
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructure, reactivity and luminescence studies of triphenylsiloxide complexes of tetravalent lanthanides†‡
Aurélien R.
Willauer
 a,
Iskander
Douair
c,
Anne-Sophie
Chauvin
*a,
Farzaneh
Fadaei-Tirani
a,
Jean-Claude G.
Bünzli
ab,
Laurent
Maron
*c and
Marinella
Mazzanti
*a
a,
Iskander
Douair
c,
Anne-Sophie
Chauvin
*a,
Farzaneh
Fadaei-Tirani
a,
Jean-Claude G.
Bünzli
ab,
Laurent
Maron
*c and
Marinella
Mazzanti
*a
aInstitut des Sciences et Ingénierie Chimiques, École Polytechnique Fédérale de Lausanne (EPFL), 1015 Lausanne, Switzerland. E-mail: marinella.mazzanti@epfl.ch
bDepartment of Biomedical Engineering, Southern University of Science and Technology (SUSTech), Shenzhen, China
cLaboratoire de Physique et Chimie des Nano-objets, Institut National des Sciences Appliquées, 31077, Toulouse, France
First published on 16th December 2021
Abstract
Among the 14 lanthanide elements (Ce–Lu), until recently, the tetravalent oxidation state was readily accessible in solution only for cerium while Pr(IV), Nd(IV), Dy(IV) and Tb(IV) had only been detected in the solid state. The triphenylsiloxide ligand recently allowed the isolation of molecular complexes of Tb(IV) and Pr(IV) providing an unique opportunity of investigating the luminescent properties of Ln(IV) ions. Here we have expanded the coordination studies of the triphenylsiloxide ligand with Ln(III) and Ln(IV) ions and we report the first observed luminescence emission spectra of Pr(IV) complexes which are assigned to a ligand-based emission on the basis of the measured lifetime and computational studies. Binding of the ligand to the Pr(IV) ion leads to an unprecedented large shift of the ligand triplet state which is relevant for future applications in materials science.
Introduction
Among the 14 lanthanide elements (Ce–Lu), until recently, the tetravalent oxidation state was readily accessible only for cerium while Pr(IV), Nd(IV), Dy(IV) and Tb(IV) had only been detected in solid inorganic compounds, double fluorides, oxides, perovskites,1 or in concentrated aqueous carbonate solutions.2 Seminal attempts by various groups to isolate molecular complexes of Tb(IV) and Pr(IV) were unsuccessful,3 and only recently Pr(IV)4 and Tb(IV)5 complexes were finally successfully isolated in organic solvents by taking advantage of the tuning of the +III/+IV oxidation potential by ligand field effects and by a careful choice of the chemical oxidizing agents. These landmark results open a whole new field in lanthanide coordination chemistry and related photonic and magnetic applications.The first molecular complexes of terbium(IV) were isolated using the bulky σ and π donor supporting ligands tris(tertbutoxy)siloxide5a and tris(amidyl)imidophosphorane.5b Both complexes display a coordination sphere of the Tb ion fully saturated by the supporting ligand. Despite the very different redox potentials reported for the two Tb(IV) complexes (the imidophosphorane complex has a 1.13 V more negative reduction potential than the tris(tertbutoxy)siloxide complex), no other Ln(IV) complexes, besides Ce(IV),6 were so far isolated using these ligands.
Tris(triphenylsiloxide) complexes of Ln(III) were extensively studied after being first reported 30 years ago,7–10 and the structure of several [MIII(OSiPh3)3(THF)3] complexes (M = La,7b Ce,8 Sm,9 Dy,10 and Er11) is known. The triphenylsiloxide ligand was also reported to stabilize the Ce(IV) complex [Ce(OSiPh3)4(DME)]12 more than thirty years ago but no attempts were made until recently4,5c to use the same ligand for stabilizing other Ln(IV) ions.
Only in 2020 our group isolated the third example of a Tb(IV) molecular complex using the triphenylsiloxide ligand, [TbIV(OSiPh3)4(MeCN)2], 3-TbMeCN.5c The triphenylsiloxide ligand also allowed the isolation and characterization of the first molecular complex of Pr(IV) that proved to be isomorphous to the Tb(IV) one [Pr(OSiPh3)4(MeCN)2], 3-PrMeCN.4 These complexes provide the opportunity of investigating for the first time the luminescent properties of molecular complexes of lanthanides in the +IV oxidation state.
For most Ln(III) complexes luminescence emission arises from parity-forbidden 4f → 4f transitions,13 but Ce(III) complexes show broad absorption and emission bands in the UV/visible range which are due to electric dipole allowed 4f ↔ 5d transitions.14 Ce(III) emission has shorter lifetimes compared to other lanthanides but high emission intensities. These features have led to the application of Ce(III) in light emitting diodes and in photocatalysis.15 Similar applications were identified for 4f ↔ 5d transitions of divalent lanthanides ions, such as Sm2+ and Eu2+.16 Considering that Pr(IV) has formally the electronic structure of Ce(III), 4f1, but with significantly higher energy and possibly non accessible 5d orbitals, we set out to investigate if it could display luminescence emission. It should be noted that there are no previous reports of luminescence emission from Pr(IV) ions in solid state or in solution. Complexes of Ce(IV) can display intense ligand-to-metal charge transfer transition (LMCT) and highly reactive LMCT excited states that can be tuned by ligand environment17,15b,15c,15g and can lead to photocatalytic application.18 However, examples of emissive Ce(IV) complexes are extremely rare and were interpreted in term of an emission of Ce(III) from a metal-centred fd excited state, which is generated by electron transfer from the ligand to Ce(IV).19 Here we have expanded the coordination studies of the triphenylsiloxide ligand with Ln(III) and Ln(IV) ions and we report the first observed luminescence emission spectra of Pr(IV) complexes which are assigned to a ligand-based emission. Binding of the ligand to Pr(IV) leads to an unprecedented large shift of the ligand triplet state. These findings are in line with theoretical prediction.
Results and discussion
Syntheses of lanthanide(III) complexes
It was previously shown that tetrakis Ln(III) complexes with siloxide ligands are easily prepared using the salt metathesis route20a,6a,5a,20b and we recently reported that the potassium-tetrakis-(triphenylsiloxide) Tb(III) complex [KTb(OSiPh3)4(THF)], 1-Tb, is obtained in 76% yield by direct reaction of TbI3 with 4 equivalents KOSiPh3 (KLPh) (Scheme 1).5c However, when we attempted to synthesize the analogous Pr(III) complex using the same salt metathesis route, the reaction between PrCl3 and 4 equivalents KLPh resulted in a mixture of the tris- and tetrakis(triphenylsiloxide) complexes of Pr(III).4 In contrast, we recently showed that the previously reported tris(triphenylsiloxide) Ln(III) complexes [LnIII(OSiPh3)3(THF)3] (Ln = Ce,8 Pr,8) could be used as precursors to access analytically pure tetrakis analogues [KLnIII(OSiPh3)4(THF)2+x], 1-Ln, (Ln = Ce, x = 0; Pr, x = 1) upon addition of 1 equivalent KLPh (Scheme 2).4 Using this route, tetrakis(triphenylsiloxide) complexes of trivalent Nd and Gd were also prepared.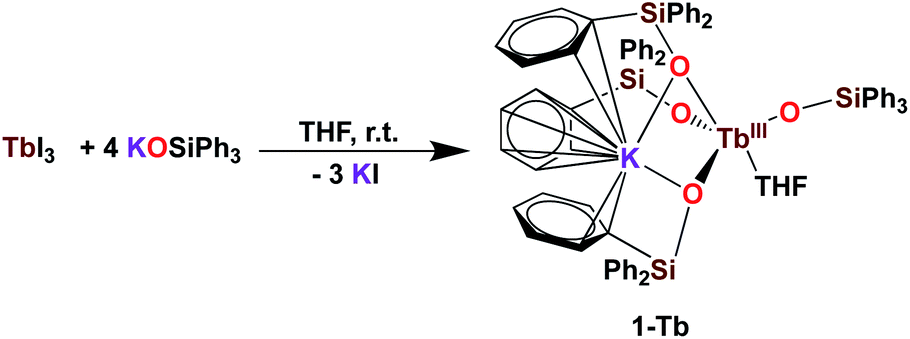 | ||
| Scheme 1 Synthesis of 1-Tb.5c | ||
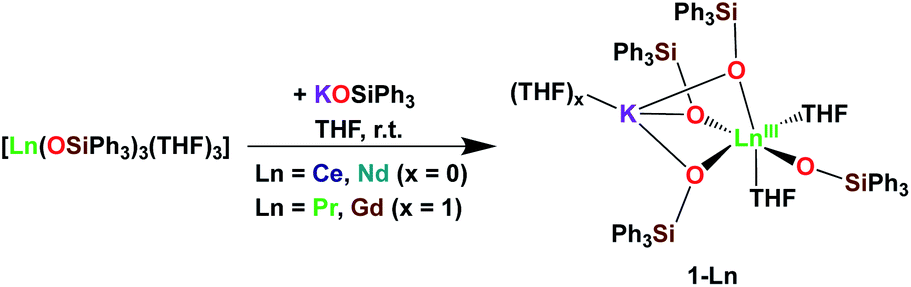
The [KNdIII(OSiPh3)4(THF)2], 1-Nd, and [KGdIII(OSiPh3)4(THF)3], 1-Gd, complexes were obtained in 75% and 82% yield respectively, by addition of 1 equivalent KLPh to their [LnIII(OSiPh3)3(THF)3] precursors in THF (Scheme 2).
