
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe identification and characterization of an oxalyl-CoA synthetase from grass pea (Lathyrus sativus L.)†
Moshe
Goldsmith
 *a,
Shiri
Barad
a,
Yoav
Peleg
b,
Shira
Albeck
b,
Orly
Dym
b,
Alexander
Brandis
b,
Tevie
Mehlman
b and
Ziv
Reich
*a
*a,
Shiri
Barad
a,
Yoav
Peleg
b,
Shira
Albeck
b,
Orly
Dym
b,
Alexander
Brandis
b,
Tevie
Mehlman
b and
Ziv
Reich
*a
aDept. of Biomolecular Sciences, Weizmann Institute of Science, Rehovot, 7610001, Israel. E-mail: moshe.goldsmith@weizmann.ac.il; ziv.reich@weizmann.ac.il; Fax: +972-8-9344118; Tel: +972-8-9343278 Tel: +972-8-9342982
bDept. of Life Science Core Facilities, Weizmann Institute of Science, Rehovot, 7610001, Israel
First published on 8th February 2022
Abstract
Oxalic acid is a small metabolite found in many plants. It serves as protection from herbivores, a chelator of metal ions, a regulator of calcium levels, and additional tasks. However, it is also a strong di-carboxylic acid that can compromise plant viability by reducing cellular pH. Several metabolic pathways have evolved to control oxalate levels in plants by enzymatic degradation. Among them is the pathway that utilizes oxalyl-CoA synthetase (OCS, EC 6.2.1.8) and ATP to convert oxalate to oxalyl-CoA. Oxalyl-CoA can then be degraded to CO2 or utilized as a precursor for the synthesis of other compounds. In grass pea (Lathyrus sativus L.), a grain legume grown in Asia and Africa for human and animal consumption, the neurotoxic compound β-N-oxalyl-L-α,β-diaminopropionic acid (β-ODAP) is synthesized from oxalyl-CoA and L-α,β-diaminopropionic acid (L-DAPA). Here, we report on the identification and characterization of oxalyl CoA-synthetase from grass pea (LsOCS). The gene encoding LsOCS was amplified from grass pea, and then expressed and purified from E. coli cells as an untagged, monomeric protein of 56 kDa. Its catalytic efficiency with oxalate, KoxalateM = 71.5 ± 13.3 μM, Vmax = 8.2 ± 0.8 μmole min−1 mg−1, was similar to that of OCS homologs from Arabidopsis thaliana (AtAAE3) and Medicago truncatula (MtAAE3). The enzyme was crystalized in complex with AMP and is the first OCS whose structure was determined in the thioester-forming conformation. Finally, we propose that substituting LsOCS with an oxalate oxidase or decarboxylase may reduce the levels of β-ODAP in grass pea.
Introduction
Oxalate is an abundant plant metabolite that serves diverse functions including pH homeostasis,1 ion balance and regulation of calcium levels,2 metal detoxification and tolerance,1,3–5 defense against insects and herbivores,6 tissue support,7 nutrient sharing with bacterial symbionts,8 and regulation of light distribution to chloroplasts in shade plants.9 In plants, it is produced from glyoxylate, glycolate, oxaloacetate or ascorbate during photorespiration and the glyoxylate cycle.10 Despite its beneficial functions, oxalate is also highly acidic and a strong chelator that may reduce the availability of essential ions. Consequently, its levels in plants need to be tightly regulated. This is achieved by mechanisms that control its synthesis, precipitation (predominantly into calcium-based crystals), and degradation.2,10,11 The latter proceeds mainly by oxidation or by CoA-dependent decarboxylation,12 in plants, and by direct decarboxylation, in bacteria and fungi.13 Decarboxylation of oxalyl-CoA is catalyzed by the enzyme oxalyl-CoA decarboxylase (OXC; 4.1.1.8), which catalyzes the thiamine diphosphate-dependent conversion of oxalyl-CoA to formyl-CoA and carbon dioxide.14 Formyl-CoA can then be further converted to formate and, subsequently, to carbon dioxide in reactions catalyzed by formyl-CoA hydrolase and formate dehydrogenase, respectively.11 The initial step in this pathway is performed by the enzyme oxalyl-CoA synthetase (OCS).Oxalyl-CoA synthetase (EC 6.2.1.8) is an ATP-dependent enzyme that catalyzes the ligation of oxalate to CoA, forming oxalyl-CoA (Scheme 1a and b). It has been identified in plants such as Arabidopsis thaliana,15Medicago truncatula,16Vigna umbellate,17Oryza sativa18 and Glycine soja5 and in the yeast Saccharomyces cerevisiae,19 where it plays a central role in oxalate catabolism. Its encoding gene, AAE3, is induced by oxalate and its deletion in dicots, such as Arabidopsis thaliana and Medicago truncatula, in which there is no known oxalate oxidase,20 was shown to inhibit oxalate degradation to carbon dioxide and to increase the intracellular concentration of oxalate.15,16 The subsequent increase in oxalate concentration was shown to interfere with seed coat-development and lead to reduced and delayed seed germination.15,21 An important role of OCS in plants is to metabolize exogenous oxalate secreted by certain pathogenic fungi, e.g., Sclerotinia sclerotiorum, as a virulence factor that facilitates infection.22,23 Indeed, inactivation of OCS both in A. thaliana and in M. truncatula was shown to increase their susceptibility to infection by S. sclerotiorum.15,16
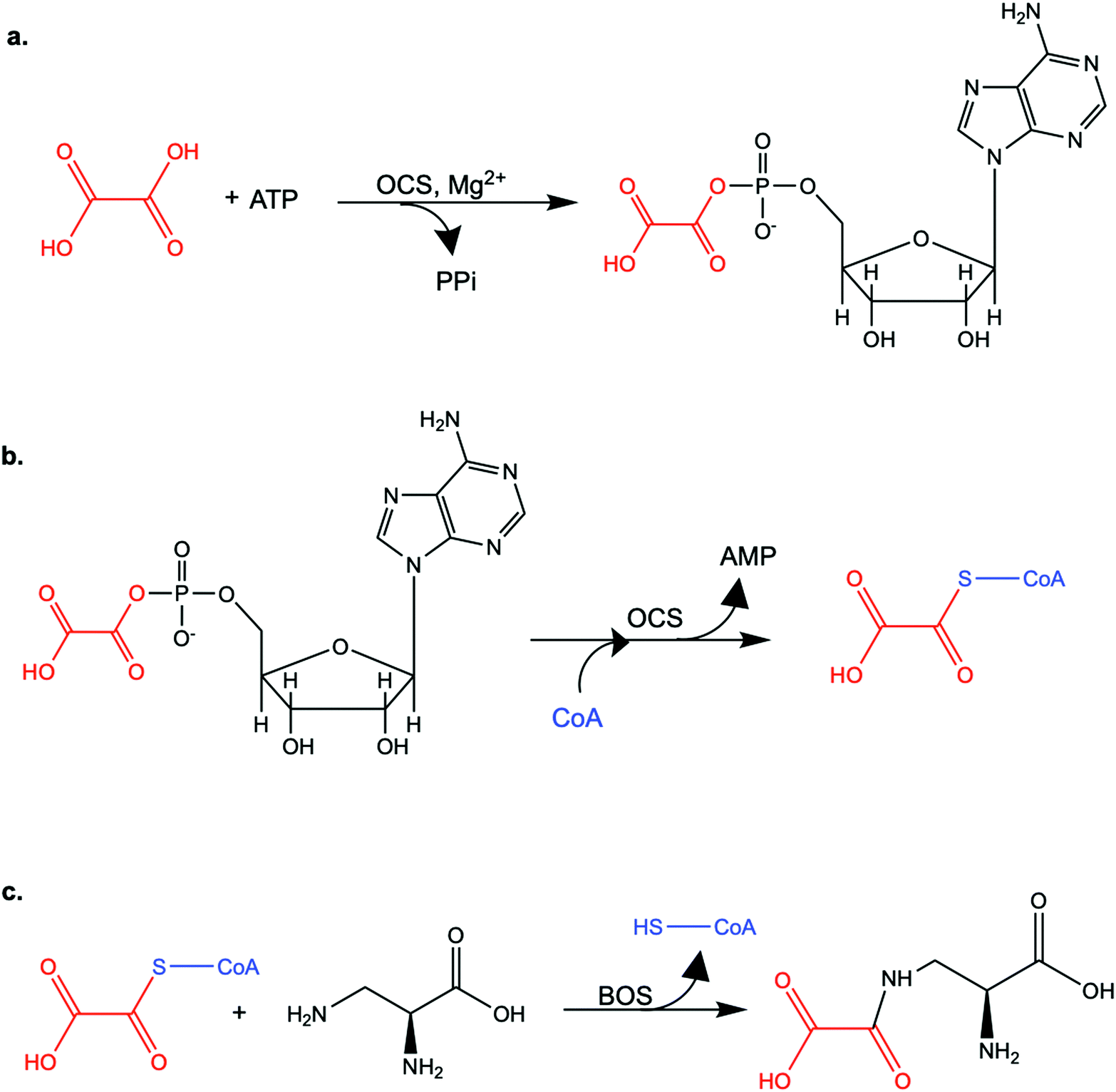 | ||
| Scheme 1 The enzymatic reactions catalyzed by LsOCS and LsBOS. a. In the first step, LsOCS catalyzes the ligation of oxalate (red) to ATP in the presence Mg2+ ions. A high-energy oxalyl-adenylate intermediate is formed and di-phosphate (PPi) is released. b. In the second step, the pantetheine thiol group of coenzyme A (CoA, blue) attacks the carboxylate carbon of oxalyl-adenylate, releasing AMP and oxalyl-CoA. c. The ligation of oxalyl-CoA to L-α,β-diaminopropionic acid is catalyzed by β-ODAP synthase (BOS) and produces β-N-oxalyl-L-α,β-diaminopropionic acid (β-ODAP), releasing CoA (blue). | ||
OCS was identified more than 50 years ago24 in grass pea (Lathyrus sativus), a legume crop that is grown for food and forage purposes in parts of South Asia and Africa.25 It was suggested to be part of a metabolic network that synthesizes a neurotoxic compound β-N-oxalyl-L-α,β-diaminopropionic acid (β-ODAP, Scheme 1c) using an enzyme that ligates oxalyl-CoA and L-α,β-diaminopropionic acid.24,26 The compound β-ODAP may cause lathyrism, a neurological disorder characterized by spastic paralysis of the lower limbs,27–29 if consumed as a primary diet component over a prolonged period of time. Since OCS in grass pea uses oxalate as a substrate and is likely involved in the synthesis of β-ODAP, an attempt has been made to eliminate β-ODAP production in grass pea by the introduction of a constitutively expressed fungal oxalate decarboxylase gene.30 Indeed, a reduction of up to 75% in oxalate levels and 73% in β-ODAP levels were obtained in the seeds of the transgenic grass pea relative to the wild type plant. However, despite the significant reduction in β-ODAP concentration, this genetically modified crop was still not devoid of the neurotoxin.
