
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemically driven C–N bond formation from CO2 and ammonia at the triple-phase boundary†
Junnan
Li
and
Nikolay
Kornienko
 *
*
Department of Chemistry, Université de Montréal, 1375 Avenue Thérèse-Lavoie-Roux, Montréal, QC H2V 0B3, Canada. E-mail: nikolay.kornienko@umontreal.ca
First published on 28th February 2022
Abstract
Electrosynthetic techniques are gaining prominence across the fields of chemistry, engineering and energy science. However, most works within the direction of synthetic heterogeneous electrocatalysis focus on water electrolysis and CO2 reduction. In this work, we moved to expand the scope of small molecule electrosynthesis by developing a synthetic scheme which couples CO2 and NH3 at a gas–liquid–solid boundary to produce species with C–N bonds. Specifically, by bringing in CO2 from the gas phase and NH3 from the liquid phase together over solid copper catalysts, we have succeeded in forming formamide and acetamide products for the first time from these reactants. In a subsequent complementary step, we have combined electrochemical analysis and a newly developed operando spectroelectrochemical method, capable of probing the aforementioned gas–liquid–solid boundary, to extract an initial level of mechanistic analysis regarding the reaction pathways of these reactions and the current system's limitations. We believe that the development and understanding of this set of reaction pathways will play significant role in expanding the community's understanding of on-surface electrosynthetic reactions as well as push this set of inherently sustainable technologies towards widespread applicability.
Introduction
With the increased focus on attaining global sustainability as a means to mitigate climate change and environmental degradation, the development of green technologies to enable the transition is increasingly important. Within this context, renewable electricity-powered electrosynthetic routes towards generating the fuels and chemicals that drive modern society stand to play a significant role if they manage to displace currently used fossil-fuel dependent methods.1–4 While the recent decade of academic research has largely focused on water electrolysis5 and CO2 reduction6 to generate H2 and carbon-based fuels, respectively, there is no reason that the scope of heterogeneous electrosynthesis needs to be limited to these reactions. In principle, almost any commodity chemical can be synthesized from abundant small molecule building blocks (N2, H2O, CH4, biomass…) if the proper catalytic system would be developed.7 The difficulty in realizing this ambitious aim is that at this point, only relatively simple electrosynthetic reactions over heterogeneous catalysts are well-understood and can be carried out at high rates and selectivity.To this end, we moved to develop electrosynthetic routes to C–N bond formation using CO2 and NH3 as model building blocks. In general, despite the biological, societal and technological importance of many chemicals containing C–N bonds,8–11 the area of electrochemical C–N bond formation is very nascent. While biological12–14 and chemical8,15 routes are established, only few examples exist in carrying out C–N coupling on heterogeneous electrocatalysts. Thus, new reaction schemes and mechanistic insights in this context stand to provide a significant boost to the community.16–22 In the context of heterogeneous catalysis, urea has previously been synthesized from co-electrolysis of N2 or nitrate together with CO2.17,19,20,22 α-Keto acids have been converted into amino acids with hydroxylamine as a N-source.23 