Upon addition of 1 equivalent 2.2.2-cryptand to THF-d8 solutions of 1-Ln (Ln = Ce, Pr,4 Nd and Tb5c) a shift in the signals assigned to the siloxide protons can be identified in the 1H NMR spectra of the reaction mixtures (Fig. S9 and S10‡). The shift is induced by the removal of the potassium cation from the coordination sphere of the complexes to form an outer-sphere [K(2.2.2-cryptand)]+ counterion and confirms that in absence of 2.2.2-cryptand, the potassium cation in 1-Ln remains bound to the complexes in solution.
Further addition of 1 equivalent KLPh to the tetrakis complex 1-Nd in toluene resulted in the formation of the pentakis [K2NdIII(OSiPh3)5], 2-Nd, in 78% yield (Scheme 3 and Fig. S11‡). Single crystals of 2-Nd suitable for X-ray diffraction analysis were isolated from a saturated toluene solution stored at −40 °C overnight. Complex 2-Nd provides the first example of a Ln(III) complex containing five siloxide ligands21 demonstrating that electron-rich triphenylsiloxide ligands allow the synthesis of pentakis Ln(III) complexes. In contrast, performing the ligand addition in coordinating solvent such as THF, or redissolving 2-Nd in THF, only resulted in the presence in solution of the tetrakis 1-Nd and 1 equivalent KLPh as shown by 1H NMR spectroscopy (Fig. S12‡). These results show that the existence, in solution, of the pentakis 2-Nd complex is limited to non-coordinating solvent and its decomposition in THF can be explained by a competitive binding between the THF and the fifth anionic siloxide ligand.
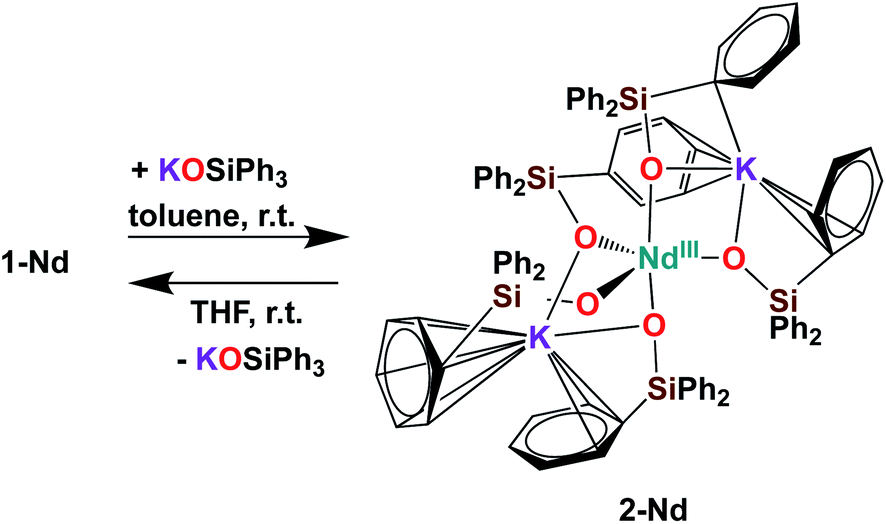 | ||
| Scheme 3 Synthesis of 2-Nd. | ||
Syntheses of lanthanide(IV) complexes
With the electron-rich tetrakis-siloxide complexes on hand we pursued their oxidation to yield the tetravalent analogues. The complex 1-Ce can be oxidized with 1.1 equivalent of AgBPh4 in THF resulting in the formation of the cerium(IV) complex [CeIV(OSiPh3)4(THF)2], 3-CeTHF (Scheme 4, Fig. S13 and S14‡). Slow diffusion at −40 °C of n-hexane into the concentrated toluene filtrate of the reaction mixture resulted in the isolation of colorless crystals of 3-CeTHF in 61% yield. Interestingly, once isolated, the colorless crystals of 3-CeTHF do not redissolve in toluene. This differs from the rather high solubility of its precursor 1-Ce in toluene. Single crystal of 3-CeTHF suitable for X-ray diffraction analysis were grown from storage of a concentrated THF solution of 3-CeTHF at −40 °C overnight. The synthesis and the crystal structure of the DME analogue [Ce(OSiPh3)4(DME)] were reported more than 30 years ago.8,12 The [Ce(OSiPh3)4(DME)] was previously prepared either by salt metathesis, from [(NH4)2Ce(NO3)6],8 or by protonolysis from [Ce(OiPr)4].12 The present results show that we can also access the tetrakis(triphenylsiloxide) Ce(IV) complex, 3-CeTHF, in good yield, by direct oxidation of the potassium-tetrakis-(triphenylsiloxide) Ce(III) complex, 1-Ce.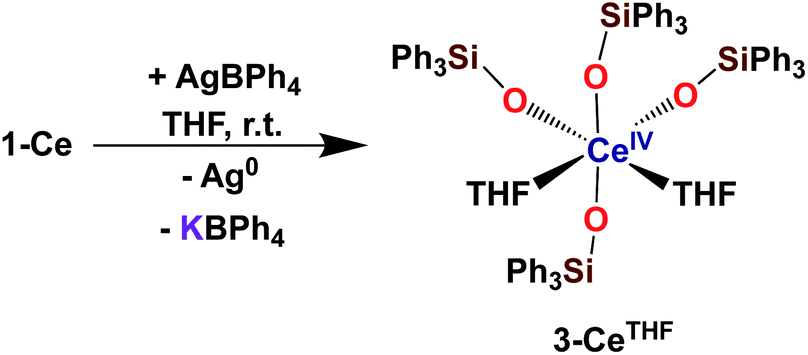 | ||
| Scheme 4 Synthesis of 3-CeTHF. | ||
Differently from 1-Ce, the oxidation of 1-Pr and 1-Tb has proven impossible with a mild oxidizing agent such as silver salts.4,5c However, we recently showed that the use of a stronger oxidizing agent such as tris(4-bromophenyl)aminium salts (E0 = 0.67 V vs. Fc compared to E0(Ag+) = 0.04 V vs. Fc in MeCN)22 leads to the oxidation of the 1-Pr and 1-Tb complex,4,5c yielding the third Tb(IV) molecular complex and the first example of a molecular complex of praseodymium in the +IV oxidation state, [LnIV(OSiPh3)4(MeCN)2], 3-LnMeCN (Ln = Pr, Tb) (Scheme 5).4,5c Complex 3-TbMeCN is insoluble in n-hexane, has a very limited solubility in toluene and acetonitrile, but is completely soluble in THF. Complex 3-PrMeCN shows similar solubility behaviour as 3-TbMeCN except that it is also completely soluble in toluene. Single crystals of the Tb(IV) THF adduct, [TbIV(OSiPh3)4(THF)2], 3-TbTHF (Scheme 5), could be grown in this work from a concentrated THF solution of 3-TbMeCN stored at −40 °C overnight. The molecular structures of the MeCN adduct 3-TbMeCN and the THF adduct 3-TbTHF have the same coordination environment except for the nature of the bound solvent molecules (vide infra). It is noticeable that the bound solvent molecules can be easily exchanged without significant changes in the structure.
