state to 5Li, which is primarily mJ = 5/2, and that the first excited state is primarily mJ = 3/2 in both complexes. The differences in U–Nnitride bonding are most clearly seen in the energy of the highest 5f excited state, which is ca. 1000 cm−1 greater in 5Livs.4. Since this excited state is primarily 5f-Nnitride σ-antibonding, the greater energy in 5Li is consistent with a stronger interaction between its nitride ligand and uranium center, which is presumably a consequence of its shorter U–Nnitride distance.
| States | Orbitals | |||
|---|---|---|---|---|
| |Wavefunction|2 as Σ|mJ〉a | 4 (cm−1) | 5Li (cm−1)8 | |Wavefunction|2 | cm−1 |
| a Only one of the Kramers doublets is shown. The other has the same coefficients, but mJ has the opposite sign. | ||||
| 0.88|5/2〉 + 0.10|−1/2〉 + 0.02|−7/2〉 | 0 | 0 | 0.87fϕ + 0.13fσ | 0 |
| |3/2〉 | 426 | Not observed | fδ (degenerate) | 635 |
| 0.36|5/2〉 + 0.37|−1/2〉 + 0.26|−7/2〉 | 4848 | 4700 | fϕ | 2656 |
| 0.70|5/2〉 + 0.19|−1/2〉 + 0.11|−7/2〉 | 6269 | 6060 | fπ (degenerate) | 5985 |
| 0.50|−1/2〉 + 0.50|−7/2〉 | 7291 | 6900 | 0.13fϕ + 0.87fσ | 13756 |
| |3/2〉 | 9816 | 8900 | ||
| 0.05|5/2〉 + 0.84|−1/2〉 + 0.11|−7/2〉 | 16852 |
18000 |
||
The effect of the ligands on the uranium center in 4 is more easily seen in the energies of the orbitals (Table 4) rather than the energies of the 5f states, the latter of which include the effect of spin–orbit coupling. The orbital energies of 4 were determined by setting the spin–orbit coupling to a small value and performing the calculation with the same crystal field parameters (Bqk). Unlike the 5f states, in which |mJ〉 are extensively mixed by the crystal field and spin–orbit coupling, the f-orbitals in 4 show little mixing as a result of the interactions with the ligands. For example, the fδ and fπ orbitals are mixed by the crystal field, but the magnitude is less than 1%. The 5f orbital interactions in 4 are similar to those of 5Li with some differences arising from the slightly different geometries imposed by the supporting ligands. In both complexes, the nitride ligands strongly destabilize the fσ and fπ orbitals. However, in 4, the highest energy f-orbital is a mixture of fσ and fϕ orbitals due to the pseudo-tetrahedral symmetry about the U(V) center (the relationship between tetrahedral symmetry and the structure of 4 is illustrated in Fig. S22†). As a result, it is destabilized by σ interactions with both the nitride and the N(SiMe3)2 ligands.
Conclusions
We have synthesized and characterized a novel hetero-bimetallic actinide nitride complex, [(NR2)3UV(μ-N)ThIV(NR2)3] (4). This complex was characterized by a variety of techniques, including EPR spectroscopy, SQUID magnetometry, and NIR-visible spectroscopy. A crystal field analysis of 4 reveals a predominantly mJ = 5/2 ground state. Moreover, the highest energy 5f excited state is primarily 5f-Nnitride σ-antibonding in character, in accord with the strong ligand field expected for the nitride ligand. Both findings are consistent with an orbital picture that is dominated by the uranium-nitride interaction. Importantly, this analysis was enabled by the 5f1 electronic configuration of the U(V) center in 4, coupled with the overall C3v symmetry and closed-shell configuration of the capping Th(IV) ion, demonstrating the important role that synthesis can play in advancing our understanding of An–L bonding.Going forward, we plan to target the synthesis of a hetero-bimetallic transuranic nitride, which we could potentially access using the same synthetic methodology used to generate complex 4. Transuranic nitride complexes are currently unknown, but their isolation would greatly advance non-aqueous transuranic coordination chemistry and provide excellent benchmarking opportunities for theory.52–57 Our recent synthesis of a Np bis(metallacycle), [Na(DME)3][Np{N(R)(SiMe2CH2)}2(NR2)],58 could be particularly useful in this regard.