Further, CO was co-electrolyzed with a series of different amines to generate amide products.18 Finally, a host biomass-derived furans were reductively aminated to produce to amine derivatives.21 To expand the scope of possibilities of heterogeneously catalyzed C–N bond formation, we have developed a novel electrosynthetic scheme. Here, NH3 from the liquid phase would react with CO2 from the gas phase over a heterogeneous Cu catalyst at a gas–liquid–solid boundary (Fig. 1). As such, we generated formamide and acetamide from CO2 and NH3 for the first time, opening up a new avenue to the research community. Through quantitative reaction analysis and newly-developed infrared spectroelectrochemical investigations, we have built up a set of mechanistic insights in terms of elucidating reaction pathways and performance limitations, thus enabling the rational design of next-generation electrosynthetic systems.
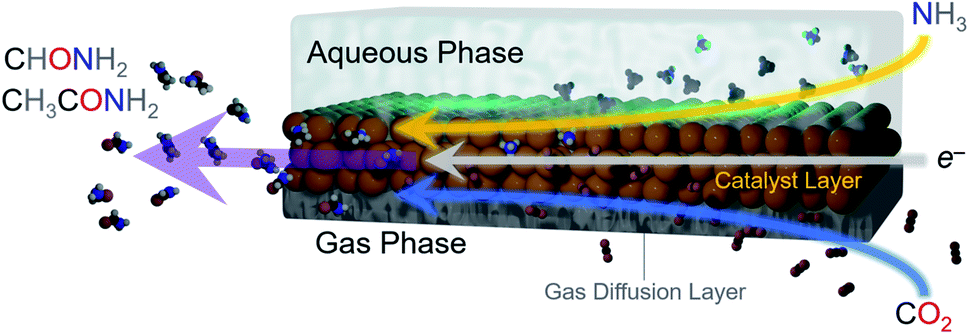 | ||
| Fig. 1 Illustration of electrosynthetic strategy for on-surface C–N bond formation. A gas diffusion electrode was employed in which the reactants were simultaneously brought in from the gas phase (CO2) and from the liquid phase (NH3) and reacted over a solid Cu catalyst onto which an electrochemical potential was applied. This configuration enabled the generation of formamide and acetamide C–N bond containing products. | ||
Catalyst construction
As a starting point, we selected two types of commercially available copper catalysts, Cu and CuO nanoparticles, for use in our study. Copper was selected as the element of choice because it has an intermediate binding energy to many carbon-based species.24 This is a favorable metric in CO2 reduction because it enables the retention of surface intermediates en route to the formation of highly reduced products like ethylene. At the same time, the binding strength to the intermediates is not too high to poison the surface. Thus, we reasoned that the same argument would apply in retaining CO2 reduction intermediates long enough for their coupling with NH3 would hold. While there is a plethora of studies of Cu-based catalysts and how defects, surface crystallographic facets, ligands, oxygen species and more dictate reaction pathways, we chose to leave such catalyst modifications for future follow-up works given the novelty of this reaction path.24 The one variable that we did choose to investigate was the use of CuO as a starting material, which, when reduced to Cu under cathodic potentials, would likely contain additional binding sites in the form of defects. As such, Cu (Fig. 2a) and CuO (Fig. 2b) with no deliberate surface or structural modifications and size around 100 nm were used. The catalysts were mixed with a Nafion binder to generate an ink which was then drop cast onto a gas diffusion electrode. This type of electrode featured a gas-permeable gas diffusion layer and microporous layer through which CO2 could reach the catalyst layer on top (Fig. 2c). The goal here was to drive the C–N coupling reaction at this interfacial gas–liquid–solid boundary. This type of electrode geometry is particularly beneficial in overcoming the limited solubility of CO2 in aqueous electrolyte through the use of alkaline electrolytes that minimize the competing hydrogen evolution reaction at industrially relevant current densities (hundreds of mA cm−2).25,26 The reaction cell was a modified one from those commonly employed in the field in order to minimize reaction volume and consequently maximize sensitivity for products. In particular, we employed an open cell design in which approximately 1 mL volume of electrolyte was used (Fig. S2†).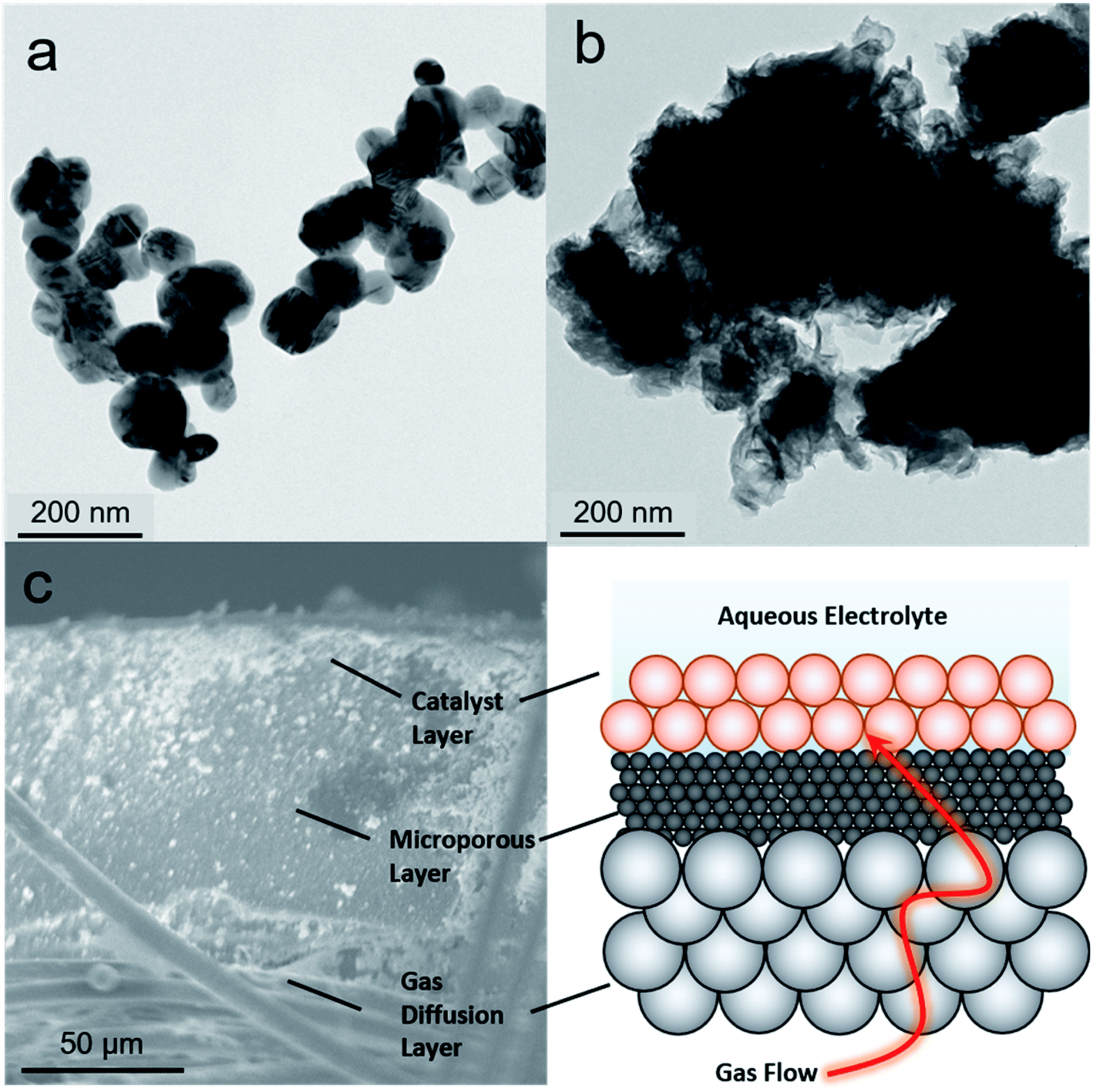 | ||