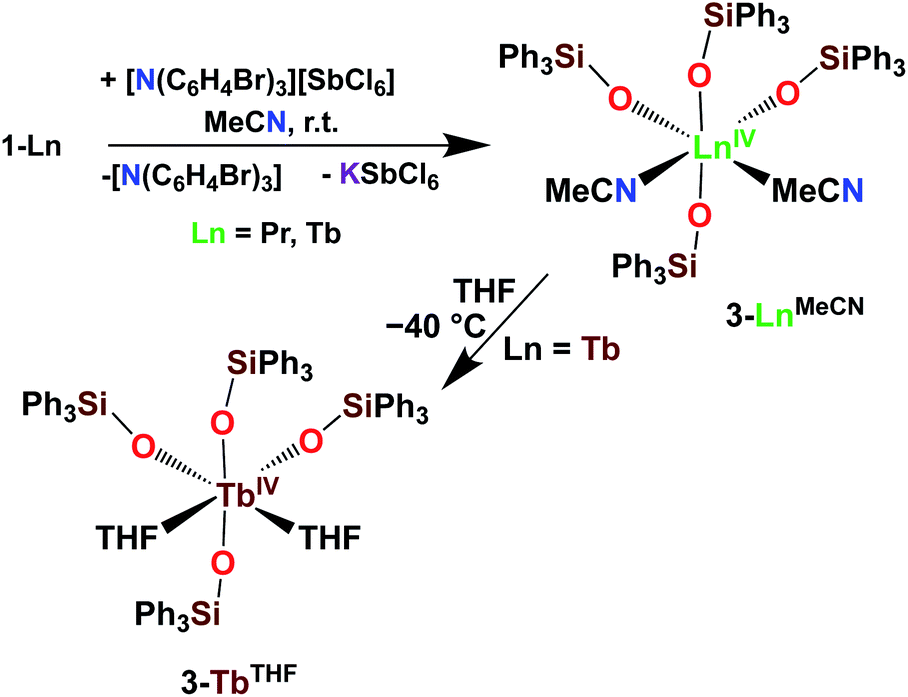 | ||
| Scheme 5 Synthesis of 3-LnMeCN (Ln = Pr, Tb)4,5c and isolation of 3-TbTHF. | ||
The addition of [N(C6H4Br)3][SbCl6] to a MeCN suspension of 1-Nd did not result in the isolation of any Nd(IV) oxidation products. It should be noted that 1-Nd is not soluble in MeCN. Furthermore, cyclic voltammetry experiments performed on 2 mM THF solutions of 1-Nd with [NBu4][B(C6F5)4] as the supporting electrolyte remained silent. This also differs from 1-Tb and 1-Pr which displayed very well-defined oxidation events at Epa = 0.41 V vs. Fc and Epa = 0.65 V vs. Fc, respectively.4,5c This can be explained by the large difference in the measured aqueous Ln(IV)/Ln(III) reduction potentials between the free Nd and the Tb/Pr ions (Nd(IV)/Nd(III) − Tb(IV)/Tb(III) = 1.9 V; Nd(IV)/Nd(III) − Pr(IV)/Pr(III) = 1.8 V).23
The addition of 1 equivalent OPR3 (R = Et, Ph) to 3-TbMeCN in THF was previously shown to yield the Tb(IV) complexes [TbIV(OSiPh3)4(OPPh3)], 4-TbPh and [TbIV(OSiPh3)4(OPEt3)], 4-TbEt (Scheme 6).5c These complexes demonstrated the relevance of the two open coordination sites in 3-TbMeCN for reactivity studies and the possibility of replacement of the labile solvent molecules. Interestingly, the phosphinoxide adducts 4-TbR displayed a much higher solubility in toluene compared to the acetonitrile adduct 3-TbMeCN.
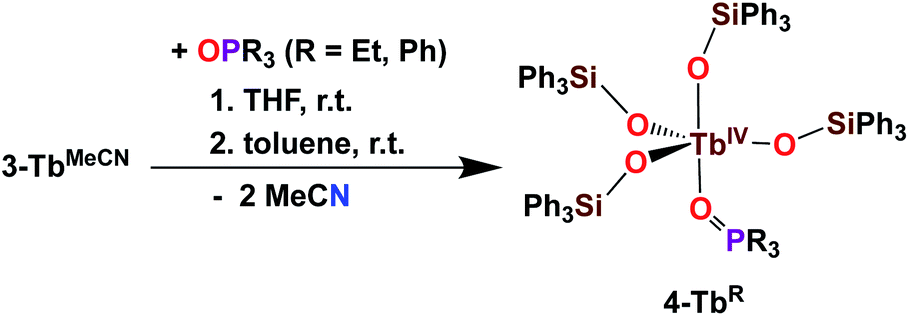 | ||
| Scheme 6 Synthesis of 4-TbR (R = Et, Ph).5c | ||
Here we have explored the reactivity of the previously reported4 Pr(IV) complex 3-PrMeCN with phosphinoxide and we found that the complex [PrIV(OSiPh3)4(OPPh3)(MeCN)], 4-PrPh, can be synthetized by treating 3-PrMeCN with 1 equivalent triphenylphosphine oxide in toluene at room temperature (Scheme 7). The slow diffusion of n-hexane into the toluene reaction mixture at −40 °C allowed the isolation of dark brown crystals of 4-PrPh in 90% yield. The 1H NMR spectrum of 4-PrPh in toluene-d8 shows four signals at δ = 7.2 ppm, 7.1 ppm, 6.9 ppm, and 6.7 ppm corresponding to the 75 protons of the siloxide and the phosphinoxide ligands (Fig. S15‡). The binding of the OPPh3 moiety is reversible in coordinating solvent such as THF, resulting in the formation of 3-PrTHF and free OPPh3 immediately after the dissolution of 4-PrPh in THF-d8 (Fig. S17‡).
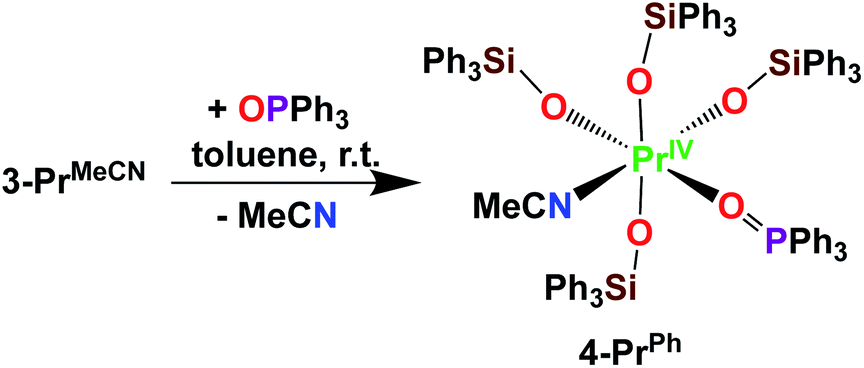 | ||
| Scheme 7 Synthesis of 4-PrPh. | ||
Interestingly, the phosphinoxide adduct 4-PrPh shows a higher solution stability than 3-PrMeCN. Notably, UV-visible stability studies showed that after storing a 3 mM solution of 4-PrPh in toluene at room temperature for 24 hours, 80% of 4-PrPh is still present in solution (Fig. S22‡). In contrast, UV-visible stability studies performed at room temperature on a 3 mM toluene solution of 3-PrMecN only showed 40% of the initial absorbance after 24 hours.4
A similar high increase in solution stability was observed for the phosphinoxide adduct of Tb 4-TbR (R = Et, Ph) compared to the THF and MeCN complexes. Notably, 3 mM toluene solutions of 4-TbR (R = Et, Ph) display an unchanged absorbance (<2%) in the UV-vis spectra for up to 24 hours (Fig. S23 and S24‡). After 4 days at room temperature, 80% of 4-TbPh (Fig. S23‡) and 70% of 4-TbEt (Fig. S24‡) are still present in solution.
