
Designing the electronic and geometric structures of single-atom and nanocluster catalysts
Tianxiang
Chen
ab,
Lin
Ye
*c and
Tsz Woon Benedict
Lo
 *ab
*ab
aState Key Laboratory of Chemical Biology and Drug Discovery, Department of Applied Biology and Chemical Technology, The Hong Kong Polytechnic University, Hong Kong, China. E-mail: twblo@polyu.edu.hk
bThe Hong Kong Polytechnic University Shenzhen Research Institute, Shenzhen 519700, China
cDepartment of Chemistry and Shanghai Key Laboratory of Molecular Catalysis and Innovative Materials, Fudan University, Shanghai 200438, China. E-mail: yelin@fudan.edu.cn
First published on 7th June 2021
Abstract
This article highlights some recent advances in the electronic and geometric structures of single-atom and nanocluster catalysts, which play an inevitably important role in modern catalysis. The combined use of modern characterisation techniques enables surface, solid-state and coordination chemists to develop more extensive, more cohesive and more appropriate frameworks to rationalise the structure–reactivity functions of these atomically dispersed entities, which further provides the necessary details for the identification of chemical trends and benchmark quantum sciences. We also offer an outlook on the challenges and opportunities of this research area.
 Tsz Woon Benedict Lo | Lo is a solid-state chemist who has interest in the fundamental aspects of microporous materials and their catalytic applications. He developed a reliable methodology for using state-of-the-art synchrotron X-ray techniques to reveal molecular behaviours, based upon scattering and spectroscopic evidence, for the subsequent design of novel catalytic systems. He obtained his MChem and DPhil degrees from St John's College, the University of Oxford (UK). Since 2017, he has been a Research Assistant Professor at the Hong Kong Polytechnic University. |
Introduction
In nature, enzymes are biological catalysts that have been evolving over millions of years. Most enzymes exhibit ultra-high catalytic turnover frequency and product specificity from an optimised combination of metal centre(s) and protein scaffolds. It has often been debated whether these combinations of metals and proteins are ‘perfect’. Some may argue that certain metal(s) and proteins are chosen or used due to regional abundance, where there could be better combinations that exist theoretically. The molecular and catalytic specificities of enzymes come from their unique electronic and geometric (three-dimensional) structures.1,2 Thanks to the extensively variable oxidation states, many redox-active enzymes contain mid-to-late 3d transition metal ions. The unique electronic (redox of metal centres) and geometric properties (inert protein scaffold) lead to exceptional biological functionality and product specificity in bio-catalysis. The electronic structures of the metal centres are related to the highest occupied molecular orbital–lowest unoccupied molecular orbital (HOMO–LUMO) work functions that control the binding preferences and redox properties; the metal co-factors are optimised to facilitate adsorption and activate the catalytic processes subsequently. In contrast, the geometric properties, such as coordination and surrounding environments of the metal centre(s), confinement and wall effects, can affect the adsorption kinetics, hence regulating the molecular specificity.3 A notable example is the Cu centre in plastocyanin where the Cu(II)/Cu(I) redox couple is facilitated by the pseudo-tetrahedral coordination environment of the Cu centre where minimal structural reorganisation and rapid electron transfer can be achieved.The phenomenal cooperative actions between the electronic and geometric structures in many biological species shed light on the design of atomically precise metal-containing co-factors for catalysis. Due to the high surface-to-volume ratio, single-atoms (SAs) and multinuclear nanoclusters (NCs) are highly reactive and exhibit colloidal instability when compared to their bulk counterparts.4,5 Researchers have accordingly designed many SA and NC metal-containing inorganic compounds for highly chemo-, regio-, and enantio-selective catalytic processes.
The concept of supported SA and NC chemistry has been long discussed. The progress in the development of modern characterisation techniques, notably aberration-corrected scanning transmission electron microscopy (AC-STEM) and extended X-ray absorption fine structure (EXAFS) spectroscopy, has driven the recent blossom in the research of these materials, where these sub-nanometric species can be more reliably determined.
In 1999, Iwasawa et al. determined the structure of atomically dispersed Pt single-atoms on MgO by primarily employing EXAFS.6 In 2003, a simple cyanide leaching method was proposed by Stephanopoulos et al. for the synthesis of atomically dispersed single-atoms on metal oxide, where the single-atom Au,Pt/CeO2 catalysts exhibit much higher catalytic activity toward water-gas shift than Au and Pt nanoparticles.7 During the 1990s–2000s, the role of single-atoms in catalytic reactions was extensively discussed.8–13 Isolated Pd on Al2O3 and Ir on MgO have been observed by Lee14 and Gates15 by AC-STEM. Although Lee et al. proposed that ‘single-site catalysts’ can effectively promote the catalytic performance, the precise synthesis of well-defined ‘single-site catalysts’ is still a challenge to be extensively realised in a heterogeneous system. In 2011, Zhang et al. fabricated isolated Pt atoms on an FeOx support and further introduced the concept of ‘single-atom catalysis’ to describe the catalysis by SACs.16
As for NCs, Cotton first defined the term ‘metal atom cluster’ after investigating the chemistry of low-valent polynuclear rhenium halides of [Re3Cl12]3− in 1964.17,18 In the following decades, cluster chemistry has made substantial progress. Various transition metal clusters have emerged and exhibited remarkable catalytic potential.19–21 Among them, supported Cu NCs have received a lot of attention due to their potential in offering a practical solution for the utilisation of methane by selectively oxidising methane molecules to methanol.22–25 Microporous materials, such as zeolites and metal–organic frameworks, have been widely employed to encapsulate SAs and NCs thanks to the unique chemical functionality and framework confinement effect. However, to practically realise selective methane oxidation is extremely challenging primarily due to two reasons, (i) the high activation barrier to activate the non-polar C–H bond, and (ii) the difficulty in stabilising methanol at the intermediate step but not over-oxidising to yield CO2. Besides selective methane oxidation, Cu NCs have also been found useful in various catalytic applications. For example, Solomon et al. reported trinuclear Cu NCs in 1997 and revealed the role of trinuclear copper nanoclusters in the hydroperoxide reduction process.26 In 2003, Schoonheydt et al. reported bis(μ-oxo)dicopper species encapsulated in ZSM-5 zeolite for the decomposition of NO.27 Different zeolite topologies, such as Y and MOR, have also been used to host Cu NCs for catalytic applications.25,28,29
In general, there are two common and effective approaches for the fabrication of these atomically precise species, namely, (1) the conventional inorganic chemistry approach and (2) the solid-state chemistry approach.
Certainly, the inorganic chemistry approach (metal complexes) is well-established, where the electronic and geometric structures of the SAs and NCs can be finely tuned by elemental variation, nuclearity control and ligand design. Through the solid-state approach, SAs and NCs can also be prepared by immobilisation on the surface functionalities of various support materials.30,31 Recent advances in the design and fabrication of SAs and NCs have been focused on their stabilisation against sintering/leaching and achieving a high loading ratio. The successful immobilisation of these species over solid-state supports brings heterogeneous catalysis towards the molecular frontier and delivers substantial impact towards the catalysis and functional materials community. The surface free energy per metal atom increases significantly when the ensemble of nuclearity decreases from bulk to nanoparticles, to the NC and SA level. The high surface free energies from a lowered coordination make the supported SAs and NCs achieve higher chemical reactivity.32 In some cases, the assembly of surface metal atoms as NCs is essential to provide an optimal geometry for the activation of substrate molecules with the required surface bond formation.33
For these kinetically stable sub-nm species used in catalysis where their surface becomes more dominant, the properties of the chemical bonds on the surface can be altered because of various external microenvironmental factors.3,34 The bonding coordination, when compared to that in the bulk, is closely associated with local geometry that is affected by surface relaxation and reconstruction. The electronic properties at the surface can also differ substantially from that of the bulk. For example, the existence of a surface can induce additional electronic states or surface states.35 It should be noted that it is challenging to obtain precise atomic information about the local structure, leading to a general lack of understanding of the geometric properties and surface chemistry. Also, the asymmetric coordination to metal or ligand in a crystal can generate higher surface energetic facets that alter the local surface polarity in the structure.36,37
The support approach is a realistic alternative in offering extensive possibilities for the rational design and precise control of the electronic and geometric structures of SAs and NCs. The choice of support is critical; the support not only acts as physical carriers to immobilise SAs and NCs but also influences the electronic configurations and local geometric structures, as well as the molecular specificities through steric effects.32 Besides some open surface supports (e.g., TiO2 and MgO), micro/mesoporous supports (e.g., zeolites and metal–organic frameworks (MOFs)) can additionally provide a relatively inert framework to offer spatial properties.22,38–42
The advantages and disadvantages of using metal complexes (homogeneous) and solid-state materials (heterogeneous) for catalysis have long been discussed.35,43,44 However, a reliable characterisation of the atomically precise SAs and NCs over solid-state supports was difficult to achieve in the past due to technical limitations, in stark contrast to the more mature characterisation of inorganic metal complexes. The main limitation is that the local electronic and geometric environments of the metal sites (e.g., the coordination of the metal atom, the spatial structure of metal–ligand complexes, etc.) have not been well understood, which hinders the development of this class of highly promising solid-state materials. With the impetus in modern characterisation techniques, even single metal species can now be directly observed using suitable high-resolution electron microscopes.45,46 By combining with other advanced techniques, a more accurate and precise determination of the atomic and structural parameters can be achieved.16,47,48 Researchers can more confidently design sub-nm materials with highly precise atomicity based upon the crucial structural information.