We identified and cloned the gene encoding OCS from L. sativus (LsOCS), expressed the protein recombinantly, characterized its catalytic activity and determined its structure. We show that LsOCS is a bonne fide oxalyl-CoA synthetase that catalyzes the ligation of oxalate to CoA, yet exhibits low promiscuous activity with glyoxalate. We determine that LsOCS has a moderate thermal stability, which is not significantly enhanced by binding of its substrates. We then describe the crystal structure we obtained of LsOCS, captured in a thioester-forming conformation, and compare it with the structure of its homolog from Arabidopsis thaliana (AtAAE3) – the only other plant OCS crystalized so far, and with structures of other members of the ANL superfamily of enzymes.
Results
Identification of the gene encoding LsOCS in grass pea
In order to identify the ORF encoding LsOCS in grass pea we searched sequence databases for homologous sequences in other legumes. Specifically, we chose four related legumes: Arachis duranensis (XR_001592575.1), Cicer arietinum (XM_004514691.2), Vigna radiata (XM_014659057.1) and Glycine max (XM_014764165.1). Using multiple sequence alignments, we identified a consensus sequence region of ∼1 kb and used it to design gene-specific primers. The region was then PCR-amplified from purified genomic grass pea DNA (Fig S1, ESI†) and sequenced. The sequence of the amplified region was compared to a published grass pea transcriptome,31 enabling the identification of the entire transcript. The identified transcript sequence has been deposited in the GenBank database (GenBank: MK492104.1) as the sequence of LsOCS.We performed a database search using this sequence and found OCS homologs from >90 different plants that had a relatively high sequence identity (>76%) to LsOCS (Fig. S2, ESI†). The most similar sequence, of an oxalyl-CoA ligase from Medicago truncatula (GenBank: XP_003599555.1), displayed ∼88% amino-acid identity to LsOCS. During the course of this work, a putative OCS sequence from grass pea was deposited by Kushwah, N. S. et al., in GenBank (GenBank: MH469748.1). There are, however, two differences between the protein sequence we derived and the one deposited by Kushwah, N. S. et al. While the sequence we identified contains Ala at position 474 and Thr at position 497, the deposited sequence lists Pro at both positions. In the multiple sequence alignment of OCS homologs we made, none of them possesses a Pro residue at these positions.
LsOCS was expressed and purified from E. coli cells as a soluble, monomeric protein
To ensure a high expression level of the recombinant protein, the gene encoding LsOCS was N-terminally fused to a cleavable His-bdSUMO tag.32,33 The expressed protein was purified from bacterial cells using a Ni-NTA column followed by on-column cleavage using a bdSUMO protease. An additional size-exclusion chromatography step resulted in >95% pure, untagged protein (Fig. S4, ESI†), which migrated as a single band with an apparent molecular weight of ∼56 kDa (data not shown), consistent with the expected size of a monomeric protein.LsOCS displays its highest catalytic efficiency with oxalic acid
We examined the ability of the purified LsOCS protein to ligate CoA and oxalate in the presence of ATP and Mg2+ (Scheme 1). Enzymatic activity was measured using a coupled enzyme assay (see Materials and methods). The turnover number (kcat) and Michaelis (KoxalateM) constant derived for the reaction were 7.6 ± 0.7 s−1 and 71.5 ± 13.3 μm respectively, giving rise to a catalytic efficiency (kcat/KoxalateM) of 1.1 × 105 M−1 s−1 (Fig. 1 and Table 1). The corresponding specific activity was 8.2 ± 0.8 μmole min−1 mg−1. To verify that ATP hydrolysis is coupled to the ligation of CoA to oxalic acid we measured the concentration of unligated CoA at different time points along the reaction, using a DTNB assay that detects free thiol groups (Materials and ethods, Fig. S5, ESI†). We found that free CoA levels were reduced by ∼80% within 10 min following the addition of substrates (Fig. S5, ESI†). Control reactions lacking the enzyme, ATP, or oxalic acid resulted in no reduction in the concentration of free CoA (Fig. S5, ESI†). Finally, we examined samples of the reaction mixture before and after incubation with the enzyme using LC-MS, and found that >95% of the initial CoA substrate had been converted to oxalyl-CoA under the reaction conditions used (Fig. S6, ESI†).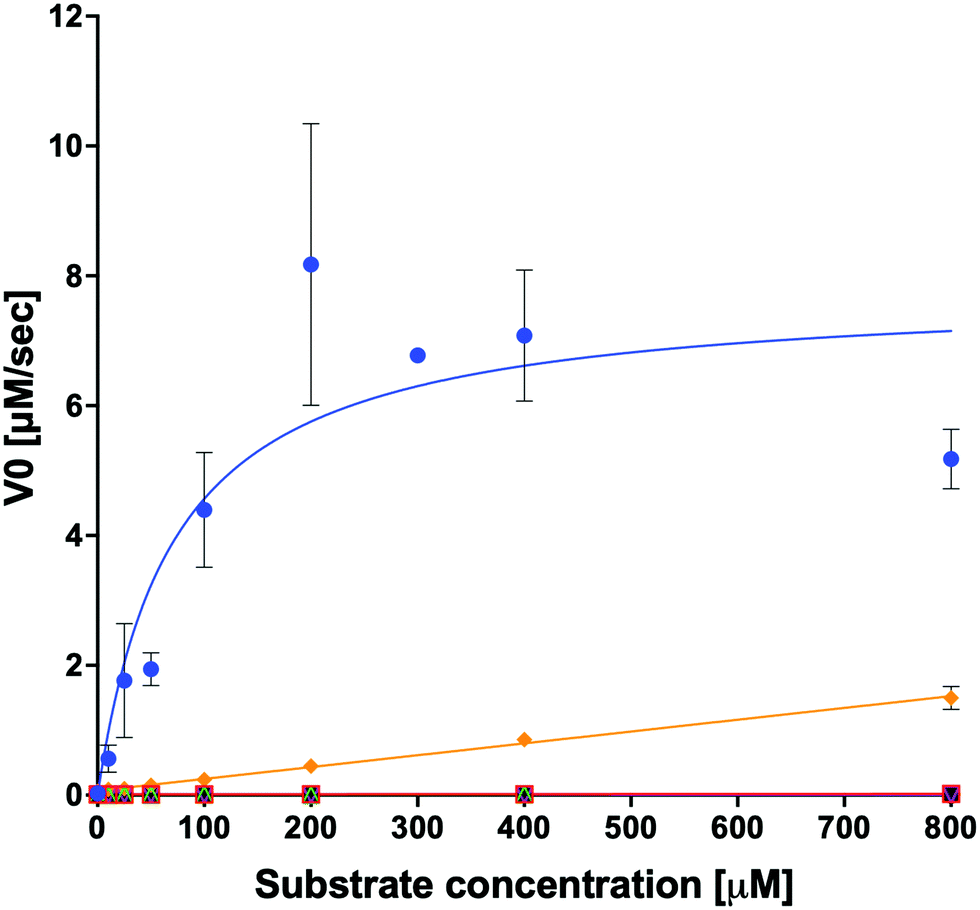 | ||
Fig. 1 Kinetics of organic acid ligation to CoA by LsOCS. The rates of CoA ligation to: oxalate ( ), malonate ( ), malonate ( ), glyoxalate ( ), glyoxalate ( ), succinate ( ), succinate ( ), lactate (○) and glycolate ( ), lactate (○) and glycolate ( ) were assayed using purified LsOCS (1 μM) and excess concentrations of ATP, CoA, and Mg+2 (≥1 mM). The data for oxalate was fitted to the Michaelis–Menten equation; error bars denote SD of three independent biological replicates. Data for all other substrates was fitted to a linear regression equation; error bars denote SE of the fit. ) were assayed using purified LsOCS (1 μM) and excess concentrations of ATP, CoA, and Mg+2 (≥1 mM). The data for oxalate was fitted to the Michaelis–Menten equation; error bars denote SD of three independent biological replicates. Data for all other substrates was fitted to a linear regression equation; error bars denote SE of the fit. | ||
| Substrate | k cat [s−1] | ±[s−1] | K M [μM] | ±[μM] | k cat/KM [s−1 M−1] | ±[s−1 M−1]ab |
|---|---|---|---|---|---|---|
| a Errors derived from two independent measurements per substrate except for oxalate, for which 3 independent measurements were performed. All measurements were preformed using excess amounts of CoA, ATP and Mg2+ over the carboxylate substrates. b The error in kcat/kM for oxalate was calculated using the individual errors in kcat and KM. c ND – not detectable. | ||||||
| Oxalate | 7.6 | 0.7 | 71.5 | 13.3 | 1.1 × 105 | 2.2 × 104 |
| Glyoxalate | — | — | — | — | 1.8 × 103 | 3.8 × 101 |
| Malonate | — | — | — | — | 1.2 × 101 | 2.1 |
| Succinate | — | — | — | — | NDc | ND |
| Glycolate | — | — | — | — | ND | ND |
| Lactate | — | — | — | — | ND | ND |
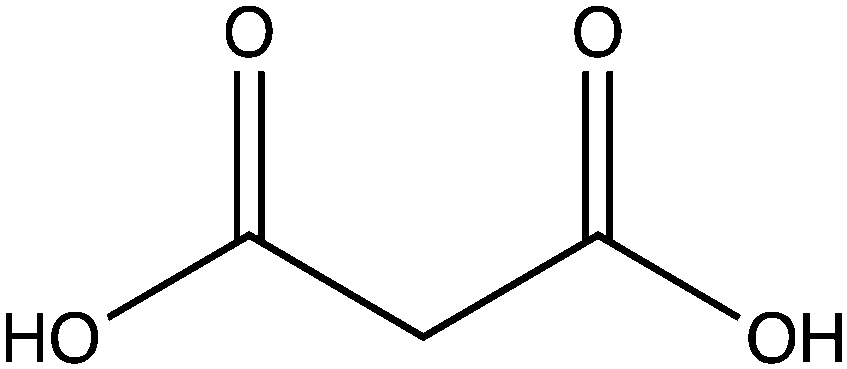
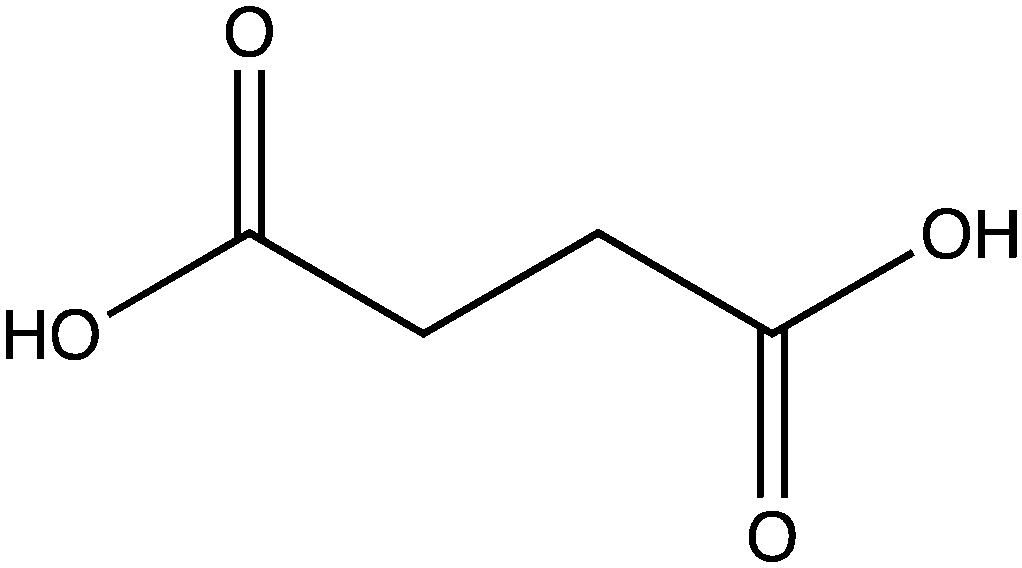
In order to examine its substrate specificity, we measured the activity of LsOCS with a number of carboxylates, all of which are prevalent plant metabolites that have similar structures to oxalate (Table 2). Two of these, malonate and succinate, are 3- and 4-carbon chain dicarboxylic acid analogues of oxalate (Table 2). Moving from the 2-carbon chain oxalate to malonate and to the longer succinate substrate, resulted in a 4-order of magnitude decrease in the enzyme's catalytic efficiency (kcat/KM) for malonate (Table 1) and the activity with succinate was bellow detection limit. With glyoxylate, a C2 monocarboxylic acid, in which one of the carboxylic groups present in oxalate was reduced to an aldehyde, a sharp 61-fold decrease in catalytic efficiency (kcat/KM) was observed (Table 1). This is likely due to loss of planarity of the hydrated species (the aldehyde form is converted to a geminal-diol in aqueous solutions). The complete loss of planarity probably underlies the lack of detectable catalytic efficiencies observed with another C2 monocarboxylate, glycolate, and with the 3-carbon monocarboxylate, lactate (Fig. 1 and Table 1).