Solid state structures of lanthanide(III) complexes
The solid-state structures of 1-Ln (Ln = Ce, Nd and Gd) as determined by X-ray diffraction studies present a six-coordinated Ln(III) metal center in a distorted octahedral geometry (Fig. 1). Four coordination sites are occupied by an oxygen atom from the –OSiPh3 ligands while the two others are filled by oxygen atoms from THF solvent molecules. An analogous coordination environment of the Ln ion is found in the 1-Ln (Ln = Ce, Pr,4 Nd and Gd) complexes. The only difference in these structures is the presence of one THF bound to the potassium counterion in 1-Pr and 1-Gd which is not present in 1-Ce and 1-Nd. Differently, the solid state structure of the 1-Tb complex contains only one Tb-bound THF molecule with a five-coordinated Tb(III) metal center in a distorted trigonal-bipyramidal molecular geometry.5c The lower coordination number found for the Tb(III) ion is consistent with its decreased ionic radius compared to the Gd(III) ion.24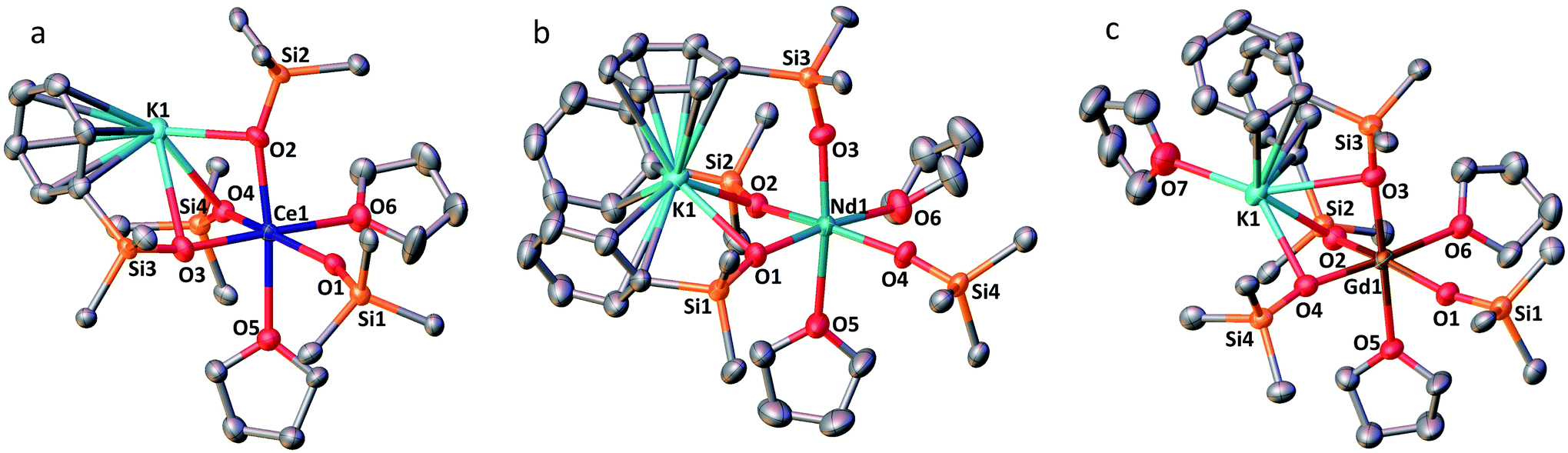 | ||
| Fig. 1 Solid-state molecular structures of 1-Ln (50% probability ellipsoids); hydrogen atoms, phenyl groups not bound to the potassium ions and residual solvent molecules are omitted for clarity. (a) 1-Ce, selected distances (Å): Ce1–Osiloxide range = 2.268(5)–2.351(5); mean Ce1–Osiloxide = 2.30(3); Ce1–OTHF = 2.598(6)–2.605(6); Ce1–K1 = 3.6305(16). (b) 1-Nd, selected distances (Å): Nd1–Osiloxide range = 2.231(2)–2.3300(19); mean Nd1–Osiloxide = 2.27(4); Nd1–OTHF = 2.573(2)–2.574(2): Nd1–K1 = 3.6925(6). (c) 1-Gd, selected distances (Å): Gd1–Osiloxide range = 2.212(3)–2.253(3); mean Gd1–Osiloxide = 2.22(2); Gd1–OTHF = 2.488(3)–2.491(3); Gd1–K1 = 3.5143(12). | ||
The decrease of the mean value of the Ln1–Osiloxide bond distances identified along the series 1-Ce (2.30(3) Å), 1-Pr (2.27(2) Å),41-Nd (2.27(4) Å) and 1-Gd (2.22(2) Å) (Table 1) is consistent with the difference in Shannon's ionic radii of these 6-coordinate Ln(III) ions (Ce(III), 1.01 Å; Pr(III), 0.99 Å; Nd(III), 0.983 Å and Gd(III), 0.938 Å).25 The mean value of the Ce1–Osiloxide bond distances in 1-Ce is larger than the one found in the six-coordinate tris-complex [CeIII(OSiPh3)3(THF)3] (2.222 (4) Å).8 The observed bond elongation is explained both in terms of the increased steric hindrance and increased negative charge provided by the additional siloxide ligand bound to the cerium ion in the tetrakis 1-Ce complex compared to the THF ligand in the tris-complex. In all the 1-Ln complexes (Ln = Ce, Pr, Nd, Gd and Tb), the K+ ion lies in close proximity of the lanthanide center (3.5143 (12)–3.6921 (7) Å; see Table 2) and connects the siloxide ligands through the anionic oxygen and through cation–π interactions with the phenyl groups.
| Ln(III) | 1-Ce | 1-Pr | 1-Nd | 2-Nd | 1-Gd | 1-Tb |
|---|---|---|---|---|---|---|
| Ln–Osiloxide range | 2.268(5)–2.351(5) | 2.248(9)–2.304(9) | 2.231(2)–2.3300(19) | 2.206(2)–2.336(2) | 2.212(3)–2.253(3) | 2.138(2)–2.194(2) |
| (Ln–Osiloxide)avg | 2.30(3) | 2.27(2) | 2.27(4) | 2.28(4) | 2.22(2) | 2.17(3) |
| Ln(IV) | 3-CeTHF | 3-PrMecN | 4-PrPh | 3-TbMeCN | 3-TbTHF | 4-TbPh | 4-TbEt |
|---|---|---|---|---|---|---|---|
| Ln–Osiloxide range | 2.109(3)–2.154(3) | 2.088(4)–2.121(4) | 2.113(2)–2.180(2) | 2.028(5)–2.087(5) | 2.047(2)–2.087(2) | 2.015(8)–2.149(7) | 2.014(3)–2.047(3) |
| (Ln–Osiloxide)avg | 2.13(3) | 2.10(1) | 2.15(3) | 2.06(2) | 2.07(2) | 2.06(5) | 2.03(1) |
| 1-Gd | 1-Tb | 2-Nd | |
|---|---|---|---|
| Ln1–K | 3.5143(12) | 3.6921(7) | 3.6808(8), 3.7774(8) |
The complex 2-Nd crystallizes in the monoclinic P21/n space group. The solid-state structure of 2-Nd shows a five-coordinated Nd(III) metal center bound by five monodentate triphenylsiloxide ligands in a distorted trigonal bipyramidal geometry (Fig. 2). The triphenylsiloxide ligands bind the two potassium in close proximity to the Nd(III) metal center through the anionic oxygen and cation–π interactions with the phenyl groups. The two potassium cations lie at a distance from the Nd(III) center of 3.6808(8) Å for K1 and 3.7774(8) Å for K2. The K1 potassium is at a similar distance from the metal center compared to the Nd1–K1 distance in 1-Nd (3.6925(6) Å). However, the second potassium (K2) is found at a larger distance, suggesting a weaker binding. The presence of five triphenylsiloxide ligands around the Nd(III) ion in 2-Nd does not significantly affect the Nd–Osiloxide distances ((Nd1–Osiloxide)avg = 2.28(4) Å) compared to those found in the tetrakis complex 1-Nd ((Nd1–Osiloxide)avg = 2.27(4) Å).
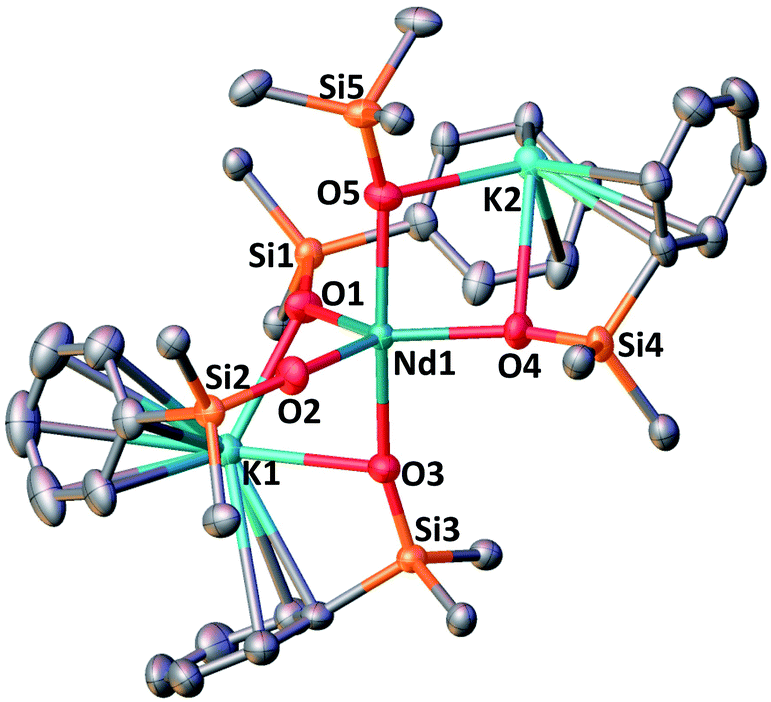 | ||
| Fig. 2 Solid-state molecular structure of 2-Nd (50% probability ellipsoids). Hydrogen atoms, phenyl groups not bound to the potassium ions and residual solvent molecules are omitted for clarity. Selected distances (Å): Nd1–Osiloxide range = 2.206(2)–2.336(2); mean Nd1–Osiloxide = 2.28(4); Nd1–K1 = 3.6808(8); Nd1–K2 = 3.7774(8). | ||
Solid state structures of lanthanide(IV) complexes