With increasing attention to the atomically precise SAs and NCs with low nuclearity, this perspective article will primarily highlight some recent progress in the fundamental study of their electronic and geometric structures, and their corresponding influence on catalytic science.
Electronic properties
For species that are in the regime of SAs and multinuclear NCs, their electronic structures approach the atomic/molecular frontier. The classical HOMO–LUMO theory is often considered a more appropriate description of the energetics of SAs and NCs (see Fig. 1), which can be applied to both metal complexes and supported SAs and NCs.49 The binding energies and the ligand-field splitting are the two key parameters of the HOMO–LUMO work functions of d-metal complexes.50 The former is attributed to effective nuclear charges (Zeff) as well as oxidation states,51 while the latter is related to coordination and interactions between the metal nuclei and the ligands.52 The fine adjustment of the HOMO–LUMO work functions, based upon the factors mentioned above, could directly affect the catalytic properties by altering the redox properties and binding strengths to incoming reaction substrates. Consequently, the electronic structures of the metal-containing co-factors can be collectively engineered by varying the elemental nature, nuclearity, topology, and coordination/ligation system.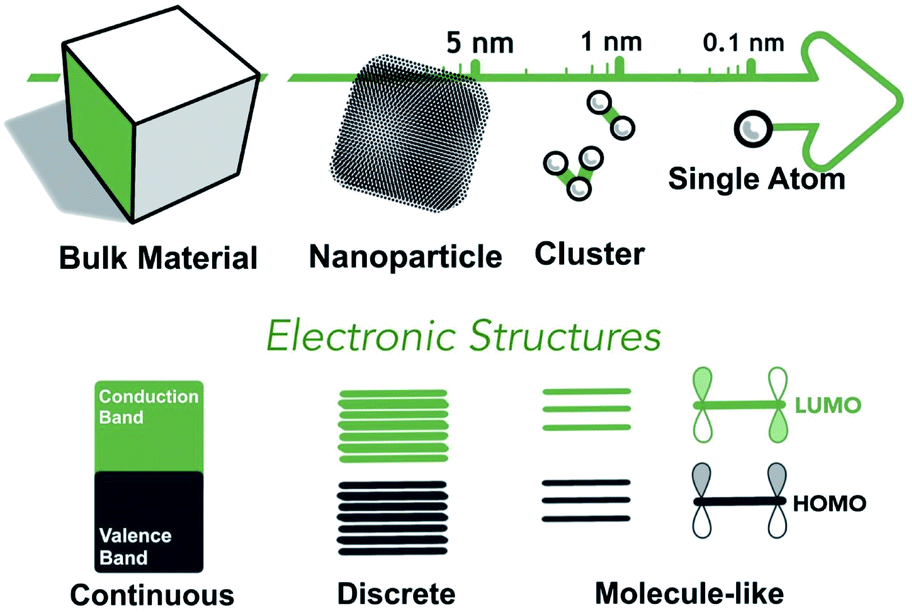 | ||
| Fig. 1 Electronic and geometric structures of SAs, NCs, nanoparticles, and bulk materials. | ||
Through elemental variation
The effective nuclear charge (Zeff) and the outermost electron configuration of different d-metal species are pivotal in governing the electronic structures, leading to notably different redox properties and binding preferences. A slight alternation in the binding energy of reaction substrates and intermediates could notably influence the catalytic properties. In many recent studies, many catalytic reactions, including CO oxidation, CO2 reduction, the hydrogen evolution reaction (HER), oxygen evolution reaction (OER), oxygen reduction reaction (ORR), and nitrogen reduction reaction (NRR), have been used as model reactions to probe the electronic structures of SAs due to their more defined adsorbate and intermediate structures.53–59 In this sub-section, some strategies to rationalise the electronic structures and catalytic properties as a function of elemental variation will be highlighted.The electronic structures are often elucidated and expressed in terms of density of states of specific metal sites, based upon simulations. Li et al. have compared the catalytic performance for CO oxidation with respect to the electronic structures of a series of d-metal SAs (Au, Rh, Pd, Co, Cu, Ru, and Ti) supported on iron oxide (FeOx) under the Langmuir–Hinshelwood mechanism.60 It has been found that the d-band centres of supported Pt, Rh, Pd and Ru SAs were shifted to higher energy when compared to the inactive supported Au SAs. Higher d-band metal centres render more empty d states and stronger adsorption interaction with the reaction substrates (CO and O2). Yang and co-workers have employed a similar approach to study the relationship between the electronic structures and catalytic activities of a series of SAs (Pd, Fe, Co, and Ni) supported on γ-Al2O3.61 Zheng et al. reported that the conversion of CO2 to CO is preferred over Au, Ag and Pd SA electrocatalysts as they can bind the COOH* intermediate strongly, but bind CO* weakly.62 Back et al. have predicted the electrochemical CO2 reduction reaction (eCO2RR) properties of a series of SAs on defective graphene based on theoretical calculations, where the electronic interactions between the d-orbital of the SAs and the p-orbital of graphene govern the density of states and hence the catalytic behaviours.63 Also, Li et al. have investigated the electronic structure–reactivity functions of a series of SA M–Nx moieties (M = Mn, Fe, Co, Ni, Cu) with respect to the eCO2RR.64 A volcano trend has been found between their catalytic activities toward CO formation and the nature of the transition metal in M–Nx sites, with Fe and Co at the top of the volcano. To correctly model the active sites under in operando conditions, operando X-ray absorption near edge structure spectroscopy was performed to study the changes in the metal oxidation state with electrochemical potential. The oxidation states of Co and Mn remained unchanged with potential, while Fe and Ni were partially reduced and Cu mostly reduced to Cu(0). M2+N4–H2O was identified as the most active centre in Fe–Nx and Co–Nx, while Ni1+N4 was predicted to be the most active one in Ni–Nx. The experimental activity and selectivity have been rationalised from the difference between the binding energies for CO2* and H* as a descriptor of selectivity toward CO.
The experimental measurements of the electronic structures of supported species have often relied on probing the oxidation states via a study of the metal binding energies. Wu et al. prepared a series of 3d-metal SAs supported on graphene oxide (Fe, Co, Ni and Cu).65 The electronic structures of these supported SAs were determined by X-ray absorption spectroscopy (XAS), where a charge transfer from the metal to oxygen (M–O bond formation) and the oxidation states of the isolated metal SAs Mδ+ within 0 < δ < 3 have been determined.
Besides oxidation states, the study of the electronic communication via redox hopping between neighbouring SAs can also offer critical information on the catalytic properties, where the extent of necessary electrons supplied can be probed. Xue et al. have employed voltammetric measurements to probe the redox hopping properties of a series of 3d-metal SAs (Fe, Co, Ni, Cu, and Zn) supported on UiO-66-NH2.66 It has been found that Cu2+ is the most active modifier that allows a rapid electron movement between neighbouring metal sites in the form of Cu2+/Cu+. It is consistent with their model eCO2RR reaction, which has been attributed to the enhanced redox hopping properties, in combination with optimum CO* binding, that facilitates multielectron (>2e−) products.
Bimetallic (or multimetallic) NCs particularly with well-defined alloy structures of noble metals, like Au–Ag, Au–Cd and Hg–Au, provide model examples for the influence of elemental composition and electronic structures on catalytic properties.67,68 The elemental variation effect has been commonly studied in colloidal species, such as Au25, where Zhu and co-workers utilised Au25(SR)18 (SR = SPhMe2) as a template to investigate this effect by employing the metal-exchange method.67 The doping effect has been investigated with a single foreign atom (Au, Pd, or Pt) in the core of an Ag25 NC on the catalytic properties, where the foreign atom is encapsulated by 24 Ag atoms (Au@Ag24, Pd@Ag24, Pt@Ag24) (see Fig. 2).69 It has been found that the catalytic performance in the carboxylation reaction of CO2 was influenced substantially due to the alternation in the electronic structures. Also, Kobayashi et al. systematically studied the electronic structures of three bimetallic Au–Ag NCs, namely Au12.2Ag2.8, Au14.4Ag3.6, and Au17.6Ag7.4.68 The optical spectroscopy results suggested that the electronic structures of Au NCs were modulated by the Ag heteroatoms.