| Name | Structure |
|---|---|
| Oxalate |

|
| Malonate |
|
| Succinate |
|
| Glyoxylate |
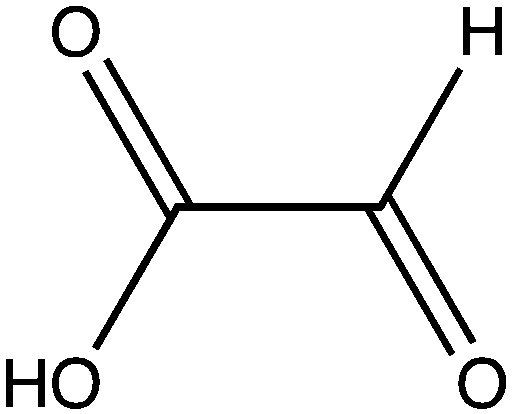
|
| Glycolate |

|
| Lactate |
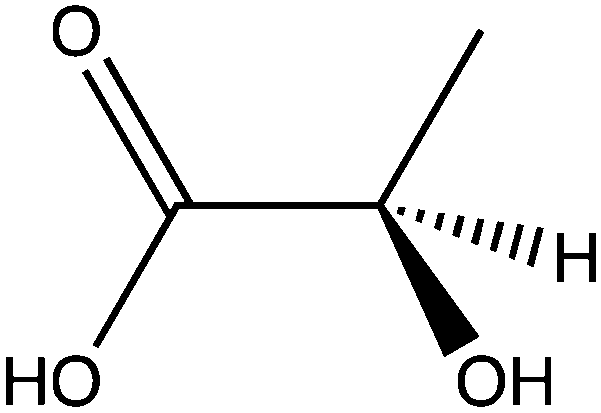
|
LsOCS is most active at pH 8
The activity of LsOCS was assayed at different pHs, ranging from 6 to 8.5 (Fig. S7, ESI†). The highest activity was measured at pH 8. Similar results were reported for the AtAAE315 and MtAAE316 homologues of LsOCS.The thermal stability of LsOCS is slightly increased by CoA or ATP binding
We used differential scanning fluorimetry (DSF) to assess the effect of ligand binding on the stability of LsOCS. In its Apo state, LsOCS was found to have moderate thermal stability, with an apparent midpoint temperature of unfolding (Tappm) of 43.9 °C (Table 3). Addition of CoA at 100-fold or ATP + Mg2+ at 10-fold molar excess over LsCOS, led to an increase of 1.8 °C and 2.7 °C in the Tappm of the protein, respectively (Table 3). The addition of either oxalic acid at 100-fold, CoA at 10-fold or Mg2+ at 70-fold excess, on the other hand, had no effect the thermal stability of the enzyme (Table 3). Thus, the thermal stability of OCS was only slightly increased in the presence of a large, 100-fold, excess of CoA but not by a similar concentration of oxalic acid. A greater increase was obtained using only a 10-fold excess of ATP + Mg2+ but not by a large excess of magnesium. This may indicate that in its Apo form, LsOCS prefers binding the combination of ATP and magnesium over the binding of CoA or oxalate.| Molar ratioa |
T
appm![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b (°C) b (°C) |
±(°C)c | ΔTappm relative to LsOCS-Apo (°C) | |
|---|---|---|---|---|
| a Molar ratio of LsOCS to the added ligand. b T appm is the apparent midpoint temperature of melting determined by differential scanning fluorimetry (DSF). Results represent the mean of two independent measurements. c Experimental error from two independent measurements. | ||||
| LsOCS – apo | 43.9 | 0.02 | — | |
| LsOCS + CoA | 1:100 |
45.7 | 0.03 | 1.8 |
| LsOCS + CoA | 1:10 |
44.2 | 0.01 | 0.3 |
| LsOCS + Oxalate | 1:100 |
44.1 | 0.02 | 0.2 |
| LsOCS + Mg2+ | 1:70 |
44.2 | 0.01 | 0.3 |
| LsOCS + ATP+ Mg2+ | 1:10 |
46.6 | 0.02 | 2.7 |
The crystal structure of LsOCS complexed with AMP
LsOCS was co-crystallised with Mg·ATP and sodium oxalate and the structure of the enzyme, bound to AMP, was obtained at 2.7 Å resolution (Table 4). No electron density was observed for the oxalyl moiety or for a Mg2+ ion. The crystals appeared after a few weeks, at which time the phosphoester bond between the oxalyl and AMP may have been hydrolyzed and the oxalate could have dissociated from the active site. Similar incidents of hydrolysis of the adenylate intermediate were reported for AtAAE3,34 and for the phenylalanine-adenylating domain of gramicidin S non-ribosomal peptide synthetase, GrsA.35| Data collection | |
|---|---|
| a Values in parentheses refer to the data of the corresponding upper resolution shell. | |
| PDB code | 6QJZ |
| Space group | P22121 |
| Cell dimensions: | |
| a,b,c (Å) | 61.99 74.26 101.68 |
| α,β,γ (°) | 90, 90, 90 |
| No. of copies in a.u. | 1 |
| Resolution (Å) | 47.59–2.7 |
| Upper resolution shell (Å) | 2.797–2.7 |
| Unique reflections | 13429 (1294)a |
| Completeness (%) | 99.97 (100.00) |
| Multiplicity | 19.7 (20.2) |
| Average I/σ(I) | 21.78 (6.76) |
| R-pim | 0.1858 (0.2636) |
| CC1/2 | 0.851 (0.782) |
| Refinement | |
| Resolution range (Å) | 47.59–2.7 |
| No. of reflections (I/σ(I) > 0) | 13429 |
| No. of reflections in test set | 649 |
| R-working/R-free | 0.2152/0.2306 |
| No. of protein atoms | 3797 |
| No. of water molecules | 117 |
| Overall average B factor (Å2) | 22.37 |
| Root mean square deviations: | |
| – Bond length (Å) | 0.014 |
| – Bond angle (°) | 1.69 |
| Ramachandran plot | |
| Most favored (%) | 96.2 |
| Additionally allowed (%) | 3.6 |
| Disallowed (%) | 1.0 |
By virtue of its activity and sequence, LsOCS belongs to the acyl CoA-synthetase enzyme family, which is a subfamily of the ANL superfamily of adenylating enzymes.36 Similar to other members of this superfamily (Fig. 2), LsOCS exhibits a two-domain architecture, comprising a large N-terminal domain and a smaller C-terminal domain (Fig. 3a). The N-terminal domain consists of residues 1–415 and contains three β-sheets and 12α-helices arranged in an αβαβα domain structure. The C-terminal domain (residues 420–521) contains two β-sheets: an antiparallel, two stranded β-sheet and a central three-stranded β-sheet packed between three helices. The N- and C-terminal domains are connected via a small hinge-loop region 417IKEL420 (Fig. 3a). This loop contains a central hinge residue, Lys418, which is commonly replaced by an Asparagine or even a Serine residue in other homologs of the ANL superfamily (Fig. 2). In homologous enzymes, the hinge residue serves as a pivot for the rotation of the C-terminal domain upon the transition between the adenylate- and thioester-forming reactions.36 The hinge loop is preceded by Arg416, a highly conserved residue known to function in the positioning and stabilization of the ATP substrate and the resulting AMP moiety in the binding pocket through interactions with the nucleotide ribose hydroxyls.36 In the structure of LsOCS, Lys418 was found bound to the ribose of AMP, however, Arg416 was detached from it (Fig. 3b).