The complexes 3-CeTHF and 3-TbTHF are isomorphous and crystallize in the monoclinic C2/c space group while 3-LnMeCN (Ln = Pr, Tb) was reported to crystallize in the orthorhombic Pca21 space group.4,5c Their molecular structures are presented in Fig. 3 and show Ln(IV) metal centers coordinated by two THF molecules and four triphenylsiloxide ligands in a distorted octahedral geometry. The Ce1–Osiloxide distances in 3-CeTHF (2.109(3)–2.154(4) Å) are consistent with the Ce1–Osiloxide bond length found in the previously reported DME analogue [CeIV(OSiPh3)4(DME)] (2.10(1)–2.13(1) Å).12 Furthermore, the shorter Ce1–Osiloxide bond distances in 3-CeTHF (mean value = 2.13(3) Å) compared to those of the Ce(III) complex 1-Ce (2.30(3) Å) corroborate the +IV oxidation state of the cerium metal center. This shortening can also be identified in the Ce1–OTHF bond distances (2.501(4) Å in 3-CeTHF; 2.598(6) and 2.605(6) Å in 1-Ce). The presence of THF instead of MeCN coordinated to the Tb(IV) metal center does not significantly affect the mean Tb1–Osiloxide bond distance in 3-TbTHF (2.07(2) Å) compared to 3-TbMeCN (2.06(2) Å).5c The 0.03 Å difference in the mean Ln1–Osiloxide bond distances between 3-CeTHF and 3-PrMeCN (Table 1) is consistent with the difference in the 6-coordinate Shannon radii of Ce(IV) (0.87 Å) and Pr(IV) (0.85 Å) ions.25 However, the 0.06–0.07 Å difference in Ln1–Osiloxide bond distances between 3-CeTHF and 3-TbMeCN or 3-TbTHF (Table 1) is smaller than what is expected from the difference in the Shannon ionic radii of Ce(IV) (0.87 Å) and Tb(IV) (0.76 Å).25 A smaller than expected difference was already noticed for 3-PrMeCN compared to 3-TbMeCN and an increased covalency in the Ln(IV)–O bonds in 3-PrMeCN was proposed on the basis of DFT studies.4 This can be correlated with studies of the tetravalent inorganic Cs2RbLnF7 materials (Ln = Ce, Pr, Nd, Tb, Dy) where an increases in the 4f–ligand–p orbital hybridization was discovered in the order Tb < Dy < Ce < Pr < Nd.26 | ||
Fig. 3 (a) Solid-state molecular structure of 3-CeTHF (a) and 3-TbTHF (b) (50% probability ellipsoids). Hydrogen atoms, phenyl groups and one residual solvent molecule are omitted for clarity. Selected distances (Å): Ce1–Osiloxide range = 2.109(3)–2.154(3); mean Ce1–Osiloxide = 2.13(3); Ce1–OTHF = 2.501(4); Tb1–Osiloxide range = 2.047(2)–2.087(2); mean Tb1–Osiloxide = 2.07(2); Tb1–OTHF = 2.394(3). Symmetry transformation used to generate equivalent atoms:1 1 − x, +y;  . . | ||
The complex 4-PrPh crystallizes in the monoclinic P21/c space group and shows a six-coordinated Pr(IV) metal center in a distorted octahedral geometry (Fig. 4). The Pr(IV) ion is coordinated to four monodentate triphenylsiloxide ligands, one acetonitrile molecule and one triphenylphosphinoxide. Compared to 3-PrMeCN, one MeCN moiety is replaced with a phosphinoxide ligand while the second acetonitrile molecule remains bound retaining the distorted octahedral geometry of the complex and the same coordination number (CN = 6). This is different from what was previously observed with the smaller Tb(IV) ion. Indeed, the addition of 1 equivalent OPPh3 to 3-TbMeCN resulted in the removal of the two bound acetonitrile molecules. As such, the solid-state structure of the phosphinoxide complex 4-TbR (R = Ph, Et) does not retain the distorted octahedral geometry found in 3-TbMeCN but, instead, displays a distorted trigonal bipyramidal molecular geometry with a five-coordinated Tb(IV) metal center.5c The mean value of the Pr1–Osiloxide bond length in 4-PrPh (2.15(3) Å) is slightly larger than in 3-PrMeCN (2.10(1) Å).4 This can be rationalized in terms of the increased steric hindrance provided by the bulky triphenylphosphine oxide ligand compared to the MeCN molecule. In the case of the smaller Tb(IV) ion, the lower coordination number of the phosphinoxide complex 4-TbPh (2.06(5) Å) compared to the 3-TbMeCN results in similar values of the Tb(IV)–Osiloxide bond distance in the two complexes (4-TbPh:2.06(5) Å and 3-TbMeCN:2.06(2) Å).5c Anyhow, the mean value of the Pr(IV)–Osiloxide bond length in 4-PrPh (2.15(3) Å) is considerably smaller than the mean Pr(III)–Osiloxide bond length in 1-Pr (2.27(2) Å). Furthermore, the Pr1–Ophosphinoxide bond distance in 4-PrPh (2.280(2) Å) is shorter than those found in the literature for Pr(III)–OPPh3 bond lengths (2.333(3)–2.409(5) Å;27,28) and is consistent with a Pr metal center in the +IV oxidation state.
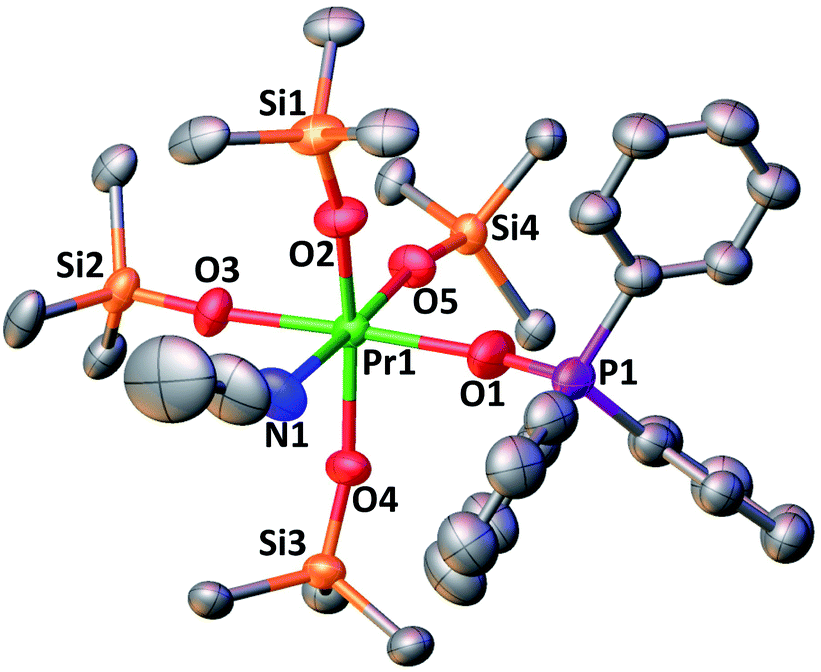 | ||
| Fig. 4 Solid-state molecular structure of 4-PrPh (50% probability ellipsoids). Hydrogen atoms and phenyl groups of the siloxide ligands are omitted for clarity. Selected distances (Å): Pr1–Osiloxide range = 2.113(2)–2.180(2); mean Pr1–Osiloxide = 2.15(3); Pr1–O1 = 2.280(2); Pr1–N1 = 2.636(3). | ||
Infrared spectra
The IR spectra are reported in Fig. S18 and S19.‡ The IR spectra of the free ligand HOSiPh3, the deprotonated ligand (KLPh) and of the lanthanide complexes show similar features with aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) H stretching and bending absorptions at 3060–2995 cm−1 and 700 cm−1, respectively, and the Si–O stretching at 1121 cm−1, as reported in the literature.29 Upon deprotonation or binding to a Ln cation, the peak at 855 cm−1 assigned to the Si–O–H stretching disappears30 and a new intense band appears at 964 cm−1 for the Ln(III) complexes 1-Ln (Ln = Ce, Pr, Gd) which is assigned to the Ln–O–Si linkage vibration. This value compares well with the range 960–1000 cm−1 reported for tin(IV) aryl- and alkyl siloxyl complexes. In the Ln(IV) complexes 3-CeTHF, 3-PrMeCN and 4-PrPh the Ln–O–Si stretch is significantly shifted to lower frequencies (883/899 cm−1) compared to Ln(III) complexes but reasonably consistent with the values reported for the tetrakis(triphenylsiloxide)-titanium(IV) complex (926 cm−1)29a and with the M–O–Si stretch observed for group IV and group V metals (920–906 cm−1).29b Two weak features are present at 2276 and 2304 cm−1 for 3-PrMeCN and 4-PrPh that are assigned to the ν2 and (ν3 + ν4) modes of coordinated acetonitrile molecules, as reported previously31 (2281 and 2310 cm−1, respectively, for [NdIII(ClO4)x(MeCN)y]). This observation confirms the complex formulae reported above.