| Fig. 2 The Cu (a) and CuO (b) catalyst particles were first characterized through transmission electron microscopy to probe their size and morphology. They were drop-cast onto a gas-diffusion electrode, which was characterized through scanning electron microscopy (c). The gas diffusion electrode consisted of several layers, illustrated with the graphic as a simplified representation. This electrode enabled gaseous reactants (CO2 in this case) to reach a solid electrocatalyst (Cu/CuO) and circumvent the limited solubility of CO2. | ||
Electrosynthetic studies
We employed 1 M KOH as an electrolyte for this work as highly alkaline electrolytes tend to minimize the hydrogen evolution reaction (HER) and thus favor CO2 reduction. NH3 was set to be a model nitrogen source. In the long term, NH3 would ideally be replaced directly by N2 as an abundant feedstock, though at this stage, electrochemically activating N2 not yet a well-established reaction.27,28 Formamide (Fig. 3a) and acetamide (Fig. 3b) primary amines were two likely C–N coupled products that could be formed from NH3 and C1 and C2 surface intermediates via several proton and electron transfer steps. In a gas-diffusion based electrochemical cell, both Cu and CuO featured an onset of catalytic current around 0 V vs. the reversible hydrogen electrode (RHE) and reached 100 mA cm−2 by −1.0 VRHE (Fig. S3a and b†). The addition of NH4OH (present as mainly NH3 in alkaline solutions) to the electrolyte did not significantly alter the current density. Product quantification with gas chromatography (GC) and NMR revealed formate and H2 to be the two main products from the reaction (Fig. S4†). However, on both Cu and CuO, formamide and acetamide were detected and were formed with partial current densities of ranging from 0.1 to 1.2 mA cm−2, depending on the applied potential (Fig. 3c and d). While the faradaic efficiency for their formation was rather modest, peaking at approximately 1% (Fig. 3e and f), this study constitutes the first report of their synthesis from CO2 and NH3 building blocks. In addition, performing the same measurements in a standard 3-electrode setup with the working electrode completely immersed in the aqueous phase did not result in any detectible C–N products, even after 24 h of electrolysis. This is likely due to a lower CO2 concentration and lack of an alkaline environment that together promote a high degree of strongly bound C-based intermediates needed to couple with NH3. As a control experiment, CO2 electrolysis alone only resulted in formate (Fig. 3g) and acetate (Fig. 3h) products that gave rise to NMR peaks in the range of interest. Interestingly, while the formate selectivity was very high (up to 90%) without NH3, NH3 addition to the electrolyte decreased this value by a factor of 2–3 (Fig. S5†) while promoting hydrogen evolution. While this performance is not yet sufficient for economically competitive electrosynthesis, improving the initial system should certainly be feasible as one could point to the rapid maturation of CO2 electrosynthetic technologies over the last decade.2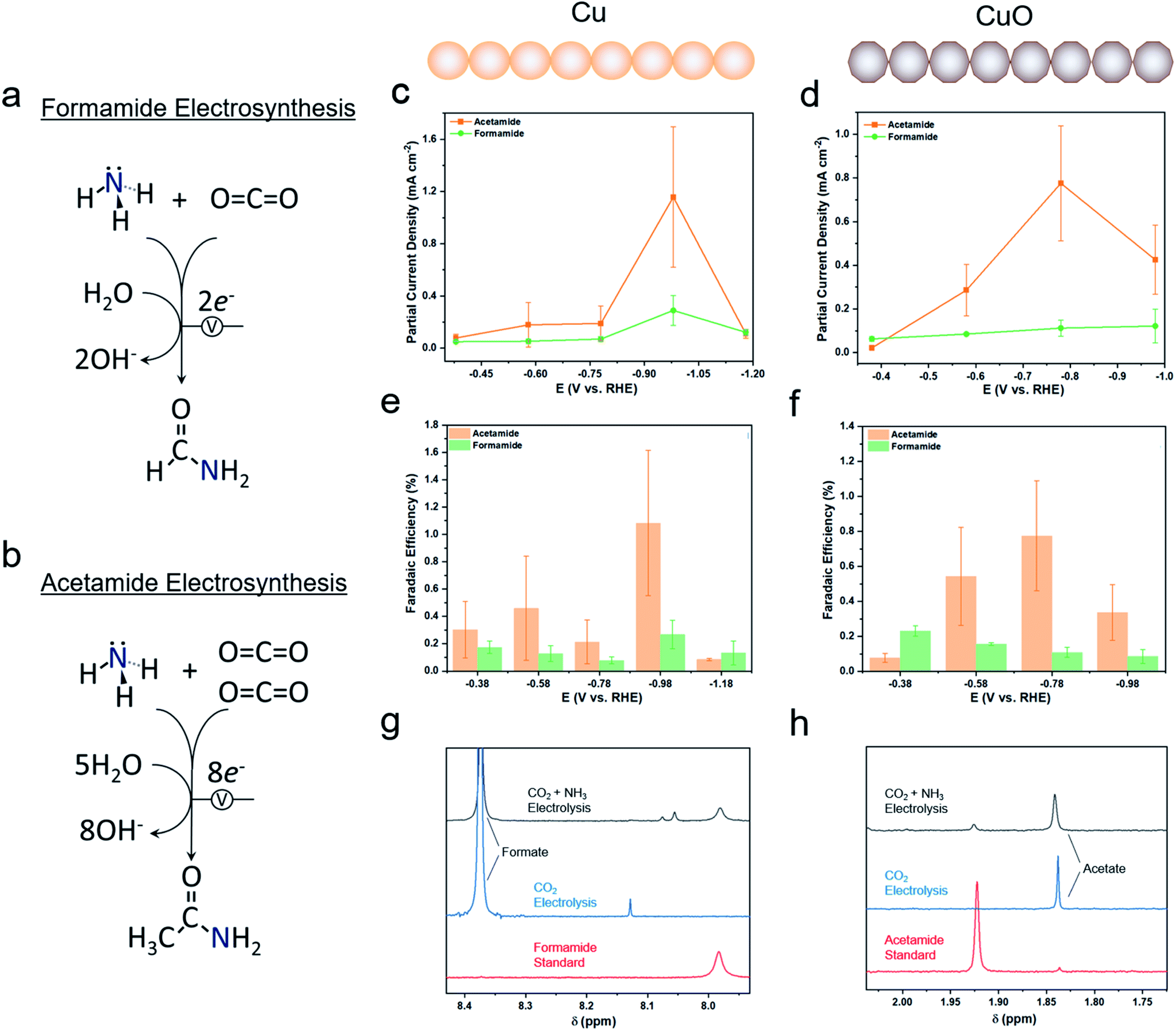 | ||
| Fig. 3 The overall reaction is depicted for formamide involves 2 electrons and 1 CO2 molecule (a) while acetamide electrosynthesis entails 8 electrons and 2 CO2 molecules (b). In the gas diffusion electrode cell with a 1 M KOH electrolyte, 6SCCM CO2 flow, and the optimized concentration of NH3, formamide (c) and acetamide (d) were quantified and their partial current densities derived from the resulting concentrations. The faradaic efficiencies for both products were similarly obtained for Cu (e) and CuO (f) catalysts. Representative NMR spectra are shown for formamide (g) and acetamide (h) from which the concentrations are quantified. | ||