Data availability
Raw experimental data will be made available free of charge upon request.Author contributions
S. L. S., W. W. L., and T. W. H. performed various portions of the synthesis, characterization, and data analysis. G. W. assisted with the crystallography. All authors jointly wrote the manuscript. W. W. L. and T. W. H. secured funding for the research.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by the U.S. Department of Energy, Office of Basic Energy Sciences, Chemical Sciences, Biosciences, and Geosciences Division, under contract DE-SC0001861. The MRL Shared Experimental Facilities are supported by the MRSEC Program of the NSF under award no. DMR 1720256; a member of the NSF-funded Materials Research Facilities Network (www.mrfn.org). Measurement of magnetic susceptibility and analysis of the electronic structure of 4 was supported by U.S. Department of Energy, Basic Energy Sciences, Chemical Sciences, Biosciences, and Geosciences Division, Heavy Element Chemistry Program and was performed at Lawrence Berkeley National Laboratory under contract no. DE-AC02-05CH11231. WWL gratefully thanks Jan van Leusen for detailed instructions on using CONDON 3.0. We also thank Shamon Walker (UCSB MRL) for help with the EPR measurements.References
- L. Barluzzi, R. Scopelliti and M. Mazzanti, J. Am. Chem. Soc., 2020, 142, 19047–19051 CrossRef CAS PubMed.
- D.-C. Sergentu, G. T. Kent, S. L. Staun, X. Yu, H. Cho, J. Autschbach and T. W. Hayton, Inorg. Chem., 2020, 59, 10138–10145 CrossRef CAS PubMed.
- R. G. Denning, J. Phys. Chem. A, 2007, 111, 4125–4143 CrossRef CAS PubMed.
- T. W. Hayton, J. M. Boncella, B. L. Scott, E. R. Batista and P. J. Hay, J. Am. Chem. Soc., 2006, 128, 10549–10559 CrossRef CAS PubMed.
- T. W. Hayton, J. M. Boncella, B. L. Scott and E. R. Batista, J. Am. Chem. Soc., 2006, 128, 12622–12623 CrossRef CAS PubMed.
- T. W. Hayton, J. M. Boncella, B. L. Scott, P. D. Palmer, E. R. Batista and P. J. Hay, Science, 2005, 310, 1941–1943 CrossRef CAS PubMed.
- L. Chatelain, R. Scopelliti and M. Mazzanti, J. Am. Chem. Soc., 2016, 138, 1784–1787 CrossRef CAS PubMed.
- D. M. King, P. A. Cleaves, A. J. Wooles, B. M. Gardner, N. F. Chilton, F. Tuna, W. Lewis, E. J. L. McInnes and S. T. Liddle, Nat. Commun., 2016, 7, 13773 CrossRef CAS PubMed.
- D. M. King and S. T. Liddle, Coord. Chem. Rev., 2014, 266–267, 2–15 CrossRef CAS.
- D. M. King, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Science, 2012, 337, 717 CrossRef CAS PubMed.
- D. M. King, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Nat. Chem., 2013, 5, 482 CrossRef CAS PubMed.
- N. H. Anderson, S. O. Odoh, Y. Yao, U. J. Williams, B. A. Schaefer, J. J. Kiernicki, A. J. Lewis, M. D. Goshert, P. E. Fanwick, E. J. Schelter, J. R. Walensky, L. Gagliardi and S. C. Bart, Nat. Chem., 2014, 6, 919–926 CrossRef CAS PubMed.
- N. H. Anderson, H. Yin, J. J. Kiernicki, P. E. Fanwick, E. J. Schelter and S. C. Bart, Angew. Chem., Int. Ed., 2015, 54, 9386–9389 CrossRef CAS PubMed.
