
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancing proline-rich antimicrobial peptide action by homodimerization: influence of bifunctional linker†
Wenyi
Li
 *ab,
Feng
Lin
c,
Andrew
Hung
d,
Anders
Barlow
e,
Marc-Antoine
Sani
af,
Rita
Paolini
b,
William
Singleton
b,
James
Holden
b,
Mohammed Akhter
Hossain
cf,
Frances
Separovic
af,
Neil M.
O'Brien-Simpson
*ab and
John D.
Wade
*cf
*ab,
Feng
Lin
c,
Andrew
Hung
d,
Anders
Barlow
e,
Marc-Antoine
Sani
af,
Rita
Paolini
b,
William
Singleton
b,
James
Holden
b,
Mohammed Akhter
Hossain
cf,
Frances
Separovic
af,
Neil M.
O'Brien-Simpson
*ab and
John D.
Wade
*cf
aThe Bio21 Institute of Molecular Science and Biotechnology, Australia
bMelbourne Dental School, Centre for Oral Health Research, Australia. E-mail: wenyi.li@unimelb.edu.au; neil.obs@unimelb.edu.au
cFlorey Institute of Neuroscience and Mental Health, University of Melbourne, Australia. E-mail: john.wade@florey.edu.au
dSchool of Science, RMIT University, Australia
eMaterials Characterization and Fabrication Platform, Australia
fSchool of Chemistry, University of Melbourne, Victoria 3010, Australia
First published on 1st February 2022
Abstract
Antimicrobial peptides (AMPs) are host defense peptides, and unlike conventional antibiotics, they possess potent broad spectrum activities and, induce little or no antimicrobial resistance. They are attractive lead molecules for rational development to improve their therapeutic index. Our current studies examined dimerization of the de novo designed proline-rich AMP (PrAMP), Chex1-Arg20 hydrazide, via C-terminal thiol addition to a series of bifunctional benzene or phenyl tethers to determine the effect of orientation of the peptides and linker length on antimicrobial activity. Antibacterial assays confirmed that dimerization per se significantly enhances Chex1-Arg20 hydrazide action. Greatest advantage was conferred using perfluoroaromatic linkers (tetrafluorobenzene and octofluorobiphenyl) with the resulting dimeric peptides 6 and 7 exhibiting potent action against Gram-negative bacteria, especially the World Health Organization's critical priority-listed multidrug-resistant (MDR)/extensively drug-resistant (XDR) Acinetobacter baumannii as well as preformed biofilms. Mode of action studies indicated these lead PrAMPs can interact with both outer and inner bacterial membranes to affect the membrane potential and stress response. Additionally, 6 and 7 possess potent immunomodulatory activity and neutralise inflammation via nitric oxide production in macrophages. Molecular dynamics simulations of adsorption and permeation mechanisms of the PrAMP on a mixed lipid membrane bilayer showed that a rigid, planar tethered dimer orientation, together with the presence of fluorine atoms that provide increased bacterial membrane interaction, is critical for enhanced dimer activity. These findings highlight the advantages of use of such bifunctional tethers to produce first-in-class, potent PrAMP dimers against MDR/XDR bacterial infections.
Introduction
Since the discovery of penicillin and subsequent antibiotics, the increasing global consumption of such wonder drugs has played a critical role in driving antimicrobial resistance (AMR) at an alarming rate in the last two decades as assessed by the World Health Organization (WHO).1,2 The O'Neill report estimates deaths caused by AMR will exceed 10 million by 2050.3 The ongoing COVID-19 pandemic is further constricting the pipeline of antimicrobials to handle bacterial infections and highlights the critical need for novel treatments by antibacterial agents.4 A recent economic model for evaluating the global cost of a newly developed active antibiotic with FDA approval was estimated at US$1.58 billion, excluding the marketing and surveillance after approval.5 With limited revenues from antibiotic investment, many big pharmaceutical firms have redirected their antimicrobial research programs to more profitable fields such as anticancer drugs. To date, the antibiotic market remains stocked with classical antibacterial drugs with slight chemical variations on existing antibiotic classes.6 In 2018, the WHO identified a critical priority list calling for the development of new antibiotics against three Gram-negative bacteria, including Acinetobacter baumannii, Pseudomonas aeruginosa and Enterobacteriaceae7 Various strategies, including peptides, vaccines, probiotics, engineered bacteriophages and adjuvants, have been investigated to identify and develop novel antibacterial therapies.8Due to their multimodal action and broad potency antimicrobial peptides (AMPs), primarily host defense peptides, have demonstrated significant antibacterial activity against a range of multi-drug resistant (MDR) pathogens labelled as “superbugs”.9 Since their discovery a century ago, various chemical modifications of AMPs have been applied to increase or enhance their potency against target microbes. These have been comprehensively summarised in our recent review.10 Proline-rich AMPs (PrAMPs) are considered a promising class of candidates for rational design to target Gram-negative pathogens due to their low toxicity and multiple intracellular targets, such as DnaK and the bacterial ribosome.11–13 We previously applied several chemical modifications on the de novo designed PrAMP, Chex1-Arg20, with different terminal functional moieties to broaden its antibacterial spectrum.14,15 Additionally, multimerization of Chex1-Arg20 can further broaden its spectrum of activity as well as increase its antibacterial activity.16,17 Based on these previous studies, we observed that the bifunctional cross linkage can modulate the antimicrobial activities of the synthetic multimers. However, the syntheses required to form these linked tetrameric analogues with high potency against nosocomial bacteria, involve multistep purification and are time-consuming.16,17 Consequently we have revisited the simpler dimer entity and, following a screen of numerous bifunctional linkers, have focused on a class of perfluoroaromatic compounds. In the current report, we developed a series of new dimeric PrAMPs to target the WHO priority critical Gram-negative bacterial pathogens, such as A. baumannii and its MDR and extensively drug-resistant (XDR) strain. The aim was to determine the optimum length of linker and orientation of the linked PrAMPs and found that tetrafluorobenzene- and octofluorobiphenyl-linked dimers displayed significantly enhanced activity. A comprehensive study of bactericidal kinetics, membrane interaction and binding, as well as stress response, combined with imaging analysis and molecular dynamics has provided further mechanistic details for the development of such next generation PrAMPs.
Results
Dimeric PrAMP Chex1-Arg20 preparation
Synthetic Chex1-Arg20 containing a C-terminal hydrazide (–NHNH2) modification was prepared via Fmoc/tBu solid phase peptide synthesis (SPPS) on chloro-(2′-chloro)trityl polystyrene resin. Due to the efficiency of thiol chemistry, thiol bioconjugation provides multiple pathways to construct biomolecules, including thiol-disulfide, thiol-halogeno and thiol-parafluoro.18 To facilitate the thiol chemistry, a single Cys residue was incorporated into the C-terminus of monomeric PrAMP Chex1-Arg20. A control, monomeric PrAMP with Cys, was thiol-capped via the treatment of iodoacetamide to form the PrAMP analogue 1. Each monomeric Chex1-Arg20 analogue bearing a C-terminal Cys was subjected to bioconjugation to different bifunctional linkers in biological buffer (Fig. 1). The disulfide bond-linked homodimeric PrAMP 2 was also obtained via thiol-pyridyl activation. For the thiol-halogeno reaction, ortho-, meta- and para-dibromo-xylene were each used to produce the thiol-tethered homo dimeric PrAMPs 3, 4 & 5. Hexafluorobenzene and decafluorobiphenyl were also chosen to react with C-terminal thiol of monomeric PrAMP to obtain PrAMPs 6 & 7. Each homodimer PrAMP was obtained in modest overall yield and subjected to chemical characterization by analytical reversed-phase high performance liquid chromatography (RP-HPLC) and matrix-assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF MS) (Table S1 and Fig. S1†).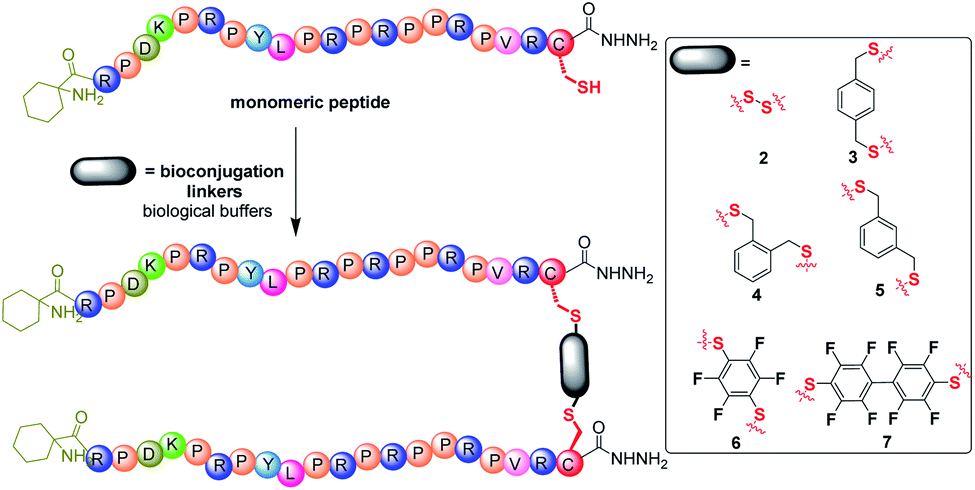 | ||
| Fig. 1 The preparation of homodimers of the PrAMP, Chex1-Arg20 bearing a C-terminal hydrazide moiety. The N-terminal of the peptides were modified with 1-aminocyclohexanecarboxylic acid. The bioconjugation linkers are shown in the box as disulfide dimer-NHNH22, p-xylene dimer-NHNH23, o-xylene dimer-NHNH24, m-xylene dimer-NHNH25, tetrafluorobenzene dimer-NHNH26, and octofluorobiphenyl dimer-NHNH27. | ||
Antimicrobial activity
Each PrAMP homodimer analogue (1–7) was subjected to antibacterial testing against a panel of Gram-negative pathogens, including Escherichia coli ATCC 25922, Klebsiella pneumoniae ATCC 13883, A. baumannii ATCC 19606, as well as FADDI-KP028 (MDR) and FADDI-AB156 (colistin-resistant, rifampin-resistant & MDR, XDR), in undiluted Mueller-Hinton broth (MHB) as previously reported.19 The minimum inhibitory concentration (MIC) that measures their efficacy in inhibiting bacterial growth (Table 1) showed the dimeric Chex1-Arg20 (2–7) had significantly improved activity against MDR/XDR pathogens. In comparison with our tetrameric analogues (MIC 20 μg ml−1 to 140 μg ml−1),17 tetrafluorobenzene dimer-NHNH26 and octofluorobiphenyl dimer-NHNH27 had particularly low MIC values (5–13 μg ml−1) on both A. baumannii and MDR/XDR-FADDI-AB156, bacterial species that are associated with a high mortality rate in hospital-acquired infections20 and are WHO priority critical listed pathogens. It has been shown that A. baumannii is responsible for diverse serious infections such as septicaemia, pneumonia, postsurgical meningitis and skin infections.21 With respect to the influence of the linkers alone, it is interesting to note that both the hexafluorobenzene and decafluorobiphenyl showed no antibacterial activity themselves (Table S2†). The membrane disruption assay further showed the potent membrane activity against Gram-negative bacteria (Table S3†) by the lead candidates, tetrafluorobenzene dimer-NHNH26 and octofluorobiphenyl dimer-NHNH27, and absence of cytotoxicity (Fig. S2†). Their high therapeutic index (Table S4†) reflects their significant potential for future therapeutic investigation. Additionally, a trimeric PrAMP via a 1,3,5-tris(methyl)benzene linkage was prepared but did not confer any improvement in activity (unpublished data).| No. | Peptides | E. coli 25922 | K. pneumoniae 13883 | A. baumannii 19606 | FADDI-KP028 | FADDI-AB156 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a d.a., antibacterial test not performed. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | Monomer-NHNH2 | 2.5 ± 1.1 (0.9) | 3.2 ± 0.1 (1.2) | >250 (>94.3) | 199.8 ± 0.2 (75.4) | >250 (>94.3) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | Disulfide dimer-NHNH2 | 3.1 ± 0.1 (0.6) | 5.1 ± 0.8 (1.0) | 30.8 ± 3.3 (5.9) | 111.3 ± 0.3 (21.5) | 59.2 ± 0.2 (11.4) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | p-Xylene dimer-NHNH2 | 8.1 ± 0.4 (1.5) | 3.5 ± 0.6 (0.7) | 13.1 ± 0.7 (2.5) | 59.6 ± 0.3 (11.3) | 54.4 ± 1.4 (10.3) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | o-Xylene dimer-NHNH2 | 8.5 ± 0.7 (1.6) | 3.7 ± 0.6 (0.7) | 16.2 ± 2.8 (3.1) | 67.5 ± 4.1 (12.8) | 55.4 ± 0.9 (10.5) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | m-Xylene dimer-NHNH2 | 8.1 ± 0.6 (1.5) | 3.6 ± 0.7 (0.7) | 13.1 ± 0.6 (2.5) | 70.1 ± 3.9 (13.3) | 53.9 ± 2.1 (10.2) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | Tetrafluorobenzene dimer-NHNH2 | 6.7 ± 0.9 (1.3) | 3.6 ± 0.1 (0.7) | 6.5 ± 0.1 (1.2) | 120.9 ± 2.9 (22.7) | 13.4 ± 0.9 (2.5) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | Octofluorobiphenyl dimer-NHNH2 | 2.7 ± 0.7 (1.5) | 2.8 ± 0.9 (0.5) | 4.9 ± 1.3 (0.9) | 51.8 ± 1.5 (9.4) | 13.7 ± 0.1 (2.5) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | Gentamicin | 0.9 ± 0.1 (1.88) | 0.1 ± 0.05 (0.2) | 2.7 ± 0.2 (5.7) | >250 (>523.5) | >250 (>523.5) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 | Colistin | d.a. | d.a. | 0.5 ± 0.04 (0.4) | d.a. | 12.6 ± 1.6 (10.9) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The leading candidates, tetrafluorobenzene dimer-NHNH26 and octofluorobiphenyl dimer-NHNH27 (Table 1), with their potent antibacterial inhibition activity against both A. baumannii and MDR/XDR-FADDI-AB156, were further assessed for minimum bactericidal concentration (MBC) as previously reported19. In comparison with the conventional antibiotics, gentamicin and the last resort antibiotic, colistin, both the lead PrAMP candidates showed very strong killing activity against A. baumannii and MDR/XDR-FADDI-AB156 (Table 2).