 | ||
| Fig. 2 Structural based sequence alignment of LsOCS and homologous proteins. Depicted is a protein sequence alignment of OCS from Lathyrus Sativus (LsOCS) to homologs with known structures from the ANL superfamily: OCS from Arabidopsis thaliana (AtAAE3), 4-chlorobenzoyl-CoA ligase/synthetase (CBL) from Alcaligenes sp. (AsCBL), methylmalonyl Coenzyme A synthetase (MatB) from Rhodopseudomonas palustris (RpMatB), long chain fatty acyl-CoA synthetase from Thermus thermophillus (TtFattyAcs), 4-coumarate CoA ligase from Populus tomentosa (Pt4CL), and o-succinylbenzoate CoA synthetase (MenE) from Bacillus subtilis (BsMenE). LsOCS secondary structure elements are labeled above the corresponding sequence; α-helices are depicted as spirals, and β-strands as arrows. Residues conserved in all proteins are shown as red blocks. P-loop residues are marked by a green arrow. Residues of LsOCS that interact with AMP are marked by green asterisks. The hinge residue Lys418 is marked by an inverted, green triangle. The alignment was done using MultAlin61 and the figure was created using ESPript.62 | ||
 | ||
| Fig. 3 Structural features of LsOCS. a. The crystal structure of LsOCS with the N- (residues 1–411) and C-terminal (residues 420–521) domains shown in grey and cyan, respectively. The P-loop (177SGTTSRP183) is shown in red and the hinge-loop region (417IKEL420) is shown in purple. AMP is shown as a stick model with carbon atoms in orange, oxygen in red and nitrogen in blue. b. Plot of the interactions between LsOCS and the bound AMP. Residues making hydrogen bonds with the ligand are represented in ball and stick, whereas residues making hydrophobic interactions are shown as red crowns. The figure was created using LigPlus. c. The structures of LsOCS (PDB ID: 6QJZ), AsCBL in its thioester forming conformation (PDB ID: 3CW9) and AtAAE3 in its adenylate-forming confirmation (PDB ID: 5IE3) were superimposed, yet only their P-loops and AMP ligands are shown for simplicity. The P-loops of LsOCS (red), AtAAE3 (blue) and AsCBL (yellow) are shown next to their respective AMP ligands: LsOCS (magenta sticks), AtAAE3 (blue sticks) and AsOCS (yellow sticks). d. The oxalyl-binding pocket of AtAAE3 is shown as a gray surface. The oxalate and AMP ligands (blue sticks) are surrounded by the pocket forming residues (yellow sticks). The pocket forming residues of LsOCS (pink sticks) were overlaid. Figures a, c and d were created using PyMOL.63 | ||
The active site of LsOCS, like that of other ANL enzymes, is located at the interface between the N- and C-terminal domains. The AMP moiety resides in a cleft on the surface of the N-terminal domain (Fig. 3a and Fig. S8, ESI†). This position is the same as that occupied by AMP in other structures, including its Arabidopsis homolog (PDB ID: 5IE3)34 and a chlorobenzoyl-CoA ligase (CBL) homolog from Alcaligenes sp. (AsCBL, PDB ID: 3CW9)37 (Fig. 3c). Within the cleft, AMP interacts with eleven residues (Fig. 2 and 3b), nine of which are located in the N-terminal domain (His221, Ser296, Ala297, Ser298, Ala318, Ala320, Met321, Thr322, Asp401), one in the hinge region (Lys418) and one in the C-terminal domain (Lys427). Of these nine residues, Thr322 and Asp401 are highly conserved amongst all ANL family members, with the former residue interacting with the phosphate oxygens of the nucleotide and the latter with its ribose hydroxyls. As discussed below, the P-loop (177SGTTSRP183; marked with a green arrow in Fig. 2 and in red in Fig. 3a), which plays a critical role in the binding of ATP, points away from the binding site, similar to its orientation in AsCBL crystalized in its thioester-forming conformation (Fig. 3c).
Oxalyl CoA-synthetase from Arabidopsis thaliana (AtAAE3) is a close homolog (74.7% sequence identity) of LsOCS. Currently, it is the only oxalyl CoA-synthetase whose structures have been deposited in the Protein Data Bank (PDB).34 As mentioned above, the oxalyl moiety was not detected in the crystal structure of LsOCS. However, a binding pocket for oxalate was observed in the structure of AtAAE3, which was co-crystalized with oxalate (PDB ID: 5IE3). The pocket is shaped by eight residues in AtAAE3 (Val215-His216, Cys288-Ser289, Ala313-Met314, His319 and Lys500), three of which (Ser289, His319 and Lys500) directly interact with the substrate.34 While Lys500 is present in all ANL enzymes and is essential for the adenylation reaction,36 Ser289 and His319 are conserved only amongst oxalyl-CoA synthetases.34 The latter two residues were therefore proposed to serve primarily in the binding and positioning of oxalate, governing the substrate specificity of this subset of acyl-CoA synthetases.34 We found that the conformations of the corresponding residues in LsOCS (Val222-His223, Cys295-Ser296, Ala320-Met321, and His326) overlapped with those of AtAAE3 (Fig. 3d). The only oxalyl binding residue missing from the binding pocket structure of LsOCS was Lys507, which corresponds to Lys500 in AtAAE3. This is because no electron density was observed for the loop region 502KTATGKIL509, within which Lys507 resides in LsOCS (Fig. 3a). In addition, since Lys507 is part to the C-terminal region of the enzyme, it moves away from the active site upon transition to the thioester binding conformation (see below), thus losing its binding contacts with the oxalate substrate.
The LsOCS structure exhibits a thioester forming conformation
Acyl-CoA synthetases catalyze the ligation of carboxylate containing substrates to CoA via a two-step reaction mechanism (Scheme 1). In the first step, the substrate's carboxylate reacts with ATP, forming an adenylated substrate and releasing inorganic phosphate (PPi). In the second, the adenylated substrate reacts with CoA, forming a CoA-thioester and releasing AMP. The enzymes employ two different conformations, the adenylate- and thioester-forming conformations, to catalyze the two reaction steps. The transition between the two conformations is mediated by changes in the position of the small C-terminal domain relative to the large N-terminal domain.36 In order to obtain structures of LsOCS in both conformations, we attempted to crystalize the enzyme with its oxalate substrate and ATP, with CoA only, and without substrates. Unfortunately, we obtained a structure of the enzyme in only one conformation. To determine in which conformation we crystalized LsOCS, we compared its structure to structures of other members of the superfamily, crystalized in either one or both conformations.Currently, there are at least 200 crystal structures from 79 different proteins of the ANL superfamily of adenylating enzymes deposited in the Protein Data Bank (http://www.acsu.buffalo.edu/~amgulick/RANLChart.html). The enzymes have been crystallised in different liganded states, providing detailed insights into the catalytic mechanism they employ. We compared the conformation of LsOCS with the adenylate- and thioester-forming conformations of six CoA synthetases/ligases: 4-coumarate CoA ligase from Populus tomentosa (Pt4CL), o-succinylbenzoate CoA synthetase from Bacillus subtilis (BsMenE), 4-chlorobenzoyl-CoA ligase/synthetase from Alcaligenes sp (AsCBL), the long chain fatty acyl-CoA synthetase from Thermus thermophillus (TtFattyAcs), oxalyl CoA-synthetase from Arabidopsis thaliana (AtAAE3), and methylmalonyl CoA synthetase from Rhodopseudomonas palustris (RpMatB) (Fig. 4). Three of these enzymes had been crystalized in both their adenylating and thioester forming conformations (AsCBL, BsMenE, TtFattyAcs).
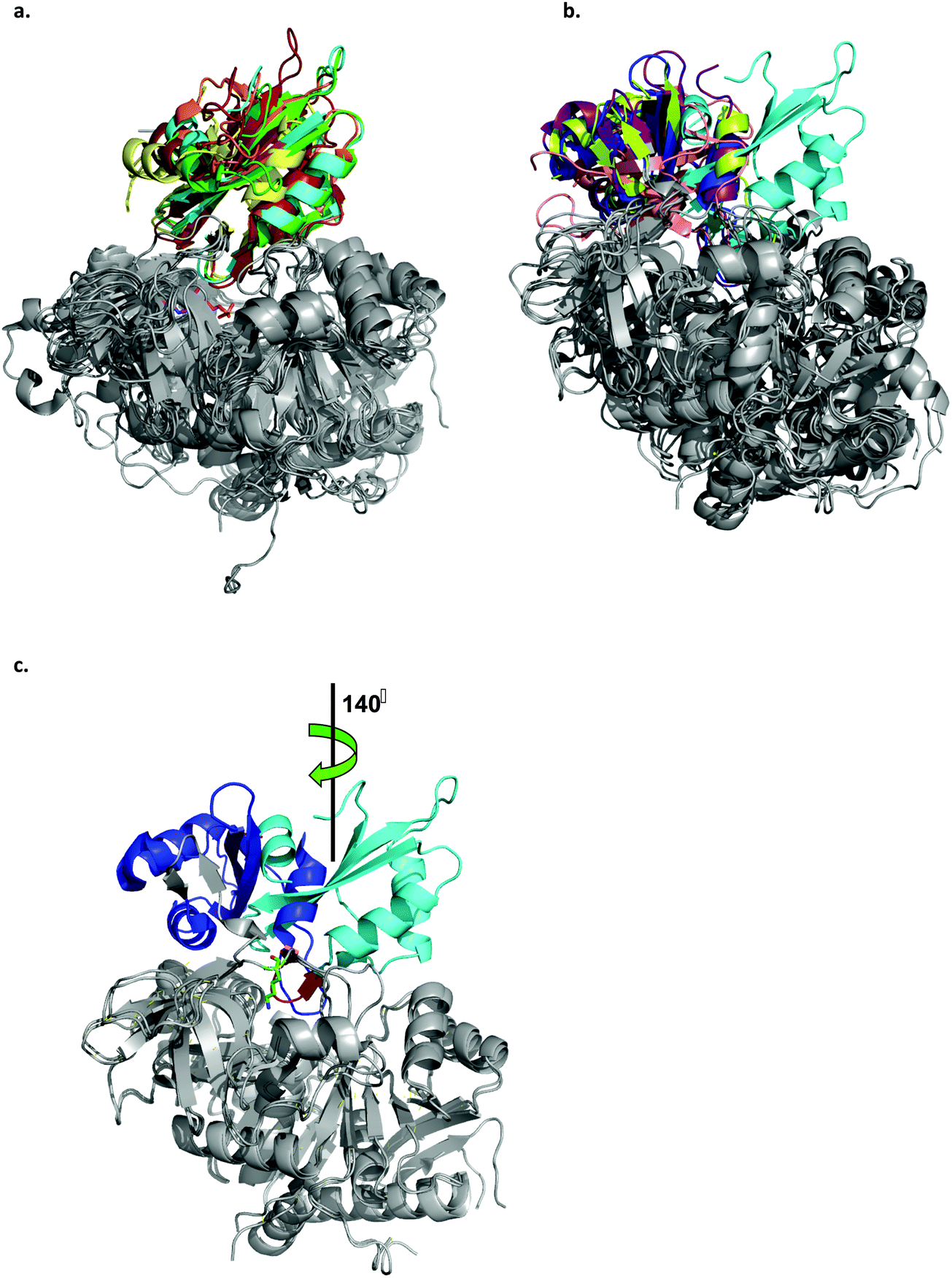 | ||
| Fig. 4 Ribbon representation of the LsOCS crystal structure and related ANL family members. a. Structural overlay of LsOCS (PDB ID: 6QJZ) and the thioester-forming conformations of Pt4CL (PDB: 3A9V), BsMenE (PDB ID: 5X8G), AsCBL (PDB ID: 3CW9) and TtFattyAcs (PDB ID: 1V25). The N-terminal domains are in grey and the C-terminal domain are shown in cyan, green, orange, red and yellow, respectively. AMP is shown as stick model with the carbon atoms in pink, oxygen in red and nitrogen in blue. b. Structural overlay of LsOCS (PDB ID: 6QJZ) and the adenylate-forming conformations of AtAAE3 (PDB ID: 5IE3), RpMatB (PDB ID:4FUT), BsMenE (PDB ID: 5BUS) and AsCBL (PDB ID:3CW8). The N-terminal domains are in grey and the C-terminal domains are shown in cyan, blue, purple, pink and lemon-green respectively. c. Structural overlay of LsOCS (PDB ID: 6QJZ) and the adenylate-forming conformation of AtAAE3 (PDB ID: 5IE3). The N-terminal domains are in grey and the C-terminal domains are shown in cyan and blue, respectively. Also highlighted are the symmetry rotation axis between the conformations of the two C-terminal regions, the pivot residue of LsOCS, Lys418 (green sticks) and the beta hairpin following the hinge region in LsOCS (red). The figure was created using PyMOL.63 | ||
A structural overlay of LsOCS with the thioester-forming conformations of AsCBL, Pt4CL, BsMenE and TtFattyAcs, revealed a very good alignment of both N- and C-terminal domains (RMSD = 1.8 ± 0.7 Å), despite a low amino-acid sequence identity of 27 ± 2% (Fig. 4a). However, when we overlaid the structure of LsOCS with the adenylate-forming conformation of AsCBL, BsMenE, RpMatB and AtAAE3 (29 ± 1% aa seq. identity to LsOCS except for AtAAE3 that is 75% identical), only the N-terminal domains aligned well (Fig. 4b). The C-terminal domain of LsOCS was shifted by 138–150 degrees with respect to those of the other structures (Fig. 4c).