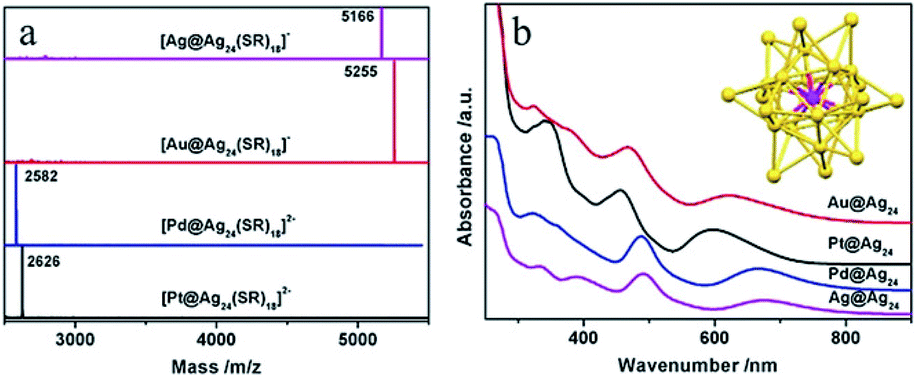 | ||
| Fig. 2 (a) ESI-MS and (b) UV/vis spectra of Ag@Ag24, Au@Ag24, Pd@Ag24, and Pt@Au24 clusters capped by thiolates. Inset: the structural framework of Au/Pd/Pt/Ag@Ag24. SR = SPhMe2; the SR moiety is omitted for clarity.67 Reprinted with permission from ref. 67. Copyright (2015) American Chemical Society. | ||
Through nuclearity control
When compared to typical SAs, multinuclear NCs often show remarkably different catalytic properties because of the general tunability of the electronic and geometric structures. Multinuclear NCs bridge between bulk (classical chemistry) and atomic/molecular species (quantum chemistry), where the concept of discrete quantum levels in atomic/molecular structures can be fine-tuned based on the nuclearity at the active sites. The effect of nuclearity on the electronic structures has been extensively exemplified in various Aux macro NC structures.70,71 Meanwhile, atom-by-atom electrodeposition was employed to assemble supported Ptn and CoxOy clusters and the size effect of the cluster has been comprehensively studied in Ptn (ref. 72) and CoxOy clusters.73 The size-dependent catalytic performance was evaluated in the HER and OER, respectively. The potential of the HER was shifted positively with increasing size and the OER exhibits a higher relative rate on smaller clusters, where both the minimum-ensemble sized catalysts (i.e., individual Pt and modular Co1Ox) show more superior activity than the bulk nanoparticles.Yang and co-workers have employed first-principles calculations to probe the electronic structures of Con (n = 1–5) NCs supported on γ-Al2O3 (100) and (110) surfaces (see Fig. 3),74 and Pdn (n = 1–5) NCs supported on rutile TiO2 (100), (001) and (110) surfaces.75 It has been found that the stability of the NCs varies as a function of nuclearity and surface support, as the surface binding preferences and nucleation energies of the NCs are notably different. Based upon charge analysis, both metal clusters and support are critical in the charge state of the NCs. The charge of the NCs is highly dependent on the balance between the preferential binding to the surface atoms. The density of states of Pdn/TiO2 revealed that the 4d orbitals of Pd are close to the Fermi level in some cases (such as Pd4/TiO2 and Pd5/TiO2), which can be correlated with catalytic activity.
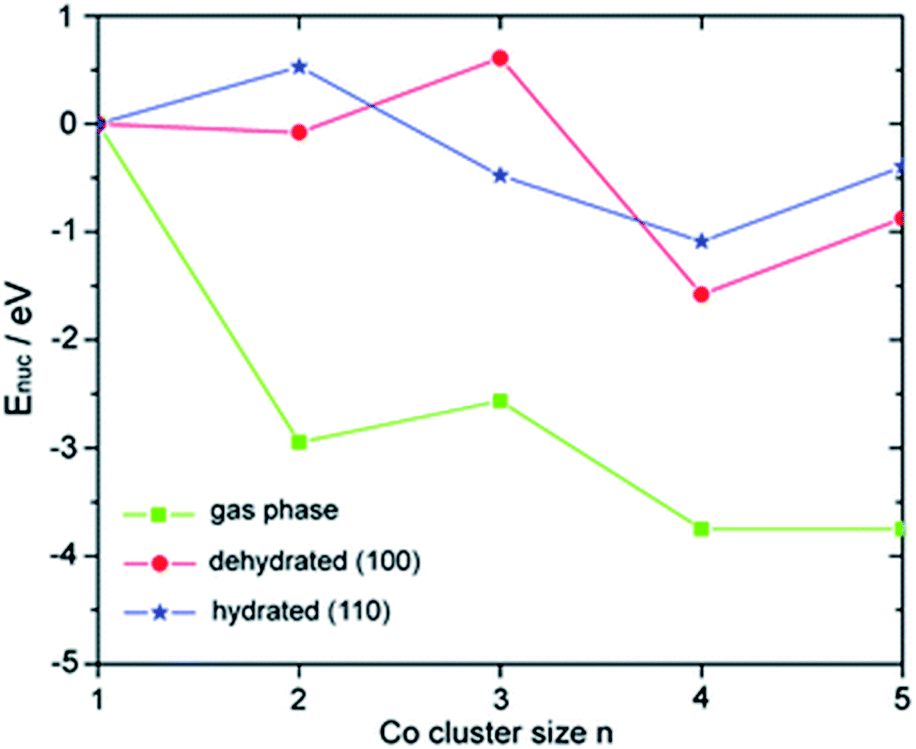 | ||
| Fig. 3 Nucleation energies Enuc of Con clusters in the gas phase and on the dehydrated (100) and hydrated (110) surfaces.74 Reproduced from ref. 74 with permission from The Royal Society of Chemistry. | ||
Through metal–support interaction
As seen in the examples above, the electronic structures of Con and Pdn NCs are affected by their interactions with the supports.74 Different local structures of these support substrates could provide alternative coordination environments and electronic correlations. Hence, supports not only act as physical carriers to immobilise the highly reactive SAs and NCs by offering kinetic stabilisation that prevents them from sintering, but can also perturb the electronic structures of the supported SAs and NCs through chemical bonding, generally known as metal–support interaction.35,76 The metal–support interaction can subtly alter the charge state of the SAs and NCs by electron transfer between SAs and NCs and the support, which can offer activation for reaction substrates and enhance catalytic properties.77 As a result, the optimisation of the metal–support interaction by choosing an appropriate support material has attracted great research attention which leads to the rational modification of the electronic structures to achieve more superior catalytic performance.78Yao et al. have engineered the electronic structures of Ru SAs on a series of PtCu alloys via compressive strain to boost the electrocatalytic oxygen evolution reaction.79 A volcano relation has been reported between the catalytic activity and the lattice constant of the PtCu alloys. According to density functional theory calculations, the compressive strain of the outer Pt shell can influence the electronic structures of the Ru SAs, which optimises the binding strengths of oxygen species. As discussed, Yang and co-workers have shown that the electronic structures of Co3 on dehydrated γ-Al2O3 (100) and hydrated γ-Al2O3 (110) surfaces are notably different (see Fig. 4).74
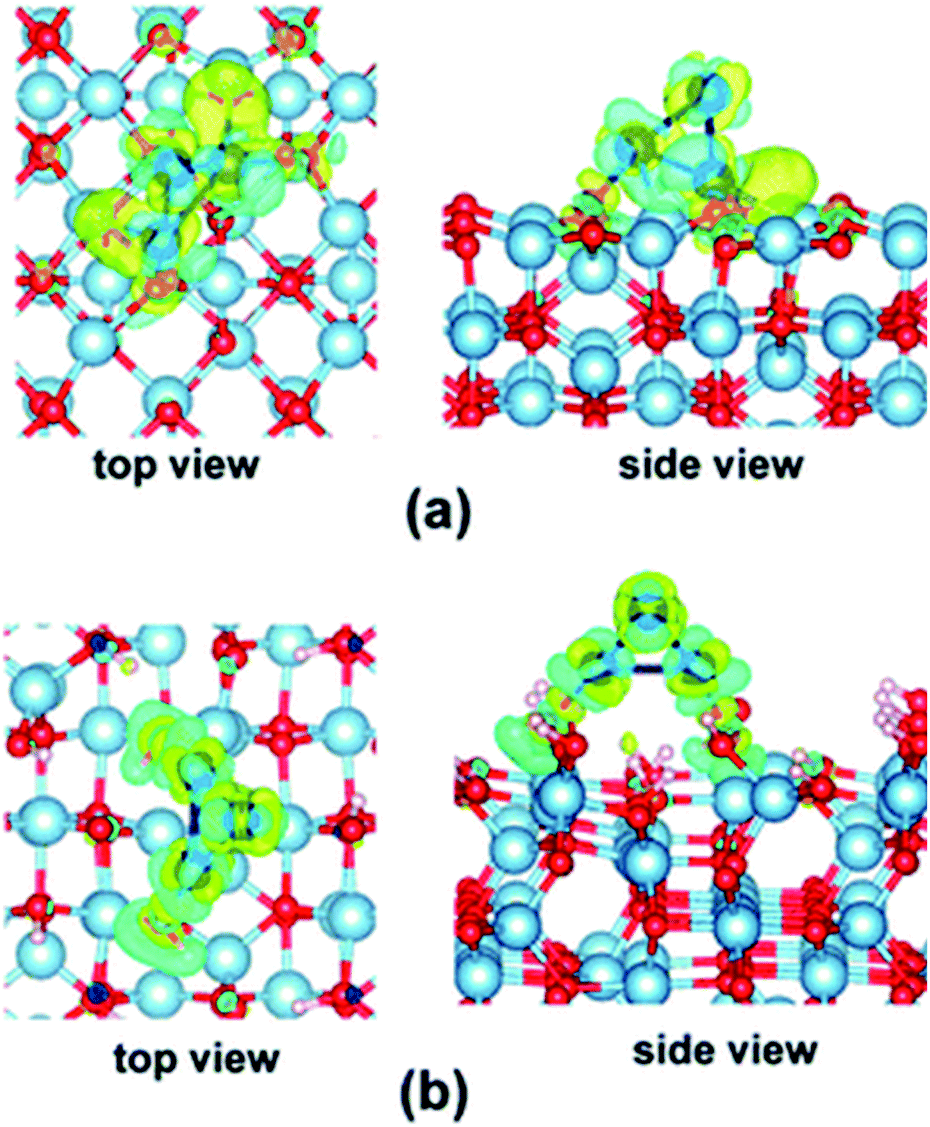 | ||
| Fig. 4 Electron density difference maps (isovalue = 0.04) for the Co3 cluster adsorbed on the (a) dehydrated γ-Al2O3 (100) surface and (b) hydrated γ-Al2O3 (110) surface. The charge depletion and accumulation regions are shown in blue and yellow, respectively.74 Reproduced from ref. 74 with permission from The Royal Society of Chemistry. | ||
Through ligand engineering
One of the most important factors that affect electronic structures is the ligand effect. The ligand-field theory has been well established and can elucidate the electronic structures of d-metal complexes where the key parameters include σ-bonding, π-bonding, and the symmetry of the metal centres. There have been extensive research efforts to investigate the electronic structures of d-metal complexes, which offers substantial insight into the subsequent modifications and further design of the supported counterpart for the solid-state community. The ligand effect can promote activation of reaction substrates and provide a mean for activity and selectivity control.The systematic study of the ligand effect of d-metal complexes is undeniably more mature than that of supported SAs and NCs. Desnoyer et al. systematically studied the electronic structures of a series of square planar d10 Ni diphosphine π-complexes that vary as a function of the degree of π-backbonding of alkene or carbonyl-containing π-ligand.80 The degree of π-backbonding has been quantitatively correlated between J-coupling (from nuclear magnetic resonance (NMR)) and the binding energy of Ni (from the 1s → 4pz pre-edge in Ni K-edge X-ray absorption spectroscopy (XAS)), which is determined by the energy of the π* ligand acceptor orbital (see Fig. 5). In addition, He et al. have also utilised the binding energy of Ni (Ni L-edge XAS) to quantify the degree of π-backbonding in a series of Ni diphosphine π-complexes.81
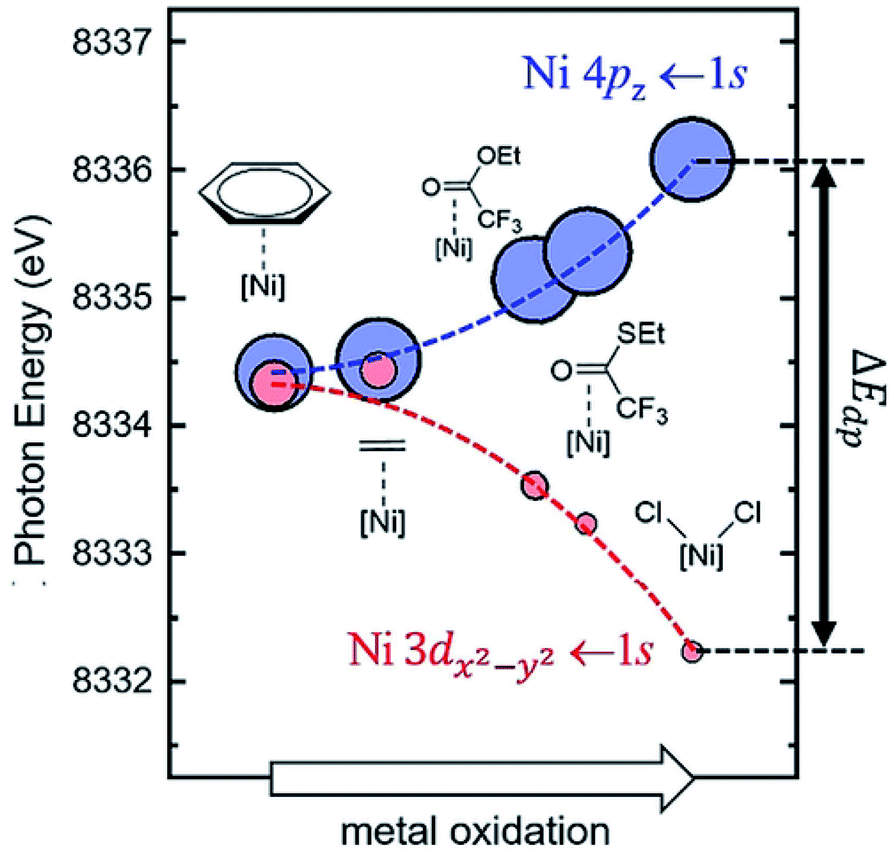 | ||