| Peptides | A. baumannii 19606 | MDR-FADDI-AB156 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a n.d., no measurable bactericidal activity. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | Tetrafluorobenzene dimer-NHNH2 | 14.1 ± 0.9 (2.6) | 22.5 ± 2.5 (4.2) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | Octofluorobiphenyl dimer-NHNH2 | 30.6 ± 0.6 (5.6) | 32.5 ± 2.5 (5.9) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | Gentamicin | 4.6 ± 0.3 (9.6) | n.d.a | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 | Colistin | 0.8 ± 0.02 (0.7) | >31.25 (>27.1) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Killing kinetic assay
To determine the killing efficiency of the two lead PrAMP dimers, tetrafluorobenzene dimer-NHNH26 and octofluorobiphenyl dimer-NHNH27, against A. baumannii and MDR-FADDI-AB156, we designed a time-killing kinetic assay by measuring the death rate constant under PrAMPs 6 & 7 treatment. By choosing different concentrations (4 × MIC, 2 × MIC, 1 × MIC, 0.5 × MIC, 0.25 × MIC) and a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log(99.9%) reduction in survival and zero colony forming unit (CFU) as a basis, both the PrAMPs 6 & 7 (4 × MIC ∼ MBC) clearly showed potent bactericidal activity and sterility, respectively, to kill A. baumannii and MDR-FADDI-AB156 at the 75 min to 90 min incubation time points (Fig. S3†). These results are consistent with the MBC killing of all the bacteria after the full-course incubation (Table 2). More specifically, at the 4 × MIC concentration, PrAMP 6 reduced A. baumannii and MDR-FADDI-AB156 survival within 60 minutes by 2.38log(>99%) and 2.78log(>99%), respectively (Fig. S3a and c†), while PrAMP 7 reduced the survival by 1.81log(>98%) and 2.72log(>99%) (Fig. S3b and d†). At 2 × MIC of 1 hour incubation, PrAMP 6 still displayed strong bactericidal activity by survival reduction of 1.18log(>93%) and 2.09log(>99%) towards A. baumannii and MDR-FADDI-AB156 (Fig. S3a and c†). In contrast, PrAMP 7 at 2 × MIC showed less killing activity within 60 minutes with a CFU reduction of 0.68log(>79%) and 0.94log(>88%) (Fig. S3b and d†).
log(99.9%) reduction in survival and zero colony forming unit (CFU) as a basis, both the PrAMPs 6 & 7 (4 × MIC ∼ MBC) clearly showed potent bactericidal activity and sterility, respectively, to kill A. baumannii and MDR-FADDI-AB156 at the 75 min to 90 min incubation time points (Fig. S3†). These results are consistent with the MBC killing of all the bacteria after the full-course incubation (Table 2). More specifically, at the 4 × MIC concentration, PrAMP 6 reduced A. baumannii and MDR-FADDI-AB156 survival within 60 minutes by 2.38log(>99%) and 2.78log(>99%), respectively (Fig. S3a and c†), while PrAMP 7 reduced the survival by 1.81log(>98%) and 2.72log(>99%) (Fig. S3b and d†). At 2 × MIC of 1 hour incubation, PrAMP 6 still displayed strong bactericidal activity by survival reduction of 1.18log(>93%) and 2.09log(>99%) towards A. baumannii and MDR-FADDI-AB156 (Fig. S3a and c†). In contrast, PrAMP 7 at 2 × MIC showed less killing activity within 60 minutes with a CFU reduction of 0.68log(>79%) and 0.94log(>88%) (Fig. S3b and d†).
Based on the CFU count at various time points, the first order death rate constant was determined by plotting log(CFU) vs. time ranging from 5–60 min,22 the slope of linear fit providing the death rate constant (Fig. S4†). On comparing the death rate constant at various concentrations (Fig. 2), we observed a rapid killing rate for both the leading PrAMPs, 6 & 7. However, the activity of PrAMP 7 at 1 × MIC was significantly (P <0.05) weaker than that of PrAMP 6 (1 × MIC), reducing survival by 0.74log(>81%) and 0.19log(>35%), respectively (Fig. S3†). Taken together, the tetrafluorobenzene dimer-NHNH26 possesses a faster killing rate for both A. baumannii and MDR-FADDI-AB156 compared to the octofluorobiphenyl dimer-NHNH27. Those death rate constants and killing efficiency are critical parameters for further clinical pharmacological kinetics and drug development.
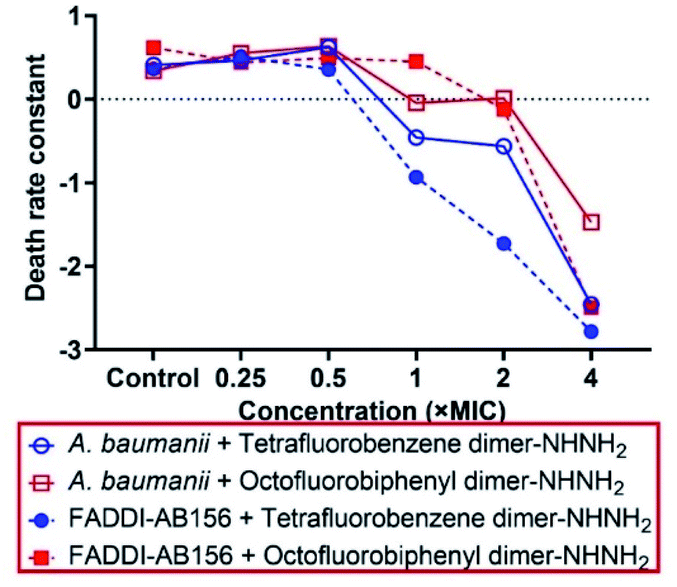 | ||
| Fig. 2 Death rate constant of A. baumannii and MDR-FADDI-AB156 caused by the lead dimeric PrAMPs, tetrafluorobenzene dimer-NHNH26 and octofluorobiphenyl dimer-NHNH27. The death rate constants were determined by the first order of the linear regression of log(CFU count) vs. various time points (Fig. S4†). | ||
Interaction with the outer and inner membranes
The membrane permeability of the two lead PrAMPs was further determined by using 1-N-phenylnaphthylamine (NPN). NPN is a small molecule that can become strongly fluorescent in contact with a phospholipid bilayer and is widely used to indicate the outer membrane permeabilization of Gram-negative bacteria.23 Polymyxin B, as a positive control, is well known to permeabilise the outer membrane, as reflected by a high fluorescent intensity of NPN signal in Fig. 3.24 As shown in Fig. 3, both the lead PrAMPs displayed a similar trend to promote NPN uptake across the outer membrane of A. baumannii (Fig. 3a) and MDR-FADDI-AB156 (Fig. 3b) as the concentration of peptide increased. Compared to the positive control of polymyxin treated bacterial samples, tetrafluorobenzene dimer-NHNH26 showed significant NPN permeability over the concentration of 0.1 × MIC or 0.25 × MIC. Notably at the 1 × MIC or 2 × MIC, the PrAMP 6 treatment increased significantly (***P ≤0.001) 1.5-fold to 2-fold NPN fluorescence intensity over the NPN bacteria sample in the absence of peptide/polymyxin treatment (Fig. 3a and b). The octofluorobiphenyl dimer-NHNH27 showed slightly lower permeability than tetrafluorobenzene dimer-NHNH26, PrAMP 7 at the 1 × MIC or 2 × MIC can significantly induce 1.3–1.7-fold (***P ≤0.001) NPN fluorescence intensity over the NPN bacteria sample without peptide/polymyxin treatment.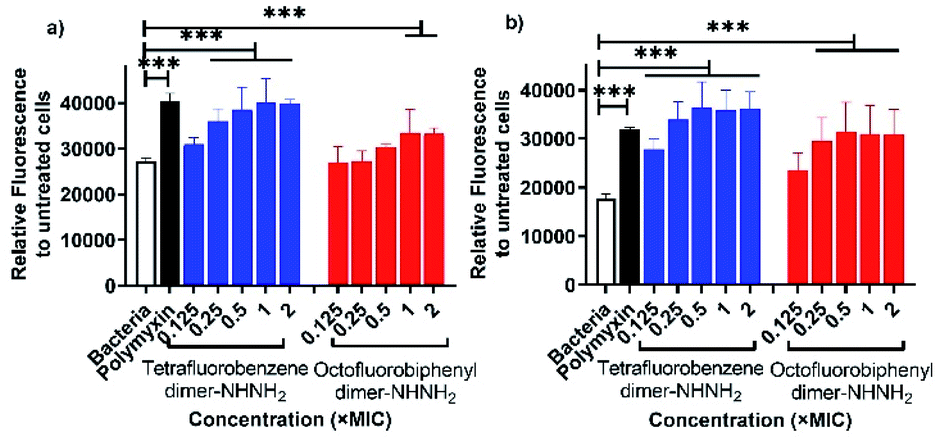 | ||
| Fig. 3 NPN permeability assay in presence of lead PrAMPs, tetrafluorobenzene dimer-NHNH26 and octofluorobiphenyl dimer-NHNH27 for (a) A. baumannii, and (b) MDR-FADDI-AB156. Polymyxin was used as a positive control for its strong NPN permeability. All data are expressed as mean ± standard deviation as indicated by the error bars (n = 4). *P ≤0.05, **P ≤0.01, ***P ≤0.001. Student's t-test, a significant difference from the bacteria only with NPN control to indicate less potent NPN permeability. | ||
Given the observation that the effective concentration for outer membrane permeability (Fig. 3) caused by the PrAMPs is much lower than their bacterial inhibition or killing activity (Table 1), we further assessed the inner membrane permeability by using the DNA binding dyes, propidium iodide (PI) and SYTO 9 via flow cytometry. As SYTO 9 being a membrane permeable dye will label every bacterium (thus discriminating bacteria from background), while PI, a membrane impermeable dye, can only enter the cytoplasm when the inner membrane has been disrupted by peptide treatment. By measuring the percentage of PI-labelled bacteria via flow cytometry, we can show the ability of the lead PrAMPs to permeabilise the inner membrane. As shown in Fig. 4, both tetrafluorobenzene dimer-NHNH26 and octofluorobiphenyl dimer-NHNH27 gradually increased the portion of PI-labelled bacteria to >80% of the whole bacterial population. At the bacterial inhibition (MIC) or killing concentration (MBC) (Table 2), we observed 50–60% of A. baumannii are PI-positive labelled bacteria population at 1 × MBC (Fig. 4a), and that 50–60% of MDR-FADDI-AB156 are PI+ at 2 × MBC (Fig. 4b) for both dimers, 6 and 7. The results strongly correlated their inner membrane permeability with their bacterial inhibition or killing activity.