We concluded that, while LsOCS was crystallised in the presence of ATP and oxalate, the substrates of the adenylation reaction, and in the absence of CoA, its crystal structure had adopted the thioester-forming conformation of the enzyme. Similar results had been obtained with the D-alanine-D-alanyl carrier protein ligase (DltA) from Bacilus subtilis,38 and with the medium chain acyl-CoA synthetase (AAE) from Methanosarcina acetivorans;39 Likewise, they both crystalized in the thioester-forming conformation in the absence of the coenzyme in the crystallization buffer.
Structural features of LsOCS indicate a partial transition to the thioester-forming conformation
As was mentioned earlier, the transition between the two partial reactions of acyl-CoA synthetases involves a large rotation of their C-terminal domain, which pivots, in LsOCS, around the hinge region residue Lys418 (Fig. 2 and 4c). This movement, termed “domain alternation”, brings the bulk of the C-terminal domain over the N-terminal domain, giving rise to multiple interactions between the two.36 The structure of LsOCS exhibits many of the expected features of such a rotation. Lys507, a highly conserved residue that is likely involved in the catalysis of the adenylation reaction, and the P-loop are removed from the active site, whereas, the first two strands of the C-terminal (β21, β22 Fig. 2) have moved into the active site (Fig. 3d). The removal of the P-loop from the active site is essential for the initiation of the thioesterification reaction because it enables the rotation of the C-terminal domain, which is otherwise hindered by steric clashes between the P-loop and the hinge region.34,36 Similarly, the side chain of Arg416 points away from the nucleotide rather than pointing towards its ribose hydroxyls with which it probably interacts in the ATP- and oxalyl-AMP-bound states (Fig. S9, ESI†).Another structural prerequisite for thioesterification is the opening of the pantetheine tunnel, by which the pantetheine moiety of CoA passes into the active site. This tunnel is obstructed by a histidine or by another aromatic residue, when it binds the α-phosphate group of ATP during the adenylation reaction. Upon transitioning into the thioester conformation, the obstructing residue detaches from the α-phosphate group and moves away from the tunnel.36 In AtAAE3, this residue, His214, had been crystalized both in its ATP bound, tunnel-obstructing rotamer (PDB ID: 5IE2) and in its perpendicular, tunnel-opening rotamer (PDB ID: 5IE3, 5IE2).34 In the structure of LsOCS, the corresponding residue (His221) is bound to AMP and blocks the pentathein tunnel (Fig. S10, ESI†). It therefore appears, that while LsOCS has been crystalized in the thioester-forming conformation, its transition to this conformation has not been fully completed.
Discussion
The activity of the enzyme oxalyl-CoA synthetase was first identified 60 years ago in extracts of germinated pea seeds (Pisum sativum) during an investigation into the metabolism of oxalic acid in plants.40 OCS itself was later partially purified and characterized from extracts of pea seeds41 and grass pea (Lathyrus sativus L.), in which it was first suggested to produce oxalyl-CoA, and that the latter is a precursor to the neurotoxin β-ODAP.24,26 Grass pea seeds have high nutritional value42,43 and serve as a source of food and forage mostly in South Asia and Sub-Saharan Africa.25,44,45 Unfortunately, if consumed as a primary diet component over a prolonged period of time, the β-ODAP they contain may cause neurolathyrism, a disorder characterized by spastic paralysis of the lower limbs.27–29 Since β-ODAP is synthesized by the ligation of oxalyl-CoA and L-α,β-diaminopropionic acid, the concentration of oxalyl-CoA in grass pea is key to regulating the amount of β-ODAP it produces. This was exemplified by the concomitant reduction in both oxalate and β-ODAP concentrations in grass pea following the introduction of a constitutively expressed fungal oxalate decarboxylase gene into its genome.30 It also indicates the role of LsOCS in grass pea, which may serve not only to reduce the levels of oxalate but also to promote the synthesis of β-ODAP.The properties of LsOCS we characterized in this work highlight its resemblance to previously characterized OCS homologs. The specific activity of LsOCS with oxalate, 8.2 ± 0.2 μmole min−1 mg−1, is similar to that of several plant homologs such as AtAAE3, MtAAE3 and GsAAE3 (11.4 ± 1, 19 ± 0.9 and 12.64 ± 0.34 μM min−1 mg−1), respectively,5,15,16 as well as to that of a yeast homolog ScAAE3 (12 ± 1 μM min−1 mg−1).19 The KM we determined for oxalate binding by LsOCS (71.5 ± 13.28 μM) is also similar to those reported for AtAAE3, MtAAE3 and GsAAE3 (149 ± 12.7, 81 ± 8.1 and 105.1 ± 12.3 μM), but ∼3 fold greater than that reported for ScAAE3 (20 ± 2.7 μM). All five OCS's have similar molecular weights ∼55–60 kDa and display a maximal activity at pH 8. Additionally, the two enzymes crystalized, LsOCS and AtAAE3, present similar structures15 and display a strong preference for oxalate as a substrate over other carboxylic acids such as malonate, succinate, glycolate, glyoxalate and lactate.15 The main difference between LsOCS and AtAAE3 was the higher catalytic efficiency we found with glyoxalate (Table 1) with respect to the low activity found by Foster et al.15 This difference may result from intrinsic differences in the substrate promiscuity of the two orthologs or may be attributed to differences in the experimental systems used.
The ANL superfamily of adenylating enzymes consists of acyl- and aryl-CoA synthetases, the adenylation domains of the non-ribosomal peptide synthetases (NRPSs) and firefly luciferase. These enzymes are highly diverse, share little overall sequence identity and catalyze different overall reactions. Nonetheless, all of them contain two independent domains: a large N-terminal domain and a small C-terminal domain, connected by a short hinge region, and all of them employ a two-step reaction mechanism, in which the active site alternates between two main active conformations. The first conformation relates to the initial partial reaction, in which the carboxylate substrate is activated by interaction with ATP, forming an adenylate intermediate (e.g. acyl/aryl-AMP) followed by the release of inorganic phosphate (PPi) (Scheme 1). The second conformation is formed by a rotation of the C-terminal domain around the hinge region (Fig. 3). This conformation is suitable for catalyzing the second step; the interaction of a nucleophile (e.g. an amine, an alcohol or a thiol) with the adenylated substrate and the release of AMP (Scheme 1). The adenylate intermediate, a high-energy acid anhydride, provides the activation energy for a diverse set of second partial reactions.36 In the acyl-CoA synthetase family, to which LsOCS belongs, during the second part of the reaction the carboxylate carbon of the acyl-AMP intermediate undergoes a nucleophilic attack by the pantetheine thiol group of CoA, displacing AMP and forming the acyl-CoA product (Scheme 1). Thus, the two distinct active conformations of acyl-CoA synthetases are those promoting the adenylate intermediate and the thioester product. While AtAAE3 adapted the adenylate forming conformation in its crystal structure, LsOCS had crystalized in the thioester conformation. The similarity between LsOCS and AtAAE3 in sequence, structure and activity enables their crystal structures to be seen to complement each other and provides information on the rotation of their C-terminal domain between its two conformational states.
The inactivation of AtAAE3 in Arabidopsis thaliana resulted in the accumulation of 3-fold higher oxalate levels in its seeds, the formation of oxalate crystals and seed coat defects that reduced their germination substantially.15 Similarly, the inactivation of MtAAE3 in Medicago truncatula resulted in reduced vegetative growth and seed germination, as well as, increased calcium levels, calcium-oxalate crystal number and permeability of its seeds.21 In both cases, the reduction in oxalate degradation capabilities had also increased the susceptibility of their host plants to infection by the pathogenic fungus Sclerotinia sclerotiorum.15,16 In view of this, we suspect that simply inactivating LsOCS in grass pea using genetic engineering may serve to reduce the biosynthesis of β-ODAP, but may also result in the accumulation of toxic oxalate in its cells and increase its susceptibility to pathogens such as S. sclerotiorum. Thus, we suggest that replacing the gene encoding LsOCS in grass pea with an exogenous oxalate-oxidase or decarboxylase may enable the plant to regulate cellular oxalate levels while reducing the levels of β-ODAP in the plant.