| Fig. 5 Calculated Ni K-edge XAS time dependent-DFT results for the pre-edge region of the spectrum. Each complex is represented by a blue circle (Ni 4pz ← 1s) and a red circle (Ni 3dx2−y2 ← 1s). The area of each circle is proportional to the calculated oscillator strength (fosc) for each transition. All calculated time dependent-DFT energies at the Ni K-edge were shifted by −98.55 eV.80 Reproduced from ref. 80 with permission from Wiley, copyright 2019. | ||
The influence of ligands on the electronic structures of d-metal inorganic NC complexes has been shown to be similar to those of traditional d-metal complexes. In recent work by Cirri et al., the electronic structures of precise Au NCs with different ligands, namely Au9(P(p-X–Ph)3)83+ and Au8(P(p-X–Ph)3)72+, where X is –H, –CH3, or –OCH3, at the para position, have been elucidated.82 The electronic structures have been computed to generalise the unique observation from the mass-selective UV/visible absorption spectra. Using chemical derivatisation, the electronic structures of these complexes are related to the electron-donating or -withdrawing strength of the ligands. Wan et al. have also investigated the ligand effect in catalysis over Au38 NCs with different ligands.83
In brief, the ligands can affect the electronic structures of the NCs from two major perspectives, (1) by the manipulation of the energies of the ligand orbitals (which dominates the metal-to-ligand charge transfer transitions), and (2) by the redistribution of charge density in the NCs (which controls transitions local to the metal core). However, the surface coordination species (or ‘ligand’) in supported SAs and NCs are more difficult to probe accurately and precisely due to a lack of appropriate techniques from the atomic perspective.
Geometric structures
Nature is the master architect in designing the geometric structures of SAs and NCs, where metalloprotein is an excellent masterpiece example. Metalloproteins contain metal co-factors at the active sites where the co-factors are surrounded by an extensive pocket of protein scaffold substrates that can offer unique molecular specificities to incoming substrates.Metal–protein environment in enzymes
In natural metalloenzymes, the combination of metal nuclei and protein scaffolds is pivotal for the unique activity of enzymes.84 Since many transition metals can exist in multiple oxidation states, metalloenzymes usually construct unique environments to immobilise metal ions to allow a minimal structural reorganisation for the redox couples. Adjusting the geometry structures can ensure that the electrons transfer rapidly at the desired metal sites, and specifically targeted molecules can be adsorbed. For example, the Cu active site in plastocyanin is coordinated with two N atoms from two histidine residues and an S atom from cysteine to form a distorted trigonal pyramidal geometry, which leads to a rigid coordination environment for the Cu(II)/Cu(I) redox couple.85 In haemoglobin, the pseudo-tetrahedral Fe-containing heme group in the hydrophobic protein pocket can selectively bind O2.86 In rubredoxin, the Fe site is coordinated with four cysteine residues forming a pseudo-tetrahedron.87 The superoxide dismutase (SOD), Cu,Zn-SOD, contains a binuclear NC co-factor where the Cu and Zn sites are bridged by a histidine residue.88 There are also other SOD isozymes that contain Fe, Mn, or Ni metal co-factors.89,90 Ni-SOD is an interesting example as it involves Ni(III) which is an unusual oxidation state of Ni. The geometry of the Ni active site transforms between square planar Ni(II), with thiolate (Cys2 and Cys6) and backbone nitrogen (His1 and Cys2) ligands, and square pyramidal Ni(III) with an added axial His1 side-chain ligand.91 There are also many other textbook examples, such as carbonic anhydrase and cobalamin.92,93 Briefly, the mechanistic discussion of metalloenzymatic activity relies heavily on the elucidation of the interactions between the metal-containing co-factors and the surrounding protein scaffolds, in terms of coordination geometry, coordination number, bonding distances and angles, and so forth.In metal complexes
As most enzymes exhibit ultra-high catalytic turnover frequency and product specificity thanks to their optimised electronic and geometric structures, this has inspired the design and development of many inorganic mimicking systems. Taking the artificial particulate form of methane monooxygenase as an example, numerous studies have reported on the design and the subsequent elucidation of the geometric structures with respect to the catalytic properties. For instance, Karlin et al. reported a copper–dioxygen complex mimicking methane monooxygenase,25 Stack and co-workers reported a series of Cu2-bis(μ-oxo) (pseudo-trigonal bipyramidal with the O atoms occupying the axial positions) and Cu3-bis(μ-oxo) complexes,94,95 and Chan and co-workers synthesised a series Cu3 complexes.24,96 Another well-studied example is superoxide dismutase mimetics; Fukuzumi et al. reported an imidazolate-bridged Cu–Zn binuclear active site model of Cu,Zn-SOD.97 Meanwhile, various Mn-based metal complexes, such as Mn(III)/(II) porphyrin,98 Mn(II) penta-azamacrocyclic,99 and Mn(III) salen,100 have been designed as Mn-SOD mimics.101–103There are also many multinuclear NC systems; for instance, Zhang et al. reported a series of related binuclear Cu2 complexes to mimic carboxylesterase.104 The Cu–Cu interatomic distance was determined to be ca. 3.0 Å, which agrees with the typical M–M interatomic distances in most multinuclear NC enzymes. Also, Jiang and co-workers have reported binuclear Cu2 complexes as carboxylesterase mimics which employed an alkoxo/acetato-bridged moiety as a model to promote the hydrolytic cleavage of p-nitrophenyl picolinate.105,106 Beloglazkina et al. synthesised a mixed-valence Cu complex with a tridentate N2S type organic ligand as an N2O reductase mimic, where it is often regarded as a mimic model for N2O reductase.107 Generally, the geometric structures about the metal-containing active sites can be readily elucidated by a combination of single-crystal XRD and spectroscopic techniques (NMR and electron paramagnetic resonance (EPR)).108,109 The prosperous development of this research area has benignly guided the design of the solid-state counterpart.