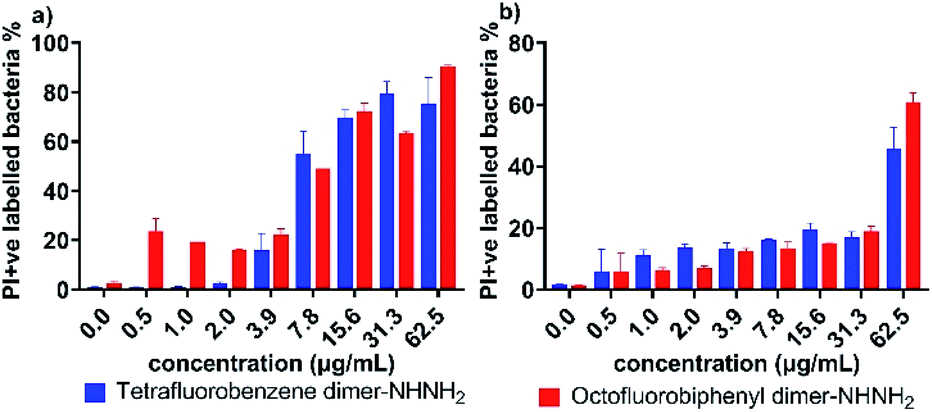 | ||
| Fig. 4 Inner membrane permeability of tetrafluorobenzene dimer-NHNH26 and octofluorobiphenyl dimer-NHNH27 for (a) A. baumannii, and (b) MDR-FADDI-AB156. The flow cytometric graphs are provided in Fig. S5.† | ||
A major energy source for nutrient uptake, waste material excretion and micro-environment upregulation is the trans-cytoplasmic membrane ion motive force driven by an electrical potential gradient.25 Based on the NPN and PI/SYTO 9 assays (Fig. 3 and 4), the observed membrane interaction with the tetrafluorobenzene dimer-NHNH26 and octofluorobiphenyl dimer-NHNH27 may further modulate the membrane potential. Thus, we then examined ion permeability by using a membrane potential-sensitive fluorescent probe, DiOC2(3),26 which can be taken up through the inner membrane by the electrical potential gradient and fluoresces green in the cytoplasm under normal membrane potential conditions. When any AMP or compound interact and depolarise the inner membrane, DiOC2(3) self assembles and produces a red shift in fluorescence, with the strength in the shift indicating the strength of the change in membrane potential. As Fig. 5 shows, both the tetrafluorobenzene dimer-NHNH26 and octofluorobiphenyl dimer-NHNH27 caused almost 100% depolarization at their 1 × MBC concentration, the same as the positive control sample treated with CCCP. Even at 0.1 × MBC (A. baumannii) or 0.5 × MBC (MDR-FADDI-AB156) peptide treatment resulted in more than 50% depolarization (p** <0.01). Bacterial membranes are composed of roughly equal proportions of membrane-associated proteins and phospholipids forming a classical bilayer structure.27 To further investigate the phospholipid bilayer membrane interaction, large unilamellar vesicles (LUVs) were further used to examine pore formation property and showed no leakage of calcein (Fig. S7†). Such an observation is consistent with our previous finding for PrAMPs in the presence of a negatively charged model membranes, for which the PrAMP did not induce sufficiently large pore formation to release the calcein28 but did release ions.29 Those results suggest both the PrAMPs can permeabilise the outer and inner membranes at low concentrations (0.25 × MIC or 0.5 × MIC) and induce smaller pore formation enabling PI or ions exchange but not calcein (larger molecules). The surprising weaker PI-positive labelling (60–80%) at higher concentration (>31.3 μg ml−1, Fig. 4) further indicated the small pore size formed by dimers 6 and 7, with only some PI getting into the bacteria. Together with the capability of strong membrane depolarization, this contributes to their potent antibacterial activity towards A. baumannii and MDR-FADDI-AB156.
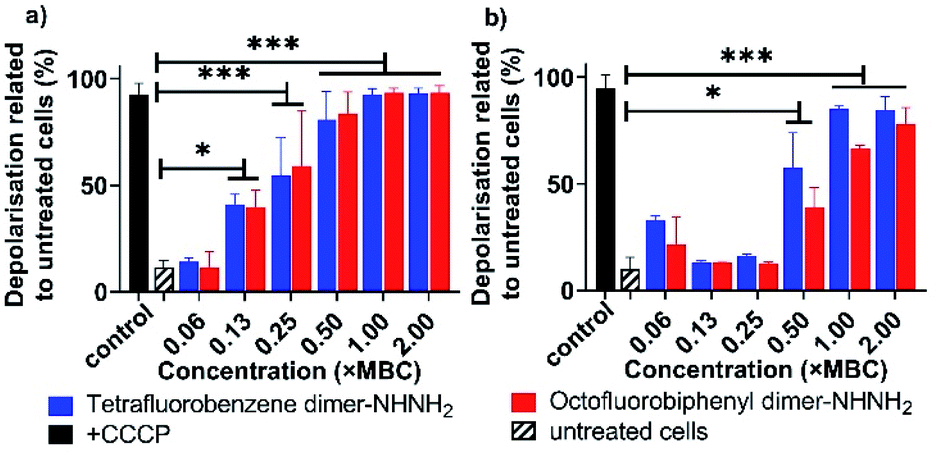 | ||
| Fig. 5 Membrane potential analysis of (a) A. baumannii, and (b) MDR-FADDI-AB156 with treatment of tetrafluorobenzene dimer-NHNH26 and octofluorobiphenyl dimer-NHNH27. The flow cytometric graphs are provided in Fig. S8 and S9.† All data are expressed as mean ± standard deviation as indicated by the error bars (n = 4). *P ≤0.05, **P ≤0.01, ***P ≤0.001. Student's t-test, a significant difference from the untreated bacteria control. | ||
Stress response by generation of reactive oxygen species under PrAMP treatment
Disturbance of membrane integrity and potential can further lead to a stress response and the generation of reactive oxygen species (ROS), thus resulting in oxidative damage then cell death via the oxidative damage-mediated pathway.30,31 To investigate the effect of our lead PrAMPs for ROS production, the microbial flow cytometry method19 was applied to determine the ROS level for A. baumannii and MDR-FADDI-AB156 treated with the PrAMPs, tetrafluorobenzene dimer-NHNH26 and octofluorobiphenyl dimer-NHNH27, by using CellROX® Deep Red fluorescence. The addition of thiourea, a ROS scavenger,32 was used to quench ROS to a level of the untreated bacterial sample, which further verified the induction of ROS production under PrAMP treatment. As shown in Fig. 6, both the lead PrAMPs induced significant (p** <0.01) amounts of ROS as compared to the untreated bacteria and thiourea-quenched ROS control. At 2 × MBC, tetrafluorobenzene dimer-NHNH26 triggered a 4.8-fold and 3.9-fold increase of ROS generation for A. baumannii and MDR-FADDI-AB156, respectively, while the octofluorobiphenyl dimer-NHNH27 induced 2.2-fold and 2.7-fold increase (Fig. 6). At the lethal dose level (1 × MBC), both PrAMPs displayed very similar effects on the ROS generation with a 2.4–3.0-fold difference over the control samples (Fig. 6). Compared to octofluorobiphenyl dimer-NHNH27, the tetrafluorobenzene dimer-NHNH26 could trigger a high level of ROS at the lower concentrations (0.25–0.5 × MBC) with a 1.5-fold and 2.7-fold increase for A. baumannii and MDR-FADDI-AB156, respectively. As ROS includes multiple species such as hydroxyl radicals and superoxide anion radicals, we further compared the peptide disulfide linked dimer 2 and peptide 7 after the incubation with A. baumannii culture for 90 min (the full-time course of MBC determination) (Fig. S12†). As the RP-HPLC trace (Fig. S12†) indicated, the disulfide linked dimer 2 was reduced, but peptide 7 remained intact during the same period. Taken together, both the lead PrAMPs, 6 and 7, significantly induced the generation of ROS at the lethal and sublethal doses, which contributed to the death of A. baumannii and MDR-FADDI-AB156.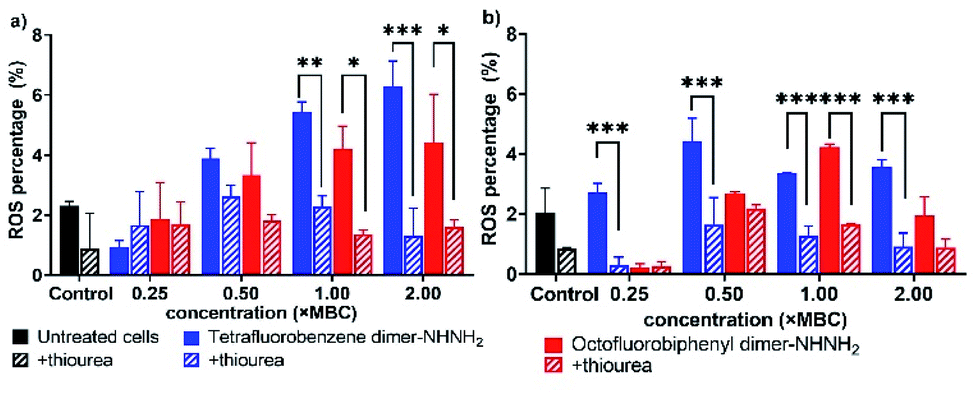 | ||