Infrared spectroscopy
To extract a further level of insights into the formamide and acetamide electrosynthetic pathways, we turned to infrared spectroscopy.29 This technique measures the vibrational modes of species within the electrolyte and on the catalyst surface. Typically, measurements are carried out in difference mode, using the system at open circuit as a reference and subtracting this from the spectra under applied bias, thus detecting the appearance of new species (positive bands) and disappearance of others (negative bands). The spectroscopic measurements were carried out in an attenuated-total reflection (ATR) mode using a home-built spectroelectrochemical setup (Fig. 4a). Briefly, a thin layer of aqueous electrolyte (KOH or KOH + NH3) was on top of the diamond-coated ZnSe ATR crystal. The Cu catalyst layer/microporous layer/gas diffusion layer composite electrode was placed overtop so that the gas/liquid/solid boundary could be spectroscopically probed. In this configuration, the liquid was in static mode while the gas flowed above. The ability to probe this region was evident when measuring the difference spectrum between Ar flow and CO2 flow in this configuration, which shows the presence of both gaseous and dissolved CO2 and carbonate species (Fig. S12†).30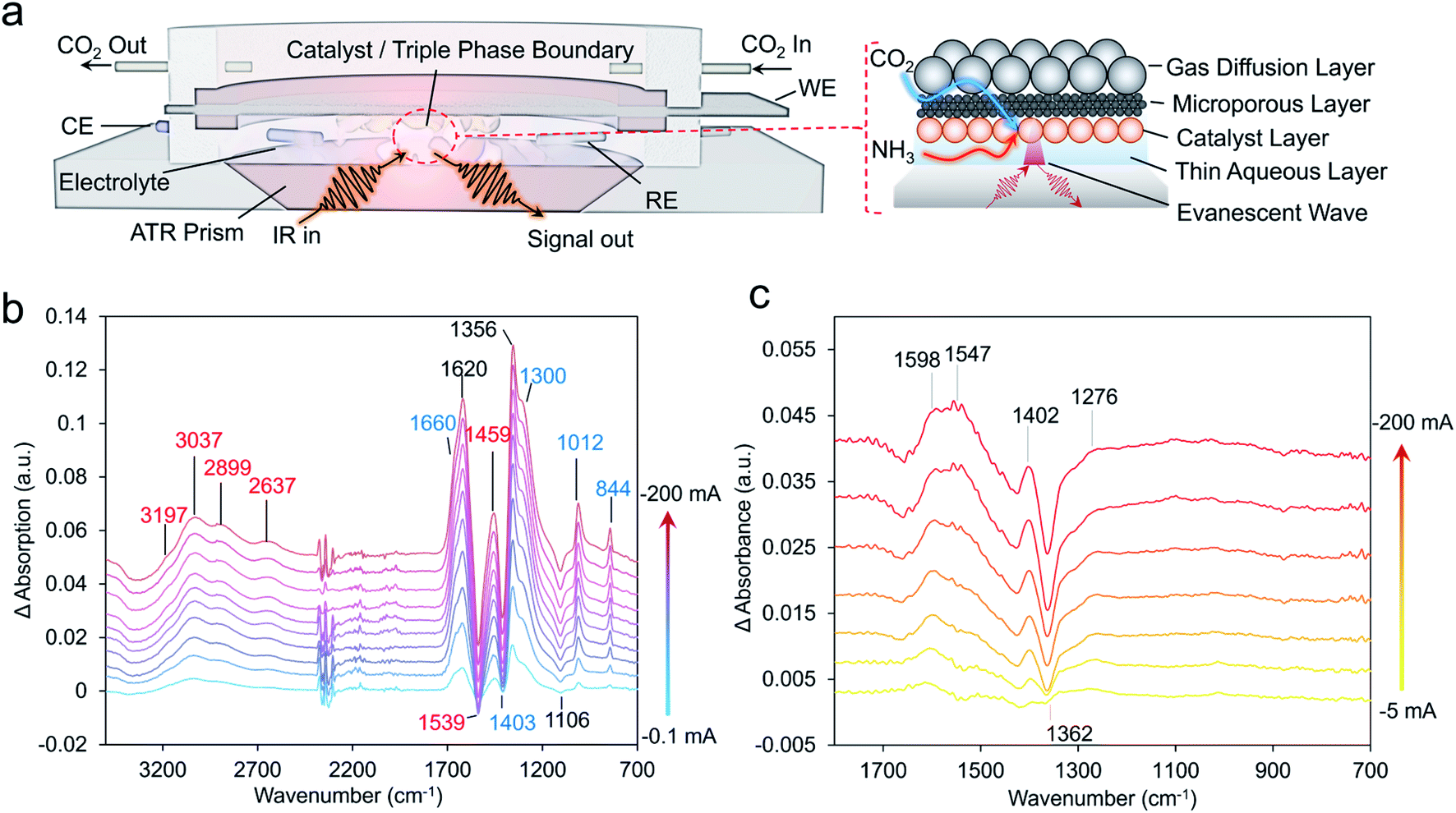 | ||
| Fig. 4 Spectroelectrochemical setup enabling operando infrared spectroscopic probing of the electrosynthetic reactions in a gas-diffusion electrode cell (a). This setup used a thin electrolyte window with the GDE just overtop to probe both liquid, gas and solid phases. The gas was flowing through while the liquid was static. Using the spectrum at open circuit as the background, spectra under select operating current densities with CO2 and NH3 present were recorded (b). Subtracting out the bi(carbonate) contributions using the spectra at 1 mA cm−2 as a background, enables the identification of additional species present on the CuO surface (c). | ||
Under an argon flow with NH3 and no CO2, the main spectral features corresponded mainly to that of water and to that of NH3 (Fig. S13–S15†).30 Under the same conditions but with CO2 flowing in place of Ar, a new set of positive bands appeared which can primarily be assigned to carbonate and bicarbonate species coming from CO2 reacting with the KOH electrolyte and changes in pH (Fig. S14†).31 Such species been shown to spontaneously appear at the gas–liquid–solid interface in similar conditions with Raman measurements.32 While spectral features in the 1800–2100 cm−1 are noted where the C–O stretch of *CO is located, the inherent absorbance of our diamond-coated ATR crystal makes this region rather noisy rendering bands here more difficult to fit and explicitly assign.
In the presence of both CO2 and NH3, new bands appeared in the region containing N–H bonds from the generation of NH4+ (Fig. 4b).33 As a method of validation, spectra were also acquired with 15NH3 instead of 14NH3 (Fig. S13†). Indeed, the isotope effect was noted via a red-shift around 30 cm−1 for bands at both spectral regions. Interestingly, the intensity of the bicarbonate band at 1300 cm−1 saturated very early with only CO2 present, but continually gained intensity under increasingly higher currents when NH3 was present (Fig. S13†). A possible explanation for this could that be the presence of NH3 diminishes the concentration of CO2/carbonate reactants near the interface at low currents.