H stretching and bending absorptions at 3060–2995 cm−1 and 700 cm−1, respectively, and the Si–O stretching at 1121 cm−1, as reported in the literature.29 Upon deprotonation or binding to a Ln cation, the peak at 855 cm−1 assigned to the Si–O–H stretching disappears30 and a new intense band appears at 964 cm−1 for the Ln(III) complexes 1-Ln (Ln = Ce, Pr, Gd) which is assigned to the Ln–O–Si linkage vibration. This value compares well with the range 960–1000 cm−1 reported for tin(IV) aryl- and alkyl siloxyl complexes. In the Ln(IV) complexes 3-CeTHF, 3-PrMeCN and 4-PrPh the Ln–O–Si stretch is significantly shifted to lower frequencies (883/899 cm−1) compared to Ln(III) complexes but reasonably consistent with the values reported for the tetrakis(triphenylsiloxide)-titanium(IV) complex (926 cm−1)29a and with the M–O–Si stretch observed for group IV and group V metals (920–906 cm−1).29b Two weak features are present at 2276 and 2304 cm−1 for 3-PrMeCN and 4-PrPh that are assigned to the ν2 and (ν3 + ν4) modes of coordinated acetonitrile molecules, as reported previously31 (2281 and 2310 cm−1, respectively, for [NdIII(ClO4)x(MeCN)y]). This observation confirms the complex formulae reported above.
In inorganic matrices, four f–f transitions have been reported for Ce(III) in the range 2100–3800 cm−1, in line with the crystal field splitting of 2F7/2 (ref. 32) while transitions to the PrIV (2F7/2) level have been detected around 2900 cm−1 for fluorides.27 Close scrutiny of the IR spectra up to 6000 cm−1 for the Pr(IV) complexes did not reveal any feature that could be assigned to an f–f transition. This might be due to very weak oscillator strengths and/or overlap with ligand vibrations, particularly in the 2800–3000 cm−1 range. On the other hand, the spectrum of a more concentrated sample of 1-Ce revealed a very weak and sharp band at 2163 cm−1 (see insert, Fig. S18‡) not seen in any other spectra including the blank, that might possibly be assigned to the most intense f–f transition of CeIII identified at 2128 cm−1 in YGdO3:CeIII.32
UV-visible absorption spectra
The UV-vis spectra of KLPh, 1-Ln (Ln = Ce, Pr, Gd) and 3-PrMeCN were recorded in THF (Fig. S20‡). The spectrum of KOSiPh3 features two main bands located at 206 and 223 nm and assigned to π → π* transitions; in addition, a very weak feature is observed in the range 250–275 nm. The same bands are present for all the complexes, with negligible red shifts of 1–2 nm. The ligand absorptions overlap with the 4f–5d absorptions of Ce(III), usually in the range 200–270 nm, and have much larger oscillator strengths so that these 4f–5d transitions could not be identified in 1-Ce. For the Ln(IV) complexes 3-PrMeCN4 and 3-TbMeCN,5c an additional broad signal has been reported with maximum at 363 nm (ε = 3800 M−1 cm−1) and 386 nm (ε = 3000 M−1 cm−1), respectively. These bands can be assigned to ligand-to-metal charge transfers (LMCT) on the basis of previous UV studies on a range of Ln(IV) oxides.2,33 Since 4-PrPh reverts to the 3-PrTHF complex in THF, the UV-visible spectrum of 4-PrPh was measured in toluene (Fig. S21‡), as well as the spectra of 4-TbR5c and of the previously reported 3-PrMeCN complex4 for comparison. All compounds display a similar broad absorption peak, assigned to a LMCT transition, around 374–379 nm with ε = 4500 for 4-PrPh, 3300 for 4-TbPh,5c and 3150 M−1 cm−1 for 4-TbEt,5c while data for 3-PrMeCN are similar to those in THF: λmax = 365 nm and ε = 4140 M−1 cm−1.4 As reported in the synthesis description above, time-dependent spectra for 4-PrPh and 4-TbR evidence a slow decomposition of the complexes with time. To ensure reliable photophysical data, samples were measured immediately after their preparation.Ligand-centered luminescence of KLPh and 1-Gd
Data are reported in Table S1.‡ In toluene at room temperature, the KOSiPh3 (KLPh) ligand has one excitation band with maximum at 289 nm and one structured emission band with several components and main maxima at 325 and 339 nm (Fig. S25‡). In frozen solution, the excitation is blue shifted at 276 nm while the emission band at 325/339 nm disappears to the benefit of a broad feature with complicated vibrational structure (Fig. 5 and S25‡). The luminescence decay in frozen solution could be fitted with a biexponential model resulting in an average lifetime of 1.8 ± 0.1 μs (Table 3 and S2‡). These data are consistent with this band corresponding to emission from the triplet state, with the 0-phonon component located at 403 nm (24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 815 cm−1).
815 cm−1).
| Sample | λ ex/nm | λ em/nm | τ 1/μs | τ av /μs |
|---|---|---|---|---|
| a Biexponential decay, amplitude average lifetime: τav = Σaiτi. | ||||
| KLPh | 276 | 431 | — | 1.8 ± 0.1 |
| 1-Gd | 280 | 427 | — | 0.9 ± 0.2 |
| 1-Ce | 276 | 410 | 1.5 ± 0.2 | — |
| 1-Pr | 277 | 450 | — | 5.80 ± 0.2 |
| 3-PrMeCN | 307 | 611 | 3.4 ± 0.2 | — |
| 4-PrPh | 345 | 599 | — | 3.45 ± 0.17 |
The energy of the lowest lying excited f-level of Gd(III) (6P7/2) is 32150 cm−1, therefore at higher energy than the highest component of the ligand states so that negligible ligand-to-metal energy transfer is expected. The complex is then useful for identifying the singlet and triplet levels of the coordinated ligands. In toluene at room temperature (Fig. S25‡), 1-Gd has excitation and emission envelopes with maxima close to those recorded for the ligand, although their components have different intensities. In particular, the component at 317 nm (31545 cm−1), assigned to the 0-phonon transition of the lowest lying singlet state 1ππ* has a much larger intensity. As for the ligand, in frozen solution this band disappears and a structured feature emerges with well resolved vibrational components; the 0-phonon component is identified at 401 nm (24940 cm−1). The average lifetime, 0.90 ± 0.2 μs is compatible with the assignment to 3ππ* emission, and shorter than the ligand alone average lifetime due to the paramagnetic effect of Gd(III).