- N. H. Anderson, J. Xie, D. Ray, M. Zeller, L. Gagliardi and S. C. Bart, Nat. Chem., 2017, 9, 850 CrossRef CAS PubMed.
- S. S. Rudel, H. L. Deubner, M. Müller, A. J. Karttunen and F. Kraus, Nat. Chem., 2020, 12, 962–967 CrossRef CAS PubMed.
- L. Barluzzi, F.-C. Hsueh, R. Scopelliti, B. E. Atkinson, N. Kaltsoyannis and M. Mazzanti, Chem. Sci., 2021, 12, 8096–8104 RSC.
- L. Chatelain, E. Louyriac, I. Douair, E. Lu, F. Tuna, A. J. Wooles, B. M. Gardner, L. Maron and S. T. Liddle, Nat. Commun., 2020, 11, 337 CrossRef CAS PubMed.
- M. Yadav, A. Metta-Magaña and S. Fortier, Chem. Sci., 2020, 11, 2381 RSC.
- M. Falcone, L. Chatelain, R. Scopelliti, I. Živković and M. Mazzanti, Nature, 2017, 547, 332 CrossRef CAS PubMed.
- M. Falcone, L. Barluzzi, J. Andrez, F. F. Tirani, I. Zivkovic, A. Fabrizio, C. Corminboeuf, K. Severin and M. Mazzanti, Nat. Chem., 2019, 11, 154–160 CrossRef CAS PubMed.
- M. Falcone, L. N. Poon, F. F. Tirani and M. Mazzanti, Angew. Chem., Int. Ed., 2018, 57, 3697–3700 CrossRef CAS PubMed.
- M. Falcone, C. E. Kefalidis, R. Scopelliti, L. Maron and M. Mazzanti, Angew. Chem., Int. Ed., 2016, 55, 12290–12294 CrossRef CAS PubMed.
- N. Edelstein, D. Brown and B. Whittaker, Inorg. Chem., 1974, 13, 563–567 CrossRef CAS.
- W. W. Lukens, N. M. Edelstein, N. Magnani, T. W. Hayton, S. Fortier and L. A. Seaman, J. Am. Chem. Soc., 2013, 135, 10742–10754 CrossRef CAS PubMed.
- L. A. Seaman, G. Wu, N. Edelstein, W. W. Lukens, N. Magnani and T. W. Hayton, J. Am. Chem. Soc., 2012, 134, 4931–4940 CrossRef CAS PubMed.
- M. A. Boreen, G. Rao, D. G. Villarreal, F. A. Watt, R. D. Britt, S. Hohloch and J. Arnold, Chem. Commun., 2020, 56, 4535–4538 RSC.
- S. Fortier, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2010, 132, 6888–6889 CrossRef CAS PubMed.
- A. R. Fox, P. L. Arnold and C. C. Cummins, J. Am. Chem. Soc., 2010, 132, 3250–3251 CrossRef CAS PubMed.
- I. Korobkov, S. Gambarotta and G. P. A. Yap, Angew. Chem., Int. Ed., 2002, 41, 3433–3436 CrossRef CAS PubMed.
- C. T. Palumbo, L. Barluzzi, R. Scopelliti, I. Zivkovic, A. Fabrizio, C. Corminboeuf and M. Mazzanti, Chem. Sci., 2019, 10, 8840–8849 RSC.
- C. R. Graves and J. L. Kiplinger, Chem. Commun., 2009, 3831–3853 RSC.
- S. L. Staun, D.-C. Sergentu, G. Wu, J. Autschbach and T. W. Hayton, Chem. Sci., 2019, 10, 6431–6436 RSC.
- N. L. Bell, L. Maron and P. L. Arnold, J. Am. Chem. Soc., 2015, 137, 10492–10495 CrossRef CAS PubMed.
- S. J. Simpson, H. W. Turner and R. A. Andersen, Inorg. Chem., 1981, 20, 2991–2995 CrossRef CAS.
- G. Zi, L. L. Blosch, L. Jia and R. A. Andersen, Organometallics, 2005, 24, 4602–4612 CrossRef CAS.