Finally, as (bi)carbonate species dominate the IR spectra, we opted to subtract spectra of the catalyst systems operating at −1 mA from those at −200 mA, as the (bi)carbonate species are mostly saturated and those with smaller spectral contributions could be visualized (Fig. 4c). Indeed, for the CuO catalyst in the presence of CO2 and NH3, the evolution of positive spectral features (1645, 1598, 1547 1402 and 1096 cm−1) and negative bands (1660, 1425 and 1362 cm−1) were noted. While a fully unambiguous assignment at this stage is not yet possible, we note that these spectral features correlate well with those previously assigned to *COO− and *COOH and these species are thus our tentative assignments.34–36 The complete set of plausible band assignments is compiled in Table 1.
| Band position (cm−1) | Species | Figure | Possible assignment |
|---|---|---|---|
| 1660 | HCO3− | 4b | ν as(–C–O)30 |
| 1356 | HCO3− | 4b | ν(–C–O)30 |
| 1300 | HCO3− | 4b | δ(C–OH)30 |
| 1012 | HCO3− | 4b | ν as(C–OH)30 |
| 844 | HCO3− | 4b | ν s(C–OH)38 |
| 1459 | CO32− | 4b | δ(–NH)30 |
| 3197 | NH4+ | 4b | ν(N–H)33 |
| 3037 | NH4+ | 4b | ν(N–H)33 |
| 2899 | NH4+ | 4b | ν(N–H)33 |
| 1459 | NH4+ | 4b | ν(N–H)33 |
| 1645 | H2O | 4c | δ(H–O–H) |
| 1598 | *COO− | 4c | ν as(COO−)37 |
| 1547 | *COO− | 4c | ν as(COO−)35 |
| 1402 | *COO− | 4c | ν s(COO−)35,36 |
| 1276 | *COOH | 4c | νCOOH, OH-deformation35 |
As formate is the dominant product in each of these systems, it would seem reasonable to have a substantial *COO− surface coverage and thus this is our tentative assignment. While the precise mechanism of formate electrosynthesis is still under debate, on copper surfaces, it has been argued through a combination of surface-enhanced Raman spectroscopy and DFT modelling that the all CO2 reduction pathways share a common first intermediate in a μ2, –C, –O bound CO2*− that subsequently gets hydrogenated en route to formate or protonated to *COOH en route to CO and other C2 downstream products.37 Thus, the observation of *COO− and *COOH intermediates being the main ones on the surface would support our product distribution.
While this study used NH3 as a starting point for generating C–N bond containing products, ideally, the nitrogen source would be gaseous N2 as the technology matures and scales. To this end, we have explored as an intermediate step the co-reduction of nitrate and nitrite ions in place of NH3 to generate the same products. The experimental procedure was the same except that the nitrate/nitrite was in place of NH3 in the electrolyte, with optimized concentrations of NO2/NO3 (Fig. S15 and S16†). Over a Cu catalyst at −0.98 V vs. RHE, formamide and acetamide were indeed formed, albeit at reduced faradaic efficiencies and partial current densities (Fig. 5). While a comprehensive set of electroanalytical and spectroscopic experiments is outside of the scope of this initial work, the results indicate that there is much to discover in optimizing the reduction pathways of both C and N sources en route to C–N bond formation. A likely reaction pathway that would explain our results would be the reduction of nitrate/nitrite on the electrode surface to NH3 or *NH2, which then couples with intermediates from CO2 reduction.
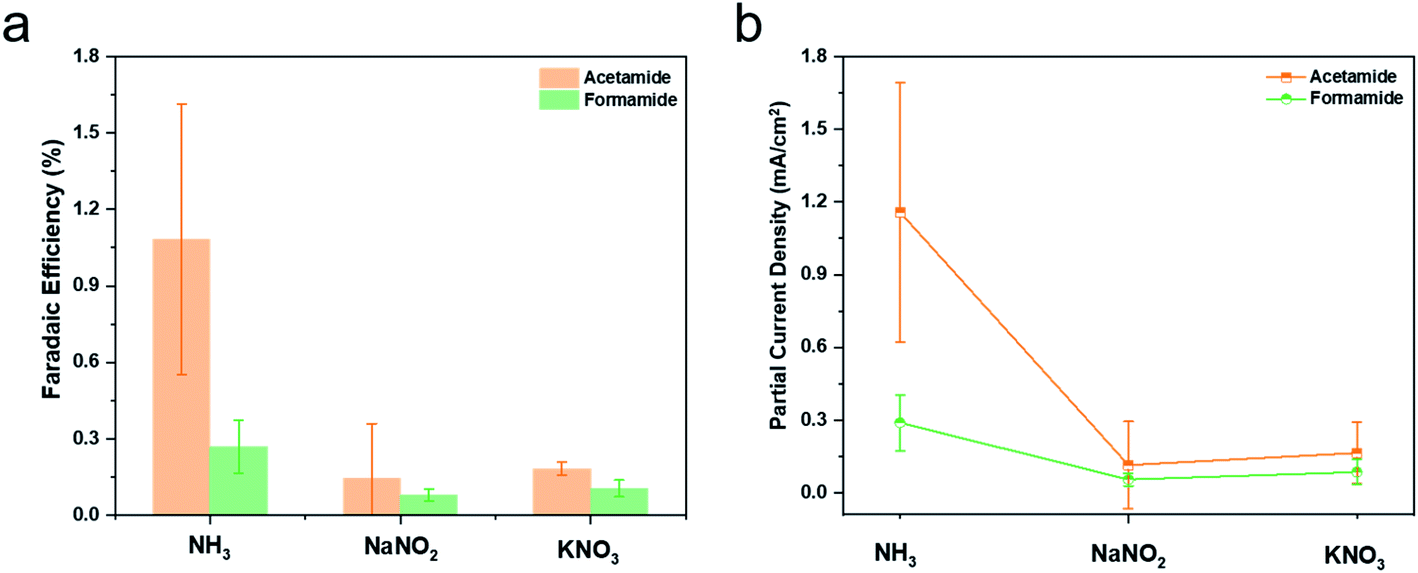 | ||
| Fig. 5 In the equivalent reaction setup as in Fig. 3 with Cu catalysts and NO2− (0.5 M) or NO3− (1 M) in place of NH3 at −0.98 V vs. RHE, formamide and acetamide were generated. The faradaic efficiencies (a) and partial current densities (b) for their generation were compared to those from using NH3. | ||
When using NH3 as the starting point we believe that it is not directly bound to the surface but rather located in the near-surface region (Fig. S18†). Its presence seems to hinder both hydrogen evolution and CO2 reduction, likely through inhibiting reactant diffusion to active sites on the catalyst. The tendency to enhance hydrogen evolution likely stems through an enhanced hindrance of CO2 diffusion as opposed to that of water molecules.
Finally, we moved to optimize the C–N product generation of our system. We first screened the parameters of KOH concentration, Cu catalyst loading, and cation identity (Fig. S21†). The biggest enhancement in acetamide generation came from a higher Cu loading (10 mg cm−2vs. 2 mg cm−2). We attribute this to the propensity of the catalyst layer to generate a higher amount of highly reduced C2 intermediates through the thicker Cu film (Fig. 6a and b). Further, a lower KOH concentration and a change from K+ to Cs+ also yielded selectivity enhancements for acetamide, possible due to a more favorable near-surface reaction environment to stabilize reaction intermediates en route to the C2 species that can couple with NH3.39 Interestingly, the activity enhancements were not realized for formamide. This can be rationalized as the factors necessary for favorizing a CO2 reduction pathway to C2 species as not being necessary for formamide, which is more dependent on the initial *COO− intermediate coupling with NH3.