Metal centered luminescence of 1-Ln (Ln = Ce, Pr), 3-PrMeCN and 4-PrPh
Pr(IV) having the same electronic configuration than Ce(III), 4f1, we set out to identify transitions specific to this configuration, for instance d–f transitions that are well known for Ce(III). We therefore compare the luminescence spectra obtained for the Pr(IV) complexes with those of the Ce(III) and Pr(III) complexes. 3-PrMeCN being not stable enough in toluene at room temperature the following discussion mainly involves spectra recorded at 77 K, see Fig. 5, S26–S29 and Tables 3 and S2‡ list the corresponding lifetimes.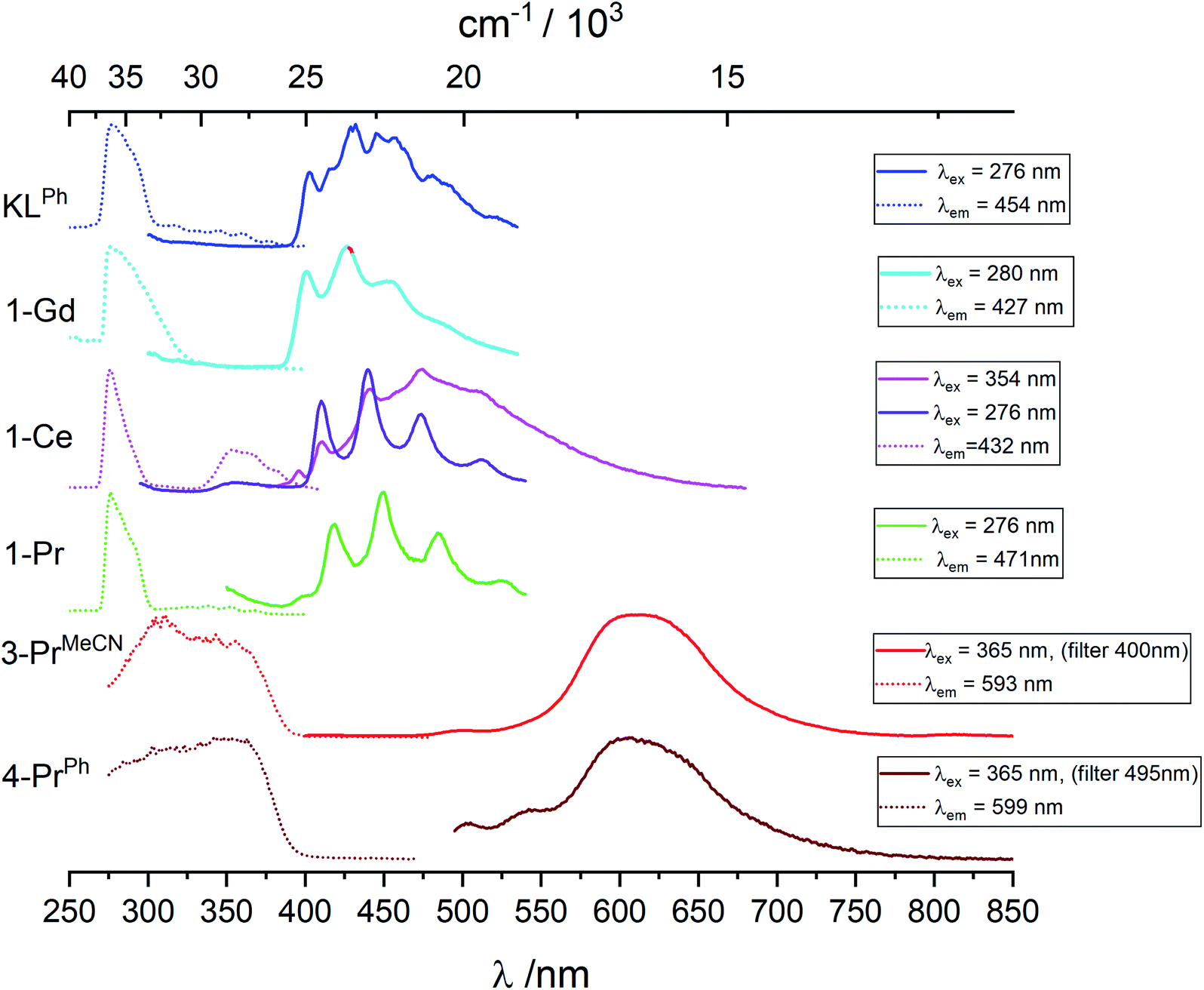 | ||
| Fig. 5 Excitation and emission spectra of KOSiPh3, 1-Ln (Ln = Gd, Ce, Pr), 3-PrMecN and 4-PrPh in 5 mM toluene solution at 77 K. Spectra are normalized and their intensities cannot be directly compared. | ||
Upon excitation in the ligand levels at 276 nm, the emission spectrum of 1-Ce (Fig. S26‡) features a weak emission band at 360 nm and an intense, well-structured band between 400 and 535 nm with a vibrational progression of ∼1500 cm−1; the lifetime determined by monitoring the 410 nm component, 1.5 μs, is in line with a 3ππ* emission. Excitation at 354 nm results in a composite spectrum with the same structured band featuring a 0-phonon transition at 395 nm (25320 cm−1) superimposed onto a broad emission centered around 471 nm. We assign this broad band to d–f transitions of Ce(III) which are the basis of its spectroscopy with tuneable bands ranging from the blue to the red.34,16a,15c,15g
The Pr(III) ion has two emitting levels, 3P0 (21390 cm−1) and 1D2 (17335 cm−1), that generate blue and red lines.35 In 1-Pr, the triplet state energy level is higher than that of the 3P0 level, so that both emitting levels might be populated. However, as shown on Fig. S28,‡ upon excitation at 352 nm, a spectrum identical to that of 1-Ce is obtained, corresponding again to 3ππ* emission from the ligand. No f–f transitions specific to Pr(III) are detected, which implies negligible energy transfer from the ligand and/or efficient back transfer and quenching. Indeed, it is known that Pr(III) complexes tend to have very low quantum yields.36
Upon excitation at 307 or 365 nm, 3-PrMeCN (Fig. 5 and S29‡) exhibits one broad and symmetrical emission band centered at 611 nm as well as a very weak band at 500 nm and a shoulder at ∼550 nm. The luminescence decay at 611 nm can be fitted with a single exponential, leading to a lifetime of 3.4 μs. This differs markedly from the emission spectrum of 1-Pr and, as anticipated, the excitation spectrum is also different with, in addition to the ligand-centered transitions, a broad band around 350 nm corresponding to the LMCT identified in the absorption spectrum. These spectra represent the unique signature of the tetravalent complex.
The luminescence spectrum of 4-PrPh is similar to the one of 3-PrMeCN (Fig. S29‡) with a maximum at 608 nm, except that 4-PrPh is much less luminescent and that the two weak features at 500 and 545 nm are proportionally more intense. No residual signal attributed to the triphenylphosphine oxide was detected even when both ligands were excited at 365 nm. The average lifetime corresponding to the 608 nm band is 3.45 μs and its onset is estimated to be at 19000 cm−1. The origin of the broad emission band of the Pr(IV) complexes is discussed in the section below.
Electronic structure of the Pr(IV) complexes
The ≈600 nm emission of the two Pr(IV) complexes, could originate either from d–f transitions red-shifted with respect to 1-Ce or from emission from the 3ππ* state of the coordinated ligand. Although in a few cases longer lifetimes have been reported,37 lifetimes of d–f transitions are usually in the ns range. The measured decays for the Pr(IV) complexes are in the μs range, which could favour the second explanation. In order to substantiate this point, we have performed calculations at the DFT level (B3PW91 functional) for both Ce(III) and Pr(IV) complexes. In order to look for the d–f excitation, UV-visible spectra were computed using the TDDFT method. As expected for Ce(III), the d–f excitation appears as the first significant absorption band whereas no trace of such an absorption was found for Pr(IV) (see ESI‡).Therefore, the hypothesis of an emission derived from a d–f transition in the Pr(IV) complex is quite unlikely and we thus investigated the possibility of an emission from the ligand triplet state. In order to do so, both the ground state and an excited state were optimized. To make sure that the excitation arises from the ligand, a doublet and a quartet spin states were computed for the two complexes and we verified that in both cases the quartet excited state is located on the ligand. One outcome is that the quartet state of 1-Ce is calculated as lying at 27420 cm−1, well in line with the observed value of 24630 cm−1. The corresponding calculated level for Pr(IV) is largely red shifted by 12100 cm−1; the shift observed experimentally in going from Ce(III) to Pr(IV), is smaller, but remains sizable at ≈5100 cm−1. The results are sketched in the electronic diagram of Fig. 6. It should be noted that the calculated absorption spectra (see ESI‡) also show a LMCT transition at 506 nm but the associated energy of 2.44 eV (19680 cm−1) is larger than that of the triplet state at 1.9 eV (15325 cm−1, 625 nm) so that we have ruled out a potential contribution to the emission spectrum.
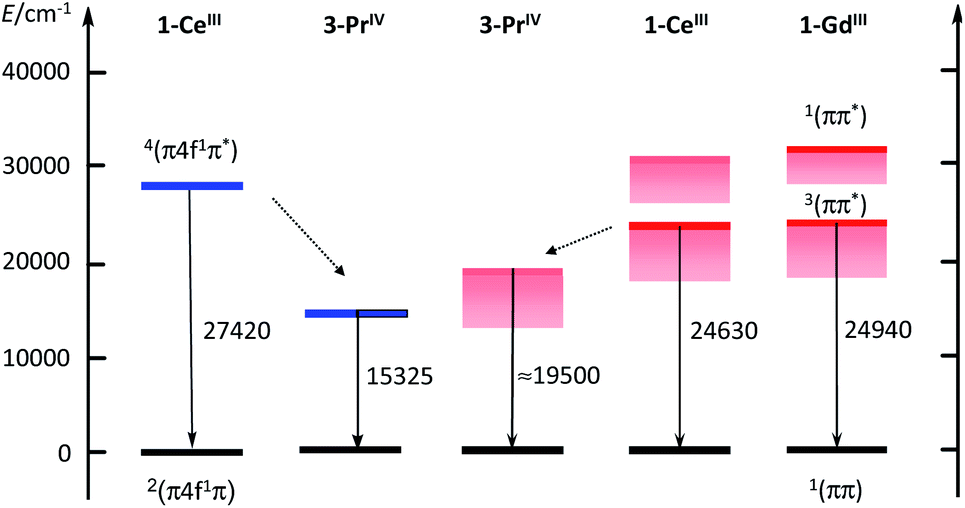 | ||
| Fig. 6 Partial electronic diagram for 1-Ce, 3-PrMecN, and 1-Gd. Calculated levels in blue, experimental 0-phonon levels in red or rose when estimated from the onset of the emission band. Energies of the transitions in cm−1. | ||
Conclusions
In conclusion, we have synthesised and crystallographically characterized tetrakis-complexes of several Ln(III) ions (Ce, Pr, Nd, Gd, Tb), 1-Ln, with the triphenylsiloxide ligand and the first example of a siloxide pentakis-complex of Nd(III), 2-Nd.Oxidation of the 1-Ce complex with silver tetraphenylborate allowed the synthesis of the Ce(IV) analogue 3-CeTHF. The oxidation of 1-Nd or 2-Nd to yield Nd(IV) was proven impossible, but the oxidation of 1-Pr and 1-Tb with the strong oxidizing agent “magic blue” allowed the synthesis of the Ln(IV) complexes 3-TbMeCN and 3-PrMeCN. The two molecules of bound acetonitrile in complexes 3-LnMeCN (Ln = Pr, Tb) could be easily replaced by THF or phosphinoxides OPR3 (R = Et, Ph) leading to isolation of 3-TbTHF and the phosphinoxide adducts [TbIV(OSiPh3)4(OPR3)], 4-TbR (R = Et, Ph) and [PrIV(OSiPh3)4(OPPh3)(MeCN)], 4-PrPh. Complex 4-PrPh is the second ever reported example of a Pr(IV) molecular complex and shows significantly increased solution stability compared to 3-PrMeCN suggesting that the synthesis of other Pr(IV) molecular complexes should be in reach. The infrared, UV-visible and luminescence spectra of the isolated complexes were measured and the assignment of the luminescence spectra was corroborated by computational studies. The luminescence spectra of Gd(III), Pr(III) and Ce(III) all show emission from the coordinated ligand 3ππ* state which is only slightly displaced with respect to the free ligand. The Ce(III) ion also shows a broad emission assigned to d–f transition. The first luminescence spectrum of the Pr(IV) was measured and shows a broad intense emission. On the basis of the computational studies and of the measured lifetime in the microsecond range the emission is assigned to the 3ππ* state of the coordinated ligand. The coordination of the ligands to the Pr(IV) ion results in an unprecedented large shift of the ligand triplet state probably due to an increased covalency in the Ln(IV)–O bonds compared with Ln(III)–O bonds. The observed large Stokes' shift anticipates the possibility of applications of Ln(IV) in material science.