- X. Yu and Z.-L. Xue, Inorg. Chem., 2005, 44, 1505–1510 CrossRef CAS PubMed.
- G. T. Kent, X. Yu, C. Pauly, G. Wu, J. Autschbach and T. W. Hayton, Inorg. Chem., 2021, 60, 15413–15420 CrossRef CAS PubMed.
- A.-G. D. Nelson, T. H. Bray and T. E. Albrecht-Schmitt, Angew. Chem., Int. Ed., 2008, 47, 6252–6254 CrossRef CAS PubMed.
- A.-G. D. Nelson, T. H. Bray, F. A. Stanley and T. E. Albrecht-Schmitt, Inorg. Chem., 2009, 48, 4530–4535 CrossRef CAS PubMed.
- E. J. Schelter, R. Wu, B. L. Scott, J. D. Thompson, D. E. Morris and J. L. Kiplinger, Angew. Chem., Int. Ed., 2008, 47, 2993–2996 CrossRef CAS PubMed.
- O. Benaud, J.-C. Berthet, P. Thuery and M. Ephritikhine, Inorg. Chem., 2010, 49, 8117–8130 CrossRef CAS PubMed.
- S. Fortier, B. C. Melot, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2009, 131, 15512–15521 CrossRef CAS PubMed.
- J. D. Sears, D.-C. Sergentu, T. M. Baker, W. W. Brennessel, J. Autschbach and M. L. Neidig, Angew. Chem., Int. Ed., 2020, 59, 13586–13590 CrossRef CAS PubMed.
- X. Yu, S. Bi, I. A. Guzei, Z. Lin and Z.-L. Xue, Inorg. Chem., 2004, 43, 7111–7119 CrossRef CAS PubMed.
- K. Marc, H. Klaus, S. Gerd, M. Werner, F. Stefan, F. Gernot and D. Kurt, Z. Anorg. Allg. Chem., 1999, 625, 2055–2063 CrossRef.
- C. R. Graves, E. J. Schelter, T. Cantat, B. L. Scott and J. L. Kiplinger, Organometallics, 2008, 27, 5371–5378 CrossRef CAS.
- S. J. Simpson, H. W. Turner and R. A. Andersen, J. Am. Chem. Soc., 1979, 101, 7728–7729 CrossRef CAS.
- S. Fortier, J. L. Brown, N. Kaltsoyannis, G. Wu and T. W. Hayton, Inorg. Chem., 2012, 51, 1625–1633 CrossRef CAS PubMed.
- B. G. Wybourne, Spectroscopic Properties of Rare Earths, Interscience Publishers, New York, 1965 Search PubMed.
- M. Speldrich, J. van Leusen and P. Kögerler, J. Comput. Chem., 2018, 39, 2133–2145 CrossRef CAS PubMed.
- B. R. Judd, Proc. R. Soc. A, 1955, 232, 458–474 CAS.
- J. Ibers, Nat. Chem., 2010, 2, 996 CrossRef CAS PubMed.
- P. L. Arnold, M. S. Dutkiewicz and O. Walter, Chem. Rev., 2017, 117, 11460–11475 CrossRef CAS PubMed.
- T. W. Hayton, Dalton Trans., 2010, 39, 1145–1158 RSC.
- T. W. Hayton, Chem. Commun., 2013, 49, 2956–2973 RSC.
- M. B. Jones and A. J. Gaunt, Chem. Rev., 2013, 113, 1137–1198 CrossRef CAS PubMed.
- S. T. Liddle, Angew. Chem., Int. Ed., 2015, 54, 8604–8641 CrossRef CAS PubMed.
- S. L. Staun, L. M. Stevens, D. E. Smiles, C. A. P. Goodwin, B. S. Billow, B. L. Scott, G. Wu, A. M. Tondreau, A. J. Gaunt and T. W. Hayton, Inorg. Chem., 2021, 60, 2740–2748 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, crystallographic details (as CIF files), computational results, and spectral data for complexes 1–4. CCDC 2108832–2108835. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc05072a |
| This journal is © The Royal Society of Chemistry 2021 |