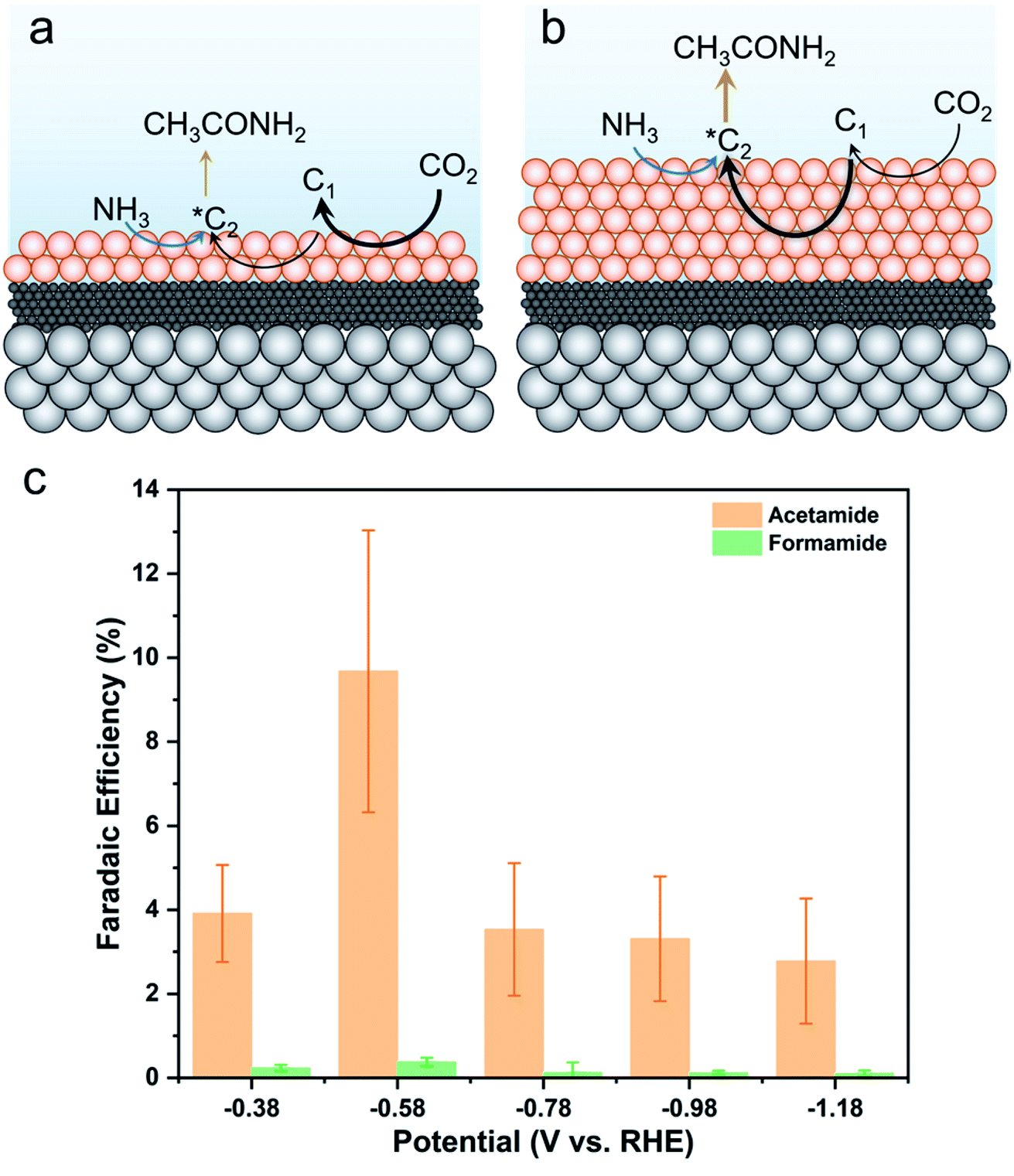 | ||
| Fig. 6 A thicker catalyst loading was found to promote acetamide formation. Thin layers tend to form C1 products at a greater rate (a) while increasing the layer thickness leads to further reduction and accumulation of C2 intermediates that can be used for C–N bond formation (b). As such, an optimized Cu loading of 10 mg cm−2 resulted in up to 10% acetamide selectivity (c). | ||
Then, with all three optimized parameters incorporated (0.1 M CsOH electrolyte, 10 mg cm−1 Cu loading), we compared the faradaic efficiency and partial current densities for the optimized system to that of the original from Fig. 2 across the experimental potential range (Fig. 6c).
We believe that the formate and formamide electrosynthetic pathways are linked on the Cu surface in that they share a common intermediate. This belief is backed by their similarity in chemical structure. Considering that the formation of C–N containing products involves the nucleophilic attack of a carbon atom by the lone pair on the nitrogen atom of ammonia, an activated, yet exposed carbon species that could couple with ammonia for formamide generation could be that of the μ2, –C, –O bound *CO2−.37 There would then be a competition between hydrogenation of this species to produce formate or a nucleophilic attack to eventually form formamide (Fig. 7a). This branching point is also supported by the fact that using 1 M formate instead of CO2 as the C-source in otherwise identical conditions did not lead to any detectable formamide and only a small amount of acetamide (Fig. S14†).