Over supported systems
Two-dimensional metal oxides and porous materials (e.g., zeolites, MOFs, etc.) are great supports to immobilise SAs and NCs, where their topological properties can offer unique molecular specificities to incoming substrates. The recent progress in modern characterisation instrumentation has promoted a more reliable elucidation to allow the subsequent design of supported SA and NC systems in atomic resolution.Here are many studies on the precise design of supported SAs and NCs in recent years. Notable zeolite-support examples include the trinuclear [Cu3(μ-O)3]2+ NC supported on MOR zeolite that exhibits a high reactivity towards C–H activation of methane (that mimics methane monooxygenase) by Lercher et al.22 The structure of the NC was determined by combined XAS analysis and density functional theory (DFT) calculation. The [Cu3(μ-O)3]2+ NC is stabilised by two anionic centres at the entrance of the MOR side pocket. It is proposed that the C–H activation can be facilitated by the unique atomic arrangement of the extra-framework O atoms directed towards the main MOR channel. Tsang et al. have also employed zeolite (MFI type) to host a nucleophilic ‘Zn–OH’ on the negatively charged framework (see Fig. 6), where the Zn/ZSM-5 catalyst was highly active for the catalytic conversion of γ-valerolactone to p-xylene.110 The atomic parameters were determined by combined XAS and synchrotron X-ray powder XRD (SXRD). The formation of the framework ‘Zn–OH’ site has been found to be responsible for the selective decarboxylation of the lactone group to butene through a new hydrolysis pathway, while the adjacent Brønsted acid sites can catalyse the subsequent dimerisation to yield aromatic products. The structure and basic pathway of nucleophilic attack at the terminal Zn–OH site are comparable to those of Zn-containing enzymes in biological systems.110
 | ||
| Fig. 6 The refined structures of the Zn/ZSM-5 catalyst derived from SXRD and refinements. (a) Zn/ZSM-5; (b) gamma-valerolactone (GVL) adsorbed on Zn/ZSM-5; (c) a close-up view of Zn-2-OH interacting with the carbonyl of GVL; (d) a published crystal structure showing a complex intermediate of Zn–OH of T199A-CA II enzyme with adsorbed CO2 for comparison (PDB: 1CAM) with permission for reproduction.110 Ball-and-stick model: O = red, Si = grey, Al = green, Zn = blue, and C = black. Reproduced from ref. 110 with permission from Wiley, copyright 2017. | ||
Recent MOF-supported examples include the binuclear Cu2 co-factor in a Ti-MOF (MIL-125, in the form of Ti8–Cu2; see Fig. 7) as an efficient artificial monooxygenase by Lin et al.111 It was found that the Cu2 site in Ti8–Cu2 exhibits much higher stability than the mononuclear Cu1 analogue (Ti8–Cu1), which has been attributed to the much promoted catalytic activities to a range of mono-oxidation reactions, such as epoxidation, hydroxylation, Baeyer–Villiger oxidation, and sulphoxidation. Zr-based MOF-808 was employed to post-synthetically install imidazole-based ligand units for subsequent Cu(I)-metalation. The Cu atoms were found coordinated to N atoms from imidazole and O2 to form N–Cu2O2–N species, and the interatomic distance between the Cu sites was found at a distance of 2.51 Å with a coordination number of 0.6 (assigned as bis(μ-oxo) dicopper species). Extra-framework binuclear Fe2 species have also been successfully incorporated into a Zr-based MOF by a post-synthetic exchange method by Ott et al.112 The catalyst has high structural similarities to the active site of [FeFe]-hydrogenase. From combined single-crystal XRD and XAS analysis, the Fe centres occupy a distorted octahedral geometry with three C atoms from carbonyl groups and two S atoms bridging the binuclear Fe2 centre at bond distances of 1.796–1.814 and 2.283–2.285 Å, respectively. A similar post-synthetic metalation approach has been reported by Bien et al. to generate Zn–OH within a Zn-based MOF, CFA-1.113 The Zn–OH site can promote trace CO2 capture due to the CO2/HCO3− chemisorption mechanism and cooperative intercluster H-bonding interactions, which is similar to the secondary coordination sphere interactions in α-carbonic anhydrases.
 | ||
| Fig. 7 Cu2 active sites with O2 bonding in (a) the natural enzyme tyrosinase (PDB code: 1WX2) and (b) the MOF-based artificial enzyme Ti8–Cu2.111 Reprinted with permission from ref. 111. Copyright (2021) American Chemical Society. | ||
Besides porous materials, the catalytic properties of SAs and NCs can also be modified by interactions with open-surface supports from the modulation of the geometric structures. The syntheses of SAs and NCs on g-C3N4 (ref. 114–116), porous SiO2 (ref. 117 and 118) and carbon-based materials119–122 have been shown to offer exciting catalytic performance in many reports. However, it has been challenging to systematically elucidate the geometric structures due to the lack of appropriate characterisation techniques, which hence hinders the establishment of structure–activity relationships.
Comprehensive elucidation of the electronic/geometric structures of SAs and NCs is key to studying their electronic/geometric structure–activity relationships. Various advanced techniques have been developed for the elucidation of the physical characteristics of SAs and NCs, such as high-resolution transmission electron microscopy (HRTEM), AC-STEM, CO-absorbed Fourier-transform infrared spectroscopy (CO-IR), XAS, X-ray photoelectron spectroscopy (XPS), ultraviolet-visible spectroscopy (UV-vis), and SXRD as well as DFT calculation, as summarised in Table 1.
| Items | Characterisation contents | Examples | Ref. |
|---|---|---|---|
| HRTEM | Atomic dispersion | Pt clusters/γ-Al2O3, Pt/FeOx | 16 and 125 |
| AC-STEM | Metal particle size distribution | Pt@MCM-22, 0.2 Pt/m-Al2O3 | 20 and 126 |
| CO-IR | Surface properties | Pt/MFI, Pt/CeO2 | 127 and 128 |
| XAS | Oxidation state, bond length, and average coordination numbers | Pt/θ-Al2O3, Pt1δ+/TiO2 | 47 and 129 |
| XPS | Chemical and compositional properties of metal species | Pt1@Fe–N–C, Pt@PCM | 130 and 131 |
| UV-vis | Geometric structure | Cu2(μ-O)2/MFI, [Cu3(μ-O)3]2+/MOR | 28 and 132 |
| SXRD | Atomic and crystallographic information | Cu/UiO-66-NH2, Zn/MFI | 66 and 110 |
| DFT | Binding nature to the supports, adsorption properties, and reaction energetics | pfSAC-Fe, PtSn/MFI | 133 and 134 |
Conclusion and outlook
In brief, we have summarised some recent advances in the relationships between electronic and geometric structures, and catalytic activity of selected SAs and NCs. The progress in elucidating the electronic and geometric structures using modern characterisation techniques is extraordinary. It has enabled surface, solid-state and coordination chemists to develop more extensive, more cohesive and more appropriate frameworks to rationalise the electronic and geometric structures of these atomically synthesised entities, which further provides the necessary details for the identification of chemical trends and benchmark quantum sciences.70From the extensive collection of studies, it can be seen that the catalytic properties are highly dependent on the electronic and geometric structures of the NCs. On the one hand, the electronic structures are highly related to four key parameters, namely elemental variation, nuclearity control, metal–support interaction, and ligand coordination. They all affect the electronic structures by influencing the HOMO–LUMO work functions that ultimately influence the thermodynamics when the NCs interact with incoming reaction substrates. On the other hand, the geometric structures, as extensively illustrated in multiple examples from metal complexes, enzymes, and over different supported materials, play pivotal roles in providing suitable adsorption configurations and stabilising particular reaction intermediates, as well as affecting the kinetics of catalytic reactions.
We anticipate that the precise synthesis of multi-metallic multinuclear NC active sites with tuneable electronic and geometric structures will be a key future research direction that shares significant coherence with the SA systems. By taking advantage of the synergistic effect between the neighbouring metal nuclei, many long-standing challenges that require the use of more than one catalytic centre can be tackled. For instance, multinuclear NCs can be highly effective catalysts in cross-coupling reactions, due to the presence of multiple neighbouring metal nuclei where reaction substrates can be co-adsorbed for the subsequent co-activation. However, it is important to first fill the critical knowledge gap of the difficulty in the atomic design of solid-state catalysts for the control of the structure sensitivity of catalytic reactions.
Although it is well known that the traditional inorganic chemistry approach can enable the precise synthesis of multinuclear complexes, as far as we are aware, directly preparing well-defined NCs on solid-state support materials is inevitably more challenging. This is due to various intrinsic limitations, such as direct metal aggregation and the combination of metal species and multi-basic species that often yield various reticular products.