Finally, the importance of identifying new OCS enzymes extends beyond the need to generate improved grass pea cultivars. Oxalate-degrading enzymes have potential use not only for crop improvement,10 and for human therapeutic purposes,46,47 but also in other areas. For example, in the pulp and paper industry and in forest bio-refineries, oxalate-degrading enzymes are used in the prevention of scaling, the formation of calcium oxalate incrusts.48 Similarly, in the brewery industry, the use of these enzymes reduces calcium oxalate deposits in beer production. Thus, the identification of LsOCS and additional homologous may serve a broad range of research fields and applications.
Materials and methods
Materials
Grass Pea (Lathyrus sativus L.) seeds were obtained from Fratelli Ingengnoli Milano© (https://www.ingegnoli.it) and from Plant World Seeds© (http://www.plant-world-seeds.com). Protease inhibitor cocktail for plant cells and for bacterial cells, HEPES, EDTA, DTT, IPTG, TRIS, DTT, NADH sodium phosphate monobasic and dibasic, magnesium chloride, coenzyme A sodium salt, 5,5′-dithio-bis-(2-nitrobenzoic acid) (DTNB), phosphoenol-pyruvate, Myokinase (rabbit muscle), Pyruvate kinase, Lactate dehydrogenase, REDTaq Ready mix PCR reaction mix, DNA primers and sodium salts of: oxalate, malonate, succinate, glyoxalate, glycolate, lactate were all obtained from Sigma-Aldrich© and MERCK©. DNeasy plant mini kit and QIAquick Gel Extraction kit were obtained from Qiagen©. PiColorLock™ from Novus Biologicals© 96-well ELISA plates were obtained from Greiner®. UPLC/MS grade solvents were used for all chromatographic steps.Amplification of LsOCS from L. sativus
A BLASTn search was performed using the following gene sequences: Arachis duranensis (XR_001592575.1), Cicer arietinum (XM_004514691.2), Vigna radiata (XM_014659057.1) and Glycine max (XM_014764165.1). Multi sequence alignment was performed using MultiAlign (http://multalin.toulouse.inra.fr/multalin/).49 Genomic DNA from grass pea was isolated from 100 mg of fresh leaf tissue using the Plant DNeasy™ Kit according to the manufacturer protocol (Qiagen©, GmbH, Germany). The LsOCS gene was PCR amplified as follows: 20 ng of genomic DNA were added to a 50 μl PCR mixture containing: 25 μl of REDTaq Ready mix PCR reaction mix (Sigma-Aldrich©) and 0.5 μM of the primers F393-LsOCS and R1366-LsOCS, and performed as follows: (1) 2 min at 95 °C, (2) 35 cycles of: 30 s at 95 °C, 1 min at 54 °C, and 2 min at 72 °C, (3) 5 min at 72 °C in a SimpliAmp thermal cycler (Thermo Fisher Scientific©). Primer sequences: F393-LsOCS 5′-GAAGARTTCGAGTTTTAYTTATC-3′, R1366-LsOCS 5′-CACRAGRTRCAAATANCCRTCAGA-3′ (Sigma-Aldrich©, Israel). The PCR product was purified from 1% agarose gel by QIAquick Gel Extraction kit (Qiagen GmbH©, Hilden, Germany), and sequenced.Cloning expression and purification of LsOCS
The gene encoding LsOCS was PCR amplified from genomic DNA using the primers specified bellow and cloned into a pET28-bdSumo expression vector. This vector was constructed by transferring the His14-bdSUMO cassette from the expression vector (designated K151) generously obtained from Prof. Dirk Görlich from the Max-Planck-Institute, Göttingen, Germany32 into the expression vector pET28-TevH.50 Cloning was performed by the Restriction-Free (RF) method51 using primers 28Sumo_LsOCS_F (5′-ATCGACGCAATGCTTCATCAGACTGGTGGCGAAACCGCAACCACCCTCACC) and 28Sumo_LsOCS_R (5′-GATCTCAGTG GTGGTGGTGGTGGTGCTCGAGTTAAACTTTAGAAACAAAGTGTTCTGCTAC). For LsOCS expression, a 5 L culture of BL21(DE3) was induced with 200 μM IPTG and grown at 15 °C overnight. The cells were harvested and lysed by cell disrupter (Constant Systems) in lysis buffer (50 mM Tris 7.5, 0.5 M NaCl, 20 mM Imidazole) containing 200 KU/100 ml lysozyme, 20 μg ml−1 DNase, 1 mM MgCl2, 1 mM phenylmethylsulfonyl fluoride (PMSF) and protease inhibitor cocktail. After clarification of the soup by centrifugation, the lysate was incubated with 5 ml washed Ni beads (Adar Biotech©) for 1 h at 4 °C. After removing the supernatant, the beads were washed 4 times with 50 ml lysis buffer. LsOCS (without tags) eluted from the beads by incubation with 5 ml cleavage buffer (50 mM Tris 7.5, 0.5 M NaCl containing 0.4 mg bdSumo protease) for 2 h at RT. The soup containing the cleaved LsOCS was removed and additional 5 ml cleavage buffer was added for 2 h at RT. The two elutions were combined, concentrated and applied to a size exclusion (SEC) column (HiLoad_16/60_Superdex200 prep-grade, GE Healthcare) equilibrated with 20 mM Tris pH 8, 500 mM NaCl and 5 mM DTT. Pure LsOCS migrating as a single peak at 85 ml was pooled, concentrated to 22 mg ml−1 and frozen in aliquots at −80 °C. For crystallization experiments the following were added to a final concentration: 10 mM ATP, 20 mM MgCl2, and 10 mM NaOxalate.Determination of enzymatic activity and kinetic parameters of LsOCS
The enzymatic activity of purified LsOCS was assayed using a coupled enzymatic assay.15,52 The assay mixture contained: HEPES buffer (50 mM, pH 8), MgCl2 (5 mM), phosphoenol-pyruvate (3 mM), NADH (1 mM), ATP (5 mM), DTT (2 mM), CoA (2 mM), purified LsOCS (1 μM), myokinase (5U), pyruvate kinase (5U), lactate dehydrogenase (6U) and the assayed substrate (Oxalate, Malonate, Succinate, Glyoxalate, Glycolate or Lactate, 10–800 μM). The reaction was initiated by the addition of the buffered substrate solution (10–800 μM, pH 8) to the reaction mix containing the LsOCS enzyme and above described compounds, in UV-transparent 96-well plates (final volume of 200 μl) at room temperature. The rate of NADH oxidation was then determined by measuring absorbance at 340 nm for 20 min using an Infinite M Plex microplate reader (TECAN©). The initial rates of NADH oxidation at different substrate concentrations were used to derive the catalytic rate parameters (i.e. kcat, KM and kcat/KM) of LsOCS by fitting them to a Michaelis–Menten or linear regression equations (Prism 7, GraphPad©). The concentration of free CoA in the ligation reaction was measured as follows: a buffered solution (HEPES 125 mM, pH 8) of MgCl2 (2 mM), ATP (10 mM), CoA (1.5 mM) and purified LsOCS (0.25 μM) was mixed with a buffered solution of NaOxalate (5 mM, pH 8) and incubated at room temperature. At different time points, solution samples (100 μl) were mixed with a DTNB solution (100 μl, 2 mM) inside 96-well plates. Sample absorption was measured at 412 nm using a Cytation 5 microplate reader (BioTek©), and the concentration of free (i.e. unligated) CoA was calculated. Control reactions, lacking either ATP, CoA, LsOCS or NaOxalate were measured in a similar manner.Determination of oxalyl-CoA using HPLC and LC-MS
HPLC analysis of oxalyl-CoA was done as follows: oxalyl-CoA was synthesized in vitro by incubating a reaction mixture containing: Na-CoA (4 mM), NaOxalate (40 mM, pH 8), ATP (50 mM), MgCl2 (4 mM) and purified LsOCS (0.4 μM) in HEPES buffer (125 mM, pH 8) for 1 h at 37 °C. Samples of the reaction mixture before and after incubation with LsOCS, as well as samples of pure CoA and ATP were filtered through 0.2 μm PTFE filters (StarTech®) prior to HPLC analysis, and separated by reversed-phased chromatography on a C18 column (Gemini C18, 4.6 × 150 mm, 5 μM, Phenomenex©). The column was attached to a Prominence UFLC LC-20AD system (Shimadzu©) consisting of a SIL-20AC autosampler (Shimadzu©), CTO-20AC column oven (Shimadzu©) and a SPD-M20A diode array detector (Shimadzu©). Elution was done using a gradient of 0–50% solvent B (100% acetonitrile) in solvent A (13 mM Sodium Phosphate buffer, pH 7.4) at a flow rate of 1 mL per minute for 20 minutes at 25 °C, while monitoring at 260 nm. Data analysis was performed using LabSolutions ver. 5.97 (Shimadzu©). LC-MS analysis was done as follows: a 2.5 μl aliquot of the reaction mixture (with or without enzyme) was diluted with 1 ml of 50%-aqueous acetonitrile, then 100 μl of the resulted solution was placed in nanofilter vial (Thomson, 0.2 μm PES) for LC-MS analysis. The LC-MS/MS instrument consisted of an Acquity I-class UPLC system and Xevo TQ-S triple quadrupole mass spectrometer (both Waters) equipped with an electrospray ion source. LC was performed using a 150 × 2.1 mm i.d., 1.7 μm UPLC BEH Amide column (Waters) using gradient 10 mM ammonium bicarbonate, pH 10 in acetonitrile. Mass spectrometry was performed for signal monitoring in two ways: for visualization – as MS scan in 50–1200 m/z range, by analysis of total ion current or extracted ion chromatograms; for quantitation – as MS/MS transitions for CoA, 768.0 → 428.0 and 768.0 → 261.2 m/z, collision energy 25 and 35 eV, respectively, in positive ion mode; for oxalate 89.0 → 61.0 and 89.0 → 45.0 m/z, collision energy 3 eV, and for oxalyl-CoA 837.5 → 766.1 m/z, with collision energy 19 eV (all in negative ion mode).