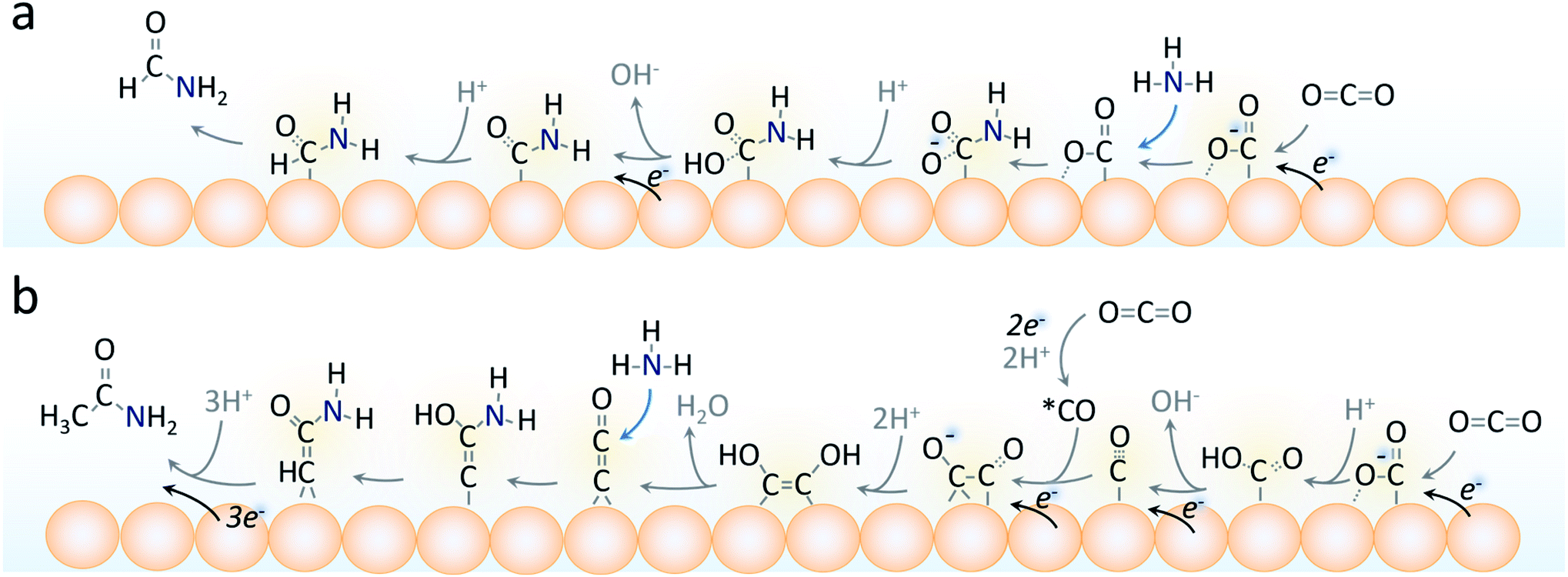 | ||
| Fig. 7 Plausible surface reaction pathways in the electrosynthetic process of formamide (a) and acetamide (b) generation. Formamide generation depends on NH3 coupling with the first reaction intermediate while acetamide generation requires highly reduced C2 intermediates to be present on the catalyst surface. For simplicity, the donation of a proton to surface intermediates via H2O → OH− is depicted as →H+. | ||
On the other hand, acetamide synthesis likely shares a reaction pathway with acetate and thus requires a C2 intermediate to already be present.18 The branching for this step also plausible occurs at the *CO2− intermediate, which is converted to *CO. The coupling of 2 *CO molecules is thought to be a key step to generating C2 products (acetate, ethanol, ethylene). The *CCO intermediate was recently proposed as a likely candidate for this through a DFT analysis of acetamide synthesis via CO and NH3 building blocks and would be a plausible candidate for our work as well.18 The middle carbon would thus be subject to nucleophilic attack by the NH3 in this pathway where it diverges from the acetate pathway as previously postulated.18 The enhancement of C2 intermediates such as this with a thick catalyst layer is postulated to be the main driving factor for its generation.
The two pathways presented here are not so different than what occurs in enzymatic catalytic pockets, where an electron rich amine couples with an electron poor carbon12–14 and one can imagine that generating on-surface catalytic pockets in a synthetic system to promote this reaction would lead to further enhancements of electrosynthetic selectivity. As the nucleophilic attach is by the ammonia nitrogen is a thermodynamically downhill step, reduction potential for both of these reactions is still dictated by that necessary to reduce CO2.
Concluding remarks
While two new reaction pathways have been discovered in formamide and acetamide electrosynthesis using CO2 and NH3 building blocks, many avenues are now opened for further understanding and improving the efficiency of these reactions. First, while we used commercially purchased Cu and CuO nanoparticles as a readily available model system, they feature a diversity of active sites, defects, (sub)surface oxygen species, and exposed crystallographic facets. It is entirely possible that each of these factors may influence the reaction like they do in the electrosynthesis of carbon-based products via CO2 reduction. A rational way forward would be the precise study of well-defined copper catalysts in which the nature surface-active sites are known and with complementary theoretical modelling of likely reaction pathways on these surfaces. Further, it is not known if Cu is the only catalyst capable of carrying out this reaction and if formate-selective metals like Sn and Bi would thus be more effective at formamide synthesis. In addition, we have developed an operando infrared spectroscopic method for the first time that was used to help understand this reaction pathway but additional complementary techniques such as Raman and X-ray absorption would contribute immensely valuable pieces to this puzzle.40This principal significance to this work is the electrosynthetic reaction discovery which we envision will accelerate the adoption of this methodology at large in both the academic and industrial domains. While NH3 is used as the model nitrogen source and nitrate and nitrite as further examples, eventually, this may be replaced by N2 in a fully sustainable nitrogen cycle. In general, the capacity to drive heteroatomic surface coupling reactions with renewable-electricity powered systems stands to open up an abundance of decentralized green synthetic routes in place of heavy-infrastructure requiring fossil fuel based thermochemical approaches. In parallel, there is much more fundamental chemistry to be discovered through the use of new interfaces, spectroscopic methodology, and catalytic systems.
Data availability
Experimental data available upon reasonable request.Author contributions
N. K. and J. L. both designed the project, carried out experiments, processed data, contributed intellectual insights and wrote the manuscript.Conflicts of interest
None to declare.Acknowledgements
N. K. and J. L acknowledge NSERC Discovery Grant RGPIN-2019-05927.References
- J. Masa, C. Andronescu and W. Schuhmann, Angew. Chem., Int. Ed., 2020, 59, 15298–15312 CrossRef CAS PubMed.
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef CAS PubMed.
- M. Wang, M. A. Khan, I. Mohsin, J. Wicks, A. H. Ip, K. Z. Sumon, C.-T. Dinh, E. H. Sargent, I. D. Gates and M. G. Kibria, Energy Environ. Sci., 2021, 14, 2535–2548 RSC.
- Y. Zhang, J. Li and N. Kornienko, Cell Rep. Phys. Sci., 2021, 2, 100682 CrossRef.
- J. Kibsgaard and I. Chorkendorff, Nat. Energy, 2019, 4, 430–433 CrossRef.
- M. B. Ross, P. De Luna, Y. Li, C.-T. Dinh, D. Kim, P. Yang and E. H. Sargent, Nat. Catal., 2019, 2, 648–658 CrossRef CAS.
- Z. J. Schiffer and K. Manthiram, Joule, 2017, 1, 10–14 CrossRef.