| Fig. 6 Reactive oxygen species generated from (a) A. baumannii, and (b) MDR-FADDI-AB156 under treatment of tetrafluorobenzene dimer-NHNH26 and octofluorobiphenyl dimer-NHNH27. The flow cytometric graphs are provided in Fig. S10 and S11.† All data are expressed as mean ± standard deviation as indicated by the error bars (n = 4). *P ≤0.05, **P ≤0.01, ***P ≤0.001. Student's t-test, a significant difference from thiourea quenched ROS control. | ||
Microscopic visualization of PrAMP treated bacteria
Although fluorescence imaging methods are widely used to visualise the interaction between antibacterial agents and bacteria, the addition of the fluorescent dyes can reduce the bioactivity of the target compounds.16,19 The advanced development of surface scanning microscopy, such as the helium ion microscope (HIM), enables direct analysis of the biological samples at super-high resolution.33 To directly observe the morphological changes of bacterial cells due to the PrAMPs, HIM was used to investigate the morphologies of A. baumannii and MDR-FADDI-AB156 under the treatment of tetrafluorobenzene dimer-NHNH26 and octofluorobiphenyl dimer-NHNH27 at different concentrations (2 × MIC, 1 × MIC, 0.5 × MIC, 0.25 × MIC, 0.125 × MIC). The tested bacterial samples with PrAMPs were transferred onto glass coverslips after 90 min for HIM measurement, as previously described.34 The HIM images showed clear visualization of the morphological changes of A. baumannii and MDR-FADDI-AB156 with PrAMP treatment (Fig. 7). The multiple morphological changes with the PrAMPs at the lethal dose (2 × MIC) can be explained by destabilization of the outer membrane, followed by membrane fragmentation and disruption (Fig. 7). Interestingly, the tetrafluorobenzene dimer-NHNH2 6 treated bacteria led to compaction or shrinkage (Fig. 7a and c), while the octofluorobiphenyl dimer-NHNH27 caused the membrane to burst (Fig. 7b and d). Such observations of the morphological charges clearly showed the mechanism for membrane interaction via destabilization in the outer membrane, followed by disruption of the inner membrane.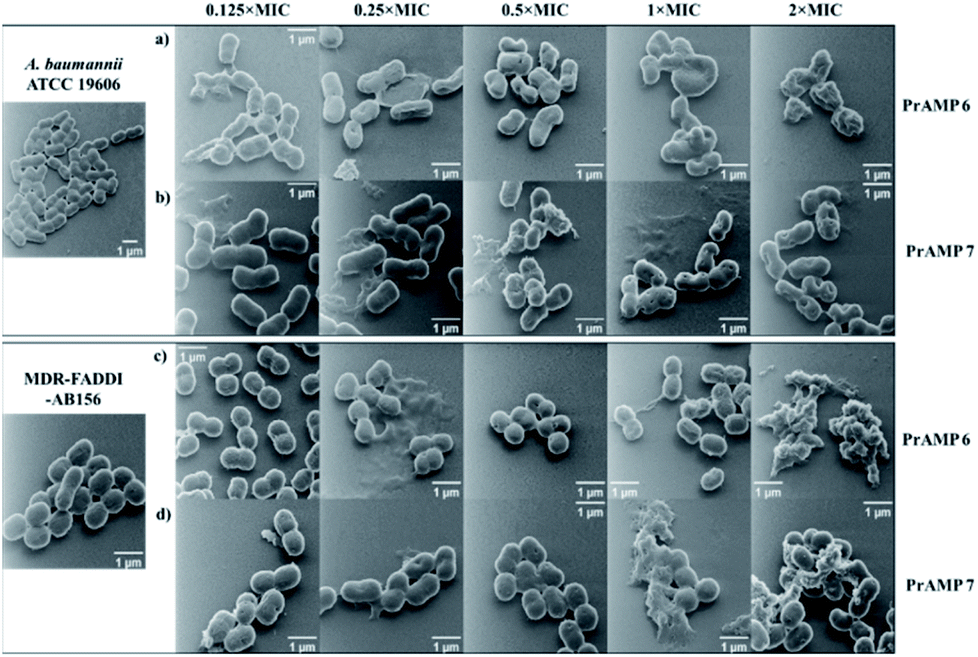 | ||
| Fig. 7 HIM showing the morphologies of A. baumannii and MDR-FADDI-AB156 under treatment of (a and c) tetrafluorobenzene dimer-NHNH26 and (b and d) octofluorobiphenyl dimer-NHNH27 at different concentrations (2 × MIC, 1 × MIC, 0.5 × MIC, 0.25 × MIC, 0.125 × MIC). The untreated A. baumannii and MDR-FADDI-AB156 were used as controls. | ||
Eradicating preformed biofilms by the lead PrAMP dimers
Biofilms are an accumulation of microbes forming a self-produced matrix of extracellular polymeric substances (EPS) which are more resistant to abrupt environmental changes and antibiotics than planktonic bacteria.35 Due to biofilms comprising over 65% of human infections, antibiotics used to treat planktonic bacterial infections are commonly prescribed to combat bacterial biofilms.36 The recent development of AMPs has provided a therapeutic application against bacterial biofilms by preventing their formation or by eradicating preformed biofilms.37,38 For our leading PrAMPs, their biofilm disruption properties were investigated. In the microtiter plate preformed biofilm assay, the biofilm biomass was measured after crystal violet staining which quantified detaching bacteria after PrAMP treatment at various concentrations (0.5 × MBC, 1 × MBC, 2 × MBC, 4 × MBC, 8 × MBC). Both the lead PrAMPs eradicated the preformed biofilm with a similar trend, that generally destroyed the biofilm with increased peptide concentration (Fig. 8). With 0.5 × MBC and 1 × MBC peptide treatment, both PrAMPs had little effect on eradicating the preformed biofilm with only 10–15% reduction in the crystal violet absorption (Fig. 8). At a higher lethal dose for the planktonic bacteria (2 × MBC), almost 50% of the preformed biofilm was significantly (P <0.05) destroyed during the 90 min peptide treatment and suggested potent antibiofilm activity (Fig. 8). However, a higher dose of PrAMPs (4 × MBC, 8 × MBC) did not further eradicate higher percentage (over 50%) of biofilm (Fig. 8), which reflects the difficulty in combating biofilms.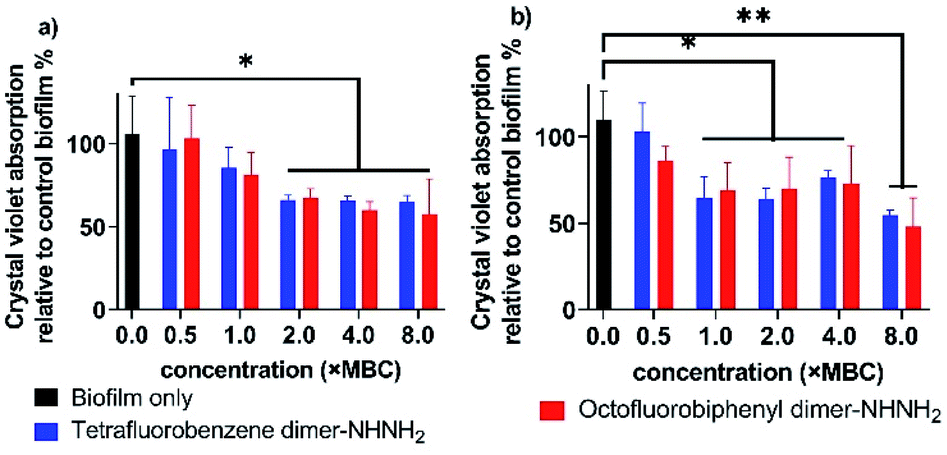 | ||
| Fig. 8 Eradication of the preformed biofilms of (a) A. baumannii, and (b) MDR-FADDI-AB156 by the treatment of tetrafluorobenzene dimer-NHNH2 6 and octofluorobiphenyl dimer-NHNH2 7 at different concentrations (0.5 × MBC, 1 × MBC, 2 × MBC, 4 × MBC, 8 × MBC). The crystal violet was used to stain bacterial biofilm and percentages were calculated with the untreated biofilm as the basis. *P ≤0.05, **P ≤0.01. Student's t-test, a significant difference from the untreated preformed biofilm control. | ||
To further visualise their action on the preformed biofilms, we used HIM to assess the effect of tetrafluorobenzene dimer-NHNH26 and octofluorobiphenyl dimer-NHNH27 on the biofilm formed on glass coverslips after 90 min peptide treatment (Fig. 9). The untreated performed bacterial biofilm sample clearly showed the EPS matrix and the connections among bacteria. With the increased concentration of PrAMPs (0.5 × MBC, 1 × MBC, 2 × MBC, 4 × MBC, 8 × MBC), the EPS matrix was gradually disrupted or demolished, especially at the higher concentrations (4 × MBC, 8 × MBC) with destroyed EPS matrix and disrupted bacteria both observed (Fig. 9). In addition to the EPS matrix disturbance, the bacteria underwent a similar trend as the planktonic bacteria (Fig. 7) with destabilization of the outer membrane, followed by membrane fragmentation and disruption (Fig. 9). In addition to the direct action on the preformed biofilm, the induction of ROS by the lead PrAMPs (Fig. 6) also contributes to their antibiofilm activity.39 Given the increasing antibiotic resistance of biofilms, this observation strongly indicated that our PrAMP dimers can be considered as an effective new class of antibiofilm agents for further investigation.