On the other hand, the characterisation of the electronic and geometric structures will be essential where it is important for the understanding of the structure–reactivity functions. For the elucidation of the supported SAs and NCs, a combination of techniques will be required, depending on the nature of the materials. For instance, for SAs and NCs supported on amorphous materials (such as graphene oxide and amorphous carbon), electron microscopy and XAS have been the two primary techniques to reveal the microenvironment. The lack of long-range order hinders the use of whole-pattern analysis of XRD data to reveal the crystal structure due to the absence of a crystalline phase. In contrast, high-resolution powder XRD can be used to offer essential crystal information of SAs and NCs supported on crystalline support materials (such as zeolites and MOFs). Synchrotron-based XAS and powder XRD techniques, alongside other spectroscopic techniques such as solid-state NMR and EPR, can be combined to determine the coordination information and to elucidate the atomic parameters of supported SAs and NCs. Meanwhile, the Rietveld refinement of powder XRD data can be used to obtain the atomic parameters (to derive the bonding information around the metal sites hereafter), and XAS can provide direct coordination information. We hope that, in the near future, a combination of different in situ/operando synchrotron-based techniques will be applied to further elucidate the structure–reactivity functions, which will consequently make the preparation of more superior SA and NC catalysts more sophisticated and rational. Finally, we provide an outlook of several possible methods based upon our experience that could be effective in the preparation of atomically precise supported NCs where their electronic and geometric structures can be carefully engineered.
Utilising the chemical and directional functionalities of the templates
A possible preparation method could utilise the inherent molecular chemical, structural and directional specificities of templates such as zeolites and covalent/metal–organic frameworks. The symmetry operations of these templates should aid the preparation of precise NCs by post-synthetic methods. By subsequent treatments, atomically precise NCs could be formed and supported on the framework.Combining solid-state and coordination chemistries
Another possible preparation method could be a modular approach that directly utilises the underlying principles of the solid-state and coordination chemistries. Lewis acidic metal ions/complexes and Lewis di-basic linker molecules can be applied in a modular manner, so that electronic and geometric properties can be rationally tuned based upon critical parameters, such as nuclearity control, elemental variation, and ligand design. The use of chiral linker molecules could further result in the formation of peptide-like secondary structures for asymmetric catalysis.Synergetic catalysis between neighbouring SAs
The synergetic effect between various neighbouring SAs has been shown to be promising to promote catalytic performance.123,124 A desirable distribution of active sites, such as Lewis and Brønsted acid sites, with optimal strengths should lead to cooperative and/or tandem reactions. Multiple atomically dispersed SAs within molecular distances could be prepared as a result of a combination of preparation methods.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Hong Kong Research Grants Council (15300819 and 25300918), the National Natural Science Foundation of China (21902139), and the Natural Science Fund of Guangdong Province (2021A1515010218) (TWBL).References
- J. R. Knowles, Nature, 1991, 350, 121–124 CrossRef CAS PubMed.
- S. J. Benkovic and S. Hammes-Schiffer, Science, 2003, 301, 1196–1202 CrossRef CAS PubMed.
- T. Chen, Q. Xue, K. C. Leung and B. T. W. Lo, Chem.–Asian J., 2020, 15, 1819–1828 CrossRef CAS.
- C. Gao, J. Low, R. Long, T. Kong, J. Zhu and Y. Xiong, Chem. Rev., 2020, 120, 12175–12216 CrossRef CAS PubMed.
- J. Cai, R. Javed, D. Ye, H. Zhao and J. Zhang, J. Mater. Chem. A, 2020, 8, 22467–22487 RSC.
- K. Asakura, H. Nagahiro, N. Ichikuni and Y. Iwasawa, Appl. Catal., A, 1999, 188, 313–324 CrossRef CAS.
- Q. Fu, H. Saltsburg and M. Flytzani-Stephanopoulos, Science, 2003, 301, 935–938 CrossRef CAS PubMed.
- V. Vidal, A. Théolier, J. Thivolle-Cazat and J. M. Basset, Science, 1997, 276, 99–102 CrossRef CAS PubMed.
- C. Lecuyer, F. Quignard, A. Choplin, D. Olivier and J.-M. Basset, Angew. Chem., Int. Ed., 1991, 30, 1660–1661 CrossRef.
- P. S. Kirlin and B. C. Gates, Nature, 1987, 325, 38–40 CrossRef CAS.
- E. A. Wovchko and J. T. Yates, J. Am. Chem. Soc., 1998, 120, 10523–10527 CrossRef CAS.
- S. Abbet, U. Heiz, H. Häkkinen and U. Landman, Phys. Rev. Lett., 2001, 86, 5950–5953 CrossRef CAS PubMed.
- S. Abbet, A. Sanchez, U. Heiz, W. D. Schneider, A. M. Ferrari, G. Pacchioni and N. Rösch, J. Am. Chem. Soc., 2000, 122, 3453–3457 CrossRef CAS.
- S. F. J. Hackett, R. M. Brydson, M. H. Gass, I. Harvey, A. D. Newman, K. Wilson and A. F. Lee, Angew. Chem., Int. Ed., 2007, 46, 8593–8596 CrossRef CAS PubMed.
- A. Uzun, V. Ortalan, N. D. Browning and B. C. Gates, Chem. Commun., 2009, 4657–4659 RSC.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS.
- F. A. Cotton and T. E. Haas, Inorg. Chem., 1964, 3, 10–17 CrossRef CAS.
- F. A. Cotton, Inorg. Chem., 1964, 3, 1217–1220 CrossRef CAS.
- L. Liu, M. Lopez-Haro, C. W. Lopes, C. Li, P. Concepcion, L. Simonelli, J. J. Calvino and A. Corma, Nat. Mater., 2019, 18, 866–873 CrossRef CAS PubMed.
- L. Liu, U. Díaz, R. Arenal, G. Agostini, P. Concepción and A. Corma, Nat. Mater., 2017, 16, 132–138 CrossRef CAS PubMed.
- S. Tian, Q. Fu, W. Chen, Q. Feng, Z. Chen, J. Zhang, W. C. Cheong, R. Yu, L. Gu, J. Dong, J. Luo, C. Chen, Q. Peng, C. Draxl, D. Wang and Y. Li, Nat. Commun., 2018, 9, 1–7 CrossRef PubMed.
- S. Grundner, M. A. C. Markovits, G. Li, M. Tromp, E. A. Pidko, E. J. M. Hensen, A. Jentys, M. Sanchez-Sanchez and J. A. Lercher, Nat. Commun., 2015, 6, 1–9 Search PubMed.
- J. Zheng, J. Ye, M. A. Ortuño, J. L. Fulton, O. Y. Gutiérrez, D. M. Camaioni, R. K. Motkuri, Z. Li, T. E. Webber, B. L. Mehdi, N. D. Browning, R. L. Penn, O. K. Farha, J. T. Hupp, D. G. Truhlar, C. J. Cramer and J. A. Lercher, J. Am. Chem. Soc., 2019, 141, 9292–9304 CrossRef PubMed.
- P. P. Y. Chen, R. B. G. Yang, J. C. M. Lee and S. I. Chan, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 14570–14575 CrossRef CAS PubMed.
- R. A. Himes and K. D. Karlin, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18877–18878 CrossRef CAS PubMed.
- U. M. Sundaram, H. H. Zhang, B. Hedman, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 1997, 119, 12525–12540 CrossRef CAS.
- M. H. Groothaert, J. A. Van Bokhoven, A. A. Battiston, B. M. Weckhuysen and R. A. Schoonheydt, J. Am. Chem. Soc., 2003, 125, 7629–7640 CrossRef CAS PubMed.
- J. S. Woertink, P. J. Smeets, M. H. Groothaert, M. A. Vance, B. F. Sels, R. A. Schoonheydt and E. I. Solomon, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18908–18913 CrossRef CAS PubMed.
- P. Vanelderen, R. G. Hadt, P. J. Smeets, E. I. Solomon, R. A. Schoonheydt and B. F. Sels, J. Catal., 2011, 284, 157–164 CrossRef CAS PubMed.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- F. Liao, T. W. B. Lo, J. Qu, A. Kroner, A. Dent and S. C. E. Tsang, Catal. Sci. Technol., 2015, 5, 3491–3495 RSC.
- Q. Xue, Z. Zhang, B. K. Y. Ng, P. Zhao and B. T. W. Lo, Top. Curr. Chem., 2021, 379, 11 CrossRef CAS PubMed.
- W. H. Lai, Z. Miao, Y. X. Wang, J. Z. Wang and S. L. Chou, Adv. Energy Mater., 2019, 9, 1900722 CrossRef CAS.
- H. T. Kreissl, M. M. J. Li, Y. K. Peng, K. Nakagawa, T. J. N. Hooper, J. V. Hanna, A. Shepherd, T. S. Wu, Y. L. Soo and S. C. E. Tsang, J. Am. Chem. Soc., 2017, 139, 12670–12680 CrossRef CAS.
- Y. K. Peng, Y. Hu, H. L. Chou, Y. Fu, I. F. Teixeira, L. Zhang, H. He and S. C. E. Tsang, Nat. Commun., 2017, 8, 675 CrossRef PubMed.
- W. Zang, T. Yang, H. Zou, S. Xi, H. Zhang, X. Liu, Z. Kou, Y. Du, Y. P. Feng, L. Shen, L. Duan, J. Wang and S. J. Pennycook, ACS Catal., 2019, 9, 10166–10173 CrossRef CAS.
- X. Y. Dong, R. Wang, J. Bin Li, S. Q. Zang, H. W. Hou and T. C. W. Mak, Chem. Commun., 2013, 49, 10590–10592 RSC.
- F. Wang, R. M. Yu, X. Y. Wu and C. Z. Lu, Inorg. Chem. Commun., 2012, 19, 70–72 CrossRef CAS.
- X. Meng, S. Y. Song, X. Z. Song, M. Zhu, S. N. Zhao, L. L. Wu and H. J. Zhang, Chem. Commun., 2015, 51, 8150–8152 RSC.
- C. H. Lee, T. S. Lin and C. Y. Mou, Nano Today, 2009, 4, 165–179 CrossRef CAS.