- O. I. Afanasyev, E. Kuchuk, D. L. Usanov and D. Chusov, Chem. Rev., 2019, 119, 11857–11911 CrossRef CAS PubMed.
- J. R. Dunetz, J. Magano and G. A. Weisenburger, Org. Process Res. Dev., 2016, 20, 140–177 CrossRef CAS.
- X. Guo, A. Facchetti and T. J. Marks, Chem. Rev., 2014, 114, 8943–9021 CrossRef CAS PubMed.
- M. Höhne and U. T. Bornscheuer, ChemCatChem, 2009, 1, 42–51 CrossRef.
- M. D. Patil, G. Grogan, A. Bommarius and H. Yun, ACS Catal., 2018, 8, 10985–11015 CrossRef CAS.
- O. Mayol, K. Bastard, L. Beloti, A. Frese, J. P. Turkenburg, J.-L. Petit, A. Mariage, A. Debard, V. Pellouin, A. Perret, V. de Berardinis, A. Zaparucha, G. Grogan and C. Vergne-Vaxelaire, Nat. Catal., 2019, 2, 324–333 CrossRef CAS.
- G. A. Aleku, S. P. France, H. Man, J. Mangas-Sanchez, S. L. Montgomery, M. Sharma, F. Leipold, S. Hussain, G. Grogan and N. J. Turner, Nat. Chem., 2017, 9, 961–969 CrossRef CAS PubMed.
- H. Kim and S. Chang, ACS Catal., 2016, 6, 2341–2351 CrossRef CAS.
- J. E. Kim, S. Choi, M. Balamurugan, J. H. Jang and K. T. Nam, Trends Chem., 2020, 2, 1004–1019 CrossRef CAS.
- C. Chen, X. Zhu, X. Wen, Y. Zhou, L. Zhou, H. Li, L. Tao, Q. Li, S. Du, T. Liu, D. Yan, C. Xie, Y. Zou, Y. Wang, R. Chen, J. Huo, Y. Li, J. Cheng, H. Su, X. Zhao, W. Cheng, Q. Liu, H. Lin, J. Luo, J. Chen, M. Dong, K. Cheng, C. Li and S. Wang, Nat. Chem., 2020, 12, 717–724 CrossRef CAS PubMed.
- M. Jouny, J.-J. Lv, T. Cheng, B. H. Ko, J.-J. Zhu, W. A. Goddard and F. Jiao, Nat. Chem., 2019, 11, 846–851 CrossRef CAS PubMed.
- N. Meng, Y. Huang, Y. Liu, Y. Yu and B. Zhang, Cell Rep. Phys. Sci., 2021, 2, 100378 CrossRef CAS.
- Y. Feng, H. Yang, Y. Zhang, X. Huang, L. Li, T. Cheng and Q. Shao, Nano Lett., 2020, 20, 8282–8289 CrossRef CAS PubMed.
- J. J. Roylance and K.-S. Choi, Green Chem., 2016, 18, 5412–5417 RSC.
- M. Shibata, K. Yoshida and N. Furuya, J. Electrochem. Soc., 1998, 145, 2348–2353 CrossRef CAS.
- T. Fukushima and M. Yamauchi, Chem. Commun., 2019, 55, 14721–14724 RSC.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- D. Higgins, C. Hahn, C. Xiang, T. F. Jaramillo and A. Z. Weber, ACS Energy Lett., 2019, 4, 317–324 CrossRef CAS.
- F. P. García de Arquer, C.-T. Dinh, A. Ozden, J. Wicks, C. McCallum, A. R. Kirmani, D.-H. Nam, C. Gabardo, A. Seifitokaldani, X. Wang, Y. C. Li, F. Li, J. Edwards, L. J. Richter, S. J. Thorpe, D. Sinton and E. H. Sargent, Science, 2020, 367, 661–666 CrossRef PubMed.
- S. L. Foster, S. I. P. Bakovic, R. D. Duda, S. Maheshwari, R. D. Milton, S. D. Minteer, M. J. Janik, J. N. Renner and L. F. Greenlee, Nat. Catal., 2018, 1, 490–500 CrossRef.
- D. Liu, M. Chen, X. Du, H. Ai, K. H. Lo, S. Wang, S. Chen, G. Xing, X. Wang and H. Pan, Adv. Funct. Mater., 2021, 31, 2008983 CrossRef CAS.
- N. Heidary, K. H. Ly and N. Kornienko, Nano Lett., 2019, 19, 4817–4826 CrossRef CAS PubMed.
- F. Milella and M. Mazzotti, React. Chem. Eng., 2019, 4, 1284–1302 RSC.
- R. Kas, O. Ayemoba, N. J. Firet, J. Middelkoop, W. A. Smith and A. Cuesta, ChemPhysChem, 2019, 20, 2904–2925 CrossRef CAS PubMed.
- X. Lu, C. Zhu, Z. Wu, J. Xuan, J. S. Francisco and H. Wang, J. Am. Chem. Soc., 2020, 142, 15438–15444 CrossRef CAS PubMed.
- P. P. Sethna, H. D. Downing, L. W. Pinkley and D. Williams, J. Opt. Soc. Am., 1978, 68, 429–431 CrossRef CAS.
- M. F. Baruch, J. E. Pander, J. L. White and A. B. Bocarsly, ACS Catal., 2015, 5, 3148–3156 CrossRef CAS.
- N. J. Firet and W. A. Smith, ACS Catal., 2017, 7, 606–612 CrossRef CAS.
- S. Zhu, B. Jiang, W.-B. Cai and M. Shao, J. Am. Chem. Soc., 2017, 139, 15664–15667 CrossRef CAS PubMed.
- I. V. Chernyshova, P. Somasundaran and S. Ponnurangam, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E9261 CrossRef CAS PubMed.
- E. Garand, T. Wende, D. J. Goebbert, R. Bergmann, G. Meijer, D. M. Neumark and K. R. Asmis, J. Am. Chem. Soc., 2010, 132, 849–856 CrossRef CAS PubMed.
- J. Resasco, L. D. Chen, E. Clark, C. Tsai, C. Hahn, T. F. Jaramillo, K. Chan and A. T. Bell, J. Am. Chem. Soc., 2017, 139, 11277–11287 CrossRef CAS PubMed.
- Y. Zhu, J. Wang, H. Chu, Y.-C. Chu and H. M. Chen, ACS Energy Lett., 2020, 5, 1281–1291 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc06590d |
| This journal is © The Royal Society of Chemistry 2022 |