Author contributions
M. M. conceived the concepts and supervised the project. A. R. W. performed the majority of the experiments. A.-S. C. conceived and measured the luminescence experiments. A.-S. C. and J.-C. G. B. analysed the luminescence data. I. D. and L. M. carried out and analysed the computational data. F. F.-T. carried out and analysed the X-ray data. All authors contributed to the analysis and writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge support from the Swiss National Science Foundation and the Ecole Polytechnique Fédérale de Lausanne (EPFL). We thank Dr R. Scopelliti for important contributions to the X-ray single crystal structure analyses. L. M. is a senior member of the Institut Universitaire de France. CalMip is acknowledged for a generous grant of computing time.Notes and references
-
(a) Y. Doi, K. Ninomiya, Y. Hinatsu and K. Ohoyama, J. Phys. Condens. Matter, 2005, 17, 4393–4401 CrossRef CAS
; (b) S. Cotton, Lanthanides and Actinides Chemistry, John Wiley & Sons, Chichester, 2006 CrossRef
- D. E. Hobart, K. Samhoun, J. P. Young, V. E. Norvell, G. Mamantov and J. R. Peterson, Inorg. Nucl. Chem. Lett., 1980, 16, 321–328 CrossRef CAS
-
(a) W. Noh and G. S. Girolami, Polyhedron, 2007, 26, 3865–3870 CrossRef CAS
- A. R. Willauer, C. T. Palumbo, F. Fadaei-Tirani, I. Zivkovic, I. Douair, L. Maron and M. Mazzanti, J. Am. Chem. Soc., 2020, 142, 5538–5542 CrossRef CAS PubMed
-
(a) C. T. Palumbo, I. Zivkovic, R. Scopelliti and M. Mazzanti, J. Am. Chem. Soc., 2019, 141, 9827–9831 CrossRef CAS PubMed
-
(a) R. P. Kelly, L. Maron, R. Scopelliti and M. Mazzanti, Angew. Chem., Int. Ed., 2017, 56, 15663–15666 CrossRef CAS PubMed
-
(a) W. J. Evans, R. E. Golden and J. W. Ziller, Inorg. Chem., 1991, 30, 4963–4968 CrossRef CAS
- P. S. Gradeff, K. Yunlu, T. J. Deming, J. M. Olofson, R. J. Doedens and W. J. Evans, Inorg. Chem., 1990, 29, 420–424 CrossRef CAS
- Z. W. Xie, K. Chui, Q. C. Yang, T. C. W. Mak and J. Sun, Organometallics, 1998, 17, 3937–3944 CrossRef CAS
- T. J. Boyle, S. D. Bunge, P. G. Clem, J. Richardson, J. T. Dawley, L. A. M. Ottley, M. A. Rodriguez, B. A. Tuttle, G. R. Avilucea and R. G. Tissot, Inorg. Chem., 2005, 44, 1588–1600 CrossRef CAS PubMed
- T. J. Boyle, L. A. M. Ottley, L. N. Brewer, J. Sigman, P. G. Clem and J. J. Richardson, Eur. J. Inorg. Chem., 2007, 3805–3815 CrossRef CAS
- P. S. Gradeff, K. Yunlu, A. Gleizes and J. Galy, Polyhedron, 1989, 8, 1001–1005 CrossRef CAS
-
(a) J.-C. G. Bünzli, S. Comby, A. S. Chauvin and C. D. B. Vandevyver, J. Rare Earths, 2007, 25, 257–274 CrossRef
-
S. Cotton, Lanthanides and Actinides, MacMillan Education, London, 1991 Search PubMed
-
(a) L. Armelao, S. Quici, F. Barigelletti, G. Accorsi, G. Bottaro, M. Cavazzini and E. Tondello, Coord. Chem. Rev., 2010, 254, 487–505 CrossRef CAS
-
(a) X. Qin, X. W. Liu, W. Huang, M. Bettinelli and X. G. Liu, Chem. Rev., 2017, 117, 4488–4527 CrossRef CAS PubMed
- J. R. Levin, W. L. Dorfner, P. J. Carroll and E. J. Schelter, Chem. Sci., 2015, 6, 6925–6934 RSC
-
(a) A. H. Hu, J. J. Guo, H. Pan, H. M. Tang, Z. B. Gao and Z. W. Zuo, J. Am. Chem. Soc., 2018, 140, 1612–1616 CrossRef CAS PubMed
-
(a) H. Kunkely and A. Vogler, J. Photochem. Photobiol., A, 2001, 146, 63–66 CrossRef CAS
-
(a) J. Andrez, J. Pecaut, P.-A. Bayle and M. Mazzanti, Angew. Chem., Int. Ed., 2014, 53, 10448–10452 CrossRef CAS PubMed
- T. J. Boyle and L. A. M. Ottley, Chem. Rev., 2008, 108, 1896–1917 CrossRef CAS PubMed
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed
- P. Dorenbos, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 165107 CrossRef
- A. Habenschuss and F. H. Spedding, J. Chem. Phys., 1980, 73, 442–450 CrossRef CAS
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef
- Z. W. Hu, G. Kaindl and B. G. Muller, J. Alloys Compd., 1997, 246, 177–185 CrossRef CAS
- A. W. G. Platt, J. Fawcett, R. S. Hughes and D. R. Russell, Inorg. Chim. Acta, 1999, 295, 146–152 CrossRef CAS
- Y. Ma, S. Xu, X. Wang, M. Liu, Y. X. Li, X. L. Xin and Q. H. Jin, Z. Anorg. Allg. Chem., 2017, 643, 780–788 CrossRef CAS
-
(a) V. A. Zeitler and C. A. Brown, J. Phys. Chem., 1957, 61, 1174–1177 CrossRef CAS
- I. S. Ignatyev, F. Partal, J. J. L. Gonzalez and T. Sundius, Spectrochim. Acta, Part A, 2004, 60, 1169–1178 CrossRef PubMed
- J.-C. G. Bünzli and C. Mabillard, Inorg. Chem., 1986, 25, 2750–2754 CrossRef
- G. A. Slack, S. L. Dole, V. Tsoukala and G. S. Nolas, J. Opt. Soc. Am. B, 1994, 11, 961–974 CrossRef CAS
- H. E. Hoefdraad, J. Inorg. Nucl. Chem., 1975, 37, 1917–1921 CrossRef CAS
-
(a) P. N. Hazin, C. Lakshminarayan, L. S. Brinen, J. L. Knee, J. W. Bruno, W. E. Streib and K. Folting, Inorg. Chem., 1988, 27, 1393–1400 CrossRef CAS
-
(a) H. Dornauf and J. Heber, J. Lumin., 1979, 20, 271–281 CrossRef CAS
- E. V. Salerno, S. V. Eliseeva, B. L. Schneider, J. W. Kampf, S. Petoud and V. L. Pecoraro, J. Phys. Chem. A, 2020, 124, 10550–10564 CrossRef CAS PubMed
-
(a) T. Hoshina, J. Phys. Soc. Jpn., 1980, 48, 1261–1268 CrossRef CAS
Footnotes |
| † Dedicated to Professor Peter Junk on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2104729–2104735. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc05517h |
| This journal is © The Royal Society of Chemistry 2022 |