- X. Cui, W. Li, P. Ryabchuk, K. Junge and M. Beller, Nat. Catal., 2018, 1, 385–397 CrossRef CAS.
- Q. H. Xia, H. Q. Ge, C. P. Ye, Z. M. Liu and K. X. Su, Chem. Rev., 2005, 105, 1603–1662 CrossRef CAS PubMed.
- J. Jones, H. Xiong, A. T. DeLaRiva, E. J. Peterson, H. Pham, S. R. Challa, G. Qi, S. Oh, M. H. Wiebenga, X. I. P. Hernández, Y. Wang and A. K. Datye, Science, 2016, 353, 150–154 CrossRef CAS PubMed.
- V. Ortalan, A. Uzun, B. C. Gates and N. D. Browning, Nat. Nanotechnol., 2010, 5, 506–510 CrossRef CAS PubMed.
- M. Moses-Debusk, M. Yoon, L. F. Allard, D. R. Mullins, Z. Wu, X. Yang, G. Veith, G. M. Stocks and C. K. Narula, J. Am. Chem. Soc., 2013, 135, 12634–12645 CrossRef CAS PubMed.
- J. Gu, C. S. Hsu, L. Bai, H. M. Chen and X. Hu, Science, 2019, 364, 1091–1094 CrossRef CAS PubMed.
- G. Li, H. Abroshan, C. Liu, S. Zhuo, Z. Li, Y. Xie, H. J. Kim, N. L. Rosi and R. Jin, ACS Nano, 2016, 10, 7998–8005 CrossRef CAS PubMed.
- P. Atkins, T. Overton, J. Rourke, M. Weller and F. Armstrong, Shriver and Atkins' Inorganic Chemistry, Oxford University Press, USA, 2009 Search PubMed.
- G. Silversmit, D. Depla, H. Poelman, G. B. Marin and R. De Gryse, J. Electron Spectrosc. Relat. Phenom., 2004, 135, 167–175 CrossRef CAS.
- J. S. Griffith and L. E. Orgel, Q. Rev., Chem. Soc., 1957, 11, 381–393 RSC.
- Z. Pu, I. S. Amiinu, R. Cheng, P. Wang, C. Zhang, S. Mu, W. Zhao, F. Su, G. Zhang, S. Liao and S. Sun, Nano-Micro Lett., 2020, 12, 1–29 CrossRef.
- Y. Qu, Z. Li, W. Chen, Y. Lin, T. Yuan, Z. Yang, C. Zhao, J. Wang, C. Zhao, X. Wang, F. Zhou, Z. Zhuang, Y. Wu and Y. Li, Nat. Catal., 2018, 1, 781–786 CrossRef CAS.
- P. Li, Z. Jin, Y. Qian, Z. Fang, D. Xiao and G. Yu, Mater. Today, 2020, 35, 78–86 CrossRef CAS.
- D. Qi, Y. Liu, M. Hu, X. Peng, Y. Qiu, S. Zhang, W. Liu, H. Li, G. Hu, L. Zhuo, Y. Qin, J. He, G. Qi, J. Sun, J. Luo and X. Liu, Small, 2020, 16, 2004855 CrossRef CAS PubMed.
- P. Li, Z. Jin, Y. Qian, Z. Fang, D. Xiao and G. Yu, ACS Energy Lett., 2019, 4, 1793–1802 CrossRef CAS.
- R. Zhang, L. Jiao, W. Yang, G. Wan and H. L. Jiang, J. Mater. Chem. A, 2019, 7, 26371–26377 RSC.
- P. Li, Z. Jin, Z. Fang and G. Yu, Energy Environ. Sci., 2021, 14, 3522–3531 RSC.
- F. Li, Y. Li, X. C. Zeng and Z. Chen, ACS Catal., 2015, 5, 544–552 CrossRef CAS.
- T. Yang, R. Fukuda, S. Hosokawa, T. Tanaka, S. Sakaki and M. Ehara, ChemCatChem, 2017, 9, 1222–1229 CrossRef CAS PubMed.
- W. Zhang, Y. Hu, L. Ma, G. Zhu, Y. Wang, X. Xue, R. Chen, S. Yang and Z. Jin, Adv. Sci., 2018, 5, 1700275 CrossRef PubMed.
- S. Back, J. Lim, N. Y. Kim, Y. H. Kim and Y. Jung, Chem. Sci., 2017, 8, 1090–1096 RSC.
- J. Li, P. Pršlja, T. Shinagawa, A. J. Martín Fernández, F. Krumeich, K. Artyushkova, P. Atanassov, A. Zitolo, Y. Zhou, R. García-Muelas, N. López, J. Pérez-Ramírez and F. Jaouen, ACS Catal., 2019, 9, 10426–10439 CrossRef CAS.
- Y. Qu, L. Wang, Z. Li, P. Li, Q. Zhang, Y. Lin, F. Zhou, H. Wang, Z. Yang, Y. Hu, M. Zhu, X. Zhao, X. Han, C. Wang, Q. Xu, L. Gu, J. Luo, L. Zheng and Y. Wu, Adv. Mater., 2019, 31, 1904496 CrossRef CAS PubMed.
- Q. Xue, Y. Xie, S. Wu, T. S. Wu, Y. L. Soo, S. Day, C. C. Tang, H. W. Man, S. T. Yuen, K. Y. Wong, Y. Wang, B. T. W. Lo and S. C. E. Tsang, Nanoscale, 2020, 12, 23206–23212 RSC.
- S. Wang, Y. Song, S. Jin, X. Liu, J. Zhang, Y. Pei, X. Meng, M. Chen, P. Li and M. Zhu, J. Am. Chem. Soc., 2015, 137, 4018–4021 CrossRef CAS PubMed.
- R. Kobayashi, Y. Nonoguchi, A. Sasaki and H. Yao, J. Phys. Chem. C, 2014, 118, 15506–15515 CrossRef CAS.
- W. Hou, Acta Phys.-Chim. Sin., 2019, 35, 453–454 Search PubMed.
- T. Higaki, Y. Li, S. Zhao, Q. Li, S. Li, X. S. Du, S. Yang, J. Chai and R. Jin, Angew. Chem., Int. Ed., 2019, 58, 8291–8302 CrossRef CAS.
- Y. Du, H. Sheng, D. Astruc and M. Zhu, Chem. Rev., 2020, 120, 526–622 CrossRef CAS.
- M. Zhou, J. E. Dick and A. J. Bard, J. Am. Chem. Soc., 2017, 139, 17677–17682 CrossRef CAS PubMed.
- Z. Jin and A. J. Bard, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 12651–12656 CrossRef CAS PubMed.
- T. Yang and M. Ehara, Phys. Chem. Chem. Phys., 2017, 19, 3679–3687 RSC.
- X. Zhao, C. Kong, Z. Yang and T. Yang, ChemistrySelect, 2020, 5, 6939–6945 CrossRef CAS.
- S. J. Tauster, S. C. Fung and R. L. Garten, J. Am. Chem. Soc., 1978, 100, 170–175 CrossRef CAS.
- H. Stach, The Surface Science of Metal Oxides, Cambridge University Press, 1995, vol. 191 Search PubMed.
- Y. Yun, H. Sheng, J. Yu, L. Bao, Y. Du, F. Xu, H. Yu, P. Li and M. Zhu, Adv. Synth. Catal., 2018, 360, 4731–4743 CrossRef CAS.
- Y. Yao, S. Hu, W. Chen, Z. Q. Huang, W. Wei, T. Yao, R. Liu, K. Zang, X. Wang, G. Wu, W. Yuan, T. Yuan, B. Zhu, W. Liu, Z. Li, D. He, Z. Xue, Y. Wang, X. Zheng, J. Dong, C. R. Chang, Y. Chen, X. Hong, J. Luo, S. Wei, W. X. Li, P. Strasser, Y. Wu and Y. Li, Nat. Catal., 2019, 2, 304–313 CrossRef CAS.
- A. N. Desnoyer, W. He, S. Behyan, W. Chiu, J. A. Love and P. Kennepohl, Chem.–Eur. J., 2019, 25, 5259–5268 CrossRef CAS.
- W. He and P. Kennepohl, Faraday Discuss., 2019, 220, 133–143 RSC.
- A. Cirri, H. Morales Hernández, C. Kmiotek and C. J. Johnson, Angew. Chem., Int. Ed., 2019, 58, 13818–13822 CrossRef CAS PubMed.
- X. K. Wan, J. Q. Wang, Z. A. Nan and Q. M. Wang, Sci. Adv., 2017, 3, 1701823 CrossRef PubMed.
- A. J. Thomson and H. B. Gray, Curr. Opin. Chem. Biol., 1998, 2, 155–158 CrossRef CAS.
- M. Seth and T. Ziegler, Adv. Inorg. Chem., 2010, 62, 41–109 CrossRef CAS.
- N. Abbaspour, R. Hurrell and R. Kelishadi, J. Res. Med. Sci., 2014, 19, 164–174 Search PubMed.
- J. R. Chipperfield, in Encyclopedia of Food Sciences and Nutrition, Elsevier, 2003, pp. 3367–3373 Search PubMed.
- A. K. Boal and A. C. Rosenzweig, Chem. Rev., 2009, 109, 4760–4779 CrossRef CAS PubMed.
- G. E. O. Borgstahl, H. E. Parge, M. J. Hickey, W. F. Beyer, R. A. Hallewell and J. A. Tainer, Cell, 1992, 71, 107–118 CrossRef CAS.