DSF measurements of LsOCS
The solution buffer of purified LsOCS was exchanged to phosphate buffer (PBS 20 mM, pH 8) by repeated (4×) concentration (4000 × g, 20 min) and washing cycles, using an Amicon® Ultra-2 centrifugal filter unit (10 kDa, MWCO). Protein concentration was determined using solution absorbance at 280 nm. Solutions of purified LsOCS (final concentration 0.5 mg ml−1 in PBS) with CoA (0.1, 1 mM), Oxalate (1 mM), MgCl2 (0.7 mM), ATP + MgCl2 (0.1 mM) or without any added compounds (Apo), were incubated for 15–20 min on ice. Differential scanning fluorometry (DSF) was measured for duplicate samples at 330 and 350 nm using a NanoDSF Prometheus NT.48™ instrument.pH range profile of LsOCS
The enzymatic activity of purified LsOCS was assayed using an ATPase colorimetric assay kit (Novus Biologicals©), at different pH's, in the following manner: the assay mixture contained: purified LsOCS (10 μM), MgCl2 (5 mM), ATP (0.5 mM), CoA (1 mM), Tris–HCl buffer (50 mM, pH 6–8.5) and oxalate (400 μM). The reaction was initiated by the addition of the buffered oxalate solution (100 mM, pH 6–8.5) at room temperature, to the reaction mix containing the enzyme and above described compounds. At two time points (2′, 20′), samples of the assay mixture (200 μl) were removed and mixed with the PiColorLock™ stop solution mix (50 μl) in 96-well, flat-bottom ELISA plates (Novusbio©). Following a 2 min R.T incubation, a stabilizer solution (20 μl) was added and the sample mixture was incubated for another 30 min at R.T. Sample absorbance was then measured at 620 nm using a Cytation 5 microplate reader (BioTek©). The concentration of PPi per sample was determined using a PPi standard calibration curve according to the manufacturer's protocol. The initial rates of ATP hydrolysis at different pH's were plotted to derive the pH at which the initial rate is maximal.Crystallization, data collection, and refinement
We co-crystallised LsOCS in the presence of ATP, MgCl2 and oxalate, the substrates of the adenylate-forming half-reaction. LsOCS complexes formed rod-like crystals using the hanging drop vapor diffusion method and a Mosquito robot (TTP LabTech) at 19 °C utilizing the precipitants 0.2 M NaCl and 25% Polyethylene glycol (PEG) 3350 in 100 mM Tris pH 8.5. The LsOCS complex crystals formed in the orthorhombic space group P22121, with one monomer per asymmetric unit and diffracted to 2.7 Å resolution. Data collection was performed under cryo conditions (100 K), in-house, using a Rigaku RU-H3R X-ray instrument. All diffraction images were indexed and integrated using the Mosflm program,53 and the integrated reflections were scaled using the SCALA program.54 Structure factor amplitudes were calculated using TRUNCATE55 from the CCP4 program suite. The structure of LsOCS was solved by molecular replacement with the program PHASER,56 using the refined homologous (75% sequence identity) structure of the LsOCS from Arabidopsis thaliana (AtAAE3), PDB-ID 5IE2.34 All steps of the atomic refinements were performed with the PHENIX.refine program.57 The model was built into 2mFobs-DFcalc, and mFobs – DFcalc maps using COOT.58 Refinement movements were accepted only when they produced a decrease in the Rfree value. The model was optimized using PDB_REDO,59 and was evaluated with MOLPROBIDITY.60 Details of the data collection and refinement statistics of the LsOCS structure are described in Table 4.Abbreviations
| CoA | Coenzyme A |
| LsOCS | Lathyrus sativus oxalyl-CoA synthetase |
| AtAAE3 | Arabidopsis thaliana oxalyl-CoA synthetase |
| MtAAE3 | Medicago truncatula oxalyl-CoA synthetase |
| AAE3 | Acyl activating enzyme 3 |
| β-ODAP | L-β-N-Oxalyl-α-β-diaminopropionic acid. |
Author contributions
M. G. performed the kinetic analyses, the thermal stability assays, and the HPLC analysis, analyzed the data and wrote the paper with contributions from all authors. S. B. identified and isolated the gene from grass pea. A. B. and T. M. performed the LC-MS analysis. Y. P. cloned and expressed the protein for purification. S. A. purified the protein. O. D. crystalized the protein and determined its structure. Z. R. initiated and supervised the study and wrote the manuscript with contributions from all authors.Data and materials availability
Refined coordinates were deposited in the Protein Data Bank Database under accession code 6QJZ. The identified transcript sequence has been deposited in the GenBank database (GenBank MK492104.1) as the sequence of LsOCS. Materials are available from the authors upon request.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by grants from the Israel Ministry of Agriculture and Rural Development (712884), Israel Science Foundation (1082/17), and the National Science Foundation-United States-Israel Binational Science Foundation (NSF-BSF) Molecular and Cellular Biosciences Program (2015839). Dr Shira Albeck is the incumbent of Prof. David Casson Research Fellowship. We would like to thank Dr Yael Fridmann-Sirkis for her assistance with DSF measurements.References
- R. P. Walker and F. Famiani, Organic Acids in Fruits: Metabolism, Functions and Contents, Hortic. Rev., 2018, 371 Search PubMed
.
- V. R. Franceschi and P. A. Nakata, Calcium oxalate in plants: formation and function, Annu. Rev. Plant Biol., 2005, 56, 41–71 CrossRef CAS PubMed
- M. C. de Souza, M. C. Scalon, C. Poschenrieder, R. Tolra, T. Venancio and S. P. Teixeira,
et al., Aluminium detoxification in facultative (Passovia ovata (Pohl ex DC.) Kuijt and Struthanthus polyanthus Mart. – Loranthaceae) and dependent (Psittacanthus robustus (Mart.) Marloth – Loranthaceae) Al-accumulating mistletoe species from the Brazilian savanna, Phytochemistry, 2018, 153, 58–63 CrossRef CAS PubMed
- B. Klug and W. J. Horst, Oxalate exudation into the root-tip water free space confers protection from aluminum toxicity and allows aluminum accumulation in the symplast in buckwheat (Fagopyrum esculentum), New Phytol., 2010, 187(2), 380–391 CrossRef CAS PubMed
- P. Xian, Z. Cai, Y. Cheng, R. Lin, T. Lian and Q. Ma,
et al., Wild Soybean Oxalyl-CoA Synthetase Degrades Oxalate and Affects the Tolerance to Cadmium and Aluminum Stresses, Int. J. Mol. Sci., 2020, 21(22), 8869 CrossRef CAS PubMed
- R. Prasad and Y. S. Shivay, Oxalic acid/oxalates in plants: from self-defence to phytoremediation, Curr. Sci., 2017, 112(8), 1665–1667 CrossRef CAS
- V. R. Franceschi and H. T. Horner, Calcium-Oxalate Crystals in Plants, Bot. Rev., 1980, 46(4), 361–427 CrossRef CAS
- M. Koch, N. Delmotte, C. H. Ahrens, U. Omasits, K. Schneider and F. Danza,
et al., A link between arabinose utilization and oxalotrophy in Bradyrhizobium japonicum, Appl. Environ. Microbiol., 2014, 80(7), 2094–2101 CrossRef PubMed
- V. Franceschi, Calcium oxalate in plants, Trends Plant Sci., 2001, 6(7), 331 CrossRef CAS PubMed
- V. Kumar, M. Irfan and A. Datta, Manipulation of oxalate metabolism in plants for improving food quality and productivity, Phytochemistry, 2019, 158, 103–109 CrossRef CAS PubMed
- X. Cai, C. Ge, C. Xu, X. Wang, S. Wang and Q. Wang, Expression Analysis of Oxalate Metabolic Pathway Genes Reveals Oxalate Regulation Patterns in Spinach, Molecules, 2018, 23(6), 1286 CrossRef PubMed
- D. Svedruzic, S. Jonsson, C. G. Toyota, L. A. Reinhardt, S. Ricagno and Y. Lindqvist,
et al., The enzymes of oxalate metabolism: unexpected structures and mechanisms, Arch. Biochem. Biophys., 2005, 433(1), 176–192 CrossRef CAS PubMed
- M. R. Makela, K. Hilden and T. K. Lundell, Oxalate decarboxylase: biotechnological update and prevalence of the enzyme in filamentous fungi, Appl. Microbiol. Biotechnol., 2010, 87(3), 801–814 CrossRef PubMed
- T. Werther, A. Zimmer, G. Wille, R. Golbik, M. S. Weiss and S. Konig, New insights into structure-function relationships of oxalyl CoA decarboxylase from Escherichia coli, FEBS J., 2010, 277(12), 2628–2640 CrossRef CAS
- J. Foster, H. U. Kim, P. A. Nakata and J. Browse, A previously unknown oxalyl-CoA synthetase is important for oxalate catabolism in Arabidopsis, Plant Cell, 2012, 24(3), 1217–1229 CrossRef CAS PubMed
- J. Foster, B. Luo and P. A. Nakata, An Oxalyl-CoA Dependent Pathway of Oxalate Catabolism Plays a Role in Regulating Calcium Oxalate Crystal Accumulation and Defending against Oxalate-Secreting Phytopathogens in Medicago truncatula, PLoS One, 2016, 11(2), e0149850 CrossRef PubMed
- H. Q. Lou, W. Fan, J. M. Xu, Y. L. Gong, J. F. Jin and W. W. Chen,
et al., An Oxalyl-CoA Synthetase Is Involved in Oxalate Degradation and Aluminum Tolerance, Plant Physiol., 2016, 172(3), 1679–1690 CrossRef CAS PubMed
- C. Peng, X. Liang, E. E. Liu, J. J. Zhang and X. X. Peng, The oxalyl-CoA synthetase-regulated oxalate and its distinct effects on resistance to bacterial blight and aluminium toxicity in rice, Plant Biol., 2017, 19(3), 345–353 CrossRef CAS PubMed
- J. Foster and P. A. Nakata, An oxalyl-CoA synthetase is important for oxalate metabolism in Saccharomyces cerevisiae, FEBS Lett., 2014, 588(1), 160–166 CrossRef CAS PubMed
- N. Membre, F. Bernier, D. Staiger and A. Berna, Arabidopsis thaliana germin-like proteins: common and specific features point to a variety of functions, Planta, 2000, 211(3), 345–354 CrossRef CAS PubMed
- N. Cheng, J. Foster, K. S. Mysore, J. Wen, X. Rao and P. A. Nakata, Effect of Acyl Activating Enzyme (AAE) 3 on the growth and development of Medicago truncatula, Biochem. Biophys. Res. Commun., 2018, 505(1), 255–260 CrossRef CAS PubMed
- R. L. Guimaraes and H. U. Stotz, Oxalate production by Sclerotinia sclerotiorum deregulates guard cells during infection, Plant Physiol., 2004, 136(3), 3703–3711 CrossRef CAS PubMed
- D. D. Hegedus and S. R. Rimmer, Sclerotinia sclerotiorum: when “to be or not to be” a pathogen?, FEMS Microbiol. Lett., 2005, 251(2), 177–184 CrossRef CAS PubMed
- G. A. Johnston and H. J. Lloyd, Oxalyl-coenzyme A synthetase and the neurotoxin beta-n-oxalyl-l-alpha, beta-diaminopropionate, Aust. J. Biol. Sci., 1967, 20(6), 1241–1244 CrossRef CAS PubMed
- F. Lambein, S. Travella, Y. H. Kuo, M. Van Montagu and M. Heijde, Grass pea (Lathyrus sativus L.): orphan crop, nutraceutical or just plain food?, Planta, 2019, 250(3), 821–838 CrossRef CAS PubMed
- K. Malathi, G. Padmanaban, S. L. Rao and P. S. Sarma, Studies on the biosynthesis of beta-N-oxalyl-L-alpha, beta-diaminopropionic acid, the Lathyrus sativus neurotoxin, Biochim. Biophys. Acta., 1967, 141(1), 71–78 CrossRef CAS
- R.-Y. Tan, G.-Y. Xing, G.-M. Zhou, F.-M. Li, W.-T. Hu and F. Lambein,
et al., Plant toxin β-ODAP activates integrin β1 and focal adhesion: a critical pathway to cause neurolathyrism, Sci. Rep., 2017, 7, 40677 CrossRef CAS PubMed
- P. S. Spencer and H. H. Schaumburg, Lathyrism: a neurotoxic disease, Neurobehav. Toxicol. Teratol., 1983, 5(6), 625–629 CAS
- D. D. Ngudi, Y. H. Kuo, M. Van Montagu and F. Lambein, Research on motor neuron diseases konzo and neurolathyrism: trends from 1990 to 2010, PLoS Neglected Trop. Dis., 2012, 6(7), e1759 CrossRef PubMed
- V. Kumar, A. Chattopadhyay, S. Ghosh, M. Irfan, N. Chakraborty and S. Chakraborty,
et al., Improving nutritional quality and fungal tolerance in soya bean and grass pea by expressing an oxalate decarboxylase, Plant Biotechnol. J., 2016, 14(6), 1394–1405 CrossRef CAS PubMed
- N. F. Almeida, N. Krezdorn, B. Rotter, P. Winter, D. Rubiales and M. C. Vaz Patto,
Lathyrus sativus transcriptome resistance response to Ascochyta lathyri investigated by deepSuperSAGE analysis, Front. Plant Sci., 2015, 6, 178 Search PubMed
- S. Frey and D. Gorlich, A new set of highly efficient, tag-cleaving proteases for purifying recombinant proteins, J. Chromatogr. A, 2014, 1337, 95–105 CrossRef CAS PubMed
- R. J. Peroutka Iii, S. J. Orcutt, J. E. Strickler and T. R. Butt, SUMO fusion technology for enhanced protein expression and purification in prokaryotes and eukaryotes, Methods Mol. Biol., 2011, 705, 15–30 CrossRef PubMed
- M. Fan, Y. Xiao, M. Li and W. Chang, Crystal Structures of Arabidopsis thaliana Oxalyl-CoA Synthetase Essential for Oxalate Degradation, Mol. Plant, 2016, 9(9), 1349–1352 CrossRef CAS PubMed
- E. Conti, T. Stachelhaus, M. A. Marahiel and P. Brick, Structural basis for the activation of phenylalanine in the non-ribosomal biosynthesis of gramicidin S, EMBO J., 1997, 16(14), 4174–4183 CrossRef CAS PubMed
- A. M. Gulick, Conformational dynamics in the Acyl-CoA synthetases, adenylation domains of non-ribosomal peptide synthetases, and firefly luciferase, ACS Chem. Biol., 2009, 4(10), 811–827 CrossRef CAS PubMed
- A. S. Reger, R. Wu, D. Dunaway-Mariano and A. M. Gulick, Structural characterization of a 140 degrees domain movement in the two-step reaction catalyzed by 4-chlorobenzoate:CoA ligase, Biochemistry, 2008, 47(31), 8016–8025 CrossRef CAS PubMed
- H. Yonus, P. Neumann, S. Zimmermann, J. J. May, M. A. Marahiel and M. T. Stubbs, Crystal structure of DltA. Implications for the reaction mechanism of non-ribosomal peptide synthetase adenylation domains, J. Biol. Chem., 2008, 283(47), 32484–32491 CrossRef CAS PubMed
- M. B. Shah, C. Ingram-Smith, L. L. Cooper, J. Qu, Y. Meng and K. S. Smith,
et al., The 2.1 A crystal structure of an acyl-CoA synthetase from Methanosarcina acetivorans reveals an alternate acyl-binding pocket for small branched acyl substrates, Proteins, 2009, 77(3), 685–698 CrossRef CAS PubMed
- J. Giovanelli and N. F. Tobin, Adenosine triphosphate- and coenzyme A-dependent decarboxylation of oxalate by extracts of peas, Nature, 1961, 190, 1006–1007 CrossRef CAS PubMed
- J. Giovanelli, Oxalyl-coenzyme A synthetase from pea seeds, Biochim. Biophys. Acta., 1966, 118(1), 124–143 CrossRef CAS
- M. M. A. Mondal and A. B. Puteh, Spectrum of Variability in Seed Size and Content of Protein and Odap in Grass Pea (Lathyrus sativus L.) Germplasm, Legume Res, 2014, 37(5), 479–482 CrossRef
- M. Arslan, Diversity for vitamin and amino acid content in grass pea (Lathyrus sativus L.), Legume Res., 2017, 40(5), 803–810 Search PubMed
- M. C. Vaz Patto and D. Rubiales, Lathyrus diversity: available resources with relevance to crop improvement–L. sativus and L. cicera as case studies, Ann. Bot., 2014, 113(6), 895–908 CrossRef CAS PubMed
- A. H. Rizvi, A. Sarker and A. Dogra, Enhancing grasspea (Lathyrus sativus L.) production in problematic soils of South Asia for nutritional security, Indian J. Genet. Plant Breed., 2016, 76(4), 583–592 CrossRef
- J. D. Watterson, P. A. Cadieux, D. T. Beiko, A. J. Cook, J. P. Burton and R. R. Harbottle,
et al., Oxalate-degrading enzymes from Oxalobacter formigenes: a novel device coating to reduce urinary tract biomaterial-related encrustation, J. Endourol., 2003, 17(5), 269–274 CrossRef PubMed
- L. Mogna, M. Pane, S. Nicola and E. Raiteri, Screening of different probiotic strains for their in vitro ability to metabolise oxalates: any prospective use in humans?, J. Clin. Gastroenterol., 2014, 48(Suppl 1), S91–S95 CrossRef CAS PubMed
- P. Cassland, A. Sjode, S. Winestrand, L. J. Jonsson and N. O. Nilvebrant, Evaluation of oxalate decarboxylase and oxalate oxidase for industrial applications, Appl. Biochem. Biotechnol., 2010, 161(1–-8), 255–263 CrossRef CAS PubMed
- F. Corpet, Multiple sequence alignment with hierarchical clustering, Nucleic Acids Res., 1988, 16(22), 10881–10890 CrossRef CAS PubMed
- Y. Peleg and T. Unger, Application of high-throughput methodologies to the expression of recombinant proteins in E. coli, Methods Mol. Biol., 2008, 426, 197–208 CrossRef CAS PubMed
- T. Unger, Y. Jacobovitch, A. Dantes, R. Bernheim and Y. Peleg, Applications of the Restriction Free (RF) cloning procedure for molecular manipulations and protein expression, J. Struct. Biol., 2010, 172(1), 34–44 CrossRef CAS PubMed
- K. Ziegler, K. Braun, A. Bockler and G. Fuchs, Studies on the Anaerobic Degradation of Benzoic-Acid and 2-Aminobenzoic Acid by a Denitrifying Pseudomonas Strain, Arch. Microbiol., 1987, 149(1), 62–69 CrossRef
- in Processing diffraction data with mosflm. Evolving Methods for Macromolecular Crystallography, ed. Leslie A. G. W. and Powell H. R., Dordrecht, Springer Netherlands, 2007 Search PubMed
- P. Evans, Scaling and assessment of data quality, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2006, 62(Pt 1), 72–82 CrossRef PubMed
- S. French and K. Wilson, Treatment of Negative Intensity Observations, Acta Crystallogr., Sect. A: Found. Crystallogr., 1978, 34(Jul), 517–525 CrossRef
- A. J. McCoy, Solving structures of protein complexes by molecular replacement with Phaser, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2007, 63, 32–41 CrossRef CAS PubMed
- P. V. Afonine, R. W. Grosse-Kunstleve, N. Echols, J. J. Headd, N. W. Moriarty and M. Mustyakimov,
et al., Towards automated crystallographic structure refinement with phenix.refine, Acta Crystallogr., Sect. D: Struct. Biol., 2012, 68, 352–367 CrossRef CAS PubMed
- P. Emsley and K. Cowtan, Coot: model-building tools for molecular graphics, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 2126–2132 CrossRef
- R. P. Joosten, K. Joosten, S. X. Cohen, G. Vriend and A. Perrakis, Automatic rebuilding and optimization of crystallographic structures in the Protein Data Bank, Bioinformatics, 2011, 27(24), 3392–3398 CrossRef CAS
- V. B. Chen, W. B. Arendall, J. J. Headd, D. A. Keedy, R. M. Immormino and G. J. Kapral,
et al., MolProbity: all-atom structure validation for macromolecular crystallography, Acta Crystallogr., Sect. D: Struct. Biol., 2010, 66, 12–21 CrossRef CAS PubMed
- F. Corpet, Multiple Sequence Alignment with Hierarchical-Clustering, Nucleic Acids Res., 1988, 16(22), 10881–10890 CrossRef CAS PubMed
- X. Robert and P. Gouet, Deciphering key features in protein structures with the new ENDscript server, Nucleic Acids Res., 2014, 42(W1), W320–W4 CrossRef CAS PubMed
-
L. L. C. Schrodinger, The PyMOL Molecular Graphics System, Version 1.8. 2015 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cb00202c |
| This journal is © The Royal Society of Chemistry 2022 |