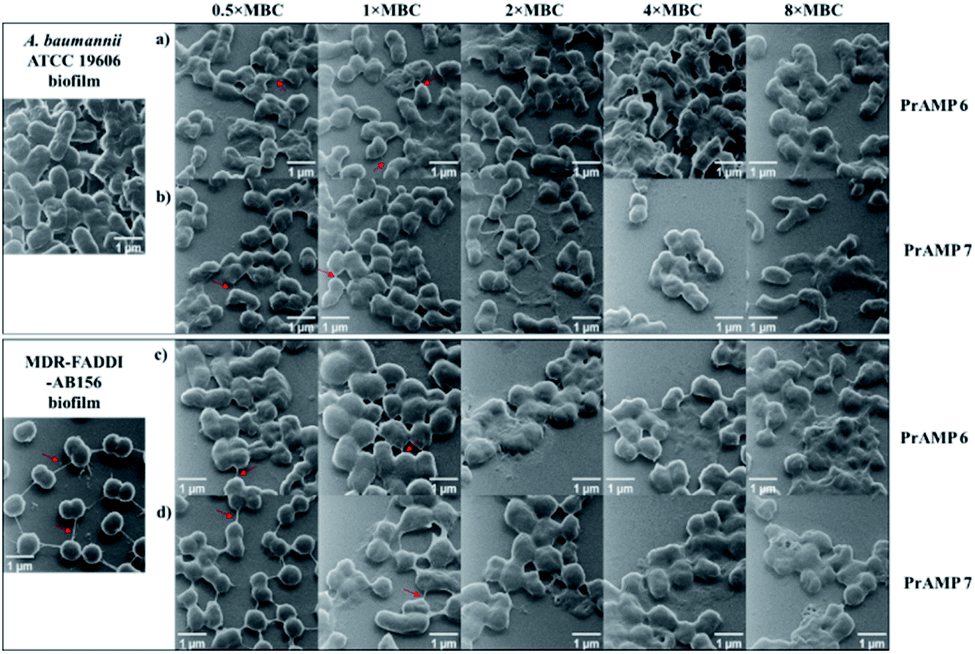 | ||
| Fig. 9 HIM of the preformed biofilms of A. baumannii and MDR-FADDI-AB156 under treatment of (a and c) tetrafluorobenzene dimer-NHNH26 and (b and d) octofluorobiphenyl dimer-NHNH27 at different concentrations (8 × MBC, 4 × MBC, 2 × MBC, 1 × MBC, 0.5 × MBC). The untreated A. baumannii and MDR-FADDI-AB156 biofilms were used as controls. The red arrow indicated the EPS in preformed biofilm. | ||
Immunomodulatory activity determination
AMPs, as host defense peptides, can modulate the host innate immunity by promoting protective responses, such as cytokine tumour necrosis factor-α (TNF-α), and suppressing inflammation, like nitric oxide (NO).40 Interestingly, PrAMPs, most of which have been isolated from insects,13,41 have been found to have potent immunomodulatory properties that modulate cell function and cytokine secretion to up-regulate the recovery from bacterial infections.12,42 This phenomenon raises the potential of our two lead PrAMPs, tetrafluorobenzene dimer-NHNH26 and octofluorobiphenyl dimer-NHNH27, to also perform as immune-modulating and anti-inflammation agents. Using freshly purified lipopolysaccharide (LPS) from A. baumannii and MDR-FADDI-AB156, the potential anti-inflammation effect of those PrAMPs on LPS-stimulated NO production from macrophages was assessed by using the Griess assay. As shown in Fig. S13,† the NO concentration gradually increased in RAW 264.7 cell cultures as the purified LPS concentration increased, while the NO production was substantially suppressed by 2–3-fold at the higher concentration of the tested PrAMPs (Fig. 10). Through their dose-dependent manner to neutralize the endotoxin (bacterial LPS) and NO release in macrophages (via well-recognized TLR4-dependant pathway43), the lead PrAMPs 6 and 7 indicate a synergistic approach by PrAMP dimers resulting in enhanced direct killing and modulation of the adaptive immune response.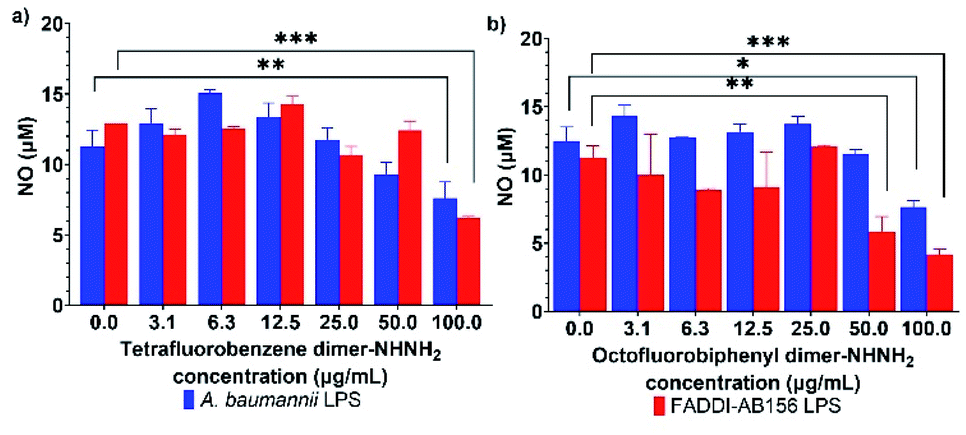 | ||
| Fig. 10 Nitric oxide release from LPS stimulated RAW264.7 cells under treatment of (a) tetrafluorobenzene dimer-NHNH26, and (b) octofluorobiphenyl dimer-NHNH27. All data are expressed as mean ± standard deviation as indicated by the error bars (n = 4). *P ≤0.05, **P ≤0.01, ***P ≤0.001. Student's t-test, a significant difference from LPS stimulated RAW264.7 cells control. | ||
Molecular dynamics simulations
Given the effects of the PrAMPs on membrane permeability (Fig. 3 and 4) and potential (Fig. 5), as well as phospholipid bilayer membrane interactions (Fig. S7†), we further elucidated the atomic-level details of a lead PrAMP, octofluorobiphenyl dimer-NHNH27, adsorbed on and permeating through a mixed phospholipid membrane bilayer via molecular dynamics simulations. Although phosphatidylethanolamine (PE), rather than neutral phosphatidylcholine (PC), is more commonly found in bacterial membranes, palmitoyloleoylphosphatidylcholine (POPC) and palmitoyloleoylphosphatidylglycerol (POPG) mixtures are frequently used as proxies for bacterial membranes in experimental studies and molecular dynamics (MD) simulations of protein-44 and peptide45-lipid interactions, as the use of PC confers the advantage of producing a membrane with less curvature stress46 compared to PE. For each bilayer adsorption simulation, a pre-equilibrated mixed bilayer composed of POPC and POPG in a 7:3 ratio serves as a model of Gram negative inner membranes.
The initial adsorption mechanism of the PrAMP towards the membrane surface was investigated using equilibrium simulations of the diffusion (Fig. 11). Fig. 11a shows MD simulation snapshots of the initial adsorption of the PrAMP dimer 7 to the model membrane, with persistent binding of the peptide to the bilayer surface from 60 ns to the end of the trajectory at 230 ns. The relative preference for binding zwitterionic neutral PC or negatively charged PG was quantified by measuring the total number of contacts between the PrAMP and each of the two lipid components in the mixed bilayer. Fig. 11b shows time series plots of the compositions of PC and PG lipids whose atoms lie within 0.4 nm of the PrAMP. These plots indicate that PC comprises an average of ∼60% of the lipids in the vicinity of the dimer, while PG comprises an average of the remaining ∼40%. These values indicate a relative enrichment of PG in vicinity of the PrAMP relative to the nominal bulk composition value of 30%, suggesting marginally higher preference of the peptide for binding PG at the membrane surface. To elucidate the specific residues responsible for this enrichment, the average numbers of contacts between each AMP residue with PC and PG for each of the two PrAMP chains in the octofluorobiphenyl dimer-NHNH27 is shown in Fig. 11c, which indicated that only one monomer (chain B) is relatively enriched with PG contacts, especially the basic residues Lys5, Arg7 and Arg14. The predominant involvement of a single chain of the dimer in forming preferential interactions with PG suggests that a favourable conformation for initial adsorption is one in which the binding monomer (chain B) remains relatively unencumbered by the other, less-strongly binding monomer (chain A). Such a conformation is adopted by PrAMP 7, in which the cross-helix angle between the monomers remains relatively obtuse (approximately 90°), likely as a result of the rigidity imposed by the octofluorobiphenyl linker (Fig. S16a†). A similar conformation is expected to be the adopted by other dimers with similar sterically rigid linkers, including PrAMPs 3 and 6, which may contribute to their comparable antibacterial activity to PrAMP 7.
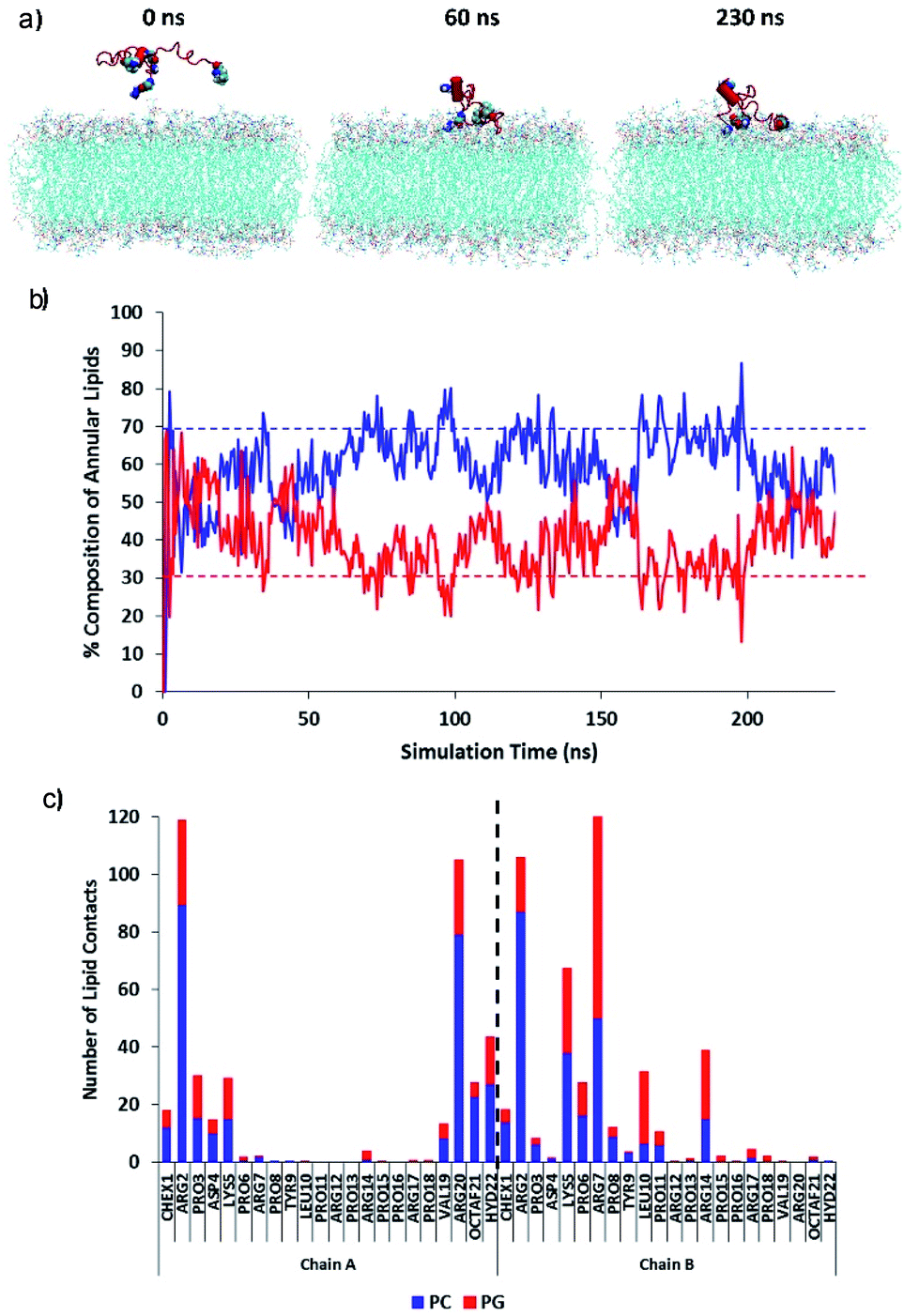 | ||
| Fig. 11 (a) MD simulation snapshots of the initial adsorption of octofluorobiphenyl dimer-NHNH27 to the model membrane. The PrAMP backbone is shown as a red ribbon. Chex, hydrazide (–NHNH2) and octofluorobiphenyl groups are shown as large spheres. (b) Time series plots of the compositions of PC (blue line) and PG lipids (red line) whose atoms lie within 0.4 nm of the PrAMP. Nominal bulk percentages for PC (70%) and PG (30%) are shown as dotted lines. (c) Average numbers of contacts between each PrAMP residue with PC (blue) and PG (red) for each of the two PrAMP chains of the octofluorobiphenyl dimer-NHNH27. | ||
Then, the permeation mechanism of the lead PrAMP dimer-NHNH27 was further studied by using non-equilibrium simulations whereby an external force was exerted on the peptide to slowly drive it through the bilayer. Fig. 12a shows MD simulation snapshots of the permeation of the octofluorobiphenyl dimer-NHNH27 into the model membrane, indicating significant disruption of the lipid chain order as the PrAMP enters (65 ns) and exits (78 ns) the bilayer centre. Based on MD study, annular lipids in direct contact with the peptide exhibit tails which splay and adopt roughly perpendicular orientations with respect to the membrane normal. In contrast, a similar permeation trajectory for the monomer showed negligible disruption to the lipid chains in the vicinity of the peptide, with the PC and PG tails remaining roughly parallel to the bilayer normal (Fig. S17†).
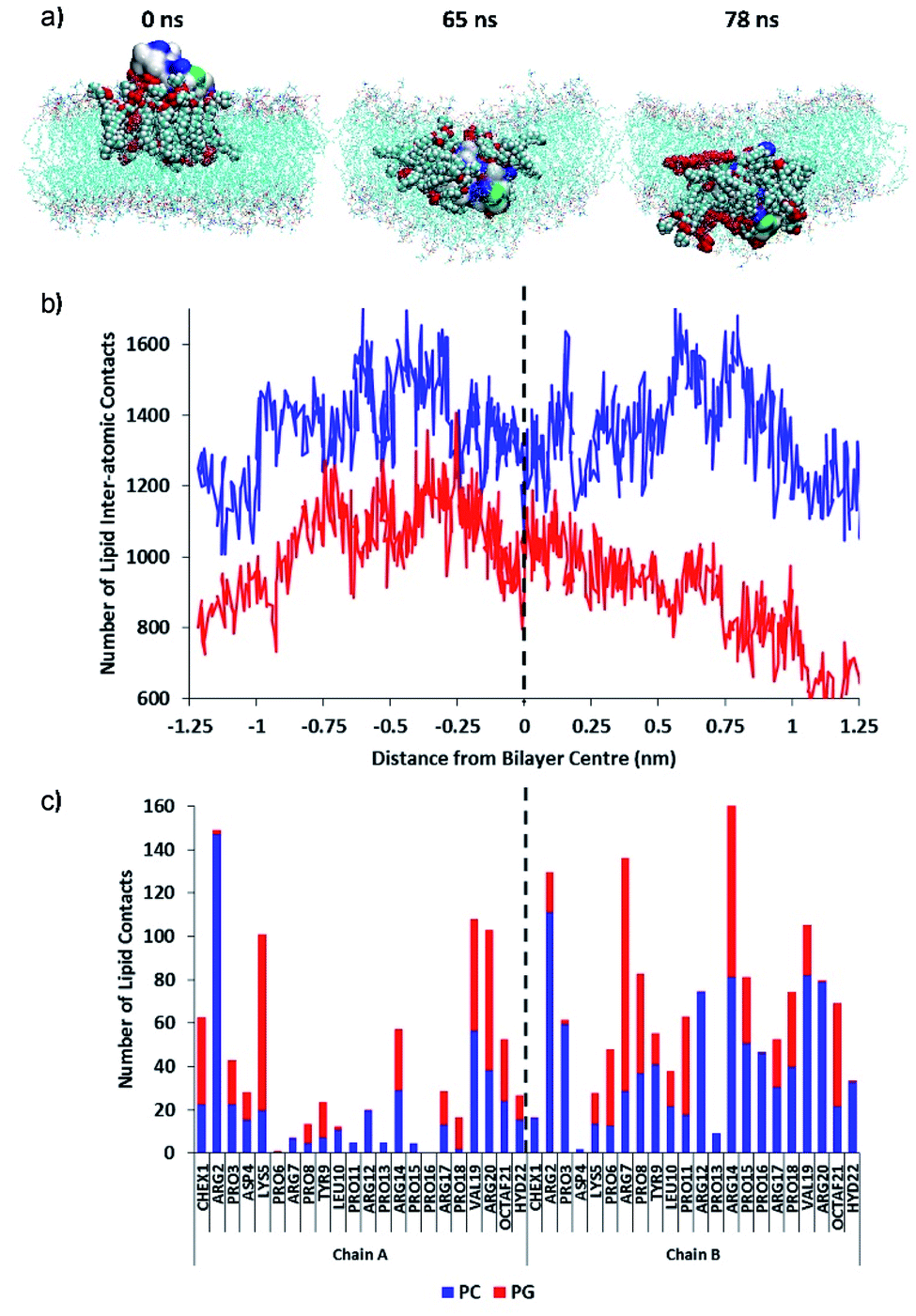 | ||
| Fig. 12 (a) MD simulation snapshots of the permeation of octofluorobiphenyl dimer-NHNH27 into the model membrane. The PrAMP is shown as a surface and colour-coded according to residue charge (blue = Arg, green = neutral polar, white = non-polar). The annular lipids are also shown as large spheres, with PG in red. (b) Total number of contacts between the PrAMP with PC (blue line) and PG lipids (red line) with respect to approximate position of the peptide centre-of-mass relative to the bilayer centre (dotted black line). (c) Average numbers of contacts between each PrAMP residue with PC (blue) and PG (red) for each of the two AMP chains in the octobiphenyl-dimer during membrane permeation. | ||
Inside the membrane core, the dimer, together with its annular lipids, adopted a supramolecular complex resembling an inverse micelle, with polar head-groups of the lipids in persistent contact with AMP residues, while lipid tails face outwards (Fig. 12). This inverse-micellar configuration facilitates dimer transport across the membrane. Additionally, the adoption of this peptide-lipid configuration suggests that the dimer is far more effective at disrupting bilayer integrity and may be a contributing factor to the markedly higher antibacterial activity of the octofluorobiphenyl dimer-NHNH27 compared to the monomeric PrAMP. Fig. 12b shows the total number of contacts between the dimer with PC and PG lipids with respect to approximate position of the peptide centre-of-mass relative to the bilayer centre. This plot indicates increasing relative enrichment of PG (depletion of PC) in the annular shell of the PrAMP as it approached the bilayer centre (dotted black line); and suggests increasing contribution of PG to the inverse-micelle complex as the peptide permeates and disrupts the bilayer. Consistent with this mechanism, Fig. 12c shows the average numbers of contacts between each PrAMP residue with PC and PG during membrane permeation. While only one PrAMP chain was enriched with PG at the membrane surface, upon permeation both chains of the dimer became relatively enriched with PG contacts, with Lys5, Arg7 and Arg14 forming the most persistent contacts. These results highlight the importance of PG in facilitating the surface binding, bilayer permeation, inverse-micelle formation and membrane disruption capacity of this dimeric PrAMP and may relate to its specific antibacterial activity against Gram-negative bacteria.
The higher potency of PrAMP 7 relative to PrAMP 2 may be explained by the molecular dynamics simulations of these dimeric PrAMPs in solution, which suggest subtle linker-dependent differences in conformation. As shown in Fig. S16a,† the octofluorobiphenyl linker confers greater steric hindrance and structural rigidity to the dimer, resulting in a more extended structure compared to the Cys-linked dimer, 2 (Fig. S16b†). The broader molecular dimensions of 7 may enable it to adsorb to, and permeate through, membranes with a larger molecular ‘footprint’, causing a greater degree of bilayer disruption compared to that of Cys-linked dimer 2 and other less active dimers (Table 1). To further probe the origins of the differential antibacterial activity of dimer 7 compared to other active, but less potent, dimers, a permeation simulation of the disulfide-linked dimer 2 through a model bilayer was performed. Dimer 2 was selected for this purpose as it was the least active dimer amongst all of those examined in this work, as shown in Table 2. Thus, its bilayer interaction mechanism is most likely to exhibit the same stark contrast as its bioactivity relative to the highest ranked dimer 7, rendering this comparison more likely to help illuminate the roles of the linker in PrAMP dimer structure and permeability. Fig. S18A† shows simulation snapshots of the permeation of dimer 2 into the model membrane, indicating disruption of the lipid chain order as the PrAMP enters (40 ns) and begins to exit (75 ns) the bilayer centre in a similar manner to that of dimer 7. This membrane disruption is consistent with the higher antibacterial activity exhibited by dimer 2 relative to monomer 1; in the latter case, the integrity of the bilayer is maintained (Fig. S17B†).
More substantial differences are evident, however, between dimers 7 and 2 in the number of PC and PG lipid contacts upon bilayer insertion. Fig. S18B† shows the total number of contacts between dimer 2 with PC and PG lipids with respect to approximate position of the peptide centre-of-mass relative to the bilayer centre. While this plot also indicates increasing relative enrichment of PG in the vicinity of dimer 2 as it approached the bilayer centre (dotted black line), the total number of dimer-lipid contacts for both PC and PG are overall lower than that of dimer 7 (average values for the latter are indicated by horizontal dashed lines), particularly within the lipid core region at −0.5 to +0.5 nm relative to the bilayer centre, despite the similarity of the molecular masses of the two dimers. The lower number of lipid contacts for dimer 2 indicates a reduced capacity for peptide-lipid interactions at the bilayer centre, consistent with its lower molecular footprint (Fig. S16A and B†) and this may contribute to its lower antibacterial potency compared to dimer 7.
Fig. S18C† shows the average numbers of contacts between each dimer 2 residue with PC and PG during membrane permeation, indicating that one chain (labelled chain A) becomes broadly enriched with PG contacts, along with the N-terminus of chain B. This is in contrast with dimer 7 (Fig. 12c), in which both chains are broadly enriched with PG contacts. These results once again highlight the importance of PG in facilitating bilayer permeation and disruption, with the greater association between dimer 7 and PG proposed to play a role in this dimer's membrane disruption capacity. Furthermore, Fig. 12c and S17C† show that the linkers (residue position 21) form relatively low numbers of contacts with the surrounding lipids. Thus, simulations suggest that the influences of different linkers on the activities of the peptides are likely related to the respective linkers' modulatory effects on PrAMP dimer conformation and size, rather than via direct contacts between the linkers with the membranes.
Conclusions
In summary, a series of dimeric PrAMPs were prepared via bioconjugation linkers to optimise their antibacterial activities against a panel of Gram-negative bacteria, including E. coli ATCC 25922, K. pneumoniae ATCC 13883, A. baumannii ATCC 19606, as well as the MDR FADDI-KP028 and FADDI-AB156 (MDR/XDR). In comparison to our tetrameric analogues,17 the lead dimeric PrAMPs analogues, tetrafluorobenzene dimer-NHNH2 6 and octofluorobiphenyl dimer-NHNH2 7, were easy to prepare and displayed very potent antibacterial activity (3–10 fold increase) against WHO priority critical listed pathogens, A. baumannii and MDR/XDR-FADDI-AB156. Perfluoroaromatic linkers have proved useful in bioconjugated reactions, including peptide stapling and macrocyclization, and a means of introducing fluorine into organic molecules with pharmaceutical potential.4719F NMR showed that such linkers possess significant rigidity and lipophilicity.48 By using fluorescent reporters,49,50 we further assessed their modes of action involving outer and inner membrane permeability and change in membrane potential. The HIM imaging analysis further identified the bacterial morphological changes caused by PrAMPs 6 and 7. The effect on bacterial membrane depolarization and membrane integrity, thus result in membrane fragmentation, rupture and cell lysis. MD simulations elucidated the potential adsorption and permeation mechanisms of the PrAMPs on a mixed phospholipid membrane bilayer, predicting the formation of an inverse-micelle-like structure and marked disruption of membrane integrity upon insertion of the octofluorobiphenyl dimer-NHNH27 inside the bilayer centre.Additional analysis of ROS production from stress response due to the lead PrAMP treatment also contributed to the bacterial disruption, further leading to cell death. The reduction of inflammation factor, NO, further indicated that our lead PrAMPs can neutralise the effect of LPS from A. baumannii ATCC 19606 and FADDI-AB156 on macrophages for NO production in vitro, which is a critical element for anti-inflammatory activity to modulate the immune response. Such an effect may further contribute to neutralizing inflammation induced by bacterial infections and enhance the host innate immunity to the bacteria. The reduction in preformed biofilms by over 50% under treatment by the lead PrAMPs further strengthen the potential for therapeutic application of these PrAMPs. Interestingly, the two lead PrAMPs displayed remarkable difference in stability in human serum (Fig. S15†) with tetrafluorobenzene dimer-NHNH26 half-life time over 8 hours while the octofluorobiphenyl dimer-NHNH27 around 1 hour half-life time. Given the potent activity and long-lasting human serum stability of the PrAMP lead candidates, 6 and 7, their in vivo activities and tolerance against MDR A. baumannii bacterial infection will be evaluated in wound infection and blood-stream infection mouse models. Due to the high cost of total peptide synthesis and restrictions of recombinant DNA methods, peptidomimetics strategies51 are needed to address those limitations for translational application towards clinical use. Overall, our study of novel linked dimer PrAMPs highlight the advantages of bioconjugation application to enhance their antibacterial and antibiofilm activity together with their immunomodulatory effects for therapeutic applications to combat the WHO priority critical pathogens A. baumannii and MDR/XDR strains.
Materials and methods
General procedure for the synthesis of dimeric PrAMPs
To a solution of monomeric C-terminal Cys-containing PrAMP-NHNH2 in dimethylformamide was added dropwise a 20 mM solution of linker (1,2-dibromomethyl-benzene, 1,3-dibromomethyl-benzene, 1,4-dibromomethyl-benzene, hexafluorobenzene or decafluorobiphenyl) dissolved in dimethylformamide. The mixture was reacted overnight at room temperature and the resulting dimeric PrAMPs were purified by reversed-phase high performance liquid chromatography (RP-HPLC) in water and acetonitrile containing 0.1% TFA. The final products were characterized by both RP-HPLC and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) (Fig. S1†). The detailed information and steps are provided in the ESI.†Antibacterial assays
Each synthetic PrAMP was tested for antibacterial activity in 100% Mueller Hinton broth (MHB) against a panel of Gram-negative bacteria, E. coli ATCC 25922, K. pneumoniae ATCC 13883, A. baumannii ATCC 19606, as well as the multi-drug resistant FADDI-KP028 (MDR) and FADDI-AB156 (colistin-resistant, rifampin-resistant & MDR, XDR). A series of 100 μl solutions diluted from 250 μg ml−1 to 4 μg ml−1 PrAMPs, antibiotics or bifunctional linkers in MHB were added to a 96-well plate followed by the addition of 100 μl of 2 × 106 cells per ml with 90 min incubation at 37 °C. The minimum inhibitory concentration (MIC) was determined by plotting maximal growth versus peptide concentration, according to the Lambert and Pearson analysis method,19 while minimum bactericidal concentration (MBC) was confirmed via colony-forming unit (CFU) measuring assay.Antibiofilm assay
The microtiter plate preformed biofilms were prepared by adding 100 μl of the diluted bacterial culture of A. baumannii ATCC 19606 or FADDI-AB156 (OD600 = 0.5–0.6, 10 μl into 8 ml MHB) into each well of a 96-well plate. After incubating for 24 h at 37 °C, the biofilm formation was confirmed by crystal violet staining under microscopy. At 24 h incubation, the media and planktonic bacteria were removed gently followed by two washing steps (200 μl phosphate buffered saline, PBS). Then 100 μl PrAMPs in MHB of different concentration (0.5 × MBC, 1 × MBC, 2 × MBC, 4 × MBC, 8 × MBC) in triplicate and 100 μl MHB as control were added to each well. After 90 min incubation at 37 °C, the peptides were removed and washed with 200 μl PBS (two times), followed by crystal violet (100 μl, 2% w/v in 20% v/v ethanol/water) staining for 30 min. The crystal violet was removed and washed with 200 μl PBS (two times). The crystal violet stained samples were dissolved with 100 μl acetic acid (30% in MilliQ water) and absorption measured at 540 nm. In comparison to the untreated control biofilms, the absorption of each sample indicated the efficiency of the PrAMPs, tetrafluorobenzene dimer-NHNH26 and octofluorobiphenyl dimer-NHNH27, to eradicate preformed biofilms.Helium ion microscopy for planktonic cells and biofilms
The morphologies of planktonic bacteria A. baumannii ATCC 19606 and FADDI-AB156 was assessed after treatment with tetrafluorobenzene dimer-NHNH26 and octofluorobiphenyl dimer-NHNH27 at different concentration (2 × MIC, 1 × MIC, 0.5 × MIC, 0.25 × MIC, 0.125 × MIC) by using helium ion microscopy (HIM, Zeiss, Germany). The samples incubated with PrAMPs (90 min) and the PBS treated control sample were added to clean glass cover slides, followed by evaporation at 37 °C for 20–30 min. The dried samples on5terminationAs shown in Fig. S13,† 12.5 μg ml−1 of LPS originated from A. baumannii ATCC 19606 and FADDI-AB156 can fully stimulate RAW264.7 cells for nitric oxide (NO) release. To assess the effect of the lead PrAMPs on suppressing LPS stimulation, PrAMPs, tetrafluorobenzene dimer-NHNH26 and octofluorobiphenyl dimer-NHNH27, were incubated with RAW264.7 cells at different concentrations in the presence of 12.5 μg ml−1 of the extracted LPS. In brief, 100 μl of 1 × 106 RAW264.7 cells per ml was seeded into a flat-bottom 96-well plate for 24 h at 37 °C with 5% CO2 in Dulbecco's Modified Eagle Medium (DMEM). Then, a series of 50 μl per well PrAMPs diluted from 100 μg ml−1 to 3 μg ml−1 in DMEM were added into each well of RAW264.7 cells, followed by the addition of 50 μl extracted LPS (12.5 μg ml−1 in DMEM) and 50 μl DMEM. After overnight incubation at 37 °C, the culture supernatant (50 μl) was collected for NO concentration determination using a Promega Griess Reagent Kit to measure the absorption with a 540 nm filter. The concentration of NO in μM was further converted via a nitrate standard curve.
Data availability
All experimental supporting data and procedures are available in the ESI.†Author contributions
W. L., N. M. O. and J. W. D designed the research; W. L., F. L., A. H., A. B., M. A. S., R. P., W. S. and J. H. performed the research; W. L., A. H., M. A. H, F. S., N. M. O. and J. W. D analyzed the data, and W. L., A. H., M. A. S., M. A. H, F. S., N. M. O. and J. W. D. wrote the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We thank Professor Jian Li, Monash Biomedicine Discovery Institute, Monash University, Australia, for critical advice during the course of this work. We acknowledge the Melbourne Cytometry Platform (Melbourne Dental School, The University of Melbourne) for the provision of flow cytometry services. The HIM work was performed in part at the Materials Characterization and Fabrication Platform (MCFP) at the University of Melbourne and the Victorian Node of the Australian National Fabrication Facility (ANFF). Computational resources were provided by the National Computational Infrastructure (NCI), which is supported by the Australian Government, and the Pawsey Supercomputing Centre with funding from the Australian Government and the Government of Western Australia. This work was supported by grants to W. L. (University of Melbourne Early Career Researcher Grant, Weary Dunlop Foundation Grant and Australian Dental Research Foundation Grant 2545-2020), to J. D. W. (NHMRC Project grant APP1158841, NHMRC Principal Research Fellowship APP1117483); to N. M. O-S (NHMRC Project grants APP1142472, APP1158841, APP1185426, ARC funding DP210102781, DP160101312, LE200100163) and Australian Dental Research Funding in antimicrobial materials. W. L. acknowledges NHMRC funding (APP1185426) to support his salary. Studies at the FINMH were supported by the Victorian Government's Operational Infrastructure Support Program.Notes and references
- E. Y. Klein, M. Milkowska-Shibata, K. K. Tseng, M. Sharland, S. Gandra, C. Pulcini and R. Laxminarayan, Lancet Infect. Dis., 2021, 21, 107–115 CrossRef PubMed.
- S. C. Roberts and T. R. Zembower, Lancet Infect. Dis., 2020, 21, 10–11 CrossRef PubMed.
- J. O'Neill, Tackling a crisis for the health and wealth of nations, 2014 Search PubMed.
- F. Zhou, T. Yu, R. Du, G. Fan, Y. Liu, Z. Liu, J. Xiang, Y. Wang, B. Song, X. Gu, L. Guan, Y. Wei, H. Li, X. Wu, J. Xu, S. Tu, Y. Zhang, H. Chen and B. Cao, Lancet, 2020, 395, 1054–1062 CrossRef CAS.
- A. Towse, C. K. Hoyle, J. Goodall, M. Hirsch, J. Mestre-Ferrandiz and J. H. Rex, Health Policy, 2017, 121, 1025–1030 CrossRef PubMed.
- P. Fernandes and E. Martens, Biochem. Pharmacol., 2017, 133, 152–163 CrossRef CAS PubMed.
- E. Tacconelli, E. Carrara, A. Savoldi, S. Harbarth, M. Mendelson, D. L. Monnet, C. Pulcini, G. Kahlmeter, J. Kluytmans, Y. Carmeli, M. Ouellette, K. Outterson, J. Patel, M. Cavaleri, E. M. Cox, C. R. Houchens, M. L. Grayson, P. Hansen, N. Singh, U. Theuretzbacher, N. Magrini, A. O. Aboderin, S. S. Al-Abri, N. Awang Jalil, N. Benzonana, S. Bhattacharya, A. J. Brink, F. R. Burkert, O. Cars, G. Cornaglia, O. J. Dyar, A. W. Friedrich, A. C. Gales, S. Gandra, C. G. Giske, D. A. Goff, H. Goossens, T. Gottlieb, M. Guzman Blanco, W. Hryniewicz, D. Kattula, T. Jinks, S. S. Kanj, L. Kerr, M.-P. Kieny, Y. S. Kim, R. S. Kozlov, J. Labarca, R. Laxminarayan, K. Leder, L. Leibovici, G. Levy-Hara, J. Littman, S. Malhotra-Kumar, V. Manchanda, L. Moja, B. Ndoye, A. Pan, D. L. Paterson, M. Paul, H. Qiu, P. Ramon-Pardo, J. Rodríguez-Baño, M. Sanguinetti, S. Sengupta, M. Sharland, M. Si-Mehand, L. L. Silver, W. Song, M. Steinbakk, J. Thomsen, G. E. Thwaites, J. W. M. van der Meer, N. Van Kinh, S. Vega, M. V. Villegas, A. Wechsler-Fördös, H. F. L. Wertheim, E. Wesangula, N. Woodford, F. O. Yilmaz and A. Zorzet, Lancet Infect. Dis., 2018, 18, 318–327 CrossRef PubMed.
- L. Czaplewski, R. Bax, M. Clokie, M. Dawson, H. Fairhead, V. A. Fischetti, S. Foster, B. F. Gilmore, R. E. W. Hancock, D. Harper, I. R. Henderson, K. Hilpert, B. V. Jones, A. Kadioglu, D. Knowles, S. Ólafsdóttir, D. Payne, S. Projan, S. Shaunak, J. Silverman, C. M. Thomas, T. J. Trust, P. Warn and J. H. Rex, Lancet Infect. Dis., 2016, 16, 239–251 CrossRef CAS PubMed.
- M. Magana, M. Pushpanathan, A. L. Santos, L. Leanse, M. Fernandez, A. Ioannidis, M. A. Giulianotti, Y. Apidianakis, S. Bradfute, A. L. Ferguson, A. Cherkasov, M. N. Seleem, C. Pinilla, C. de la Fuente-Nunez, T. Lazaridis, T. Dai, R. A. Houghten, R. E. W. Hancock and G. P. Tegos, Lancet Infect. Dis., 2020, 20, e216–e230 CrossRef CAS PubMed.
- W. Li, F. Separovic, N. M. O'Brien-Simpson and J. D. Wade, Chem. Soc. Rev., 2021, 50, 4932–4973 RSC.
- A. Krizsan, C. Prahl, T. Goldbach, D. Knappe and R. Hoffmann, ChemBioChem, 2015, 16, 2304–2308 CrossRef CAS PubMed.
- W. Li, J. Tailhades, N. O'Brien-Simpson, F. Separovic, L. Otvos Jr, M. A. Hossain and J. Wade, Amino Acids, 2014, 46, 2287–2294 CrossRef CAS PubMed.
- M. Graf, M. Mardirossian, F. Nguyen, A. C. Seefeldt, G. Guichard, M. Scocchi, C. A. Innis and D. N. Wilson, Nat. Prod. Rep., 2017, 34, 702–711 RSC.
- W. Li, N. M. O'Brien-Simpson, J. A. Holden, L. Otvos, E. C. Reynolds, F. Separovic, M. A. Hossain and J. D. Wade, Pept. Sci., 2018, 110, e24059 Search PubMed.
- W. Li, J. Tailhades, M. A. Hossain, N. M. O'Brien-Simpson, E. C. Reynolds, L. Otvos, F. Separovic and J. D. Wade, Aust. J. Chem., 2015, 68, 1373–1378 CrossRef CAS.
- W. Li, N. M. O'Brien-Simpson, J. Tailhades, N. Pantarat, R. M. Dawson, L. Otvos Jr, E. C. Reynolds, F. Separovic, M. A. Hossain and J. D. Wade, Chem. Biol., 2015, 22, 1250–1258 CrossRef CAS PubMed.
- W. Li, N. M. O'Brien-Simpson, S. Yao, J. Tailhades, E. C. Reynolds, R. M. Dawson, L. Otvos, M. A. Hossain, F. Separovic and J. D. Wade, Chem.–Eur. J., 2017, 23, 390–396 CrossRef CAS PubMed.
- M. H. Stenzel, ACS Macro Lett., 2013, 2, 14–18 CrossRef CAS.
- S. J. Lam, N. M. O'Brien-Simpson, N. Pantarat, A. Sulistio, E. H. H. Wong, Y.-Y. Chen, J. C. Lenzo, J. A. Holden, A. Blencowe, E. C. Reynolds and G. G. Qiao, Nat. Microbiol., 2016, 1, 16162 CrossRef CAS PubMed.
- M. E. Falagas and E. A. Karveli, Clin. Microbiol. Infect., 2007, 13, 117–119 CrossRef CAS PubMed.
- F. Perez, A. M. Hujer, K. M. Hujer, B. K. Decker, P. N. Rather and R. A. Bonomo, Antimicrob. Agents Chemother., 2007, 51, 3471–3484 CrossRef CAS PubMed.
- F. H. Waghu, S. Joseph, S. Ghawali, E. A. Martis, T. Madan, K. V. Venkatesh and S. Idicula-Thomas, Frontiers in Microbiology, 2018, 9, 325 CrossRef PubMed.
- I. M. Helander and T. Mattila-Sandholm, J. Appl. Microbiol., 2000, 88, 213–219 CrossRef CAS PubMed.
- L. Zhang, P. Dhillon, H. Yan, S. Farmer and R. E. W. Hancock, Antimicrob. Agents Chemother., 2000, 44, 3317–3321 CrossRef CAS PubMed.
- J. M. Benarroch and M. Asally, Trends Microbiol., 2020, 28, 304–314 CrossRef CAS PubMed.
- H. M. Shapiro, Methods, 2000, 21, 271–279 CrossRef CAS PubMed.
- H. Strahl and J. Errington, Annu. Rev. Microbiol., 2017, 71, 519–538 CrossRef CAS PubMed.
- W. Li, M.-A. Sani, E. Jamasbi, L. Otvos Jr, M. A. Hossain, J. D. Wade and F. Separovic, Biochim. Biophys. Acta, 2016, 1858, 1236–1243 CrossRef CAS PubMed.
- N. M. O'Brien-Simpson, W. Li, N. Pantarat, M. A. Hossain, F. Separovic, J. D. Wade and E. C. Reynolds, Aust. J. Chem., 2017, 70, 220–228 CrossRef.
- X. Zhao and K. Drlica, Curr. Opin. Microbiol., 2014, 21, 1–6 CrossRef CAS PubMed.
- M. P. Brynildsen, J. A. Winkler, C. S. Spina, I. C. MacDonald and J. J. Collins, Nat. Biotechnol., 2013, 31, 160–165 CrossRef CAS PubMed.
- A. Novogrodsky, A. Ravid, A. L. Rubin and K. H. Stenzel, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 1171–1174 CrossRef CAS PubMed.
- M. S. Joens, C. Huynh, J. M. Kasuboski, D. Ferranti, Y. J. Sigal, F. Zeitvogel, M. Obst, C. J. Burkhardt, K. P. Curran, S. H. Chalasani, L. A. Stern, B. Goetze and J. A. J. Fitzpatrick, Sci. Rep., 2013, 3, 3514 CrossRef PubMed.
- T. Huang, J. A. Holden, D. E. Heath, N. M. O'Brien-Simpson and A. J. O'Connor, Nanoscale, 2019, 11, 14937–14951 RSC.
- L. Dieltjens, K. Appermans, M. Lissens, B. Lories, W. Kim, E. V. Van der Eycken, K. R. Foster and H. P. Steenackers, Nat. Commun., 2020, 11, 107 CrossRef CAS PubMed.
- C. de la Fuente-Núñez, F. Reffuveille, L. Fernández and R. E. W. Hancock, Curr. Opin. Microbiol., 2013, 16, 580–589 CrossRef PubMed.
- A. Di Somma, A. Moretta, C. Canè, A. Cirillo and A. Duilio, Biomolecules, 2020, 10, 652 CrossRef PubMed.
- H. Etayash, E. F. Haney and R. E. W. Hancock, Nat. Protoc., 2021, 16, 1850–1870 CrossRef CAS PubMed.
- C. de la Fuente-Núñez, F. Reffuveille, E. F. Haney, S. K. Straus and R. E. W. Hancock, PLoS Pathog., 2014, 10, e1004152 CrossRef PubMed.
- R. E. W. Hancock, E. F. Haney and E. E. Gill, Nat. Rev. Immunol., 2016, 16, 321–334 CrossRef CAS PubMed.
- L. Otvos, J. Pept. Sci., 2000, 6, 497–511 CrossRef CAS PubMed.
- E. Ostorhazi, M. C. Holub, F. Rozgonyi, F. Harmos, M. Cassone, J. D. Wade and L. Otvos Jr, Int. J. Antimicrob. Agents, 2011, 37, 480–484 CrossRef CAS PubMed.
- S. M. Zughaier, W. M. Shafer and D. S. Stephens, Cell. Microbiol., 2005, 7, 1251–1262 CrossRef CAS PubMed.
- B. Reddy, N. Bavi, A. Lu, Y. Park and E. Perozo, eLife, 2019, 8, e50486 CrossRef CAS PubMed.
- L. Sessa, S. Concilio, P. Walde, T. Robinson, P. S. Dittrich, A. Porta, B. Panunzi, U. Caruso and S. Piotto, Membranes, 2020, 10, 294 CrossRef CAS PubMed.
- R. M. Venable, F. L. H. Brown and R. W. Pastor, Chem. Phys. Lipids, 2015, 192, 60–74 CrossRef CAS PubMed.
- S. Alapour, B. G. de la Torre, D. Ramjugernath, N. A. Koorbanally and F. Albericio, Bioconjugate Chem., 2018, 29, 225–233 CrossRef CAS PubMed.
- A. M. Spokoyny, Y. Zou, J. J. Ling, H. Yu, Y.-S. Lin and B. L. Pentelute, J. Am. Chem. Soc., 2013, 135, 5946–5949 CrossRef CAS PubMed.
- Y. Hong and W. Li, Aust. J. Chem., 2022 DOI:10.1071/CH21070.
- A. Boaro, L. Ageitos, M. Torres, F. H. Bartoloni and C. de la Fuente-Nunez, Cell Rep. Phys. Sci., 2020, 1, 100257 CrossRef CAS PubMed.
- E. Lenci and A. Trabocchi, Chem. Soc. Rev., 2020, 49, 3262–3277 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Materials, dimeric PrAMP synthesis, antibacterial test of the biofunctional linker, determination of minimum membrane disruption concentration, N-phenylnaphthylamine (NPN) outer membrane permeability, cytoplasm membrane permeability, cell proliferation test, LDH cytotoxicity, therapeutic index (TI), bacterial time killing measurement, dye leakage assay, membrane potential, ROS production determination, LPS extraction from A. baumannii and MDR-FADDI-AB156, titrated LPS of NO release, serum stability and monomeric PrAMP MD simulation snapshots, can be found with this article. See DOI: 10.1039/d1sc05662j |
| This journal is © The Royal Society of Chemistry 2022 |