- J. Wuerges, J. W. Lee, Y. I. Yim, H. S. Yim, S. O. Kang and K. D. Carugo, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 8569–8574 CrossRef CAS PubMed.
- D. P. Barondeau, C. J. Kassmann, C. K. Bruns, J. A. Tainer and E. D. Getzoff, Biochemistry, 2004, 43, 8038–8047 CrossRef CAS PubMed.
- S. Lindskog, Pharmacol. Ther., 1997, 74, 1–20 CrossRef CAS.
- M. T. Croft, A. D. Lawrence, E. Raux-Deery, M. J. Warren and A. G. Smith, Nature, 2005, 438, 90–93 CrossRef CAS PubMed.
- A. P. Cole, D. E. Root, P. Mukherjee, E. I. Solomon and T. D. P. Stack, Science, 1996, 273, 1848–1850 CrossRef PubMed.
- C. Citek, B. L. Lin, T. E. Phelps, E. C. Wasinger and T. D. P. Stack, J. Am. Chem. Soc., 2014, 136, 14405–14408 CrossRef CAS.
- S. I. Chan and S. S. F. Yu, Acc. Chem. Res., 2008, 41, 969–979 CrossRef CAS.
- H. Ohtsu, Y. Shimazaki, A. Odani, O. Yamauchi, W. Mori, S. Itoh and S. Fukuzumi, J. Am. Chem. Soc., 2000, 122, 5733–5741 CrossRef CAS.
- B. Gonzalez, J. Kouba, S. Yee, J. F. Kirner and W. R. Scheldt, J. Am. Chem. Soc., 1975, 97, 3247–3249 CrossRef CAS.
- C. M. Weekley, I. Kenkel, R. Lippert, S. Wei, D. Lieb, T. Cranwell, J. L. Wedding, A. S. Zillmann, R. Rohr, M. R. Filipovic, I. Ivanović-Burmazović and H. H. Harris, Inorg. Chem., 2017, 56, 6076–6093 CrossRef CAS PubMed.
- S. R. Doctrow, K. Huffman, C. B. Marcus, G. Tocco, E. Malfroy, C. A. Adinolfi, H. Kruk, K. Baker, N. Lazarowych, J. Mascarenhas and B. Malfroy, J. Med. Chem., 2002, 45, 4549–4558 CrossRef CAS PubMed.
- M. Baudry, S. Etienne, A. Bruce, M. Paluck, E. Jacobsen and B. Malfroy, Biochem. Biophys. Res. Commun., 1993, 192, 964–968 CrossRef CAS PubMed.
- R. O. Costa, S. S. Ferreira, C. A. Pereira, J. R. Harmer, C. J. Noble, G. Schenk, R. W. A. Franco, J. A. L. C. Resende, P. Comba, A. E. Roberts, C. Fernandes and A. Horn, Front. Chem., 2018, 6, 491 CrossRef CAS PubMed.
- I. Batinić-Haberle, J. S. Rebouças and I. Spasojević, Antioxidants Redox Signal., 2010, 13, 877–918 CrossRef.
- Q. Zhang, J. Shu, Y. Zhang, Z. Xu, J. Yue, X. Liu, B. Xu, Z. Chen and W. Jiang, Dalton Trans., 2020, 49, 10261–10269 RSC.
- W. Jiang, B. Xu, Z. Xiang, S. Huang, F. Liu and Y. Wang, J. Chem. Sci., 2013, 125, 1145–1149 CrossRef CAS.
- B. Xu, W. Jiang, X. Liu, F. Liu and Z. Xiang, J. Biol. Inorg Chem., 2017, 22, 625–635 CrossRef CAS PubMed.
- A. G. Majouga, E. K. Beloglazkina, A. A. Moiseeva, O. V. Shilova, E. A. Manzheliy, M. A. Lebedeva, E. S. Davies, A. N. Khlobystov and N. V. Zyk, Dalton Trans., 2013, 42, 6290–6293 RSC.
- C. Esmieu, P. Raleiras and G. Berggren, Sustainable Energy Fuels, 2018, 2, 724–750 RSC.
- X. Zhao, S. Q. Zang and X. Chen, Chem. Soc. Rev., 2020, 49, 2481–2503 RSC.
- L. Ye, Q. Song, B. T. W. Lo, J. Zheng, D. Kong, C. A. Murray, C. C. Tang and S. C. E. Tsang, Angew. Chem., Int. Ed., 2017, 56, 10711–10716 CrossRef CAS.
- X. Feng, Y. Song, J. S. Chen, Z. Xu, S. J. Dunn and W. Lin, J. Am. Chem. Soc., 2021, 143, 1107–1118 CrossRef CAS PubMed.
- S. Pullen, H. Fei, A. Orthaber, S. M. Cohen and S. Ott, J. Am. Chem. Soc., 2013, 135, 16997–17003 CrossRef CAS PubMed.
- C. E. Bien, K. K. Chen, S. C. Chien, B. R. Reiner, L. C. Lin, C. R. Wade and W. S. W. Ho, J. Am. Chem. Soc., 2018, 140, 12662–12666 CrossRef CAS PubMed.
- Z. Zhang, W. Liu, Y. Zhang, J. Bai and J. Liu, ACS Catal., 2021, 11, 313–322 CrossRef CAS.
- J. Wang, T. Heil, B. Zhu, C. W. Tung, J. Yu, H. M. Chen, M. Antonietti and S. Cao, ACS Nano, 2020, 14, 8584–8593 CrossRef CAS PubMed.
- W. Liu, W. Hu, L. Yang and J. Liu, Nano Energy, 2020, 73, 104750 CrossRef CAS.
- A. Samanta, B. B. Dhar and R. N. Devi, New J. Chem., 2012, 36, 2625–2629 RSC.
- S. Gao, H. Lin, H. Zhang, H. Yao, Y. Chen and J. Shi, Adv. Sci., 2019, 6, 1801733 CrossRef PubMed.
- L. Huang, J. Chen, L. Gan, J. Wang and S. Dong, Sci. Adv., 2019, 5, eaav5490 CrossRef CAS PubMed.
- F. He, L. Mi, Y. Shen, T. Mori, S. Liu and Y. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 35327–35333 CrossRef CAS PubMed.
- C. Zhao, C. Xiong, X. Liu, M. Qiao, Z. Li, T. Yuan, J. Wang, Y. Qu, X. Q. Wang, F. Zhou, Q. Xu, S. Wang, M. Chen, W. Wang, Y. Li, T. Yao, Y. Wu and Y. Li, Chem. Commun., 2019, 55, 2285–2288 RSC.
- Y. Wang, Z. Zhang, G. Jia, L. Zheng, J. Zhao and X. Cui, Chem. Commun., 2019, 55, 5271–5274 RSC.
- H. Li, Y. Wen, M. Jiang, Y. Yao, H. Zhou, Z. Huang, J. Li, S. Jiao, Y. Kuang and S. Luo, Adv. Funct. Mater., 2021, 2011289 CrossRef CAS.
- Y. Wang, M. Li, J. K. Chang, D. Aurelio, W. Li, B. J. Kim, J. H. Kim, M. Liscidini, J. A. Rogers and F. G. Omenetto, Nat. Commun., 2021, 12, 1–9 CrossRef PubMed.
- P. D. Nellist and S. J. Pennycook, Science, 1996, 274, 413–415 CrossRef CAS.
- Z. Zhang, Y. Zhu, H. Asakura, B. Zhang, J. Zhang, M. Zhou, Y. Han, T. Tanaka, A. Wang, T. Zhang and N. Yan, Nat. Commun., 2017, 8, 1–10 CrossRef PubMed.
- K. Ding, A. Gulec, A. M. Johnson, N. M. Schweitzer, G. D. Stucky, L. D. Marks and P. C. Stair, Science, 2015, 350, 189–192 CrossRef CAS PubMed.
- L. Nie, D. Mei, H. Xiong, B. Peng, Z. Ren, X. I. P. Hernandez, A. DeLaRiva, M. Wang, M. H. Engelhard, L. Kovarik, A. K. Datye and Y. Wang, Science, 2017, 358, 1419–1423 CrossRef CAS PubMed.
- Y. Chen, S. Ji, W. Sun, W. Chen, J. Dong, J. Wen, J. Zhang, Z. Li, L. Zheng, C. Chen, Q. Peng, D. Wang and Y. Li, J. Am. Chem. Soc., 2018, 140, 7407–7410 CrossRef CAS PubMed.
- X. Zeng, J. Shui, X. Liu, Q. Liu, Y. Li, J. Shang, L. Zheng and R. Yu, Adv. Energy Mater., 2018, 8, 1701345 CrossRef.
- H. Zhang, P. An, W. Zhou, B. Y. Guan, P. Zhang, J. Dong and X. W. Lou, Sci. Adv., 2018, 4, eaao6657 CrossRef PubMed.
- T. Ikuno, S. Grundner, A. Jentys, G. Li, E. Pidko, J. Fulton, M. Sanchez-Sanchez and J. A. Lercher, J. Phys. Chem. C, 2019, 123, 8759–8769 CrossRef CAS.
- P. Peng, L. Shi, F. Huo, C. Mi, X. Wu, S. Zhang and Z. Xiang, Sci. Adv., 2019, 5, eaaw2322 CrossRef CAS PubMed.
- L. Liu, M. Lopez-Haro, C. W. Lopes, S. Rojas-Buzo, P. Concepcion, R. Manzorro, L. Simonelli, A. Sattler, P. Serna, J. J. Calvino and A. Corma, Nat. Catal., 2020, 3, 628–638 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |